

Abstract
Repeated exposure to toll-like receptor 4 (TLR4) ligands, such as lipopolysaccharide (LPS), reduces responses of monocytes/macrophages to LPS (LPS/endotoxin tolerance). Microglial exposure to Aβ deposits, a TLR4 ligand, may cause “Aβ/LPS tolerance”, leading to decreased Aβ clearance. We demonstrated that microglial activation by LPS is diminished in Aβ deposit-bearing 12-month-old model mice of Alzheimer’s disease (AD), compared with non-AD mice and Aβ deposit-free 2-month-old AD mice. Because miR-146a plays a predominant role in inducing TLR tolerance in macrophages and because miR-146a in extracellular vesicles (EVs) shed by inflammatory macrophages increases in circulation, we investigated potential roles of miR-146a and inflammatory EVs in inducing TLR tolerance in microglia and in altering expression of inflammatory AD risk genes. We found that miR-146a upregulation induces TLR tolerance and alters expression of inflammatory AD risk genes in response to LPS treatment in BV2 microglia. LPS brain injection altered expression of the AD risk genes in 12-month-old AD mice but not in non-AD littermates. EVs from inflammatory macrophages polarize BV2 microglia to M1 phenotype and induce TLR tolerance. Microglia exposed to Aβ in the brain show reduced cytokine responses to systemic inflammation due to peripheral LPS injection, indicating TLR/Aβ tolerance in microglia. Our results suggest that increased miR-146a induces microglial Aβ/LPS tolerance and that circulating EVs shed by inflammatory macrophages contribute to microglial Aβ/LPS tolerance, leading to reduced Aβ clearance. Our study also suggests that altered expression of inflammatory AD risk genes may contribute to AD development via the same molecular mechanism underlying LPS tolerance.
Keywords: Alzheimer’s disease, microglia, microRNA, toll-like receptor, endotoxin tolerance
Introduction
Patients with Alzheimer’s disease (AD) develop deposits of aggregated amyloid β-protein (Aβ) in neuritic plaques and cerebral vessels. According to the amyloid cascade hypothesis, accumulation of Aβ in the brain is postulated to be a causal event in the etiology of AD (Selkoe and Hardy 2016). Aβ deposits in the brain are accompanied by innate immune responses such as activated microglia and increased levels of cytokines (Akiyama et al. 2000). Moreover, there is a broad range of evidence, which supports the notion that peripheral inflammation promotes AD progression and initiates altered activation of microglia and neurodegeneration (Fulop, Itzhaki et al. 2018, Holmes 2013, Perry and Holmes 2014). Aging is the largest known risk factor for AD and characterized by chronic, systemic low-grade inflammation, referred to as “inflamm-aging” (Fulop, Witkowski et al. 2018, Hebert et al. 2013). Additionally, almost all highly ranked, modifiable risk factors for AD such as depression, hypertension, diabetes, obesity, hyperlipidemia and cardiovascular disease are characterized by a chronic, systemic low-grade inflammation (Deckers et al. 2015, Kennedy et al. 2014). Furthermore, the importance of immune responses in the pathogenesis of AD is strongly supported by the genome-wide association studies on patients with late-onset AD, which identified a dozen genetic risk variants that are involved in immune/inflammatory responses (Pimenova et al. 2018). Likewise, certain TLR4 gene variants are also associated with AD risk (Chen et al. 2012, Miron et al. 2019). However, the molecular mechanisms by which these genetic variants and inflammatory diseases lead to increased AD risk remain to be elucidated.
Toll-like receptors (TLRs) are a class of pattern-recognition receptors in the innate immune system. TLR ligation initiates a signaling cascade leading to activation of transcription factors that upregulate expression of cytokines, chemokines, growth factors and other inflammatory mediators. One of the important roles of TLRs is to activate phagocytes/microglia in response to pathogens and damaged host cells, and to clear pathogens, damaged tissues, and accumulated wastes. Indeed, an acute (one-time) injection of lipopolysaccharide (LPS), a TLR4 ligand, into the brains of AD mouse models activates microglia and decrease brain Aβ deposits (DiCarlo et al. 2001, Herber et al. 2004, Malm et al. 2005). However, repeated exposure to certain TLR ligands such as LPS induces hyporesponsiveness to subsequent LPS or TLR ligand challenge (endotoxin/TLR tolerance). Because Aβ aggregates activate microglia via TLR4 (Capiralla et al. 2012, Tahara et al. 2006), chronic exposure of microglia to Aβ aggregates may induce “Aβ tolerance” similar to TLR tolerance, possibly leading to decreased clearance of Aβ aggregates and reduced neuronal survival and plasticity. In agreement with this notion, we previously demonstrated that LPS brain injection activates less microglia in 12-month-old AD model (TgAPP/PS1) mice with Aβ deposition than in 12-month-old wild-type littermates (non-Tg mice) but activates more microglia in 2-month-old TgAPP/PS1 mice without Aβ deposition than in 2-month-old wild-type littermates (Go et al. 2016).
TLR tolerance has been intensively investigated for decades and several regulatory molecules in the TLR signaling pathways have been identified as modulators of TLR tolerance. Particularly, miR-146a plays a predominant role in development of TLR tolerance (Saba et al. 2014) and miR-146a expression in several different AD mouse models increases around the onset of Aβ deposits (Arena et al. 2017, Li et al. 2011). Moreover, circulating miR-146a increases during peripheral inflammation associated with certain AD risk factors including aging, diabetes, cardiovascular disease and periodontitis and miR-146a belongs to inflammation-related miRs (inflamma-miRs) and senescence-associated miRs (SA-miRs) (Han et al. 2020, Olivieri et al. 2013, Rippo et al. 2014). These findings suggest the potential role of miR-146a in Aβ/TLR tolerance in AD, which results in decreased clearance of Aβ aggregates. However, the molecular mechanisms underlying TLR tolerance appear to be complex and specific to cell/tissue types and induction conditions. To investigate the molecular mechanism involved in Aβ- and inflammation-induced TLR tolerance in AD, we determined expression levels of signaling molecules that are known to regulate TLR signaling as well as expression levels of immune-related AD risk genes in 2- and 12-month-old TgAPP/PS1 mice treated with hippocampal injection of LPS. Furthermore, because miR-146a plays a central role in negative regulation of TLR signaling in monocytes/macrophages and can regulate expression of multiple genes (Chan et al. 2013, Quinn et al. 2012, Saba et al. 2014), we have tested if up- or down-regulation of miR-146a in BV2 mouse microglial cells can modulate LPS tolerance, M1 and M2 phenotype, and expression of AD risk genes.
Methods
Animals and LPS treatment
B6.Cg-Tg(APPswe,PSEN1dE9) 85Dbo/J mice (here referred to as TgAPP/PS1 mice) were purchased from Jackson Laboratory (Bar Harbor, ME) and used as a transgenic mouse model of AD. TgAPP/PS1 mice express a chimeric mouse/human APP with the double mutations (K670N and M671L) and human presenilin 1 (PS1) with an exon 9 deletion found in familial AD patients. TgAPP/PS1 mice develop Aβ deposits in the brain as early as 4 months of age. The same experimental mice that had been reported in (Go et al. 2016) were used for this study. To determine the effect of LPS on gene expression in young mice with no Aβ deposits, 2-month-old TgAPP/PS1 mice and their non-transgenic littermates (non-Tg mice) received 4μg LPS injection into their right hippocampus. To determine the effect of LPS on gene expression in old mice with Aβ deposits, 12-month-old TgAPP/PS1 and non-Tg mice received of 2μg LPS due to decreased survival with the original higher LPS dose (4μg). Control groups similarly received phosphate-buffered saline (PBS) injection. Because microglial activation peaks 7 days after intrahippocampal injection (Herber et al. 2004), experimental mice were euthanized 7days after LPS injection and their brains were quickly removed and stored in −80° C. Additionally, 13-month-old TgAPP/PS1 mice and their non-transgenic littermates (non-Tg mice) were intraperitoneal injected with LPS (0.5 mg/Kg bodyweight) or PBS. Mice were euthanized 24 hours after intraperitoneal injection and their brains were quickly removed and hippocampi were separated and stored in −80°C. The animal protocols described here were prospectively reviewed and approved by the Institutional Animal Care and Use Committee of the University of Illinois College of Medicine at Peoria. Methods of euthanasia were consistent with the recommendations of the Panel on Euthanasia of the American Veterinary Medical Association.
BV2 cell culture and microRNA mimic transfection
BV2 cells were plated in 6-well plates (3.5 × 105cells/ml). Cells were transfected with 25 nM of negative control-mimic, miR-146a mimic, or miR-146a inhibitor using Lipofectamine RNAiMax Transfection reagent (Life Technologies, Grand Island, NY) and, then 24 h later, treated with LPS (100ng/ml). Culture media were collected for TNF-α ELISA at 3, 6 and 24 h after LPS treatment and messenger RNA (mRNA) for qPCR at 24 h (n = 6).
