

Abstract
Background and aims:
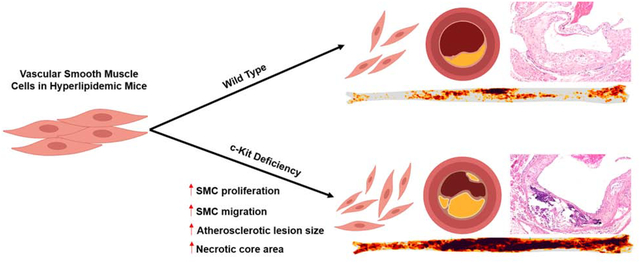
Increased receptor tyrosine kinase (RTK) activity has been historically linked to atherosclerosis. Paradoxically, we recently found that global deficiency in c-Kit function increased atherosclerosis in hyperlipidemic mice. This study aimed to investigate if such unusual atheroprotective phenotype depends upon c-Kit’s function in smooth muscle cells (SMC).
Methods:
We studied atherosclerosis in a SMC-specific conditional knockout mice (KitSMC) and control littermate. Tamoxifen (TAM) and vehicle treated mice were fed high fat diet for 16 weeks before atherosclerosis assessment in the whole aorta using oil red staining. Smooth muscle cells were traced within the aortic sinus of conditional c-Kit tracing mice (KitSMC eYFP) and their control littermates (KitWT eYFP) by immunofluorescent confocal microscopy. We then performed RNA sequencing on primary SMC from c-Kit deficient and control mice, and identified significantly altered genes and pathways as a result of c-Kit deficiency in SMC.
Results:
Atherosclerosis significantly increased in KitSMC mice with respect to control groups. In addition, the loss of c-Kit in SMC increased plaque size and necrotic core area in the aortic sinus of hyperlipidemic mice. Smooth muscle cells from KitSMC eYFP mice were more prone to migrate and express foam cell markers (e.g., Mac2 and MCAM) than those from control littermate animals. RNAseq analysis showed a significant upregulation in genes associated with cell proliferation, migration, lipid metabolism, and inflammation secondary to the loss of Kit function in primary SMCs.
Conclusions:
Loss of c-Kit increases SMC migration, proliferation, and expression of foam cell markers in atherosclerotic plaques from hyperlipidemic mice.
Keywords: atherosclerosis, smooth muscle cells, c-Kit, foam cell, receptor tyrosine kinase, atheroprotective
Graphical Abstract
1. INTRODUCTION
Atherosclerosis is the most common underlying condition behind cardiovascular complications and deaths worldwide [1]. Atherogenesis begins upon increased endothelium permeability, intimal inflammation due to lipoprotein accumulation and oxidation, and smooth muscle cells (SMC) migration [2–4]. Specifically, SMC provide strength to the plaque cap and actively participate in arterial remodeling by secreting extracellular matrix (ECM) and other structural proteins [5]. However, these proliferating and migrating synthetic SMC are pro-inflammatory in nature and add to the lesion size after up-taking oxidized low density lipoprotein (ox-LDL) and transforming into foam cells [6]. In late stage atherogenesis, SMC death leads to cap thinning and eventual rupture [7].
Multiple studies indicate that phenotypic switching of SMC is influenced by the activity of receptor tyrosine kinases (RTKs), like PDGFR and VEGFR [8, 9]. The release of these receptors’ ligands by endothelial cells, platelets, and macrophages during vascular disease potentially promotes SMC differentiation and migration [10]. For instance, increased PDGFRβ or VEGFR activity promotes plaque formation and intimal hyperplasia in hyperlipidemic animal models compared to littermate controls [8, 11]. However, translation of these findings has been problematic. Inhibition of RTKs has failed to benefit patients in their fight against SMC proliferative diseases such as restenosis in a Phase III clinical trial [12].
c-Kit is a member of the RTK family that regulates differentiation, proliferation and survival of hematopoietic stem cells [13]. This receptor is also commonly used to identify stem cells in the heart [14]. Gain and loss of c-Kit function have been implicated in carcinogenesis [15]. Recent studies have shown that the receptor tyrosine kinase c-Kit (CD117) and its ligand stem cell factor (SCF) are present in arterial endothelial cells and SMC [16]. Evidence for an involvement of c-Kit in vascular disease is presently limited to descriptive data with progression of neointimal hyperplasia after balloon injury [17]. Bone marrow cells expressing c-Kit were described as part of the pool of circulating progenitors that contribute to post injury wall thickness in mice [18]. However, the pathogenic character of circulating c-Kit+ cells in neointimal hyperplasia contradicts recent studies demonstrating the lack of contribution from circulating progenitor cells to the neointimal lesion [19]. Consequently, the contribution of c-Kit in vascular disease remains unclear and not sufficiently explored.
We have recently identified c-Kit in the cap and media of atherosclerotic lesions of patients and hyperlipidemic mice [20]. We showed that global c-Kit deficiency in mutant mice leads to a significant increase in atherosclerotic burden compared to control littermates [20]. In addition, we determined that c-Kit deficient primary SMC show an increase in proliferation, migration, and foam cell formation compared to wild type (WT) controls in vitro [20]. Our previous results indicate that c-Kit signaling has an atheroprotective role in vivo, and that this role may be dependent on SMC. Using state-of-the-art animal models, this work investigates whether inactivation of c-Kit in SMCs is sufficient to increase atherosclerotic burden in hyperlipidemic mice. We also aim at identifying the signaling pathways responsible for the atheroprotective role of c-Kit in SMC.
2. MATERIALS AND METHODS
2.1. Animal models
2.1.1. KitSMC:
The c-Kit conditional knockout mouse (Kitlox66−71/lox66−71) from Dr. Rafii’s laboratory (Weill Cornel Medical College, Ithaca, NY) [21] was crossed with ApoE−/− animals (Stock #002052, The Jackson Laboratories, Bar Harbor, ME) [22] until homozygous double transgenic mice were obtained. Simultaneously, male mice carrying the Myh11-CreERT2 from Dr. Offermanns’ lab (Max-Planck-Institute, Berlin, Germany) [23], which allows Cre mediated recombination in SMC after tamoxifen (TAM) injections, were bred with female ApoE−/− animals. Then, male Myh11-CreERT2 ApoE−/− mice were cross-bred with female Kitlox66−71/lox66−71 ApoE−/− to obtain the Kitlox66−71/lox66−71 Myh11-CreERT2 ApoE−/− triple transgenic mouse (KitSMC). The experimental colony was maintained by crossing male Kitlox66−71/+ Myh11-CreERT2 ApoE−/− mice with female Kitlox66−71/+ ApoE−/−. Genotyping was performed by tail-DNA PCR as previously described [21, 23]. To inactivate the c-Kit gene specifically in SMCs, we randomly assigned male KitSMC mice into two groups at 8 weeks of age and fed one group TAM-rich diet (TD 130858, Harlan Laboratories) for 4 weeks to inactivate the c-Kit gene, and the other group normal chow for four weeks (VEH) as the control. Afterwards, all mice were fed high fat diet (HFD, Harlan Laboratories, Indianapolis, IN) for 16 weeks to accelerate the development of atherosclerosis.
2.1.2. KitSMC eYFP:
Male Myh11-CreERT2 Kitlox66−71/+ ApoE−/− eYFP+ mice that were obtained using a Myh11-CreERT2 ApoE−/− eYFP+ breeder from Owen’s lab [24] were cross-bred with female Kitlox66−71/+ ApoE−/− to obtain the KitSMC eYFP transgenic (Kitlox66−71/lox66−71 Myh11-CreERT2 eYFP+/+ ApoE−/−) and control littermate mice KitWT eYFP (Kit+/+ Myh11-CreERT2 eYFP+/+ ApoE−/−). To inactivate the c-Kit gene specifically in SMCs and activate the eYFP gene, male KitSMC eYFP and KitWT eYFP mice were fed a TAM-rich diet for 4 weeks starting at 8 weeks of age. Afterwards, all mice were fed HFD for 16 weeks to accelerate the development of atherosclerosis.
2.1.3. KitMut:
KitW/+ and KitW-v/+ mice (Stock #100410, The Jackson Laboratories) were cross-bred to generate KitW/W-v (KitMut) mice and wild type littermate controls. KitMut mice carry a c-Kit null mutation (W) and a hypomorphic allele (W-v), and represent the severest c-Kit mutant that survives gestation [25]. This model was only used for cell culture and in vitro experiments. All animal procedures were approved by the Institutional Animal Care and Use Committee of the University of Miami Miller School of Medicine.
