

Abstract
Methylation of cytosine in CpG dinucleotides and histone lysine and arginine residues is a chromatin modification that critically contributes to the regulation of genome integrity, replication, and accessibility. A strong correlation exists between the genome‐wide distribution of DNA and histone methylation, suggesting an intimate relationship between these epigenetic marks. Indeed, accumulating literature reveals complex mechanisms underlying the molecular crosstalk between DNA and histone methylation. These in vitro and in vivo discoveries are further supported by the finding that genes encoding DNA‐ and histone‐modifying enzymes are often mutated in overlapping human diseases. Here, we summarize recent advances in understanding how DNA and histone methylation cooperate to maintain the cellular epigenomic landscape. We will also discuss the potential implication of these insights for understanding the etiology of, and developing biomarkers and therapies for, human congenital disorders and cancers that are driven by chromatin abnormalities.
Keywords: cancer, chromatin, developmental disorder, DNA methylation, histone methylation
Subject Categories: Cancer; Chromatin, Epigenetics, Genomics & Functional Genomics; Molecular Biology of Disease
Methylation of cytosines and histones regulates genome integrity, replication and chromatin accessibility. This review summarizes how DNA and histone methylation cooperate to maintain cellular epigenomic landscapes and provides links to congenital disorders and cancers.
Glossary
- ADD
ATRX‐DNMT3‐DNMT3L
- ALL
acute lymphoblastic leukemia
- AML
acute myeloid leukemia
- ChIP
chromatin immunoprecipitation
- cryo‐EM
cryogenic electron microscopy
- DIPGs
diffuse intrinsic pontine gliomas
- DLBCLs
diffuse large B‐cell lymphomas
- DNMTs
DNA methyltransferases
- ERVs
endogenous retroviruses
- H3Kx
histone H3 lysine number x
- hm‐DNA
hemi‐methylated DNA
- ICF
immunodeficiency, centromeric instability, facial anomalies syndrome
- MDSs
myelodysplastic syndromes
- MEFs
mouse embryonic fibroblasts
- mESCs
mouse embryonic stem cells
- MPNs
myeloproliferative neoplasms
- MPNST
malignant peripheral nerve sheath tumor
- OGID
overgrowth and intellectual disability
- PMDs
partially methylated domains
- PRC1
polycomb repressive complex 1
- PRC2
polycomb repressive complex 2
- RFTS
replication foci targeting sequence
- SRA
SET‐ and RING‐associated
- TBRS
Tatton‐Brown–Rahman syndrome
- TCL
T‐cell lymphoma
- TET
ten‐eleven translocation
- TTD
tandem Tudor domain
- UBL
ubiquitin‐like
- UHRF
ubiquitin‐like, containing PHD and RING finger domains
- UIM
ubiquitin‐interacting motif
- WGBS
whole‐genome bisulfite sequencing
Introduction
Methylation of the 5‐position of cytosine is a highly conserved chromatin modification among vertebrates. The majority of cytosine methylation occurs in the context of CpG dinucleotides. Nevertheless, in certain tissue and cell types such as brain and embryonic stem cells, non‐CpG methylation is readily detectable (He & Ecker, 2015). In mammals, 60–80% of CpGs are methylated (Ehrlich et al, 1982; Lister et al, 2009), which are non‐randomly distributed in repetitive sequences, gene bodies, and intergenic regions (Suzuki & Bird, 2008; Jones, 2012). At these regions, CpG methylation is believed to play a primary role in restricting chromatin accessibility, leading to silencing of retrotransposon elements, prevention of cryptic transcription (Neri et al, 2017), and regulation of transcription factor binding (Zhu et al, 2016). In contrast, genomic regions containing high density of CpGs, known as CpG islands, are normally free of methylation. CpG islands are often located within gene promoters and cis‐regulatory elements, and their low methylation levels are thought to facilitate a transcriptionally permissive state for their target genes.
The landscape of DNA methylation is shaped by the collective action of DNA methyltransferases (DNMTs), including DNMT1, DNMT3A, and DNMT3B, and enzymes involved in DNA demethylation, including the TET family of methylcytosine dioxygenases. The two de novo DNA methyltransferases DNMT3A and DNMT3B catalyze CpG methylation at previously unmethylated CpGs. Once established, during replication DNMT1 serves to copy pre‐existing CpG methylation to newly synthesized daughter strand with high fidelity, thus preserving the patterns of CpG methylation through cell division (Goll & Bestor, 2005). However, it should be noted that in certain contexts, DNMT3A/B could facilitate maintenance DNA methylation (Jones & Liang, 2009), while DNMT1 could mediate de novo DNA methylation (Yarychkivska et al, 2018; Li et al, 2018b); therefore, their functional distinctions are not univocal. On the other hand, TET1‐3 enzymes catalyze stepwise oxidation of methylcytosine, generating 5‐hydroxymethylcytosine, 5‐formylcytosine, and 5‐carboxylcytosine that contribute to either active or passive DNA demethylation (Wu & Zhang, 2014).
In the cycle of mouse development, there are two major waves of global genome demethylation and re‐methylation: one occurring following germ cell specification and the other during early embryonic development after fertilization (Zeng & Chen, 2019). While the bulk levels of CpG methylation are otherwise relatively stable, it is evident that DNMTs and TETs continuously act to shape focal CpG methylation to yield tissue‐ and development‐specific DNA methylomes that are strongly correlative with chromatin accessibility and enhancer activation (He et al, 2020). Accordingly, genetic knockout of DNMTs arrests embryonic or neonatal mouse development (Li et al, 1992; Okano et al, 1999), and tissue‐specific ablation of DNMTs and TETs affects organ homeostasis and regeneration (Challen et al, 2011; Rinaldi et al, 2016; Bowman & Levine, 2017). Moreover, germline and somatic mutations in DNA methylation modifiers are associated with human developmental disorders (Hansen et al, 1999; Tatton‐Brown et al, 2014; Heyn et al, 2019) and cancers (Abdel‐Wahab et al, 2009; Ley et al, 2010). These findings suggest that precise regulation of the dynamics of CpG methylation is key to proper cell fate specification and represents a barrier to neoplastic transformation.
A central question in the field is how DNA methylation machineries interact with other chromatin components to ensure faithful establishment and maintenance of genome‐wide CpG methylation. Whereas the roles of transcription factors and non‐coding RNAs in shaping cellular methylome have been excellently reviewed elsewhere (Zhu et al, 2016; Zhao et al, 2016b), in this review we focus on the interplay between DNA and histone arginine and lysine methylation. Indeed, early genome‐wide profiling of CpG methylation noted a strong correlation between DNA methylation and histone methylation, including a positive correlation with histone H3K9 methylation and a negative correlation with H3K4 methylation (Meissner et al, 2008). Furthermore, recent studies have uncovered that various regulatory domains within DNA methylation modifiers and their associated factors possess intrinsic affinity to histones in a modification‐dependent manner (Table 1). We will summarize the progress made toward a better understanding of the molecular mechanisms underlying the crosstalk between DNA and histone methylation and discuss their implications for human diseases driven by dysregulation of these chromatin marks.