RAW 264.7 cell culture, extracellular vesicle (EV) preparation and EV treatment of BV2 cells
RAW 264.7 (ATCC® TIB-71™) cells were purchased from ATCC and cultured in DMEM containing 10% FBS. RAW cells were seeded in 6-well plates (3.5 × 105cells/ml) in 10% FBS DMEM. Forty hour later, the cells were treated with PBS or LPS (100 ng/ml) for 24 hours, washed with PBS three times to remove LPS and, then, cultured in DMEM without FBS for 20 hours. EVs from the media were collected and purified using the Total Exosome Isolation Reagent kit (Life Technologies, Carlsbad, CA) according to the manufacturer’s instructions. Expression levels of miR-146a in EVs were calculated by the 2−ΔΔCt method of qPCR using ExiLENT SYBR® Green Master Mix and microRNA specific microRNA LNATM PCR primer sets (Exiqon) after normalization to the endogenous U6 small RNA and RNU1A1. EVs suspended in DMEM were added to BV2 cell cultures. For the first set of experiments (outlined in Fig. 3A), the BV2 culture media were collected for TNF-α ELISA at 3, 6 and 24 h after EV treatment and BV2 cells were harvested for isolation of mRNA for qPCR at 24 h (n = 6/group). For the second set of experiments (outlined in Fig. 4A), BV2 cells were incubated with EVs for 24 h, washed with PBS three times and treated with LPS (100 ng/ml). The BV2 culture media were collected for TNF-α ELISA at 3, 6 and 24 h after LPS addition and the BV2 cells were harvested for isolation of mRNA for qPCR at 24 h (n=6/group).
Fig. 3.
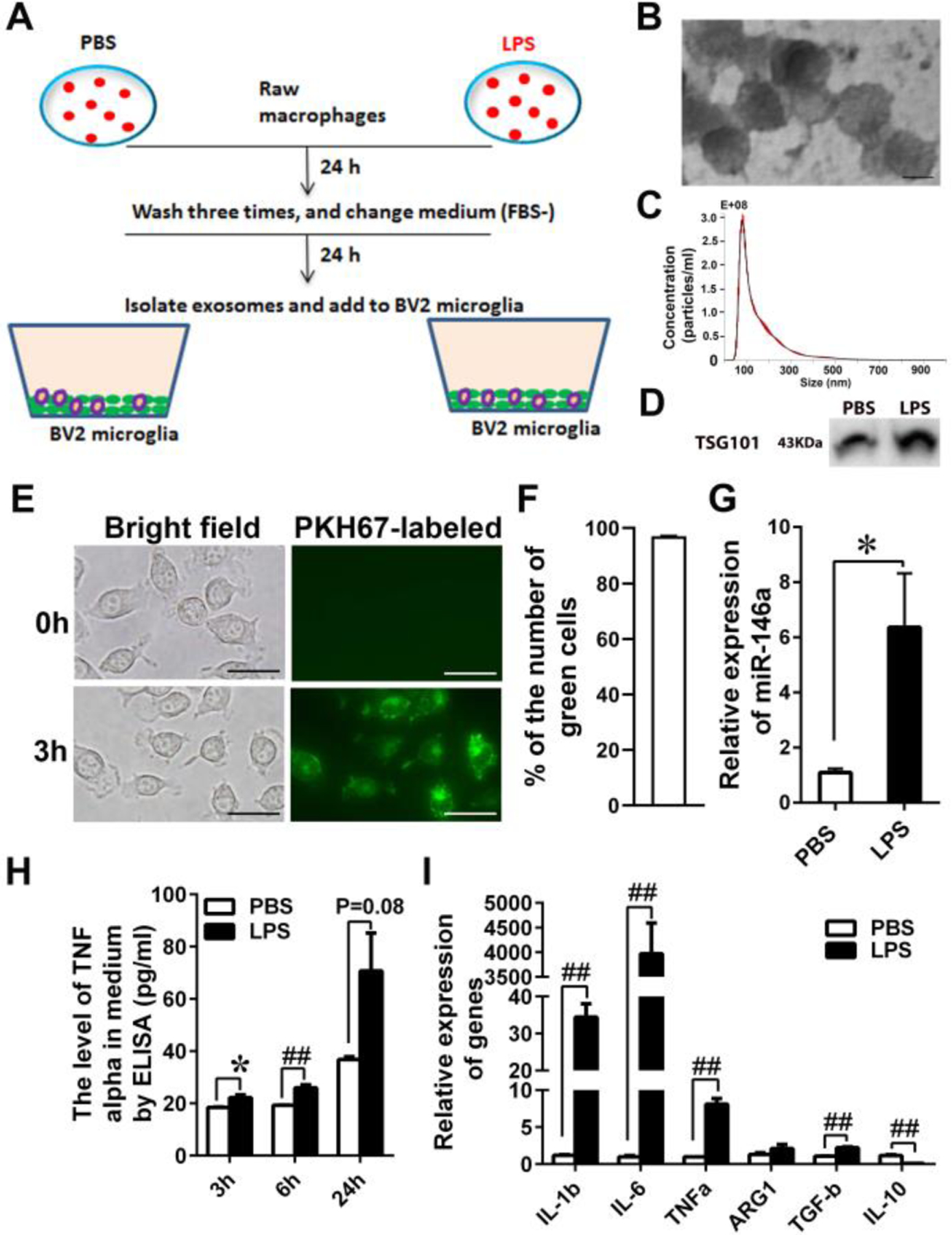
EVs derived from LPS-treated RAW cells induce inflammation and M1 polarization in BV2 microglia. (A) Schematic diagram of RAW and BV2 cultures. RAW cells were treated with PBS or LPS (100 ng/ml) for 24 hours, washed three times with PBS to remove LPS, and further incubated in FBS-free DMEM for 24 hours to isolate EVs from the medium. The EVs suspended in DMEM were added to BV2 cell cultures. (B) EVs isolated from the media of RAW cell cultures range from 50 – 100 nm in diameter by transmission electron microscopy (bar size: 50nm). (C) The mean size and mode of the isolated EVs are 154.2 ± 1.2 nm and 79 ± 2.3 nm, respectively, by nanoparticle tracking analysis (NTA). (D) TGS101, an exosomal marker, in EVs was identified by western blotting. (E and F) EV uptake by BV2 cells is confirmed by fluorescence microscopy 3 hours after EV addition to the culture (bar size: 15μm) and quantified by counting the number of green particle positive and negative cells. The percentages of green-positive cells are shown. (G) The expression levels of miR-146a in EVs derived from PBS- and LPS-treated RAW cells were determined by qPCR. LPS treatment (LPS) increases miR-146a levels in EVs shed by RAW cells compared to PBS treatment (PBS) (* P<0.05). (H) EVs from LPS-treated RAW cells (LPS) increase TNF-α secretion from BV2 cells compared to EVs from PBS-treated RAW cells (PBS) at 3h, 6h and 24h after EV addition (* P<0.05, ## P<0.001). (I) Expression levels of M1 and M2 marker genes were determined by qPCR. EVs from LPS-treated RAW cells polarizes BV2 cells towards an M1 phenotype (LPS) (n=6) (## P<0.001).
Fig. 4.
