

Abstract
Age-related cataracts (ARC) are the primary cause of blindness worldwide, and oxidative stress is considered the central pathogenesis of age-related cataractogenesis. Interestingly, ample evidence suggests that there is no remarkable apoptosis present in aged and cataractous human lenses despite the profound disruption of redox homeostasis, raising an essential question regarding the existence of other cell death mechanisms. Here we sought to explore the lens epithelial cell’s (LEC) susceptibility to ferroptosis after documentation has concluded that aged and cataractous human lenses manifest with increased reactive oxygen species (ROS) formation, elevated lipid peroxidation, and accumulative intracellular redox-active iron, constituting the three hallmarks of ferroptosis during aging and cataractogenesis. Here we show that very low concentrations of system Xc− inhibitor Erastin (0.5μM) and glutathione peroxidase 4 (GPX4) inhibitor RSL3 (0.1 μM) can drastically induce human LEC (FHL124) ferroptosis in vitro and mouse lens epithelium ferroptosis ex vivo. Depletion of intracellular glutathione (GSH) in human LECs and mouse lens epithelium significantly sensitizes ferroptosis, particularly under RSL3 challenge. Intriguingly, both human LECs and the mouse lens epithelium demonstrate an age-related sensitization of ferroptosis. Transcriptome analysis indicates that clusters of genes are up- or down-regulated in aged LECs, impacting cellular redox and iron homeostases, such as downregulation of both cystine/glutamate antiporter subunits SLC7A11 and SLC3A2 and iron exporter ferroportin (SLC40A1). Here, for the first time, we are suggesting that LECs are highly susceptible to ferroptosis. Moreover, aged and cataractous human lenses may possess more pro-ferroptotic criteria than any other organ in the human body.
Keywords: Ferroptosis, age-related cataract, lipid peroxidation, glutathione homeostasis, iron homeostasis, aging, reactive oxygen species (ROS), cataractogenesis
Graphical Abstract

1. Introduction
Cataracts are a very common cause of blindness worldwide, especially in the aged population. In the US, more than 70% of Caucasians, 53% of African Americans, and 61% of Hispanic Americans develop cataracts by the age of 80[1]. Age-related cataracts are considered to be the result of accumulative damage to lens proteins, i.e., lens crystallins. Among various other risk factors, growing evidence suggests that accumulative reactive oxygen species (ROS) formation and disruption of lens redox homeostasis play a key role in the pathogenesis of age-related cataractogenesis[2–4]. The aging process brings substantial changes to the human lens.
The declined GSH de novo synthesis enzyme activity in the aged human lenses in conjunction with the increased oxidation and usage of GSH significantly decreases lens GSH content[5, 6]. The barrier formation further impedes GSH diffusion to the lens nucleus and in turn makes the lens nucleus more susceptible to oxidation and protein aggregation[7]. The lens conditional gamma-glutamyl cysteine ligase, catalytic subunit (Gclc) deletion (LEGSKO) mouse we created a few years ago suppressed over 60% of its lens GSH concentration and developed an age-related nuclear cataract within a few months under a C57BL6/FVB hybrid genetic background[8].
Impaired GSH homeostasis also directly shifts the equilibrium of ascorbic acid (ASA, 3-5mM level in the human lens) to dehydroascorbic acid (DHA), an oxidized form of ascorbic acid. Especially with the presence of redox active iron (Fe2+) and copper (Cu2+), which have been found to be increased in aged and cataractous human lenses[9]. Ascorbic acid oxidation profoundly promotes hydroxyl free radical (HÔ) production via a Fenton-like reaction[10, 11]. This directly impacts lens aging because UV exposure is one of the major risk factors in age-related cataractogenesis. In addition, several metabolic intermediates from ascorbic acid oxidation and metabolism are highly reactive and can cause lens protein damage via non-enzymatic post-synthetic modification, also known as ascorbylation [12]. Recently, we discovered that ascorbylation might be a primary source of methylglyoxal-derived hydroimidazolone-1 (MG-H1) formation[13], the most abundant glycation product identified in the aged and cataractous human lens[14, 15].
Furthermore, aged human lenses demonstrate a reduced activity of antioxidative defense enzymes. For example, decreased activity of thioltransferase (TTase), thioredoxin (TRx), and thioredoxin reductase (TR) is found in the aged human lenses, which is also reflected by both increased protein-thiol mixed disulfide and oxidized GSH (GSSG)/GSH ratios in the aged human lenses[4, 16]. The total activity of glutathione peroxidase (GPX) continuously decreases during aging [17] and is remarkably declined in cataractous human lenses[18]. Of note, Gpx1 deficient mice demonstrate increased lens nuclear light scattering and develop age-related cataracts[19]. Accumulative ROS production and an impaired antioxidative defense system are also accompanied by increased lipid peroxidation[20–22], which has been found to have increased linearly in the human lens during ageing [23]. A growing consensus postulates that lipid peroxidation is a pathogenic factor in cataractogenesis[24–27].
Ferroptosis is a new type of regulated cell death first proposed by Dixon et al. in 2012[28] and defined as excessive phospholipid (PLs) peroxidation catalyzed by intracellular redox-active iron. Ferroptosis has a distinct cell death mechanism from apoptosis or necrosis. For instance, ferroptosis-induced cell death can be prevented by iron-chelating and anti-lipid peroxidation reagents but not caspase inhibitors[29]. Morphologically, ferroptosis displays shrinkage of mitochondria and loss of mitochondrial cristae, but not chromatin condensation and apoptotic body formation[30]. Ferroptosis is an emerging research area attracting great interest in almost all biomedical fields[31–33], notably in the cancer field, which extensively studies ferroptosis as a therapeutic approach[34]. At the same time, its existence as a pathophysiological process of diseases, such as neurodegenerative diseases, stroke, traumatic brain injury, ischemia-reperfusion associated kidney, heart, and liver injuries, as well as liver fibrosis and acute kidney injury, further supports the importance of this unique type of cell death[35]. A growing number of genes and pathways have been linked to ferroptosis. However, pathways that directly or indirectly impact glutathione peroxidase 4 (GPX4) activity, redox-active iron content, and GSH homeostasis, are considered essential to trigger polyunsaturated fatty acid (PUFA)-containing phospholipid oxidation mediated cell death[29, 35]. GPX4 is currently believed to be the only peroxidase that detoxifies hydroperoxides of PUFA-containing phospholipids (PLs) that are excessively present in mammalian cell membranes[36]. RSL3, a GPX4 inhibitor, has been reported to sensitize cell ferroptosis significantly [29]. GSH is a co-factor in GPX4 mediated detoxification of hydroperoxides. Disrupting intracellular cysteine homeostasis by blocking system Xc−, a cystine glutamate antiporter, with agents such as Erastin enhances cell ferroptosis and indirectly impairs GPX4 function[36]. Aberrant iron regulation and metabolism, such as increased iron import via upregulation of transferrin (TF) or decreased iron export via downregulation of ferroportin, can accumulate the intracellular labile iron pool (LIP) which will facilitate hydroxyl free radical production via Fenton-like reactions[37].
Aging human lens and cataractous human lens display disrupted redox homeostasis, decreased GSH concentration, impaired GPX activity, increased redox iron content, and elevated lipid oxidation. These manifestations fit the crucial criteria of ferroptosis. In the present study, we examined the sensitivity of lens epithelial cell (LEC) ferroptosis using both in vitro and ex vivo models and further confirmed the aging impact.
2. Materials and method
2.1. Reagents
All chemicals used were of analytical reagent grade. Milli-Q water was used for the preparation of standards and reagents. 0.4% trypan blue solution (Cat. 15250061), C11-Bodipy 581/591 (Cat. D3861), and Hoechst33342 (Cat. H3570) were purchased form Thermo Fisher (Waltham, MA). Erastin (Cat. B1524), RSL3 (Cat. B6095), and ferrostatin-1 (Cat. A4371) were obtained from APExBIO (Boston, MA). Buthionine sulfoximine (Cat. 83730-53-4), dimethyl fumarate (Cat. 242926-25G), TNF-α (Cat. H8916), and cycloheximide (Cat. C7698) propidium iodide (Cat. p4170) were purchased from Sigma-Aldrich (St. Louis, MO). ZVAD-FMK (Cat. FMK001), was purchased from R&D Systems (Minneapolis, MN). Antibody to glutamate-cysteine ligase, catalytic subunit (GCLC) (Cat. EP13475) was obtained from Abcam. Pro-caspase3 (Cat. MA1-91637) and GAPDH (Cat. PA1-987) antibodies were purchased from Thermo Fisher. Cleaved caspase 3 antibody (Cat. #9664S) was purchased from Cell Signaling Technology (Danvers, MA). Iron assay kit (Cat. I7504-60) was obtained from Pointe Scientific (Canton, MI). All other chemicals were obtained from Sigma-Aldrich and Fisher Scientific.
2.2. Cell Culture
The human lens epithelial cell line, FHL124 cells [38], established by Prof. John Reddan at Oakland University, was supplied by Prof. Michael Wormstone at the University of East Anglia, UK and was grown in MEM with 5% FBS, 100 U/ml of penicillin and streptomycin at 35□ in a humidified 5% CO2 incubator. HeLa cells and HEK293T cells were maintained in DMEM with 10% FBS, 100 U/ml of penicillin and streptomycin at 37□ in a humidified 5% CO2 incubator.
2.3. Animals
All animal experiments were performed in accordance with procedures approved by the Augusta University Animal Care and Use Committee and conformed to the Association for Research in Vision and Ophthalmology Statement for the Use of Animals in Ophthalmic and Vision Research. Animals were housed under a diurnal lighting condition and allowed free access to food and water. The lens conditional gamma glutamyl-cysteine ligase, catalytic subunit (Gclc), knockout (KO) mice, named LEGSKO mice, were generated by our group, as described in our previous report [8]. The systemic gamma glutamyl-cysteine ligase, modifier subunit (Gclm), KO [39] mice were kindly provided by Dr. Terrence J. Kavanagh ( University of Washington, Seattle, WA).
2.4. Ex vivo explant culture and treatment
3-month-old and 20-month-old male mice were used in this experiment. To prepare mouse lens explants, the mouse lens was dissected and placed in a 3.5 cm petri-dish with 4 ml HBSS containing 100 U/ml of penicillin and streptomycin and 2.5 μg/ml amphotericin B (Cat. 15290026, Thermo Fisher). The peripheral tissue attached to the lens was removed, and the lens was then washed three times by 10 ml HBSS before being transferred to a new 3.5 cm petri-dish containing 2 ml HBSS in the orientation of the posterior side face up. A small nick was made at the posterior pore of the lens capsule, 4 to 6 flaps were peeled from the posterior pore to the equator of the lens with small tweezers. The fiber mass was then removed. Subsequently, the epithelial explant was flattened and pinned to the dish. The explant was then gently washed twice with HBSS and cultured in MEM containing 100 U/ml of penicillin and streptomycin and 2.5 μg/ml amphotericin B at 37□ in a humidified 5% CO2 incubator for 24hr before treatment.
2.5. Cell Viability Assay by CCK8 and trypan blue exclusion
5000 cells/well were seeded in 96-well plates 16hr before treatment. Cells were treated with Erastin or DMSO (control) for 24hr, and cell viability was measured by CCK8 kit (Cat. HY-K0301, MedChem Express, Monmouth Junction, NJ) following the manufacturer’s instruction. In brief, the CCK8 reagent was mixed with DMEM in a 1:10 ratio, a 100 μl mixture was added to each well to replace the medium. After 2hr incubation, the optical absorbance at 450 nm was measured, and cell viability for a given treatment was expressed relative to the non-treated cells.
For Trypan blue exclusion assay, 1×105 cells/well were seeded in a 6-well plate 16hr before treatment, and viable cells after 24hr treatment by Erastin and DMSO were counted by LUNA-II Automated Cell Counter (Logos Biosystems, Annandale, VA) after staining with 0.4% trypan blue.
