

Abstract
Background:
The selective reduction of memory Th2 responses could be key to affording tolerance and protection from the reoccurrence of damaging allergic pathology.
Objective:
We asked whether TNF family costimulatory molecules co-operated to promote accumulation and reactivity of effector memory CD4 T cells to inhaled complex allergen, and if their neutralization could promote airway tolerance to subsequent re-exposure to allergen.
Methods:
Mice were sensitized intraperitoneally or intranasally with house dust mite (HDM), and challenged with intranasal allergen after memory had developed. We assessed whether single or combined blockade of OX40L/CD252 and CD30L/CD153 inhibited memory T cells from driving acute asthmatic lung inflammation, and protected mice upon exposure to allergen at a later time.
Results:
OX40- or CD30-deficient animals showed strong or partial protection against allergic airway inflammation, however neutralizing either molecule alone during the secondary response to allergen had little effect on the frequency of effector memory CD4 T cells formed and acute lung inflammation. In contrast, a significant reduction in eosinophilic inflammation was observed when OX40L and CD30L were simultaneously neutralized, with dual blockade inhibiting effector memory Th2 expansion in the lungs while formation of peripherally-induced Treg remained intact. Moreover, dual blockade during the secondary response resulted in a tolerogenic state such that mice did not develop a normal tertiary memory Th2 cell and lung inflammatory response when challenged weeks later with allergen.
Conclusion:
Memory T cell responses to complex allergens are controlled by several TNF costimulatory interactions, and their combination targeting might represent a strategy to reduce the severity of inflammatory reactions upon re-exposure to allergen.
Keywords: allergen, Th2, TNF family, OX40L, CD30L, memory, asthma
Graphical Abstract

Capsule Summary:
We report that combination targeting of two TNF family costimulatory molecules, OX40L and CD30L, can suppress memory Th2 driven acute allergic lung inflammation and reduce the severity of subsequent responses to inhaled allergen.
Introduction
T cell tolerance in the lungs is important for normal homeostasis, but tolerance also may limit development or maintenance of airway inflammatory diseases such as asthma. 300 million people worldwide suffer from asthma, including ~25 million Americans, and it is estimated that ~65–75% of asthma is driven by adaptive immune responses to allergens. While efforts to block immune-driven allergic lung inflammation are increasing, they have resulted in only moderate success clinically with a small number of biologics approved to date. These target effector molecules (e.g. omalizumab against IgE, and mepolizumab against IL-5) or effector cells (benralizumab against IL-5R for depleting eosinophils). Importantly, these biologics are unlikely to affect persistence of memory Th2 cell populations that are acknowledged to drive and maintain asthmatic lung inflammation1–4, or to alter the ratio of effector memory T cells to CD4+ Treg that can recognize allergen which may be a desired outcome of any effective asthma therapy5, 6. Recent findings of asthmatics with Th1 and Th17 signatures7–9 suggest that immune deviation is less likely a mechanism that can lead to allergic tolerance, implying that focusing on a reduction in the frequency of any effector memory T cell population and/or enhancing Treg numbers is favorable. Periodic and long-term exposure to allergens is used as a current tolerogenic therapy (SCIT/SLIT), and data from these trials have substantiated the idea that deletion, or reduction in numbers, of pathogenic CD4 T cells, and expansion or a bias towards more Treg, are important10–13. The newly approved biologic dupilumab (against IL-4Rα) does have the potential to modify or limit Th2 differentiation driven by IL-4. However, other strategies and targets that may afford specific hypo-responsiveness in the T cell compartment and prevent recurrence of allergic lung inflammation are needed. A major hurdle for therapy though is the notion that memory T cells (i.e. those present in subjects with ongoing disease) are difficult to inhibit, potentially explaining why current approaches may not be universally successful.
Costimulatory molecules in the TNF superfamily can control activation, expansion, differentiation and/or survival of T cells14–17. Specifically with regard to asthma, we showed some time ago in mouse models with OVA as the allergen18, 19, with knockouts and blocking studies, that OX40 (CD134/TNFRSF4), which is inducible on naïve and memory T cells after activation, was required to drive formation as well as reactivation of memory Th2 populations that induce lung inflammation. Additional work from us supported that OX40 signaling enhances expansion, survival, and differentiation of Th2 cells, while suppressing development of peripherally induced Treg20–24. Further interest came from finding that its ligand (OX40L/CD252/TNFSF4), which is inducible on APCs, could be upregulated by TSLP and IL-3324–27 in addition to signals from other molecules relevant to allergens like TLR416; and studies from other groups in various model systems and with human T cells further confirmed the idea of targeting this interaction in human asthma27–32. Phase I trials of an antibody to OX40L (Oxelumab) showed safety, and a phase II trial was conducted in mild allergic asthmatics reactive with several allergens, including HDM and cat. However, the end-point of this short-term treatment (change in FEV1) was not met, even though a trend to decreased eosinophilia was observed33. Several possibilities exist for this failure, including the wrong end-point being used for a biologic targeting T cells, the reagent and dose used not being optimal, as well as the length of the trial being too short. Additionally, a severe asthmatic population, or treatment during asthma exacerbations where OX40L has been observed at elevated levels34, 35, may have been more appropriate. However, an alternate possibility exists, which is that neutralizing OX40L in isolation is simply not sufficient to strongly modify the memory T cell response that exists in asthma patients. If correct, targeting other molecules and the use of combination therapies will be needed to produce the desired effect. In this manuscript we tested the notion that two TNF family costimulatory molecules may co-operate to control accumulation, reactivity, and persistence of memory CD4 T cells to allergen. We show that upon re-exposure of sensitized mice to house dust mite allergen, therapeutic blocking of both OX40L and CD30L (CD153/TNFSF8) during the recall response of memory T cells is required to induce a tolerogenic-like state whereby animals do not respond strongly to future insults with allergen.
Methods
Mice
C57BL/6 (8–10 weeks old) mice were purchased from The Jackson Laboratory (Bar Harbor, ME). OX40-deficient36 and CD30-deficient mice37, OT-II TCR transgenic mice38 or Foxp3-GFP OT-II transgenic mice, and CD45.1/2+ mice on a BL/6 background were bred in house. Only female mice were used due to the propensity for aggression of male mice, resulting in injuries which could have affected the results due to the long-term housing required for many experiments. All experiments complied with the regulations of the La Jolla Institute for Immunology Animal Care Committee, in accordance with guidelines of the Association for the Assessment and Accreditation of Laboratory Animal Care.
Experimental protocols
Mice were sensitized systemically (i.p.) on day 0 with 20 μg of HDM extract (Greer Labs, Lenoir, NC) mixed with 2 mg alum (250 μl in 1x PBS) and then challenged with 10 μg of HDM given i.n. on days 21, 22, 23, and 24 (40 μl in 1x PBS each time). In some experiments, mice were rested after the secondary response for 3 weeks, and then rechallenged with i.n. HDM (10 μg) in a tertiary response given on days 45, 46, 47, and 48. As a comparison to i.p. sensitization, mice were sensitized i.n. with 25 μg HDM given on days 0, 1, and 2, and challenged with 10 μg HDM i.n. on days 14, 15, 16, and 17 (40 μl in 1x PBS each time). Mice were anesthetized with Isoflurane gas prior to administration of intranasal HDM.
