

Abstract
Global DNA hypomethylation is a most common epigenetic alteration in human neoplasia. However, accumulative evidence shows that global DNA hypomethylation impacts tumorigenesis in a tissue-specific manner, promoting tumorigenesis in some but suppressing tumorigenesis in others including colorectal cancer. The underlying mechanisms, especially how DNA hypomethylation suppresses tumorigenesis, remain largely unknown. Here, we investigate how DNA hypomethylation affects intestinal tumorigenesis by using an Uhrf1 tandem tudor domain knockin mutant mouse model (Uhrf1ki/ki) that exhibits a moderate ~10% reduction of global DNA methylation. We found that both chemical-induced colorectal carcinogenesis and Apc loss of heterozygosity (LOH)-induced intestinal tumorigenesis are substantially suppressed in the Uhrf1 mutant mice. Furthermore, unlike Dnmt1 hypomorphic mice in which DNA hypomethylation suppresses the incidence of macroscopic intestinal tumors but promotes the formation of microadenoma in ApcMin/+ background, Uhrf1ki/ki/ApcMin/+ mice have markedly reduced incidence of both microadenoma and macroadenoma. DNA hypomethylation does not appear to affect Apc LOH, activation of the Wnt or Hippo pathway, or tumor cell proliferation, but acts cooperatively with activated Wnt pathway to enhance the caspase-3 gene expression, activation, and apoptosis. Furthermore, increased caspase-3 expression correlates with DNA hypomethylation within the caspase-3 enhancer regions. Taken together, we present a new mouse model for investigating the role of and the molecular mechanisms by which DNA hypomethylation suppresses intestinal tumorigenesis. Our finding that a moderate DNA hypomethylation is sufficient to suppress intestinal tumorigenesis by promoting caspase-3 expression and apoptosis sheds new light on DNA-methylation inhibitor-based colorectal cancer therapeutics.
Subject terms: Epigenetics, Epigenetics
Introduction
Genome-wide DNA hypomethylation and concomitant locus-specific DNA hypermethylation are among the most common molecular alterations in human neoplasia1–5. These DNA-methylation alterations often occur in the early stage of cancer and is believed to promote cancer pathogenesis through tumor-suppressor gene silencing via promoter hypermethylation and/or through proto-oncogene activation and increased chromosomal instability as a result of global hypomethylation5–10. A causal role for global DNA hypomethylation in tumorigenesis was demonstrated by using Dnmt1 hypomorphic mouse models, which shows genome-wide DNA hypomethylation in all tissues due to a substantially reduced Dnmt1 expression11. These mutant mice developed aggressive T-cell lymphomas at age 4–8 months11,12. However, accumulative evidence with various Dnmt1 hypomorphic mouse models indicate that, while DNA hypomethylation promotes T- and B-cell lymphomas11,12, brain tumors13, the early stage of prostate cancer14, micro-adenomas of intestine and live tumors15, it also strongly suppresses overall tumorigenesis in prostate cancer14, pancreatic acinar cell tumors16, and the macroadenoma of intestine tumors12,15,17–19. In fact, weekly treatment with low doses of DNA-methylation inhibitor 5-azaC was shown to almost completely suppress the intestinal adenomas in ApcMin/+ mice harboring only one wild-type Dnmt1 allele20, providing the first evidence that DNA hypomethylation can actually suppress tumorigenesis.
How global DNA hypomethylation promotes tumorigenesis in some but suppresses in other tissues has become a subject of intensive studies. Because global DNA hypomethylation has been linked to an increased mutation rate21, increased genome instability22, impaired epigenetic silencing3,8,23,24, and activation of interferon pathways25,26, it has been proposed that global DNA hypomethylation promotes tumorigenesis in cells that rely on genome instability and suppresses tumorigenesis in cells that are dependent on epigenetic silencing of tumor-suppressor genes. However, exactly how global DNA hypomethylation suppresses intestinal tumorigenesis remains largely unknown. Furthermore, because all global DNA hypomethylation studies reported so far rely on the use of Dnmt1 hypomorphic mice with substantially reduced levels of DNMT1 proteins, a DNMT1-dependent but DNA-methylation-independent function in tumorigenesis could not be completely ruled out. Thus, a new Dnmt1-independent DNA hypomethylation model is needed and can be valuable for testing if the observed tissue-specific effect on tumorigenesis is indeed attributed to global DNA hypomethylation and/or reduced DNMT1 proteins.
Ubiquitin-like with PHD and RING finger domains 1 (UHRF1) has been recognized as a key accessory factor essential for DNMT1-mediated DNA maintenance methylation27,28. As a multi-structural and functional domain protein, UHRF1 contains at least five distinct domains from the N- to C-terminus: ubiquitin-like domain, tandem tudor domain (TTD), plant homeo-domain (PHD), SET and RING-associated domain, and RING domain. These domains act cooperatively in binding hemi-methylated CpG sites in newly replicated DNA and histones in targeting DNMT1 to DNA replication foci to carry out DNA maintenance methylation27,29–34. The TTD domain shows high affinity of binding histone H3 with di- or tri-methylated lysine 9 (H3K9me2/3)32,35–37 and methylated DNA ligase 138. We previously reported the generation of a mutant mouse strain that carried mutations changing tyrosine 187 and proline 188 in TTD to alanine39. These mutations abolished UHRF1 binding to H3K9me2/3 and DNA ligase 1. The resulting mutant mice that we named Uhrf1-TTD-KI or Uhrf1ki/ki hereafter are viable and grossly normal. However, the mutant mice were found to exhibit an ~10% reduction of global DNA methylation in various tissues39. Thus, the Uhrf1-TTD-KI mice may serve as a new model for investigating the role of DNA hypomethylation in tumorigenesis.
In this study, we used Uhrf1-TTD-KI mice to investigate and demonstrate that DNA hypomethylation substantially suppresses intestinal tumorigenesis induced by chemical carcinogens or genetic mutation of the Apc gene in Apcmin/+ mice. While previous studies with Dnmt1 hypomorphic mice conclude that DNA hypomethylation suppresses macroadenoma but promotes microadenoma in Apcmin/+ background, we find that both intestinal macroadenoma and microadenoma are markedly suppressed in Uhrf1ki/ki/ApcMin/+ mice. Importantly, we present evidence that DNA hypomethylation in Uhrf1-TTD-KI suppresses intestinal tumorigenesis by inducing caspase-3 expression and apoptosis.
Results
Uhrf1-TTD-KI mice have normal intestinal tissues despite of global DNA hypomethylation
We previously reported the Uhrf1-TTD-KI mouse model with both 187 tyrosine and 188 proline residues in TTD mutated to alanine39. The homozygous mutant mice are viable and grossly normal, despite an ~10% reduction of global DNA methylation in liver, brain, and heart tissues. To examine if this mouse model is suitable for investigating how DNA hypomethylation affects intestinal tumorigenesis, we first obtained wild-type, heterozygous and homozygous mutant mice by crossing between heterozygote mice. The genotypes of the resulting mice were determined by both DNA sequencing and PCR-based genotyping (Supplementary Fig. 1A, B). Through observing a large number of resulting littermate mice, we found no significant difference between the wild-type, heterozygous (data not shown), and homozygous Uhrf1-TTD-KI mice in gross morphology and the growth rate within 14 months of observation (Fig. 1A, B). At the end of experiments, all mice were examined and found to be free of intestinal tumors and tumors in other tissues. However, measurement of DNA-methylation (5mC) levels from three pairs of littermates by quantitative high-pressure liquid chromatography (HPLC) analysis revealed an average of 8.4% reduction of 5mC in intestines and 11.5% reduction of 5mC in colons of mutant mice compared to the corresponding wild-type littermates (Fig. 1C), a result consistent with our previous measurement of 5mC levels in other tissues39. The same analysis revealed no difference in DNA methylation in both small intestine and colon tissues between wild-type and heterozygote Uhrf1-TTD-KI mice (data not shown). The defect in DNA methylation could be attributed to impaired binding of H3K9me2/3 and/or methylated DNA ligase 1 by the mutated Uhrf1 protein, which consequently led to reduced Dnmt1 DNA maintenance activity, because Western blot analysis detected no difference between wild-type and mutant mice in the protein levels of Uhrf1, Dnmt1 and Dnmt3a, and H3K9me3 in the small intestine and colorectal tissues (Fig. 1D). Furthermore, hematoxylin and eosin (H&E) staining of tissue sections showed that Uhrf1-TTD-KI mice had normal intestinal crypts and villi as observed in the wild-type mice (Fig. 1E, left panel). BrdU incorporation assay showed normal proliferation of intestinal stem and transient cells of mutant mice (Supplementary Fig. 1C, left panel), as indicated by equal BrdU labeling of the intestinal crypt cells of both wild-type and Uhrf1-TTD-KI mice 5 h after BrdU injection. Furthermore, at 50 h post BrdU injection, the BrdU-labeled cells were found to account for ~1/2 of the villi in both the wild-type and the Uhrf1-TTD-KI mice, indicating normal proliferation and migration of the intestinal epithelial cells in Uhrf1-TTD-KI mice (Supplementary Fig. 1C, right panel). Consistent with the BrdU labeling result, quantitative real-time PCR (qRT-PCR) analysis revealed a similar level of stem-cell-specific transcripts of Lgr5 in both Uhrf1-TTD-KI and wild-type mice (Supplementary Fig. 1D). Moreover, the alcian blue staining showed that the intestinal epithelium of Uhrf1-TTD-KI mice was able to differentiate into mature goblet cells as effective as the wild-type mice (Fig. 1E, right panel and Supplementary Fig. 1E). qRT-PCR analysis also revealed a similar level of Lyz1 mRNA, a marker of mature Paneth cells, in wild-type and Uhrf1-TTD-KI mice (Supplementary Fig. 1F). Taken together, we conclude that, despite of an ~10% reduction of global DNA methylation, the small intestine and colorectal tissues in Uhrf1-TTD-KI mice develop and differentiate normally and are free of spontaneous tumors.