2.6. Immunoblot Assay
Immunoblot assays were performed as previously described [40]. In brief, the protein concentration from the supernatant was measured by protein bicinchoninic acid (BCA) assay (Thermo Fisher). Equal amounts of protein were subjected to appropriate SDS-PAGE gel electrophoresis and transferred to a 0.45 μm pore size PVDF membrane. Detection was done using the ECL western blotting detection system.
2.7. Lipid Peroxidation Assay
Cells were seeded on a round coverslip in a 24-well-plate at a density of 3×104 cells/well 16hr before treatment. At various time points, C11-Bodipy 581/591 at a final concentration of 5 μM was added and incubated for another 30min at 37□ in a humidified 5% CO2 incubator. Subsequently, cells were washed before being fixed with 4% paraformaldehyde containing 1 μg/ml Hoechst 33342 for 10 min. The coverslip was then gently washed twice with 2ml Mill-Q water and mounted with ProLong Gold Antifade Mountant (P10144, ThermoFisher) for imaging by confocal microscopy.
The lipid peroxidation product malondialdehyde (MDA) was measured by TBARS colorimetric assay using the TBARS Assay kit (Cat. 10009055, Cayman Chemical, Ann Arbor, MI) following the manufacturer’s instruction. The MDA concentration was calculated according to the standard MDA curve and expressed as nanomole per milligram protein.
The lipid peroxidation product 4-hydroxynonenal (4-HNE) was determined by immunofluorescence (IF) stain using a 4-HNE antibody (Cat. HNE11s, Alpha Diagnostic, San Antonio, TX). In brief, FHL124 cells were seeded onto coverslips 16hr before treatment, the cells were fixed with 4% formaldehyde after treatment, and the immunofluorescent stain was performed following the published protocol [40].
2.8. Cell death analysis by propidium iodide staining
After treatment, cells or lens epithelial explants were fixed by 4% paraformaldehyde containing 1 μg/ml hoechst33342 and 1 μg/ml propidium iodide for 15 min at room temperature. The samples were then mounted with ProLong Gold Antifade Mountant for imaging by confocal microscope after being gently washed twice by PBS.
2.9. Confocal Microscopy
All images were captured by a Zeiss LSM 780 inverted confocal microscopy (Carl Zeiss AG, Oberkochen, Germany) and analyzed by a ZEN lite software.
2.10. Transmission electron microscopy (TEM)
FHL124 cells were seeded at a density of 8×105 cells in a 10 cm culture dish 16hr before Erastin treatment. Cells were treated with 2 μM Erastin for 24 hrs. After treatment, cells were fixed by media/fixative (2% glutaraldehyde in 0.1M sodium cacodylate buffer, pH 7.4 (1:1) for 10 min at room temperature followed by a complete fixative for another 30 min at room temperature. Cells were harvested by scraping and pelleting by a 600xg spin in a 15 ml conical tube. The supernatant was replaced by fresh fixative and continuously fixed overnight at 4°C. Cells were then subjected to transmission electron microscopy (TEM) analysis using JSM 1400-Flash TEM machine.
2.11. Creating GCLC knockout FHL124 cells by CRISPR/Cas9 editing
sgRNA was cloned into lentiCRISPR v2 vector (lentiCRISPR V2 was a gift from Dr. Feng Zhang, Addgene plasmid # 52961). The lentiviral particles were produced from HEK293T cells by co-transfecting lentiCRISPR v2, VSV-G, BH10 ø−envࢤ, and pRev c (kindly provided by Dr. Jacek Skowronski at Case Western Reserve University). The FHL124 cells were infected by lentiviral particles for 48hr. The stably infected cells were grown out of 2 ng/ml puromycin. Single-cell expansion was achieved by dilution plating in 96-well-plate, and the individual clonal cell line with GCLC deletion was validated by immunoblot analysis. Two sets of GCLC gRNA were used. GCLC gRNA1: AGGCCAACATGCGAAAACGC, and GCLC gRNA2: AGAAATATCCGACATAGGAG.
2.12. GSH Assay
Cells were washed twice by cold PBS and collected by 0.05% trypsin-EDTA digestion. The cell pellet was homogenized in 200 μl ice-cold 0.6% sulfosalicylic acid mixture in freshly made 0.1 M potassium phosphate buffer, pH 7.5 containing 5 mM EDTA followed by 15 min centrifugation at 21,000g. The supernatant was subjected to GSH assay using glutathione reductase (GR), and β-NADPH enzymatic recycle colorimetric assay after derivatization by 5, 5’-dithio-bis (2-nitrobenzoic acid) (DTNB) as described [41].
2.13. Iron assay
7×105 FHL124 cells were seeded in a 10 cm culture plate 16hr before treatment. Cells were then treated with Erastin (2μM), RSL3 (1μM), and DMSO (control) for 12 hours. After treatment, cells were collected by tryptic digestion and washed twice by 5 ml PBS. Subsequently, cells were lysed by two freeze-thaw cycles at room temp/−80 □ in 50 μl Mill-Q water. The supernatant of cell lysate after 21,000g centrifugation at 4□ was subjected to iron assay using the iron assay kit (Point Scientific, Canton MI) following the manufacturer’s instruction. The amount of protein in the supernatant was measured by BCA assay (ThermoFisher). The iron level of each sample was calculated according to the standard iron curve and expressed as nanomole per milligram protein.
2.14. Lens epithelial cell (LEC) aging model
A LEC aging model was established by continuously culturing FHL124 cells until cell senescence was reached. The cell senescence was considered as failing to reach cell confluency in one week after seeding the same number of cells[42, 43]. We found that FHL124 cells reached their senescence around passage 55 (p55). To avoid completely senescent cells, less than p55 FHL124 cells were used for ferroptosis analysis for the present study.
2.15. RNAseq and data analysis
Passage 15 (p15) and 42 (p42) FHL124 cells were used for next-generation deep sequencing, and three biological replicates of each passage were sequenced. The RNAseq was performed following a similar procedure as our previous study[44]. In brief, for differential gene expression profiling, RNA-seq was performed by the Genomics, Epigenomics and Sequencing Core at the University of Cincinnati. The RNA quality was determined by Bioanalyzer (Agilent, Santa Clara, CA) QC analysis. To enrich polyA RNA from 200ng QC passed total RNA, NEBNext Poly(A) mRNA Magnetic Isolation Module (NEB, Ipswich, MA) combined with SMARTer Apollo NGS library prep system (Takara Bio USA, Mountain View, CA) was used. The library from enriched polyA RNA was created by using the NEBNext Ultra II Directional RNA Library Prep Kit (New England BioLabs). The library was indexed and amplified under 11 PCR cycle number. After library Bioanalyzer QC analysis and quantification via real-time PCR (NEBNext Library Quant Kit, NEB), the proportionally pooled libraries were sequenced using NextSeq 550 sequencer (Illumina, San Diego, CA). Under the sequencing setting of single read 1x85 bp, about 25 million pass filter reads per sample were generated.
To analyze differential gene expression, sequence reads were aligned to the human genome using standard Illumina sequence analysis pipeline, and the deferentially expressed genes (DEGs) and pairwise comparison of p15 and p42 were extrapolated and normalized using a negative binomial model in DESeq2. The significantly DEGs between groups were expressed as log2 fold change with p value<0.05 and false discovery rate (FDR<0.1).
2.16. RNA Extraction and Real-time PCR Analysis for Gene Expression
Mice at 2 and 20 months of age were used for the study. The lens capsule was separated from the lens body and gently rinsed by HBSS to remove attached fiber tissue before RNA extraction. Eight capsules from 4 mice were pooled in each age group. The DNA-free total RNA was extracted by RNeasy Micro Kit (Cat. 74004, Qiagen, Germantown, MD) following the manufacturer’s instructions. The RNA concentration and quality were determined by DS-11 FX+ NanoDrop. Only RNA samples with UV 260nm/280nm ratio range of 1.9-2.0 and UV 260nm/230 nm ratio >1.8 were used. RNA was reverse transcribed to cDNA with oligo(dT) random primer mix (Cat. S1330S, New England Biolabs, Ipswich, MA) and M-MuLV reverse transcriptase (Cat. M0253S, New England Biolabs). Real-time PCR was performed using the SYBR Green probe and a QuantStudio 3 Real-Time PCR system (ThermoFisher). Relative expression was calculated using the ΔΔCt: method normalized to the housekeeping gene GAPDH. Specific primer sequences are: Slc7a11, Fwd: CGTAATACGCCCTGGAGCTA, Rev: CAGCTGTCACGAGCTTGATT; Slc3a2, Fwd: GGCTTGATGTCCAGGTTGTC, Rev: TCTCCGGTTCTAGCTCGTTC; Slc40a1, Fwd: GCTGTCTATGGACTGGTGGT, Rev: CTGGGCCACTTTAAGTCTGG; Gpx4, Fwd: TTACGAATCCTGGCCTTCCC, Rev: CGGCTGCAAACTCCTTGATT. All reactions were performed in duplicate. Minimum Information for Publication of Quantitative Real-Time PCR Experiments (MIQE) guidelines[45] was followed for all real-time PCR experiments.
2.17. Statistical analysis
Statistical analyses were performed by the unpaired two-tailed Student’s t-test or One-Way ANOVA test. P-value is showing as below: * meaning p<0.05, ** meaning p<0.01, *** meaning p<0.001, **** meaning p<0.0001. All results are expressed as mean ± SD. Significance was considered P<0.05. All experiments were repeated at least twice, and representative results were shown.
3. Results
3.1. LECs manifest ferroptotic cell death by Erastin and RSL3 treatment
To test whether LECs are susceptible to ferroptosis, we treated FHL124 cells, a spontaneously immortalized cell line derived from the human lens epithelium, with Erastin, an inhibitor of the cystine/glutamate antiporter (xCT), and RSL3, an inhibitor of glutathione peroxidase 4 (GPX4) (Fig.1A). As shown in Fig.1B, the phase-contrast image, FHL124 cells demonstrated round cell bodies after 24hr treatment with 0.5 to 10μM Erastin as compared to the control cells with the same volume of DMSO added. The viable cell number counted by the trypan blue dye exclusion assay (Fig. S1A) and the cell viability measured by cell counting kit 8 (CCK8) (Fig.S1B) all showed more than 95% cell death after 24hr treatment with 2 μM Erastin. Similarly, 24hr treatment with 0.1 to 2 μM RSL3 also triggered massive FHL124 cell death compared to untreated control cells (Fig. 1C), and the cell death was also reflected in viable cell counts (Fig S1C) and cell viability assays (Fig. S1D) after 24hr challenge by 1 μM RSL3. As shown in Fig. 1D, Erastin (2μM) and RSL3 (1 μM) mediated FHL124 cell death at 24hr treatment was also evidenced by propidium iodide (PI) stain, a cell membrane-impermeable DNA dye that only stains dead cells.
Figure 1.