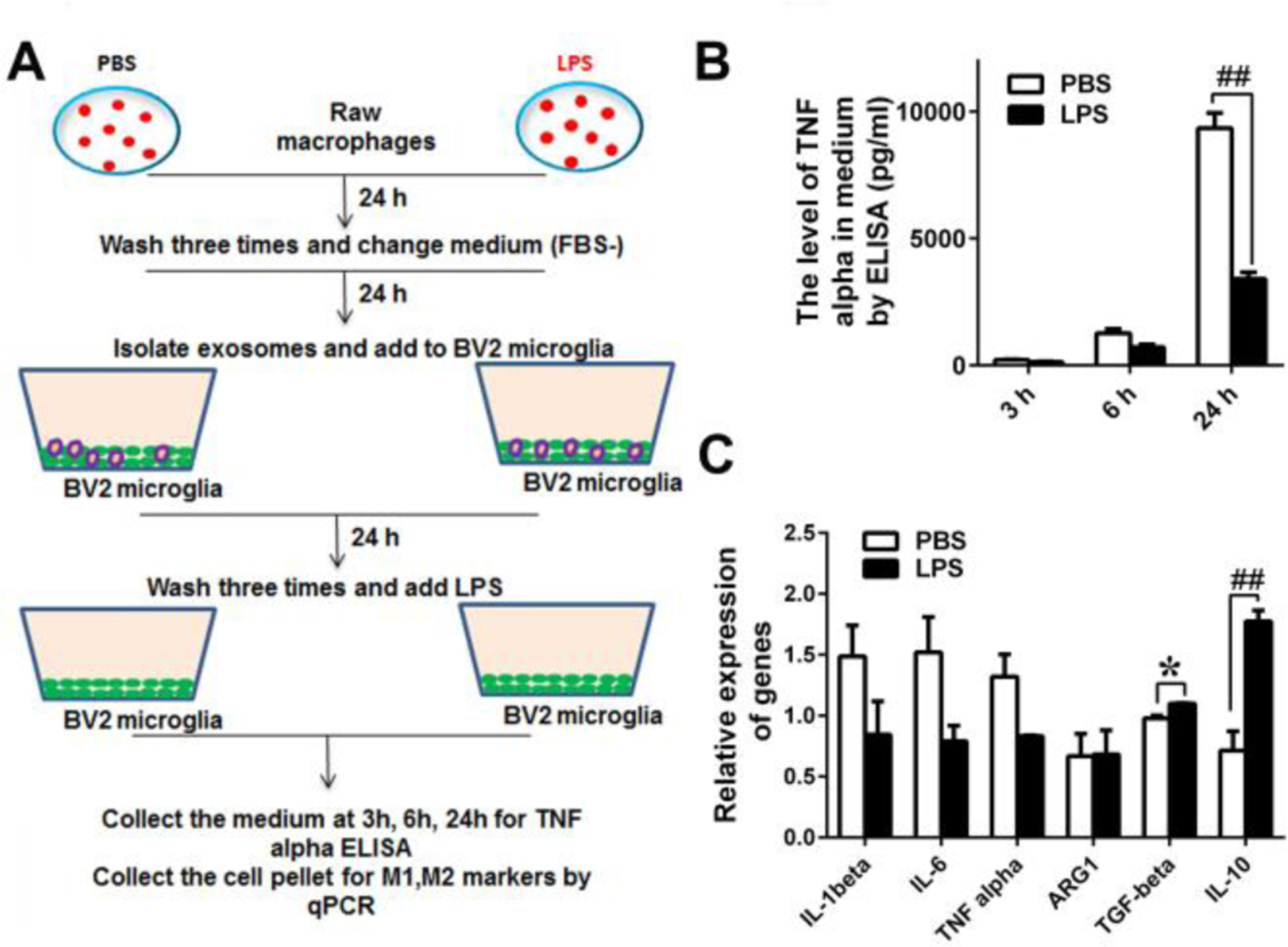
EVs derived from LPS-treated RAW cells induce LPS tolerance in BV2 microglia. (A) Schematic diagram of RAW and BV2 cultures. RAW cells were treated with PBS or LPS (100 ng/ml) for 24 hours, washed three times with PBS to remove LPS, and further incubated in FBS-free DMEM for 24 hours to isolate EVs from the medium. BV2 cells were treated with the EVs for 24 hours, washed with PBS three times, and stimulated with LPS (100ng/ml). (B) TNF-α levels in the BV2 culture media were determined by ELISA at 3h, 6h and 24h after LPS addition. In response to LPS stimulation, BV2 cells treated with EVs derived from PBS-treated RAW cells (PBS) secrete higher levels of TNF-α than BV2 cells treated with EVs derived from LPS-treated RAW cells (LPS) at 24h (## P<0.001). (C) Expression levels of M1 and M2 marker genes were determined by qPCR. In response to LPS stimulation, BV2 cells treated with EVs derived from LPS-treated RAW cells (LPS) express higher levels of M2 markers (TGF-β and IL-10) than BV2 cells treated with EVs derived from PBS-treated RAW cells (PBS) (n=6, * P<0.05, ## P<0.001).
The EV samples were verified by transmission electron microscopy (TEM), nanoparticle tracking analysis (NTA) and expression of an EVs marker, TGS101. As for TEM, purified EVs were treated with 3% phosphotungstic acid for 5 min, placed on formvar grids, dried and scanned by a transmission electron microscope (JEOL, JEM100C). NTA was conducted by System Bioscience (SBI, Palo Alto, CA). For immunoblot analysis, purified EVs were homogenized in RIPA lysis buffer (Sigma, St. Louis, MO) containing complete miniprotease-inhibitor and phosphatase inhibitors cocktail tablets (Roche Diagnostics Corporation, Indianapolis, IN) and centrifuged at 12,000 × g for 10 min 4°C for collecting supernatants. The proteins were electrophoresed under reducing conditions in 10% SDS-PAGE gels and transferred to PVDF membranes. The membranes were incubated overnight at 4°C with anti-TSG101 antibody (1:500 diution) (Abcam, Cambridge, UK) and specific bands were visualized by an enhanced chmiluminescence system (Amershan, Arlington Heights, IL). EVs were labeled with PKH67 Green Fluorescent Cell Linker Mini Kit (Sigma, St. Louis, MO) according to the manufacturer’s instructions and EV uptake by BV2 cells was visualized by fluorescence microscopy. The number of green cells were counted from three different experiments.
mRNA and microRNA Expression
Total RNA from the cerebra (n = 6/group), hippocampus and cultured BV2 cells was isolated using the Direct-zolTM RNA kit (Zymo Research Corporation, Irvine, CA). For quantification of mRNA expression, one microgram of total RNA from each sample was reverse transcribed using oligo (dT)15 primer and SuperScript® III Reverse Transcriptase (Life Technologies). Primer pairs used for real-time PCR are presented in Table I. The levels of mRNA expression were measured by real-time PCR using a CFX96 Real-Time PCR detection system (Bio-Rad) with FastStart SYBR Green Master Mix (Roche, Indianapolis, IN). Amplification conditions consisted of a 10-min preincubation at 95°C, followed by 40 cycles of denaturation at 95°C for 15 s and primer annealing and extension for 1 min at 60°C. PCR product melting curves were examined to confirm the homogeneity of PCR products. mRNA levels of each gene were normalized by subtracting cycle threshold (Ct) values obtained with beta-actin mRNA and expressed as 2 −ΔΔCt. The fold changes in hippcampus for each genotype were expressed as 2 −ΔΔCt (LPS) - 2 −ΔΔCt (PBS)avg.
Table I.
Primer DNA Sequences
| Gene | Forward primer (5’ to 3’) | Reverse primer (5’ to 3’) |
|---|---|---|
| TGF-β | CTCCCGTGGCTTCTAGTGC | GCCTTAGTTTGGACAGGATCTG |
| IL-1β | GCCCCTGCATGGGAAATCAA | ATCTTTTGGGGTCCGTCAACT |
| A20 | GAACAGCGATCAGGCCAGG | GGACAGTTGGGTGTCTCACATT |
| p50 | ATGGCAGACGATGATCCCTAC | TGTTGACAGTGGTATTTCTGGTG |
| TLR4 | ATGGCATGGCTTACACCACC | GAGGCCAATTTTGTCTCCACA |
| ApoE | CTGACAGGATGCCTAGCCG | CGCAGGTAATCCCAGAAGC |
| TREM2 | CTGGAACCGTCACCATCACTC | CGAAACTCGATGACTCCTCGG |
| INPP5D | GCCCCTGCATGGGAAATCAA | TGGGTAGCTGGTCATAACTCC |
| CD33 | CCGCTGTTCTTGCTGTGTG | AAGTGAGCTTAATGGAGGGGTA |
| ABCA1 | GCTTGTTGGCCTCAGTTAAGG | GTAGCTCAGGCGTACAGAGAT |
| CR1 | AACACATGGTTACCAGGTGTACC | CGTGCCTCTCCAGCCATAAG |
| MEF2C | GTCAGTTGGGAGCTTGCACTA | CGGTCTCTAGGAGGAGAAACA |
| SOCS1 | CTGCGGCTTCTATTGGGGAC | AAAAGGCAGTCGAAGGTCTCG |
| EPHA1 | GTGTGGCGCTAGTGTCTGTAA | CACGTTCCAGCTACTTCAACC |
| IL-1beta | GCCCCTGCATGGGAAATCAA | ATCTTTTGGGGTCCGTCAACT |
| IL-6 | GAGGATACCACTCCCAACAGACC | AAGTGCATCATCGTTGTTCATACA |
| TNF alpha | CCCTCACACTCAGATCATCTTCT | GCTACGACGTGGGCTACAG |
| ARG1 | CACTCCCCTGACAACCAGCT | AAGGACACAGGTTGCCCATG |
| IL-10 | GGTTGCCAAGCCTTATCGGA | ACCTGCTCCACTGCCTTGCT |
| CD14 | CTCTGTCCTTAAAGCGGCTTAC | GTTGCGGAGGTTCAAGATGTT |
| ABCA7 | AATTACACCTATCGACGGAGACA | TGACGGACAGCCACTAGGA |
| COX2 | TTCAACACACTCTATCACTGGC | AGAAGCGTTTGCGGTACTCAT |
| TSC1 | ATGGCCCAGTTAGCCAACATT | CAGAATTGAGGGACTCCTTGAAG |
| IkBα | TGAAGGACGAGGAGTACGAGC | TTCGTGGATGATTGCCAAGTG |
| RELB | CCGTACCTGGTCATCACAGAG | CAGTCTCGAAGCTCGATGGC |
| CCL2 | TTAAAAACCTGGATCGGAACCAA | GCATTAGCTTCAGATTTACGGGT |
| IRF3 | GAGAGCCGAACGAGGTTCAG | CTTCCAGGTTGACACGTCCG |
| TRAF6 | AAAGCGAGAGATTCTTTCCCTG | ACTGGGGACAATTCACTAGAGC |
| IRAK4 | CATACGCAACCTTAATGTGGGG | GGAACTGATTGTATCTGTCGTCG |
| beta-actin | GGCTGTATTCCCCTCCATCG | CCAGTTGGTAACAATGCCATGT |
Statistical analysis
SPSS version 19 was used for statistical analysis. Intergroup differences were assessed by a repeated measure ANOVA. A level of p≤ 0.05 was accepted as significant.