Table 1.
Annotated regulatory domains of DNMTs and associated proteins.
| Gene (‐Domain) | Interaction | Location | Functional significance |
|---|---|---|---|
| DNMT1‐ RFTS | H3K18ub (+) H3K23ub (+) | Replication fork | Maintenance methylation |
| DNMT1‐ RFTS | H3K9me3 (+) | Heterochromatin | Maintenance methylation |
| UHRF1‐TTD/PHD | H3K9me3 (+) | Heterochromatin | Replication‐uncoupled maintenance methylation |
| UHRF1‐PHD | H3R2me2a (−) | Active genes | Prevent aberrant maintenance methylation |
| DNMT3L/A/B‐ADD | H3K4me3 (−) | Active gene promoter | Prevent aberrant de novo methylation |
| DNMT3B‐PWWP | H3K36me3 (+) | Active gene body | Genic de novo methylation |
| DNMT3A‐PWWP | H3K36me2 (+) | Intergenic region | Intergenic de novo methylation |
Interaction between DNMT1/UHRF1 and histone methylation facilitates maintenance DNA methylation
DNMT1 plays an indispensable function in propagating patterns of CpG methylation across cell cycles by reinstalling hemi‐methylated CpG to full methylation state. UHRF1, a RING E3 ubiquitin ligase also known as NP95 in mouse or ICBP90 in human, is essential for DNMT1‐mediated maintenance of DNA methylation. UHRF1 is required for DNMT1’s loading to replicating heterochromatin and genetic knockout of Uhrf1 phenocopied loss of Dnmt1, leading to DNA hypomethylation and early developmental arrest (Sharif et al, 2007). Furthermore, UHRF1 specifically recognizes hemi‐methylated DNA through its SET‐ and RING‐associated (SRA) domain, therefore bridging DNMT1 to its substrate to maintain DNA methylation (Bostick et al, 2007; Sharif et al, 2007). The recruitment can occur directly as UHRF1 physically interacts with DNMT1 during the S phase. Moreover, the E3 ubiquitin ligase activity of UHRF1 catalyzes the ubiquitination of histone H3K18 and H3K23, which can be recognized and bound by DNMT1 via its ubiquitin‐interacting motif (UIM) within the replication foci targeting sequence (RFTS) domain (Nishiyama et al, 2013; Qin et al, 2015). Recent crystal structures revealed that the RFTS domain of DNMT1 binds simultaneously to both H3K18 and H3K23 mono‐ubiquitination, which not only facilitates DNMT1 chromatin targeting but also stimulates its methyltransferase activity (Ishiyama et al, 2017; Li et al, 2018a). Therefore, through direct and indirect mechanisms, UHRF1 can target DNMT1 onto newly synthesized DNA substrates during semi‐conservative DNA replication.
In addition to SRA domain and RING domain, UHRF1 contains multiple functional domains including a tandem Tudor domain (TTD) and PHD finger connected by a linker region. When analyzed individually in vitro, the TTD of UHRF1 was reported to preferentially recognize histone H3K9 methylation (Rottach et al, 2010; Nady et al, 2011), while the PHD finger specifically recognizes the N‐terminal region of histone H3 with unmodified arginine 2 (H3R2) (Hu et al, 2011; Rajakumara et al, 2011; Wang et al, 2011). Interestingly, the TTD recognition of H3K9 methylation is independent of H3S10 phosphorylation (H3S10P) (Rothbart et al, 2012). H3S10 phosphorylation (H3S10P) is a mitotic “phospho‐methyl switch” and acts to antagonize H3K9 methylation readers such as HP1 during cell cycle (Fischle et al, 2003, 2005). Structural analysis suggested that compared to other H3K9me3‐interacting domains, UHRF1 TTD, enabled by Asn147, readily accommodates H3S10P. Indeed, mutating Asn147 to negatively charged glutamate abolishes UHRF1’s insensitivity toward H3K9me3‐to‐S10P switch, which is important for UHRF1’s association with chromatin during mitosis and maintenance of DNA methylation especially at late‐replicating genomic regions (Rothbart et al, 2012).
Tandem Tudor domain and PHD finger of UHRF1 are only separated by a 17‐aa linker region, raising the possibility that the two domains may operate as a single functional unit to recognize combinatorial histone modifications. Structural and biochemical studies of the linked TTD‐PHD finger module demonstrated that while the recognition of N‐terminal histone H3 by PHD seems to be independent of TTD, the binding to H3K9 methylation by TTD is markedly enhanced by the presence of PHD finger (Arita et al, 2012; Xie et al, 2012; Cheng et al, 2013; Rothbart et al, 2013). The linker region also appears to play a role, as its mutation and phosphorylation disrupt the higher order structure and the binding to H3K9me3 (Arita et al, 2012). The coordinated recognition of histone tails by TTD‐PHD finger module in vitro suggests that UHRF1’s recruitment to chromatin is dependent on histone engagement through cooperation between multiple reader domains. Indeed, point mutations in either the TTD, PHD, or linker regions reduce UHRF1’s association with chromatin in cells and compromise UHRF1’s ability to maintain DNA methylation (Rothbart et al, 2013). This is further illustrated by a recent study that identified a splicing variant of mouse UHRF1, which has additional 9 amino acids inserted in the TTD‐PHD finger linker region compared to canonical mouse and human UHRF1 (Tauber et al, 2020). As a result, the variant UHRF1 had distinct H3K9me3 binding profiles and displayed enhanced ubiquitination activity toward H3K9me3‐modified nucleosomes.
Beyond TTD and PHD finger, recent reports also pointed to the importance of SRA domain, which recognizes hemi‐methylated DNA (hm‐DNA), in directing the histone engagement and substrate specificity of UHRF1 (Fang et al, 2016; Harrison et al, 2016). It was shown that prior to binding histones and DNA, UHRF1 adopts a “closed” conformation, in which the histone‐binding (e.g., PHD finger) and DNA‐binding (e.g., SRA) modules are physically associated. Upon chromatin recruitment, the inter‐domain rearrangement of UHRF1 allows it to transit into “open” conformation in which the SRA binds to the hm‐DNA and TTD‐PHD finger to H3K9me2/3 via a positive feedback mechanism. Furthermore, the recognition of hm‐DNA by SRA domain allosterically activates and directs the ubiquitin ligase activity of UHRF1 toward H3K18 and H3K23. Collectively, these biophysical and structural data suggest a highly sophisticated mechanism involving extensive inter‐domain communication and cooperation in directing UHRF1’s chromatin engagement and activating its enzymatic activity (Fig 1A). Indeed, yet another domain—ubiquitin‐like domain (UBL)—was found to interact with other UHRF1 domains to facilitate histone/DNA binding and H3 ubiquitination (DaRosa et al, 2018; Foster et al, 2018).