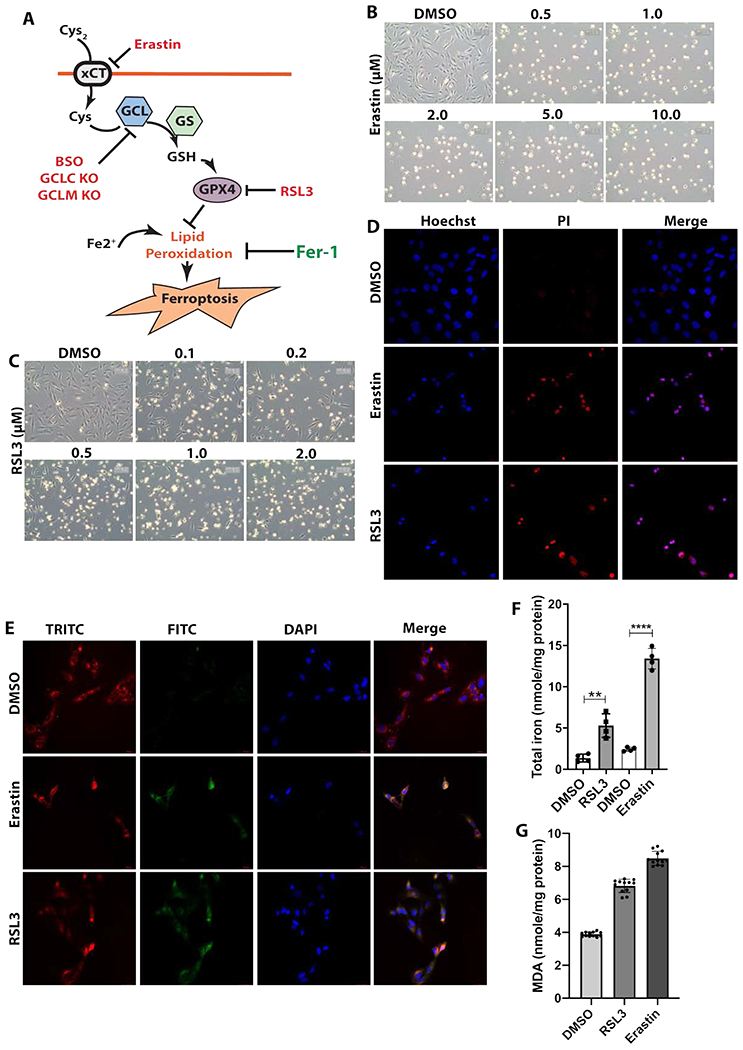
Erastin and RSL3 induce FHL124 cell death that display ferroptosis characteristics. FHL124, human lens epithelial cells, were stimulated by various concentration of Erastin and RSL3, and cell death was examined. (A) Regulatory pathways of ferroptosis and inhibitory targets used in this study. xCT, the cystine/glutamate antiporter; GCL, gamma glutamate-cysteine ligase, the key enzyme for glutathione (GSH) synthesis; GS, glutathione synthase, the second enzyme for GSH synthesis, but not a speed limiting enzyme; GPX4, Glutathione peroxidase 4; Fer-1, ferrostin-1, lipid peroxidation inhibitor; RSL3, GPX4 inhibitor; Erastin, xCT inhibitor. GCLC, catalytic subunit of GCL; GCLM, modifier subunit of GCL. (B) Optical microscope images of FHL124 cell morphology treated with various concentration of Erastin for 24hr. (C) Optical microscope images of FHL124 cell morphology treated with various concentration of RSL3 for 24hr. (D) FHL124 cell death probed by propidium iodide (PI) stain after 24hr treatment by 2μM Erastin and 1 μM RSL3. (E) Lipid peroxidation of FHL124 cells measured by C11-Bodipy 581/591 probe after 12hr treatment with 2μM Erastin and 2hr treatment with 1μM RSL3. (F) Intracellular total iron concentration w/ or w/o 1μM RSL3 treatment for 12hr. (G) Malondialdehyde (MDA) levels in FHL124 cells after treated with 2μM Erastin for 20hr and 1μM RSL3 for 9hr. For B, C, D, and E, n=3, and at least 10 images were collected in each treatment, for F, n=4, and for G, n=12. Unpaired two-tailed Student’s t-test was used to compare two groups, and only p<0.05 is considered significant (**<0.01).
We measured the cell membrane lipid peroxides by using Bodipy 581/591-C11, a fluorescence lipid peroxidation probe with a ~595 nm emission when not oxidized while having a ~520 nm emission when oxidized[46]. With triple-band excitation for DAPI, FITC (oxidized), and TRITC (non-oxidized), we were able to trace the membrane lipid peroxides by confocal microscopy. As shown in Fig. 1E, both Erastin and RSL3 induced a significantly high-level membrane lipid peroxidation compared to non-treated cells after 12hr Erastin (2μM) and 2hr RSL3 (1 μM) treatment, respectively. To confirm the lipid peroxidation, we also measured two common lipid peroxidation products, malondialdehyde (MDA) and 4-hydroxynonenal (4-HNE). The MDA was measured by TBARS colorimetric method and the results were shown in Fig. 1G. Around 1.8-fold and 2-fold increase of MDA was detected in FHL124 cells after 9hr treatment with 1μM RSL3, and 20hr treatment with 2μM Erastin, respectively, compared to non-treated cells. The 4-HNE levels measured by the immunofluorescence (IF) stain also showed more than a 2-fold increase after the treatment with either 1μM RSL3 for 3hr or 2μM Erastin for 12hr (Fig. S1E&F).
To further confirm that FHL124 cell death is through ferroptosis, we determined the intracellular iron levels reflected by the total iron-binding capacity (TIBC) assay shown in Fig. 1F. The intracellular iron level increased 5 and 7-fold in FHL124 cells treated by RSL3 (1 μM) and Erastin (2μM) for 12hr, respectively. These results of increased cell death, increased lipid peroxidation, and elevated intracellular iron levels under very low concentrations of Erastin (0.5μM) and RSL3 (0.1μM)[47, 48] indicate that the lens epithelial cells (FHL124) are highly susceptible to ferroptosis.
3.2. Apoptosis is not involved in Erastin and RSL3 mediated cell death
To verify whether programmed cell death/apoptosis was involved in the Erastin and RSL3 mediated FHL124 cell death, we first pre-treated FHL124 cells with 10 μM ZVAD-FMK, a pan-caspase inhibitor for 2hr before adding Erastin or RSL3 for 24hr. To block ferroptosis, we cotreated FHL124 cells with ferrostatin-1 (Fer-1), a lipid peroxidation inhibitor. As shown in Fig. S2A of the cell phase-contrast images, the pan-caspase inhibitor did not block Erastin (2μM), and RSL3 (1 μM) induced cell death at 24hr. Conversely, Fer-1 (0.1 μM) significantly attenuated cell death compared to cells without Fer-1 (Fig. S2A). The RSL3 induced cell death (Fig. 2A) and Erastin induced cell death (Fig. S2B) probed by propidium iodide (PI) all demonstrated no remarkable decrease of PI-positive cells by ZVAD-FMK, but were almost entirely prevented by Fer-1 compared to proper controls. Similarly, the Bodipi-C11 based cell membrane lipid peroxidation determination shown in Fig. 2B (RSL3 treatment) and Fig. S3A (Erastin treatment) indicated that Fer-1 but not ZVAD-FMK was able to block both Erastin and RSL3 induced lipid peroxidation and subsequent cell death. This was further confirmed by CCK8 cell viability determination. As shown in Fig. 2D, the profound cell death triggered by Erastin and RSL3 could be completely blocked by Fer-1 but not ZVAD-FMK. Furthermore, only pro-caspase 3 was detected in FHL124 cells with and without Erastin (Fig. S3B) and RSL3 treatment (Fig. 2C). To validate the ZVAD-FMK’s anti-caspases activation and anti-apoptosis function, we used Hela cells that were treated by a 30 ng/ml tumor necrosis factor-α (TNFα) plus 10 γg/ml cycloheximide (CHX) for 7hr, a well-established cell apoptotic system[49]. As shown in Fig. 2C, S2A, and S3B, ZVAD-FMK, but not Fer-1 blocked TNFα/CHX induced caspase 3 cleavage and apoptosis in Hela cells.
Figure 2.
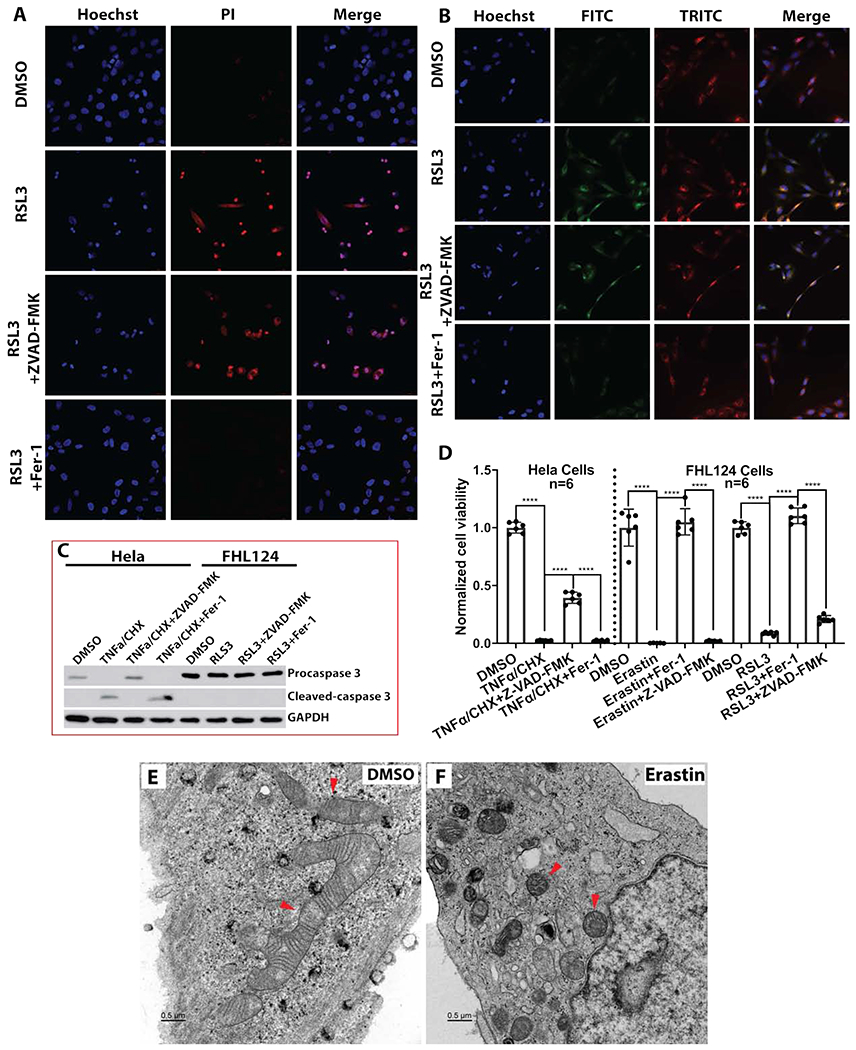
Erastin and RSL3 mediated cell ferroptosis is independent of apoptosis. Pan-caspase inhibitor ZVAD-FMK and lipid peroxidation inhibitor ferrostatin-1 (Fer-1) were used to elucidate each cell death pathway. (A) FHL124 cell death probed by propidium iodide. FHL124 cells were either treated 24hr with 1μM RSL3 alone, co-treated with 10μM ZVAD-FMK, or co-treated with 0.1μM Fer-1. (B) Lipid peroxidation measured by C11-Bodipy 581/591 probe. FHL124 cells were treated 2hr with either 1μM RSL3 alone, co-treated with 10μM ZVAD-FMK, or co-treated with 0.1μM Fer-1. (C) RSL3 inhibition did not induce caspase 3 activation in FHL124 cells. FHL124 cells were treated with 1μM; RSL3 for 24hr with or without 10μM ZVAD-FMK and 0.1μM Fer-1. The Hela cells treated 7hr with 30ng/ml TNFα and 10μg/ml cycloheximide (CHX) apoptosis system served as a positive control. The pro-caspase 3 and cleavage caspase 3 were determined by immunoblot. (D) Fer-1 but not ZVAD-FMK rescued FHL124 cells’ viability after 24hr treatment by 2μM Erastin and 1μM RSL3. The Hela cells treated 7hr with 30ng/ml TNFα and 10μg/ml cycloheximide (CHX) apoptosis system served as a positive control. (E) The ultrastructure of FHL124 cells was determined by transmission electron microscopy after 24hr treatment with 2μM Erastin. The red arrows point to the mitochondria. The scale bar is 0.5μm. One-way ANOVA with Tukey’s Honest post-hoc analysis was used to compare between groups, and only p<0.05 is considered significant. *<0.05, **<0.01, ***<0.001, ****<0.0001.