2.2. Atherosclerosis assessment
Murine aortas in neutral buffered 10% formalin (Sigma, St Louis, MO) were submitted to Eehscience LLC (Pickerington, OH) for Oil Red-O staining and independent quantification of atherosclerotic burden. Eehscience remained blinded to experimental groups during atherosclerosis assessment (Supplementary Methods). Serial cryosections of 5 μm thickness were cut through the aortic sinus (AV). Two sections separated by 100 μm distance were examined from each mouse, corresponding to two different depths inside the valve. Sections were stained with hematoxylin and eosin and Oil Red O, and whole aortic valve area, lesion area, and necrotic core area were quantified by digital imaging [26].
2.3. Blood chemistry and lipid profile
Blood chemistry, lipid profile, and complete blood counts (CBC) were determined in heparinized venous blood collected from each mouse before euthanasia. Blood samples were submitted to the University of Miami Comparative Pathology lab for automated analysis.
2.4. Confocal immunofluorescence microscopy
Co-detection of 1) c-Kit and smooth muscle actin (SMA), 2) eYFP and SMA, and 3) eYFP, SMA, and Mac2 was performed in paraffin embedded mouse arterial tissues. These included the aorta, carotid artery, and aortic sinus. After antigen retrieval, blocking, and staining with antibodies (Supplementary Methods), sections were counter stained with 300 μM DAPI solution (cat# D1306, Invitrogen) in PBS for 2 minutes, and mounted in polyvinyl alcohol mounting media with DABCO antifading (Cat#10981, Sigma-Aldrich). Tissue sections were examined with a confocal scanning laser microscope Zeiss LSM 510 META (Carl Zeiss, Thornwood, NY) in an inverted configuration. Data were captured and analyzed with Zeiss LSM 510 Meta and Image Browser software (Carl Zeiss,_Oberkochen, Germany). For aortic sinus tissues, images were taken from existing atherosclerotic plaques at two depths (160 and 260 um) in the valve, and quantification was done in two lesions per depth. Two independent investigators blinded to the study analyzed the cellular composition of the plaques. Image acquisition and quantification details are found in the Supplementary Methods.
2.4. Cell culture
Murine primary aortic SMCs were isolated from KitMut mice and control wild type littermates using the explant technique and cultured in DMEM-F12-FBS (5:3:2; Thermo Fisher Scientific) supplemented with 100 μg/ml penicillin, 100 μg/ml streptomycin, 0.1 mM glutamine, 10 mM sodium pyruvate, and 0.75% sodium bicarbonate [27]. Primary cells were used for three passages to avoid extensive loss of contractile features. For mechanistic studies, c-Kit expression was rescued in KitMut SMC using a lentiviral vector (Supplementary Methods).
2.5. Western blot
Protein lysates were prepared in RIPA buffer supplemented with 200 mM phenylmethylsulfonyl fluoride, 100 mM sodium orthovanadate (Santa Cruz Biotechnology, Dallas, TX), and a complete protease inhibitor cocktail (Roche Life Science, IN). Protein concentration was determined using a commercial Bradford’s protein assay kit (BioRad). Proteins were resolved in 4–12% SDS-polyacrylamide gels (NuPAGE, Thermo Fisher Scientific) before transferring onto PVDF membranes (GE Healthcare, Marlborough, MA). Specific proteins were detected using antibodies against c-Kit (1:1000, Cat# sc-1494, Santa Cruz Biotechnology), MCAM (1:1000, Cat#ab75769, Abcam), and β-actin (1:5000, Cat# A5316 and Cat# A1978, Sigma). Bound antibodies were detected after sequentially incubating the membranes with HRP-conjugated secondary antibodies, and Amersham ECL Western Blotting Detection Reagent (GE Healthcare) or Supersignal West Femto Maximum Sensitivity Substrate (Cat# 34095 Thermo Fisher). For immunoprecipitation (IP), c-Kit was pulled down from 200 μg of arterial or cellular lysate using 1 μg of anti-c-Kit antibody (Cat# A4502, Dako) and 20 μl of Protein A/G PLUS-Agarose microbeads (Cat# sc-2003, Santa Cruz Biotechnology). Microbeads were washed with pre-cold RIPA buffer before analysis by Western blot.
2.6. RNA sequencing
Total RNA was isolated from KitWT (n=5) and KitMut (n=5) primary SMC using the Quick-RNA MiniPrep kit (Zymo Research, Irvine, CA, USA). RNA quality was validated in the Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA). Sequencing was carried out on the Illumina NovaSeq 6000 NGS platform at the University of Miami’s Hussman Institute for Human Genomics (Supplementary Methods).
2.7. Quantitative Real-Time PCR (qPCR)
Cell and tissue RNAs were isolated using the Quick-RNA MiniPrep kit (Zymo Research, Irvine, CA). cDNAs were generated with a high capacity cDNA reverse transcription kit (Thermo Fisher Scientific). The relative abundance of c-Kit (Kit) and select differentially expressed genes was assessed using TaqMan Gene Expression Assays (Thermo Fisher Scientific) and normalized with respect to GAPDH. Real-time PCR was performed on an ABI Prism 7500 Fast Real-Time PCR System (96-well plate) (Applied Biosystems). Relative gene expression was determined using the ΔΔCT method [28].
2.8. Statistics
Results were presented as the mean ± S.E.M, or median [interquartile range] for non-parametric values. A two-tailed Student t-test was used to compare the difference between two groups, and a one-way ANOVA followed by a Tukey’s or Newman-Keuls test was applied to compare the difference between multiple groups. Non-parametric values were compared using the Mann-Whitney test. A p-value <0.05 was considered significant.
3. RESULTS
3.1. c-Kit expression in SMCs is vasculoprotective
To evaluate the role of SMC-derived c-Kit in atherogenesis, we developed an SMC-targeted c-Kit conditional knockout mouse (KitSMC). The re-arrangement of the c-Kit gene that leads to inactivation was demonstrated in the aorta of triple transgenic mice fed a TAM–rich diet but not in those fed normal chow (Supplementary Figure 1A). Gene inactivation of c-Kit was further confirmed by IP Western blot. c-Kit protein levels decreased 68% after gene inactivation in the vasculature of TAM treated mice compared to controls (Supplementary Figure 1B, 1.00 ± 0.22 vs. 0.32 ± 0.05, p=0.041). As expected, TAM did not affect splenic c-Kit levels in conditional mice (Supplementary Figure 1C, 1.00 ± 0.16 vs. 1.06 ± 0.04, p=0.89). In addition, immunofluorescent stained images for c-Kit and SMA in the aorta of these mice showed almost complete elimination of c-Kit in the media of TAM fed KitSMC mice compared to vehicle fed animals (Supplementary Figure 2).
To further investigate the role of SMC-derived c-Kit in atherosclerosis, KitSMC mice were fed TAM-rich diet or normal chow for one month followed by HFD for 16 additional weeks. Supplementary Figure 3 presents a heat map of atherosclerotic burden in harvested aortas stained with Oil-Red-O (Supplementary Figure 4). Tamoxifen fed KitSMC mice showed a 2.4-fold increase in percent plaque burden compared to chow fed controls (Supplementary Figure 3B, VEH 12.34 [7.29 – 13.35] vs. TAM 29.03 [23.02 – 32.23] %, p=0.0014). Tamoxifen treatment was also associated with a significant increase in lipid content in aortic lesions (Supplementary Figure 5; VEH 1120.00 ± 54.08 vs. TAM 1371.00 ± 57.74 μm2, p=0.0081). Interestingly, when we examined the plasma cholesterol levels of TAM fed mice, they were significantly lower than the controls; however, both groups were above the threshold to induce atherosclerosis (Supplementary Table 1). Next, we ruled out the potential effects of TAM on atherosclerosis. Tamoxifen has shown to reduce atherosclerotic lesion size in the coronary arteries of cynomolgus macaques, by degrading and preventing the entry of LDL products into the artery [29]. To control the effects of TAM on atherosclerotic burden, both KitSMC and their control littermates KitWT were fed TAM chow for 4 weeks, followed by 16 weeks on HFD (Figure 1 and Supplementary Figure 6). The KitSMC group showed a 2.2-fold increase in percent atherosclerotic burden in the overall aorta compared to their control littermates (Figure 1B, KitWT 16.30 [9.72 – 19.11], KitSMC 36.25 [28.56 −54.67] %, p=0.0003). Again, the plasma cholesterol levels of TAM treated KitSMC mice showed a significant decrease compared to controls, even though they were above the threshold for causing atherosclerosis (Supplementary Table 2). We further demonstrated increased atherosclerosis in the aortic sinus (AV) of mice with deficient c-Kit expression in SMC (Figure 2; lesion area, KitSMC 642698 ± 65742 vs. KitWT 411612 ± 39121 μm2, p=0.0071; necrotic core area, KitSMC 302401 ± 45771 vs. KitWT 137099 ± 28386 μm2, p=0.0070). This increment in atherosclerosis severity in c-Kit deficient mice occurred in the absence of quantitative changes in collagen content with respect to control animals (Supplementary Figure 7; KitSMC 31.28 ± 3.60 vs. KitWT 25.48 ± 3.20 %, p=0.27). The outcomes from this transgenic mouse model confirm that loss of c-Kit in SMC exacerbates atherogenesis.
