

Abstract
Metabolic transformation is a hallmark of cancer and a critical target for cancer therapy. Cancer metabolism and behavior are regulated by cell-intrinsic factors, as well as metabolite availability in the tumor microenvironment (TME). This metabolic niche within the TME is shaped by four tiers of regulation, including: 1) intrinsic tumor cell metabolism, 2) interactions between cancer and non-cancerous cells, 3) tumor location and heterogeneity, and 4) whole-body metabolic homeostasis. Here, we will define these modes of metabolic regulation and review how distinct cell types contribute to the metabolite composition of the TME. Finally, we will connect these insights to understand how each of these tiers offers a unique therapeutic potential to modulate the metabolic profile and function of all cells inhabiting the TME.
Keywords: tumor microenvironment, cell heterogeneity, metabolism, tumor niches, mitochondria, cancer, immunity
1. Introduction
Metabolic rewiring of tumor cells is essential for the initiation, proliferation and progression of cancer1. Tumors remodel metabolite consumption in order to generate molecular products such as co-factors and building blocks2. For example, the early studies by Otto Warburg were based on the observations that tumors consume glucose and secrete lactate even in the presence of oxygen. Warburg attributed this phenomenon to a defective oxidative metabolism3. Subsequent studies have shown that respiration is required for tumor growth and have contributed significantly to our understanding of the molecular underpinnings of these observations4–7. Historically, cell-intrinsic factors, such as the genomic landscape and cell signaling were considered key drivers of cancer cell metabolism, proliferation and survival8–10. However, emerging studies have highlighted an exciting role for the unique niche formed by the tumor microenvironment (TME) in shaping the metabolic landscape of a tumor. Yet, we still do not fully appreciate all of the elements that contribute to the metabolic composition within the TME. Furthermore, we are only beginning to understand how altered metabolite levels in the TME contributes to cancer metabolism and behavior11–14.
The TME represents the immediate niche surrounding the tumor and is composed of a variety of cell types in a unique metabolic landscape. Whereas blood vessels supply oxygen and nutrients and are responsible for the removal of waste products, stromal and immune cells modulate tumor growth through the secretion of signaling molecules and extracellular matrix (ECM) components15. Due to the high metabolic activity of cancer cells, dysfunctional blood flow, and increased inflammation, the TME is characterized by deregulated physiological properties. Indeed, the tumor niche has been described as hypoxic, acidic, nutrient-deprived, and characterized by electrolyte imbalance and elevated oxidative stress16–22. These constraints lead to a constant struggle for adaptation in which tumor cells often have an advantage over non-cancerous cell types. As metabolites are major determinants of cancer, immune, and stromal cell fate, modulation of the local metabolite availability represents a novel aspect to influence tumor progression.
In this review, we focus on four modes of regulation that may contribute to the metabolite niche within the TME (Figure 1), including: 1) intrinsic tumor cell metabolism, 2) competition and cross-talk between different cell types, 3) tumor location and heterogeneity, and 4) whole-body metabolic homeostasis. Throughout this review, we will discuss the potential for these insights to improve therapies. Understanding how metabolism influences and is affected by the complexity of cellular interactions within the TME will provide a deeper understanding of the mechanisms that control cancer and may identify new opportunities to improve patient care.
Figure 1. Schematic representation of the metabolic fluctuations/niches that influence the metabolite composition of the TME.
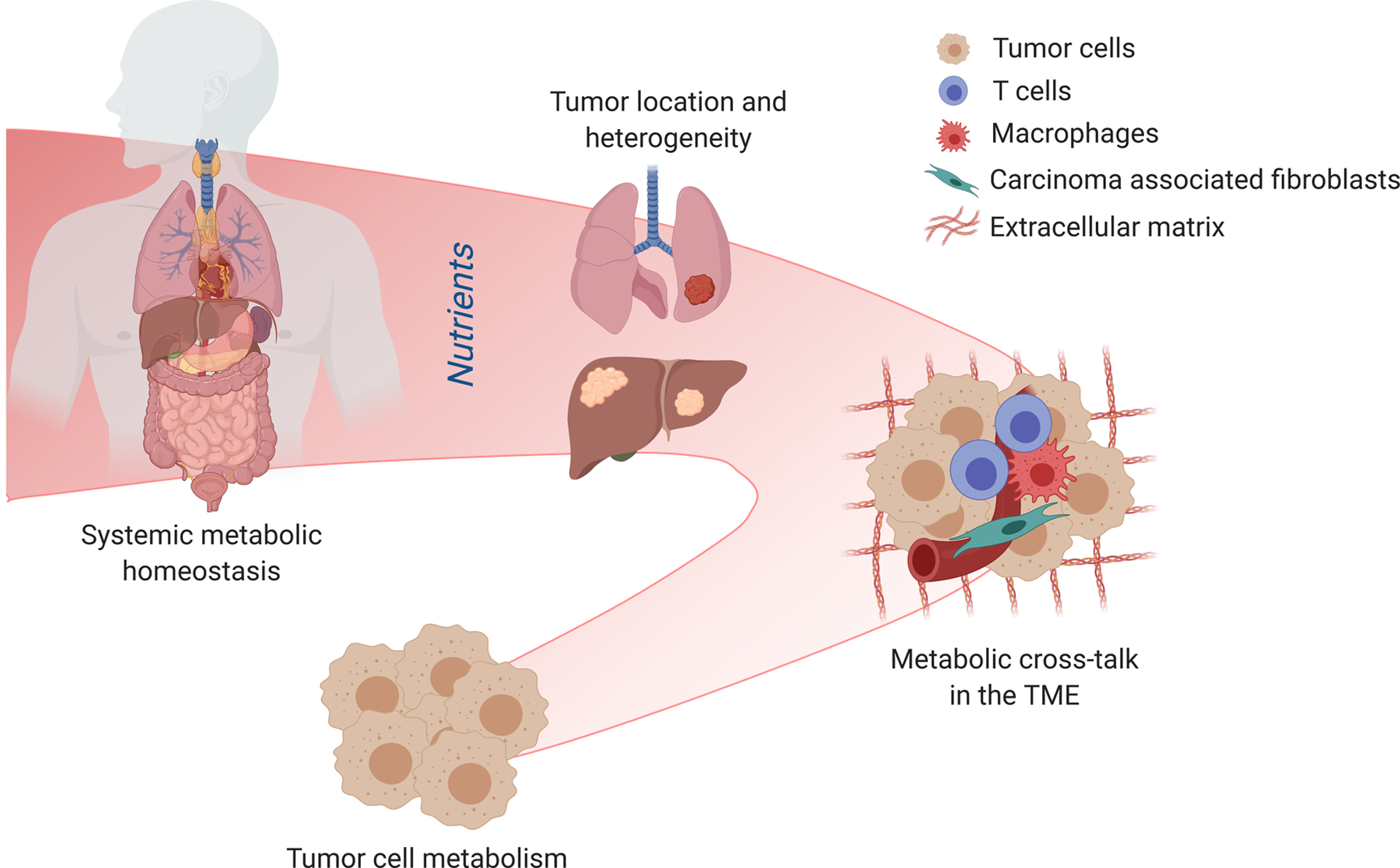
The metabolite composition of the TME is determined by different levels of regulation. Primarily, the local nutrient availability is defined by tumor cell metabolism and through the metabolic cross-talk of tumor cells, infiltrating immune cells and supporting stromal cells. All these cell types change the TME through the consumption and secretion of metabolites, and are influenced by the resulting conditions. Nevertheless, the metabolic microenvironment is also determined by the anatomical location of the tumor. Metabolite heterogeneity has been found in different organs and even in sublocal organ and tumor locations, due to tissue structure, levels of perfusion and function. Lastly, differences in metabolite availability can also arise from a change in systemic metabolism. This could be dietary interventions, function of the metabolic organs or metabolic syndromes. Although each of these levels contribute to the heterogeneity of the TME, they all possess specific therapeutic potential.
2. Tumor cell metabolism
Cancer cells possess the ability to adjust fuel utilization in order to support their requirements of cellular function, proliferation, and survival even under dynamic conditions2. Indeed, metabolic reprogramming is a hallmark of cancer, and several metabolic enzymes have been found to be valuable drug targets for cancer therapy23. Yet, no uniform tumor-specific metabolic signature has been identified. Instead, different tumors display distinct and heterogeneous metabolic programs, making it challenging to target metabolism in different tumor types. What features contribute to distinct patterns in metabolic usage? Although some common themes exist, cancer cells seem to be highly opportunistic and adapt to local metabolite levels of the TME. How do local metabolite levels determine tumor growth? In this section, we will touch on these questions and describe the influence of the TME on tumor cell metabolism.
2.1. Tumor metabolism exploits and shapes metabolite availability in the TME
Tumor cells must adapt their metabolism to meet the increased demands for energy, biomass precursors, and co-factors. Enhanced aerobic glycolysis, also known as the Warburg effect, has emerged as the most obvious and widespread metabolic adaptation sustaining cancer progression3. Indeed, most tumors are characterized by a massive increase in glucose consumption and lactate secretion, as exemplified by the wide use of PET imaging in tumors24. Aerobic glycolysis enables cancer cells, even in the presence of oxygen, to convert large amounts of glucose-derived pyruvate to lactate through the activity of lactate dehydrogenase (LDH). High levels of lactate secretion allow for the regeneration of NAD+ necessary to sustain glycolysis. In most somatic cells, pyruvate is mainly converted into mitochondrial acetyl-CoA or oxaloacetate by the enzymes pyruvate dehydrogenase (PDH) and pyruvate carboxylase (PC), respectively. These metabolites are oxidized to CO2 in a process known as oxidative phosphorylation (OXPHOS), while consuming oxygen and generating NADH and FADH2, which are utilized by the electron transport chain (ETC) to produce ATP. Therefore, aerobic glycolysis is a low-efficiency process for energy generation compared with mitochondrial ATP generation; synthesizing 2 or 36 molecules of ATP via glycolysis or OXPHOS, respectively25. Finally, glycolysis is utilized to generate large amounts of biomolecules for biomass production6,26. In tumors, glycolytic intermediates are diverted to generate precursors for the biosynthesis of anabolic molecules, and molecules controlling the cellular redox state. For example, serine synthesis from glucose through phosphoglycerate dehydrogenase (PHGDH) sustains protein, heme, and nucleotide biosynthesis even when serine availability is low27–29. Additionally, PHGDH activity results in the conversion of glutamate to α-ketoglutarate in order to replenish the tricarboxylic acid (TCA) cycle, thus allowing pyruvate-derived acetyl-coA to be used for fatty acid synthesis30,31. Furthermore, the glycolytic intermediate glucose-6-phosphate is sidetracked into the pentose phosphate pathway for the production of nucleotides and reducing power under the form of NADPH, which is in turn used for both lipid synthesis and modulation of the redox balance32–34. In summary, these studies demonstrate that tumor cells directly exploit glucose metabolism within the TME35.
The ability of cancer cells to reprogram their metabolism is especially important in environments with diverse metabolite composition. Studies in both mice and humans have actually identified lactate to be recycled by cancer cells, and to promote proliferation through fueling of mitochondrial metabolism in non-small-cell lung carcinoma (NSCLC)36 (Figure 2). Therefore, it would be interesting to examine whether this phenotype extends to other tumor cell types and whether lactate recycling is dependent on the biochemical composition of the TME as a whole37.
Figure 2. Tumor metabolism influences and is influenced by the metabolite composition of the TME.