To investigate the ultrastructural changes of FHL124 cells before and after Erastin treatment, transmission electron microscope (TEM) was performed in FHL124 cells with and without Erastin (2μM) treatment for 24hr. As illustrated in Fig. 2E&F, condensed mitochondrial membrane and a significantly reduced mitochondrial size was observed in Erastin treated cells compared to non-treated cells. Shrinkage of mitochondrial volume and condensation of mitochondrial membrane are strong indicators of ferroptosis [50].
Taken all together, these results imply that lens epithelial cells are susceptible to Erastin and RSL3 induced ferroptosis independent of apoptosis.
3.3. Blocking GCL activity exacerbates Erastin and RSL3 induced ferroptosis in FHL124 cells
As illustrated in Fig.1A, glutathione (GSH) synthesis and its homeostasis are one of the key players in ferroptosis. To test the role of GSH homeostasis in Erastin and RSL3 induced FHL124 cell ferroptosis, we used both chemical intervention and gene deletion approaches to block the GSH de novo synthesis. For chemical intervention, FHL124 cells were treated with 500 μM buthionine sulfoximine (BSO) or 25 μM dimethyl fumarate (DMF). For gene targeted intervention, gamma glutamate-cysteine ligase, catalytic subunit (GCLC) knockout FHL124 cells were employed. Notably, despite a large number of selected colonies, we could still detect a weak band of GCLC after a long exposure (Fig. 3J), which suggested that lens epithelial cells might not survive under complete GCLC deletion. Nevertheless, FHL124 cells with GCLC deletion showed more than 95% depletion of the intracellular GSH levels (Fig.3I). The intracellular GSH levels of the chemically treated cells were also shown in Fig. 3I. All approaches significantly blocked cell GSH synthesis and reduced intracellular GSH concentration compared to non-treated wild-type (WT) cells. Of note, BSO and GCLC KO cells showed more profound intracellular GSH depletion than DMF treatment. Both BSO and DMF treatment did not change GCLC protein expression analyzed by immunoblot (Fig. 3K).
Figure 3.
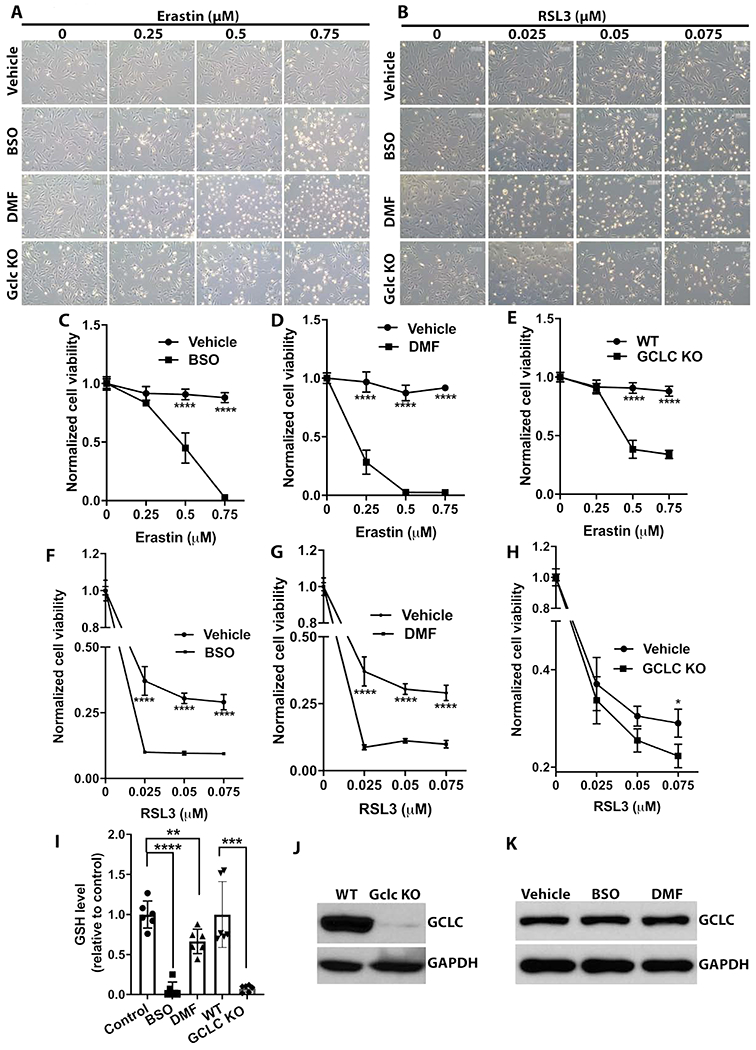
Depleting intracellular glutathione (GSH) sensitizes FHL124 ferroptosis. Intracellular GSH was depleted by either 500μM buthionine sulfoximine (BSO), 25μM dimethyl fumarate (DMF), or GCLC knockout FHL124 cells (GCLC KO). (A) Optical microscope image of FHL124 cell morphology treated with various concentration of Erastin under different GSH depletion conditions for 12hr. (B) Optical microscope image of FHL124 cell morphology treated with various concentration of RSL3 under different GSH depletion conditions for 12hr. (C, D, E) FHL124 cells’ viability was determined by CCK8 after 24hr treatment with increased concentration of Erastin w/ or w/o GSH depletion by BSO, DMF or GCLC KO. (F, G, H) FHL124 cells’ viability was determined by CCK8 after 24hr treatment with increased concentration of RSL3 w/ or w/o GSH depletion by BSO, DMF or GCLC KO. (I) Intracellular GSH concentration of FHL124 cells by different depletion methods. (J) Immunoblot confirmed GCLC deletion in FHL124 cells. (K) BSO and DMF treatment did not impact FHL124 cell GCLC expression assayed by immunoblot. Unpaired two-tailed Student’s t-test and One-way ANOVA with Tukey’s Honest post-hoc analysis was used to compare between groups, and only p<0.05 is considered significant. *<0.05, **<0.01, ***<0.001, ****<0.0001.
In order to differentiate the response of these GSH deficient cells to Erastin and RSL3 induced ferroptosis, we lowered the concentration of both Erastin (0.25, 0.5, and 0.75μM) and RSL3 (0.025, 0.05, and 0.075μM). As shown in Fig. 3A, GSH depletion by BSO, DMF, and GCLC KO significantly exacerbated the Erastin (0.25-0.75 μM) induced cell death via ferroptosis compared to GSH undepleted cells. Similarly, a very low dosage of RSL3 (25 to 75nM) could trigger massive cell death via ferroptosis in the GSH depleted cells compared to undepleted cells (Fig. 3B). The cell death was also confirmed by the cell viability assay. The Erastin treated cells were shown in Fig. 3 C, D, & E, and RSL3 treated cells were shown in Fig. 3F, G & H. These results suggest that impaired GSH de novo synthesis and decreased intracellular GSH concentration will greatly sensitize FHL124 cell ferroptosis susceptibility.
3.4. Erastin and RSL3 induces ferroptosis in ex vivo explant cultured mouse lens epithelium
Given the exciting findings above, we intended to see whether this also applied to ex vivo cultured lens epithelium. A face-up anterior lens capsule explant culture was performed following a previous report[40]. The lens epithelium explant was first adapted to the MEM medium’s culture condition for 24hr before an additional 24hr of Erastin (5, 10, and 20μM) and RSL3 (0.5, 1.0, 2.0 μM) treatment. To test the pan-caspase inhibitor effect, 10 μM ZVAD-FMK was pre-incubated with the lens epithelium before adding Erastin and RSL3. To test the intervention of lipid peroxidation by Fer-1, the lens epithelium explant was co-treated by Erastin or RSL3 along with 0.1 μM Fer-1 for 24hr. The cell death was determined by propidium iodide (PI) stain, and the results of different dosages of RSL3 induced cell death were shown in Fig. 4A. The positive PI stain was seen in lens epithelium treated by 1.0 and 2.0μM RSL3 for 24hr. Furthermore, the RSL3 mediated cell death could be attenuated by the lipid peroxidation inhibitor Fer-1 but not the caspase inhibitor ZVAD-FMK (Fig. 4B). Interestingly, a higher concentration of Erastin was required to trigger mouse lens epithelium ferroptosis than FHL124 cells. As shown in Fig. S4A and Fig. S4B, a positive PI stain was seen in lens epithelium treated by 20 μM Erastin, but no remarkable positive PI stain was observed in lower concentration Erastin treated epithelium (Fig. S4A). Similar to RSL3, Fer-1 but not ZVAD-FMK was able to block Erastin (20 μM) induced ferroptosis (Fig. S4B). These results clearly demonstrate that the lens epithelium is also susceptible to ferroptosis, whereby blocking GPX4 activity is a primary target.
Figure 4.

Ex vivo mouse lens epithelium is susceptible to RSL3 induced ferroptosis. (A) Mouse lens capsular explants were treated with various concentration of RSL3 for 24hr, and cell death/ferroptosis was probed by propidium iodide. The same volume of DMSO treatment served as a control. Hoechst 33342 was used for the cell nucleus stain. (B) Fer-1 but not ZAVD-FMK blocked RSL3 mediated mouse lens epithelium ferroptosis. Mouse lens capsule explants were treated with 1μM RSL3 for 24hr w/ or w/o the presence of 10μM ZVAD-FMK and 0.1μM Fer-1. The cell death was determined by propidium iodide stain and the cell nucleus was visualized by Hoechst 33342 stain. Each assay was repeated three times and at least 10 images were collected for each stain and the representative images were shown.
3.5. Disruption of GSH biosynthesis enzymes Gclc and Gclm sensitize lens epithelium ferroptosis
It has been well documented that lens GSH levels decline continuously during aging [6], and this has inspired us to examine whether impaired GSH biosynthesis also impacts lens epithelium ferroptosis. Again, we used an ex vivo lens explant culture model for the investigation. Besides the WT mouse lens epithelium, we also cultured lens epithelium from lens conditional gamma-glutamylcysteine ligase, catalytic subunit (Gclc) knockout mouse lens (LEGSKO), and gamma-glutamylcysteine ligase, modifier subunit (Gclm) somatic knockout mouse lens. Lens specific Gclc KO and somatic Gclm KO mice all showed a more than 50% reduction of lens GSH concentration, closely mimicking the aged human lens condition [8]. As shown in Fig. 5A & B, 0.5 μM RSL3 could trigger a significantly higher level of cell ferroptosis reflected by positive PI-stain in both LEGSKO and Gclm KO lens epithelium compared to wt after 24hr of treatment. Similar to GSH deficient FHL124 cells, we only observed a mild level of ferroptosis in Gclm KO lens epithelium but not in LEGSKO lens epithelium after 48hr of treatment with 10 μM Erastin (Fig.S5). These results demonstrate that an impaired GSH biosynthesis and decreased GSH concentration will sensitize lens epithelium ferroptosis susceptibility.
Figure 5.

GSH deficient lens epithelium sensitizes RSL3 induced ferroptosis. Mouse explants from wild-type, Gclc knockout (LEGSKO), and Gclm knockout were treated with 0.5μM RSL3 and cell ferroptosis was visualized by propidium iodide stain. (A) WT and Gclc knockout explants were treated with 0.5μM RSL3 for 8hr. (B) WT and Gclm knockout explants were treated with 0.5μM RSL3 for 24hr. The cell nucleus was visualized by Hoechst 33342 stain. Each assay was repeated three times and at least 10 images were collected for each stain and the representative images were shown.