Figure 1:
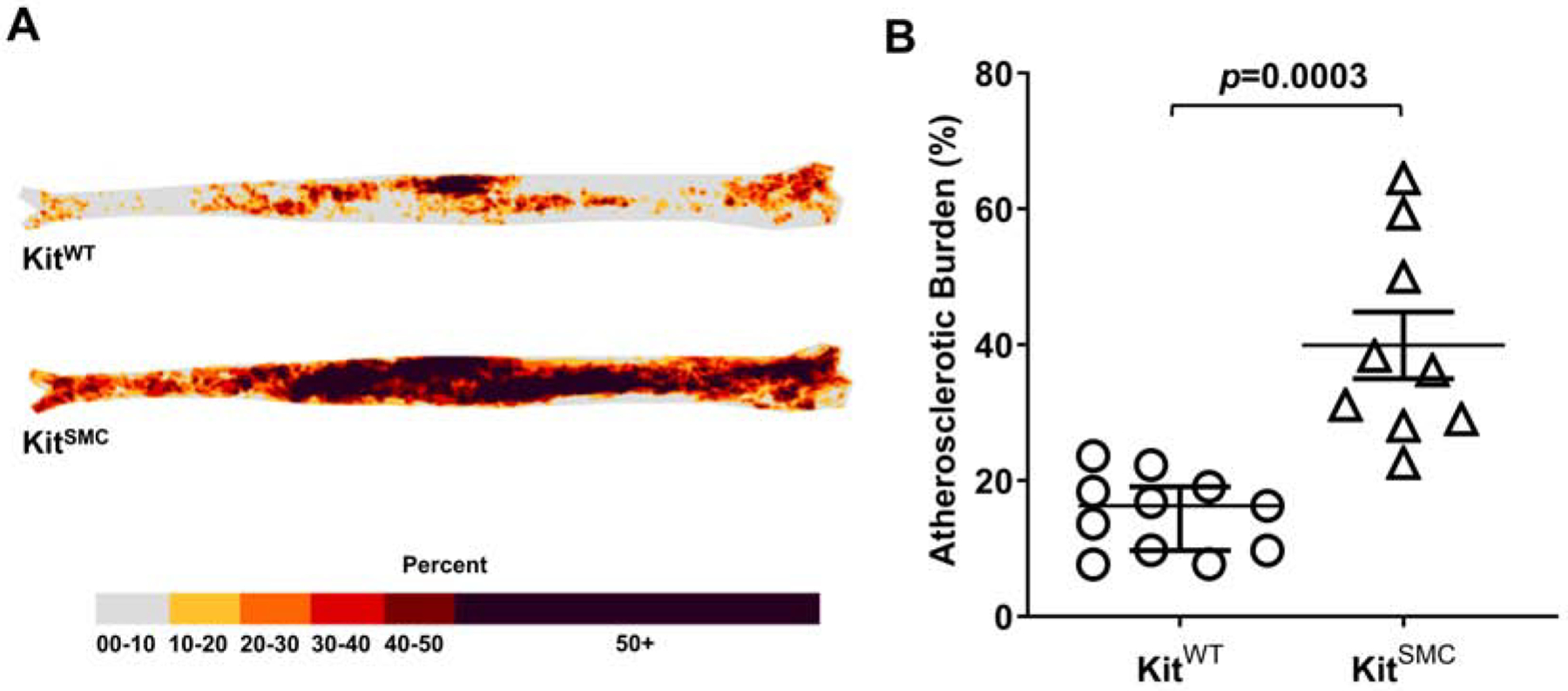
Atherosclerosis development in c-Kit conditional knockout (KitSMC) and wild type (KitWT) mice treated with tamoxifen.
(A) Heat map of plaque occurrence in whole aortas from and KitWT (Kit+/+ Myh11-CreERT2 ApoE−/−, n=11) and KitSMC (Kitlox66−71/ lox66−71 Myh11-CreERT2 ApoE−/−, n=9) mice after feeding a tamoxifen rich diet for 4 weeks, followed by 16 weeks of high fat diet. (B) Quantification of percent atherosclerosis burden in the aorta. Error bars indicate the groups’ median ± interquartile range. Groups were compared using the Mann-Whitney test.
Figure 2:
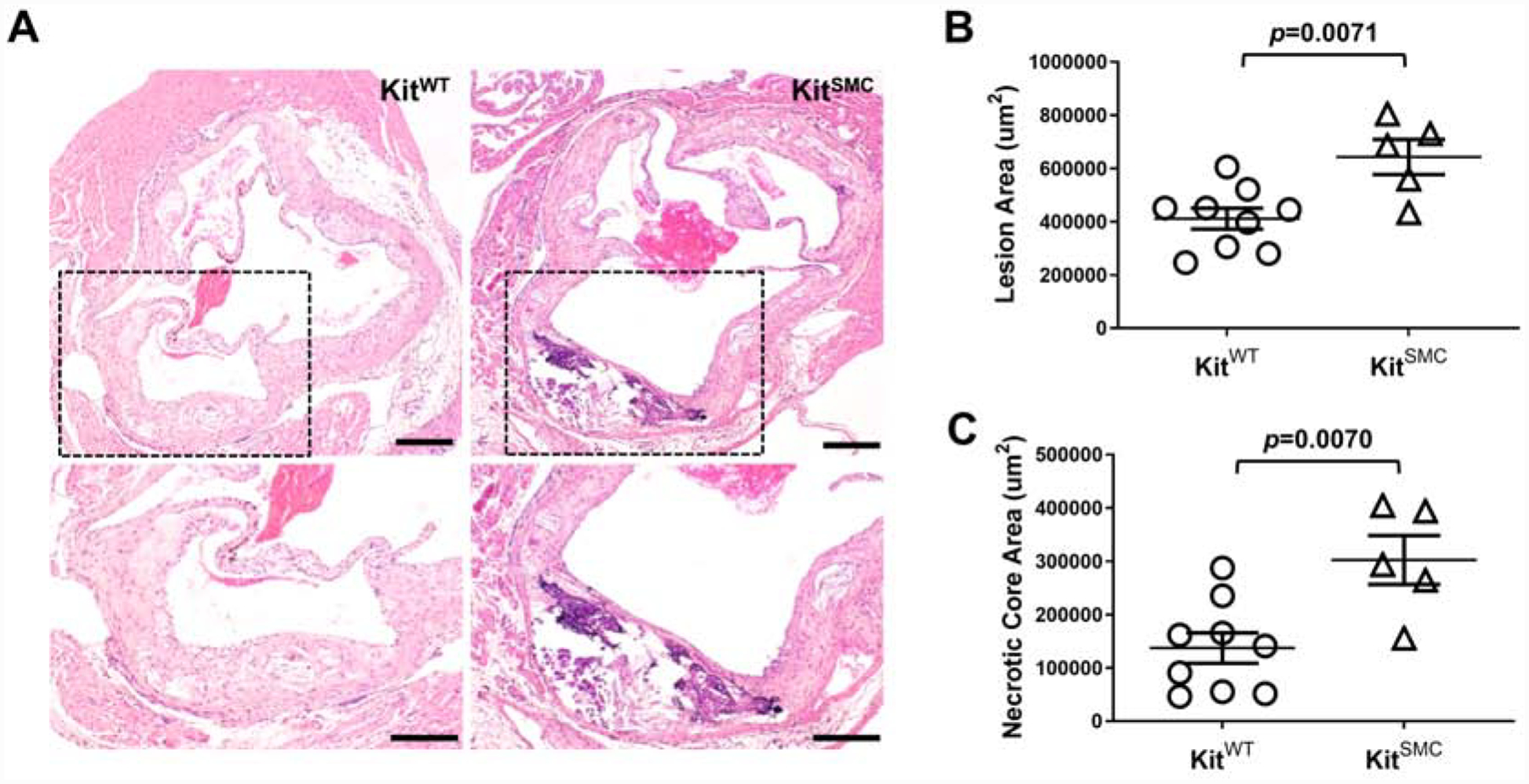
c-Kit inactivation in SMC increases atherosclerotic disease in the aortic valve (AV).