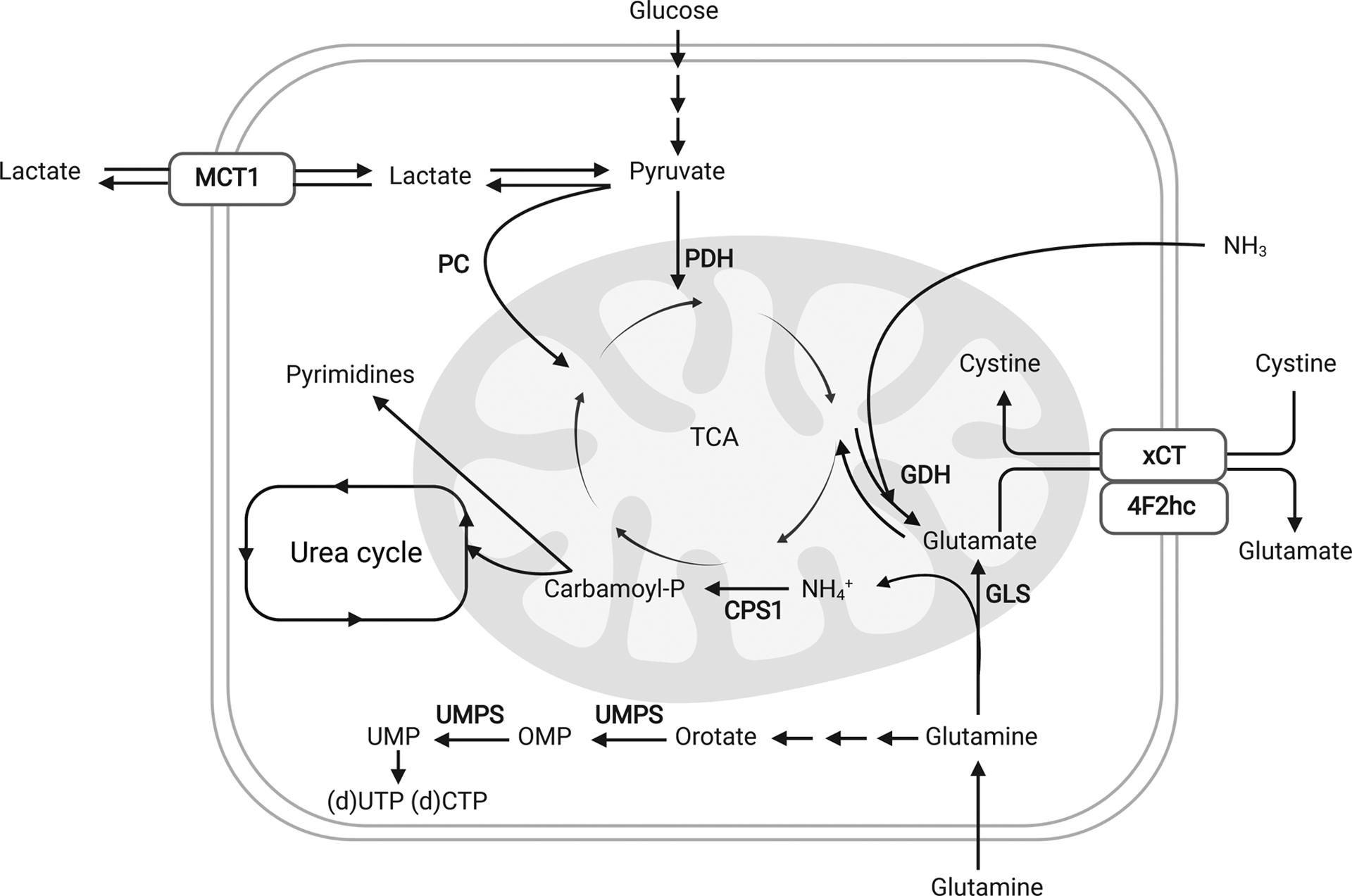
Tumor cells have been shown to adapt to the TME and to take advantage of local metabolite compositions in order to sustain tumor growth, proliferation and survival. Aside from the well-studied Warburg effect, tumor cells also recycle waste products such as ammonia and lactate in order to sustain biomass production. Furthermore, specifically in vivo, low levels of cystine reduce the activity of the antiporter xCT/SLC7A11 and thus glutamate export. The high intracellular glutamate pool limits glutamine catabolism and anaplerosis. Finally, high uric adic levels in the plasma inhibit UMP synthase (UMPS) and pyrimidine synthesis.
Beyond glucose and lactate, tumors utilize numerous fuels such as fatty acids, amino acids, and proteins (reviewed in DeBerardinis R.J. et al)4. The role of glutamine in cancer has been extensively studied. Glutamine is converted into glutamate and ammonia by the enzyme glutaminase (GLS) in a process known as glutaminolysis38. Glutamate has essential roles in supporting biomass production, contributing to redox homeostasis and modulating signaling pathways39,40. For example, glutamate can be used in many cancers for the production of aspartate which allows cancer cells with a severely dysfunctional ETC to proliferate. Interestingly, aspartate is limiting in tumors and its deficiency impairs protein and nucleotide synthesis. Thus aspartate availability could be targeted for therapy41–43.
The process of glutaminolysis generates the metabolic byproduct ammonia, which has been less investigated, but is emerging as an important metabolite40,44. In accordance with the lactate studies, high levels of extracellular ammonia accumulate within the TME and were shown to support amino amino acid production in estrogen receptor (ER)-positive breast cancers via recycling through glutamate dehydrogenase (GDH) to produce glutamate and downstream amino acids38 (Figure 2). Ammonia can also be utilized by carbamoyl phosphate synthetase-1 (CPS1) in KRAS/LKB1 lung cancer to maintain pyrimidine pools and DNA synthesis45 (Figure 2). Thus, ammonia can fuel tumor cell growth via multiple mechanisms. In total, these studies demonstrate that cancer cells intrinsically rewire fuel utilization to develop metabolic flexibility in order to fulfill their energy and biomass requirements.
2.2. Metabolic Adaptation of Cancers
As tumors are so dependent on metabolites for growth and survival, they have evolved mechanisms to adjust to changes in the nutrient composition of their environment. This rapid metabolic adaptation of cancer cells to the TME highlights the importance of studying tumor biology in an appropriate and physiological nutritional context
Therefore, several studies have focused on understanding how the levels of major carbon sources such as glucose and glutamine affect metabolic reprogramming and finally cancer proliferation and survival. In this regard, metformin treatment of cancer cells in low glucose conditions enhances cancer cell death by decreasing ATP levels both in vitro and in vivo46. Furthermore, high glucose concentrations were found to mask the importance of mitochondrial activity and susceptibility to OXPHOS inhibitors such as biguanides47. In accordance, RAS-driven NSCLC seems to sustain OXPHOS through glutaminolysis in glutamine high in vitro conditions, while the same cells fuel mitochondrial metabolism through glucose-dependent PDH and PC activity in vivo12. Moreover, although glutaminase inhibition was identified as a viable therapy to halt cell proliferation in culture, most inhibitors have failed to provide therapeutic benefit in in vivo cancer models48. Mechanistically, glutamine addiction was shown to be dependent on local levels of cystine. As the antiporter xCT/SLC7A11 mediates the transport of glutamate and cystine across the plasma membrane, cystine import allows for glutamate secretion and glutamine catabolism. Low levels of cystine in vivo thus make cancer cells less dependent on glutamine49 (Figure 2). These changes may have implications for tumor survival and proliferation in vivo, as low glutamine conditions may impair cancer cell survival after DNA damage56. Collectively, these studies indicate that modulation of cell culture conditions greatly influence cellular identity and recapitulate metabolic dependencies that converge with the observation made in animal models51–54.
In accordance, the development of plasma-like media has led to the discovery of unexpected carbon and nitrogen sources that hold relevance in vivo. For example, a high concentration of uric acid, normally absent in standard culture media, inhibits UMP synthase and pyrimidine synthesis making cancer cells more sensitive to the chemotherapeutic agent 5-fluorouracil55 (Figure 2). In contrast, supraphysiological pyruvate and arginine levels in standard culture media were shown to stabilize a hypoxia-inducible factor 1-alpha (HIF1α)-dependent transcriptional program or reverse the urea cycle reaction catalyzed by the enzyme argininosuccinate lyase, respectively. Notably, these cellular processes were not recapitulated when the cells were cultured in physiological nutrient conditions53. Taken together, these studies demonstrate that physiological metabolite levels allow for the identification of new vulnerabilities that would be otherwise difficult to uncover by using standard culture media.
Besides modulating metabolite levels, cell culture systems can be utilized to recapitulate unique and distinct cellular phenotypes of cancer cells important in vivo. Although many insights are based on two dimensional (2D) monolayer cell culture, several studies have shown the physiological relevance of three dimensional (3D) organoid cultures56–59 in which 2D cultures promoted proliferation, while 3D cultures displayed migration and invasion phenotypes. 3D culture systems identified proline and pyruvate as key metabolites essential for lung metastasis colonization but not primary breast cancer growth56,60. Furthermore, 3D organotypic culture systems were used as a model to measure metabolic differences between proliferating and quiescent epithelial cells. While proliferating cells utilize glutamate to produce non-essential amino acids, quiescent cells upregulate GDH expression57. Notably, a full recapitulation of the cancer phenotype also requires physiological metabolite levels. For example, breast cancer spheroids cultured in physiological conditions better recapitulate the metabolic profile of mammary tumors53.
In conclusion, the tumor phenotype is shaped by the metabolite composition of the local environment. Novel culture systems capable of recapitulating physiological metabolite conditions as well as novel approaches to quantify cellular metabolism in vivo are of key importance to investigate metabolic adaptation to the TME.
3. The metabolic cross-talk between cells in the TME
The TME is inhabited by both cancer and non-cancer cells, all of which have potential to be influenced by local metabolites61–65. The close proximity of multiple cell types within this niche raises a unique question – how does the metabolism of nearby, non-cancerous cells affect cancer cell metabolism and growth in the TME? In this section, we will describe the influence of the TME on the cross-talk between cancer cells and the surrounding immune or stromal cells.
3.1.1. T cells and tumor cells
The TME is comprised of a complex milieu of immune cells, including cells of the innate immune response such as natural killer (NK) cells, macrophages, and dendritic cells, and cells of the adaptive immune response such as CD8+ and CD4+ T cells. While NK cells and CD8+ T cells are classified as cytotoxic lymphocytes, CD4+ T cells support (Th1, Th17) or repress (Treg) the activity of other immune cells including CD8+ T cells. Cytotoxic CD8+ T cells offer a natural defense against tumor progression through the specific killing of tumor cells after recognition of tumor-associated antigens64. Therefore, researchers are now aiming to harness T cells for cancer therapy through potentiating adoptively transferred T cell responses.
In the last decade, metabolic pathways have emerged as unexpected regulators of T cell fate, function and differentiation65–67. When T cells interact with an antigen and induce signaling through the T cell receptor (TCR), a cascade of events is initiated that triggers rapid metabolic remodeling. These metabolic alterations are not simply bystander events, but altering metabolism can directly alter immune cell fate and function (reviewed in Buck M.D. et al)65. Thus, metabolic pathways and the response to metabolite availability represent unexpected arms of regulation that may have profound impacts on T cell activity. As T and tumor cells both rely on metabolism, it is important to define similarities and differences between metabolism of immune cells and cancers to discern potential metabolic cross-talk or competition within the TME. This mechanistic insight may reveal novel strategies to inhibit tumor growth while maximizing anti-tumor immunity.