3.6. Aged FHL124 cells and lens epithelium from aged mouse lens are more susceptible to ferroptosis
The above findings inspired us to examine whether there is an age-associated sensitization of ferroptosis in lens epithelial cells. First, we established an in vitro cell aging model by continuously culturing FHL124 cells to a high passage although before reaching their senescence stage, which was defined as cell confluency that could not be achieved one week after seeding the same number of cells. For instance, the 15th passage FHL124 cells (p15) reached their confluency in less than three days when 1×106 cells were seeded in a 10cm culture dish. FHL124 cells reached their senescence beyond passage 55 (p55). We tested the sensitivity of ferroptosis by comparing low and high passage cells after RSL3 treatment. As shown in Fig. 6A, FHL124 cells at 52nd passage (p52) demonstrated significantly lower cell viability than the 15th passage (p15) after 24hr challenge by RSL3 at both 0.5 and 0.75 μM dosages. Similar results were also seen from cell morphological changes via phase-contrast imaging (Fig. S6). Second, we tested the aging response using an ex vivo culturing system by comparing lens explants cultured from 3-and 20-month old mouse lenses after RSL3 treatment. The cell death was probed by propidium iodide (PI) stain. As shown in Fig. 6B, 24hr treatment with 0.5 μM RSL3 could profoundly induce lens epithelial cell death only in 20-month-old lens explants but not in 3-month-old lens explants. These results strongly suggest that aged lens epithelial cells are highly vulnerable to ferroptosis.
Figure 6.

Aged FHL124 cells and mouse lens epithelium is more susceptible to ferroptosis. FHL124 cells at 19th and 52nd passage were used as in vitro cell aging model, and 3 months and 20 months mouse lens capsular explants were used as ex vivo aging model. (A) FHL124 cells’ viability at low and high passage was measured by CCK8 after 24hr treatment with various concentration of RSL3. (B) 2- and 20-months old lens epithelium was treated with 0.5μM RSL3 for 24hr. The cell death/ ferroptosis was visualized by propidium iodide stain and the cell nucleus was visualized by Hoechst 3342 stain. Each assay was repeated three times and at least 10 images were collected for each stain and the representative images were shown. Unpaired two-tailed Student’s t-test analysis was used to compare between groups, and only p<0.05 is considered significant. *<0.05, **<0.01, ***<0.001, ****<0.0001.
3.7. Transcriptome analysis indicates that aged lens epithelial cells express pro-ferroptotic genes
To further explore the mechanisms of age-related sensitization of the lens epithelial cells to ferroptosis, we performed gene transcriptome analysis by comparing p15 and p41 FHL124 cells. Intriguingly, 18 pro-ferroptosis genes were significantly upregulated (>1.2-fold, p<0.05), and 11 anti-ferroptosis genes were significantly downregulated (>1.2-fold, p<0.05) in p41 FHL124 cells as compared to 15th passage (p15) (Fig. 7A). These genes have been reported to be directly or indirectly regulating pathways that impact the cell susceptibility to ferroptosis. They were clustered in two major pathways:
Figure 7.
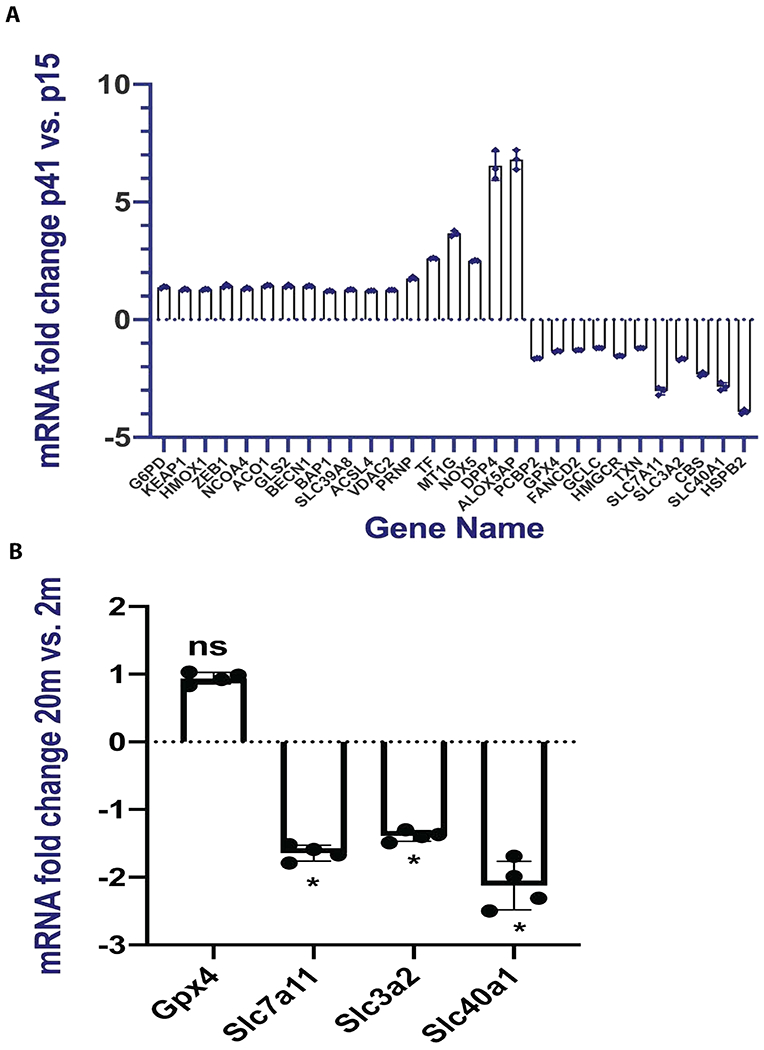
Gene transcriptome clusters of upregulated pro-ferroptosis and downregulated anti-ferroptosis genes in FHL124 aging model and aging mouse lens epithelium. (A) 18 pro-ferroptosis genes were upregulated and 11 anti-ferroptosis genes were downregulated in 41st passage vs. 15th passage FHL124 cells. All genes listed in the panel were significant with p value less than 0.05 and false discovery rate (FDR<0.1). (B) Real-time PCR analysis of 2- and 20-months old lens epithelium. Unpaired two-tailed Student’s t-test analysis was used to compare between groups, and only p<0.05 is considered significant. *<0.05, ns, not significant.
1). redox homeostasis.
P41 FHL124 cells had a mild but significant reduction of GPX4(1.4-fold) expression while also having increased NADPH oxidase NOX5 (2.5-fold) and arachidonate 5-lipoxygenase activating protein (ALOX5AP, 6.8-fold) expression in comparison to p15 cells. We also found greater KEAP1 mRNA expression in p41 FHL124 cells compared to p15, which suggested that the nuclear factor erythroid 2 (NRF2) activation might be suppressed. This was reflected by mild but significant downregulation of GCLC (1.2-fold, p<0.05), the downstream gene of NRF2. Interestingly, multiple genes involved in the cystine/glutamate antiporter system Xc− were found either up or down-regulated, which would impede intracellular cystine shuttling and GSH biosynthesis. For instance, both subunits of system Xc−, i.e., SLC7A11 (3-fold) and SLC3A2 (1.7-fold) were significantly downregulated in aged FHL124 cells as compared to lower passage cells. In addition, glutaminase 2 (GLS2, 1.4-fold), beclin 1 (BECN1, 1.4-fold), and BRCA1 associated protein 1 (BAP1, 1.3-fold) were upregulated in aged FHL124 cells compared to lower passage cells. These genes are known to either block system Xc− activity or repress Xc− subunit expression[51, 52].
2). iron homeostasis.
Iron trafficking and uptake related genes, such as transferrin (TF, 2.g-fold)[53], aconitase 1 (ACO1, 1.5-fold)[54], metallothionein 1G (MT1G, 3.7-fold)[55], nuclear receptor coactivator 4 (NCOA4, 1.4-fold)[56], zip transporter 8 (SLC39A8, 1.3-fold)[57], prion protein (PRNP, 1.7-fold)[58], and heme oxygenase-1 (HMOX1, 1.3-fold)[59] were all significantly upregulated in aged FHL124 cells relative to p15 cells. Conversely, iron export-related genes, such as ploy (RC) binding protein 2 (PCBP2, 1.7-fold)[60] and ferroportin (SLC40A1, 2.9-fold)[61] were significantly downregulated in aged FHL124 cells.
We also observed several genes that have been reported to dictate cell ferroptosis sensitivity. For example, the heat shock protein family B2(HSPB2, 4-fold) was significantly downregulated[62]. Interestingly, acyl-CoA synthetase long-chain family member 4 (ACSL4, 1.3-fold), a key gene responsible for enriching cellular membrane PUFA and also a key gene that regulates cell ferroptosis sensitivity[63], was mildly but significantly upregulated in aged FHL124 cells.
To validate our in vitro human lens epithelial cell aging model findings, we selected 4 key ferroptosis regulatory genes and compared their mRNA expression between 2- and 20-months old lens epithelium. As shown in Fig 7B, both system Xc− subunits Slc7a11 and Slc3a2 were significantly downregulated in 20-month-old lens epithelium compared to the 2-month-old. The iron exporter ferroportin (SLC40A1) was also significantly downregulated in 20-month vs 2-month-old lens epithelium. However, we did not see a remarkable Gox4 mRNA expression change when comparing 2-and 20-months old lens epithelium.
4. Discussion
Oxidation is recognized as one of the primary causes of age-related cataracts (ARC)[4, 6, 23]. This is reflected by an age-related decline of lens GSH concentration[6], decreased antioxidant defense system[16], increased protein disulfide crosslink, mixed disulfide formation[64–66], elevated lipid oxidation[67], and accumulated posttranslational protein modification[13]. Interestingly, whether the lens epithelial cells (LECs) undergo apoptosis in the mature and cataractous human lenses remains an enigma. Although, despite early report finds high levels of apoptosis in cataractous human lenses[68], following studies suggest otherwise[69–72]. It is fair to say that growing consensus indicates that minimal level of apoptosis is present in aged and cataractous human lenses despite the profound disruption of lens redox homeostasis, therefore raising the question of whether other cell death mechanisms besides apoptosis may exist in lens aging and cataractogenesis[69, 72]. In the present study, we revealed that LECs were highly susceptible to ferroptosis induced by either system Xc− inhibitor Erastin or GPX4 inhibitor RSL3 from both in vitro LECs culture and ex vivo lens epithelium explant culture system studies. The LECs’ ferroptosis was further sensitized when GSH de novo synthesis was disrupted. Intriguingly, we found that the LECs and lens epithelium manifested an age-associated increase of sensitivity to ferroptosis. Gene transcriptome analysis suggested that cluster genes orchestrated the disruption of redox homeostasis and iron homeostasis, which were primary mechanisms of ferroptosis. To the best of our knowledge, this is the first evidence that shows LECs are susceptible to ferroptosis and sensitizing during the aging process. This study has the following implications:
4.1. Aging lens meets all criteria of cell ferroptosis
The human lens membrane is enriched with lipids, including sphingolipids, e.g., dihydrosphingomyelin, and glycerophospholipids, e.g., phosphatidylethanolamine (PE) and phosphatidylcholine (PC)[67]. Substantial evidence supports the presence of lipid peroxidation in the human lens. Babizhayev [18] was able to detect lipid peroxidation products in the aqueous humor of samples obtained from patients with senile and complicated cataracts, while Borchman et al.[23] measured lipid oxidation via infrared spectroscopy in human lenses ranging from 1 to 85 years of age, and found that lipid oxidation increased linearly with age vs. total phospholipids. Hughes[73] and Huang et al.[25] reported a rapid age-related decline of lens glycerophospholipids content, especially phosphatidylethanolamine (PE)-like lipids, and suggested that the loss was because of the selective lipid oxidation of these higher unsaturated PUFA-glycerophospholipids. Of note, studies have indicated that lipid peroxidation of PUFA conjugated PE (PUFA-PE) is the most crucial phospholipid inducing cell ferroptosis[74, 75].