(A) Representative hematoxylin and eosin stained sections of the aortic valve in KitWT (Kit+/+ Myh11-CreERT2 ApoE−/−, n=11) and KitSMC mice (Kit66/71 / 66/71 Myh11-CreERT2 ApoE−/−, n=9) after feeding tamoxifen rich diet for 4 weeks, followed by 16 weeks of high fat diet. Scale bars=200 um. The area within the box was digitally amplified at the bottom. (B) Percentage of plaque accumulation in the aortic valve in hyperlipidemic KitWT and KitSMC mice. Data are presented as mean ± S.E.M. Groups were compared using a two-tailed Student’s t-test.
3.2. c-Kit controls SMC identity and migration into the plaque
In atherogenesis, SMC undergo differentiation from their quiescent contractile phenotype to a more proliferative synthetic phenotype. c-Kit expression in SMC of c-Kit wild type mice decreases during the course of atherosclerosis (Supplementary Figures 8 and 9). In order to trace the effect of c-Kit loss on SMC differentiation in vivo, we bred a reporter transgenic mouse [KitSMC eYFP] and its control littermates [KitWT eYFP]. When exposed to TAM, this mouse constitutively expresses YFP in cells of SMC origin. Labeling of SMCs with eYFP after TAM exposure happens independently of the c-Kit genotype (Supplementary Figure 10; percent eYFP+ cells 84.63 ± 1.85 in KitWT vs. 84.01 ± 0.48 in KitSMC, p=0.76), while loss of c-Kit in the vasculature depended upon the presence of the conditional allele (Supplementary Figure 11; percent c-Kit+ cells 77.75 ± 1.00 in KitWT vs. 3.07 ± 1.10 in KitSMC, p=0.0001). Immunofluorescent staining of the atherosclerotic AV detected a significant increase in the total percentage of SMC-derived (YFP+) cells in lesions from KitSMC animals compared to wild type controls (Figure 3; percent of total YFP+ cells 64.58 ± 3.26 in KitSMC vs. 49.40 ± 1.57 in KitWT, p=0.0030). Interestingly, the two subpopulations responsible for this increase were fully dedifferentiated YFP+ SMCs (Supplementary Table 3). Both YFP+ cells that had lost expression of the SMA marker (SMA− Mac2−; 42.58 ± 6.77% in KitSMC vs. 19.84 ± 5.80% in KitWT, p=0.034) and those that had fully transformed into SMC-derived foam cells (SMA− Mac2+; 36.99 ± 8.02% in KitSMC vs. 12.92 ± 1.80% in KitWT, p=0.019) were significantly increased in lesions from conditional knockout animals (Supplementary Table 3 and Figure 4). These results indicate that c-Kit expression plays a role in maintaining SMC identity during atherogenesis and decreases the migration and/or proliferation of dedifferentiated SMC into the plaque.
Figure 3:
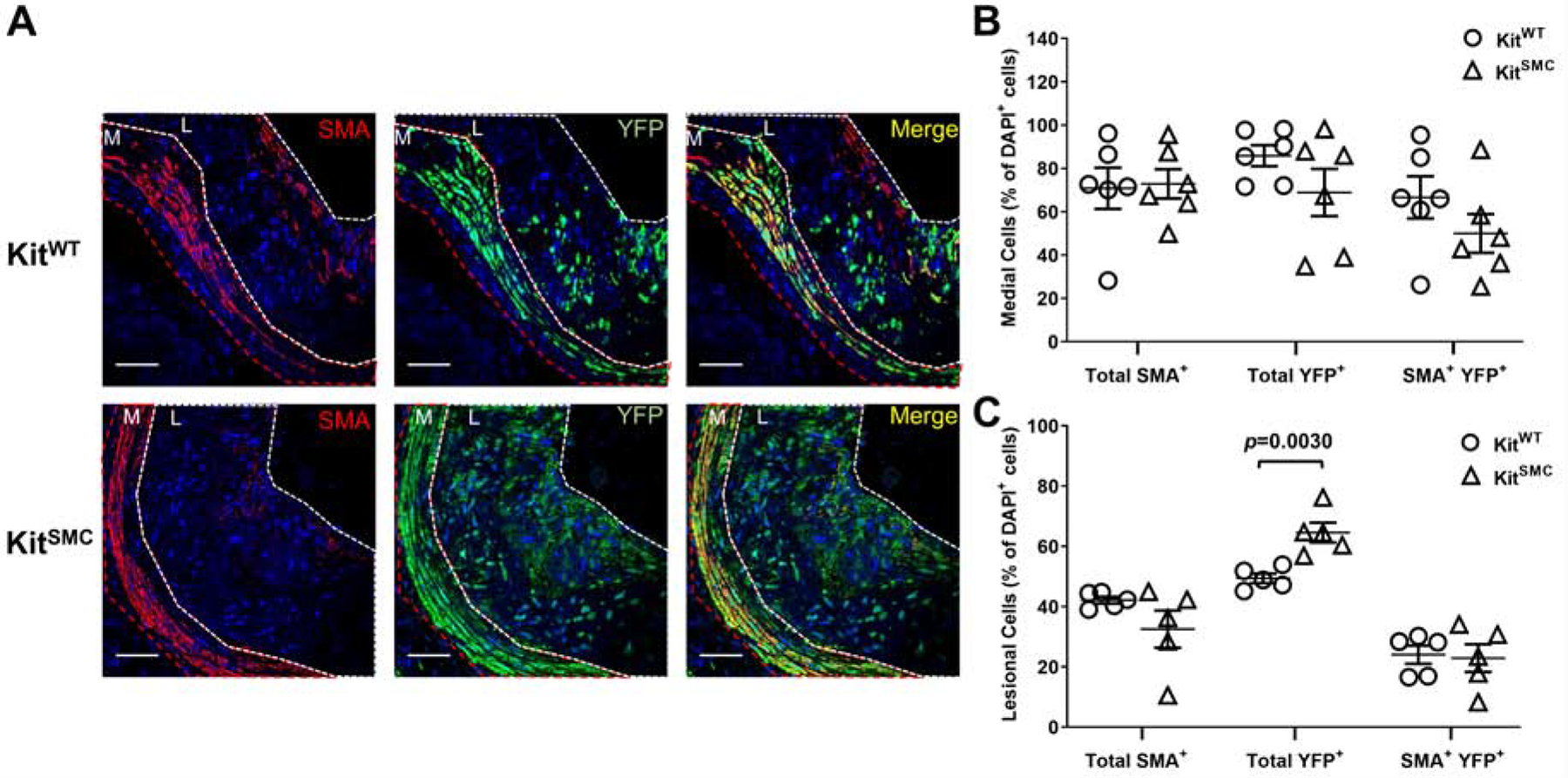
c-Kit inactivation in SMC increases migration of SMC-derived cells into plaques.
(A) Representative immunofluorescent images of aortic sinus lesions from KitWT eYFP (Kit+/+ Myh11-CreERT2 eYFP ApoE−/−, n=6) and KitSMC eYFP mice (Kit66/71 / 66/71 Myh11-CreERT2 eYFP ApoE−/−, n=6). Mice fed tamoxifen-rich diet for 4 weeks, followed by high fat diet for 16 weeks. Tamoxifen inactivates c-Kit in KitSMC mice while allowing the tracing of SMC-derived cells in both experimental groups. Images were stained for smooth muscle actin (SMA, red) and eYFP (green). Nuclei were counterstained with DAPI (blue). The media (M) is outlined in red, while the lesion (L) is outlined in white. Scale bars=100 um. (B and C) Quantification of labeled cells in the media (B) and lesion (C) as percentage of total DAPI+ cells. Data represent the mean values of two plaques per depth at two different depths (160 and 260 um) in the aortic sinus. Data are presented as mean ± S.E.M. and compared using a two-tailed Student’s t-test.