Results
LPS alters expression levels of AD risk genes and TLR signaling-related genes in 12-month-old TgAPP/PS1 mice
We determined expression levels of selected genes that are involved in TLR signaling. Such genes included TLR4, CD14, IRAK4, TRAF6, IRF3, CCL2, p50 (NFkB), RELB, IkBα, A20, TSC1, COX2, SOCS1, TGF-β and IL-1β. No difference was found in expression levels of these genes between TgAPP/PS1 and non-Tg mice 7 days after LPS injection at 2 months of age (data not shown). Similarly, no difference was also found between TgAPP/PS1 and non-Tg mice 7 days after PBS injection at 12 months of age except that IL-1β was elevated in TgAPP/PS1 mice compared to non-Tg mice (Fig. 1A). At 12 months, expression levels of p50 (P < 0.05), A20 (P < 0.05), TGF-β (P < 0.05) and TLR4 (P < 0.05) in LPS-injected TgAPP/PS1 mice were higher than those in non-Tg mice (Fig. 1B).
Fig. 1.
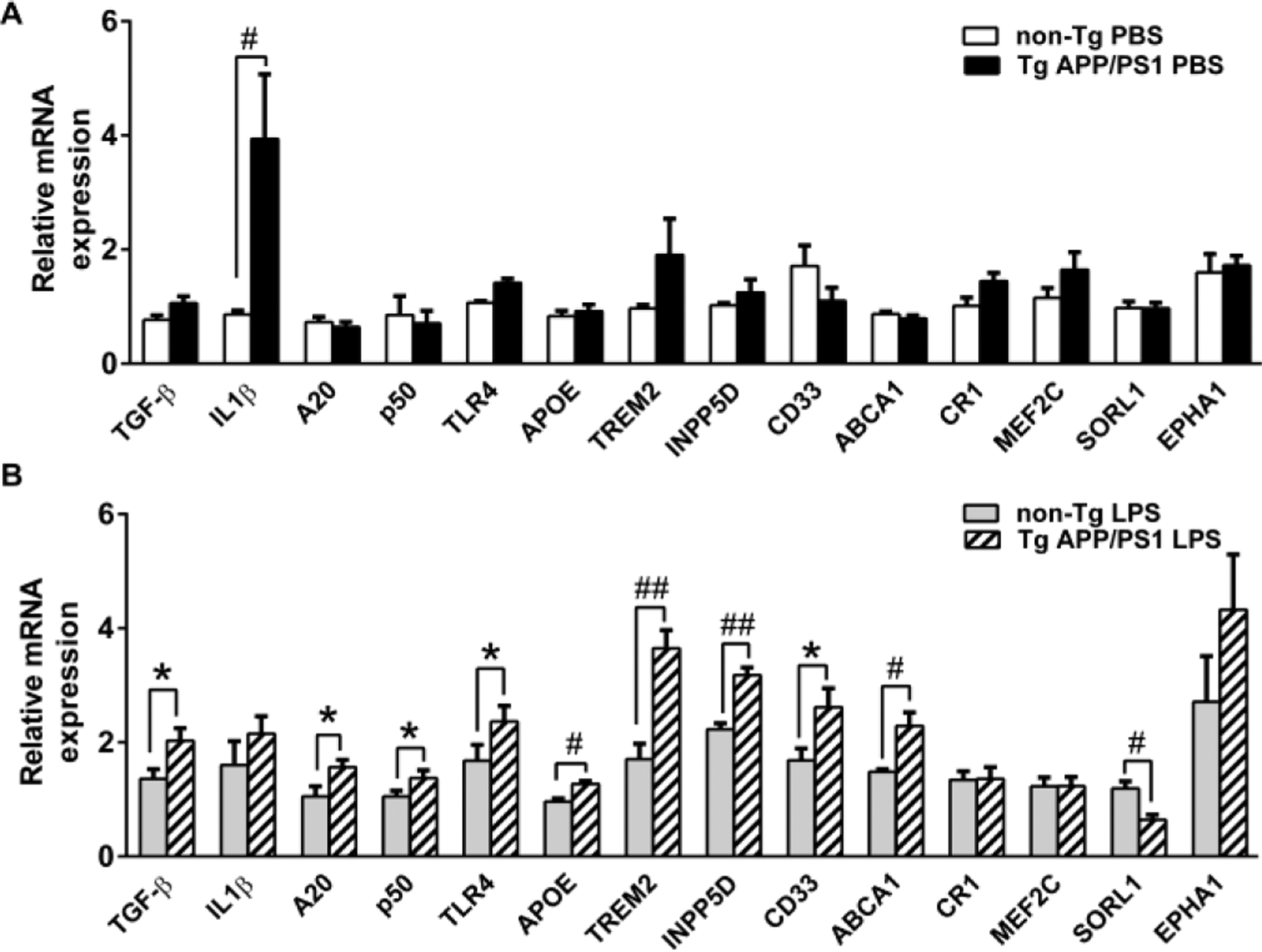
LPS injection increases mRNA expression levels of certain AD risk genes and TLR4 signaling-related genes in TgAPP/PS1 mice at age 12 months. TLR4 signaling-related genes and mRNA expression levels of certain AD risk genes in the cerebral tissues from 12-month-old TgAPP/PS1 and non-Tg mice injected with PBS (A) or LPS (B) were determined by quantitative real-time PCR using cDNA prepared by reverse-transcription of mRNA. mRNA levels were normalized by subtracting cycle threshold (Ct) values obtained with β-actin mRNA and are shown as 2−ΔΔCt [ΔCt = Ct (gene of interest) - Ct (β-actin)] (means ± SEM). *P < 0.05, #P < 0.01, and ##P < 0.001.
To investigate the possible involvement of immune-related AD risk genes in the development of altered TLR4 signaling or LPS tolerance in microglia, expression levels of such genes in the cerebra were determined by real-time PCR. The genes examined were APOE, TREM2, CLU, INPP5D, CD33, ABCA7, ABCA1, CR1, MEF2C, SORL1 and EPHA1, which are known to modulate immune responses. Particularly, TREM2, CD33, CR1, INPP5D, APOE, ABCA7 and MEF2C are highly expressed in microglia and activation of these genes are involved in neuroprotective and/or neurotoxic activities of microglia (Malik et al. 2015, Verheijen and Sleegers 2018). There was no significant difference in expression levels of these genes between TgAPP/PS1 and non-Tg mice subjected to LPS injection at 2 months of age (data not shown). There was no significant difference in these genes between TgAPP/PS1 and non-Tg mice subjected to PBS injection at 12 months, also (Fig. 1A). At 12 months, however, expression levels of APOE (P = 0.01), TREM2 (P = 0.001), INPP5D (P = 0.001), CD33 (P < 0.05) and ABCA1 (P < 0.01) were higher, and those of SORL1 (P < 0.01) were lower in LPS-injected TgAPP/PS1 mice than those in non-Tg mice (Fig. 1B). Thus, altered expression of certain AD risk and TLR signaling genes in response to LPS was observed in 12-month-old TgAPP/PS1 mice after Aβ deposition.