Both enzymatic and non-enzymatic mediated reactions can trigger lipid peroxidation. Lipoxygenases, such as 15-lipoxygenase (15-LOX) and 5-lipoxygenase (5-LOX), can catalyze lipid peroxidation and induce peroxyl radical formation[76]. 5-LOX activity is tightly regulated by the 5-LOX activating protein (ALOX15AP)[77]. Interestingly, we found an approximately 7-fold increase in ALOX15AP expression in aged LECs as opposed to early passage cells, which suggested that enzymatically catalyzed PUFA lipid oxidation is enhanced in aged LECs. However, more research is required to determine the intensity of lipoxygenase activity and its contribution to lipid peroxidation in aged LECs and lens epithelium relative to non-enzymatic mediated lipid peroxidation. The human lens frequently undergoes stresses such as UV light, smoking, and numerous other stressors in the circulating aqueous humor, e.g., hydrogen peroxide[78, 79] and cytokines[80, 81], which will either directly generate reactive oxygen species (ROS) or indirectly facilitate ROS production, especially during aging. Free radicals, such as hydroxy free radical (HOߦ), can initiate lipid peroxidation by abstracting electrons from PUFA, facilitating peroxyl radical formation. The highly reactive peroxyl radicals can promote autocatalytic propagation of chain reactions to produce a substantial amount of hydrogenperoxides, a telltale sign of ferroptosis[29].
The human lens is equipped with glutathione peroxidase (GPX) to cope with lipid peroxidation. GPX4, a phospholipid hydroperoxidase, is considered the only selenoperoxidase to detoxify lipid peroxidation, while GPX1 is believed to detoxify hydrogen peroxide[82]. Interestingly, we found a ~1.4-fold reduction of GPX4 mRNA expression in aged human LECs, but no remarkable change was seen in mouse lens epithelium between 2- and 20- months of age. Most importantly, the GPX activity was reported to be profoundly decreased in cataractous human lens compared to healthy individuals[83]. Markedly, GPX4 reduces lipid hydrogenperoxides at the expense of GSH, and GPX activity has been found to be closely correlated with GSH concentrations in human lenses[83]. Our previous study found a significant decrease in GPX activity when we compared 9-months vs. 1-month old mouse lenses[8]. So, in the scenario of an aged human lens, a remarkably decreased GSH concentration would further impede GPX4 function, which will hasten lipid peroxidation. Impaired GPX4 activity is another signifier of ferroptosis[29].
Iron is an essential element and the most abundant transition metal in the human body. Iron homeostasis is tightly regulated, and disruption of iron homeostasis has been found to be directly involved in various diseases[84, 85]. An age-associated increase in iron accumulation was reported in rabbit lenses[86], and several reports showed increased iron content in age-related cataracts compared to healthy individuals[9, 87]. Smoking has also been found to be involved in increasing the lens iron and calcium content[88]. Our present study found several folds increase of the intracellular iron content in FHL124 cells after Erastin and RSL3 treatment. The increased iron content might come from increased iron uptake from the medium or iron storage protein degradation, i.e., ferritin. This was also echoed by our transcriptome analysis that cluster genes were up-or down-regulated in aged human LECs and mouse lens epithelium to increase intracellular iron trafficking while preventing cellular iron export. Garner et al. [9] found that the level of redox-active iron, also known as labile iron pool (LIP), was significantly higher in cataractous lens compared to non-cataractous lenses. LIP has been revealed to promote ROS formation, such as hydroxyl free radical formation via Fenton-like reactions. Interestingly, the human lens has an unusually high concentration (3-5mM) of vitamin C (ascorbic acid, ASA)[12], and accumulative redox-active iron can also switch ASA’s antioxidative role to its pro-oxidative role[13]. ASA can readily reduce ferric (Fe3+) ions to ferrous (Fe2+) irons while oxidizing itself into dehydroascorbic acid (DHA) (see reaction 1). The ferrous (Fe2+) iron can then convert hydrogen peroxide into hydroxy free radical (HO•) (see reaction 2).
As we and others have demonstrated, ASA oxidation is a common phenomenon in the aging human lens and brain[13, 89]. The available redox-active metals in the tissue can significantly amplify ASA oxidation[90]. Accumulation of redox-active iron is another clear sign of ferroptosis[29].
In sum, an aged human lens can be identified with increased lipid peroxidation, impaired GPX activity, and accumulative redox-active iron, which are three hallmarks of ferroptosis. Needless to say, the aged human lens possesses a unique condition that is more susceptible to ferroptosis than any other organ in the human body.
4.2. Does ferroptosis occur in aged and cataractous lens?
The obvious question here is whether ferroptosis is occurring in the human lens during aging and cataractogenesis. There are no specific biomarkers available that allow explicit verification of ferroptosis in vivo. However, compelling evidence, such as Alzheimer’s disease (AD)[91], Huntington’s disease (HD)[92], and ischemia-reperfusion (I/R) related tissue injuries[93], supports the existence of ferroptosis in vivo reflected by their elevated lipid peroxidation, accumulative redox-active iron, and neuronal tissue degeneration.
Similarly, sufficient evidence of elevated lipid peroxidation, impaired GPX activity, and accumulative redox-active iron taking place in aged and cataractous human lenses strongly support the existence of ferroptosis during lens aging and cataractogenesis. First, growing evidence indicates a strong association between increased lipid peroxidation and age-related cataract formation[20–24]. Babizhayev[18] reported that intravitreal administration of phospholipid lipid peroxides induced posterior subcapsular cataract in a rabbit model study. Mounting evidence has led researchers in the field to postulate that lipid peroxidation is playing a pathophysiological role in the initial phase of cataractogenesis[24]. Second, iron overload induces cataract formation. For example, patients with ocular siderosis, regarded as excessive iron deposition in the ocular tissue by iron-containing foreign bodies, develop cataracts[94]. Third, low serum selenium levels, the essential element for GPX catalytic function, have been significantly associated with both nuclear and cortical senile cataracts[95]. Conversely, Dai et al.[96] found that selenite could attenuate galactose induced sugar cataracts in a rat model.
Our present study provides crucial evidence that LECs and the epithelium are susceptible to Erastin and RSL3 induced ferroptosis in an age-related manner. Furthermore, gene transcriptome analysis demonstrates that aging LECs display remarkable changes affecting intracellular GSH homeostasis, GPX4, and iron content, which will favor lipid peroxidation, a critical component of ferroptosis. However, we are fully aware that we cannot explicitly conclude the existence of ferroptosis in the aged or cataractous human lens without a specific ferroptosis biomarker. Moreover, the present study has focused on ferroptosis in LECs. The relevance of LECs’ ferroptosis to age-related cataracts, particular age-related nuclear cataracts, remains to be elucidated. Arguably, lens fiber cells may be more susceptible to ferroptosis. Fiber cells are tightly packed, therefore, are enriched with plasma membrane containing PUFA-glycerophospholipids that are readily available subjecting to oxidation. Furthermore, age-related decline of lens GSH has a more profound impact on the lens nucleus than epithelium[4], which is expected to impair GPX4 activity in detoxifying the lipid peroxidation. More studies are needed to delineate the existence of ferroptosis during lens aging and cataract formation. For instance, understanding the role of GPX4 in cataractogenesis will provide us valuable information regarding lipid peroxidation, ferroptosis, and cataractogenesis.
4.3. Potential therapeutic approach for posterior capsule opacification (PCO)
Ferroptosis is a unique type of regulated cell death mechanism. Its discovery was originated from cancer therapy and was considered a novel approach to preventing tumor growth. Interestingly, studies have suggested that tumor cells in their mesenchymal state are more sensitive to RSL3 and Erastin induced ferroptosis[97]. Our discovery of the LECs’ high sensitivity to ferroptosis may also be a therapeutic approach to the posterior capsule opacification (PCO), the most common post-cataract surgery complication, also known as the secondary cataract. For cataract patients, surgery is currently the only treatment option. During cataract surgery, the anterior lens capsule is removed via capsulorhexis followed by phacoemulsification to remove the fiber mass before intraocular lens (IOL) implantation. However, there will always be some residual lens epithelial cells left during the surgery, and these LECs often survive, proliferate, migrate, and differentiate at the posterior capsule. This results in light blockage and capsular wrinkling. Compelling evidence suggests that lens epithelial-mesenchymal transition (EMT) plays a crucial role in the pathogenesis of PCO formation[98, 99]. There is, to date, no satisfying pharmacological PCO intervention available. The highly activated EMT process during initiation and progression of PCO and the significant vulnerability of LECs to ferroptosis inducing reagents let us speculate that inducing ferroptosis may be a novel therapeutic approach to preventing lens epithelial cell survival and PCO formation. Future study is required to test the efficacy of ferroptosis inducing drugs and their potential toxicity to other ocular tissues.
Taken all together, this is the first time that LECs have been shown to be highly susceptible to Erastin and RSL3 induced ferroptosis, and the sensitivity is closely associated with aging. The aged and cataractous human lens may manifest possibly more hallmarks of ferroptosis including more profound lipid peroxidation, impaired GPX activity, accumulative redox-active iron, and disrupted GSH homeostasis than any other organ in the human body.
Supplementary Material
Highlights.
Lens epithelial cells (LECs) are highly susceptible to ferroptosis
Disruption of glutathione homeostases exacerbates LECs ferroptosis
The aging lens sensitizes the LECs’ susceptibility to ferroptosis
Acknowledgement
This research was supported by grants from EY028158(XF) and NEI Center Core Grant for Vision Research (P30EY031631) at Augusta University. We are very grateful to Rachel Cui at Augusta University Cell Imaging Core for helping in microscopy image collections. We are very grateful to Dr. Brendan Marshall, Tania Green, and Libby Perry at Augusta University Electron Microscopy and Histology Core for their help in TEM experiment.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of competing interest
ZW, CH, JH, RS, XZ, and XF declare they have no conflict of interest.