Figure 4:
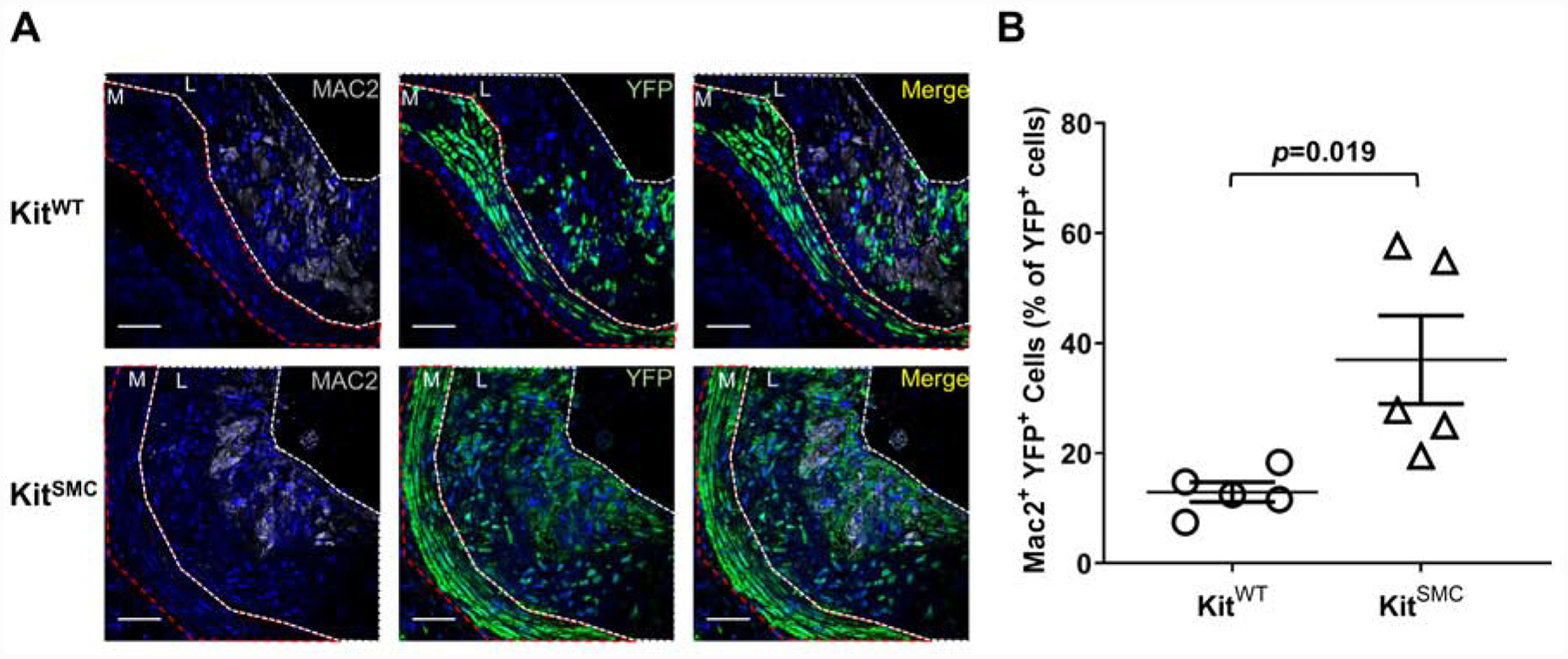
c-Kit inactivation increases SMC-derived foam cells in lesions of hyperlipidemic mice.
(A) Representative immunofluorescent images of aortic sinus lesions from KitWT eYFP (Kit+/+ Myh11-CreERT2 eYFP ApoE−/−, n=5) and KitSMC eYFP mice (Kit66/71 / 66/71 Myh11-CreERT2 eYFP ApoE−/−, n=5). Mice were fed tamoxifen-rich diet for 4 weeks, followed by high fat diet for 16 weeks. Tamoxifen inactivates c-Kit in KitSMC mice while allowing the tracing of SMC-derived cells in both experimental groups. Images were stained for Mac2 (gray) and eYFP (green). Nuclei were counterstained with DAPI (blue). The media (M) is outlined in red, while the lesion (L) is outlined in white. Scale bars=100 um (B) Quantification of Mac2+ YFP+ cells as percentage of total SMC-derived (YFP+) cells. Data represent the mean values of two plaques per depth at two different depths (160 and 260 um) in the aortic sinus. Data are presented as mean ± S.E.M. and compared using a two-tailed Student’s t-test.
3.3. Loss of c-Kit signaling leads to transcriptional upregulation of genes associated with SMC phenotypic switch and foam cell formation
In order to elucidate the pre-existing changes in c-Kit deficient SMC that might predispose to atherogenesis, gene expression profiles of primary SMC from c-Kit deficient (KitMut, n=5) and littermate wild type mice (n=5) were generated by bulk RNA sequencing. KitMut mice (also known as KitW/W-v) carry a c-Kit null mutation (W) and a hypomorphic allele (W-v), and represent the severest c-Kit mutant that survives gestation [25]. We use this model to isolate c-Kit deficient SMC because the activity of the Myh11 promoter is lost quickly in cell culture [30], preventing us from efficiently deleting c-Kit in KitSMC cells. In addition, it avoids the potential introduction of chimerism in culture if SMC are isolated from tamoxifen-treated KitSMC animals. Both c-Kit deficient and wild type controls for this experiment are generated by crossing KitW/+ and KitW-v/+ mice.
A total of 12,891 genes were differentially expressed between KitMut and littermate wild type cells (FDR<0.05; Supplementary Figure 12). Of these, 1,136 were upregulated over two folds, while 1,892 were downregulated at least two folds in KitMut vs. wild type cells. Supplementary Table 4 lists the genes with the highest expression differences between c-Kit deficient and wild type SMC. Primary SMC from KitMut mice have increased expression of genes associated with SMC proliferation such as Igfbp5, Htr1b, Creb5, and Sost [31–34]. In addition, c-Kit deficiency results in downregulation of migration inhibitors such as Lum and Stc1 [35, 36]. Lipid metabolism and processing are decreased in these cells likely secondary to reduced levels of Stc1 [35], and upregulation of Cspg4 and Mcam [37, 38]. These transcriptional differences may explain the increase in foam cell formation we have previously shown in vitro [20], and the higher number of SMC-derived foam cells in lesions of KitSMC mice (Supplementary Table 3 and Figure 4). c-Kit deficient SMC also show an upregulation of the inflammatory mediators Mcam and Mgll [37, 39], and a decrease in the inflammation repressors Il1rn and A2m [40, 41].
The 3,028 differentially expressed genes identified by RNAseq and with log2FC >ǀ1ǀ were used to predict the molecular pathways affected by loss of c-Kit signaling in SMC. Supplementary Table 5 shows the most significantly affected pathways detected by IPA. Pathways with predicted activation in KitMut compared to wild type cells covered cellular processes such as cell proliferation and migration, apoptosis signaling, inflammation, and production of ECM proteins. In contrast, pathways with predicted inhibition included those contributing to lipid metabolism and efflux, SMC contractile gene expression, and vasodilation.
Using qRT-PCR, we confirmed upregulation in KitMut cells of four differentially expressed genes (Mcam, Mgll, Smpd3, and Sost) related to foam cell formation, inflammation, or SMC migration (Supplementary Figure 12B). Expression of these genes was brought back to wild type levels after transducing KitMut cells with a lentivirus carrying the c-Kit gene under the PGK promoter. Up-regulation of MCAM in c-Kit deficient cells was confirmed at the protein level (Figure 5A and B). In addition, there was a higher percentage of total MCAM+ cells (26.14 ± 1.95 vs. 11.89 ± 1.83%, respectively; p=0.0007) and SMA+/MCAM+ cells (19.30 ± 0.88 vs. 5.24 ± 1.38%, respectively; p=0.0001) in valve lesions from SMC-specific c-Kit knockout animals (KitSMC) compared to littermate controls (KitWT; Figure 5C and D). Higher rates of proliferation and migration were also observed in vitro in c-Kit deficient cells (KitMut) SMC compared to wild type controls (Supplementary Figure 13). These results confirm the role of c-Kit signaling in the transcriptional regulation of pro-inflammatory and proliferation- and migration-related genes in primary SMC.
Figure 5.