3.1.2. T and cancer cells compete for glucose within the TME
Highly proliferative activated T cells and cancer cells both rely heavily on glucose metabolism. Limiting availability of glucose within the TME causes cellular competition, as some glycolytic tumors exhibit very low CD8+ T cell infiltration and proliferation68,69. Glycolysis is also important to promote T cell effector function by sustaining interferon-gamma (IFNγ) production. Mechanistically, the glycolytic enzyme glyceraldehyde 3-phosphate dehydrogenase (GAPDH) binds the AU-rich region in the 3’ UTR of cytokine mRNA and reduces protein translation70. Nonetheless, in a separate study, Peng and colleagues have demonstrated that LDHA maintains high levels of acetyl coenzyme A that in turn regulates histone acetylation and transcription of Ifng gene independently of its 3’ UTR71. Beside GAPDH, low levels of glucose also impair T cell function through the regulation of mammalian target of rapamycin (mTOR). Reduced activity of mTOR and phosphorylation of ribosomal protein S6 kinase beta-1 (p70S6K) diminish the transcription of IFNγ in CD8+ T cells72. Glucose deprivation does not only inhibit T cell proliferation and function, but also regulates T cell fate73. For example, CD4+ T cells utilize glucose to sustain the production of the glycolytic intermediate phosphoenolpyruvate (PEP) which restores Ca2+ - nuclear factor of activated T cells (NFAT) signaling and support the anti-tumor response74. Last, in addition to glucose limitation, accumulation of the glycolytic product lactate was shown to also be deleterious for T cell effector function and anti-tumor response by suppressing proliferation and cytokine production75 (Figure 3). Moreover, the accumulation of lactate in B16 melanoma tumors strongly impairs CD8+ T and NK cell infiltration and activity in vitro and in vivo76.
Figure 3. The metabolic cross-talk between tumor cells and immune or stromal cells within the TME.
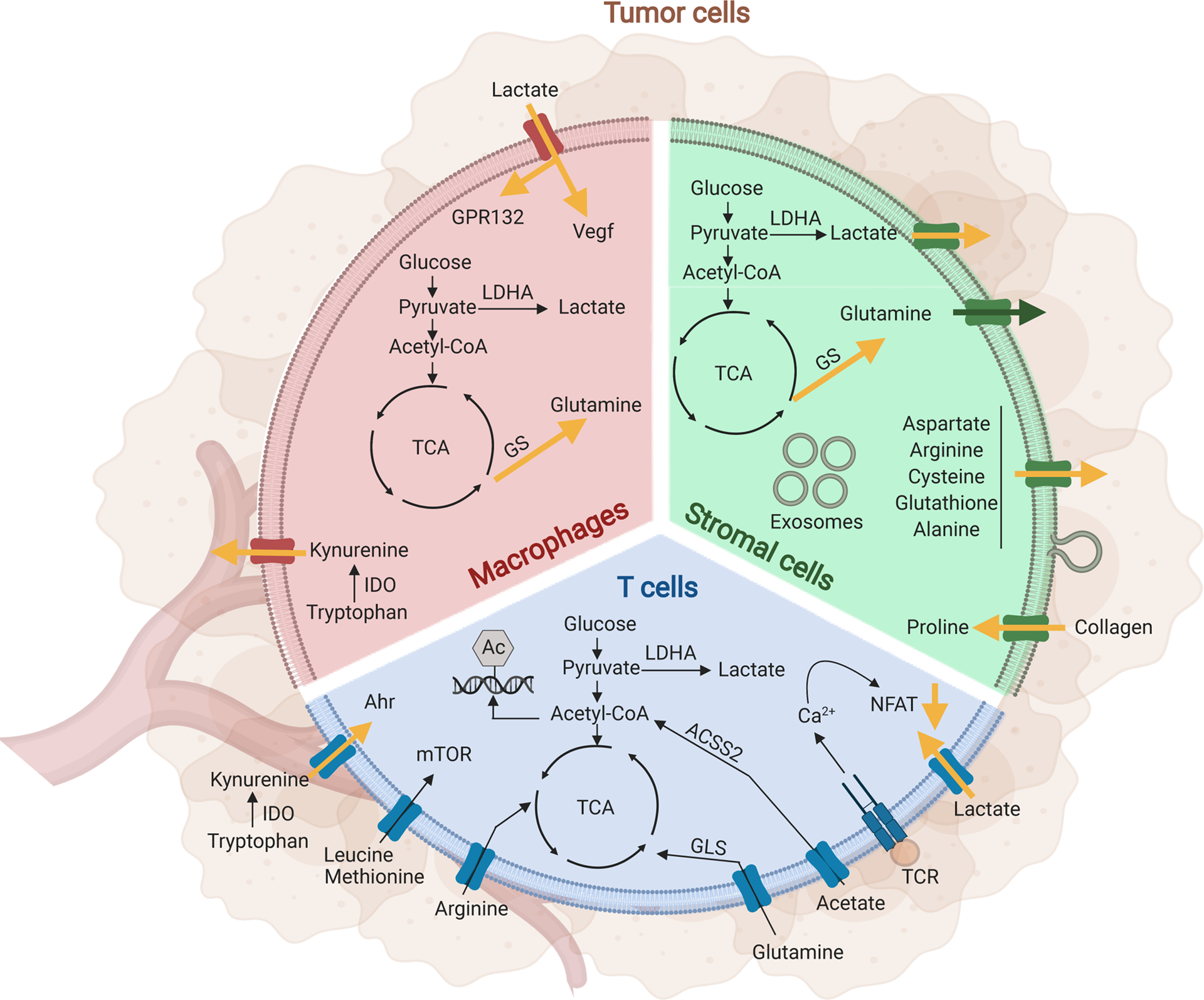
The metabolite composition of the TME is shaped by stromal and immune cells, each with unique metabolic profiles, dependencies, and vulnerabilities compared to tumor cells. Here we depict the metabolic interactions of T cells (blue), macrophages (red) or stromal cells (green) with tumor cells (brown) in the TME. Yellow arrows indicate upregulation.
Despite the importance of glucose and lactate levels, T cells appear to exhibit some degree of metabolic flexibility. In TMEs with low levels of glucose, CD8+ T cells upregulate fatty acid catabolism to provide energy in order to preserve their effector function. Importantly, the pharmacological activation of fatty acid catabolism through the use of peroxisome proliferator-activated receptor (PPAR) agonists has been found to potentiate T cell function in this hostile environment and to synergize with immunotherapy strategies to delay tumor growth77. Additionally, acetate can rescue IFNγ production in glucose-restricted T cells by promoting histone acetylation and chromatin accessibility in an acetyl-coenzyme A synthetase (ACSS)-dependent manner78 (Figure 3). Thus, these studies show that metabolic targets can be utilized to rescue T cell function in a metabolically hostile environment.
3.1.3. T and cancer cells compete for amino acids within the TME
Aside from glucose, amino acids have now also been identified to drive and fuel T cell function and differentiation. For example, glutamine uptake and catabolism are needed to sustain T cell activation79,80, and are involved in the regulation of CD4+ T cell differentiation towards either a Th1 or Treg phenotype through the modulation of intracellular α-ketoglutarate levels81. Interestingly, although GLS deficiency impairs T cell activation and Th17 differentiation, it simultaneously potentiates Th1 and CD8+ T effector function82. Furthermore, a dynamic proteome and metabolome analysis of activated T cells in an in vivo melanoma model has recently identified L-arginine as a key metabolite to induce OXPHOS, boost T cell survival, and generate central memory-like T cells that possess strong anti-tumor activity83 (Figure 3). Last, tracing studies have also identified methionine metabolism to fuel the epigenetic adaptations required for sustaining Th17 cell proliferation and cytokine production84.
The specific influence of amino acid availability on immune cell fate and function thus raises the possibility of immunomodulation within the TME through the manipulation of amino acid levels. Indeed, as described for glucose in the previous section, arginine uptake and catabolism have also been shown to primarily shift towards cancer cells in solid tumors such as melanoma and ovarian cancer, whereby the neighboring immune cells are outcompeted85. The balance of this competition has been linked to the expression and activity of amino acid transporters and metabolic enzymes that act as rate-limiting factors for metabolite uptake and conversion86. For example, in leukemia, antigen-activated T cells upregulate the Scl7a5 transporter that can simultaneously carry the mTOR-activating methionine and leucine (Figure 3). Indeed, both the unavailability of these nutrients and the limited expression of the transporter strongly compromise CD4+ and CD8+ T cell activity86,87. In accordance, many tumors, including melanoma, pancreatic ductual adenocarcinoma (PDAC) and ovarian cancer, show high levels of expression of the enzyme indoleamine 2,3-dioxygenase (IDO), important for the catabolism of tryptophan88–90. IDO activity boosts tryptophan uptake from the TME and generates the inhibitor of tryptophan import kynurenine. Therefore, both the scarcity of tryptophan and the accumulation of kynurenine have been found to suppress T cell activation and promote a Treg phenotype91,92 (Figure 3).
Because cancer cells seem to outcompete immune cells in several tumor models, potentiating immune cell function through modulation of amino acid catabolism represents a unique opportunity to shift this balance. Interestingly, a recent study was indeed able to delineate the effects of pharmacological modulation of glutamine catabolism on both T and cancer cell behavior in an MC38 colon cancer model. Here, the authors show that inhibition of glutamine metabolism, by using the glutamine antagonist 6-diazo-5-oxo-L-norleucine (DON), blocks both glycolytic and oxidative metabolism in tumor cells, but drives a switch from glutamine- to glucose-driven oxidative metabolism in T effector (Teff) cells. As this switch was able to potentiate Teff killing, modulation of glutamine catabolism was found to shift the balance in favor of T cell function within the TME93.
In summary, metabolite constraints and composition within the TME provide an added layer of control for anti-tumor immunity. This additional layer will likely vary with the tumor type and need to overcome chronic TCR signaling that can ultimately lead to T cell exhaustion. Thus, future studies based on clinically relevant mouse models, T cell engineering and single-cell analysis will be essential in order to overcome these limitations and help elucidate the mechanisms to improve immunotherapy strategies.
3.2. Macrophages and tumor cells
Tumor-associated macrophages (TAMs) possess distinct phenotypes. While M1 macrophages have a pro-inflammatory (anti-tumoral) function, M2 macrophages have an anti-inflammatory (pro-tumoral) function. These two states are characterized by different markers and gene expression94. Studies have shown that the polarization of TAMs towards an M1 or M2 phenotype is driven by environmental factors such as cytokines, chemokines and other soluble factors secreted by the neighboring cells95. TAM polarization can also be dictated by the genetic background96. While induction of p53 in hepatic stellate cells drives M1 polarization, lacking p53 induces the M2 state97. Last, also cellular metabolism was identified to control TAM polarization and thus determine pro-and anti-tumoral responses63.
Like T cells, TAMs also compete with their neighboring cells for glucose. Glycolytic activity in TAMs has mainly been associated with tumor regression. Accordingly, GAPDH activity is decreased in human M2 TAMs compared to M1 TAMs98. Moreover, hypoxic TAMs have increased expression of the mTOR negative regulator REDD1 and consequently display decreased glycolysis. Interestingly, REDD1 knockout TAMs outcompete endothelial cells for glucose utilization and therefore promote tumor vessel normalization and impair metastatic spread99. Conversely, culturing human blood monocytes with conditioned media derived from PDAC cell lines promoted the formation of TAMs with high glycolytic activity and increased metastatic potential. Pharmacological inhibition of glycolysis abrogated this effect, suggesting that the nutrient environment shaped by cancer cells phenotypically and functionally affects TAMs behavior100.
Lactate produced by tumor cells has a critical function in signaling and TAM polarization. Specifically, lactate induces a pro-tumoral M2 phenotype by inducing vascular endothelial growth factor (Vegf) through HIF1α stabilization101 and through activation of the G protein-coupled receptor 132 (GPR132), which enhances breast cancer metastasis102 (Figure 3). Recent studies have demonstrated that lactate can directly promote epigenetic modification in bacterially challenged M1 macrophages. This phenomenon, known as histone lactylation, induces the expression of genes involved in homeostasis. Because lactate levels are high in the TME, further studies will need to elucidate the role of histone lactylation in the cross-talk between tumor cells and macrophages103.
Glutamine metabolism in TAMs has been linked to a pro-tumoral phenotype through the production of α-ketoglutarate, which promotes FAO and epigenetic activation of M2 genes104. Furthermore, GS expression and activity are increased in the M2 phenotype and it becomes relevant in starvation conditions. Inhibition of macrophagic GS activity promotes a switch from an M2 to an M1 phenotype and ultimately prevents metastatic spread (Figure 3). Interestingly, the switch from a pro- to an anti-tumoral phenotype also allows vascular normalization and accumulation of cytotoxic T cells105.