MiR-146a can alter expression of certain AD risk genes and TLR signaling-related genes in response to LPS stimulation
Because miR-146a is thought to be a major driver behind TLR tolerance (Saba et al. 2014) and increase in several different AD mouse models including TgAPP/PS1 mice around the onset of Aβ deposits (Arena et al. 2017, Li et al. 2011), we investigated whether increased levels of miR-146a in cultured microglial cells can induce such alterations in gene expression in response to LPS treatment. BV2 cells were transfected with miR-146a mimic, miR-146a inhibitor or miR-negative control and then treated with LPS. As expected, LPS treatment induced increases in TNF-α levels in the media of BV2 cells treated with miR control (Fig. 2A). TNF-α levels in the media of miR control treated BV2 cells were conspicuously higher than those in miR-146a mimic-treated BV2 cells (P < 0.001) and consistently lower than those in miR-146a inhibitor-treated BV2 cells (P < 0.05) (Fig. 2A), indicating that increased miR-146a can inhibit LPS-induced inflammation in BV2 cells. Up- and down-regulation of miR-146a in BV2 cells by miR-146a mimic and inhibitor, respectively, was confirmed by qPCR (Fig. 2B, P < 0.001). LPS treatment decreased mRNA expression levels of IL-1β, IL-6 and TNF-α and increased that of IL-10 in miR-146a mimic-transfected BV2 cells compared with those in miR control-transfected BV2 cells (Fig. 2B, P < 0.001). LPS treatment increased mRNA expression levels of IL-1β, IL-6 and TNF-α and decreased that of IL-10 in miR-146a inhibitor-transfected BV2 cells compared with those in miR control-transfected BV2 cells (Fig. 2B, P < 0.05). Thus, in response to LPS treatment, miR-146a mimic shifted BV2 cells toward an M2 phenotype and its inhibitor shifted them toward an M1 phenotype. LPS treatment increased INPP5D, CD33, MEF2C and TREM2 expression and decreased ABCA1 and CR1 expression in miR-146a mimic-transfected BV2 cells compared with those in miR control-transfected BV2 cells (Fig. 2C, P < 0.05). Such alterations in expression levels of the AD risk genes were not observed by miR-146a inhibitor transfection (Fig. 2C).
Fig. 2.
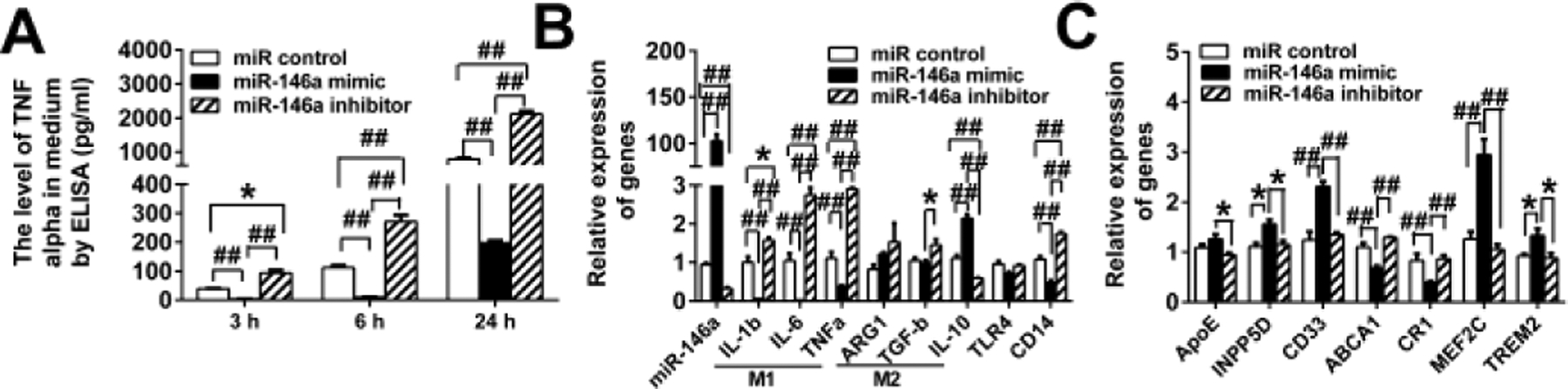
MiR-146a regulates TLR signaling, microglial M1 and M2 phenotypes and AD risk gene expression in BV2 microglia. BV2 cells transfected with miR-negative control, miR-146a mimic, or miR-146a inhibitor were treated with LPS. MiR-146a mimic decreases and its inhibitor increases TNF-α in the BV2 cell media compared to miR control at the indicated time points after LPS treatment (A). MiR-146a mimic and its inhibitor increases and decreases expression levels of miR-146a, respectively (B, column at the extremely left). In response to LPS treatment, miR-146a mimic and its inhibitor shift BV2 cells toward an M2 and M1 phenotype, respectively (B). MiR-146a mimic increases INPP5D, CD33, MEF2C and TREM2 and decreases ABCA1 and CR1 expression compared to miR control (C). MiR-146a inhibitor revokes such expression changes (C). *P < 0.05 and ##P < 0.001.
Circulating exosomal miR-146a may contribute to microglial LPS/Aβ tolerance
Circulating microRNAs (miRNAs) are abundantly found in extracellular vesicles (EVs) including exosomes and microvesicles, which are shed by or released from many types of cells. Particularly, miRNAs in EVs have been repeatedly shown to be capable of inducing phenotypic changes in the recipient cells, resulting in modulation of immune/inflammatory response and metabolism (Fernandez-Messina et al. 2015, Robbins and Morelli 2014). Because peripheral EVs are capable of crossing the blood-brain barrier (BBB) (Agliardi and Clerici 2020, Matsumoto et al. 2017), it is possible that the peripheral inflammatory and metabolic states are conveyed to the cells in CNS via circulating exosomal miRNAs. MiR-146a is enriched in EVs released from macrophages stimulated by an artherogenic stimulus (Nguyen et al. 2018, Zhang et al. 2019) and in circulating EVs under inflammatory conditions (Brianza-Padilla et al. 2016, Han et al. 2020). Accordingly, circulating exosomal miR-146a in patients at risk of developing AD may be involved in the induction of LPS/Aβ tolerance in microglia, leading to accumulation of Aβ aggregates. Here, we investigated whether EVs isolated from LPS-treated monocytes/macrophages can induce inflammation (outlined in Fig. 3A) and LPS tolerance (outlined in Fig. 4A) in inactive microglia. RAW 264.7 cells (mouse monocyte/macrophage cell line) were treated with PBS or LPS for 24 hours, then washed three times with PBS to remove LPS, and further incubated (without serum) for 24 hours to isolate EVs from the medium. The sizes of the isolated EVs ranged from 50 – 100 nm in diameter by transmission electron microscopy (Fig. 3B). When nanoparticle tracking analysis was performed by Nanosight, the mean size and mode of the isolated EVs are 154.2 ± 1.2 nm and 79 ± 2.3 nm, respectively (Fig. 3C). The isolated EVs were subjected to immunoblot analysis using anti-TSG-101 antibody, which is commonly used as a marker protein for exosomes. Expressed TSG-101 in the isolated EVs was identified as 43 KDa protein by immunoblotting (Fig. 3D). LPS in the isolated EVs was undetectable by Pierce LAL chromogenic endotoxin quantitation kit (Data not shown). The isolated EVs were labeled with PKH67 Green Fluorescence and readily taken up by BV2 cells within 3 hours after they were added to the culture (Fig. 3E). The average percentage of cells that were positive for green fluorescence was 97.1 ± 0.05% (Fig. 3F). The isolated EVs derived from LPS-treated RAW cells contained significantly higher levels of miR-146a than those derived from PBS-treated RAW cells (Fig. 3G). BV2 cells that were treated with the isolated EVs derived from LPS-treated RAW cells (lps-EV-BV2 cells), secreted modestly but significantly higher levels of TNF-α in the medium than BV2 cells that were treated with EVs derived from PBS-treated (control) RAW cells (pbs-EV-BV2 cells) 3 and 6 hours after the EV addition (Fig. 3H). The BV2 cells were harvested 24 hours after the EV addition and their mRNAs for M1 and M2 macrophage/microglia markers were subjected to qRT-PCR. Large increases in expression levels of M1 markers (IL-1β, IL-6, and TNF-α) were found in lps-EV-BV2 cells compared to pbs-EV-BV2 cells (Fig. 3I). In the separate experiments, 24 hours after the addition of EVs, lps-EV-BV2 and pbs-EV-BV2 cells were treated with LPS for 3, 6 and 24 hours and TNF-α levels in the media were determined by ELISA. pbs-EV-BV2 cells produced significantly more TNF-α than lps-EV-BV2 cells 24 hours after LPS treatment (Fig. 4B). No difference in cell viability was found between the two groups and absolute TNF-α levels in Fig. 4B are much higher than those in Fig. 3E. After 24 hour-treatment with LPS, expression levels of M2 macrophage/microglia markers (TGF-β and IL-10) in lps-EV-BV2 cells are significantly higher than those in pbs-EV-BV2 cells (Fig. 4C). Thus, a LPS-tolerant state of microglia that were not previously exposed to LPS can be induced by EVs derived from LPS-treated macrophages.