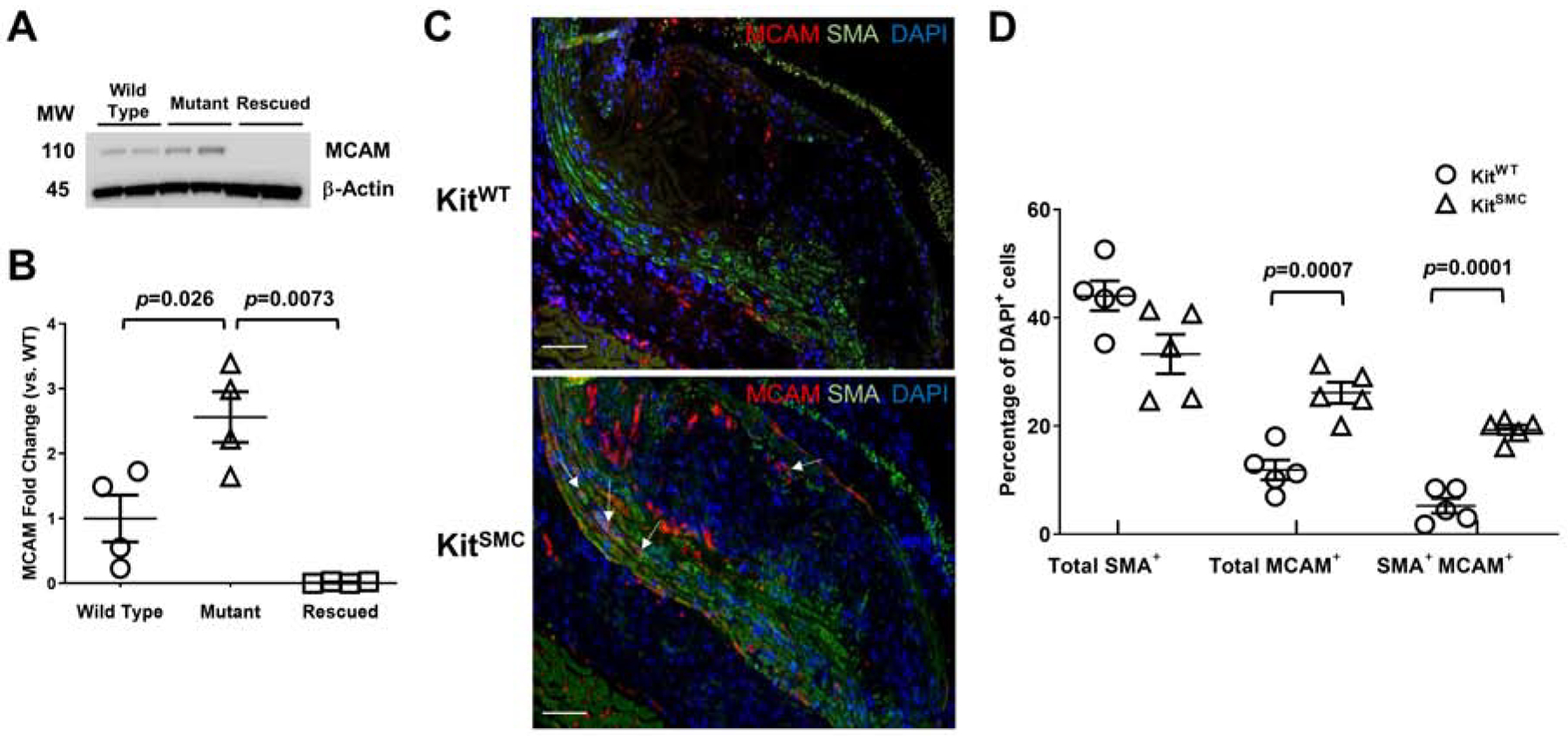
Loss of c-Kit increases protein expression of melanoma cell adhesion molecule (MCAM) in SMC.
(A and B) Protein expression and quantification of MCAM in wild type (n=4), c-Kit deficient (mutant, KitMut, n=4), and rescued KitMut primary SMC (n=4) after transduction with a c-Kit expressing lentivirus. Data are presented as mean ± S.E.M. MCAM expression was normalized using β-actin levels, then standardized against expression in the wild type group. Groups were compared using two-tailed t-tests with or without Welch’s correction for unequal variances. (C) Representative immunofluorescent images of aortic valves from KitWT (Kit+/+ Myh11-CreERT2 ApoE−/−, n=5) and KitSMC mice (Kit66/71 / 66/71 Myh11-CreERT2 ApoE−/−, n=5). Mice fed tamoxifen-rich diet for 4 weeks, followed by high fat diet for 16 weeks. Images were stained for MCAM (red) and SMA (green). Nuclei were counterstained with DAPI (blue). Scale bars=100 um. (D) Quantification of MCAM+, SMA+, and double positive cells as percentage of total (DAPI+) cells in the media and lesions from KitWT and KitSMC mice. Data represent the mean values of two plaques per depth at two different depths (160 and 260 um) in the aortic sinus. Data are presented as mean ± S.E.M. and compared using two-tailed Student’s t-tests.
4. DISCUSSION
The present work demonstrates that c-Kit expression in SMC is vasculoprotective. We have shown that c-Kit in SMC: 1) protects the murine vasculature against atherosclerosis, 2) decreases the migration of SMC-derived cells towards the plaque and their phenotypic dedifferentiation, and 3) regulates the expression of genes involved in SMC migration, proliferation, and lipid metabolism. To our knowledge, we are the first to provide evidence that a tyrosine kinase receptor in SMC plays a pivotal and protective role in the pathophysiology of atherosclerosis.
Our data demonstrate that c-Kit is an important regulator of SMC differentiation in atherogenesis, and its presence is necessary to prevent the formation of SMC-derived macrophage like cells that contribute to disease burden. These results contrast with a vast literature on the role of c-Kit as a functional marker for vascular progenitor cells [42]. In the setting of other vascular diseases, prior studies have shown reduced neointimal hyperplasia secondary to the loss of c-Kit function in progenitors [43] and SMC [44]. However, the contribution of progenitors to the atherosclerotic plaque has been controversial [45], and subsequent studies showed that the majority of plaque cells originate from pre-existing SMC in the wall [46]. This apparent discrepancy reflects the differences between neointimal hyperplasia (restenosis) and atherosclerosis in terms of etiology, natural history, culprit lesions, and progenitor cell contribution to lesion progression. The absence of mast cells in some c-Kit mutant mice (KitW-sh/W-Sh) reduces plaque burden in hypercholesterolemic mice [47]. Nonetheless, our studies with the global c-Kit mutant mouse confirm that the protective effect of vascular c-Kit expression overrides the negative role of circulating inflammatory cells [20].
Our study revealed that loss of vascular c-Kit changes the lesion composition in the plaque of hyperlipidemic mice. The conditional mouse developed plaques that were abundant in SMC-derived cells (eYFP+) with either a macrophage (SMA− Mac2+ eYFP+) or undefined phenotype (SMA− Mac2− eYFP+). It is plausible that the absence of c-Kit favors the SMC transformation towards a macrophage like phenotype during atherogenesis, reducing the number of structural SMCs within the plaque. Interestingly, loss of c-Kit increases expression of melanoma cell adhesion molecule (Mcam, CD146) in SMC. MCAM is a cell surface glycoprotein that controls adhesion and migration of inflammatory cells into and out of the lesions [37, 48]. MCAM expression in macrophages causes CD36 internalization and lysosomal degradation during lipid uptake [37]. This causes the lipid loaded macrophages to be retained in the lesion and contributes to foam cell formation [37].
In silico pathway analyses have also identified that c-Kit inhibition impacts the Smad signaling pathway. Inhibition of the interaction of the Smad2/3 complex with TGFβ prevents the transcription of contractile genes like SM22α and differentiation of SMC into their quiescent contractile phenotype [49]. Moreover, c-Kit deficiency leads to inhibition of the liver X receptor-retinoid X receptor pathway. This receptor plays a crucial role in lipid efflux through transcription of the ATP-binding cassette transporters ABCA1 and ABCG1 [50]. We have previously shown that ABCG1 is downregulated in c-Kit deficient SMC [20].
The phenotype of SMC-derived foam cells in human and mouse plaques remains elusive. Typically, they are identified by the downregulation of SMC contractile markers and expression of the macrophage markers Mac2 and CD68 [24, 51]. However, a recent single-cell RNA-seq analysis revealed that dedifferentiated SMC in murine plaques did not have a macrophage-like transcriptional phenotype, and lacked upregulation of key macrophage markers such as CD16, CD32, CD11b, CD64, CD86, and F4/80 [52]. Despite the absence of an extensive macrophage-like transformation, many dedifferentiated SMC in the lesion were positive for lipid uptake [52], suggesting that more subtle phenotypic changes may be sufficient for SMC-cell derived foam cell formation. Considering the proposed beneficial function of other dedifferentiated SMC phenotypes in plaque stability [53], it is of paramount importance to define the mechanisms that promote SMC lipid uptake and retention.