Fig. 1. The Uhrf1ki/ki mice are grossly normal with a moderate DNA hypomethylation in intestinal tissues.

A Representative images of Uhrf1+/+ and Uhrf1ki/ki mice. B Growth curves of Uhrf1+/+ and Uhrf1ki/ki mice. Data were based on 14 pairs of littermates of Uhrf1+/+ and Uhrf1ki/ki mice. ns no significant. Error bars, S.E. C Relative levels of DNA methylation in small intestine and colon tissues from Uhrf1+/+ and Uhrf1ki/ki mice. Genomic DNA was prepared from the epithelium of small intestines and colons of three pairs of Uhrf1ki/ki and Uhrf1+/+ littermate mice and subjected to measurement of 5mC by HPLC. Uhrf1+/+, n = 3; Uhrf1ki/ki, n = 3. Error bars, S.E. *p < 0.05. D Western blot analysis showing the levels of Uhrf1, Dnmt3a, and Dnmt1 proteins and H3K9me3 in small intestine and colon tissues from Uhrf1ki/ki and Uhrf1+/+ mice. The protein extracts were prepared from the epithelium cells derived from small intestines and colons of 8-week-old mice. Actin and H3 served as a loading control. E H&E staining (left) and Alcian blue staining (right) of the small intestine tissues from Uhrf1+/+ and Uhrf1ki/ki mice. Shown are samples from 8-week mice. Scale bar, 100 μm. F Volcano plot of RNA-seq data. RNA-seq analysis was performed with RNAs from intestinal epithelium cells derived from three pairs of Uhrf1+/+ and Uhrf1ki/ki mice. Sixty-five upregulated genes (red) and 33 downregulated genes (green) were identified in small intestinal epithelium from Uhrf1ki/ki mice when compared with Uhrf1+/+. G Heat maps showing differentially expressed transposable elements in small intestinal epithelium from Uhrf1+/+ and Uhrf1ki/ki mice.
To characterize the potential effect of a moderate DNA hypomethylation on gene expression in the small intestine, epithelial cells were prepared from small intestine tissues of three pairs of wild-type and Uhrf1-TTD-KI littermate mice. Total RNAs were prepared from epithelial cells and used for gene expression profiling by RNA-seq. This analysis revealed that DNA hypomethylation did not significantly alter the patterns of gene expression, because RNA-seq analysis only identified 60 upregulated genes and 26 downregulated genes in small intestinal epithelial cells from Uhrf1-TTD-KI compared to those from the wild-type control by using p value < 0.05 and fold change >1.5 as cut-off (Fig. 1F and a list of the genes in Supplementary Table 1). Six differentially expressed genes, Acer1, Ceacam10, Fmr1nb, Msr1, Dpcr1, and Slc4a11, were picked for validation by qRT-PCR and the results were in general consistent with the RNA-seq data (Supplementary Fig. 1G). We also compared the expression profile of transposon sequences, because DNA methylation is known to play a role in genome stability by suppressing various transposon elements in the mouse genome. Again, only 70 transposable elements were found to have a moderately altered expression (Fig. 1G and Supplementary Table 2). We therefore conclude that the moderate DNA hypomethylation in Uhrf1-TTD-KI has a relatively mild effect on gene expression in intestinal tissues. Furthermore, since only a small percentage of transposon elements was affected, this moderate DNA hypomethylation does not appear to significantly impact genome stability.
Moderate DNA hypomethylation in Uhrf1-TTD-KI mice is sufficient to suppress chemically induced colorectal cancer
We next tested whether moderate DNA hypomethylation in Uhrf1-TTD-KI mice is sufficient to impact colorectal tumorigenesis induced by a combined treatment of azoxymethane (AOM) and dextran sulfate sodium (DSS). AOM, the metabolite of 1,2-dimethylhydrazine, is a specific colon carcinogen, whereas DSS is a chemical that induces colitis in rodents. The combined use of AOM and DSS can result in 100% incidence of colorectal tumors in rodents40,41. We therefore employed a published protocol as illustrated in Fig. 2A to induce colorectal tumors by AOM/DSS in wild-type and Uhrf1-TTD-KI mice42. At the end of 90 days of experiments, the mice were sacrificed and colorectal tissues were dissected for analysis of tumor numbers and sizes. We found that Uhrf1-TTD-KI mice had a much lower tumor burden than wild-type mice (Fig. 2B, C). Moreover, the Uhrf1-TTD-KI mice had a significantly reduced number of large-size tumors (tumors with diameter L ≥ 3 mm and 3 > L ≥ 2 mm) than wild-type mice (Fig. 2D–E), although the difference in tumor lesions with a size smaller than 2 mm is less significant. This observation was further confirmed by H&E staining, which revealed less tumor lesions in colorectal tissues from Uhrf1-TTD-KI mice than those from the wild-type mice (Fig. 2F). In addition, H&E staining also confirmed that tumors in Uhrf1-TTD-KI mice were smaller in size than those in the wild-type mice (Fig. 2F). Thus, moderate DNA hypomethylation can substantially suppress chemically induced colorectal tumorigenesis in mice.
Fig. 2. Uhrf1ki/ki mice are resistant to AOM/DSS-induced colorectal tumorigenesis.

A Experimental scheme for induction of colorectal tumors in Uhrf1+/+ and Uhrf1ki/ki mice by AOM/DSS treatment. B Representative macroscopic images of AOM/DSS-induced colorectal tumors from Uhrf1+/+ and Uhrf1ki/ki mice. Comparison of total number of tumors per mouse (C) and sizes of tumors (D) from AOM/DSS-treated Uhrf1+/+ and Uhrf1ki/ki mice. The tumor size was measured by stereomicroscope. Error bars, S.E. Uhrf1+/+, n = 19; Uhrf1ki/ki, n = 8. **p < 0.01; *p < 0.05. E Tumors were classified according to the size ranges (L > 3 mm; 3 > L > 2 mm, and L < 2 mm) and plotted as number of tumors per mouse. L: diameter of tumor. Error bars, S.E. Uhrf1+/+ (n = 19); Uhrf1ki/ki (n = 8). **p < 0.01; *p < 0.05. F Representative H&E staining images of colon cross-sections. Tumor lesions in Uhrf1+/+ mice and Uhrf1ki/ki mice are marked in blue or red circle, respectively. Upper scale bars, 2 mm. Bottom scale bars, 50 μm. G DNA-methylation levels in colon tissues from control Uhrf1+/+ and Uhrf1ki/ki mice or tumors from AOM/DSS-treated Uhrf1+/+ and Uhrf1ki/ki mice. For control group, Uhrf1+/+, n = 3; Uhrf1ki/ki, n = 3. For AOM/DSS-treated mice, Uhrf1+/+, n = 6; Uhrf1ki/ki, n = 6. Genomic DNA was prepared from normal epithelium cells or microdissected tumor tissues, and 5mC level was measured by HPLC. Error bars, S.E.
As tumorigenesis often results in global DNA hypomethylation, we assessed if the pattern of differential DNA methylation between Uhrf1-TTD-KI and wild-type mice was persisted in tumors. Genomic DNA was prepared from AOM/DSS-untreated colorectal tissues and AOM/DSS-induced colorectal tumors that were microdissected from both wild-type and Uhrf1-TTD-KI mice, and the levels of 5mC were quantitatively measured by HPLC. The results in Fig. 2G showed that there was a reduced level of DNA methylation in tumors vs. normal colorectal tissues. Furthermore, the level of DNA methylation in tumors from Uhrf1-TTD-KI mice was substantially lower than that in tumors from the wild-type mice, indicating that reduced DNA methylation in Uhrf1-TTD-KI mice was inherited to tumor cells.
Taken together, this chemically induced colorectal tumor model demonstrates that moderate DNA hypomethylation is sufficient to substantially suppress colorectal tumorigenesis and tumor progression in mice.
Moderate DNA hypomethylation also substantially suppresses tumorigenesis and tumor progression in ApcMin/+ mice
Germline mutation of the adenomatous polyposis coli (APC) gene causes familial adenomatous polyposis, a hereditary tumorous predisposition syndrome, which is diagnosed by detection of adenomatous polyps43,44. The multiple intestinal neoplasia (Min) mice (Apcmin/+) carry a T to A transversion mutation in the Apc gene that results in a premature stop codon at codon 85045,46. Like humans with germline mutations in APC, Apcmin/+ mice are predisposed to intestinal adenoma formation and have been extensively used as a model for intestinal tumorigenesis. To investigate how Uhrf1-TTD-KI-induced DNA hypomethylation affects intestinal tumorigenesis in Apcmin/+ mice, male Apcmin/+ mice were crossed with female Uhrf1-TTD-KI mice according to a scheme illustrated in Fig. 3A to generate six different genotypes of mice. At the age of 20 weeks, mice were sacrificed and the number and size of intestinal tumors were analyzed. As expected, no intestinal and colon tumors were observed in Apc+/+ mice for Uhrf1+/+, Uhrf1+/ki and Uhrf1ki/ki genetic background (data not shown). Importantly, comparison of Uhrf1+/+/ApcMin/+ and Uhrf1ki/ki/ApcMin/+ mice revealed a substantially reduced tumor burden in Uhrf1ki/ki/ApcMin/+ mice (Fig. 3B, C). The average number of small intestinal and colorectal tumors in Uhrf1+/+/ApcMin/+ is 44.04 and this is reduced to 8.6 in Uhrf1ki/ki/ApcMin/+ mice (Fig. 3C). Tumor reduction occurred primarily in small intestines, whereas much less tumors were observed in colons (Fig. 3C). Furthermore, the sizes of tumors in Uhrf1ki/ki/ApcMin/+ mice were also much smaller in comparison to tumors in Uhrf1+/+/ApcMin/+ (Fig. 3D and Supplementary Fig. 2). We also analyzed the tumor status according to the anatomical structure (duodenum, jejunum, and ileum). This analysis revealed a more dramatic reduction of tumor numbers in the ileum and jejunum than duodenum (Fig. 3E). To more precisely define the number and sizes of intestinal tumors, we also carried out H&E staining of tissue sections. Representative results in Fig. 3F confirmed a substantial reduction of both tumor number and tumor size in the small intestines of Uhrf1ki/ki/ApcMin/+ mice. Consistent with a substantial reduction in tumor burden, the Uhrf1ki/ki/ApcMin/+ mice had a significantly prolonged lifespan than Uhrf1+/+/ApcMin/+ mice (Fig. 3G). Similar to the results from AOM/DSS-induced tumor model, measurement of 5mC levels in normal tissues from Apc+/+ background and tumors from Apcmin/+ background revealed a substantial reduction of DNA methylation in tumors, and the DNA hypomethylation phenotype is more drastic in tumors from the Uhrf1ki/ki/ApcMin/+ mice (Fig. 3H).