Figure 1. Interaction between DNMT1/UHRF1 and histone methylation during maintenance DNA methylation.
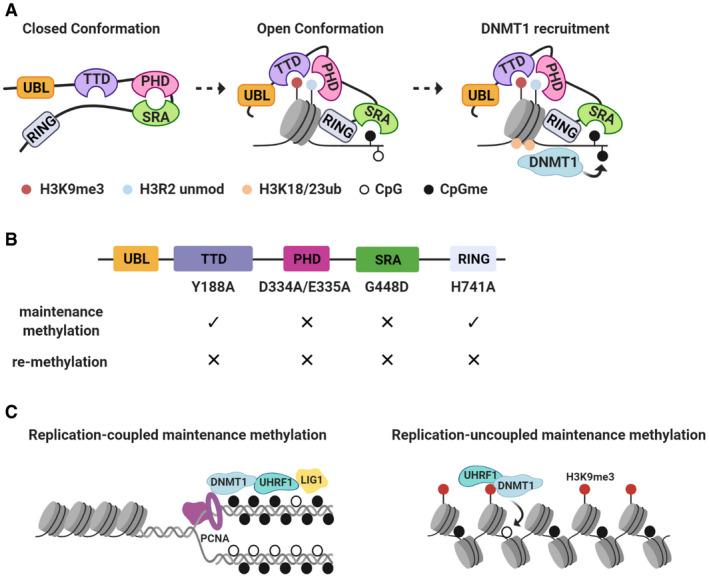
(A) In the absence of chromatin interaction, UHRF1 adopts a closed conformation, where the linker region between SRA and RING domains binds to the TTD domain and the SRA domain binds to PHD finger. Upon engagement with histone and hemi‐methylated DNA, inter‐domain conformational change will enable the TTD‐PHD module to recognize H3K9me3 and H3R2, and SRA domain to bind to hemi‐methylated DNA. This open conformation will facilitate the ubiquitination of histone H3K18 and H3K23 by UHRF1, which in turn recruits DNMT1 to catalyze maintenance DNA methylation. (B) Schematics summarizing the functional impact of various domain‐inactivating UHRF1 missense mutations on maintenance DNA methylation or re‐methylation following global demethylation. (C) Recent study suggests two distinct modes of maintenance methylation by DNMT1‐UHRF1. At the replication fork, interactions between DNMT1 and PCNA, and UHRF1‐TTD and methylated LIG1, facilitate replication‐coupled maintenance methylation. The interaction between UHRF1‐TTD and H3K9me3, on the other hand, facilitates replication‐uncoupled maintenance methylation.
In parallel to in vitro studies of the multidomain‐mediated UHRF1 binding to histones, in vivo analyses have generated interesting insight into the functional importance of these reader domains and the interacting histone modifications (Fig 1B). Veland et al (2017) identified PRMT6 as the arginine methyltransferase for histone H3R2me2a. Overexpressing PRMT6 in mouse embryonic stem cells (mESCs) resulted in dissociation of UHRF1 from chromatin and global DNA hypomethylation, supporting the notion that unmodified H3R2 facilitates UHRF1 chromatin recruitment and maintenance of CpG methylation (Veland et al, 2017). In mouse embryonic fibroblasts (MEFs) deficient for Suv39h genes encoding H3K9 methyltransferases, the decrease in H3K9 methylation at pericentric heterochromatin is correlated with a reduced localization of UHRF1 (Karagianni et al, 2008), suggesting an important role of H3K9 methylation for UHRF1 chromatin recruitment. Using the same system, it was shown that the PHD finger and SRA domain are required for UHRF1 to localize to pericentric heterochromatin. These two domains are also necessary for UHRF1 to restore or maintain DNA methylation in various cell types (Qin et al, 2015; Harrison et al, 2016; Kong et al, 2019).
In contrast, the role of TTD seems to be more context‐dependent. Whereas a point mutation (Y188A) that abolishes TTD’s interaction with H3K9me2/3 is unable to complete CpG re‐methylation following UHRF1 re‐expression in UHRF1‐depleted cells (Rothbart et al, 2012), the same mutation in TTD does not affect UHRF1’s function in maintaining CpG methylation in colorectal cancer cells (Kong et al, 2019). Moreover, a knock‐in TTD mutant mouse model has no overt phenotypes and shows modest (∼10%) reduction in CpG methylation (Zhao et al, 2016a). Similarly, the RING domain appears to be essential for de novo or re‐methylation, but dispensable for maintaining CpG methylation (Qin et al, 2015; Harrison et al, 2016; Li et al, 2018a; Kong et al, 2019).
A recent in‐depth analysis of the kinetics and fidelity of DNMT1‐mediated maintenance methylation offers critical insights into the function of UHRF1‐TTD in the process (Ming et al, 2020) (Fig 1C). By developing a novel method—Hammer‐seq—that combines EdU labeling, biotin‐mediated enrichment, and hairpin bisulfite sequencing technologies, Ming et al (2020) uncovered two distinct maintenance kinetics: replication‐coupled and replication‐uncoupled maintenance phases. The TTD is required for both phases, yet through interactions with different ligands: The interaction of TTD with methylated replication fork protein LIG1 (Ferry et al, 2017) is required for replication‐coupled maintenance phase, whereas TTD‐H3K9me2/3 for replication‐uncoupled maintenance phases. At steady state, the inactivation of TTD causes delays in replication‐coupled CpG methylation maintenance that could be compensated by other mechanisms, resulting in modest reduction in bulk CpG methylation. However, when replication‐coupled phase is compromised, or when the genome is largely devoid of CpG methylation (e.g., UHRF1 depletion), TTD mutation will significantly impact the rate of replication‐uncoupled re‐methylation. In support of this notion, genomic regions of high H3K9me2 display greater loss of CpG methylation in UHRF1‐TTD mutant cells (Ming et al, 2020). It is possible that the RING domain of UHRF1 plays a similar function in replication‐uncoupled maintenance of CpG methylation.
Finally, in addition to recognizing H3K18 and H3K23 ubiquitination catalyzed by UHRF1, a recent study demonstrates that the RFTS domain of DNMT1 also specifically binds to H3K9me3 (Ren et al, 2020). It was proposed that the direct interaction between DNMT1‐RFTS and H3K9me3 in conjunction with H3 mono‐ubiquitination could compensate for the loss of UHRF1‐TTD to maintain CpG methylation. While DNMT1 does not harbor any canonical histone‐binding domains, it would be interesting to explore whether DNMT1 directly interacts with additional histone modifications via non‐canonical mechanisms.