Besides increased expression of foam cell markers, our work indicates that loss of c-Kit signaling in primary SMC results in profound changes at the transcriptional level, including pathways involved in inflammation, proliferation, migration, and SMC differentiation. The multi-target transcriptional effects of c-Kit and, in turn, its role in SMC differentiation are likely due to this receptor’s ability to phosphorylate and activate multiple signaling pathways including PI3K-Akt [54] and the Src family of kinases [55]. Activation of Akt has been shown to protect against SMC apoptosis, promote plaque stability through ECM proteins, and reduce the formation of necrotic cores in lesions [56]. Wang et al. proposed that the SCF/c-Kit axis protects SMC from apoptosis [44]. Our previous work showed that global loss of c-Kit increased atherosclerosis and favored a synthetic phenotypic switch of SMC in vitro [20]. This transformation was characterized by activation of pro-inflammatory pathways associated with NF-κβ transcription [57].
Of note, SMC-specific c-Kit deficient animals develop lower levels of hyperlipidemia than the littermate wild type group. Despite this difference, plasma cholesterol levels in both experimental groups are above the threshold for disease development, and the former in fact develop more atherosclerosis than controls. This is a phenomenon that is also observed in constitutive c-Kit deficient ApoE−/− mice (KitMut) [20]. Previously, it was proposed that this effect was due to gastrointestinal dysfunction due to the loss of Cajal cells in KitMut mice [58]. Our study with SMC-specific models suggests a different or synergistic culprit, namely the deletion of c-Kit in the gastrointestinal smooth muscle that is innervated by Cajal cells.
In summary, this study is highly significant because: 1) it is the first study to rigorously evaluate the role of SMC-derived c-Kit in atherosclerosis, 2) it provides the first evidence that c-Kit plays a central role in determining the SMC phenotype, and 3) it furnishes new therapeutic targets to mitigate the devastating effects of atherosclerosis. We now confirm that SMC are the source of c-Kit’s vasculoprotective function in the wall, and propose that c-Kit expression specifically in SMC reduces atherosclerosis by controlling the transcription of genes associated with synthetic foam cell-like SMC differentiation. Findings from this study may also shed light on the role of c-Kit in other vascular occlusive diseases.
Supplementary Material
HIGHLIGHTS.
c-Kit, a well-known stem cell marker, plays an important role in smooth muscle cells
c-Kit deficiency increases smooth muscle cell migration to atherosclerotic lesions
c-Kit deficiency increases smooth muscle cell-derived foam-cell formation
c-Kit expression decreases atherosclerosis unlike other receptor tyrosine kinases
Financial support
This study was supported by the National Institutes of Health grants R01-HL125672 to RIVP and OCV, R01-DK121227 to RIVP, K08-HL151747 to LM, K01-HL145359 to RMLS, and the American Heart Association Predoctoral Fellowship 18PRE34030314 to ZMZ.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of competing interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
REFERENCES
- 1.Benjamin EJ, et al. , Heart Disease and Stroke Statistics—2019 Update: A Report From the American Heart Association. Circulation, 2019. 139(10): p. e56–e528. [DOI] [PubMed] [Google Scholar]
- 2.Thomas HE Jr., et al. , Cholesterol-phospholipid ratio in the prediction of coronary heart disease. The Framingham study. N Engl J Med, 1966. 274(13): p. 701–5. [DOI] [PubMed] [Google Scholar]
- 3.Hansson GK, Robertson AK, and Soderberg-Naucler C, Inflammation and atherosclerosis. Annu Rev Pathol, 2006. 1: p. 297–329. [DOI] [PubMed] [Google Scholar]
- 4.Mack CP, Signaling Mechanisms That Regulate Smooth Muscle Cell Differentiation. Arteriosclerosis, Thrombosis, and Vascular Biology, 2011. 31(7): p. 1495–1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bennett MR, Sinha S, and Owens GK, Vascular smooth muscle cells in atherosclerosis. Circulation research, 2016. 118(4): p. 692–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rong JX, et al. , Transdifferentiation of mouse aortic smooth muscle cells to a macrophage-like state after cholesterol loading. Proc Natl Acad Sci U S A, 2003. 100(23): p. 13531–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Newby AC and Zaltsman AB, Fibrous cap formation or destruction--the critical importance of vascular smooth muscle cell proliferation, migration and matrix formation. Cardiovasc Res, 1999. 41(2): p. 345–60. [PubMed] [Google Scholar]
- 8.He C, et al. , PDGFRβ signaling regulates local inflammation and synergizes with hypercholesterolemia to promote atherosclerosis. Nature communications, 2015. 6: p. 7770–7770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liao XH, et al. , VEGF-A Stimulates STAT3 Activity via Nitrosylation of Myocardin to Regulate the Expression of Vascular Smooth Muscle Cell Differentiation Markers. Sci Rep, 2017. 7(1): p. 2660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Muto A, et al. , Smooth muscle cell signal transduction: implications of vascular biology for vascular surgeons. J Vasc Surg, 2007. 45 Suppl A: p. A15–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bhardwaj S, et al. , Adventitial gene transfer of VEGFR-2 specific VEGF-E chimera induces MCP-1 expression in vascular smooth muscle cells and enhances neointimal formation. Atherosclerosis, 2011. 219(1): p. 84–91. [DOI] [PubMed] [Google Scholar]
- 12.Zohlnhöfer D, et al. , A randomized, double-blind, placebo-controlled trial on restenosis prevention by the receptor tyrosine kinase inhibitor imatinib. J Am Coll Cardiol, 2005. 46(11): p. 1999–2003. [DOI] [PubMed] [Google Scholar]
- 13.Edling CE and Hallberg B, c-Kit--a hematopoietic cell essential receptor tyrosine kinase. Int J Biochem Cell Biol, 2007. 39(11): p. 1995–8. [DOI] [PubMed] [Google Scholar]
- 14.Leri A, Kajstura J, and Anversa P, Role of cardiac stem cells in cardiac pathophysiology: a paradigm shift in human myocardial biology. Circ Res, 2011. 109(8): p. 941–61. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 15.Liang J, et al. , The C-kit receptor-mediated signal transduction and tumor-related diseases. Int J Biol Sci, 2013. 9(5): p. 435–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Miyamoto T, et al. , Expression of stem cell factor in human aortic endothelial and smooth muscle cells. Atherosclerosis, 1997. 129(2): p. 207–13. [DOI] [PubMed] [Google Scholar]
- 17.Orlandi A, et al. , Stem cell marker expression and proliferation and apoptosis of vascular smooth muscle cells. Cell Cycle, 2008. 7(24): p. 3889–97. [DOI] [PubMed] [Google Scholar]
- 18.Wang CH, et al. , Stem cell factor deficiency is vasculoprotective: unraveling a new therapeutic potential of imatinib mesylate. Circ Res, 2006. 99(6): p. 617–25. [DOI] [PubMed] [Google Scholar]
- 19.Hoofnagle MH, et al. , Origin of neointimal smooth muscle: we’ve come full circle. Arterioscler Thromb Vasc Biol, 2006. 26(12): p. 2579–81. [DOI] [PubMed] [Google Scholar]
- 20.Song L, et al. , c-Kit suppresses atherosclerosis in hyperlipidemic mice. Am J Physiol Heart Circ Physiol, 2019. 317(4): p. H867–H876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kimura Y, et al. , c-Kit-Mediated Functional Positioning of Stem Cells to Their Niches Is Essential for Maintenance and Regeneration of Adult Hematopoiesis. PLoS One, 2011. 6(10): p. e26918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang SH, et al. , Spontaneous hypercholesterolemia and arterial lesions in mice lacking apolipoprotein E. Science, 1992. 258(5081): p. 468–71. [DOI] [PubMed] [Google Scholar]
- 23.Wirth A, et al. , G12-G13-LARG-mediated signaling in vascular smooth muscle is required for salt-induced hypertension. Nat Med, 2008. 14(1): p. 64–68. [DOI] [PubMed] [Google Scholar]