Fig. 3. Uhrf1ki/ki mutation markedly suppresses intestinal tumorigenesis in ApcMin/+mice.

A Mating strategy for generation of Uhrf ki/ki mutation in Apcmin/+ mice. B Representative macroscopic images of small intestinal tumors and colorectal tumors from Uhrf1+/+/Apcmin/+ and Uhrf1 ki/ki/Apcmin/+ mice. The number of intestinal tumors per mouse (C) and the sizes of intestinal tumors (D). Uhrf1+/+/Apc+/+, n = 21; Uhrf1 ki/ki/Apc+/+, n = 16; Uhrf1+/+/Apcmin/+, n = 23; and Uhrf1 ki/ki/Apcmin/+, n = 20 mice. Tumors were classified as total number of tumor, numbers of tumors in small intestine (SI) or colon (CL). Error bars, S.E. ***p < 0.001; **p < 0.01; *p < 0.05. E The numbers of tumors per mouse in duodenum, jejunum, and ileum. Uhrf1+/+/Apc+/+, n = 21; Uhrf1ki/ki/Apc+/+, n = 16; Uhrf1+/+/Apcmin/+, n = 23; and Uhrf1ki/ki/Apcmin/+, n = 20 mice. Error bars, S.E. ***p < 0.001. F Representative H&E staining images of small intestines. Tumor lesions in Uhrf1+/+/Apcmin/+ and Uhrf1ki/ki/Apcmin/+ mice are shown in blue circle and red circle, respectively. Upper scale bars, 2 mm. Bottom scale bars, 50 μm. G Kaplan–Meier curves for overall survival. Uhrf1+/+/Apc+/+, n = 27; Uhrf1ki/ki/Apc+/+, n = 22; Uhrf1+/+/Apcmin/+, n = 37; Uhrf1ki/ki/Apcmin/+, n = 35 mice. Error bars, S.E. ****p < 0.0001. H DNA-methylation levels in normal intestinal tissues or tumors. Uhrf1+/+/Apc+/+, n = 3; Uhrf1 ki/ki/Apc+/+, n = 3; Uhrf1+/+/Apcmin/+, n = 6; and Uhrf1ki/ki/Apcmin/+, n = 3 mice. Genomic DNA was prepared from the small intestinal epithelium cells, and 5mC level was measured by HPLC. Error bars, S.E.
Taken altogether, these results demonstrate that DNA hypomethylation caused by Uhrf1-TTD-KI mutation can markedly suppress intestinal tumorigenesis in ApcMin/+ mice.
DNA hypomethylation caused by Uhrf1-TTD-KI mutation does not affect Apc loss of heterozygosity and activation of Wnt and Hippo pathways
Tumorigenesis in small intestine and colon tissues in ApcMin/+ mice is known to associate with loss of heterozygosity (LOH) of wild-type Apc allele47,48, which results in activation of both Wnt and Hippo pathways49–51. Having established that DNA hypomethylation caused by Uhrf1-TTD-KI mutation substantially suppresses colorectal tumorigenesis induced by AOM/DSS and intestinal tumorigenesis in ApcMin/+ mice, we next investigated the potential mechanisms. To this end, we first analyzed if DNA hypomethylation affected LOH of Apc. According to previous publications, wild-type Apc allele and ApcMin allele could be distinguished by PCR amplification followed by restriction enzyme digestion by HindIII, which generated a 144-bp DNA fragment for the ApcMin allele and a 123-bp fragment for the wild-type Apc allele (Supplementary Fig. 3A). Using this assay, we detected LOH of the Apc allele in small intestine tumors from both Uhrf1+/+/ApcMin/+ and Uhrf1ki/ki/ApcMin/+ mice (Supplementary Fig. 3B). Consistent with the observed LOH of Apc allele, Western blot analysis revealed elevated levels of β-catenin protein in tumors compared to the normal tissues in both Uhrf1+/+/ApcMin/+ and Uhrf1ki/ki/ApcMin/+ mice (Supplementary Fig. 3C). Similarly, Western blot analysis revealed elevated levels of YAP protein in tumors compared to the normal tissues in both Uhrf1+/+/ApcMin/+ and Uhrf1ki/ki/ApcMin/+ mice, in agreement with previous observation49 (Supplementary Fig. 3D). Furthermore, while immunohistochemistry (IHC) analysis revealed cytoplasmic localization of β-catenin in normal tissues, IHC detected obvious nuclear localization of β-catenin in tumor tissues from both Uhrf1+/+/ApcMin/+ and Uhrf1ki/ki/ApcMin/+ mice (Supplementary Fig. 4A, B). Together, these data suggest that there is no significant difference in LOH of Apc and activation of Wnt and Hippo pathways in both Uhrf1+/+/ApcMin/+ and Uhrf1ki/ki/ApcMin/+ mice.
DNA hypomethylation caused by Uhrf1-TTD-KI mutation does not affect intestinal tumor stem cells and tumor cell proliferation
To analyze if DNA hypomethylation suppresses intestinal tumorigenesis in Uhrf1ki/ki/ApcMin/+ mice by affecting tumor stem cells, we examined the levels of intestinal stem-cell marker genes Lgr5, Ascl2, and Sox9 by qRT-PCR. The results showed that compared with the normal tissues, these genes were highly expressed in the tumor tissues, but there was no significant difference between tumors from Uhrf1+/+/ApcMin/+ and Uhrf1ki/ki/ApcMin/+ mice (Supplementary Fig. 5A).
We also analyzed if potential difference in cell proliferation contributes to suppressed tumorigenesis in Uhrf1ki/ki/ApcMin/+ mice. However, IHC analysis revealed no difference in the level of PCNA protein between Uhrf1+/+/ApcMin/+ and Uhrf1ki/ki/ApcMin/+ mice, even though the levels of PCNA were increased in tumors from both mice (Supplementary Fig. 5B, C). This observation was further confirmed by Western blot analysis in Supplementary Fig. 5D, showing that although PCNA was highly expressed in tumors, its levels were similar between Uhrf1+/+/ApcMin/+ and Uhrf1ki/ki/ApcMin/+ mice regardless of normal or tumor tissues. Similarly, IHC analysis showed no significant difference of Ki67, another marker for cell proliferation, between Uhrf1+/+/ApcMin/+ and Uhrf1ki/ki/ApcMin/+ mice in both normal and tumor tissues (Supplementary Fig. 5E, F).
DNA hypomethylation caused by Uhrf1-TTD-KI mutation is not sufficient to activate interferon pathway
Recent studies provide evidence that DNMT inhibitor 5-Aza-C exerts an antitumor therapeutic effect by inducing activation of the interferon pathway25,26. This is mediated by DNA hypomethylation-induced expression of endogenous retrovirus and formation of double-stranded RNAs, which activates the interferon signaling pathway through toll-like receptor signaling and induces apoptosis of tumor cells. To test if DNA hypomethylation in Uhrf1ki/ki/ApcMin/+ mice activated the interferon pathway, we measured the transcript levels of the key members of toll-like receptor signaling and interferon pathways by qRT-PCR. Results in Supplementary Fig. 6 showed no obvious difference in expression of toll-like receptor signaling and interferon pathway components Irf3/7/9, Stat1, and Isg15 between Uhrf1+/+/ApcMin/+ and Uhrf1ki/ki/ApcMin/+ mice in either normal or tumor tissues. Thus, the moderate DNA hypomethylation observed in Uhrf1-TTD-KI mice is not sufficient to activate the interferon pathway in ApcMin/+ background. This observation is consistent with limited alteration of transposable element expression in Uhrf1-TTD-KI mice (Fig. 1F). Thus, we conclude that suppression of intestinal tumorigenesis in ApcMin/+ mice by Uhrf1-TTD-KI-induced DNA hypomethylation is unlikely due to activation of the antiviral interferon pathway.