Role of histone methylation in targeting de novo DNMTs
In mammals, de novo CpG methylation is catalyzed by DNMT3A and DNMT3B. During embryogenesis, DNMT3L, which shares partial sequence homology with DNMT3A/B yet lacks the catalytic domain, acts as an essential accessary protein for establishing genomic methylation (Bourc’his, 2001). DNMT3L is absent or expressed at extremely low levels in adult tissues. Its function, however, can be partially compensated by catalytically inactive isoforms of DNMT3B such as DNMT3B3 (Weisenberger et al, 2004; Duymich et al, 2016; Zeng et al, 2020). DNMT3A and DNMT3B have distinct mechanisms of DNA substrate engagement and show differential preference for the flanking sequence of target CpG (Handa & Jeltsch, 2005; Dukatz et al, 2020; Gao et al, 2020; Mallona et al, 2021), and we point readers to the excellent reviews of the molecular and structural basis for the catalysis of de novo methylation by DNMT3 (Jurkowska & Jeltsch, 2016; Ren et al, 2018). Furthermore, DNMT3A/B as well as DNMT3L can interact with histone tails through shared regulatory domains. Thus, pre‐existing histone methylation plays a plausible role in shaping the non‐random targeting of de novo DNMTs and distribution of CpG methylation throughout the genome. Indeed, recent meta‐analysis of whole‐genome bisulfite sequencing (WGBS) and ChIP‐seq datasets from 35 human cell types revealed that CpG methylation is negatively correlated with H3K4 and H3K27 methylation and positively correlated with H3K9 and H3K36 methylation (Fu et al, 2020).
ADD domain‐mediated interaction with H3K4 methylation
Genome‐scale profiling study revealed a strong anti‐correlation between CpG methylation and H3K4 methylation that is particularly pronounced at CpG islands (Meissner et al, 2008). Immunoprecipitation and in vitro biochemical assays found that DNMT3L directly binds to N‐terminal tail of histone H3 and the binding is abolished with methylation of H3K4 (Ooi et al, 2007). Crystal structure revealed a direct interaction between the first seven amino acids of H3 and the cysteine‐rich region within the ATRX‐DNMT3‐DNMT3L (ADD) domain of DNMT3L. As expected from the significant sequence homology, the ADD domains of DNMT3A/B interact with H3 tails in a similar manner (Otani et al, 2009; Zhang et al, 2010). In addition to H3K4 methylation, this interaction is sensitive to phosphorylation of H3T3, H3S10, or H3T11 (Zhang et al, 2010). Furthermore, unmodified but not H3K4‐methylated histone H3 peptide can stimulate the activity of DNMT3A up to 8‐fold (Li et al, 2011). Therefore, it appears that H3K4 methylation, a mark for active transcription, antagonizes the recruitment as well as the allosteric activation of DNMT3A/B‐DNMT3L complex to prevent de novo CpG methylation and gene silencing.
The opposing action between H3K4 methylation and de novo DNMTs is also supported by in vivo evidence. By introducing DNMT3A and DNMT3L into yeast, Hu et al (2009) reported that the histone H3 N‐terminal tail is required for ectopic genomic methylation (Hu et al, 2009). Notably, in yeast stains lacking H3K4 methyltransferases, the levels of de novo methylation are substantially increased in a manner dependent on the ADD domain of DNMT3L. The H3K4 demethylase KDM1B is highly expressed in growing oocytes, and its ablation results in increased H3K4 methylation and subsequent failure of de novo DNA methylation and establishment of genomic imprints (Ciccone et al, 2009). These findings were confirmed by Stewart et al (2015), which further nominated another H3K4 demethylase KDM1A/LSD1 in regulating DNA methylation during oogenesis (Stewart et al, 2015). Similarly, during the development of male germline, H3K4me2‐enriched CpG islands are protected from de novo methylation (Singh et al, 2013). A point mutation in the ADD domain of DNMT3L that abolishes its histone‐binding activity is sufficient to impair establishment of both CpG and non‐CpG methylation, spermatogenesis and fertility (Vlachogiannis et al, 2015). In the context of embryogenesis, complete loss of maternal KDM1A/LSD1 arrests embryo at the maternal‐to‐zygotic transition stage, whereas partial loss of maternal KDM1A leads to developmental and behavioral abnormality that is associated with DNA hypomethylation and misexpression of imprinted genes (Wasson et al, 2016). It should be noted, however, that the maternal effects of KDM1A during embryogenesis may be independent of H3K4 methylation, since it has been demonstrated that KDM1A also directly demethylates DNMT1 which in turn affects maintenance CpG methylation (Wang et al, 2009). Finally, through structural modeling and protein engineering, Noh et al (2015) identified point mutations that render the ADD domain of DNMT3A insensitive to either H3K4 methylation or H3T3 phosphorylation (Noh et al, 2015). Re‐expression of H3K4 methylation‐insensitive mutant DNMT3A in DNMT triple knockout mESCs results in accumulation of DNA methylation at H3K4me3/2‐enriched regions and a defect in mESC differentiation. Expression of H3T3 phosphorylation‐insensitive mutant DNMT3A, on the other hand, leads to chromosomal instability. Together, these functional analyses corroborate with in vitro studies and strongly support a pivotal role of ADD‐H3K4me3/2 antagonism for protecting transcriptionally active promoter CpG islands from de novo methylation (Fig 2A).
Figure 2. Various mechanisms underlying targeting of de novo DNMTs by histone methylation.
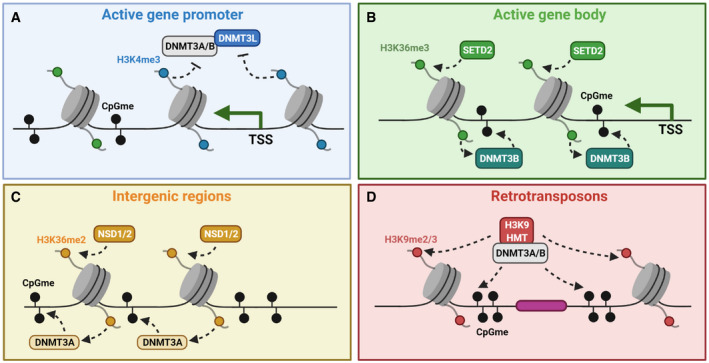
(A) At promoters of actively transcribing genes, high levels of H3K4me3 oppose ADD domain and the binding of DNMT3L‐DNMT3A/B to prevent de novo CpG methylation. (B) At gene bodies of actively transcribing genes, high levels of H3K36me3 interact with PWWP domain of DNMT3B and facilitate its genic localization. (C) A parallel pathway operates at the intergenic region, where H3K36me2 interacts with PWWP domain of DNMT3A and facilitates its intergenic localization. (D) At repetitive elements and retrotransposons, interactions between H3K9 methyltransferases and DNMT3A/B enable co‐localization of H3K9me3 and CpG methylation for transcriptional silencing.