In other experiments, naive OT-II CD4 T cells were stimulated in vitro with anti-mouse CD3 (145–2C11, ThermoFisher Scientific) and anti-mouse CD28 (37.51, ThermoFisher Scientific) as well as irradiated splenocytes under Th2 conditions by adding 20 ng/ml IL-4, 5 ng/ml IL-2 (Peprotech), 10 μg/ml anti-IL-12 (C17.8, BioXCell) and anti-IFN-γ (XMG1.2, BioXCell) for 4 days, followed by washing and rest for 3 days in complete RPMI media. 1 × 106 CD45.2+ OT-II memory-like Th2 T cells were adoptively transferred i.v. into unsensitized naive CD45.1/2+ BL/6 mice with 1 × 106 naive CD45.1+ Foxp3-negative OT-II CD4 T cells from Foxp3-GFP reporter mice. In some experiments, 3 × 106 CD45.1+ memory-like Th2 OT-II cells were labelled with CellTrace Violet (CTV, ThermoFisher Scientific, 5 μM), before adoptive transfer into BL/6 mice. Recipients were challenged 24 hours later with 20 μg OVA (Worthington Biochemicals) mixed with 10 μg HDM, given i.n. on days 1, 2, 3, and 4 (40 μl in 1x PBS).
Mice were sacrificed 24 hours after the last allergen challenge for analyses in all model systems. On the first and third day of the allergen secondary challenges, antibodies to OX40L (RM134L) or CD30L (RM153) or control Rat IgG (BioXCell) were injected i.p (250 μg for each antibody, 200 μl in 1x PBS). With combined blockade, 250 μg anti-OX40L was injected with 250 μg anti-CD30L compared to 500 μg control IgG.
Bronchoalveolar lavage analysis, lung histology and cell analysis
Bronchoalveolar lavage (BAL) fluid was examined for Th2 cytokines and for cellular content. BAL cell differentials were determined using flow cytometry. Cells were counted, and after Fc block with 2.4G2 mAb, were stained with antibodies against CD45 (30-F11), Siglec-F (E50–2440), CD11c (N418), CD11b (M1/70), CD4 (GK1.5) and CD3 (17A2). For lung histology, 5–6 μm sections were cut and stained with hematoxylin and eosin (H&E) for examining cell infiltration. Magnification x200 was used for histologic scoring. The degree of peribronchial and perivascular inflammation were evaluated as follows: severity: 0, normal; 1, <3 cell diameter thick; 2, 3–10 cells thick; 3, >10 cells thick, and extent: 0, normal; 1, <10% of sample; 2, 10–25%; 3, >25%. Scores were calculated by multiplying severity by extent (max 9). This was performed blinded, and at least 5 fields were scored to obtain the average for each mouse. In some cases, lung tissue was digested with collagenase D (3 mg/ml Worthington Biochemicals) and DNAse I (150 μg/ml, Worthington Biochemicals) for 1.5 h at 37°C with agitation. The digested lung tissue was then passed through a 70 μM cell strainer to obtain lung mononuclear cells, followed by RBC lysis. In addition to looking at OX40 (OX86) and CD30 (mCD30.1), effector memory CD4 T cells were enumerated by flow staining for CD11a (M17/4) and CD49d (R1–2) in the CD44hi (IM7) CD4+ fraction. Induction of Foxp3 was assessed by flow in OT-II T cells using GFP as a marker. Antibodies were purchased from Biolegend (San Diego, CA), eBioscience/ThermoFisher Scientific (Carlsbad, CA) and BD Bioscience (San Jose, CA). Samples were analyzed on a LSR fortessa or LSRII flow cytometer and Flow Jo software.
In vitro analysis of T cell responsiveness
Lung tissue was digested with collagenase and DNAse followed by RBC lysis as above. T cell responsiveness was assessed in vitro by culturing 2 × 106 total lung tissue cells with 10 μg HDM for 24h and proliferation was assessed by incorporation of 10 μM BrdU (BD Bioscience) added 1h after culture setup. Gated CD4+ T cells were stained with anti-BrdU (BU20A) antibody and BrdU labelling determined by flow cytometry. In other experiments with OT-II T cell transfer, lung cells were stimulated for 24h with 10 μg OVA323–339 peptide (Innopep, San Diego). Production of IL-5 and IL-13 were measured by intracellular flow in gated CD4+ T cells, after adding GolgiStop & GolgiPlug for the last 5 hours, using antibodies for mouse IL-5 (TRFK5, Biolegend) and mouse IL-13 (eBio13A, ThermoFisher Scientific).
ELISA
Murine cytokines in BALF were assayed by means of ELISA with paired antibodies, according to the manufacturers’ recommendations. IL-4 (11B11 and BVD6–2G2), IL-5 (TRFK4 and TRFK5) and IL-13 (eBio13A and eBio1316H) ELISAs were obtained from eBioscience/ThermoFisher Scientific.
Quantitative real-time PCR
Lung tissue from mice was homogenized (Tissue Master, OMNI International), followed by total RNA isolation using TRIzol reagent (Life Technologies). Total RNA (1 μg) was then reverse transcribed to cDNA by using Transcriptor First Strand cDNA Synthesis Kit (Roche Diagnostics). Real-time PCR assays were performed with LightCycler (Roche Diagnostics, Basel, Switzerland) by using LightCycler 480 SYBR Green I master (Roche Diagnostics) and RT-qPCR–specific primers for OX40L, OX40, CD30L and CD30. Data are presented as normalized to ribosomal protein housekeeping gene L32 and relative expression is evaluated using the comparative threshold cycle (ΔΔCt) method. The mouse oligonucleotide primers were as follows: OX40L forward, 5′-GGGATGCTTCTGTGCTTCATCT- 3′; OX40L reverse, 5′-TTTGGATTGGAGGGTCCTTTG- 3′, OX40 forward, 5′- CAGTGGTCACAAGTGCTGTCGT- 3′; OX40 reverse, 5′- CCCTGGTATGATCACAGCGG- 3′, CD30L forward, 5′-ATACAACTGAGAAGGCCCCC- 3′; CD30L reverse, 5′- GGAGGTAGGCCCATGACTTC- 3′, CD30 forward, 5′- CAACCCTGGCTGAGTTACTC - 3′; CD30 reverse, 5′-AGCGGCAGGTTCTCCAGGTA - 3′ and L32 forward, 5′-GAAACTGGCGGAAACCCA- 3′; L32 reverse, 5′-GGATCTGGCCCTTGAACCTT- 3′.
Airway hyperresponsiveness
Invasive lung function testing was performed by using the flexivent system (Scireq) and airway resistance was monitored. Measurements of airway resistance were taken at baseline (nebulized PBS) as well as after each increasing concentration of nebulized methacholine.
Statistical analyses
All results are presented as mean ± SEM. Statistical analyses were performed with GraphPad Prism software (GraphPad Software, La Jolla, CA): for comparison of two groups a Welch’s t test in case of normally distributed data or a Mann–Whitney U test was performed. For multiple group data, ANOVA or Kruskal-Wallis with parametric (Dunnett’s) or nonparametric (Dunn’s) post-test, respectively, was performed for comparison of groups. For airway hyperresponsiveness, statistical analysis was by 2-way ANOVA followed by Sidak’s multiple comparison test. Differences between the groups were considered as statistically significant with p<0.05.