DNA hypomethylation caused by Uhrf1-TTD-KI mutation promotes intestinal epithelium apoptosis in ApcMin/+ mice
Having excluded cell proliferation and activation of the interferon pathway as potential mechanisms for tumor suppression by Uhrf1-TTD-KI mutation, we examined if elevated apoptosis52,53 occurred in Uhrf1ki/ki/ApcMin/+ mice. We performed TUNEL assay to compare apoptosis in normal intestinal tissues from Apc+/+ and ApcMin/+ background and tumor tissues from ApcMin/+ background. Notably, we observed a marked elevation of apoptosis in normal tissues from Uhrf1ki/ki/ApcMin/+ mice in comparison to those from Uhrf1+/+/ApcMin/+ mice (Fig. 4A, B). In addition, there was also a significantly higher level of apoptosis in the tumor tissues from Uhrf1ki/ki/ApcMin/+ vs. Uhrf1+/+/ApcMin/+ mice (Fig. 4A, B). In support of the above observation, IHC analysis revealed a significantly elevated level of cleaved-caspase-3 (cl-CAPS-3, the active form of caspase-3)-positive cells in normal tissues from Uhrf1ki/ki/ApcMin/+ vs. Uhrf1+/+/ApcMin/+ mice (Fig. 4C). Mirroring the TUNEL assay results, quantitative analysis of cl-CAPS-3 revealed that despite a reduced level of cl-CAPS-3-positive cells in tumors vs. normal tissues, the levels of cl-CAPS-3-positive cells remained significantly higher in tumors from Uhrf1ki/ki/ApcMin/+ than tumors from Uhrf1+/+/ApcMin/+ mice (Fig. 4D). To ensure a correct detection of cl-CAPS-3, we prepared the normal and tumor tissues from small intestines of all four genetic backgrounds and carried out Western blot analysis for both cl-CAPS-3 and the full-length proteins. The representative results in Fig. 4E confirmed a marked increase of cl-CAPS-3 proteins in normal intestinal tissues from Uhrf1ki/ki/ApcMin/+ vs. Uhrf1+/+/ApcMin/+ mice. In accord with the IHC results, the levels of cl-CASP-3 in tumor tissues from Uhrf1ki/ki/ApcMin/+ mice were clearly lower than that in normal tissues, suggesting a suppressed apoptosis in tumors. Nevertheless, comparison between tumors from Uhrf1ki/ki/ApcMin/+ and Uhrf1+/+/ApcMin/+ mice revealed a higher level of cl-CAPS-3 in tumors from Uhrf1ki/ki/ApcMin/+ mice. Notably, Western blot analysis revealed an elevated level of full-length caspase-3 that was detected in normal small intestinal tissues of Uhrf1-TTD-KI mice in comparison to Uhrf1-WT mice (Fig. 4F). The increase of full-length caspase-3 was more obvious when compared between Uhrf1ki/ki/ApcMin/+ and Uhrf1+/+/ApcMin/+ mice (Fig. 4F). Thus, while increased full-length caspase-3 levels can be attributed to Uhrf1-TTD-KI mutation, cleaved caspase-3 and activation of apoptosis seem to occur primarily in Uhrf1ki/ki/ApcMin/+ mice, suggesting that loss of Apc somehow leads to cleavage of caspase-3 and activation of apoptosis.
Fig. 4. Elevated apoptosis and active caspase-3 in the intestinal tissues of Uhrf1ki/ki/Apcmin/+ mice.

A, B TUNEL assay showing levels of apoptosis in the intestinal sections from different mice. Representative confocal microscope images of TUNEL assay (A). Nuclei were stained with DAPI. Scale bar, 25 μm. Quantitative results of TUNEL-positive cells per 0.01 mm2 (B). Three mice were analyzed for each genotypes. Apcmin/+-Tumor, tumor samples from Uhrf1+/+/Apcmin/+ and Uhrf1ki/ki/Apcmin/+ mice. Error bars, S.E. C, D Representative immunohistochemistry images showing the cleaved-caspase-3 (cl-CASP3) levels in small intestinal tissue sections from different mice (C). Scale bar, 50 μm. The number of cl-CASP3-positive cells were analyzed by ImageJ software (D). Three mice were analyzed for each genotypes. Error bars, S.E. Western blot results showing the levels of cl-CASP3 (E) and full-length caspase-3 (F-CASP3) (F) in small intestinal epithelium cells from different mice as indicated. The relative levels of cl-CASP3 and F-CASP3 were determined by ImageJ software. G Western blot results showing the levels of cleaved PRAP (cl-PARP) and full-length PARP (F-PARP) proteins in small intestinal epithelium cells from different mice as indicated. Note elevated levels of cl-PARP proteins were observed in small intestinal epithelium cells from Uhrf1ki/ki/Apcmin/+ mice. H Western blot results showing the levels of cleaved-caspase-3 (cl-CASP3) proteins in liver tissues and small intestine epithelium cells from different mice as indicated. Note that cl-CASP3 was not detected in liver tissues from Uhrf1ki/ki/Apcmin/+ mice.
Poly ADP-ribose polymerase (PARP) is an important substrate of cleaved caspase-354. During activation of apoptosis, the 116-kDa PARP is cleaved into 31 and 85-kDa fragments by activated caspase-3. In further support of elevated apoptosis in small intestines of Uhrf1ki/ki/ApcMin/+ mice, Western blot analysis detected an increased level of cleaved PARP in small intestines from Uhrf1ki/ki/ApcMin/+ mice but not in small intestines from Uhrf1+/+/ApcMin/+ mice or Uhrf1ki/ki and Uhrf1+/+ mice (Fig. 4G).
DNA hypomethylation in Dnmt1 hypomorphic mice has been shown to impact tumorigenesis in a tissue-specific manner, suppressing tumorigenesis in small intestines and promoting tumorigenesis in livers15. Having unraveled an increased apoptosis in small intestines of Uhrf1ki/ki/ApcMin/+ mice, we wondered if increased apoptosis also occurs in liver. However, as shown in Fig. 4H, we found that the elevated cl-CAPS-3 levels were specifically detected in small intestinal tissues and were absent in liver tissues from Uhrf1ki/ki/ApcMin/+ mice.
Altogether, these data provide evidence that increased apoptosis occurs specifically in the intestinal tissues and is likely a mechanism for DNA hypomethylation-induced suppression of intestinal tumors in Uhrf1ki/ki/ApcMin/+ mice.
Enhancer DNA hypomethylation correlates with increased caspase-3 expression in Uhrf1ki/ki/ApcMin/+ mice
Having observed elevated levels of both cleaved and full-length caspase-3 in the intestinal tissues of Uhrf1ki/ki/ApcMin/+ mice, we next examined if elevated expression of caspase-3 occurs at the level of transcription. Indeed, qRT-PCR analysis revealed an elevated level of caspase-3 mRNA in the small intestinal epithelium of Uhrf1ki/ki/ApcMin/+ mice vs. that of Uhrf1+/+/ApcMin/+ mice (Fig. 5A). Elevated caspase-3 mRNA was also observed in the small intestinal epithelium of Uhrf1ki/ki mice vs. that of Uhrf1+/+ mice, although the results were not statistically significant (Fig. 5A).
Fig. 5. Activation of caspase-3 gene expression in Uhrf1ki/ki/Apcmin/+ mice correlates with reduced DNA methylation in the caspase-3 enhancer regions.

A qRT-PCR analysis showing elevated levels of caspase-3 mRNA in small intestinal epithelium cells from Uhrf1ki/ki/Apcmin/+ mice. The levels of caspase-3 mRNA were determined from three pairs of littermate mice and were normalized to Actin. *p < 0.05; ns, p > 0.05. Three mice were analyzed for each genotypes. Error bars, S.E. B Diagrams illustrating the epigenetic marks H3K4me1 and H3K27ac landscapes at the mouse caspase-3 gene. The profiles were derived from small intestinal tissue ChIP-seq data in UCSC genome browser. The putative enhancers, from enhancer 1 to 6, were assigned according to overlapping H3K27ac and H3K4me1 peaks. As a reference, also shown are the DNA-methylation status within or upstream of casepase-3 gene derived from E7.5 mouse embryos (downloaded from Methbank database). C Summary of bisulfite sequencing results of DNA-methylation status within promoter, enhancer 1 and enhancer 6 regions. Each CpG sites were sequenced over hundred or thousand times by high-throughput Illumina® (ND102-0102) sequencing and thus the data were highly accurate. Note that the caspase-3 promoter region in small intestinal epithelium cells is essentially unmethylated in both Uhrf1+/+/Apcmin/+ and Uhrf1ki/ki/Apcmin/+ mice. D Caspase-3 enhancer 1 and enhancer 6 contain multiple binding sites for Wnt-regulated transcription factors TCF1/2, AP-1, and c-jun. The two highly demethylated CpG sites are marked with triangle.