PWWP domain‐mediated interaction with H3K36 methylation
In contrast to promoters, actively transcribed genes have high levels of CpG methylation at their gene bodies (Jones, 2012). These regions are also enriched for tri‐methylation of histone H3K36 (H3K36me3), and as expected DNA methylation has been shown to be associated with the deposition of H3K36me3 (Fu et al, 2020). Interestingly, comparative studies of DNA methylomes found that H3K36me3‐rich genes in Drosophila can predict CpG methylation and content of orthologous genes in other organisms, suggesting that the crosstalk between H3K36me3 and CpG methylation is highly evolutionarily conserved (Nanty et al, 2011). Both DNMT3A and DNMT3B harbor PWWP domain, which in other proteins such as BRPF1 has been shown to bind to H3K36me3 (Vezzoli et al, 2010). Indeed, the PWWP domains of DNMT3A and DNMT3B interact with H3K36me3 peptide in vitro (Dhayalan et al, 2010; Baubec et al, 2015). Furthermore, ChIP‐seq revealed preferential localization of DNMT3B in the gene bodies of highly expressed, H3K36me3‐enriched genes in mESCs, which is dependent on its PWWP domain and the H3K36me3 “writer” enzyme SETD2 (Baubec et al, 2015). Similarly, when DNMT3B is heterologously expressed in yeast, its binding and de novo methylation correlate with H3K36me3 and are abolished upon deletion of SET2—the yeast homologue of SETD2 (Morselli et al, 2015). More recently, it has been shown that maternal depletion of SETD2 causes loss of H3K36me3, followed by decreased genic CpG methylation yet aberrant gain of intergenic methylation, which contributes to the defects in oocyte maturation and embryonic development arrest at 1‐cell stage (Xu et al, 2019). These findings agree with prior observations from growing murine oocytes and 195 human/mouse DNA methylomes, where a positive correlation between co‐transcriptionally deposited H3K36me3 and DNA methylation across the gene body has been noted (Morselli et al, 2015; Stewart et al, 2015; Salhab et al, 2018). The function of H3K36me3‐mediated gene body CpG methylation (Fig 2B) remains unclear, though one hypothesis is that it ensures the efficiency and fidelity of transcriptional elongation through preventing spurious intragenic transcription initiation (Neri et al, 2017). While further investigation is needed, this notion is consistent with the role of H3K36me3 in yeast where it prevents cryptic transcription through histone deacetylation (Carrozza et al, 2005).
Interestingly, the binding pattern of DNMT3A differs substantially from that of DNMT3B and is enriched in the intergenic regions of euchromatin (Wu et al, 2010; Baubec et al, 2015). Indeed, whereas DNMT3B‐PWWP preferentially interacts with H3K36me3, DNMT3A‐PWWP can recognize both H3K36me3 and di‐methylated H3K36 (H3K36me2) with a slightly higher affinity for H3K36me2 (Sankaran et al, 2016; Dukatz et al, 2019; Weinberg et al, 2019; Xu et al, 2020a; Xu et al, 2020b). While closely related, H3K36me2 is a highly abundant chromatin modification which is catalyzed by a distinct set of methyltransferases (NSD1‐3 and ASH1L) (Kuo et al, 2011). It is spatially separated from H3K36me3 and mainly enriched in the intergenic regions (Rao et al, 2005). Genome‐wide DNMT3A binding and DNMT3A‐mediated de novo CpG methylation show positive correlation with H3K36me2 (Weinberg et al, 2019). Genetic knockout of NSD1/2 depletes intergenic H3K36me2 and CpG methylation, and redistributes DNMT3A to H3K36me3‐enriched genic regions (Weinberg et al, 2019). Conversely, overexpression of NSD2 in multiple myeloma cells is coupled with elevated intergenic CpG methylation (Xu et al, 2020a; Xu et al, 2020b). Taken together, these findings support a model in which H3K36me2‐DNMT3A interaction complements and competes with H3K36me3‐DNMT3B to establish intergenic and genic CpG methylation at euchromatin, respectively (Fig 2C). It remains unclear what is the structural basis for the valence‐specific recognition of H3K36 methylation by DNMT3A‐PWWP versus DNMT3B‐PWWP. A crystal structure of DNMT3B‐PWWP bound to H3K36me3 is available (Rondelet et al, 2016), and it would be interesting to compare and contrast that to DNMT3A‐PWWP.
Consistently, the H3K36me2 methyltransferase NSD1 has been linked to DNA methylation in several developmental and disease contexts. NSD1‐mediated H3K36me2 is critical for guiding de novo methylation and establishing paternal imprints in the male germline (Shirane et al, 2020). Nsd1 knockout male mice exhibit defects in spermatogenesis and are infertile. This is in sharp contrast to the role of SETD2/H3K36me3 in oocyte development and de novo methylation (Xu et al, 2019), and suggests that the sex‐specific landscape of CpG methylation in the germline could result from distinct state of H3K36 methylation. Germline NSD1 mutations define Sotos syndrome which is characterized by developmental overgrowth and intellectual disability (Douglas et al, 2003). Blood samples from Sotos patients display profound genome‐wide CpG hypomethylation compared to the controls (Choufani et al, 2015). Interestingly, this pattern mimics aging‐induced loss of DNA methylation, leading to the hypothesis that NSD1 depletion accelerates the “epigenetic clock” (Martin‐Herranz et al, 2019). Similarly, somatic mutations and deletions of NSD1, which are common in squamous cell carcinomas of the head and neck and other body sites (Papillon‐Cavanagh et al, 2017), are associated with CpG hypomethylation (Brennan et al, 2017; Bui et al, 2018) that are particularly pronounced at the intergenic regions (Lee & Wiemels, 2016; Weinberg et al, 2019). The mechanism by which loss of intergenic H3K36me2 and DNA methylation affects gene regulation remains elusive, although recent reports suggests that the dosage of NSD2 and H3K36me2 is linked to the binding of methylation‐sensitive genome architecture protein CTCF, reprogramming of 3D chromatin organization and distal enhancer activation (Lhoumaud et al, 2019).