Results
OX40-deficient animals are protected from memory Th2-driven acute asthmatic inflammation
Although OX40/OX40L activity has been investigated extensively in several mouse models of allergic lung inflammation, including our initial studies of reactivation of OVA-specific memory Th2 cells18, 19, there are only a few studies showing that OX40 might be central to the response to a complex allergen. These studies used intranasal exposure to house dust mite (HDM) or peanut allergen to sensitize mice which results in a moderate T cell response, and they reported reduced eosinophilic lung inflammation if OX40/OX40L interactions were interrupted from the time of sensitization27, 31. We revisited this with a model that also results in Th2 eosinophilic airway disease but with a T cell response that might exceed that seen in some of the prior animal models, and so provides a very stringent test of factors that control reactivation of memory CD4 T cells (Fig. 1). Mice were sensitized with HDM given in alum intraperitoneally to drive a strong initial systemic primary T cell response, and then challenged repetitively with intranasal HDM to elicit a secondary airway response after 3 weeks. HDM has the ability to induce innate immune inflammation through its protease activity, that can directly affect lung airway epithelium and other cell populations, as well as to engage toll-like receptors, thereby eliciting production of alarmins and cytokines such as IL-33, TSLP and IL-1, that could influence the availability and use of OX40L and other costimulatory molecules.
Fig. 1. OX40-deficiency prevents memory Th2-driven airway inflammation.

WT or OX40−/−mice were sensitized with HDM in alum i.p., rested for three weeks, and then challenged intranasally with HDM for 4 days. A, Kinetics of mRNA for OX40L and OX40 in the lungs after allergen challenge. B, Surface expression of OX40 on CD44hi (black line) and CD44lo (shaded) CD4+ T cells in the lungs on day 5 after challenge (left panel) and MFI fold-increase (right panel). A-B, 3–6 mice/group, from 2 experiments. C-D, Numbers of BAL CD45+ leukocytes (C) and eosinophils (D). E-F, H&E stained lung tissue sections (E), and histological scoring of lung inflammation (F). G-J, BAL numbers of CD4+ T cells (G), and levels of IL-4 (H), IL-5 (I), and IL-13 (J). C, D, G, individual mice, with 10–11 mice/group from 4 experiments; F, 4–5 mice/group, from 2 experiments; and H-J, 3–8 mice/group, from 3 experiments.
Suggesting that OX40/OX40L might be active during the memory response, mRNA for both molecules was upregulated in the lungs upon airway rechallenge with HDM, and OX40 protein was expressed on a proportion of effector memory T cells (Fig. 1A and B). In line with the prior publications that used HDM allergen and inhibited OX40 from priming27, 31, OX40 knockout mice did not develop excessive lung inflammation (Fig. 1C–F) and the lung CD4 Th2 response was strongly reduced (Fig. 1G–J). We then tested whether blockade of OX40L would similarly inhibit the response when neutralizing antibody was given only at the time of reactivation of memory T cells, during HDM rechallenge 3 weeks after sensitization (Fig. 2). We previously had demonstrated in a similar model that blocking OX40L almost fully inhibited memory recall activity when OVA was used as the antigen19. In this case with HDM, neutralizing OX40L did not affect accumulation of total cells and eosinophils in BAL (Fig. 2A–B). No reduction in the accumulation of CD4 T cells was observed (Fig. 2C), although a significant effect was noted on BAL IL-4, IL-5 and IL-13 (Fig. 2D–F). These data showed that OX40 might have preferentially controlled the initial priming of effector and memory Th2 cells, with a lesser role in the recall response of memory cells to this allergen, regulating Th2 cytokine production.
Fig. 2. Inhibition of OX40L is not sufficient to block memory Th2-driven airway inflammation.

WT mice were sensitized and challenged with HDM as in Fig. 1. 2h prior to the first and third intranasal challenge, anti-OX40L or isotype control were injected i.p. A-B, BAL numbers of CD45+ leukocytes (A) and eosinophils (B). C-F, BAL numbers of CD4+ T cells (C), and levels of IL-4 (D), IL-5 (E), and IL-13 (F). A, B, C, individual mice, 8–9 mice/group, from 3 experiments; D-F, 8 mice/group, from 3 experiments.
CD30 plays a less dominant role compared to OX40 in development of memory Th2-driven asthma
One possibility existed for the mild effect of blocking OX40L during the secondary T cell response, which was that other comparable molecular interactions within the TNF/TNFR superfamily might also have participated in promoting the reactivation of the memory cells and accumulation of the effector memory cells in the lungs. We found that CD30 (TNFRSF8) was a good candidate, as CD30L and CD30 mRNA were upregulated together with OX40L/OX40 in the lungs following HDM rechallenge, and CD30 was similarly found on effector memory CD4 T cells and co-expressed with OX40 on many (Fig. 3A–C). CD30 was reported many years ago to be inducible on Th2 cells and aided development of this population of cells39, 40. CD30-deficient animals were also protected from developing acute allergic lung inflammation to OVA41–43. However, no studies have shown that CD30 is required in vivo for development or reactivation of memory T cells with a complex allergen. We initially assessed the secondary HDM response in CD30-deficient animals, and found no significant effect on total cellular infiltrates in the BAL, including eosinophilia, and no defect in inflammation scored by histology (Fig. 3D–G). No difference was seen in CD4 T cell numbers in the BAL, but a statistically significant reduction in IL-5 and IL-13, along with trend to lower IL-4, was observed (Fig. 3H–K), similar to the effect of blocking OX40L during the recall response (see Fig. 2). These data suggested that CD30 was active, but to a lesser extent than OX40, and might also primarily contribute to memory cell Th2 cytokine production.
Fig. 3. CD30-deficiency impairs memory Th2 cytokine production but not airway inflammation.

WT or CD30−/− mice were sensitized and challenged with HDM as in Fig. 1. A, Kinetics of mRNA for CD30L and CD30 in the lungs after challenge. B, Surface expression of CD30 on lung CD44hi (black line) and CD44lo (shaded) CD4+ T cells in WT mice on day 5 after challenge (top panel) and MFI fold-increase (bottom panel). C, Co-expression of CD30 and OX40 on lung CD44hi (red) and CD44lo (blue) CD4+ T cells. A-C, 3–6 mice/group, from 2 experiments. D-E, Numbers of BAL CD45+ leukocytes (D) and eosinophils (E). F-G, H&E stained lung tissue sections (F), and histological scoring of lung inflammation (G). H-K, BAL numbers of CD4+ T cells (H), and levels of IL-4 (I), IL-5 (J), and IL-13 (K). D, E, H, individual mice, 8–9 mice/group, from 3 experiments; G, 4–5 mice/group, from 2 experiments; I-K, 7–8 mice/group, from 3 experiments.