- 24.Shankman LS, et al. , KLF4-dependent phenotypic modulation of smooth muscle cells has a key role in atherosclerotic plaque pathogenesis. Nat Med, 2015. 21(6): p. 628–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bernstein A, et al. , The mouse W/c-kit locus. Ciba Found Symp, 1990. 148: p. 158–66; discussion 166–72. [PubMed] [Google Scholar]
- 26.Andres-Manzano MJ, Andres V, and Dorado B, Oil Red O and Hematoxylin and Eosin Staining for Quantification of Atherosclerosis Burden in Mouse Aorta and Aortic Root. Methods Mol Biol, 2015. 1339: p. 85–99. [DOI] [PubMed] [Google Scholar]
- 27.Metz RP, Patterson JL, and Wilson E, Vascular smooth muscle cells: isolation, culture, and characterization. Methods Mol Biol, 2012. 843: p. 169–76. [DOI] [PubMed] [Google Scholar]
- 28.Livak KJ and Schmittgen TD, Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods, 2001. 25(4): p. 402–8. [DOI] [PubMed] [Google Scholar]
- 29.Williams JK, et al. , Tamoxifen Inhibits Arterial Accumulation of LDL Degradation Products and Progression of Coronary Artery Atherosclerosis in Monkeys. Arteriosclerosis, Thrombosis, and Vascular Biology, 1997. 17(2): p. 403–408. [DOI] [PubMed] [Google Scholar]
- 30.Rovner AS, Murphy RA, and Owens GK, Expression of smooth muscle and nonmuscle myosin heavy chains in cultured vascular smooth muscle cells. J Biol Chem, 1986. 261(31): p. 14740–5. [PubMed] [Google Scholar]
- 31.Lee DH, Kim JE, and Kang YJ, Insulin Like Growth Factor Binding Protein-5 Regulates Excessive Vascular Smooth Muscle Cell Proliferation in Spontaneously Hypertensive Rats via ERK 1/2 Phosphorylation. Korean J Physiol Pharmacol, 2013. 17(2): p. 157–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hinton JM, et al. , Signalling pathways activated by 5-HT(1B)/5-HT(1D) receptors in native smooth muscle and primary cultures of rabbit renal artery smooth muscle cells. J Vasc Res, 2000. 37(6): p. 457–68. [DOI] [PubMed] [Google Scholar]
- 33.Molnar P, et al. , The cyclic AMP response element-binding protein (CREB) mediates smooth muscle cell proliferation in response to angiotensin II. J Cell Commun Signal, 2014. 8(1): p. 29–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Leto G, et al. , Sclerostin is expressed in the atherosclerotic plaques of patients who undergoing carotid endarterectomy. Diabetes/Metabolism Research and Reviews, 2019. 35(1): p. e3069. [DOI] [PubMed] [Google Scholar]
- 35.Yamamoto K, et al. , Contrasting effects of stanniocalcin-related polypeptides on macrophage foam cell formation and vascular smooth muscle cell migration. Peptides, 2016. 82: p. 120–127. [DOI] [PubMed] [Google Scholar]
- 36.Brézillon S, et al. , Lumican effects in the control of tumour progression and their links with metalloproteinases and integrins. Febs j, 2013. 280(10): p. 2369–81. [DOI] [PubMed] [Google Scholar]
- 37.Luo Y, et al. , Macrophagic CD146 promotes foam cell formation and retention during atherosclerosis. Cell research, 2017. 27(3): p. 352–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Little PJ, et al. , Proteoglycans Synthesized by Arterial Smooth Muscle Cells in the Presence of Transforming Growth Factor-β1 Exhibit Increased Binding to LDLs. Arteriosclerosis, Thrombosis, and Vascular Biology, 2002. 22(1): p. 55–60. [DOI] [PubMed] [Google Scholar]
- 39.Guillamat-prats R, et al. , Pharmacological inhibition of monoacylglycerol lipase enhances IGM plasma levels and limits atherogenesis in a CB2-dependent manner. Atherosclerosis, 2018. 275: p. e45. [DOI] [PubMed] [Google Scholar]
- 40.Dewberry R, et al. , Interleukin-1 Receptor Antagonist Expression in Human Endothelial Cells and Atherosclerosis. Arteriosclerosis, Thrombosis, and Vascular Biology, 2000. 20(11): p. 2394–2400. [DOI] [PubMed] [Google Scholar]
- 41.Stouffer GA, et al. , Activated alpha 2-macroglobulin and transforming growth factor-beta 1 induce a synergistic smooth muscle cell proliferative response. J Biol Chem, 1993. 268(24): p. 18340–4. [PubMed] [Google Scholar]
- 42.Tallini YN, et al. , c-kit expression identifies cardiovascular precursors in the neonatal heart. Proc Natl Acad Sci U S A, 2009. 106(6): p. 1808–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang C-H, et al. , Stem Cell Factor Deficiency Is Vasculoprotective: Unraveling a New Therapeutic Potential of Imatinib Mesylate. Circ Res 2006. 99(6): p. 617–625. [DOI] [PubMed] [Google Scholar]
- 44.Wang CH, et al. , Stem cell factor attenuates vascular smooth muscle apoptosis and increases intimal hyperplasia after vascular injury. Arterioscler Thromb Vasc Biol, 2007. 27(3): p. 540–7. [DOI] [PubMed] [Google Scholar]
- 45.Sata M, et al. , Hematopoietic stem cells differentiate into vascular cells that participate in the pathogenesis of atherosclerosis. Nat Med, 2002. 8(4): p. 403–9. [DOI] [PubMed] [Google Scholar]
- 46.Bentzon JF, et al. , Smooth muscle cells in atherosclerosis originate from the local vessel wall and not circulating progenitor cells in ApoE knockout mice. Arterioscler Thromb Vasc Biol, 2006. 26(12): p. 2696–702. [DOI] [PubMed] [Google Scholar]
- 47.Sun J, et al. , Mast cells promote atherosclerosis by releasing proinflammatory cytokines. Nat Med, 2007. 13(6): p. 719–24. [DOI] [PubMed] [Google Scholar]
- 48.Li G, et al. , Reciprocal regulation of MelCAM and AKT in human melanoma. Oncogene, 2003. 22(44): p. 6891–6899. [DOI] [PubMed] [Google Scholar]
- 49.Suwanabol PA, et al. , Transforming growth factor-β increases vascular smooth muscle cell proliferation through the Smad3 and extracellular signal-regulated kinase mitogen-activated protein kinases pathways. Journal of vascular surgery, 2012. 56(2): p. 446–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rasheed A and Cummins CL, Beyond the Foam Cell: The Role of LXRs in Preventing Atherogenesis. International journal of molecular sciences, 2018. 19(8): p. 2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Feil S, et al. , Transdifferentiation of vascular smooth muscle cells to macrophage-like cells during atherogenesis. Circ Res, 2014. 115(7): p. 662–7. [DOI] [PubMed] [Google Scholar]
- 52.Wirka RC, et al. , Atheroprotective roles of smooth muscle cell phenotypic modulation and the TCF21 disease gene as revealed by single-cell analysis. Nat Med, 2019. 25(8): p. 1280–1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Misra A, et al. , Integrin beta3 regulates clonality and fate of smooth muscle-derived atherosclerotic plaque cells. Nat Commun, 2018. 9(1): p. 2073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lev S, Givol D, and Yarden Y, Interkinase domain of kit contains the binding site for phosphatidylinositol 3’ kinase. Proceedings of the National Academy of Sciences of the United States of America, 1992. 89(2): p. 678–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Krystal GW, et al. , Lck associates with and is activated by Kit in a small cell lung cancer cell line: inhibition of SCF-mediated growth by the Src family kinase inhibitor PP1. Cancer Res, 1998. 58(20): p. 4660–6. [PubMed] [Google Scholar]
- 56.Fernández-Hernando C, et al. , Absence of Akt1 reduces vascular smooth muscle cell migration and survival and induces features of plaque vulnerability and cardiac dysfunction during atherosclerosis. Arteriosclerosis, thrombosis, and vascular biology, 2009. 29(12): p. 2033–2040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Song L, et al. , c-Kit modifies the inflammatory status of smooth muscle cells. PeerJ, 2017. 5: p. e3418–e3418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chi MM and Powley TL, c-Kit mutant mouse behavioral phenotype: altered meal patterns and CCK sensitivity but normal daily food intake and body weight. Am J Physiol Regul Integr Comp Physiol, 2003. 285(5): p. R1170–83. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.