TAMs also express the tryptophan catabolic enzyme IDO, which triggered M2 polarization of THP-1 cells. Genetic deletion of IDO promoted an M1 anti-tumoral phenotype and enhanced T cell response106 (Figure 3). Targeting TAMs can also indirectly affect the response of PDAC to chemotherapy. Specifically, TAMs secrete pyrimidine species including deoxycytidine which molecularly inhibit gemcitabine treatment107. Collectively, these findings indicate that sharing is reciprocal thus, modulating the metabolism of one cell type within a metabolic niche can directly or indirectly affects distinct cell populations.
In conclusion, these studies define a distinct metabolic profile associated with M1 vs. M2 TAMs, which are characterized by a metabolic profile similar to cancer cells. Thus, targeting specific metabolic pathways may be exploited by dual mechanisms: to inhibit tumor growth directly, as well as indirectly by promoting a switch to an M1 anti-tumoral phenotype.
3.3. Stromal cells and tumor cells
In addition to immune cells, stromal cells also interact and modulate tumor cell behavior. Stromal cells in the TME contribute to ECM remodeling, migration, invasion, and evasion of immunosurveillance108,109. Several studies have highlighted the importance of cellular metabolism and local nutrient composition in sustaining these cellular processes110–112. Tumor-associated stromal cells derive from different cell types that give origin to cancer-associated fibroblasts (CAFs), cancer-associated adipocytes (CAAs) or cancer-associated endothelial cells (CAECs)108. CAFs are the most abundant cell population in the TME61,113.
The metabolic cross-talk between CAFs and tumor cells is often referred to as a ‘reverse Warburg effect’, whereby the metabolites secreted from CAFs are utilized as fuels for neighboring tumor cells. CAFs engage aerobic glycolysis and secrete lactate that can fuel the metabolism of cancer cells62. CAFs are also characterized by an increased glutamine anabolic metabolism. Glutamine gets secreted in the TME and is consumed by cancer cells to sustain nucleotide generation and OXPHOS (Figure 3). Notably, the combined inhibition of GS in CAFs and GLS in tumor cells decreased tumor growth and metastasis in an ovarian cancer mouse model114.
In PDAC, stroma-associated pancreatic stellate cells can secrete alanine generated via autophagy, which PDAC cells can use as an alternative pyruvate source to fuel OHPHOS when glucose is limiting115 (Figure 3). Additionally, pancreatic stellate cells can also secrete lysophosphatidylcholine which supports the production of phosphatidylcholine by PDAC cells. By supporting cell membrane synthesis and the production of lysophosphatidic acid (LPA), phosphatidylcholine supports PDAC growth and migration116. CAFs can also secrete aspartate, which promotes nucleotide biosynthesis and proliferation in multiple tumors (Figure 3). In contrast, glutamate secreted by cancer cells can feed glutathione production to sustain redox balance and ECM remodeling in CAFs117. Furthermore, ovarian and endometrial cancer cells were shown to consume arginine secreted by the adipose stromal cells and convert it to citrulline and nitric oxide (NO). While citrulline enhances adipogenesis in adipose stromal cells, NO reduces oxidative stress and promotes glycolysis in cancer cells118.
CAFs do not only influence cancer metabolism through a local modulation of metabolites in the TME, but also support tumor cells in a more direct manner. CAF-derived exosomes have been found to directly provide pancreatic and prostate cancer cells with amino acids, lipids, and TCA intermediates to sustain central carbon metabolism119. Furthermore, stromal cells also produce macromolecules such as collagen. In nutrient restricted conditions, PDAC tumors have been shown to break down this environmental collagen to consume the amino acid proline and sustain the TCA cycle and growth through Proline Dehydrogenase (PRODH) mediated proline catabolism120 (Figure 3).
Collectively, these studies show that metabolic exchange between CAFs and tumor cells is bidirectional. This can also be compromised by neighboring cells: in ovarian cancer, CAFs provide cysteine to cancer cells for glutathione production, conferring resistance towards platinum-based chemotherapeutic drugs. Notably, CD8+ T cells abrogate this resistance through IFNγ production, which represses the xCT cystine-glutamate antiporter in CAFs via JAK/STAT1 signaling121.
In summary, stromal cells and tumor cells have profound metabolic interactions dictated by the scarcity of metabolites in the TME. Whether these metabolic interactions also affect immune cells is however not known. Thus, future studies focused on the metabolic cross-talk between stromal cells and immune cells within the TME will identify whether metabolites secreted by stromal cells would benefit the neighboring immune population.
4. The role of anatomical location in TME metabolism
Organ systems in the body are defined by distinct epigenetic regulation, gene expression, proteomes, and metabolomes, which all contribute to tissue function and organismal health. The tissue-specific metabolite composition is also dependent on extrinsic factors, nutrient status and requirements, and the ability to exchange metabolites with the circulation122. Thus, it is important to consider the role of the tissue and circulating metabolism on the TME.
4.1. Organ-specific metabolite composition
Different organ systems contain distinct gene expression, proteomic and metabolite profiles122–125. This difference in tissue metabolism raises the possibility that cancers that arise in different organs have different metabolite dependencies. A transcriptomic analysis of 22 different human tumors showed that the metabolic gene expression program remains more similar to their tissue-of-origin than to other tumors from distinct tissues125. Indeed, although cancer cells are characterized by altered gene expression, some of the tissue-specific patterns are maintained126. Additionally, oncogenes like familial succinate dehydrogenase (SDH) and fumarate hydratase (FH) induce cancer only in specific tissues indicating that tissue of origin defines mutational penetrance2. Nevertheless, the metabolic phenotype of tumor cells can evolve in order to optimally take advantage of local metabolite compositions. Early-stage PDAC and NSCLC tumors are both characterized by increased protein breakdown and release of branched-chain amino acids (BCAA). However, while advanced NSCLC upregulates the enzymes responsible for BCAA breakdown and becomes critically dependent on BCAA catabolism, advanced PDAC tumors are insensitive to BCAA inhibition and rely heavily on proteins released by the leaky vasculature10,127,128.
Evidence showing the influence of the tissue and local microenvironment on tumor metabolism and behavior is strongest when comparing primary and metastatic lesions of the same tumor of origin129. Indeed, whereas primary breast cancer rely on glutamine anaplerosis, lung metastases take advantage of the pyruvate-rich lung environment to increase PC activity11 (Figure 4). In the lung environment, this adaptation was shown to promote the proliferation of established metastases, and to be essential for ECM remodeling and subsequent transition to the macrometastasis stage60. Similar observations have been made when it comes to brain metastasis. Indeed, brain metastases have been found to mainly use acetate instead of glucose and glutamine for fueling OXPHOS130,131, whereas they are also characterized by high levels of de novo serine biosynthesis through PHGDH for coping with a scarcity of serine and glycine in the brain29 (Figure 4). In accordance, several studies have highlighted the role of metabolic reprogramming required for colorectal cancer (CR) to metastasize to the liver. Differently from primary CR, liver metastases are characterized by higher expression of brain-type creatine kinase (CKB). CKB is released in the extracellular space and generates phosphocreatine which is taken up by the transporter SLC6A8 to generate ATP. Notably targeting CKB impairs colon cancer metastasis formation, thus indicating this as a critical metabolic adaptation to the liver niche132. Additionally, metastasizing CR cells upregulate both the gluconeogenic enzyme phosphoenolpyruvate carboxykinase 1 (PCK1) and the liver and blood cell pyruvate kinase (PKLR). While PCK1 supports nucleotide synthesis, PKLR sustains glutathione synthesis. Also here, targeting these pathways specifically impairs CR derived metastasis133,134.
Figure 4. Inter-organ and intra-tumor microenvironments define the metabolic properties of tumor cells.
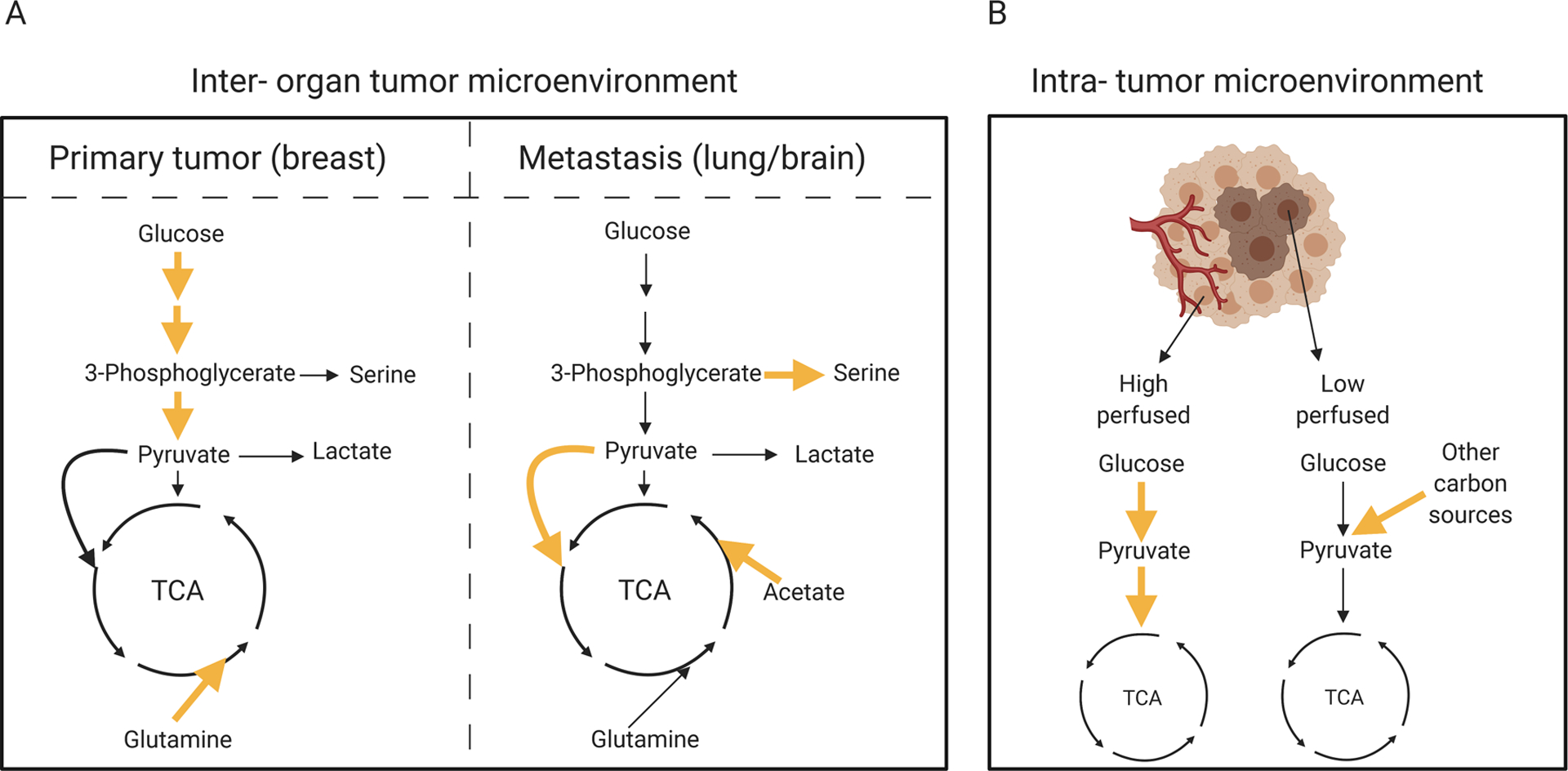
Left panel: while breast primary tumors rely on glucose and glutamine metabolism, metastatic tumors use pyruvate (lung metastasis), serine and acetate (brain metastasis) to sustain the TCA cycle. Right panel: within the same organ, a tumor can develop at different sites. The level of perfusion can dictate metabolic activity of tumor cells. Indeed, lung cancer cells in highly perfused areas consume glucose to sustain both glycolysis and OXPHOS, while cancer cells in lowly perfused areas rely on other carbon sources. Yellow arrows indicate upregulation.