Co-blocking OX40L and CD30L reduces allergen-driven memory CD4 T cell reactivation and asthmatic lung inflammation
We then blocked CD30L alone or together with OX40L during the memory recall response. Similar to blocking OX40L (Fig. 2), and to the CD30-deficient phenotype (Fig. 3), inhibiting CD30L alone had no significant effect on BAL cellularity, eosinophilia, or overall lung inflammation (Fig. 4). In contrast, co-blocking OX40L and CD30L together resulted in a marked diminution of all parameters measured. This included ~50% reduction in total BAL CD45+ infiltrates, including eosinophilia, and a comparable decrease in lung inflammation scored by histological examination of tissue sections (Fig. 4). Airway hyperresponsiveness (AHR) was also reduced by co-blockade of OX40L and CD30L to a greater extent than blockade of either molecule alone, although single antibody treatment was quite effective in this regard (Fig. 4E). Most importantly, a parallel decrease in the number of BAL CD4 T cells was found when both OX40L and CD30L were blocked together during the memory recall response to HDM, whereas no significant effect was observed blocking either CD30L alone or as shown previously OX40L alone (Fig. 5A). In contrast to the CD4 T cell and eosinophil responses, blocking each molecule separately resulted in comparable trends to reduced BAL IL-4, IL-5, and IL-13 levels, perhaps explaining the AHR results. However, co-blocking both molecules strongly reduced all Th2 cytokine levels to a greater extent than blocking either molecule alone (Fig. 5B–D).
Fig. 4. Memory Th2-driven airway inflammation is ameliorated by simultaneous blockade of OX40L and CD30L.

WT mice were sensitized and challenged with HDM as in Fig. 1. 2h prior to the first and third challenge, anti-OX40L, anti-CD30L, their combination, or isotype controls, were injected i.p. A-B, Numbers of BAL CD45+ leukocytes (A) and eosinophils (B). C-D, H&E stained lung tissue sections (C), and histological scoring of lung inflammation (D). A-B, individual mice, 10–12 mice/per group, from 4 experiments; D, 6–8 mice/group, from 3 experiments. E, AHR to methacholine. 7–8 mice/group, from 3 experiments.
Fig. 5. Dual blockade of OX40L and CD30L inhibits accumulation of lung allergen-reactive effector memory CD4+ T cells.

WT mice were sensitized, challenged, and treated as in Fig. 4. A-D, BAL numbers of CD4+ T cells (A), and levels of IL-4 (B), IL-5 (C), and IL-13 (D). E, CD11a and CD49d expression in CD44hi (black) and CD44lo (grey) CD4+ T cells (left), and numbers of CD11a+CD49d+ CD44hi CD4+ T cells (right) in the lungs. F-G, Proliferation (BrdU stain) of lung CD4+ T cells after in vitro stimulation with HDM. Representative flow plots (F) and mean % BrdU+CD4+ T cells (G). A, individual mice, 10–12 mice/group, from 4 experiments; B-D, 6–11 mice/group, from 3 experiments; E-G, 4–6 mice/group, from 2 experiments.
To further expand on the idea that both CD30 and OX40 co-operated together to drive expansion as well as reactivity of memory T cells, we monitored the number of effector memory CD4 T cells that accumulated in the lung tissue during HDM rechallenge using co-expression of CD11a and CD49d in the CD44hi fraction as surrogates for marking antigen-specific cells44. Notably, blocking OX40L or CD30L alone showed a trend to reduced accumulation of CD11a+CD49d+ CD4 T cells, but blocking both together had a much more profound effect (Fig. 5E). This was further substantiated by stimulating lung tissue cells in vitro with HDM and monitoring proliferation of antigen-reactive CD4 T cells with BrdU (Fig. 5F–G). Replicating the CD11a/CD49d results, co-blocking OX40L and CD30L in vivo led to a strong reduction in the percent of T cells that proliferated in response to HDM in vitro, likely reflecting a reduction in the number of allergen-reactive effector memory cells that had accumulated in the lungs.
We additionally tested whether co-blocking OX40L with CD30L would be effective in dampening the recall activity of memory T cells elicited when sensitization was via the intranasal route rather than systemically via intraperitoneal immunization (Fig. 6). Essentially the same results were obtained, with a strong reduction in BAL CD45+ cells, eosinophilia, and lung inflammation as visualized in tissue sections (Fig. 6A–C). This was accompanied by far fewer numbers of BAL CD4+ T cells and lower Th2 cytokine levels (Fig. 6D–G), and a much weaker accumulation of CD44hi CD11a+CD49d+ CD4 T cells in the lung tissue (Fig. 6H).
Fig. 6. Co-blockade of OX40L and CD30L inhibits recall effector memory Th2 cell accumulation following airway sensitization.
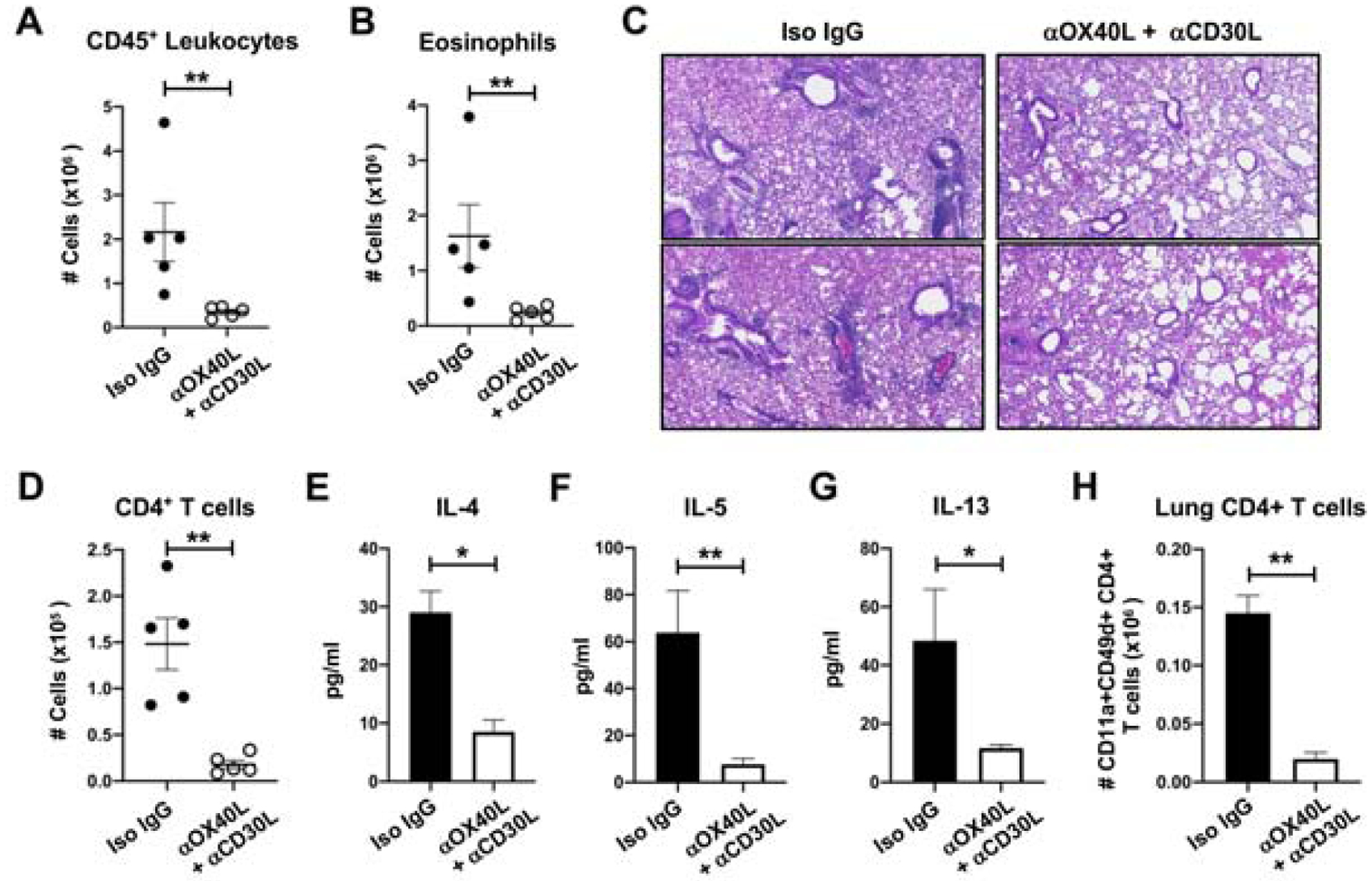
WT mice were sensitized intranasally with HDM given on days 0, 1 and 2. Mice were challenged i.n. with HDM on days 14–17 and treated with antibodies to OX40L and CD30L during this time. A-B, Numbers of BAL CD45+ leukocytes (A) and eosinophils (B). C, H&E stained lung tissue sections from two 2 individual mice. D-G, BAL numbers of CD4+ T cells (D), and levels of IL-4 (E), IL-5 (F), and IL-13 (G). H, Numbers of CD11a+CD49d+ CD44hi CD4+ T cells in the lungs. 5 mice/group.