References
- [1].Congdon N, Vingerling JR, Klein BE, West S, Friedman DS, Kempen J, O’Colmain B, Wu SY, Taylor HR, G. Eye Diseases Prevalence Research, Prevalence of cataract and pseudophakia/aphakia among adults in the United States, Arch Ophthalmol 122(4) (2004) 487–94. [DOI] [PubMed] [Google Scholar]
- [2].Truscott RJ, Age-related nuclear cataract-oxidation is the key, Exp Eye Res 80(5) (2005) 709–25. [DOI] [PubMed] [Google Scholar]
- [3].Beebe DC, Holekamp NM, Shui YB, Oxidative damage and the prevention of age-related cataracts, Ophthalmic Res 44(3) (2010) 155–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Lou MF, Redox regulation in the lens, Prog Retin Eye Res 22(5) (2003) 657–82. [DOI] [PubMed] [Google Scholar]
- [5].Sethna SS, Holleschau AM, Rathbun WB, Activity of glutathione synthesis enzymes in human lens related to age, Curr Eye Res 2(11) (1982) 735–42. [DOI] [PubMed] [Google Scholar]
- [6].Giblin FJ, Glutathione: a vital lens antioxidant, J Ocul Pharmacol Ther 16(2) (2000) 121–35. [DOI] [PubMed] [Google Scholar]
- [7].Sweeney MH, Truscott RJ, An impediment to glutathione diffusion in older normal human lenses: a possible precondition for nuclear cataract, Exp Eye Res 67(5) (1998) 587–95. [DOI] [PubMed] [Google Scholar]
- [8].Fan X, Liu X, Hao S, Wang B, Robinson ML, Monnier VM, The LEGSKO mouse: a mouse model of age-related nuclear cataract based on genetic suppression of lens glutathione synthesis, PLoS One 7(11) (2012) e50832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Garner B, Roberg K, Qian M, Eaton JW, Truscott RJ, Distribution of ferritin and redox-active transition metals in normal and cataractous human lenses, Exp Eye Res 71(6) (2000) 599–607. [DOI] [PubMed] [Google Scholar]
- [10].Chen Q, Espey MG, Sun AY, Lee JH, Krishna MC, Shacter E, Choyke PL, Pooput C, Kirk KL, Buettner GR, Levine M, Ascorbate in pharmacologic concentrations selectively generates ascorbate radical and hydrogen peroxide in extracellular fluid in vivo, Proc Natl Acad Sci U S A 104(21) (2007) 8749–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].ANTHONY EV NAPPI J, Hydroxyl Radical Production by Ascorbate and Hydrogen Peroxide Neurotoxicity Researc 2 (2000) 343–355. [Google Scholar]
- [12].Fan X, Reneker LW, Obrenovich ME, Strauch C, Cheng R, Jarvis SM, Ortwerth BJ, Monnier VM, Vitamin C mediates chemical aging of lens crystallins by the Maillard reaction in a humanized mouse model, Proc Natl Acad Sci U S A 103(45) (2006) 16912–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Fan X, Sell DR, Hao C, Liu S, Wang B, Wesson DW, Siedlak S, Zhu X, Kavanagh TJ, Harrison FE, Monnier VM, Vitamin C is a source of oxoaldehyde and glycative stress in age-related cataract and neurodegenerative diseases, Aging Cell 19(7) (2020) e13176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Fan X, Sell DR, Zhang J, Nemet I, Theves M, Lu J, Strauch C, Halushka MK, Monnier VM, Anaerobic vs aerobic pathways of carbonyl and oxidant stress in human lens and skin during aging and in diabetes: A comparative analysis, Free Radic Biol Med 49(5) (2010) 847–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Ahmed N, Thornalley PJ, Dawczynski J, Franke S, Strobel J, Stein G, Haik GM, Methylglyoxal-derived hydroimidazolone advanced glycation end-products of human lens proteins, Invest Ophthalmol Vis Sci 44(12) (2003) 5287–92. [DOI] [PubMed] [Google Scholar]
- [16].Xing KY, Lou MF, Effect of age on the thioltransferase (glutaredoxin) and thioredoxin systems in the human lens, Invest Ophthalmol Vis Sci 51(12) (2010) 6598–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Ohrloff C, Hockwin O, Olson R, Dickman S, Glutathione peroxidase, glutathione reductase and superoxide dismutase in the aging lens, Curr Eye Res 3(1) (1984) 109–15. [DOI] [PubMed] [Google Scholar]
- [18].Babizhayev MA, Failure to withstand oxidative stress induced by phospholipid hydroperoxides as a possible cause of the lens opacities in systemic diseases and ageing, Biochim Biophys Acta 1315(2) (1996) 87–99. [DOI] [PubMed] [Google Scholar]
- [19].Reddy VN, Giblin FJ, Lin LR, Dang L, Unakar NJ, Musch DC, Boyle DL, Takemoto LJ, Ho YS, Knoernschild T, Juenemann A, Lutjen-Drecoll E, Glutathione peroxidase-1 deficiency leads to increased nuclear light scattering, membrane damage, and cataract formation in gene-knockout mice, Invest Ophthalmol Vis Sci 42(13) (2001) 3247–55. [PubMed] [Google Scholar]
- [20].Bhuyan KC, Master RW, Coles RS, Bhuyan DK, Molecular mechanisms of cataractogenesis: IV. Evidence of phospholipid . malondialdehyde adduct in human senile cataract, Mech Ageing Dev 34(3) (1986) 289–96. [DOI] [PubMed] [Google Scholar]
- [21].Micelli-Ferrari T, Vendemiale G, Grattagliano I, Boscia F, Arnese L, Altomare E, Cardia L, Role of lipid peroxidation in the pathogenesis of myopic and senile cataract, Br J Ophthalmol 80(9) (1996) 840–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Huang L, Estrada R, Yappert MC, Borchman D, Oxidation-induced changes in human lens epithelial cells. 1. Phospholipids, Free Radic Biol Med 41(9) (2006) 1425–32. [DOI] [PubMed] [Google Scholar]
- [23].Borchman D, Yappert MC, Age-related lipid oxidation in human lenses, Invest Ophthalmol Vis Sci 39(6) (1998) 1053–8. [PubMed] [Google Scholar]
- [24].Babizhayev MA, Deyev AI, Linberg LF, Lipid peroxidation as a possible cause of cataract, Mech Ageing Dev 44(1) (1988) 69–89. [DOI] [PubMed] [Google Scholar]
- [25].Huang L, Grami V, Marrero Y, Tang D, Yappert MC, Rasi V, Borchman D, Human lens phospholipid changes with age and cataract, Invest Ophthalmol Vis Sci 46(5) (2005) 1682–9. [DOI] [PubMed] [Google Scholar]
- [26].Kreuzer M, Ducic T, Hawlina M, Andjelic S, Synchrotron-based FTIR microspectroscopy of protein aggregation and lipids peroxidation changes in human cataractous lens epithelial cells, Sci Rep 10(1) (2020) 15489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Borchman D, Lipid Conformational Order and the Etiology of Cataract and Dry Eye, J Lipid Res (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS, Morrison B 3rd, Stockwell BR, Ferroptosis: an iron-dependent form of nonapoptotic cell death, Cell 149(5) (2012) 1060–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].S.J.D.a.B.R. Stockwell, The Hallmarks of Ferroptosis, Annu. Rev. Cancer Biol. 3 (2019) 35–54. [Google Scholar]
- [30].Tang D, Kang R, Berghe TV, Vandenabeele P, Kroemer G, The molecular machinery of regulated cell death, Cell Res 29(5) (2019) 347–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Reichert CO, de Freitas FA, Sampaio-Silva J, Rokita-Rosa L, Barros PL, Levy D, Bydlowski SP, Ferroptosis Mechanisms Involved in Neurodegenerative Diseases, Int J Mol Sci 21(22) (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Friedmann Angeli JP, Schneider M, Proneth B, Tyurina YY, Tyurin VA, Hammond VJ, Herbach N, Aichler M, Walch A, Eggenhofer E, Basavarajappa D, Radmark O, Kobayashi S, Seibt T, Beck H, Neff F, Esposito I, Wanke R, Forster H, Yefremova O, Heinrichmeyer M, Bornkamm GW, Geissler EK, Thomas SB, Stockwell BR, O’Donnell VB, Kagan VE, Schick JA, Conrad M, Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice, Nat Cell Biol 16(12) (2014) 1180–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Jiang X, Stockwell BR, Conrad M, Ferroptosis: mechanisms, biology and role in disease, Nat Rev Mol Cell Biol (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Bebber CM, Muller F, Prieto Clemente L, Weber J, von Karstedt S, Ferroptosis in Cancer Cell Biology, Cancers (Basel) 12(1) (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Stockwell BR, Jiang X, Gu W, Emerging Mechanisms and Disease Relevance of Ferroptosis, Trends Cell Biol 30(6) (2020) 478–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Ursini F, Maiorino M, Lipid peroxidation and ferroptosis: The role of GSH and GPx4, Free Radic Biol Med 152 (2020) 175–185. [DOI] [PubMed] [Google Scholar]
- [37].Lei P, Bai T, Sun Y, Mechanisms of Ferroptosis and Relations With Regulated Cell Death: A Review, Front Physiol 10 (2019) 139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Reddan LCB J.R., Hitt AL, Bagchi M, Raphtis EM, Pena JT, Dziedzic DC, Generation of two non-transfected human lens cell lines., Invest. Ophthalmol. Vis. Sci 40 (1999) S970–S970. [Google Scholar]
- [39].Cole TB, Giordano G, Co AL, Mohar I, Kavanagh TJ, Costa LG, Behavioral Characterization of GCLM-Knockout Mice, a Model for Enhanced Susceptibility to Oxidative Stress, J Toxicol 2011 (2011) 157687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Wei Z, Caty J, Whitson J, Zhang AD, Srinivasagan R, Kavanagh TJ, Yan H, Fan X, Reduced Glutathione Level Promotes Epithelial-Mesenchymal Transition in Lens Epithelial Cells via a Wnt/beta-Catenin-Mediated Pathway: Relevance for Cataract Therapy, Am J Pathol 187(11) (2017) 2399–2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Rahman I, Kode A, Biswas SK, Assay for quantitative determination of glutathione and glutathione disulfide levels using enzymatic recycling method, Nat Protoc 1(6) (2006) 3159–3165. [DOI] [PubMed] [Google Scholar]
- [42].Chen H, Li Y, Tollefsbol TO, Cell senescence culturing methods, Methods Mol Biol 1048 (2013) 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Phipps SM, Berletch JB, Andrews LG, Tollefsbol TO, Aging cell culture: methods and observations, Methods Mol Biol 371 (2007) 9–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Whitson JA, Zhang X, Medvedovic M, Chen J, Wei Z, Monnier VM, Fan X, Transcriptome of the GSH-Depleted Lens Reveals Changes in Detoxification and EMT Signaling Genes, Transport Systems, and Lipid Homeostasis, Invest Ophthalmol Vis Sci 58(5) (2017) 2666–2684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Bustin SA, Benes V, Garson JA, Hellemans J, Huggett J, Kubista M, Mueller R, Nolan T, Pfaffl MW, Shipley GL, Vandesompele J, Wittwer CT, The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments, Clin Chem 55(4) (2009) 611–22. [DOI] [PubMed] [Google Scholar]
- [46].Drummen GP, van Liebergen LC, Op den Kamp JA, Post JA, C11-BODIPY(581/591), an oxidation-sensitive fluorescent lipid peroxidation probe: (micro)spectroscopic characterization and validation of methodology, Free Radic Biol Med 33(4) (2002) 473–90. [DOI] [PubMed] [Google Scholar]
- [47].Zhang Y, Koppula P, Gan B, Regulation of H2A ubiquitination and SLC7A11 expression by BAP1 and PRC1, Cell Cycle 18(8) (2019) 773–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Wenzel SE, Tyurina YY, Zhao J, St Croix CM, Dar HH, Mao G, Tyurin VA, Anthonymuthu TS, Kapralov AA, Amoscato AA, Mikulska-Ruminska K, Shrivastava IH, Kenny EM, Yang Q, Rosenbaum JC, Sparvero LJ, Emlet DR, Wen X, Minami Y, Qu F, Watkins SC, Holman TR, VanDemark AP, Kellum JA, Bahar I, Bayir H, Kagan VE, PEBP1 Wardens Ferroptosis by Enabling Lipoxygenase Generation of Lipid Death Signals, Cell 171(3) (2017) 628–641 e26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Franklin CC, Krejsa CM, Pierce RH, White CC, Fausto N, Kavanagh TJ, Caspase-3-Dependent Cleavage of the Glutamate-L-Cysteine Ligase Catalytic Subunit during Apoptotic Cell Death, Am J Pathol 160(5) (2002) 1887–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Wang H, Liu C, Zhao Y, Gao G, Mitochondria regulation in ferroptosis, Eur J Cell Biol 99(1) (2020) 151058. [DOI] [PubMed] [Google Scholar]