As these studies demonstrate tumor cells metabolically adapt to the diverse niches present in the body, further studies will help identify metabolic vulnerabilities of specific tumors and metastatic sites. While the metabolic differences between metastasis and primary tumor are also connected to the distinct cellular phenotypes encountered during the multi-step metastatic cascade135, further studies comparing established metastases at different organs will help to isolate and better characterize the specific role of the metabolic TME. Finally, these studies have the potential to further explore the new exciting concept of tropism that may provide new strategies to target metastasis at their weakest environment.
4.2. Tumor location and metabolic heterogeneity
Within the same organ, a tumor can develop at different sites, and thus adapt to local environments. The level of perfusion, the distinct tissue function and the cell type composition all contribute to this spatial metabolic heterogeneity. For example, single-cell expression data of human melanoma and head and neck cancers have identified a strong correlation of both glycolysis and mitochondrial metabolism with local oxygen concentrations in all cell types17. The proximity to blood vessels clearly defines distinct metabolic niches in mouse glioblastoma xenografts. In particular, perivascular tumor cells show a very high mTOR-dependent anabolic metabolism and enhanced tumorigenesis18.
Solid tumors themselves are metabolically heterogeneous. Glucose tracing studies performed in NSCLC patients have indicated that cancer cells in highly perfused areas consume glucose to sustain glycolysis and OXPHOS, while cells in lowly perfused areas rely on other carbon sources (Figure 4). Interestingly, these metabolic preferences were found to be independent of oncogenic drivers such as KRAS and EGFR, indicating an overarching influence of the metabolic TME12.
Amino acids and carbohydrates also localize in specific areas within a tumor. Indeed, solid tumors were shown to contain core regions depleted in glutamine, which can induce hypermethylation and dedifferentiation136. The amino acids serine, asparagine, and aspartate were also found to be depleted in the poorly vascularized regions136. In kidney cancer, metabolic tracer studies in tissue slices showed distinct sensitivity to metabolic drugs in different metabolic regions of tumors. Specifically, pharmacological inhibition of pyruvate transporters was shown to strongly decrease tumor growth of patient-derived kidney tumors in mice137.
These studies demonstrate that metabolite availability and thus cancer metabolism can differ significantly even within a single tumor. Nevertheless, additional studies are required to delineate the causal relationship between regional metabolic heterogeneity, differential nutrient distribution and perfusion.
5. Systemic metabolism influences the TME
It is critical to consider how systemic, organismal metabolism, defined by the overall metabolic state of an individual and by environmental factors such as diet, influences the TME. Compared with cellular metabolic studies, less is known about how systemic nutrient levels contribute to the metabolic preferences of a tumor or cells within the TME. Emerging studies have shown that both dietary interventions and modulation of hormonal signaling affect local metabolism138. Moreover, considering an individual’s metabolic state may be an important consideration in precision therapies. In this section, we will describe how dietary interventions in whole-body metabolism can affect metabolite composition and utilization by cancerous and non-cancerous cells within the TME.
In recent years, the modulation of the amino acid composition of diets has been explored in the context of cancer progression and treatment. In a variety of cancers, the restriction of serine and glycine has been found to lead to an increase in serine biosynthesis, OXPHOS, and ROS production. Interestingly, tumors lacking p53 were found to be particularly sensitive to this modulation as they were unable to counteract oxidative stress associated with the increase in ROS139. Accordingly, combining serine/glycine restriction with agents that increase ROS was found to effectively synergize and reduce tumor growth in mice140. Notably, the serine/glycine diet caused a 50% decrease in circulatory serine levels while mice remained healthy with delayed tumor development139. While serine/glycine diet seems to be detrimental for tumor cells, the absence of these nutrients strongly affects CD8+ T cell responses in vivo141. In accordance, serine/glycine diet seems to be beneficial for mantaining Treg suppressive capacity. GSH restricts serine utilization and synthesis to preserve Treg functionality. Because GSH-deficient T cells are linked to inflammatory diseases, serine/glycine diet is beneficial in these conditions to support Treg activity142. These studies thus indicate that it is possible to take advantage of the unique metabolic vulnerabilities that exist in the different cancer niches, but the immune component needs to be evaluated as well. In accordance, because of its involvement in one-carbon metabolism, dietary restriction of the essential amino acid methionine has also been exploited therapeutically. Interestingly, studies conducted in humans showed that lowering dietary methionine levels could induce changes in systemic metabolism similar to those obtained in mice, and could alter therapeutic outcome for cancer treament143. Similarly to the serine/glycine diet, methionine diet affects the immune compartment of the body. Specifically, CD4+ T cells are marked by reduced histone methylation at the levels of genes involved in proliferation and function144. Furthermore, another study has linked asparagine restriction to the inhibition of breast cancer metastasis without affecting the primary tumor145. Last, global caloric restriction is not only able to reduce the availability of lipids in plasma and in the TIF, but also to remodel PDAC lipid metabolism, inhibit the activity of the enzyme stearoyl-CoA desaturase (SCD), and thus constrain PDAC progression through the accumulation of toxic saturated lipids146.
Interestingly, dietary interventions can also synergize with more traditional pharmacological approaches. A recent study showed that phosphoinositide 3-kinases (PI3K) inhibition is able to induce a systemic glucose-insulin feedback that may reactivate the PI3K-mTOR signaling axis in tumors. Therefore, the efficacy of PI3K inhibitors is significantly increased upon ketogenic diet treatment aimed at inhibiting this feedback147. Moreover, dietary supplementation of the amino acid histidine is able to both enhance histidine catabolism and sensitivity to methotrexate in a leukemia mouse model148.
Finally, dietary intake can influence local metabolite availability in co-operation with the microbial communities that colonize the body149. The gut microbiome produces a specific set of microbial metabolites that can be altered upon diet and ultimately affect cancer cell metabolism149,150. For example, dietary nicotinamide (NAM) and nicotinamide riboside (NR) are utilized by the gut microbiota to promote NAD biosynthesis in the host cells and confer resistance to inhibitors of nicotinamide phosphoribosyl transferase (NAMPT), which is the rate-limiting enzyme of the NAD biosynthetic pathway151. Furthermore, the microbiome has a strong immunomodulatory effect and strongly affect immunotherapy strategies152.
Taken together, these observations show that systemic metabolism influences metabolite availability in the TME and further studies will determine whether dietary interventions can exploit tumor metabolic vulnerabilities. Furthermore, these studies raise the importance of understanding how systemic metabolism associated with altered physiology affects TME metabolism. For instance, how does aging or obesity affect TME metabolism? It will be interesting for future studies to dissect the heterogeneity and metabolic cross-talk in these conditions.
6. Targeting metabolism for cancer therapy
Historically, targeting metabolism has been one of the most successful ways of developing effective cancer treatments. Imaging of glucose uptake through PET scans has become a standard imaging tool to visualize tumors24, whereas antimetabolite therapy such as methotrexate represents the oldest and still one of the most effective available chemotherapeutic drug153,154. Yet, mapping cancer metabolism for therapy has, until now, mainly focused on the metabolic adaptations of uncontrolled proliferation due to genetic perturbations. Therefore, it is to be expected that the increased knowledge regarding metabolic adapatations to cancer, local, organ, and systemic inputs will lead to novel and highly specific targets. Indeed, modeling cancer metabolism within its metabolic niche will allow for a more specialized approach to oncotherapy, and will enble us to chategorize tumors based on metabolic succeptibility. Furthermore, modelling the metabolic response of additional cell types co-inhabiting the TME will also allow scientists to develop additional therapies for modulation of supporting immune cells. In this section, we will therefore describe how the metabolic adaptation of tumor, stromal, and immune cells to local nutrient levels might impact and provide novel opportunities for cancer therapy.
For example, the fatty acid transporter CD36 mediates fatty acid metabolism and promotes ovarian cancer progression and metastasis155. CD36 inhibition impairs tumor growth but also prevents M2 macrophage polarization suggesting a double benefit of using the inhibitor of this transporter in the treatment of ovarian cancer156,157. Unfortunately, many drugs designed to target tumor cells are accompanied by collateral damage to compromise immune cell function158. Although glycolytic inhibitors have been shown to affect tumor cell growth, they also abrogate the immunosurveillance of cancer cells71,159. The inhibitor of the MCT1/2 lactate transporter (AZD3965) is in phase I clinical trials for both solid tumors and large B cell lymphoma160. However, the inhibition of this transporter also has a detrimental effect on T cell proliferation161. Folate metabolism is important to transfer 1C units for biosynthetic processes including nucleotide synthesis. Notably, antifolates such as methotrexate are widely used in the treatment of cancer. Recent studies have shown that both CD4+ and CD8+ T cells require one-carbon metabolism for proliferation and survival implicating that antifolate therapy may have severe side effects on the immune system162,141.
Interestingly, some drugs seem to synergize with immunotherapy to stop tumor growth. For example, treatment with PKM2 activators reduces the expression of PD-L1 in tumors and therefore synergizes with checkpoint inhibitors163. Additionally, the PFKB3 inhibitor also targets CTLA-4 and therefore promotes an immune response164. Along similar lines, PKM2 activators enhance the conversion of PEP to pyruvate and decrease tumor cell proliferation165. Thus, both PFKB3 inhibitors and PKM2 activators represent valuable candidates to inhibit tumor growth and at the same time boost T cell function.
Additional studies are required to better define the effects of rapamycin and metformin on the immune compartment. Indeed, treatment with rapamycin can effectively inhibit tumor cell proliferation, but the effect on the immune system is controversial. Although this drug reduces the proliferation of effector T cells and promotes the expansion of Treg cells166,167, it also enhances memory T cell responses168. Interestingly, memory T cells infiltrate solid tumor and synergize with immunotherapy in order to achieve anti-tumor responses169. Finally, metformin inhibits the proliferation of cancer cells, which may be immune-mediated as metformin improves T cell function in vivo170,171.
An alternative way to therapeutically change cellular behavior in the TME is to utilize chimeric antigen receptor (CAR) T cells172. CAR T cells of second and third generations activate TCR signaling and co-stimulation that together with the cytokines present in the environment allow for a functional T cell phenotype173. Recently, fourth generation CAR T cells have been further genetically modified to counteract the challenges imposed by the TME174. For example, CAR T cells expressing the enzyme heparinase, which degrades the ECM, display enhanced T cell infiltration and tumor regression175. Although several groups have been trying to optimize CAR T cell delivery and to reduce cytotoxicity, the prospect of generating CAR T cells carrying specific metabolic substrates or cells that can thrive in specific TMEs is promising157.
Thus, these examples demonstrate that the metabolic landscape of the TME plays an important role in affecting drug sensitivity.
7. Outstanding questions and concluding remarks
The field of cancer metabolism has made great progress in describing the influence of cell-intrinsic factors on the metabolic activity of cancer cells. The ideas presented here identify major contributors to the metabolic niche within the TME. Interestingly, all these studies corroborate the notion that cellular metabolism adapts to local metabolite availability. A better understanding of differential metabolic dependencies in tumor versus immune or stromal cells could provide a unique therapeutic window for metabolic therapies. Furthermore, scientists need to study these interactions in a physiologically relevant setting and we should consider the effect of systemic metabolic state of an individual when translating metabolic targets into therapies. Taken together, this work highlights the need for a better understanding of the role of metabolic niches and cellular heterogeneity in determining the metabolic phenotype of cancer cells.
Acknowledgments
We regret that space limitations prevented the discussion of other important cell types of the tumor microenvironment such as endothelial cells and myeloid-derived suppressor cells and the citations of many other relevant studies related to this topic. We would like to thank Dr. Ruben Boon, Dr. Jiska van Der Reest and Jefte Drijvers for their valuable feedback on the manuscript. Dr. Ilaria Elia acknowledges funding from the European Molecular Biology Organization (EMBO). Prof. Marcia C. Haigis is supported by the Ludwig Center at Harvard, the Glenn Foundation for Medical Research, and National Institutes of Health grant RO1CA213062.