Additionally, in order to verify the response of Aβ-exposed microglia to peripheral inflammatory exosome, 13-month-old TgAPP/PS1 mice and their littermates were subjected to intraperitoneal injection of LPS (0.5 mg/Kg bodyweight) or PBS and, 24 hours after the LPS or PBS injection, RNA was isolated from their hippocampi. Expression levels of M1 (IL-1β, TNF-α and IL-6) and M2 (ARG1, IL-10 and TGF-β) markers were determined by real-time quantitative reverse transcription-PCR. Increases or decreases of gene expression by LPS injection are shown as fold changes after subtracting gene expression levels of PBS injection in TgAPP/PS1 mice and non-Tg littermates (Fig. 5). As expected, LPS injection increased expression levels of pro-inflammatory cytokines (IL-1β, TNF-α, and IL-6) in non-Tg littermates and expression levels of IL-1β in LPS-injected non-Tg littermates were higher than those in LPS-injected TgAPP/PS1 mice (Fig. 5). Surprisingly, LPS injection induced decreases in TNF-α and IL-6 expression in TgAPP/PS1 mice (Fig. 5). These results suggest that microglia in 13-month-old TgAPP/PS1 mice are less responsive to systemic LPS injection than microglia in their non-Tg littermates, and that Aβ deposits induce LPS tolerance in microglia.
Fig. 5.
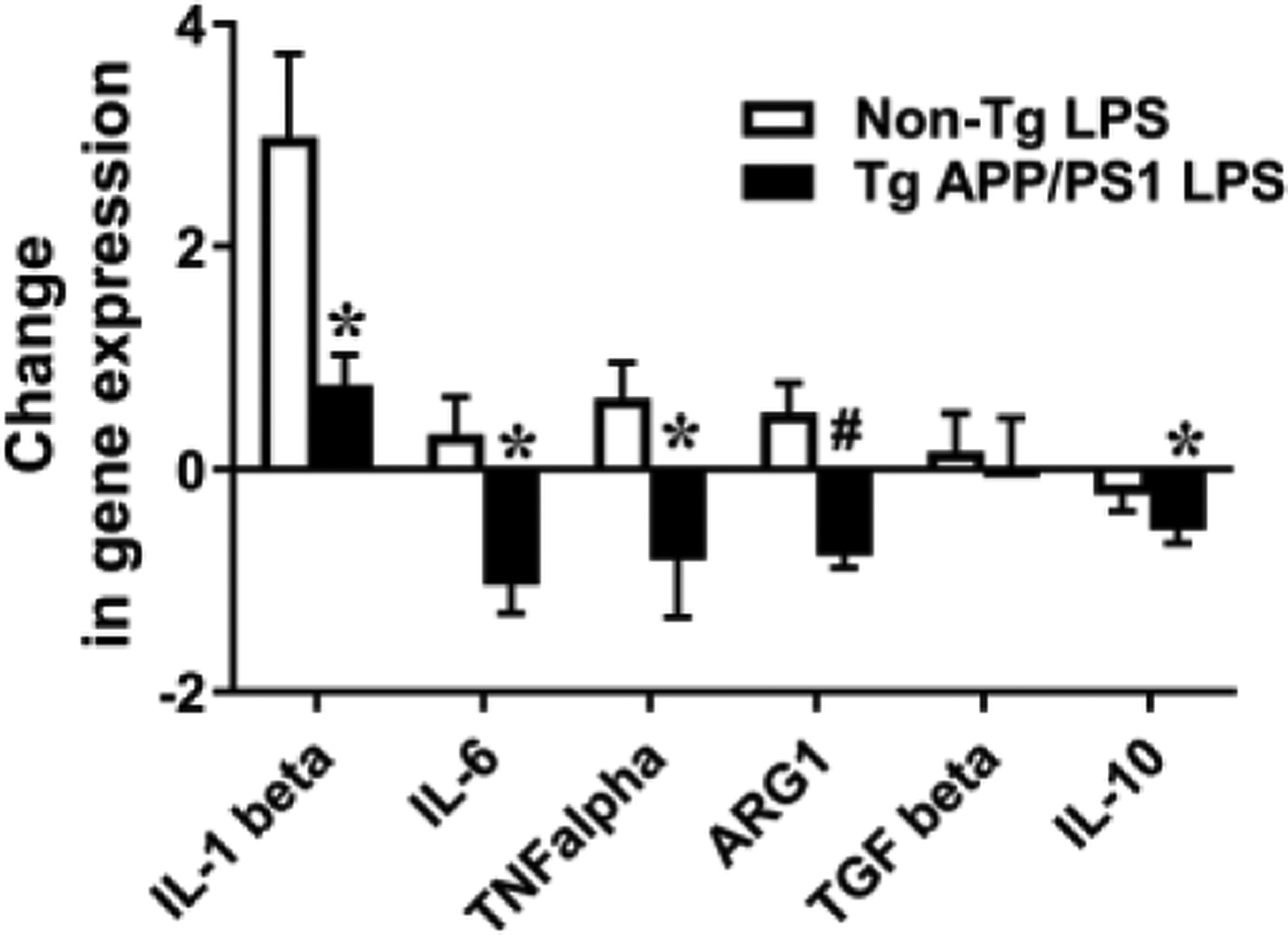
LPS tolerance in 13-month-old TgAPP/PS1 mice but not in their non-Tg littermates. LPS (0.5 mg/Kg bodyweight) or PBS was intraperitoneally injected into 13-month-old TgAPP/PS1 mice and their non-Tg littermates. RNAs were isolated from the hippocampi of the experimental mice 24 hours after LPS or PBS injection, followed by Quantitative reverse transcription PCR. mRNA expression levels of pro-inflammatory cytokines and makers for M1 and M2 macrophages were normalized by subtracting cycle threshold (Ct) values obtained with β-actin mRNA and are determined as 2 −ΔΔCt [ΔCt = Ct (genes of interest) – Ct(beta-actin), ΔΔCt = ΔCt (LPS or PBS)- ΔCt(PBS)avg]. The fold changes for each genotype are shown as 2 −ΔΔCt (LPS) - 2 −ΔΔCt (PBS)avg (*P < 0.05 and #P < 0.01).
Discussion
MicroRNAs are one class of non-coding RNAs that post-transcriptionally regulate specific gene expression by several different mechanisms. MicroRNAs are estimated to control approximately 60 % of all mRNAs and play critical roles in pathophysiological functions including immune responses and neurodegenerative diseases. Among such miRNAs, miR-146a plays an important role as a negative regulator of innate immune responses (Taganov et al. 2006, Zhao and Starczynowski 2014). LPS stimulation induces miR-146a in monocytes and macrophages, which targets mRNAs for IL-1 receptor-associated kinase 1 (IRAK1) and TNF receptor-associated factor 6 (TRAF6) and inhibits their expression. Because IRAK1 and TRAF6 are important signaling molecules of the MyD88-dependent pathways in all TLRs with the exception of TLR3, miR-146a forms a negative feedback regulation loop in TLR signaling in the MyD88 pathways (Taganov et al. 2006). MiR-146a is upregulated during LPS tolerance and counteracts overly inflammatory responses. Overexpression of miR-146a can induce both homologous and cross-tolerance and knockdown of miR-146a reduces the effects of LPS tolerance in THP-1 human monocytes (Nahid et al. 2011). Thus, miR-146a is thought to play a dominant role in LPS tolerance in monocytes/macrophages (Chan et al. 2013, Nahid et al. 2011, Quinn et al. 2012). The role of miR-146a in LPS tolerance in microglia was not investigated previously. In our study, we have demonstrated that miR-146a overexpression in BV2 microglia can induce diminished inflammatory response similar to the state of LPS tolerance and its downregulation reversed the inflammatory response.
Reported levels of miR-146a in the brain, cerebrospinal fluid and blood/plasma in AD patients are controversial (Juzwik et al. 2019). These discrepancies are attributed to insufficient sample sizes and differences in detection methods, brain regions, post-mortem intervals, and disease stages, indicating technical difficulties. However, miR-146a levels in several different lines of AD mouse models (TgAPP/PS1, Tg2576, Tg-CRND8, PSAPP, 3xTg-AD, and 5xFAD) significantly increase after development of AD-type neuropathology (Arena et al. 2017, Li et al. 2011), suggesting that Aβ deposition induces a state similar to LPS tolerance by miR-146a upregulation in microglia. Additionally, miR-146a-deficient macrophages show enhanced phagocytic activities (Lochhead et al. 2014) and increased miR-146a levels are associated with decreases in phagocytic and migratory capabilities of cultured microglia (Caldeira et al. 2014) These support our hypothesis that miR-146a induces Aβ/TLR tolerance in microglia, leading to reduced clearance of Aβ. However, the cause-effect relationships of altered TLR4 signaling and microRNA expression in AD pathogenesis and β-amyloidosis remain to be further elucidated.