Mechanisms of DNMT3A/B localization to constitutive heterochromatin
Both DNA methylation and H3K9 methylation are involved in the formation and maintenance of constitutive heterochromatin and the silencing of retrotransposons. In some organisms including Neurospora crassa and Arabidopsis thaliana, DNA methylation is strictly guided by methylation of H3K9 (Tamaru & Selker, 2001; Jackson et al, 2002). Although there is a genome‐wide positive correlation between H3K9 and CpG methylation (Meissner et al, 2008; Fu et al, 2020), the interplay between these two repressive marks seems to be complex in mammalian cells. There are three groups of methyltransferases, Suv39h1/2, G9a/GLP, and Setdb1, which catalyze H3K9 methylation at various parts of the genome. In mESCs, double knockout of Suv39h1/2 abolishes the localization of DNMT3B as well as DNA methylation at pericentric satellite repeats (Lehnertz et al, 2003). DNMT3B was also found to interact with H3K9 methylation “readers” HP1α and HP1β. Similarly, DNMT3A was co‐purified with H3K9 methyltransferase activity in HeLa cells and a direct interaction between DNMT3A and Suv39h1 and HP1β was observed (Fuks, 2003). This interaction appears to be mediated through the ADD domain of DNMT3A, although it is noteworthy that the PWWP domains of DNMT3A/B are required to direct their localization to pericentric heterochromatin (Chen et al, 2004). Therefore, additional histone‐binding domains could be involved. SETDB1, which is often complexed with KRAB‐Zinc Finger proteins and KAP1 to silence endogenous retroviruses (ERVs), also facilitates de novo DNA methylation. Depletion of either SETDB1 or KAP1 abolishes de novo methylation of natural or ectopically introduced ERVs (Rowe et al, 2013). Consistently, Leung et al (2014) observed that deletion of Setdb1 in mESCs induces hypomethylation and misexpression of LTR retrotransposons and class I and II ERVs, as well as several imprinted genes (Leung et al, 2014). This seems to be mediated through active rather than passive demethylation, as TET‐mediated 5‐hydroxymethylcytosine was transiently found prior to demethylation. Again, a direct interaction between DNMT3A (ADD domain) and SETDB1 (N‐terminal region) could be the underlying recruitment mechanism (Li et al, 2006). Finally, G9a‐dependent de novo CpG methylation has been shown in mESCs during silencing of imprinted genes (Xin et al, 2003), retrotransposons (Dong et al, 2008) and provirus (Leung et al, 2011). The proviral silencing defect in G9a knockout cells could be phenocopied by knockout of Dnmt3a, suggesting that the recruitment of de novo methylation activity is important for initiating transcriptional repression (Leung et al, 2011). In differentiating mESCs, methylation of several pluripotent genes is dependent on G9a (Epsztejn‐Litman et al, 2008), and in G9a ‐/‐ embryos, promoter methylation of germline‐specific genes is decreased which is coupled by increased gene expression (Auclair et al, 2016). This G9a‐dependent de novo methylation seems to be independent of H3K9 methylation, since catalytically inactive G9a is sufficient to restore or maintain CpG methylation (Dong et al, 2008; Epsztejn‐Litman et al, 2008; Tachibana et al, 2008). Instead, several models have been proposed: G9a’s ankyrin repeat region can directly interact with DNMT3A/B’s catalytic domain (Epsztejn‐Litman et al, 2008); alternatively, Ga9 may indirectly recruit DNMT3A/B, which can be bridged by either the HUSH complex member MPP8 (Chang et al, 2011) or UHRF1 (Meilinger et al, 2009). Collectively, these studies suggest that while DNMT3A/B do not harbor functional reader domains that recognize H3K9 methylation, they can be recruited either directly or indirectly by H3K9 methyltransferases to promote methylation and reinforce silencing at heterochromatin regions (Fig 2D).
Complex interplay between de novo CpG methylation and H3K27 methylation
Histone H3 lysine 27 methylation, established by the polycomb repressive complex 2 (PRC2), is a hallmark of facultative heterochromatin and associated with silencing of genes involved in cell cycle and differentiation (Comet et al, 2016). The enzymatic component EZH1/2, along with three core components (SUZ12, EED, and Rbbp4) and various accessary units of PRC2, catalyze the mono‐, di‐, and tri‐methylation of H3K27 (Yu et al, 2019). PRC2 closely interacts and cooperates with polycomb repressive complex 1 (PRC1) for gene silencing: Canonical PRC1 recognizes H3K27me3 to mediate chromatin compaction and transcriptional repression, while variant PRC1 complex could initiate polycomb‐mediated gene silencing through catalyzing H2AK119 ubiquitination (Holoch & Margueron, 2017).
The relationship between DNA methylation and H3K27 methylation is complex and highly dynamic. Genome‐wide studies in stem cells and cancer cells demonstrate that H3K27me3 and DNA methylation are anti‐correlated and have no obvious co‐localization (Kondo et al, 2008; Meissner et al, 2008; Lister et al, 2009; Hon et al, 2012; Fu et al, 2020). This is particularly evident at the so‐called DNA methylation valleys or canyons—large conserved genomic regions with very low (< 10%) levels of CpG methylation (Xie et al, 2013; Jeong et al, 2014). These domains are highly enriched for H3K27me3 and binding of polycomb proteins and recently have been shown to form megabase‐long chromatin contact loops (Zhang et al, 2020). The overall mutual exclusivity between DNA and H3K27 methylation could be attributed to a role of CpG methylation in antagonizing PRC2. In multiple cell types, depletion of CpG methylation via the deletion of various DNMTs leads to pervasive increases of H3K27me3 at previously methylated regions of the genome (Brinkman et al, 2012; Lynch et al, 2012; Reddington et al, 2013). Similarly, in the transitioning of mESCs from naïve to 2i‐induced ground state (Ying et al, 2008), there is a marked decrease in DNA methylation that is accompanied by a gain of H3K27me3 (van Mierlo et al, 2019). Interestingly, these genome‐wide increases and redistributions of H3K27me3 can result in a “titration” effect on the localization of PRC1 and therefore have a negative impact on polycomb‐mediated gene silencing (Reddington et al, 2013) or 3D loop formation (McLaughlin et al, 2019). Conversely, re‐expressing wild‐type but not catalytically dead DNMTs in methylation‐deficient mESCs restore patterns of H3K27me3 as well as activity of H3K27ac‐associated enhancers (King et al, 2016). Importantly, by inserting artificial sequences into the genome, two studies identified the cis‐elements that are sufficient to induce accumulation of H3K27me3 and found that CpG methylation directly counteracts H3K27me3 recruitment (Jermann et al, 2014; Wachter et al, 2014). These results are consistent with prior studies in vitro showing that CpG methylation of nucleosomal DNA inhibits PRC2 binding to H3K27me3‐marked nucleosomes (Bartke et al, 2010; Wu et al, 2010). Indirect mechanisms are also likely to be at play. For example, MTF2, an accessary subunit of PRC2, binds to CpG islands in a methylation‐dependent manner (Perino et al, 2018). Similarly, KDM2B, a component of the variant PRC1.1 that facilitates the recruitment of PRC2 (Holoch & Margueron, 2017), carries a CXXC domain that recognizes unmethylated CpGs (Blackledge et al, 2010). Finally, while less characterized, studies have suggested a reciprocal role of PRC2 in opposing CpG methylation. For example, deletion of EED leads to aberrant accumulation of CpG methylation in DNA methylation valleys in mESCs (Li et al, 2018c). This effect is recapitulated by triple knockout of TET1‐3 enzymes, consistent with the observation that PRC2 directly interacts with TET1 and regulates levels of 5‐hydroxymethylcytosine (Neri et al, 2013a).