Inhibiting OX40L with CD30L reduces expansion of effector memory T cells and biases the allergic secondary T cell response towards Treg
To support the above conclusion, we generated memory-like OVA-specific OT-II TCR transgenic CD4 T cells by stimulating in vitro under Th2 conditions followed by a rest period. These cells transition from effector to memory-like with the rest period, upregulating IL-7R and IL-33R and reverting to a more resting state (not shown). The OT-II T cells were labeled with cell trace violet (CTV), and adoptively transferred into naive hosts. The recipient mice were then repetitively challenged intranasally with a combination of OVA to provide the antigen signal for the T cells to become reactivated, and HDM, to provide the equivalent allergic inflammatory environment similar to our other experiments. Blocking OX40L or CD30L alone had minimal effect on proliferation in vivo as read out by CTV dilution, but the combined blockade restricted OT-II T cell division to an extent (Fig. 7A), and significantly reduced the number of OT-II T cells that expanded and accumulated in the lungs (Fig. 7B). The latter was confirmed by restimulating cells in vitro with OVA peptide and assessing the number of CD4 T cells that made IL-5 and IL-13 (Fig. 7C–D).
Fig. 7. Co-inhibition of OX40L and CD30L reduces expansion of antigen-specific effector memory CD4+ T cells and improves the Th2/Foxp3+ balance.

A-D, In vitro derived memory-like OT-II Th2 T cells were labelled with CTV and adoptively transferred into congenic recipients. Mice were given i.n. OVA and HDM for 4 days and treated with anti-OX40L, anti-CD30L, or their combination. A, Proliferation of donor OT-II cells determined by dilution of CTV. Rat IgG (thick grey line), anti-OX40L (dotted black line), anti-CD30L (dotted grey line) and combination (thick black line). B, Number of OT-II T cells in the lungs. C-D, Numbers of IL-5+ (C) and IL-13+ (D) OT-II cells in the lungs calculated from data in B combined with intracellular cytokine staining of CD4+ T cells after stimulation in vitro with OVA. B-D, 3–6 mice/group, from 2 experiments. E-F, Naïve Foxp3− OT-II Foxp3-GFP CD4 T cells were transferred with memory-like OT-II Th2 cells and recipients treated with allergen and antibodies as in A-D. Frequency of Foxp3/GFP+ OT-II cells in the lungs (E). Th2/Foxp3+ T cell ratio calculated for each group (F). 3–6 mice/group, from 2 experiments.
We then asked whether blocking OX40L and CD30L might lead to a more favorable balance between effector memory T cells and induced or peripheral Treg. Memory-like OT-II Th2 cells were adoptively transferred into unsensitized mice, similar to above, along with a population of naive Foxp3 negative OT-II CD4 cells purified from OT-II Foxp3-GFP reporter mice. Recipients were challenged with OVA and HDM and accumulation of OVA-reactive Foxp3+ T cells examined while blocking OX40L and CD30L. Interestingly, co-neutralization of OX40L and CD30L led to an increased percentage of these cells among transferred naïve OT-II cells (Fig. 7E). Together with fewer effector memory Th2 cells, this then led to a significant reduction in the Th2/Treg ratio towards Treg (Fig. 7F). These results in sum show that inhibiting OX40/OX40L and CD30/CD30L interactions concomitantly reduces the frequency of effector memory T cells that accumulate in the lungs in secondary responses to HDM allergen, and the production of Th2 cytokines, and may result in a more favorable balance of Treg to these effector cells, a state that could be relevant for allergen tolerance.
Co-Neutralization of OX40L and CD30L induces a tolerogenic state in the airways
Lastly, to expand on the notion of a tolerogenic effect of removing both OX40 and CD30 signaling, after intraperitoneal sensitization we treated mice with antibodies to OX40L and CD30L only during the secondary recall response to HDM, as before. We then rested mice for a further 3 weeks before rechallenging again repetitively with intranasal HDM to elicit a tertiary response. Tolerance can be functionally defined by the inability to mount a normal maximal response to a subsequent allergen challenge, and could involve reduced persistence or accumulation of allergen-specific memory T cells and/or reduced reactivity per cell. Importantly, neutralizing both interactions, but not the individual interactions, led to a substantial reduction in the ability of mice to mount such a subsequent inflammatory response to allergen, with ~50% less BAL cellular infiltration and eosinophils, and a similar lower level of lung tissue inflammation scored by histologic analyses (Fig. 8A–D). As importantly, the accumulation of CD4 T cells in the BAL with tertiary HDM recall challenge was significantly reduced by co-blockade of OX40L and CD30L during the secondary response (Fig. 8E), along with a parallel decrease in all Th2 cytokines in the BAL (Fig. 8F–H). These data show that co-neutralization of OX40L and CD30L has a long-lasting effect on the number of effector memory T cells that can develop in response to a complex allergen, and may afford a partial state of tolerance.
Fig. 8. Co-inhibition of OX40L and CD30L during secondary allergen challenge results in an inability to mount a normal tertiary recall airway inflammatory response.

WT mice were sensitized, challenged, and treated with antibodies, as in Fig. 4. Mice were then rested for 3 weeks, and subsequently challenged repetitively during a tertiary response with HDM. A-B, Numbers of BAL CD45+ leukocytes (A) and eosinophils (B). C-D, H&E stained lung tissue sections (C), and histological scoring of lung inflammation (D). E-H, BAL numbers of CD4+ T cells (E), and levels of IL-4 (F), IL-5 (G), and IL-13 (H). A, B, E, individual mice, 6–10 mice/group, from 3 experiments; D, 4–7 mice/group, from 2 experiments; F-H, 5–10 mice/group, from 3 experiments.
Discussion
In this manuscript we demonstrate that two TNF superfamily costimulatory interactions co-operate together to promote the accumulation of lung pathology-driving effector CD4 T cells derived during the memory T cell response to a complex allergen. Neutralizing either interaction in isolation inhibited Th2 cytokines and AHR, but produced a mild effect on accumulation of effector memory CD4 T cells in the lungs, and had negligible effects on reducing lung inflammation. In contrast, concomitant neutralization of both interactions strongly reduced effector memory T cell accumulation and production of Th2 cytokines and this correlated with a similar reduction in lung pathology triggered by the allergen. Moreover, this weaker memory CD4 T cell and lung inflammatory response was maintained several weeks after the therapeutic antibody treatment, such that animals treated during the secondary response did not mount a normal response to allergen when subsequently challenged with allergen. These data suggest that a combination approach to blocking TNF family costimulatory molecules, either with single agents or bispecific biologics, might have therapeutic relevance to limiting responses to allergens, including those in asthmatics.