Because greater increases in expression levels of certain AD risk genes (APOE, TREM2, CD33, INPP5D and ABCA1) by LPS injection were observed only in 12-month-old TgAPP/PS1 mice (Fig. 1) but not in 2-month-old mice (data not shown), we have tested if miR-146a upregulation in microglia can induce increases in expression levels of AD risk genes by LPS treatment. MiR-146a upregulation in BV2 cells increased expression of only TREM2, CD33 and INPP5D after LPS treatment (Fig. 2C). Thus, miR-146a upregulation in BV2 microglia partly recapitulated the changes observed in 12-month-old TgAPP/PS1 mice. Interestingly, INPP5D, an AD risk gene, suppresses TLR4-mediated LPS responses involved in endotoxin tolerance (An et al. 2005, Xiong and Medvedev 2011). Similarly, TREM2 and CD33 have been reported to downregulate TLR4 signaling (Ishida et al. 2014, Ito and Hamerman 2012). In line with these observations, systemic LPS injection diminished expression of pro-inflammatory cytokines in the hippocampus of 13-month-old TgAPP/PS1 mice (Fig. 5). Accordingly, increased levels of INPP5D, TREM2 and CD33 may reduce cytokine responses by suppressing TLR4 signaling in Aβ-exposed microglia in 13-month-old TgAPP/PS1 mice. Our results suggest that certain AD genetic risk variants may increase AD risk via the same molecular pathways that involve the altered TLR signaling.
EVs play crucial roles in cell-to-cell communication by transferring RNAs, proteins and lipids as carriers in circulation between nearby cells as well as distant cells involved in immune and inflammatory responses (Bruno et al. 2019, Robbins and Morelli 2014). Because miR-146a is enriched in circulating EVs in inflammatory conditions associated with increased risk of AD (Brianza-Padilla et al. 2016, Han et al. 2020), we have tested if EVs derived from inflammatory macrophages can induce inflammation and LPS tolerance in the recipient microglia of the EVs. The EVs polarized inactive microglia to an M1 phenotype characterized by increased expression of IL-1β, IL-6 and TNF-α and decreased expression of IL-10 (Fig. 3F). This M1 polarization by the EVs derived from inflammatory macrophages appeared to induce LPS tolerance in the recipient microglia, suggesting that peripheral inflammation found in AD risk factors such as diabetes, hyperlipidemia and periodontitis leads to increased risk of developing AD by inducing microglial Aβ/LPS tolerance via inflammatory EVs in circulation. These findings warrant further investigation of potential roles of inflammatory EVs in circulation in increasing risk of AD.
Conclusion
This study is the first to report that TLR4 signaling change is involved in altered expression of certain AD risk genes in an AD mouse model and microglial cell culture and that EVs derived from inflammatory macrophages can induce LPS tolerance in microglia. LPS tolerance and altered AD risk gene expression in an AD mouse model can be partly recapitulated in cultured microglia by upregulating miR-146a. Our results suggest that increased miR-146a induces microglial Aβ/LPS tolerance, leading to reduced Aβ clearance and that certain risk genes increase AD risk via the same molecular mechanisms underlying LPS tolerance. It will be interesting to determine if TLR4 alleles that are associated with AD risk can modulate Aβ/LPS tolerance via altering miR-146a expression.
Acknowledgements:
We thank Erika Sung for assistance in preparation of this manuscript.
Funding:
This research was supported in part by National Institutes of Health grants AG030399, AG042082, AG050854 and AG062179.
Footnotes
Publisher's Disclaimer: This Author Accepted Manuscript is a PDF file of a an unedited peer-reviewed manuscript that has been accepted for publication but has not been copyedited or corrected. The official version of record that is published in the journal is kept up to date and so may therefore differ from this version.
ETHICAL STATEMENT:
Ethics approval and consent to participate:
The submitted work is original and has not been published elsewhere in any form.
Consent for publication:
All authors are in agreement with the content of the manuscript and agree to the submission of the manuscript to Journal of Molecular Neuroscience.
Availability of data and materials:
NA
Competing interests:
All authors declare no conflict of interest.
References
- Agliardi C, Clerici M (2020) Blood extracellular vesicles (EVs) of central nervous system origin: a window into the brain. Neural Regen Res. doi: 10.4103/1673-5374.264454 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM, Cooper NR, Eikelenboom P, Emmerling M, Fiebich BL, Finch CE, Frautschy S, Griffin WS, Hampel H, Hull M, Landreth G, Lue L, Mrak R, Mackenzie IR, McGeer PL, O’Banion MK, Pachter J, Pasinetti G, Plata-Salaman C, Rogers J, Rydel R, Shen Y, Streit W, Strohmeyer R, Tooyoma I, Van Muiswinkel FL, Veerhuis R, Walker D, Webster S, Wegrzyniak B, Wenk G, Wyss-Coray T (2000) Inflammation and Alzheimer’s disease. Neurobiol Aging. doi: S019745800000124X [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- An H, Xu H, Zhang M, Zhou J, Feng T, Qian C, Qi R, Cao X (2005) Src homology 2 domain-containing inositol-5-phosphatase 1 (SHIP1) negatively regulates TLR4-mediated LPS response primarily through a phosphatase activity- and PI-3K-independent mechanism. Blood. doi: 2005-01-0191 [pii] [DOI] [PubMed] [Google Scholar]
- Arena A, Iyer AM, Milenkovic I, Kovacs GG, Ferrer I, Perluigi M, Aronica E (2017) Developmental Expression and Dysregulation of miR-146a and miR-155 in Down’s Syndrome and Mouse Models of Down’s Syndrome and Alzheimer’s Disease. Curr Alzheimer Res. doi: 10.2174/1567205014666170706112701 [doi] [DOI] [PubMed] [Google Scholar]
- Brianza-Padilla M, Carbo R, Arana JC, Vazquez-Palacios G, Ballinas-Verdugo MA, Cardoso-Saldana GC, Palacio AG, Juarez-Vicuna Y, Sanchez F, Martinez-Martinez E, Huang F, Sanchez-Munoz F, Bojalil R (2016) Inflammation Related MicroRNAs Are Modulated in Total Plasma and in Extracellular Vesicles from Rats with Chronic Ingestion of Sucrose. Biomed Res Int. doi: 10.1155/2016/2489479 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruno S, Chiabotto G, Favaro E, Deregibus MC, Camussi G (2019) Role of extracellular vesicles in stem cell biology. Am J Physiol Cell Physiol. doi: 10.1152/ajpcell.00129.2019 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caldeira C, Oliveira AF, Cunha C, Vaz AR, Falcao AS, Fernandes A, Brites D (2014) Microglia change from a reactive to an age-like phenotype with the time in culture. Front Cell Neurosci. doi: 10.3389/fncel.2014.00152 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capiralla H, Vingtdeux V, Zhao H, Sankowski R, Al-Abed Y, Davies P, Marambaud P (2012) Resveratrol mitigates lipopolysaccharide- and Abeta-mediated microglial inflammation by inhibiting the TLR4/NF-kappaB/STAT signaling cascade. J Neurochem. doi: 10.1111/j.1471-4159.2011.07594.x [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan EK, Ceribelli A, Satoh M (2013) MicroRNA-146a in autoimmunity and innate immune responses. Ann Rheum Dis. doi: 10.1136/annrheumdis-2012-202203 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YC, Yip PK, Huang YL, Sun Y, Wen LL, Chu YM, Chen TF (2012) Sequence variants of toll like receptor 4 and late-onset Alzheimer’s disease. PLoS One. doi: 10.1371/journal.pone.0050771 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deckers K, van Boxtel MP, Schiepers OJ, de Vugt M, Munoz Sanchez JL, Anstey KJ, Brayne C, Dartigues JF, Engedal K, Kivipelto M, Ritchie K, Starr JM, Yaffe K, Irving K, Verhey FR, Kohler S (2015) Target risk factors for dementia prevention: a systematic review and Delphi consensus study on the evidence from observational studies. Int J Geriatr Psychiatry. doi: 10.1002/gps.4245 [doi] [DOI] [PubMed] [Google Scholar]
- DiCarlo G, Wilcock D, Henderson D, Gordon M, Morgan D (2001) Intrahippocampal LPS injections reduce Abeta load in APP+PS1 transgenic mice. Neurobiol Aging. doi: S0197458001002925 [pii] [DOI] [PubMed] [Google Scholar]