We hypothesized that DNA hypomethylation in the caspase-3 gene promoter and/or enhancers is likely the mechanism for elevated caspase-3 gene transcription in Uhrf1ki/ki/ApcMin/+ mice. To test this hypothesis, we first identified the mouse intestinal caspase-3 enhancer regions according to the overlapping peaks of enhancer H3K4me1 and H3K27ac epigenetic marks in UCSC genome browser (Fig. 5B). We also downloaded DNA-methylation data of mouse caspase-3 locus from methbank (https://bigd.big.ac.cn/methbank/jbrowse) (Fig. 5B). This data mining showed the caspase-3 gene promoter as a CpG island with extremely low DNA methylation (Fig. 5B). In addition, six enhancer regions upstream of the caspase-3 promoter could be assigned according to overlapped H3K4me1 and H3K27ac peaks. Among these six putative enhancers, four (putative enhancers 2–5) were relatively poor in the CpG content and thus were not analyzed further. We prepared small intestinal epithelial cells from Uhrf1+/+ and Uhrf1ki/ki mice in both control and Apcmin/+ background and carried out high-throughput bisulfite sequencing analysis for enhancer 1, enhancer 6, and the promoter, and the results were shown in Fig. 5C. Consistent with the data in methbank, we found that the promoter CpG island was extremely low in methylation and there was no significant difference between the small intestinal samples from Uhrf1+/+, Uhrf1ki/ki, Uhrf1ki/ki/ApcMin/+, and Uhrf1+/+/ApcMin/+ mice. Comparison between Uhrf1+/+ and Uhrf1ki/ki in control background revealed a modest reduction of DNA methylation across enhancers 1 and 6 (Fig. 5C, lower panel), consistent with the global DNA hypomethylation phenotype in Uhrf1ki/ki mice. Interestingly, a more significant reduction of DNA methylation was observed for both enhancer 1 and enhancer 6 between Uhrf1ki/ki/ApcMin/+ and Uhrf1+/+/ApcMin/+ mice, especially at two CpG sites within the enhancer 6, which exhibit a near complete demethylation. Notably, sequence analysis revealed enhancers 1 and 6 contain multiple putative binding sites for transcription factors TCF1/2, AP-1 and c-Jun, the transcription factors that are typically activated by canonical and noncanonical Wnt pathways55–57. We thus proposed that loss of Apc-induced activation of Wnt pathways activate transcription factors TCF1/2, AP-1, and c-jun, which selectively activate caspase-3 gene transcription in the small intestinal epithelium of Uhrf1ki/ki/ApcMin/+ mice by binding more efficiently to hypomethylated caspase-3 enhancers. In addition, the binding of AP-1/c-jun may further promote DNA demethylation.
Discussion
Virtually all human cancers display aberrant patterns of DNA methylation, including global hypomethylation punctuated by regional hypermethylation of gene promoters that often lead to tumor-suppressor gene silencing1–5. Global DNA hypomethylation occurs early in tumorigenesis, implying a driving role in oncogenesis. By treating Dnmt1+/–ApcMin/+ mice weekly with a low dose of 5-azaC, Laird et al. first demonstrated that 5-azaC-induced DNA hypomethylation almost completely suppresses intestinal adenomas in Dnmt1+/–ApcMin/+ mice20. Subsequent studies with various Dnmt1 hypomorphic mice collectively demonstrated that genomic DNA hypomethylation has dual roles in tumorigenesis, strongly suppressing intestinal adenomas15,18, squamous tongue, and esophagus carcinogenesis58, and overall tumorigenesis in prostate cancer14, but promoting T-cell lymphoma11, intestinal microadenoma15, and early stages of prostate cancer and liver cancer14,15. Global DNA hypomethylation is thus proposed to promote tumorigenesis in cell types relying on genome instability and mutation for carcinogenesis and suppress tumorigenesis in cells where DNA-methylation-dependent silencing of tumor-suppressor genes is required. In this study, we used a Uhrf1-TTD-KI mouse model to investigate the role and mechanism by which DNA hypomethylation affects intestinal tumorigenesis (Fig. 1). We show that a moderate DNA hypomethylation in this mouse model is sufficient to suppress intestinal tumorigenesis (Figs. 2 and 3). Extensive mechanistic studies link the suppressed intestinal tumorigenesis to activation of caspase-3 gene expression and activity and increased apoptosis (Fig. 4 and Supplementary Figs. 3–6). The activation of caspase-3 expression is likely due to DNA hypomethylation in caspase-3 enhancers (Fig. 5). However, it remains to be tested if the activation of caspase-3 enhancers is casual to activation of apoptosis and tumor suppression observed in Uhrf1ki/ki DNA hypomethylation mouse model. Future studies are needed to elucidate further the mechanisms by which DNA hypomethylation suppresses intestinal tumorigenesis.
Previous studies of global DNA hypomethylation on tumorigenesis in mice all rely on Dnmt1 hypomorphic mouse models with a variable but substantial reduction of Dnmt1 protein. As Dnmt1 is known to interact with various proteins and thus may possess DNA-methylation-independent functions59–61, additional Dnmt1-independent models would be valuable for further defining and investigating the diverse roles of DNA hypomethylation in cancers. The Uhrf1-TTD-KI mice used in this study are an ideal and complementary model for the following reasons. First, Uhrf1 is an essential accessory protein for Dnmt1-catalyzed DNA maintenance methylation, and the Uhrf1-TTD-KI mutation drives a moderate DNA hypomethylation in mice without affecting Dnmt1 protein level (Fig. 1). Second, unlike some Dnmt1 hypomorphic mice, Uhrf1-TTD-KI mice display no observable phenotypes in development and postnatal growth (Fig. 1). Although a direct comparison between Dnmt1 hypomorphic mice and Uhrf1-TTD-KI mice in the levels of DNA hypomethylation is not yet available, the latter mice are likely less severe in DNA hypomethylation, as only ~10% reduction of global DNA methylation was detected in various tissues that we have tested (Fig. 1) and activation of interferon pathway is not observed (Supplementary Fig. 6). Nevertheless, by using chemical-induced and ApcMin/+ intestinal tumor models, we demonstrated that the moderate DNA hypomethylation caused by Uhrf1-TTD mutation is sufficient to markedly suppress intestinal tumorigenesis. Interestingly, unlike studies with Dnmt1 hypomorphic mice, both micro- and macroadenoma were suppressed in Uhrf1-TTD-KI mice (Fig. 3). This discrepancy in microadenoma formation is likely complicated and warrants for future studies. One possibility is that the degree of DNA hypomethylation may dictate whether it promotes or suppresses intestinal microadenoma. In this regard, a conditional but complete deletion of Dnmt1 in adult ApcMin/+ mice was shown to accelerate rather than suppress intestinal adenoma initiation62. Alternatively, the enhanced microadenoma observed in Dnmt1 hypomorphic mice could be due to reduced Dnmt1 protein level and not a result of DNA hypomethylation.
In defining the molecular mechanism(s) underlying suppressed intestinal tumorigenesis in Uhrf1-TTD-KI mice, we found no evidence for differences in Apc LOH, activation of Wnt and Hippo pathways, and cell proliferation (Supplementary Figs. 3–5). Instead, we detected significantly increased apoptosis in intestinal tissues of Uhrf1ki/ki/ApcMin/+ mice as compared to Uhrf1+/+/ApcMin/+ mice (Fig. 4). Consistent with elevated apoptosis, we detected significantly increased levels of activated caspase-3 as well as full-length caspase proteins in intestinal tissues of Uhrf1ki/ki/ApcMin/+ mice vs. Uhrf1+/+/ApcMin/+ mice. Notably, elevated caspase-3 expression and activation were observed in intestinal but not liver tissues, consistent with a previous report showing DNA hypomethylation suppresses intestinal tumor formation but promotes liver carcinogenesis15. It is also noteworthy that the caspase-3 levels in tumors were actually reduced when compared to normal intestinal tissues of Uhrf1ki/ki/ApcMin/+ mice, consistent with the notion that apoptosis is generally suppressed during tumorigenesis. The facts that caspase-3 and apoptosis are markedly elevated in normal intestinal tissues of Uhrf1ki/ki/ApcMin/+ mice are consistent with our observation that both micro- and macro-adenomas are suppressed in Uhrf1ki/ki/ApcMin/+ mice. Thus, elevated expression and activation of caspase-3 and the resulting elevated apoptosis are likely a mechanism for intestinal tumor suppression by DNA hypomethylation in Uhrf1ki/ki/ApcMin/+ mice. However, activation of the Casp3 enhancer may not be the sole cause of reduced tumorigenesis. Despite elevated expression of caspase 3, it still could be an effect, rather than a cause, of the increased apoptosis. In addition, it remains to be tested if elevated expression and activation of caspses-3 is sufficient to suppress intestinal tumorigenesis. Furthermore, we could not exclude that other possibilities including activation of other components in the apoptosis pathway, activation of other cell death pathway or impaired H3K9me2/3 binding of UHRF1 may also contribute to the tumor suppression phenotype.
We found that enhancer DNA hypomethylation rather than promoter demethylation is associated with increased caspase-3 gene expression. Promoter hypermethylation is widely believed to account for epigenetic silencing of tumor-suppressor genes occurring during tumorigenesis. Our extensive DNA-methylation analysis revealed that the CpG island caspase-3 promoter was unmethylated in Uhrf1ki/ki/ApcMin/+ and Uhrf1+/+/ApcMin/+ mice. However, we found substantially reduced DNA methylation in two putative caspase-3 enhancer regions (Fig. 5C). This observation points to the possibility that enhancer DNA hypomethylation is likely to contribute to elevated caspase-3 expression and apoptosis in Uhrf1ki/ki/ApcMin/+ mice. This finding is also consistent with recent genome-wide DNA-methylation studies showing widespread DNA-methylation changes in enhancer regions under physiologic and pathologic conditions63–65. Notably, there is no significant change in caspase-3 expression in the intestinal tissues between Uhrf1ki/ki and Uhrf1+/+ mice or between ApcMin/+ and Apc+/+ mice, suggesting a combinatorial effect of DNA hypomethylation and activation of Wnt signaling pathways in driving caspase-3 expression and apoptosis. In support of this hypothesis, bisulfite sequencing analysis revealed a less significant reduction of DNA methylation in caspase-3 enhancers in Uhrf1ki/ki mice as compared to Uhrf1+/+ mice (Fig. 5C, lower panel). These results suggest a working model in Fig. 6, in which activation of Wnt signaling pathways leads to activation of transcription factors TCF1/2, AP-1, and c-Jun, the downstream effectors of Wnt canonical and noncanonical pathways. The binding of one or more of these transcription factors may initiate further DNA demethylation of caspase-3 enhancer regions and activation of caspase-3 expression, which contributes to increased apoptosis and tumor suppression in intestinal tissues of Uhrf1ki/ki/ApcMin/+ mice.