Costimulatory receptors expressed on T cells can provide essential signals when they are engaged by their ligands to enhance the response triggered by antigen recognition14, 15, 45. This can include promoting expression of cell cycle proteins and driving cell division, inducing anti-apoptotic proteins that allow cell survival, as well as upregulating effector molecules such as cytokines. A number of proteins have been described to possess this function over the years, including CD28 and ICOS in the Ig superfamily, and several TNF receptor superfamily proteins, most notably OX40, CD30, 4–1BB, HVEM, CD27, GITR, DR3, and TNFR2. Some of these receptors are constitutively expressed on naive and memory T cells, whereas others such as OX40 and CD30 are induced at variable times after antigen recognition. One model that can explain their combined action or importance is that they act serially in a temporal manner to maintain a T cell response over time15, 17. Most of the costimulatory ligands, including OX40L and CD30L are inducible, further adding to the notion that their presence might be seen at varying times during inflammatory responses. However, a number of cell types have been described to express several of the ligands simultaneously, such as conventional APC like dendritic cells and macrophages46, as well as innate cells such as lymphoid tissue inducer cells (ILC3) that were described to co-express OX40L and CD30L47. This provides a second non-mutually exclusive model that costimulatory receptors co-operate together, or over a short-period of time, and their combined signals drive various stages of T cell activation or function15, 17. Which combination of receptors might be active in any give inflammatory response is not clear. A number of previous studies in virus model systems have found that several TNF family receptors act together to drive the underlying T cell response, such as OX40 and CD27 in CD8 T cell responses to vaccinia virus48, or GITR, 4–1BB, CD27 and OX40 in CD8 T cell priming to LCMV46, 49. Importantly, examples of combined blockade of costimulatory molecules have already been reported in stringent full MHC-mismatched models of transplantation, where graft acceptance and tolerance in CD4 or CD8 T cells were only afforded by neutralizing for example OX40L with CD40L and CD80/CD8650, or OX40L with CD40L and LFA-151, whereas the individual blockade of these interactions had little to no effect.
Our data now suggest that during memory responses to allergen, combined targeting of costimulatory molecules might also be needed to suppress the continued recall responses that result in large populations of pathogenic effector memory T cells. We highlight OX40L and CD30L as drivers of the memory CD4 T cell response to house dust mite allergen, which adds to the notion that these two interactions may be highly complementary for inflammatory disease. Previous studies by Lane and coworkers established co-operativity between OX40 and CD30 in regulating Th2 cells in vitro47. They expanded these observations by showing that mice deficient in both OX40 and CD30 were more compromised in generating secondary antibody responses than either OX40- or CD30-deficient mice52, and that double-deficient animals were also compromised in generating protective Th1 responses to Salmonella infection53, and memory T cells from OX40/CD30 knockout animals showed a survival defect in the gut54. Importantly, these authors further showed that Foxp3-deficient mice that normally exhibit lethal autoimmune symptoms were protected when bred onto the OX40/CD30 double-deficient background, and that by reducing the accumulation of effector CD4 T cells co-blockade of OX40L and CD30L allowed survival of Rag knockout mice when injected with Foxp3-deficient thymocytes55. These latter results correlate with our finding that combined neutralization of OX40L and CD30L led to fewer effector memory CD4 T cells accumulating in the lungs but still allowed Treg to be generated. Interestingly, blocking both molecules was required to see a strong action on limiting the number of effector memory T cells that were generated, whereas blocking only one was sufficient for a good effect on reducing production of Th2 cytokines. The latter was likely reflected in the AHR results, where blocking only one molecule strongly reduced this parameter, a functional readout thought to be strongly dependent on smooth muscle activity and cytokines such as IL-13 that can induce contractility in smooth muscle cells. The reason why single antibody treatment effectively limited Th2 cytokines but dual antibody treatment was needed to have a strong effect on T cell accumulation and persistence is unclear, but we hypothesize that the threshold level of signaling for induction of activities in memory T cells might be different. Production of Th2 cytokines might require an overall weaker signal than is needed for these cells to proliferate well and expand in numbers. There may also be differential redundancy in signaling pathways required for these activities.
Lastly, our primary model system used intraperitoneal sensitization to induce a memory T cell population. It has been debated whether i.p. sensitization with allergen is less physiological or clinically relevant than i.n. sensitization, due to differential involvement of ILC2, with some studies claiming a strong influence of ILC2 in priming naive CD4 T cells and influencing the subsequent recall lung inflammatory responses only after i.n. sensitization56, 57. In contrast, others have found that the recall lung inflammatory response following sensitization via the airways was completely dependent on memory T cells implying ILC2 were not active or played a secondary role58. Regardless of whether ILC2 do or do not participate in the T cell response, asthma in many humans has been linked to an atopic march and in particular atopic dermatitis, implying that initial sensitization of T cells may not in many cases be through the airways. Most important for our study, we are not aware of any data that have proven that memory T cells induced by systemic sensitization behave differently than those induced by airway sensitization. Although we did not investigate this in depth, we did find that blocking OX40L with CD30L was equally, if not more, effective in reducing the memory T cell response when airway sensitization was performed, supporting the idea that the route of sensitization may not be important to the reactivity of the memory T cells that result. However, we acknowledge this caveat, and that it will be of interest in future studies to fully address these issues and whether the respective contributions of costimulatory molecules like OX40L and CD30L to recall responses are different if memory T cells are initially generated via alternate routes of allergen exposure.
In summary, we report that a combination approach to inhibiting OX40/OX40L and CD30/CD30L interactions was effective in reducing the memory CD4 T cell response to a complex allergen, and resulted in a tolerogenic state that lessened allergic lung inflammation upon repeat exposure to the allergen. Previous studies had examined whether combined blockade of the innate cytokines TSLP, IL-33, and IL-25 could restrict lung inflammation when given therapeutically in a similar model with house dust mite allergen. They found no profound reduction despite reporting positive effects in models of Schistosoma mansoni type 2 immunity59. Our studies suggest that co-targeting T cell costimulatory molecules may be a good approach to limit memory Th2 immune responses to allergens and for therapy of diseases such as asthma.
Key Messages:
OX40/OX40L and CD30/CD30L interactions control the expansion and accumulation of allergen-reactive effector memory CD4 T cells
Combination blockade of OX40L and CD30L might represent a strategy to inhibit acute allergic inflammation and induce a state of T cell tolerance to allergen
Acknowledgments
This work was supported by NIH grant AI070535 to M.C.
Abbreviations:
- HDM
house dust mite
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosure of potential conflict of interest: Michael Croft has patents on OX40/OX40L. All other authors declare that they have no relevant conflicts of interest.