- Fernandez-Messina L, Gutierrez-Vazquez C, Rivas-Garcia E, Sanchez-Madrid F, de la Fuente H (2015) Immunomodulatory role of microRNAs transferred by extracellular vesicles. Biol Cell. doi: 10.1111/boc.201400081 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulop T, Itzhaki RF, Balin BJ, Miklossy J, Barron AE (2018) Role of Microbes in the Development of Alzheimer’s Disease: State of the Art - An International Symposium Presented at the 2017 IAGG Congress in San Francisco. Front Genet. doi: 10.3389/fgene.2018.00362 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulop T, Witkowski JM, Olivieri F, Larbi A (2018) The integration of inflammaging in age-related diseases. Semin Immunol. doi: S1044–5323(18)30022–8 [pii] [DOI] [PubMed] [Google Scholar]
- Go M, Kou J, Lim JE, Yang J, Fukuchi KI (2016) Microglial response to LPS increases in wild-type mice during aging but diminishes in an Alzheimer’s mouse model: Implication of TLR4 signaling in disease progression. Biochem Biophys Res Commun. doi: S0006–291X(16)31533–9 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han P, Bartold PM, Salomon C, Ivanovski S (2020) Salivary Small Extracellular Vesicles Associated miRNAs in Periodontal Status-A Pilot Study. Int J Mol Sci. doi: E2809 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebert LE, Weuve J, Scherr PA, Evans DA (2013) Alzheimer disease in the United States (2010–2050) estimated using the 2010 census. Neurology. doi: 10.1212/WNL.0b013e31828726f5 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herber DL, Roth LM, Wilson D, Wilson N, Mason JE, Morgan D, Gordon MN (2004) Time-dependent reduction in Abeta levels after intracranial LPS administration in APP transgenic mice. Exp Neurol. doi: S0014–4886(04)00292–4 [pii] [DOI] [PubMed] [Google Scholar]
- Holmes C (2013) Review: systemic inflammation and Alzheimer’s disease. Neuropathol Appl Neurobiol. doi: 10.1111/j.1365-2990.2012.01307.x [doi] [DOI] [PubMed] [Google Scholar]
- Ishida A, Akita K, Mori Y, Tanida S, Toda M, Inoue M, Nakada H (2014) Negative regulation of Toll-like receptor-4 signaling through the binding of glycosylphosphatidylinositol-anchored glycoprotein, CD14, with the sialic acid-binding lectin, CD33. J Biol Chem. doi: 10.1074/jbc.M113.523480 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito H, Hamerman JA (2012) TREM-2, triggering receptor expressed on myeloid cell-2, negatively regulates TLR responses in dendritic cells. Eur J Immunol. doi: 10.1002/eji.201141679 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juzwik CA, S Drake S, Zhang Y, Paradis-Isler N, Sylvester A, Amar-Zifkin A, Douglas C, Morquette B, Moore CS, Fournier AE (2019) microRNA dysregulation in neurodegenerative diseases: A systematic review. Prog Neurobiol. doi: S0301–0082(18)30203-X [pii] [DOI] [PubMed] [Google Scholar]
- Kennedy BK, Berger SL, Brunet A, Campisi J, Cuervo AM, Epel ES, Franceschi C, Lithgow GJ, Morimoto RI, Pessin JE, Rando TA, Richardson A, Schadt EE, Wyss-Coray T, Sierra F (2014) Geroscience: linking aging to chronic disease. Cell. doi: 10.1016/j.cell.2014.10.039 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YY, Cui JG, Hill JM, Bhattacharjee S, Zhao Y, Lukiw WJ (2011) Increased expression of miRNA-146a in Alzheimer’s disease transgenic mouse models. Neurosci Lett. doi: 10.1016/j.neulet.2010.09.079 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lochhead RB, Ma Y, Zachary JF, Baltimore D, Zhao JL, Weis JH, O’Connell RM, Weis JJ (2014) MicroRNA-146a provides feedback regulation of lyme arthritis but not carditis during infection with Borrelia burgdorferi. PLoS Pathog. doi: 10.1371/journal.ppat.1004212 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik M, Parikh I, Vasquez JB, Smith C, Tai L, Bu G, LaDu MJ, Fardo DW, Rebeck GW, Estus S (2015) Genetics ignite focus on microglial inflammation in Alzheimer’s disease. Mol Neurodegener. doi: 10.1186/s13024-015-0048-1 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malm TM, Koistinaho M, Parepalo M, Vatanen T, Ooka A, Karlsson S, Koistinaho J (2005) Bone-marrow-derived cells contribute to the recruitment of microglial cells in response to beta-amyloid deposition in APP/PS1 double transgenic Alzheimer mice. Neurobiol Dis. doi: S0969–9961(04)00218–9 [pii] [DOI] [PubMed] [Google Scholar]
- Matsumoto J, Stewart T, Banks WA, Zhang J (2017) The Transport Mechanism of Extracellular Vesicles at the Blood-Brain Barrier. Curr Pharm Des. doi: 10.2174/1381612823666170913164738 [doi] [DOI] [PubMed] [Google Scholar]
- Miron J, Picard C, Lafaille-Magnan ME, Savard M, Labonte A, Breitner J, Rosa-Neto P, Auld D, Poirier J, PREVENT-AD research group (2019) Association of TLR4 with Alzheimer’s disease risk and presymptomatic biomarkers of inflammation. Alzheimers Dement. doi: S1552–5260(19)30087–1 [pii] [DOI] [PubMed] [Google Scholar]
- Nahid MA, Satoh M, Chan EK (2011) Mechanistic role of microRNA-146a in endotoxin-induced differential cross-regulation of TLR signaling. J Immunol. doi: 10.4049/jimmunol.1002311 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen MA, Karunakaran D, Geoffrion M, Cheng HS, Tandoc K, Perisic Matic L, Hedin U, Maegdefessel L, Fish JE, Rayner KJ (2018) Extracellular Vesicles Secreted by Atherogenic Macrophages Transfer MicroRNA to Inhibit Cell Migration. Arterioscler Thromb Vasc Biol. doi: 10.1161/ATVBAHA.117.309795 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olivieri F, Rippo MR, Procopio AD, Fazioli F (2013) Circulating inflamma-miRs in aging and age-related diseases. Front Genet. doi: 10.3389/fgene.2013.00121 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry VH, Holmes C (2014) Microglial priming in neurodegenerative disease. Nat Rev Neurol. doi: 10.1038/nrneurol.2014.38 [doi] [DOI] [PubMed] [Google Scholar]
- Pimenova AA, Raj T, Goate AM (2018) Untangling Genetic Risk for Alzheimer’s Disease. Biol Psychiatry. doi: S0006–3223(17)31587–1 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinn EM, Wang J, Redmond HP (2012) The emerging role of microRNA in regulation of endotoxin tolerance. J Leukoc Biol. doi: 10.1189/jlb.1111571 [doi] [DOI] [PubMed] [Google Scholar]
- Rippo MR, Olivieri F, Monsurro V, Prattichizzo F, Albertini MC, Procopio AD (2014) MitomiRs in human inflamm-aging: a hypothesis involving miR-181a, miR-34a and miR-146a. Exp Gerontol. doi: 10.1016/j.exger.2014.03.002 [doi] [DOI] [PubMed] [Google Scholar]
- Robbins PD, Morelli AE (2014) Regulation of immune responses by extracellular vesicles. Nat Rev Immunol. doi: 10.1038/nri3622 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saba R, Sorensen DL, Booth SA (2014) MicroRNA-146a: A Dominant, Negative Regulator of the Innate Immune Response. Front Immunol. doi: 10.3389/fimmu.2014.00578 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe DJ, Hardy J (2016) The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol Med. doi: 10.15252/emmm.201606210 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taganov KD, Boldin MP, Chang KJ, Baltimore D (2006) NF-kappaB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc Natl Acad Sci U S A. doi: 0605298103 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tahara K, Kim HD, Jin JJ, Maxwell JA, Li L, Fukuchi K (2006) Role of toll-like receptor signalling in Abeta uptake and clearance. Brain. doi: awl249 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verheijen J, Sleegers K (2018) Understanding Alzheimer Disease at the Interface between Genetics and Transcriptomics. Trends Genet. doi: S0168–9525(18)30042–8 [pii] [DOI] [PubMed] [Google Scholar]
- Xiong Y, Medvedev AE (2011) Induction of endotoxin tolerance in vivo inhibits activation of IRAK4 and increases negative regulators IRAK-M, SHIP-1, and A20. J Leukoc Biol. doi: 10.1189/jlb.0611273 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang YG, Song Y, Guo XL, Miao RY, Fu YQ, Miao CF, Zhang C (2019) Exosomes derived from oxLDL-stimulated macrophages induce neutrophil extracellular traps to drive atherosclerosis. Cell Cycle. doi: 10.1080/15384101.2019.1654797 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao JL, Starczynowski DT (2014) Role of microRNA-146a in normal and malignant hematopoietic stem cell function. Front Genet. doi: 10.3389/fgene.2014.00219 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]