Fig. 6. DNA hypomethylation and Wnt pathway cooperatively activate caspase-3 expression to suppress intestinal tumorigenesis.
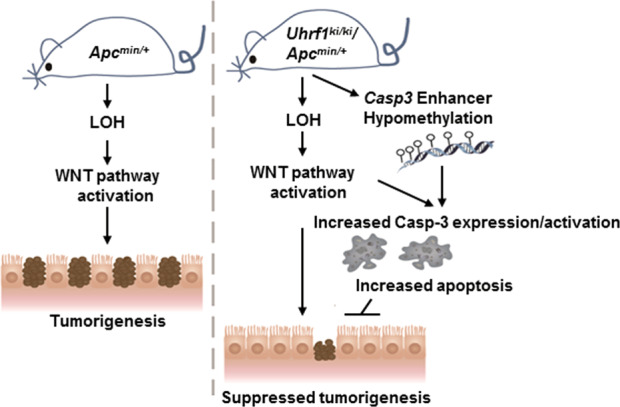
Working model illustrates how DNA hypomethylation suppresses intestinal tumorigenesis by promoting caspase-3 expression and apoptosis. In Uhrf1ki/ki mice or Apcmin/+ mice, DNA hypomethylation or activation of Wnt pathway alone is not sufficient to significantly enhance caspase-3 gene expression. However, DNA hypomethylation caused by Uhrf1-TTD-KI mutation can promote caspase-3 transcriptional activation by Wnt canonical and noncanonical pathway-activated transcription factors such as TCF1/2, AP-1, and c-jun and β-catenin, which in turn leads to elevated apoptosis and tumor suppression.
In sum, we present a novel Uhrf1-TTD-KI mouse model for investigating global DNA hypomethylation in tumorigenesis. Using this mouse model, we demonstrated that moderate global DNA hypomethylation is sufficient to markedly suppress intestinal tumorigenesis and provided evidence that enhanced caspase-3 expression driven by enhancer DNA hypomethylation and activation of Wnt pathways may lead to elevated apoptosis and suppression of intestinal tumorigenesis (Fig. 6). Targeting epigenetic modifications including DNA methylation has recently emerged as an important therapeutic approach for colorectal cancers66,67. Our finding that moderate DNA hypomethylation is sufficient to markedly suppress intestinal tumorigenesis suggests potentially using low doses of DNA-methylation inhibitors as a means of colorectal cancer prevention.
Material and methods
Animals
All mice were maintained in the Animal Research Center of East China Normal University under pathogen-free conditions at 22 ± 2 °C. All mice were maintained in the C57Bl/6J genetic background. All animal experiments were performed in accordance with guidelines and regulations drafted by the Association for Assessment and Accreditation of Laboratory Animal Care in Shanghai (Ethical number: m20141202 and m20191206). Intestinal tumors were counted by using stereomicroscope without gender selection. Littermate genotype controls were used in all experiments. The animal experiments were repeated twice at least, and the numbers of animals (at least three for each group) in the experimental groups were described in the figure legends.
Antibodies
The following primary antibodies were used in this study: c-Myc (Abcam, ab84132), GAPDH (Absci, AB41549), ACTIN (Huaan, M1210-2), UHRF1 (homemade), DNMT3A (Santa Cruz, sc-20703), DNMT1 (Huaan, ET1702-77), H3K9me3 (homemade), H3 (Abcam, ab1791), β-catenin (Huaan, ER080), Ki67 (Huaan, ER1706-46), AXIN1 (CST, 2087), GSK-3β (Klean AB, P1018 48), Caspase-3 (CST, 9662), cleaved-Caspase-3 (CST, 9664), and PARP (CST, 9532).
Uhrf1-TTD-KI, Apcmin/+ and compound mouse models
Uhrf1-TTD-KI mice were described previously39. To generate compound Uhrf1-TTD-KI and Apcmin/+ mice, Uhrf1ki/ki mice were mated with Apcmin/+ to produce F1 generations. Then male Uhrf1ki/+/Apcmin/+ were selected to cross with female Uhrf1ki/+/Apc+/+ to obtain the F2 generation with four different genotypes: Uhrf1ki/ki/Apcmin/+, Uhrf1+/+/Apcmin/+, Uhrf1ki/ki/Apc+/+, and Uhrf1+/+/Apc+/+ mice. The mice were genotyped by DNA sequencing or PCR of genomic DNA derived from mouse toe with specific primers as listed in Supplementary Table 3. The mice were sacrificed at 20 weeks of age for examining intestinal and colorectal tumors.
AOM/DSS model
Induction of colorectal tumors in wild-type and Uhrf1ki/ki mice by AOM/DSS was carried out essentially as described42. In brief, mice (2.5-month old) were treated with a single intraperitoneal dose (10 mg/kg of body weight) of AOM (sigma, A5486) followed by three 5-day cycles of 2% DSS in drinking water, with each cycle followed by a 2-week recovery. The mice were sacrificed at 90 days from initial AOM injection for analyzing colorectal tumors by both stereomicroscope and tissue section staining. The mice were sacrificed at 5.5-month old.
Paraffin tissue section, H&E staining, and Alcian blue staining
Small intestine or colon tissues were dissected and removed from the sacrificed mice. The lumens of the intestine tissues were carefully flushed with ice-cold PBS using a 50-ml syringe and intragastric needle. Cleaned intestine tissues were rolled-up and fixed in 4% paraformaldehyde, dehydrated and embedded into paraffin. Five micrometer sections were cut and stained with H&E or 1% Alcian blue (pH 2.5). The alcian blue staining should be counterstained with nuclear fast red.
Immunohistochemistry (IHC)
IHC staining were performed using paraffin-embedded tissues. Antigen retrieval was performed in citrate buffer (1.8% 0.1-M citric acid, 8.2% 0.1-M sodium citrate) for 20 min at 100 °C; sections were washed in PBS, incubated for 10 min in 3% H2O2 (for IHC), blocked in goat serum from kit (Vector PK-4001), incubated for 16 h at 4 °C in primary antibody, and washed in PBS. Secondary antibody reaction was performed using kits (Vector PK-4001 and SK-4100, VECTOR) according to manufacturer’s instruction. The slides were counterstained with hematoxylin and embedded using Entellan (Merck Millipore). The following antibodies were used: anti-mouse β-catenin (Huaan, ER080, 1:500), anti-mouse Ki67 (Huaan, ER1706-46, 1:100), anti-mouse cleaved-Caspase-3 (Cell Signaling Technology, 9664, 1:100). Images were taken using Olympus BX53 microscope.
BrdU immunofluorescence staining
Mice were intraperitoneally injected with BrdU (Sigma, 100 mg/kg) 5 h or 50 h before sacrificed. BrdU immunofluorescence staining was performed using paraffin-embedded tissues. Antigen retrieval was performed in citrate buffer (1.8% 0.1-M citric acid, 8.2% 0.1-M sodium citrate) for 20 min at 100 °C; sections were washed in PBS, incubated for 10 min in 3% H2O2, blocked in goat serum from kit (Vector PK-4001), incubated for 16 h at 4 °C with anti-BrdU (Sigma-Aldrich, B928, 1:50). Then, sections were incubated with Alexa Fluor 488 goat anti-mouse IgG (Life Technologies) and DAPI (1 μg/ml, Sigma-Aldrich) for 1 h at room temperature, washed, and embedded using FluorSave (Merck Millipore). Images were taken using Leica DM4000 microscope.
TUNEL assay
TUNEL assay was performed by the TUNEL Apoptosis Detection kit (Yeasen biotech, 40308ES20) according to manufacturer’s instruction. The slides were visualized by fluorescence confocal microscope (Leica SP8).
Intestinal epithelium isolation and polyp microdissection
Isolation of small intestinal epithelium for analysis was performed essentially as previously described68. Polyps were microdissected from small intestine under ×10–20 magnification on a dissecting microscope, and were homogenized in the RIPA buffer for Western blotting lysis and Total Trizol for RNA extraction or genomic DNA extraction.
Western blotting analysis
Small intestinal epithelial cells or tumor tissues that were first broken up by a tissue grinding apparatus were lysed with the RIPA buffer on ice for 2 h. After centrifugation at 12,000 rpm for 10 min at 4 °C, the clean extracts were obtained and subjected to Western blotting analysis essentially as described69. The immunereactive proteins were detected by the Odyssey laser digital imaging system.
Measurement of 5mC by HPLC
Genomic DNA was prepared from intestinal epithelium, mouse tissues, or tumors and dissolved in ddH2O. The measurement of 5mC levels was performed as described39.
qRT-PCR analysis
Total RNAs were extracted from the intestinal epithelium cells or microdissected fresh tumor tissues by using the Total Trizol extraction from TOROIVD (A211-01). cDNAs were synthesized using the reverse transcription kit TOYOBO (FSQ-301). Subsequent PCRs were performed using the TOROGreen® qPCR Master Mix (TOROIVD, QST-100) on the QuantStudio 3 Real-Time PCR System (Applied Biosystems) using the comparative Ct quantitation method. Results were normalized to actin B mRNA levels. PCR protocol was as follows: initial denaturing at 95 °C for 5 min, 40 cycles of 95 °C for 20 s, 60 °C for 30 s, and 72 °C for 30 s, followed by final extension at 72 °C for 5 min. Melt curve analysis was performed on a QuantStudio 3 Real-Time PCR System (Applied Biosystems) coupled with QuantStudio™ Design & Analysis Software. All primer sequences for qRT-PCR analyses are listed in Supplementary Table 3.