It should be noted, however, that despite the well‐documented antagonism between H3K27 and DNA methylation, these two marks can co‐occur in low CpG regions in more differentiated cells (Statham et al, 2012). Moreover, dynamic switch from H3K27me3‐marked to DNA‐methylated CpG islands has been observed at some imprinted genes during embryonic development (Chen et al, 2019) and at pluripotency and germline‐specific genes during mESC‐to‐neuron differentiation (Mohn et al, 2008). The molecular mechanisms underlying these transitions remain poorly understood. PRC2 may directly recruit DNMT1 and DNMT3A/B (Viré et al, 2006). It was also suggested that DNMT3L competes with DNMT3A/B for the binding to PRC2 (Neri et al, 2013b). Hence, variations in DNMT3L expression could represent a mechanism for controlling de novo methylation of PRC2 target genes. Consistently, the long isoform of DNMT3A, DNMT3A1, preferentially localizes to H3K27me3/H3K4me3 bivalent CpG islands in mESCs (Manzo et al, 2017). Interestingly, aberrant CpG methylation of polycomb target genes has been observed when the PWWP domain of DNMT3A is inactivated (Heyn et al, 2019; Sendžikaitė et al, 2019) or when DNMT3B is overexpressed (Zhang et al, 2018a). Since the levels of H3K36 methylation are low at CpG islands (Blackledge et al, 2010), it appears that the interaction between H3K36me2/3‐PWWP domain could normally serve to “trap” DNMT3A/B away from PRC2‐regulated CpG islands.
Interplay of DNA and histone methylation in disease development and therapy
Developmental disorders
DNMT3B and ICF syndrome
Consistent with the importance of CpG methylation in development, germline mutations in all three DNMTs and TET3 have been associated with human congenital disorders (Hansen et al, 1999; Klein et al, 2011; Winkelmann et al, 2012; Tatton‐Brown et al, 2014; Heyn et al, 2019; Beck et al, 2020). DNMT3B is the first DNA‐modifying enzymes implicated in human diseases (Hansen et al, 1999). Genetic alterations in DNMT3B are found in patients with immunodeficiency, centromeric instability, facial anomalies syndrome (ICF; OMIM 602900). ICF syndrome is a rare, autosomal recessive disorder characterized by distinct facial features, absence of B and plasma cells and chromosome instability, often leading to death during early childhood. The majority of ICF‐associated DNMT3B mutations are loss of function, including nonsense, splice‐site, or missense mutations within the catalytic domain, consistent with hypomethylation of pericentromeric satellite 2 and 3 repeats in the genomes of affected patients (Jeanpierre et al, 1993). Intriguingly, homozygous missense mutations (S282P) in the PWWP domain of DNMT3B have been reported (Shirohzu et al, 2002), in agreement with a role of PWWP domain in mediating DNMT3B’s localization to pericentromeric heterochromatin (Chen et al, 2004). Furthermore, the homologous (S277P) mutation in mESCs abolishes DNMT3B’s interaction with H3K36me3 and intragenic localization (Baubec et al, 2015), and recent findings have shown that ICF‐associated DNMT3B mutant cells exhibit defects in intragenic DNA methylation and mRNA splicing (Gatto et al, 2017). Therefore, DNA hypomethylation at both repeat regions and gene bodies could contribute to the etiology of ICF syndrome. In addition to DNMT3B, three genes have been linked to ICF syndrome: ZBTB24, CDCA7, and HELLS (Velasco et al, 2018). Patients carrying mutations in these genes have overlapping clinical features and share hypomethylated pericentromeric repeats with DNMT3B‐mutant patients, suggesting their involvement in de novo CpG methylation. Indeed, in MEFs or mESCs deficient for HELLS (also known as LSH), maintenance of DNA methylation is not affected but establishing methylation at retroviral transgene is impaired (Zhu et al, 2006). HELLS directly interacts with DNMT3A/B in mESCs. In more differentiated cell types, it seems that HELLS recruits and cooperates with H3K9 methyltransferase G9a/GLP to promote methylation at select loci (Myant et al, 2011). A few patients with ICF syndrome‐like feature do not have mutations in the known genes (Weemaes et al, 2013), and it would be interesting to examine whether any histone methylation modifiers are altered in these patients.
DNMT3A and Tatton‐Brown–Rahman syndrome
Germline heterozygous mutations in DNMT3A, on the other hand, define Tatton‐Brown–Rahman syndrome (TBRS, OMIM 615879) (Tatton‐Brown et al, 2014). TBRS is characterized by tall stature, macrocephaly, intellectual disability, and distinctive craniofacial features. Intriguingly, TBRS belongs to a group of genetic disorders known as overgrowth and intellectual disability (OGID) syndromes, which share many overlapping clinical features (Tatton‐Brown et al, 2017). Among them, germline deletions/mutations in NSD1 are associated with Sotos syndrome (OMIM 117550) (Kurotaki et al, 2002; Douglas et al, 2003). PRC2 complex members—EZH2, EED, and SUZ12—are associated with Weaver syndrome (OMIM 277590), Cohen–Gibson syndrome (OMIM 617561), and Weaver‐like syndrome, respectively (Tatton‐Brown et al, 2011; Gibson et al, 2012; Cohen & Gibson, 2016; Imagawa et al, 2017; Cyrus et al, 2019). Furthermore, germline duplications of 5q35 involving NSD1 and missense mutations in the PWWP domain of DNMT3A correlate with opposite developmental characteristics, including dwarfism and microcephaly (Dikow et al, 2013; Rosenfeld et al, 2013; Heyn et al, 2019). These DNMT3A PWWP domain mutations (W330R and D333N) are putatively gain of function, as they impair DNMT3A‐H3K36me2/H3K36me3 interaction yet cause DNA hypermethylation at key developmental genes enriched for H3K27me3 (Heyn et al, 2019; Sendžikaitė et al, 2019). The fact that alterations in NSD1, PRC2, and DNMT3A cause developmental disorders with considerable phenotypic overlap offers strong human genetic evidence in support of the molecular crosstalk between these chromatin enzymes (Deevy & Bracken, 2019). Indeed, consistent with the finding that H3K36me2 is required for DNMT3A targeting and intergenic DNA methylation (Weinberg et al, 2019; Xu et al, 2020a; Xu et al, 2020b), NSD1 mutations/deletions in Sotos patients are associated with a profound DNA hypomethylation phenotype in the blood (Choufani et al, 2015). Furthermore, TBRS‐associated missense mutations in DNMT3A fail to interact with chromatin and cause protein instability (Heyn et al, 2019; Weinberg et al, 2019). The DNA methylomes from the blood of TBRS patients cluster closely with that of Sotos patients (Weinberg et al, 2019) and both display signatures of accelerated aging (Jeffries et al, 2019), suggesting that impaired NSD1‐H3K36me2‐DNMT3A interplay represents a common mechanism underlying the pathogenesis of TBRS and Sotos syndrome. A similar mechanism could be at play for mutations in SETD2, which have been found in patients with “Sotos‐like” syndromes (Luscan et al, 2014).