Footnotes
Competing interests
The authors declare no competing interests.
References
- 1.Hanahan D & Weinberg RA Hallmarks of cancer: The next generation. Cell (2011). doi: 10.1016/j.cell.2011.02.013 [DOI] [PubMed] [Google Scholar]
- 2.Vander Heiden MG & DeBerardinis RJ Understanding the Intersections between Metabolism and Cancer Biology. Cell (2017). doi: 10.1016/j.cell.2016.12.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Warburg O The metabolism of carcinoma cells 1. J. Cancer Res (1925). doi: 10.1158/jcr.1925.148 [DOI] [Google Scholar]
- 4.De Berardinis RJ & Chandel NS Fundamentals of cancer metabolism. Science Advances (2016). doi: 10.1126/sciadv.1600200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Spinelli JB & Haigis MC The multifaceted contributions of mitochondria to cellular metabolism. Nature Cell Biology (2018). doi: 10.1038/s41556-018-0124-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.DeBerardinis RJ & Chandel NS We need to talk about the Warburg effect. Nature Metabolism (2020). doi: 10.1038/s42255-020-0172-2 [DOI] [PubMed] [Google Scholar]
- 7.Vyas S, Zaganjor E & Haigis MC Mitochondria and Cancer. Cell (2016). doi: 10.1016/j.cell.2016.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Elia I, Schmieder R, Christen S & Fendt SM Organ-specific cancer metabolism and its potential for therapy. in Handbook of Experimental Pharmacology (2016). doi: 10.1007/164_2015_10 [DOI] [PubMed] [Google Scholar]
- 9.Yuneva MO et al. The metabolic profile of tumors depends on both the responsible genetic lesion and tissue type. Cell Metab. (2012). doi: 10.1016/j.cmet.2011.12.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mayers JR et al. Tissue of origin dictates branched-chain amino acid metabolism in mutant Kras-driven cancers. Science (80-.) (2016). doi: 10.1126/science.aaf5171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Christen S et al. Breast Cancer-Derived Lung Metastases Show Increased Pyruvate Carboxylase-Dependent Anaplerosis. Cell Rep. (2016). doi: 10.1016/j.celrep.2016.09.042 [DOI] [PubMed] [Google Scholar]
- 12.Hensley CT et al. Metabolic Heterogeneity in Human Lung Tumors. Cell (2016). doi: 10.1016/j.cell.2015.12.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gui DY et al. Environment Dictates Dependence on Mitochondrial Complex I for NAD+ and Aspartate Production and Determines Cancer Cell Sensitivity to Metformin. Cell Metab. (2016). doi: 10.1016/j.cmet.2016.09.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Campbell SL & Wellen KE Metabolic Signaling to the Nucleus in Cancer. Molecular Cell (2018). doi: 10.1016/j.molcel.2018.07.015 [DOI] [PubMed] [Google Scholar]
- 15.Egeblad M, Nakasone ES & Werb Z Tumors as organs: Complex tissues that interface with the entire organism. Developmental Cell (2010). doi: 10.1016/j.devcel.2010.05.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nakazawa MS, Keith B & Simon MC Oxygen availability and metabolic adaptations. Nature Reviews Cancer (2016). doi: 10.1038/nrc.2016.84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xiao Z, Dai Z & Locasale JW Metabolic landscape of the tumor microenvironment at single cell resolution. Nat. Commun (2019). doi: 10.1038/s41467-019-11738-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kumar S et al. Intra-Tumoral Metabolic Zonation and Resultant Phenotypic Diversification Are Dictated by Blood Vessel Proximity. Cell Metab. (2019). doi: 10.1016/j.cmet.2019.04.003 [DOI] [PubMed] [Google Scholar]
- 19.Harris IS & Brugge JS United They Stand, Divided They Fall. Cell Metabolism (2019). doi: 10.1016/j.cmet.2019.09.008 [DOI] [PubMed] [Google Scholar]
- 20.Lyssiotis CA & Kimmelman AC Metabolic Interactions in the Tumor Microenvironment. Trends in Cell Biology (2017). doi: 10.1016/j.tcb.2017.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Eil R et al. Ionic immune suppression within the tumour microenvironment limits T cell effector function. Nature (2016). doi: 10.1038/nature19364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vodnala SK et al. T cell stemness and dysfunction in tumors are triggered by a common mechanism. Science (80-.) (2019). doi: 10.1126/science.aau0135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Luengo A, Gui DY & Vander Heiden MG Targeting Metabolism for Cancer Therapy. Cell Chemical Biology (2017). doi: 10.1016/j.chembiol.2017.08.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Witney TH et al. PET imaging of tumor glycolysis downstream of hexokinase through noninvasive measurement of pyruvate kinase M2. Sci. Transl. Med (2015). doi: 10.1126/scitranslmed.aac6117 [DOI] [PubMed] [Google Scholar]
- 25.Hsu PP & Sabatini DM Cancer cell metabolism: Warburg and beyond. Cell (2008). doi: 10.1016/j.cell.2008.08.021 [DOI] [PubMed] [Google Scholar]
- 26.Lunt SY & Vander Heiden MG Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu. Rev. Cell Dev. Biol 27, 441–464 (2011). [DOI] [PubMed] [Google Scholar]
- 27.Reid MA et al. Serine synthesis through PHGDH coordinates nucleotide levels by maintaining central carbon metabolism. Nat. Commun (2018). doi: 10.1038/s41467-018-07868-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sullivan MR et al. Increased Serine Synthesis Provides an Advantage for Tumors Arising in Tissues Where Serine Levels Are Limiting. Cell Metab. (2019). doi: 10.1016/j.cmet.2019.02.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ngo B et al. Limited Environmental Serine Confers Sensitivity to PHGDH Inhibition in Brain Metastasis. bioRxiv (2020). doi: 10.1101/2020.03.03.974980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Possemato R et al. Functional genomics reveal that the serine synthesis pathway is essential in breast cancer. Nature (2011). doi: 10.1038/nature10350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Comerford SA et al. Acetate Dependence of Tumors. Cell 159, 1591–1602 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.DeBerardinis RJ et al. Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc. Natl. Acad. Sci. U. S. A 104, 19345–19350 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Weinberg F, Ramnath N & Nagrath D Reactive oxygen species in the tumor microenvironment: An overview. Cancers (2019). doi: 10.3390/cancers11081191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sullivan LB & Chandel NS Mitochondrial reactive oxygen species and cancer. Cancer and Metabolism (2014). doi: 10.1186/2049-3002-2-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pavlova NN & Thompson CB The Emerging Hallmarks of Cancer Metabolism. Cell Metabolism (2016). doi: 10.1016/j.cmet.2015.12.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Faubert B et al. Lactate Metabolism in Human Lung Tumors. Cell (2017). doi: 10.1016/j.cell.2017.09.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.García-Canaveras JC, Chen L & Rabinowitz JD The tumor metabolic microenvironment: Lessons from lactate. Cancer Research (2019). doi: 10.1158/0008-5472.CAN-18-3726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Spinelli JB et al. Metabolic recycling of ammonia via glutamate dehydrogenase supports breast cancer biomass. Science (80-.) (2017). doi: 10.1126/science.aam9305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Csibi A et al. The mTORC1 pathway stimulates glutamine metabolism and cell proliferation by repressing SIRT4. Cell (2013). doi: 10.1016/j.cell.2013.04.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cluntun AA, Lukey MJ, Cerione RA & Locasale JW Glutamine Metabolism in Cancer: Understanding the Heterogeneity. Trends in Cancer (2017). doi: 10.1016/j.trecan.2017.01.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Birsoy K et al. An Essential Role of the Mitochondrial Electron Transport Chain in Cell Proliferation Is to Enable Aspartate Synthesis. Cell (2015). doi: 10.1016/j.cell.2015.07.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sullivan LB et al. Supporting Aspartate Biosynthesis Is an Essential Function of Respiration in Proliferating Cells. Cell (2015). doi: 10.1016/j.cell.2015.07.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Garcia-Bermudez J et al. Aspartate is a limiting metabolite for cancer cell proliferation under hypoxia and in tumours. Nat. Cell Biol (2018). doi: 10.1038/s41556-018-0118-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kurmi K & Haigis MC Nitrogen Metabolism in Cancer and Immunity. Trends in Cell Biology (2020). doi: 10.1016/j.tcb.2020.02.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kim J et al. CPS1 maintains pyrimidine pools and DNA synthesis in KRAS/LKB1-mutant lung cancer cells. Nature (2017). doi: 10.1038/nature22359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhuang Y, Chan DK, Haugrud AB & Miskimins WK Mechanisms by which low glucose enhances the cytotoxicity of metformin to cancer cells both in vitro and in vivo. PLoS One (2014). doi: 10.1371/journal.pone.0108444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Birsoy K et al. Metabolic determinants of cancer cell sensitivity to glucose limitation and biguanides. Nature (2014). doi: 10.1038/nature13110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jiang J, Srivastava S & Zhang J Starve cancer cells of glutamine: Break the spell or make a hungry monster? Cancers (2019). doi: 10.3390/cancers11060804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Muir A et al. Environmental cystine drives glutamine anaplerosis and sensitizes cancer cells to glutaminase inhibition. Elife (2017). doi: 10.7554/eLife.27713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jeong SM et al. SIRT4 has tumor-suppressive activity and regulates the cellular metabolic response to dna damage by inhibiting mitochondrial glutamine metabolism. Cancer Cell (2013). doi: 10.1016/j.ccr.2013.02.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cantor JR The Rise of Physiologic Media. Trends in Cell Biology (2019). doi: 10.1016/j.tcb.2019.08.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ackermann T & Tardito S Cell Culture Medium Formulation and Its Implications in Cancer Metabolism. Trends in Cancer (2019). doi: 10.1016/j.trecan.2019.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Voorde J Vande et al. Improving the metabolic fidelity of cancer models with a physiological cell culture medium. Sci. Adv (2019). doi: 10.1126/sciadv.aau7314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Boon R et al. Amino acid levels determine metabolism and CYP450 function of hepatocytes and hepatoma cell lines. Nat. Commun (2020). doi: 10.1038/s41467-020-15058-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cantor JR et al. Physiologic Medium Rewires Cellular Metabolism and Reveals Uric Acid as an Endogenous Inhibitor of UMP Synthase. Cell (2017). doi: 10.1016/j.cell.2017.03.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Elia I et al. Proline metabolism supports metastasis formation and could be inhibited to selectively target metastasizing cancer cells. Nat. Commun (2017). doi: 10.1038/ncomms15267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Coloff JL et al. Differential Glutamate Metabolism in Proliferating and Quiescent Mammary Epithelial Cells. Cell Metab. (2016). doi: 10.1016/j.cmet.2016.03.016 [DOI] [PubMed] [Google Scholar]
- 58.Simian M & Bissell MJ Organoids: A historical perspective of thinking in three dimensions. J. Cell Biol (2017). doi: 10.1083/jcb.201610056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Drost J & Clevers H Organoids in cancer research. Nature Reviews Cancer (2018). doi: 10.1038/s41568-018-0007-6 [DOI] [PubMed] [Google Scholar]
- 60.Elia I et al. Breast cancer cells rely on environmental pyruvate to shape the metastatic niche. Nature (2019). doi: 10.1038/s41586-019-0977-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.De Wever O, Demetter P, Mareel M & Bracke M Stromal myofibroblasts are drivers of invasive cancer growth. International Journal of Cancer (2008). doi: 10.1002/ijc.23925 [DOI] [PubMed] [Google Scholar]