Bisulfite sequencing
Bisulfite conversion was performed using the EZ DNA Methylation-GoldTM Kit (ZYMO Research) according to manufacturer’s instruction manual. Bisulfite-converted DNA was used in PCR amplification by TaKaRa Ex Taq HS (Takara, RR006A). The PCR products were gel-purified and used for Illumina® (ND102-0102) sequencing. The levels of methylation at each CpG sites were calculated based on large number (up to thousands) of sequenced DNA as described69. The primers for PCR amplification are listed in Supplementary Table 3.
RNA-sequencing analysis
Total RNAs were prepared from the small intestinal epithelial cells of three pairs of Uhrf1ki/ki and Uhrf1+/+ littermates. RNA-seq analysis was performed by Berry Genomics Co., Ltd. (Beijing, China). Gene expression analysis was performed as described70.
Statistical analysis
All statistical analysis was performed via GraphPad Prism 5.0 software. All values are depicted as the mean ± SEM. Student t test was used in analyzing the statistical significance of experiments comparing two groups. All Western blotting experiments were done at least for three times. Overall survival of different groups of mice was evaluated by the Kaplan–Meier survival analysis and the log-rank test. Differences were considered statistically significant at p < 0.05. The positive signals of β-catenin, cleaved-caspase-3, Ki67, and PCNA were analyzed by using ImageJ Analysis.
Supplementary information
Acknowledgements
We thank Dr Cheng-Ming Chiang (University of Texas Southwestern Medical Center) for critical reading of the manuscript. We thank members of Wong’s Laboratory for valuable discussion. This study is supported by grants from the Ministry of Science and Technology of China (2017YFA054201) and the National Natural Science Foundation of China (31730048 and 81530078 to J.W., 31900453 to J.L.).
Data availability
Murine small intestinal caspase-3 H3K4me1 and H3K27ac genomic profiles were downloaded from data in UCSC genome browser: http://genome.ucsc.edu/cgi-bin/hgTracks?db=mm9&lastVirtModeType=default&lastVirtModeExtraState=&virtModeType=default&virtMode=0&nonVirtPosition=&position=chr8%3A47681543-47745322&hgsid=920520527_JiUBTKq6u7USrIL8Z6vC3aEnNYBR.
Conflict of interest
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Xiaoya Duan, Yuanyong Huang
Supplementary information
The online version contains supplementary material available at 10.1038/s41389-021-00328-9.
References
- 1.Ehrlich M. DNA methylation in cancer: too much, but also too little. Oncogene. 2002;21:5400–5413. doi: 10.1038/sj.onc.1205651. [DOI] [PubMed] [Google Scholar]
- 2.Feinberg AP, Vogelstein B. Hypomethylation distinguishes genes of some human cancers from their normal counterparts. Nature. 1983;301:89–92. doi: 10.1038/301089a0. [DOI] [PubMed] [Google Scholar]
- 3.Kulis M, Esteller M. DNA methylation and cancer. Adv. Genet. 2010;70:27–56. doi: 10.1016/B978-0-12-380866-0.60002-2. [DOI] [PubMed] [Google Scholar]
- 4.Laird PW, Jaenisch R. DNA methylation and cancer. Hum. Mol. Genet. 1994;3 Spec No:1487–1495. doi: 10.1093/hmg/3.suppl_1.1487. [DOI] [PubMed] [Google Scholar]
- 5.Pfeifer, G. P. Defining driver DNA methylation changes in human cancer. Int. J. Mol. Sci. 19, 1166 10.3390/ijms19041166. (2008). [DOI] [PMC free article] [PubMed]
- 6.Akhavan-Niaki H, Samadani AA. DNA methylation and cancer development: molecular mechanism. Cell Biochem. Biophys. 2013;67:501–513. doi: 10.1007/s12013-013-9555-2. [DOI] [PubMed] [Google Scholar]
- 7.Baylin SB, Jones PA. A decade of exploring the cancer epigenome—biological and translational implications. Nat. Rev. Cancer. 2011;11:726–734. doi: 10.1038/nrc3130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jones PA, Baylin SB. The fundamental role of epigenetic events in cancer. Nat. Rev. 2002;3:415–428. doi: 10.1038/nrg816. [DOI] [PubMed] [Google Scholar]
- 9.Taberlay PC, Jones PA. DNA methylation and cancer. Prog. Drug Res. 2011;67:1–23. doi: 10.1007/978-3-7643-8989-5_1. [DOI] [PubMed] [Google Scholar]
- 10.Timp W, Feinberg AP. Cancer as a dysregulated epigenome allowing cellular growth advantage at the expense of the host. Nat. Rev. Cancer. 2013;13:497–510. doi: 10.1038/nrc3486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gaudet F, et al. Induction of tumors in mice by genomic hypomethylation. Science. 2003;300:489. doi: 10.1126/science.1083558. [DOI] [PubMed] [Google Scholar]
- 12.Trinh BN, Long TI, Nickel AE, Shibata D, Laird PW. DNA methyltransferase deficiency modifies cancer susceptibility in mice lacking DNA mismatch repair. Mol. Cell. Biol. 2002;22:2906–2917. doi: 10.1128/MCB.22.9.2906-2917.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mazor T, et al. DNA methylation and somatic mutations converge on the cell cycle and define similar evolutionary histories in brain tumors. Cancer Cell. 2015;28:307–317. doi: 10.1016/j.ccell.2015.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kinney SRM, et al. Opposing roles of Dnmt1 in early- and late-stage murine prostate cancer. Mol. Cell. Biol. 2010;30:4159–4174. doi: 10.1128/MCB.00235-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yamada Y, et al. Opposing effects of DNA hypomethylation on intestinal and liver carcinogenesis. Proc. Natl Acad. Sci. USA. 2005;102:13580–13585. doi: 10.1073/pnas.0506612102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Oghamian S, et al. Reduction of pancreatic acinar cell tumor multiplicity in Dnmt1 hypomorphic mice. Carcinogenesis. 2011;32:829–835. doi: 10.1093/carcin/bgr039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen T, et al. Complete inactivation of DNMT1 leads to mitotic catastrophe in human cancer cells. Nat. Genet. 2007;39:391–396. doi: 10.1038/ng1982. [DOI] [PubMed] [Google Scholar]
- 18.Eads CA, Nickel AE, Laird PW. Complete genetic suppression of polyp formation and reduction of CpG-island hypermethylation in Apc(Min/+) Dnmt1-hypomorphic Mice. Cancer Res. 2002;62:1296–1299. [PubMed] [Google Scholar]
- 19.Hatano Y, et al. Reducing DNA methylation suppresses colon carcinogenesis by inducing tumor cell differentiation. Carcinogenesis. 2015;36:719–729. doi: 10.1093/carcin/bgv060. [DOI] [PubMed] [Google Scholar]
- 20.Laird PW, et al. Suppression of intestinal neoplasia by DNA hypomethylation. Cell. 1995;81:197–205. doi: 10.1016/0092-8674(95)90329-1. [DOI] [PubMed] [Google Scholar]
- 21.Chen RZ, Pettersson U, Beard C, Jackson-Grusby L, Jaenisch R. DNA hypomethylation leads to elevated mutation rates. Nature. 1998;395:89–93. doi: 10.1038/25779. [DOI] [PubMed] [Google Scholar]
- 22.Howard G, Eiges R, Gaudet F, Jaenisch R, Eden A. Activation and transposition of endogenous retroviral elements in hypomethylation induced tumors in mice. Oncogene. 2008;27:404–408. doi: 10.1038/sj.onc.1210631. [DOI] [PubMed] [Google Scholar]
- 23.Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat. Rev. 2012;13:484–492. doi: 10.1038/nrg3230. [DOI] [PubMed] [Google Scholar]
- 24.Schubeler D. Function and information content of DNA methylation. Nature. 2015;517:321–326. doi: 10.1038/nature14192. [DOI] [PubMed] [Google Scholar]
- 25.Chiappinelli KB, et al. Inhibiting DNA methylation causes an interferon response in cancer via dsRNA including endogenous retroviruses. Cell. 2015;162:974–986. doi: 10.1016/j.cell.2015.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Roulois D, et al. DNA-demethylating agents target colorectal cancer cells by inducing viral mimicry by endogenous transcripts. Cell. 2015;162:961–973. doi: 10.1016/j.cell.2015.07.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bostick M, et al. UHRF1 plays a role in maintaining DNA methylation in mammalian cells. Science. 2007;317:1760–1764. doi: 10.1126/science.1147939. [DOI] [PubMed] [Google Scholar]
- 28.Sharif J, et al. The SRA protein Np95 mediates epigenetic inheritance by recruiting Dnmt1 to methylated DNA. Nature. 2007;450:908–912. doi: 10.1038/nature06397. [DOI] [PubMed] [Google Scholar]
- 29.Arita K, Ariyoshi M, Tochio H, Nakamura Y, Shirakawa M. Recognition of hemi-methylated DNA by the SRA protein UHRF1 by a base-flipping mechanism. Nature. 2008;455:818–821. doi: 10.1038/nature07249. [DOI] [PubMed] [Google Scholar]
- 30.Avvakumov GV, et al. Structural basis for recognition of hemi-methylated DNA by the SRA domain of human UHRF1. Nature. 2008;455:822–825. doi: 10.1038/nature07273. [DOI] [PubMed] [Google Scholar]