References
- 1.Walsh GM. Biologics targeting IL-5, IL-4 or IL-13 for the treatment of asthma - an update. Expert Rev Clin Immunol 2017; 13:143–9. [DOI] [PubMed] [Google Scholar]
- 2.Fajt ML, Wenzel SE. Asthma phenotypes and the use of biologic medications in asthma and allergic disease: the next steps toward personalized care. J Allergy Clin Immunol 2015; 135:299–310; quiz 1. [DOI] [PubMed] [Google Scholar]
- 3.Lambrecht BN, Hammad H. The immunology of asthma. Nat Immunol 2015; 16:45–56. [DOI] [PubMed] [Google Scholar]
- 4.Leomicronn B T Cells in Allergic Asthma: Key Players Beyond the Th2 Pathway. Curr Allergy Asthma Rep 2017; 17:43. [DOI] [PubMed] [Google Scholar]
- 5.Akdis CA, Akdis M. Mechanisms of allergen-specific immunotherapy. The Journal of allergy and clinical immunology 2011; 127:18–27; quiz 8–9. [DOI] [PubMed] [Google Scholar]
- 6.Akdis CA, Akdis M. Mechanisms of allergen-specific immunotherapy and immune tolerance to allergens. World Allergy Organ J 2015; 8:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ray A, Oriss TB, Wenzel SE. Emerging molecular phenotypes of asthma. Am J Physiol Lung Cell Mol Physiol 2015; 308:L130–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Raundhal M, Morse C, Khare A, Oriss TB, Milosevic J, Trudeau J, et al. High IFN-gamma and low SLPI mark severe asthma in mice and humans. J Clin Invest 2015; 125:3037–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chambers ES, Nanzer AM, Pfeffer PE, Richards DF, Timms PM, Martineau AR, et al. Distinct endotypes of steroid-resistant asthma characterized by IL-17A(high) and IFN-gamma(high) immunophenotypes: Potential benefits of calcitriol. J Allergy Clin Immunol 2015; 136:628–37 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee JH, Yu HH, Wang LC, Yang YH, Lin YT, Chiang BL. The levels of CD4+CD25+ regulatory T cells in paediatric patients with allergic rhinitis and bronchial asthma. Clin Exp Immunol 2007; 148:53–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.O’Hehir RE, Gardner LM, de Leon MP, Hales BJ, Biondo M, Douglass JA, et al. House dust mite sublingual immunotherapy: the role for transforming growth factor-beta and functional regulatory T cells. Am J Respir Crit Care Med 2009; 180:936–47. [DOI] [PubMed] [Google Scholar]
- 12.Radulovic S, Jacobson MR, Durham SR, Nouri-Aria KT. Grass pollen immunotherapy induces Foxp3-expressing CD4+ CD25+ cells in the nasal mucosa. J Allergy Clin Immunol 2008; 121:1467–72, 72 e1. [DOI] [PubMed] [Google Scholar]
- 13.Pereira-Santos MC, Baptista AP, Melo A, Alves RR, Soares RS, Pedro E, et al. Expansion of circulating Foxp3+)D25bright CD4+ T cells during specific venom immunotherapy. Clin Exp Allergy 2008; 38:291–7. [DOI] [PubMed] [Google Scholar]
- 14.Croft M Co-stimulatory members of the TNFR family: keys to effective T-cell immunity? Nature reviews. Immunology 2003; 3:609–20. [DOI] [PubMed] [Google Scholar]
- 15.Croft M The role of TNF superfamily members in T-cell function and diseases. Nat Rev Immunol 2009; 9:271–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Croft M Control of immunity by the TNFR-related molecule OX40 (CD134). Annu Rev Immunol 2010; 28:57–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Croft M The TNF family in T cell differentiation and function--unanswered questions and future directions. Semin Immunol 2014; 26:183–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jember AG, Zuberi R, Liu FT, Croft M. Development of allergic inflammation in a murine model of asthma is dependent on the costimulatory receptor OX40. J Exp Med 2001; 193:387–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Salek-Ardakani S, Song J, Halteman BS, Jember AG, Akiba H, Yagita H, et al. OX40 (CD134) Controls Memory T Helper 2 Cells that Drive Lung Inflammation. J Exp Med 2003; 198:315–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bansal-Pakala P, Gebre-Hiwot Jember A, Croft M. Signaling through OX40 (CD134) breaks peripheral T-cell tolerance. Nat Med 2001; 7:907–12. [DOI] [PubMed] [Google Scholar]
- 21.Rogers PR, Song J, Gramaglia I, Killeen N, Croft M. OX40 promotes Bcl-xL and Bcl-2 expression and is essential for long-term survival of CD4 T cells. Immunity 2001; 15:445–55. [DOI] [PubMed] [Google Scholar]
- 22.So T, Croft M. Cutting Edge: OX40 Inhibits TGF-beta- and Antigen-Driven Conversion of Naive CD4 T Cells into CD25+Foxp3+ T cells. J Immunol 2007; 179:1427–30. [DOI] [PubMed] [Google Scholar]
- 23.Duan W, So T, Croft M. Antagonism of airway tolerance by endotoxin/lipopolysaccharide through promoting OX40L and suppressing antigen-specific Foxp3+ T regulatory cells. J Immunol 2008; 181:8650–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Duan W, Mehta AK, Magalhaes JG, Ziegler SF, Dong C, Philpott DJ, et al. Innate signals from Nod2 block respiratory tolerance and program T(H)2-driven allergic inflammation. J Allergy Clin Immunol 2010; 126:1284–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ito T, Wang YH, Duramad O, Hori T, Delespesse GJ, Watanabe N, et al. TSLP-activated dendritic cells induce an inflammatory T helper type 2 cell response through OX40 ligand. J Exp Med 2005; 202:1213–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Besnard AG, Togbe D, Guillou N, Erard F, Quesniaux V, Ryffel B. IL-33-activated dendritic cells are critical for allergic airway inflammation. European journal of immunology 2011; 41:1675–86. [DOI] [PubMed] [Google Scholar]
- 27.Chu DK, Llop-Guevara A, Walker TD, Flader K, Goncharova S, Boudreau JE, et al. IL-33, but not thymic stromal lymphopoietin or IL-25, is central to mite and peanut allergic sensitization. J Allergy Clin Immunol 2013; 131:187–200 e1–8. [DOI] [PubMed] [Google Scholar]
- 28.Jenkins SJ, Perona-Wright G, Worsley AG, Ishii N, MacDonald AS. Dendritic cell expression of OX40 ligand acts as a costimulatory, not polarizing, signal for optimal Th2 priming and memory induction in vivo. J Immunol 2007; 179:3515–23. [DOI] [PubMed] [Google Scholar]
- 29.Seshasayee D, Lee WP, Zhou M, Shu J, Suto E, Zhang J, et al. In vivo blockade of OX40 ligand inhibits thymic stromal lymphopoietin driven atopic inflammation. J Clin Invest 2007; 117:3868–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xiao X, Balasubramanian S, Liu W, Chu X, Wang H, Taparowsky EJ, et al. OX40 signaling favors the induction of T(H)9 cells and airway inflammation. Nature immunology 2012; 13:981–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Burrows KE, Dumont C, Thompson CL, Catley MC, Dixon KL, Marshall D. OX40 blockade inhibits house dust mite driven allergic lung inflammation in mice and in vitro allergic responses in humans. Eur J Immunol 2015; 45:1116–28. [DOI] [PubMed] [Google Scholar]