- 62.Pavlides S et al. The reverse Warburg effect: Aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle (2009). doi: 10.4161/cc.8.23.10238 [DOI] [PubMed] [Google Scholar]
- 63.Geeraerts X, Bolli E, Fendt SM & Van Ginderachter JA Macrophage metabolism as therapeutic target for cancer, atherosclerosis, and obesity. Frontiers in Immunology (2017). doi: 10.3389/fimmu.2017.00289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chen DS & Mellman I Oncology meets immunology: The cancer-immunity cycle. Immunity (2013). doi: 10.1016/j.immuni.2013.07.012 [DOI] [PubMed] [Google Scholar]
- 65.Buck MD, O’Sullivan D & Pearce EL T cell metabolism drives immunity. Journal of Experimental Medicine (2015). doi: 10.1084/jem.20151159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pollizzi KN & Powell JD Integrating canonical and metabolic signalling programmes in the regulation of T cell responses. Nature Reviews Immunology (2014). doi: 10.1038/nri3701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Delgoffe GM & Powell JD Feeding an army: The metabolism of T cells in activation, anergy, and exhaustion. Mol. Immunol (2015). doi: 10.1016/j.molimm.2015.07.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Singer K et al. Warburg phenotype in renal cell carcinoma: High expression of glucose-transporter 1 (GLUT-1) correlates with low CD8+ T-cell infiltration in the tumor. Int. J. Cancer (2011). doi: 10.1002/ijc.25543 [DOI] [PubMed] [Google Scholar]
- 69.Cascone T et al. Increased Tumor Glycolysis Characterizes Immune Resistance to Adoptive T Cell Therapy. Cell Metab. (2018). doi: 10.1016/j.cmet.2018.02.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chang CH et al. XPosttranscriptional control of T cell effector function by aerobic glycolysis. Cell (2013). doi: 10.1016/j.cell.2013.05.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Peng M et al. Aerobic glycolysis promotes T helper 1 cell differentiation through an epigenetic mechanism. Science (80-.) (2016). doi: 10.1126/science.aaf6284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Cham CM & Gajewski TF Glucose Availability Regulates IFN-γ Production and p70S6 Kinase Activation in CD8 + Effector T Cells. J. Immunol (2005). doi: 10.4049/jimmunol.174.8.4670 [DOI] [PubMed] [Google Scholar]
- 73.Cham CM, Driessens G, O’Keefe JP & Gajewski TF Glucose deprivation inhibits multiple key gene expression events and effector functions in CD8+ T cells. Eur. J. Immunol (2008). doi: 10.1002/eji.200838289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ho PC et al. Phosphoenolpyruvate Is a Metabolic Checkpoint of Anti-tumor T Cell Responses. Cell (2015). doi: 10.1016/j.cell.2015.08.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Fischer K et al. Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood (2007). doi: 10.1182/blood-2006-07-035972 [DOI] [PubMed] [Google Scholar]
- 76.Brand A et al. LDHA-Associated Lactic Acid Production Blunts Tumor Immunosurveillance by T and NK Cells. Cell Metab. (2016). doi: 10.1016/j.cmet.2016.08.011 [DOI] [PubMed] [Google Scholar]
- 77.Zhang Y et al. Enhancing CD8+ T Cell Fatty Acid Catabolism within a Metabolically Challenging Tumor Microenvironment Increases the Efficacy of Melanoma Immunotherapy. Cancer Cell (2017). doi: 10.1016/j.ccell.2017.08.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Qiu J et al. Acetate Promotes T Cell Effector Function during Glucose Restriction. Cell Rep. (2019). doi: 10.1016/j.celrep.2019.04.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wang R et al. The Transcription Factor Myc Controls Metabolic Reprogramming upon T Lymphocyte Activation. Immunity (2011). doi: 10.1016/j.immuni.2011.09.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Carr EL et al. Glutamine Uptake and Metabolism Are Coordinately Regulated by ERK/MAPK during T Lymphocyte Activation. J. Immunol (2010). doi: 10.4049/jimmunol.0903586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Klysz D et al. Glutamine-dependent α-ketoglutarate production regulates the balance between T helper 1 cell and regulatory T cell generation. Sci. Signal (2015). doi: 10.1126/scisignal.aab2610 [DOI] [PubMed] [Google Scholar]
- 82.Johnson MO et al. Distinct Regulation of Th17 and Th1 Cell Differentiation by Glutaminase-Dependent Metabolism. Cell (2018). doi: 10.1016/j.cell.2018.10.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Geiger R et al. L-Arginine Modulates T Cell Metabolism and Enhances Survival and Anti-tumor Activity. Cell (2016). doi: 10.1016/j.cell.2016.09.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Roy DG et al. Methionine Metabolism Shapes T Helper Cell Responses through Regulation of Epigenetic Reprogramming. Cell Metab. 31, 250–266.e9 (2020). [DOI] [PubMed] [Google Scholar]
- 85.Mocellin S, Bronte V & Nitti D Nitric oxide, a double edged sword in cancer biology: Searching for therapeutic opportunities. Medicinal Research Reviews (2007). doi: 10.1002/med.20092 [DOI] [PubMed] [Google Scholar]
- 86.Sinclair LV et al. Control of amino-acid transport by antigen receptors coordinates the metabolic reprogramming essential for T cell differentiation. Nat. Immunol (2013). doi: 10.1038/ni.2556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Klein Geltink RI & Pearce EL The importance of methionine metabolism. Elife (2019). doi: 10.7554/eLife.47221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Brody JR et al. Expression of indoleamine 2,3-dioxygenase in metastatic malignant melanoma recruits regulatory T cells to avoid immune detection and affects survival. Cell Cycle (2009). doi: 10.4161/cc.8.12.8745 [DOI] [PubMed] [Google Scholar]
- 89.Witkiewicz AK et al. Genotyping and Expression Analysis of IDO2 in Human Pancreatic Cancer: A Novel, Active Target. J. Am. Coll. Surg (2009). doi: 10.1016/j.jamcollsurg.2008.12.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Inaba T et al. Role of the immunosuppressive enzyme indoleamine 2,3-dioxygenase in the progression of ovarian carcinoma. Gynecol. Oncol (2009). doi: 10.1016/j.ygyno.2009.07.015 [DOI] [PubMed] [Google Scholar]
- 91.Mezrich JD et al. An Interaction between Kynurenine and the Aryl Hydrocarbon Receptor Can Generate Regulatory T Cells. J. Immunol (2010). doi: 10.4049/jimmunol.0903670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Mellor AL & Munn DH IDO expression by dendritic cells: Tolerance and tryptophan catabolism. Nature Reviews Immunology (2004). doi: 10.1038/nri1457 [DOI] [PubMed] [Google Scholar]
- 93.Leone RD et al. Glutamine blockade induces divergent metabolic programs to overcome tumor immune evasion. Science (80-.) (2019). doi: 10.1126/science.aav2588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Cassetta L & Pollard JW Targeting macrophages: Therapeutic approaches in cancer. Nature Reviews Drug Discovery (2018). doi: 10.1038/nrd.2018.169 [DOI] [PubMed] [Google Scholar]
- 95.Qian BZ & Pollard JW Macrophage Diversity Enhances Tumor Progression and Metastasis. Cell (2010). doi: 10.1016/j.cell.2010.03.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Blagih J et al. Cancer-Specific Loss of p53 Leads to a Modulation of Myeloid and T Cell Responses. Cell Rep (2020). doi: 10.1016/j.celrep.2019.12.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Lujambio A et al. Non-cell-autonomous tumor suppression by p53. Cell (2013). doi: 10.1016/j.cell.2013.03.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Miller A et al. Exploring Metabolic Configurations of Single Cells within Complex Tissue Microenvironments. Cell Metab (2017). doi: 10.1016/j.cmet.2017.08.014 [DOI] [PubMed] [Google Scholar]
- 99.Wenes M et al. Macrophage Metabolism Controls Tumor Blood Vessel Morphogenesis and Metastasis. Cell Metab. (2016). doi: 10.1016/j.cmet.2016.09.008 [DOI] [PubMed] [Google Scholar]
- 100.Penny HL et al. Warburg metabolism in tumor-conditioned macrophages promotes metastasis in human pancreatic ductal adenocarcinoma. Oncoimmunology (2016). doi: 10.1080/2162402X.2016.1191731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Colegio OR et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature (2014). doi: 10.1038/nature13490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Chen P et al. Gpr132 sensing of lactate mediates tumor-macrophage interplay to promote breast cancer metastasis. Proc. Natl. Acad. Sci. U. S. A (2017). doi: 10.1073/pnas.1614035114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Zhang D et al. Metabolic regulation of gene expression by histone lactylation. Nature (2019). doi: 10.1038/s41586-019-1678-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Liu PS et al. α-ketoglutarate orchestrates macrophage activation through metabolic and epigenetic reprogramming. Nat. Immunol (2017). doi: 10.1038/ni.3796 [DOI] [PubMed] [Google Scholar]
- 105.Palmieri EM et al. Pharmacologic or Genetic Targeting of Glutamine Synthetase Skews Macrophages toward an M1-like Phenotype and Inhibits Tumor Metastasis. Cell Rep. (2017). doi: 10.1016/j.celrep.2017.07.054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Wang XF et al. The role of indoleamine 2,3-dioxygenase (IDO) in immune tolerance: Focus on macrophage polarization of THP-1 cells. Cell. Immunol (2014). doi: 10.1016/j.cellimm.2014.02.005 [DOI] [PubMed] [Google Scholar]
- 107.Halbrook CJ et al. Macrophage-Released Pyrimidines Inhibit Gemcitabine Therapy in Pancreatic Cancer. Cell Metab. (2019). doi: 10.1016/j.cmet.2019.02.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Bussard KM, Mutkus L, Stumpf K, Gomez-Manzano C & Marini FC Tumor-associated stromal cells as key contributors to the tumor microenvironment. Breast Cancer Research (2016). doi: 10.1186/s13058-016-0740-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Hanahan D & Coussens LM Accessories to the Crime: Functions of Cells Recruited to the Tumor Microenvironment. Cancer Cell (2012). doi: 10.1016/j.ccr.2012.02.022 [DOI] [PubMed] [Google Scholar]
- 110.Gouirand V, Guillaumond F & Vasseur S Influence of the tumor microenvironment on cancer cells metabolic reprogramming. Frontiers in Oncology (2018). doi: 10.3389/fonc.2018.00117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Schwörer S, Vardhana SA & Thompson CB Cancer Metabolism Drives a Stromal Regenerative Response. Cell Metabolism (2019). doi: 10.1016/j.cmet.2019.01.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Zhao X, Kwan JYY, Yip K, Liu PP & Liu FF Targeting metabolic dysregulation for fibrosis therapy. Nature Reviews Drug Discovery (2020). doi: 10.1038/s41573-019-0040-5 [DOI] [PubMed] [Google Scholar]
- 113.Leca J et al. Cancer-associated fibroblast-derived annexin A6+ extracellular vesicles support pancreatic cancer aggressiveness. J. Clin. Invest (2016). doi: 10.1172/JCI87734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Yang L et al. Targeting Stromal Glutamine Synthetase in Tumors Disrupts Tumor Microenvironment-Regulated Cancer Cell Growth. Cell Metab. (2016). doi: 10.1016/j.cmet.2016.10.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Sousa CM et al. Pancreatic stellate cells support tumour metabolism through autophagic alanine secretion. Nature (2016). doi: 10.1038/nature19084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Auciello FR et al. A stromal lysolipid–autotaxin signaling axis promotes pancreatic tumor progression. Cancer Discov. (2019). doi: 10.1158/2159-8290.CD-18-1212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Bertero T et al. Tumor-Stroma Mechanics Coordinate Amino Acid Availability to Sustain Tumor Growth and Malignancy. Cell Metab. (2019). doi: 10.1016/j.cmet.2018.09.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Rizi BS et al. Nitric oxide mediates metabolic coupling of omentum-derived adipose stroma to ovarian and endometrial cancer cells. Cancer Res. (2015). doi: 10.1158/0008-5472.CAN-14-1337 [DOI] [PubMed] [Google Scholar]