- 31.Hashimoto H, et al. The SRA domain of UHRF1 flips 5-methylcytosine out of the DNA helix. Nature. 2008;455:826–829. doi: 10.1038/nature07280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Karagianni P, Amazit L, Qin J, Wong J. ICBP90, a novel methyl K9 H3 binding protein linking protein ubiquitination with heterochromatin formation. Mol. Cell. Biol. 2008;28:705–717. doi: 10.1128/MCB.01598-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nishiyama A, et al. Uhrf1-dependent H3K23 ubiquitylation couples maintenance DNA methylation and replication. Nature. 2013;502:249–253. doi: 10.1038/nature12488. [DOI] [PubMed] [Google Scholar]
- 34.Rajakumara E, et al. PHD finger recognition of unmodified histone H3R2 links UHRF1 to regulation of euchromatic gene expression. Mol. Cell. 2011;43:275–284. doi: 10.1016/j.molcel.2011.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nady N, et al. Recognition of multivalent histone states associated with heterochromatin by UHRF1 protein. J. Biol. Chem. 2011;286:24300–24311. doi: 10.1074/jbc.M111.234104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rothbart SB, et al. Association of UHRF1 with methylated H3K9 directs the maintenance of DNA methylation. Nat. Struct. Mol. Biol. 2012;19:1155–1160. doi: 10.1038/nsmb.2391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xie S, Jakoncic J, Qian C. UHRF1 double tudor domain and the adjacent PHD finger act together to recognize K9me3-containing histone H3 tail. J. Mol. Biol. 2012;415:318–328. doi: 10.1016/j.jmb.2011.11.012. [DOI] [PubMed] [Google Scholar]
- 38.Ferry L, et al. Methylation of DNA ligase 1 by G9a/GLP recruits UHRF1 to replicating DNA and regulates DNA methylation. Mol. Cell. 2017;67:550–565.e5. doi: 10.1016/j.molcel.2017.07.012. [DOI] [PubMed] [Google Scholar]
- 39.Zhao Q, et al. Dissecting the precise role of H3K9 methylation in crosstalk with DNA maintenance methylation in mammals. Nat. Commun. 2016;7:12464–12464. doi: 10.1038/ncomms12464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bissahoyo A, et al. Azoxymethane is a genetic background-dependent colorectal tumor initiator and promoter in mice: effects of dose, route, and diet. Toxicol. Sci. 2005;88:340–345. doi: 10.1093/toxsci/kfi313. [DOI] [PubMed] [Google Scholar]
- 41.Clapper ML, Cooper HS, Chang WC. Dextran sulfate sodium-induced colitis-associated neoplasia: a promising model for the development of chemopreventive interventions. Acta Pharmacol. Sin. 2007;28:1450–1459. doi: 10.1111/j.1745-7254.2007.00695.x. [DOI] [PubMed] [Google Scholar]
- 42.Neufert C, Becker C, Neurath MF. An inducible mouse model of colon carcinogenesis for the analysis of sporadic and inflammation-driven tumor progression. Nat. Protoc. 2007;2:1998–2004. doi: 10.1038/nprot.2007.279. [DOI] [PubMed] [Google Scholar]
- 43.Groden J, et al. Identification and characterization of the familial adenomatous polyposis coli gene. Cell. 1991;66:589–600. doi: 10.1016/0092-8674(81)90021-0. [DOI] [PubMed] [Google Scholar]
- 44.Miyoshi Y, et al. Germ-line mutations of the APC gene in 53 familial adenomatous polyposis patients. Proc. Natl Acad. Sci. USA. 1992;89:4452–4456. doi: 10.1073/pnas.89.10.4452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Moser AR, et al. ApcMin, a mutation in the murine Apc gene, predisposes to mammary carcinomas and focal alveolar hyperplasias. Proc. Natl Acad. Sci. USA. 1993;90:8977–8981. doi: 10.1073/pnas.90.19.8977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Su LK, et al. Multiple intestinal neoplasia caused by a mutation in the murine homolog of the APC gene. Science. 1992;256:668–670. doi: 10.1126/science.1350108. [DOI] [PubMed] [Google Scholar]
- 47.Levy DB, et al. Inactivation of both APC alleles in human and mouse tumors. Cancer Res. 1994;54:5953–5958. [PubMed] [Google Scholar]
- 48.Luongo C, Moser AR, Gledhill S, Dove WF. Loss of Apc+ in intestinal adenomas from Min mice. Cancer Res. 1994;54:5947–5952. [PubMed] [Google Scholar]
- 49.Cai J, Maitra A, Anders RA, Taketo MM, Pan D. beta-Catenin destruction complex-independent regulation of Hippo-YAP signaling by APC in intestinal tumorigenesis. Genes Dev. 2015;29:1493–1506. doi: 10.1101/gad.264515.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Clevers H. Wnt/beta-catenin signaling in development and disease. Cell. 2006;127:469–480. doi: 10.1016/j.cell.2006.10.018. [DOI] [PubMed] [Google Scholar]
- 51.Gregorieff A, Liu Y, Inanlou MR, Khomchuk Y, Wrana JL. Yap-dependent reprogramming of Lgr5(+) stem cells drives intestinal regeneration and cancer. Nature. 2015;526:715–718. doi: 10.1038/nature15382. [DOI] [PubMed] [Google Scholar]
- 52.Hengartner MO. The biochemistry of apoptosis. Nature. 2000;407:770–776. doi: 10.1038/35037710. [DOI] [PubMed] [Google Scholar]
- 53.Porter AG, Janicke RU. Emerging roles of caspase-3 in apoptosis. Cell Death Differ. 1999;6:99–104. doi: 10.1038/sj.cdd.4400476. [DOI] [PubMed] [Google Scholar]
- 54.Tewari M, et al. Yama/CPP32 beta, a mammalian homolog of CED-3, is a CrmA-inhibitable protease that cleaves the death substrate poly(ADP-ribose) polymerase. Cell. 1995;81:801–809. doi: 10.1016/0092-8674(95)90541-3. [DOI] [PubMed] [Google Scholar]
- 55.Behrens J. Control of beta-catenin signaling in tumor development. Ann. N. Y. Acad. Sci. 2000;910:33–25. doi: 10.1111/j.1749-6632.2000.tb06698.x. [DOI] [PubMed] [Google Scholar]
- 56.Gerdes MJ, et al. Activator protein-1 activity regulates epithelial tumor cell identity. Cancer Res. 2006;66:7578–7588. doi: 10.1158/0008-5472.CAN-06-1247. [DOI] [PubMed] [Google Scholar]
- 57.Sancho R, et al. JNK signalling modulates intestinal homeostasis and tumourigenesis in mice. EMBO J. 2009;28:1843–1854. doi: 10.1038/emboj.2009.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Baba S, et al. Global DNA hypomethylation suppresses squamous carcinogenesis in the tongue and esophagus. Cancer Sci. 2009;100:1186–1191. doi: 10.1111/j.1349-7006.2009.01171.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hashimoto H, Vertino PM, Cheng X. Molecular coupling of DNA methylation and histone methylation. Epigenomics. 2010;2:657–669. doi: 10.2217/epi.10.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jeltsch A, Jurkowska RZ. Allosteric control of mammalian DNA methyltransferases—a new regulatory paradigm. Nucleic Acids Res. 2016;44:8556–8575. doi: 10.1093/nar/gkw723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Svedruzic ZM. Dnmt1 structure and function. Prog. Mol. Biol. Transl. Sci. 2011;101:221–254. doi: 10.1016/B978-0-12-387685-0.00006-8. [DOI] [PubMed] [Google Scholar]
- 62.Sheaffer KL, Elliott EN, Kaestner KH. DNA hypomethylation contributes to genomic instability and intestinal cancer initiation. Cancer Prev. Res. (Philos.) 2016;9:534–546. doi: 10.1158/1940-6207.CAPR-15-0349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Heyn H, et al. Epigenomic analysis detects aberrant super-enhancer DNA methylation in human cancer. Genome Biol. 2016;17:11. doi: 10.1186/s13059-016-0879-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ordonez, R., Martinez-Calle, N., Agirre, X. & Prosper, F. DNA methylation of enhancer elements in myeloid neoplasms: think outside the promoters? Cancers11, 1424 10.3390/cancers11101424. (2009). [DOI] [PMC free article] [PubMed]
- 65.Yang X, Gao L, Zhang S. Comparative pan-cancer DNA methylation analysis reveals cancer common and specific patterns. Brief. Bioinform. 2017;18:761–773. doi: 10.1093/bib/bbw063. [DOI] [PubMed] [Google Scholar]
- 66.Parizadeh SM, et al. Epigenetic drug therapy in the treatment of colorectal cancer. Curr. Pharm. Des. 2018;24:2701–2709. doi: 10.2174/1381612824666180730151904. [DOI] [PubMed] [Google Scholar]
- 67.Rezapour S, Hosseinzadeh E, Marofi F, Hassanzadeh A. Epigenetic-based therapy for colorectal cancer: Prospect and involved mechanisms. J. Cell Physiol. 2019;234:19366–19383. doi: 10.1002/jcp.28658. [DOI] [PubMed] [Google Scholar]
- 68.Madison BB, et al. LIN28B promotes growth and tumorigenesis of the intestinal epithelium via Let-7. Genes Dev. 2013;27:2233–2245. doi: 10.1101/gad.224659.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Li J, et al. USP7 negatively controls global DNA methylation by attenuating ubiquitinated histone-dependent DNMT1 recruitment. Cell Discov. 2020;6:58. doi: 10.1038/s41421-020-00188-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wei, et al. Class I histone deacetylases are major histone decrotonylases: evidence for critical and broad function of histone crotonylation in transcription. Cell Res. 2017;27:898–915. doi: 10.1038/cr.2017.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Murine small intestinal caspase-3 H3K4me1 and H3K27ac genomic profiles were downloaded from data in UCSC genome browser: http://genome.ucsc.edu/cgi-bin/hgTracks?db=mm9&lastVirtModeType=default&lastVirtModeExtraState=&virtModeType=default&virtMode=0&nonVirtPosition=&position=chr8%3A47681543-47745322&hgsid=920520527_JiUBTKq6u7USrIL8Z6vC3aEnNYBR.