- 32.Di C, Lin X, Zhang Y, Zhong W, Yuan Y, Zhou T, et al. Basophil-associated OX40 ligand participates in the initiation of Th2 responses during airway inflammation. J Biol Chem 2015; 290:12523–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gauvreau GM, Boulet LP, Cockcroft DW, FitzGerald JM, Mayers I, Carlsten C, et al. OX40L blockade and allergen-induced airway responses in subjects with mild asthma. Clin Exp Allergy 2014; 44:29–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ezzat MH, Imam SS, Shaheen KY, Elbrhami EM. Serum OX40 ligand levels in asthmatic children: a potential biomarker of severity and persistence. Allergy and asthma proceedings : the official journal of regional and state allergy societies 2011; 32:313–8. [DOI] [PubMed] [Google Scholar]
- 35.Lei W, Zhu CH, Zeng da X, Wang Q, Zhang XQ, Chen YB, et al. SOX40L: An Important Infl ammatory Mediator in Adult Bronchial Asthma. Annals of the Academy of Medicine, Singapore 2012; 41:200–4. [PubMed] [Google Scholar]
- 36.Pippig SD, Pena-Rossi C, Long J, Godfrey WR, Fowell DJ, Reiner SL, et al. Robust B cell immunity but impaired T cell proliferation in the absence of CD134 (OX40). J Immunol 1999; 163:6520–9. [PubMed] [Google Scholar]
- 37.Amakawa R, Hakem A, Kundig TM, Matsuyama T, Simard JJ, Timms E, et al. Impaired negative selection of T cells in Hodgkin’s disease antigen CD30-deficient mice. Cell 1996; 84:551–62. [DOI] [PubMed] [Google Scholar]
- 38.Kaye J, Hsu ML, Sauron ME, Jameson SC, Gascoigne NR, Hedrick SM. Selective development of CD4+ T cells in transgenic mice expressing a class II MHC-restricted antigen receptor. Nature 1989; 341:746–9. [DOI] [PubMed] [Google Scholar]
- 39.Del Prete G, De Carli M, D’Elios MM, Daniel KC, Almerigogna F, Alderson M, et al. CD30-mediated signaling promotes the development of human T helper type 2-like T cells. J Exp Med 1995; 182:1655–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Del Prete G, De Carli M, Almerigogna F, Daniel CK, D’Elios MM, Zancuoghi G, et al. Preferential expression of CD30 by human CD4+ T cells producing Th2-type cytokines. Faseb J 1995; 9:81–6. [PubMed] [Google Scholar]
- 41.Polte T, Behrendt AK, Hansen G. Direct evidence for a critical role of CD30 in the development of allergic asthma. J Allergy Clin Immunol 2006; 118:942–8. [DOI] [PubMed] [Google Scholar]
- 42.Nam SY, Kim YH, Do JS, Choi YH, Seo HJ, Yi HK, et al. CD30 supports lung inflammation. Int Immunol 2008; 20:177–84. [DOI] [PubMed] [Google Scholar]
- 43.Polte T, Fuchs L, Behrendt AK, Hansen G. Different role of CD30 in the development of acute and chronic airway inflammation in a murine asthma model. Eur J Immunol 2009; 39:1736–42. [DOI] [PubMed] [Google Scholar]
- 44.McDermott DS, Varga SM. Quantifying antigen-specific CD4 T cells during a viral infection: CD4 T cell responses are larger than we think. J Immunol 2011; 187:5568–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen L, Flies DB. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat Rev Immunol 2013; 13:227–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chang YH, Wang KC, Chu KL, Clouthier DL, Tran AT, Torres Perez MS, et al. Dichotomous Expression of TNF Superfamily Ligands on Antigen-Presenting Cells Controls Post-priming Anti-viral CD4(+) T Cell Immunity. Immunity 2017; 47:943–58 e9. [DOI] [PubMed] [Google Scholar]
- 47.Kim MY, Gaspal FM, Wiggett HE, McConnell FM, Gulbranson-Judge A, Raykundalia C, et al. CD4(+)CD3(−) Accessory Cells Costimulate Primed CD4 T Cells through OX40 and CD30 at Sites Where T Cells Collaborate with B Cells. Immunity 2003; 18:643–54. [DOI] [PubMed] [Google Scholar]
- 48.Salek-Ardakani S, Flynn R, Arens R, Yagita H, Smith GL, Borst J, et al. The TNFR family members OX40 and CD27 link viral virulence to protective T cell vaccines in mice. J Clin Invest 2011; 121:296–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Welten SP, Redeker A, Franken KL, Oduro JD, Ossendorp F, Cicin-Sain L, et al. The viral context instructs the redundancy of costimulatory pathways in driving CD8(+) T cell expansion. Elife 2015; 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Vu MD, Clarkson MR, Yagita H, Turka LA, Sayegh MH, Li XC. Critical, but conditional, role of OX40 in memory T cell-mediated rejection. J Immunol 2006; 176:1394–401. [DOI] [PubMed] [Google Scholar]
- 51.Xia J, Chen J, Shao W, Lan T, Wang Y, Xie B, et al. Suppressing memory T cell activation induces islet allograft tolerance in alloantigen-primed mice. Transplant international : official journal of the European Society for Organ Transplantation 2010; 23:1154–63. [DOI] [PubMed] [Google Scholar]
- 52.Gaspal FM, Kim MY, McConnell FM, Raykundalia C, Bekiaris V, Lane PJ. Mice Deficient in OX40 and CD30 Signals Lack Memory Antibody Responses because of Deficient CD4 T Cell Memory. J Immunol 2005; 174:3891–6. [DOI] [PubMed] [Google Scholar]
- 53.Gaspal F, Bekiaris V, Kim MY, Withers DR, Bobat S, MacLennan IC, et al. Critical synergy of CD30 and OX40 signals in CD4 T cell homeostasis and Th1 immunity to Salmonella. J Immunol 2008; 180:2824–9. [DOI] [PubMed] [Google Scholar]
- 54.Withers DR, Jaensson E, Gaspal F, McConnell FM, Eksteen B, Anderson G, et al. The survival of memory CD4+ T cells within the gut lamina propria requires OX40 and CD30 signals. J Immunol 2009; 183:5079–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gaspal F, Withers D, Saini M, Bekiaris V, McConnell FM, White A, et al. Abrogation of CD30 and OX40 signals prevents autoimmune disease in FoxP3-deficient mice. The Journal of experimental medicine 2011; 208:1579–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gold MJ, Antignano F, Halim TY, Hirota JA, Blanchet MR, Zaph C, et al. Group 2 innate lymphoid cells facilitate sensitization to local, but not systemic, TH2-inducing allergen exposures. J Allergy Clin Immunol 2014; 133:1142–8. [DOI] [PubMed] [Google Scholar]
- 57.Halim TY, Steer CA, Matha L, Gold MJ, Martinez-Gonzalez I, McNagny KM, et al. Group 2 innate lymphoid cells are critical for the initiation of adaptive T helper 2 cell-mediated allergic lung inflammation. Immunity 2014; 40:425–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rahimi RA, Nepal K, Cetinbas M, Sadreyev RI, Luster AD. Distinct functions of tissue-resident and circulating memory Th2 cells in allergic airway disease. J Exp Med 2020; 217: 10.1084/jem.20190865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Vannella KM, Ramalingam TR, Borthwick LA, Barron L, Hart KM, Thompson RW, et al. Combinatorial targeting of TSLP, IL-25, and IL-33 in type 2 cytokine-driven inflammation and fibrosis. Sci Transl Med 2016; 8:337ra65. [DOI] [PubMed] [Google Scholar]