- 119.Zhao H et al. Tumor microenvironment derived exosomes pleiotropically modulate cancer cell metabolism. Elife (2016). doi: 10.7554/eLife.10250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Olivares O et al. Collagen-derived proline promotes pancreatic ductal adenocarcinoma cell survival under nutrient limited conditions. Nat. Commun (2017). doi: 10.1038/ncomms16031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Wang W et al. Effector T Cells Abrogate Stroma-Mediated Chemoresistance in Ovarian Cancer. Cell (2016). doi: 10.1016/j.cell.2016.04.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Jang C et al. Metabolite Exchange between Mammalian Organs Quantified in Pigs. Cell Metab. (2019). doi: 10.1016/j.cmet.2019.06.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Mootha VK et al. Integrated Analysis of Protein Composition, Tissue Diversity, and Gene Regulation in Mouse Mitochondria. Cell (2003). doi: 10.1016/S0092-8674(03)00926-7 [DOI] [PubMed] [Google Scholar]
- 124.Nusinow DP et al. Quantitative Proteomics of the Cancer Cell Line Encyclopedia. Cell (2020). doi: 10.1016/j.cell.2019.12.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Hu J et al. Heterogeneity of tumor-induced gene expression changes in the human metabolic network. Nat. Biotechnol (2013). doi: 10.1038/nbt.2530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Gaude E & Frezza C Tissue-specific and convergent metabolic transformation of cancer correlates with metastatic potential and patient survival. Nat. Commun (2016). doi: 10.1038/ncomms13041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Mayers JR et al. Elevation of circulating branched-chain amino acids is an early event in human pancreatic adenocarcinoma development. Nat. Med (2014). doi: 10.1038/nm.3686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Kamphorst JJ et al. Human pancreatic cancer tumors are nutrient poor and tumor cells actively scavenge extracellular protein. Cancer Res. (2015). doi: 10.1158/0008-5472.CAN-14-2211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Schild T, Low V, Blenis J & Gomes AP Unique Metabolic Adaptations Dictate Distal Organ-Specific Metastatic Colonization. Cancer Cell (2018). doi: 10.1016/j.ccell.2018.02.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Maher EA et al. Metabolism of [U-13C]glucose in human brain tumors in vivo. NMR Biomed. (2012). doi: 10.1002/nbm.2794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Chen J et al. Gain of glucose-independent growth upon metastasis of breast cancer cells to the brain. Cancer Res. (2015). doi: 10.1158/0008-5472.CAN-14-2268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Loo JM et al. Extracellular metabolic energetics can promote cancer progression. Cell (2015). doi: 10.1016/j.cell.2014.12.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Yamaguchi N et al. PCK1 and DHODH drive colorectal cancer liver metastatic colonization and hypoxic growth by promoting nucleotide synthesis. Elife (2019). doi: 10.7554/eLife.52135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Nguyen A et al. PKLR promotes colorectal cancer liver colonization through induction of glutathione synthesis. J. Clin. Invest (2016). doi: 10.1172/JCI83587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Elia I, Doglioni G & Fendt SM Metabolic Hallmarks of Metastasis Formation. Trends in Cell Biology (2018). doi: 10.1016/j.tcb.2018.04.002 [DOI] [PubMed] [Google Scholar]
- 136.Pan M et al. Regional glutamine deficiency in tumours promotes dedifferentiation through inhibition of histone demethylation. Nat. Cell Biol (2016). doi: 10.1038/ncb3410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Okegawa T et al. Intratumor Heterogeneity in Primary Kidney Cancer Revealed by Metabolic Profiling of Multiple Spatially Separated Samples within Tumors. EBioMedicine (2017). doi: 10.1016/j.ebiom.2017.04.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Lien EC & Vander Heiden MG A framework for examining how diet impacts tumour metabolism. Nat. Rev. Cancer (2019). doi: 10.1038/s41568-019-0198-5 [DOI] [PubMed] [Google Scholar]
- 139.Humpton TJ, Hock AK, Maddocks ODK & Vousden KH p53-mediated adaptation to serine starvation is retained by a common tumour-derived mutant. Cancer Metab. (2018). doi: 10.1186/s40170-018-0191-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Maddocks ODK et al. Modulating the therapeutic response of tumours to dietary serine and glycine starvation. Nature (2017). doi: 10.1038/nature22056 [DOI] [PubMed] [Google Scholar]
- 141.Ma EH et al. Serine Is an Essential Metabolite for Effector T Cell Expansion. Cell Metab. (2017). doi: 10.1016/j.cmet.2016.12.011 [DOI] [PubMed] [Google Scholar]
- 142.Kurniawan H et al. Glutathione Restricts Serine Metabolism to Preserve Regulatory T Cell Function. Cell Metab. (2020). doi: 10.1016/j.cmet.2020.03.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Gao X et al. Dietary methionine influences therapy in mouse cancer models and alters human metabolism. Nature (2019). doi: 10.1038/s41586-019-1437-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Roy DG et al. Methionine Metabolism Shapes T Helper Cell Responses through Regulation of Epigenetic Reprogramming. Cell Metab. (2020). doi: 10.1016/j.cmet.2020.01.006 [DOI] [PubMed] [Google Scholar]
- 145.Knott SRV et al. Asparagine bioavailability governs metastasis in a model of breast cancer. Nature (2018). doi: 10.1038/nature25465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Lien EC, Westermark AM, Li Z, Sapp KM & Heiden M Vander. Caloric restriction alters lipid metabolism to contribute to tumor growth inhibition. bioRxiv Cancer Biol. (2020). doi: 10.1101/2020.03.09.984302 [DOI] [Google Scholar]
- 147.Hopkins BD et al. Suppression of insulin feedback enhances the efficacy of PI3K inhibitors. Nature (2018). doi: 10.1038/s41586-018-0343-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Kanarek N et al. Histidine catabolism is a major determinant of methotrexate sensitivity. Nature (2018). doi: 10.1038/s41586-018-0316-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.David LA et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature (2014). doi: 10.1038/nature12820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Schmidt TSB, Raes J & Bork P The Human Gut Microbiome: From Association to Modulation. Cell (2018). doi: 10.1016/j.cell.2018.02.044 [DOI] [PubMed] [Google Scholar]
- 151.Shats I et al. Bacteria Boost Mammalian Host NAD Metabolism by Engaging the Deamidated Biosynthesis Pathway. Cell Metab. (2020). doi: 10.1016/j.cmet.2020.02.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Cogdill AP, Gaudreau PO, Arora R, Gopalakrishnan V & Wargo JA The Impact of Intratumoral and Gastrointestinal Microbiota on Systemic Cancer Therapy. Trends in Immunology (2018). doi: 10.1016/j.it.2018.09.007 [DOI] [PubMed] [Google Scholar]
- 153.FARBER S & DIAMOND LK Temporary remissions in acute leukemia in children produced by folic acid antagonist, 4-aminopteroyl-glutamic acid. N. Engl. J. Med 238, 787–793 (1948). [DOI] [PubMed] [Google Scholar]
- 154.Locasale JW Serine, glycine and one-carbon units: cancer metabolism in full circle. Nat. Rev. Cancer 13, 572–583 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Ladanyi A et al. Adipocyte-induced CD36 expression drives ovarian cancer progression and metastasis. Oncogene (2018). doi: 10.1038/s41388-017-0093-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Huang SCC et al. Cell-intrinsic lysosomal lipolysis is essential for alternative activation of macrophages. Nat. Immunol (2014). doi: 10.1038/ni.2956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.O’Sullivan D, Sanin DE, Pearce EJ & Pearce EL Metabolic interventions in the immune response to cancer. Nature Reviews Immunology (2019). doi: 10.1038/s41577-019-0140-9 [DOI] [PubMed] [Google Scholar]
- 158.Li X et al. Navigating metabolic pathways to enhance antitumour immunity and immunotherapy. Nature Reviews Clinical Oncology (2019). doi: 10.1038/s41571-019-0203-7 [DOI] [PubMed] [Google Scholar]
- 159.Renner K et al. Metabolic plasticity of human T cells: Preserved cytokine production under glucose deprivation or mitochondrial restriction, but 2-deoxy-glucose affects effector functions. Eur. J. Immunol (2015). doi: 10.1002/eji.201545473 [DOI] [PubMed] [Google Scholar]
- 160.Renner K et al. Metabolic hallmarks of tumor and immune cells in the tumor microenvironment. Frontiers in Immunology (2017). doi: 10.3389/fimmu.2017.00248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Murray CM et al. Monocarboxylate Transporter Mctl is a Target for Immunosuppression. Nat. Chem. Biol (2005). doi: 10.1038/nchembio744 [DOI] [PubMed] [Google Scholar]
- 162.Ron-Harel N et al. Mitochondrial Biogenesis and Proteome Remodeling Promote One-Carbon Metabolism for T Cell Activation. Cell Metab. (2016). doi: 10.1016/j.cmet.2016.06.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Palsson-McDermott EM et al. Pyruvate kinase M2 is required for the expression of the immune checkpoint PD-L1 in immune cells and tumors. Front. Immunol (2017). doi: 10.3389/fimmu.2017.01300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Chesney JA, Telang S, Yaddanapudi K & Grewal JS Targeting 6-phosphofructo-2-kinase (PFKFB3) as an immunotherapeutic strategy. J. Clin. Oncol (2016). doi: 10.1200/jco.2016.34.15_suppl.e14548 [DOI] [Google Scholar]
- 165.Kung C et al. Small molecule activation of pkm2 in cancer cells induces serine auxotrophy. Chem. Biol (2012). doi: 10.1016/j.chembiol.2012.07.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Battaglia M, Stabilini A & Roncarolo MG Rapamycin selectively expands CD4+CD25+FoxP3 + regulatory T cells. Blood (2005). doi: 10.1182/blood-2004-10-3932 [DOI] [PubMed] [Google Scholar]
- 167.Battaglia M et al. Rapamycin Promotes Expansion of Functional CD4 + CD25 + FOXP3 + Regulatory T Cells of Both Healthy Subjects and Type 1 Diabetic Patients. J. Immunol (2006). doi: 10.4049/jimmunol.177.12.8338 [DOI] [PubMed] [Google Scholar]
- 168.Araki K et al. mTOR regulates memory CD8 T-cell differentiation. Nature (2009). doi: 10.1038/nature08155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Sugiura A & Rathmell JC Metabolic Barriers to T Cell Function in Tumors. J. Immunol (2018). doi: 10.4049/jimmunol.1701041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Eikawa S et al. Immune-mediated antitumor effect by type 2 diabetes drug, metformin. Proc. Natl. Acad. Sci. U. S. A (2015). doi: 10.1073/pnas.1417636112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Scharping NE, Menk AV, Whetstone RD, Zeng X & Delgoffe GM Efficacy of PD-1 blockade is potentiated by metformin-induced reduction of tumor hypoxia. Cancer Immunol. Res (2017). doi: 10.1158/2326-6066.CIR-16-0103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Schurich A, Magalhaes I & Mattsson J Metabolic regulation of CAR T cell function by the hypoxic microenvironment in solid tumors. Immunotherapy (2019). doi: 10.2217/imt-2018-0141 [DOI] [PubMed] [Google Scholar]
- 173.Sadelain M, Brentjens R & Rivière I The basic principles of chimeric antigen receptor design. Cancer Discovery (2013). doi: 10.1158/2159-8290.CD-12-0548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Jaspers JE & Brentjens RJ Development of CAR T cells designed to improve antitumor efficacy and safety. Pharmacology and Therapeutics (2017). doi: 10.1016/j.pharmthera.2017.03.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Caruana I et al. Heparanase promotes tumor infiltration and antitumor activity of CAR-redirected T lymphocytes. Nat. Med (2015). doi: 10.1038/nm.3833 [DOI] [PMC free article] [PubMed] [Google Scholar]