- [51].Lin X, Ping J, Wen Y, Wu Y, The Mechanism of Ferroptosis and Applications in Tumor Treatment, Front Pharmacol 11 (2020) 1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Kang R, Zhu S, Zeh HJ, Klionsky DJ, Tang D, BECN1 is a new driver of ferroptosis, Autophagy 14(12) (2018) 2173–2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Kawabata H, Transferrin and transferrin receptors update, Free Radic Biol Med 133 (2019) 46–54. [DOI] [PubMed] [Google Scholar]
- [54].Martelli A, Salin B, Dycke C, Louwagie M, Andrieu JP, Richaud P, Moulis JM, Folding and turnover of human iron regulatory protein 1 depend on its subcellular localization, FEBS J 274(4) (2007) 1083–92. [DOI] [PubMed] [Google Scholar]
- [55].Orihuela R, Fernandez B, Palacios O, Valero E, Atrian S, Watt RK, Dominguez-Vera JM, Capdevila M, Ferritin and metallothionein: dangerous liaisons, Chem Commun (Camb) 47(44) (2011) 12155–7. [DOI] [PubMed] [Google Scholar]
- [56].Santana-Codina N, Mancias JD, The Role of NCOA4-Mediated Ferritinophagy in Health and Disease, Pharmaceuticals (Basel) 11(4) (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Wang CY, Jenkitkasemwong S, Duarte S, Sparkman BK, Shawki A, Mackenzie B, Knutson MD, ZIP8 is an iron and zinc transporter whose cell-surface expression is up-regulated by cellular iron loading, J Biol Chem 287(41) (2012) 34032–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Singh A, Haldar S, Horback K, Tom C, Zhou L, Meyerson H, Singh N, Prion protein regulates iron transport by functioning as a ferrireductase, J Alzheimers Dis 35(3) (2013) 541–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Fraser ST, Midwinter RG, Berger BS, Stocker R, Heme Oxygenase-1: A Critical Link between Iron Metabolism, Erythropoiesis, and Development, Adv Hematol 2011 (2011) 473709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Frey AG, Nandal A, Park JH, Smith PM, Yabe T, Ryu MS, Ghosh MC, Lee J, Rouault TA, Park MH, Philpott CC, Iron chaperones PCBP1 and PCBP2 mediate the metallation of the dinuclear iron enzyme deoxyhypusine hydroxylase, Proc Natl Acad Sci U S A 111(22) (2014) 8031–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Drakesmith H, Nemeth E, Ganz T, Ironing out Ferroportin, Cell Metab 22(5) (2015) 777–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Rui Kang DT, Heat Shock Proteins: Endogenous Modulators of Ferroptosis, Ferroptosis in Health and Disease (2019) pp 61–68. [Google Scholar]
- [63].Doll S, Proneth B, Tyurina YY, Panzilius E, Kobayashi S, Ingold I, Irmler M, Beckers J, Aichler M, Walch A, Prokisch H, Trumbach D, Mao G, Qu F, Bayir H, Fullekrug J, Scheel CH, Wurst W, Schick JA, Kagan VE, Angeli JP, Conrad M, ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition, Nat Chem Biol 13(1) (2017) 91–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Wang B, Hom G, Zhou S, Guo M, Li B, Yang J, Monnier VM, Fan X, The oxidized thiol proteome in aging and cataractous mouse and human lens revealed by ICAT labeling, Aging Cell 16(2) (2017) 244–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Fan X, Zhou S, Wang B, Hom G, Guo M, Li B, Yang J, Vaysburg D, Monnier VM, Evidence of Highly Conserved beta-Crystallin Disulfidome that Can be Mimicked by In Vitro Oxidation in Age-related Human Cataract and Glutathione Depleted Mouse Lens, Mol Cell Proteomics 14(12) (2015) 3211–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Wang GM, Raghavachari N, Lou MF, Relationship of protein-glutathione mixed disulfide and thioltransferase in H2O2-induced cataract in cultured pig lens, Exp Eye Res 64(5) (1997) 693–700. [DOI] [PubMed] [Google Scholar]
- [67].Borchman D, Yappert MC, Lipids and the ocular lens, J Lipid Res 51(9) (2010) 2473–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Li WC, Kuszak JR, Dunn K, Wang RR, Ma W, Wang GM, Spector A, Leib M, Cotliar AM, Weiss M, et al. , Lens epithelial cell apoptosis appears to be a common cellular basis for non-congenital cataract development in humans and animals, J Cell Biol 130(1) (1995) 169–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Harocopos GJ, Alvares KM, Kolker AE, Beebe DC, Human age-related cataract and lens epithelial cell death, Invest Ophthalmol Vis Sci 39(13) (1998) 2696–706. [PubMed] [Google Scholar]
- [70].Andersson M, Honarvar A, Sjostrand J, Peterson A, Karlsson JO, Decreased caspase-3 activity in human lens epithelium from posterior subcapsular cataracts, Exp Eye Res 76(2) (2003) 175–82. [DOI] [PubMed] [Google Scholar]
- [71].Charakidas A, Kalogeraki A, Tsilimbaris M, Koukoulomatis P, Brouzas D, Delides G, Lens epithelial apoptosis and cell proliferation in human age-related cortical cataract, Eur J Ophthalmol 15(2) (2005) 213–20. [DOI] [PubMed] [Google Scholar]
- [72].Osnes-Ringen O, Berg KH, Moe MC, Zetterstrom C, Roger M, Nicolaissen B, Cell death pattern in lens epithelium of cataract patients, Acta Ophthalmol 94(5) (2016) 514–20. [DOI] [PubMed] [Google Scholar]
- [73].Hughes JR, Deeley JM, Blanksby SJ, Leisch F, Ellis SR, Truscott RJ, Mitchell TW, Instability of the cellular lipidome with age, Age (Dordr) 34(4) (2012) 935–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Yang WS, Stockwell BR, Ferroptosis: Death by Lipid Peroxidation, Trends Cell Biol 26(3) (2016) 165–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Li J, Cao F, Yin HL, Huang ZJ, Lin ZT, Mao N, Sun B, Wang G, Ferroptosis: past, present and future, Cell Death Dis 11(2) (2020) 88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Mashima R, Okuyama T, The role of lipoxygenases in pathophysiology; new insights and future perspectives, Redox Biol 6 (2015) 297–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Byrum RS, Goulet JL, Griffiths RJ, Koller BH, Role of the 5-lipoxygenase-activating protein (FLAP) in murine acute inflammatory responses, J Exp Med 185(6) (1997) 1065–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Spector A, Garner WH, Hydrogen peroxide and human cataract, Exp Eye Res 33(6) (1981) 673–81. [DOI] [PubMed] [Google Scholar]
- [79].Garcia-Castineiras S, Velazquez S, Martinez P, Torres N, Aqueous humor hydrogen peroxide analysis with dichlorophenol-indophenol, Exp Eye Res 55(1) (1992) 9–19. [DOI] [PubMed] [Google Scholar]
- [80].Kwon JW, Jee D, Aqueous humor cytokine levels in patients with diabetic macular edema refractory to anti-VEGF treatment, PLoS One 13(9) (2018) e0203408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Sato T, Takeuchi M, Karasawa Y, Takayama K, Enoki T, Comprehensive expression patterns of inflammatory cytokines in aqueous humor of patients with neovascular age-related macular degeneration, Sci Rep 9(1) (2019) 19447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Walshe J, Serewko-Auret MM, Teakle N, Cameron S, Minto K, Smith L, Burcham PC, Russell T, Strutton G, Griffin A, Chu FF, Esworthy S, Reeve V, Saunders NA, Inactivation of glutathione peroxidase activity contributes to UV-induced squamous cell carcinoma formation, Cancer Res 67(10) (2007) 4751–8. [DOI] [PubMed] [Google Scholar]
- [83].Bojana Kisic DM, Lepsa Zoric and Aleksandra Ilic, Role of Lipid Peroxidation in the Pathogenesis of Age-Related Cataract, Lipid Peroxidation (2012). [Google Scholar]
- [84].Gozzelino R, Arosio P, Iron Homeostasis in Health and Disease, Int J Mol Sci 17(1) (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Davidson MG, Harned J, Grimes AM, Duncan G, Wormstone IM, McGahan MC, Transferrin in after-cataract and as a survival factor for lens epithelium, Exp Eye Res 66(2) (1998) 207–15. [DOI] [PubMed] [Google Scholar]
- [86].Ishii M, Kaji K, [Studies on metal (Zn, Cu, Fe) kinetics for ageing phenomena on lens], Nihon Ronen Igakkai Zasshi 22(6) (1985) 522–9. [DOI] [PubMed] [Google Scholar]
- [87].Dawczynski J, Blum M, Winnefeld K, Strobel J, Increased content of zinc and iron in human cataractous lenses, Biol Trace Elem Res 90(1-3) (2002) 15–23. [DOI] [PubMed] [Google Scholar]
- [88].Avunduk AM, Yardimci S, Avunduk MC, Kurnaz L, Kockar MC, Determinations of some trace and heavy metals in rat lenses after tobacco smoke exposure and their relationships to lens injury, Exp Eye Res 65(3) (1997) 417–23. [DOI] [PubMed] [Google Scholar]
- [89].Ortwerth BJ, Linetsky M, Olesen PR, Ascorbic acid glycation of lens proteins produces UVA sensitizers similar to those in human lens, Photochem Photobiol 62(3) (1995) 454–62. [DOI] [PubMed] [Google Scholar]
- [90].Du J, Cullen JJ, Buettner GR, Ascorbic acid: chemistry, biology and the treatment of cancer, Biochim Biophys Acta 1826(2) (2012) 443–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Raven EP, Lu PH, Tishler TA, Heydari P, Bartzokis G, Increased iron levels and decreased tissue integrity in hippocampus of Alzheimer’s disease detected in vivo with magnetic resonance imaging, J Alzheimers Dis 37(1) (2013) 127–36. [DOI] [PubMed] [Google Scholar]
- [92].Chen J, Marks E, Lai B, Zhang Z, Duce JA, Lam LQ, Volitakis I, Bush AI, Hersch S, Fox JH, Iron accumulates in Huntington’s disease neurons: protection by deferoxamine, PLoS One 8(10) (2013) e77023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Fang X, Wang H, Han D, Xie E, Yang X, Wei J, Gu S, Gao F, Zhu N, Yin X, Cheng Q, Zhang P, Dai W, Chen J, Yang F, Yang HT, Linkermann A, Gu W, Min J, Wang F, Ferroptosis as a target for protection against cardiomyopathy, Proc Natl Acad Sci U S A 116(7) (2019) 2672–2680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Hope-Ross M, Mahon GJ, Johnston PB, Ocular siderosis, Eye (Lond) 7 ( Pt 3) (1993) 419–25. [DOI] [PubMed] [Google Scholar]
- [95].Post M, Lubinski W, Lubinski J, Krzystolik K, Baszuk P, Muszynska M, Marciniak W, Serum selenium levels are associated with age-related cataract, Ann Agric Environ Med 25(3) (2018) 443–448. [DOI] [PubMed] [Google Scholar]
- [96].Dai J, Zhou J, Liu H, Huang K, Selenite and ebselen supplementation attenuates D-galactose-induced oxidative stress and increases expression of SELR and SEP15 in rat lens, J Biol Inorg Chem 21(8) (2016) 1037–1046. [DOI] [PubMed] [Google Scholar]
- [97].Viswanathan VS, Ryan MJ, Dhruv HD, Gill S, Eichhoff OM, Seashore-Ludlow B, Kaffenberger SD, Eaton JK, Shimada K, Aguirre AJ, Viswanathan SR, Chattopadhyay S, Tamayo P, Yang WS, Rees MG, Chen S, Boskovic ZV, Javaid S, Huang C, Wu X, Tseng YY, Roider EM, Gao D, Cleary JM, Wolpin BM, Mesirov JP, Haber DA, Engelman JA, Boehm JS, Kotz JD, Hon CS, Chen Y, Hahn WC, Levesque MP, Doench JG, Berens ME, Shamji AF, Clemons PA, Stockwell BR, Schreiber SL, Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway, Nature 547(7664) (2017) 453–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].de Iongh RU, Wederell E, Lovicu FJ, McAvoy JW, Transforming growth factor-beta-induced epithelial-mesenchymal transition in the lens: a model for cataract formation, Cells Tissues Organs 179(1-2) (2005) 43–55. [DOI] [PubMed] [Google Scholar]
- [99].Eldred JA, Dawes LJ, Wormstone IM, The lens as a model for fibrotic disease, Philos Trans R Soc Lond B Biol Sci 366(1568) (2011) 1301–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.