In contrast, OGID syndrome mutations in EZH2, EED, and SUZ12 do not cause aberrant DNA hypomethylation found in TBRS and Sotos syndrome patients (Choufani et al, 2015; Martin‐Herranz et al, 2019; Weinberg et al, 2019). Instead, as described above, patterns of DNA methylation could directly or indirectly affect PRC2 activity. Moreover, H3K36 methylation has a well‐documented role in opposing H3K27me3. Nucleosomes decorated with H3K36me2/3 can directly inhibit PRC2 activity in vitro (Schmitges et al, 2011; Yuan et al, 2011). In multiple myeloma and acute lymphoblastic leukemia (ALL) with overexpression or gain‐of‐function mutations of NSD2, global increases in H3K36me2 are accompanied by a loss of H3K27me3 (Martinez‐Garcia et al, 2011; Jaffe et al, 2013). Conversely, genetic ablation of Nsd1 in mESCs or expression of an oncohistone H3.3K36M mutation leads to a reduction of H3K36me2 and a gain of H3K27me3 (Lu et al, 2016; Streubel et al, 2018). Similarly, in SETD2‐deficient oocytes and NSD1‐deficient sperms, in addition to abnormal CpG methylation there is also an “invasion” of H3K27me3 into former H3K36me2/3 territories (Xu et al, 2019; Shirane et al, 2020). Several accessary units of PRC2 contain “reader” domains for H3K36me3. PHF19 (PHD finger protein 19) and PHF1 are known to facilitate the recruitment of PRC2 through their Tudor domains (Hunkapiller et al, 2012). Both Tudor domains of PHF1 and PHF19 act as readers for H3K36me3, and it is believed that the recognition of H3K36me3 by PHF1/19 initiates PRC2 targeting, H3K27me3 deposition, and silencing of actively transcribed genes (Ballaré et al, 2012; Brien et al, 2012; Musselman et al, 2012; Cai et al, 2013). Moreover, a direct sensing mechanism of H3K36 methylation state by EZH2 has recently been reported (Jani et al, 2019). Biochemical and structural analysis shows that EZH2 contains a specific sensing pocket for H3K36 that allows the complex to distinguish between modified and unmodified H3K36 residues, altering enzymatic activity accordingly to preferentially methylate the unmodified nucleosome substrate. Interestingly, a Weaver syndrome‐associated EZH2 mutation (K634E) renders the enzyme less sensitive to the inhibition by H3K36 methylation (Jani et al, 2019). Notably, the majority of mutations affecting EZH2/EED/SUZ12 in OGID syndromes are missense. While studies have suggested that the mutations reduce the catalytic activity of PRC2 (Cohen et al, 2016; Imagawa et al, 2017), it remains to be determined if change‐of‐function mutations exist that alter the interaction between PRC2 and H3K36/DNA methylation.
Taken together, it is plausible that several OGID syndromes share a common etiology linked to the dysregulated crosstalk between NSD1, PRC2 and DNMT3A (Fig 3). Future efforts are required to validate this hypothesis and determine how the imbalance between these chromatin marks contributes to disease development. To this end, it is noteworthy that mice with heterozygous loss of Ezh2, Eed, Suz12, Nsd1, or Dnmt3a are overall developmentally normal, while homozygous knockouts lead to prenatal or neonatal lethality (Faust et al, 1995; Okano et al, 1999; O’Carroll et al, 2001; Rayasam et al, 2003; Pasini et al, 2004). Therefore, alternative models, such as tissue‐specific knockout mice or patient‐derived induced pluripotent stem cells, are needed to better model and study chromatin‐related OGID syndromes.
Figure 3. Dysregulated interplay between DNA and histone methylation in human OGID syndromes.
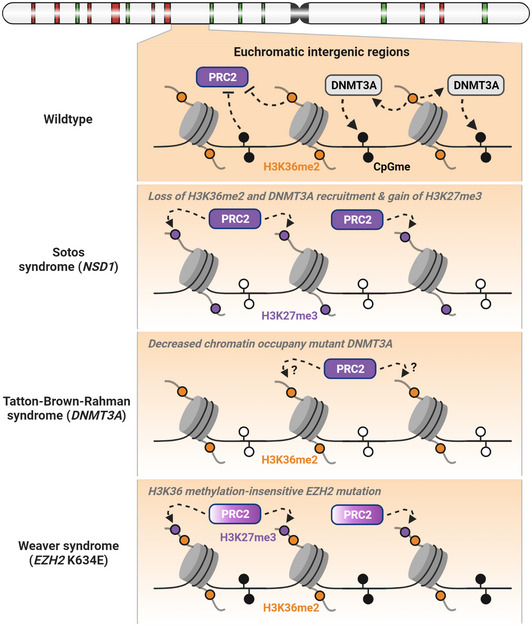
During normal development, H3K36me2 facilitates the deposition of CpG methylation by recruiting DNMT3A at euchromatic intergenic regions, and these two modifications act together to antagonize PRC2 and H3K27me3. In Soto syndrome, NSD1 mutations and deletions lead to reduced H3K36me2 and CpG methylation, and a resulting gain of H3K27me3. In TBRS, loss‐of‐function mutations in DNMT3A reduce CpG methylation, although its impact on H3K36me2 and H3K27me3 is unclear. Some Weaver syndrome patients carry missense mutation of EZH2 (e.g., K634E) that renders the PRC2 insensitive to inhibition by H3K36 methylation, which could potentially leads to accumulation of H3K27me3 at intergenic regions despite the presence of H3K36me2. The significant overlap in clinical features of Soto, Weaver, and TRBS patients suggests that an imbalance of H3K36me2, H3K27me3, and CpG methylation could represent a common pathogenic mechanism.
Additional developmental disorders linked to abnormal DNA and histone methylation
Additional histone‐modifying enzymes are linked to human congenital disorders which are associated with abnormal CpG methylation landscape. Loss‐of‐function mutations in KMT2D (also known as MLL2), an H3K4 methyltransferase, and KDM6A (also known as UTX), an H3K27 demethylase, are the primary cause of Kabuki syndrome (Ng et al, 2010; Lederer et al, 2012). Kabuki syndrome patients are characterized by developmental delays, congenital abnormalities, and globally altered CpG methylation (Sobreira et al, 2017). Mutations in H3K4 demethylase KDM5C (also known as JARID5C) are linked to X‐linked intellectual disability (Iwase et al, 2007), and blood samples from patients carrying KDM5C mutations display CpG hypomethylation at several genomic loci (Grafodatskaya et al, 2013; Schenkel et al, 2018). Frameshift mutations affecting HIST1H1E, a gene encoding histone H1, are found in another OGID syndrome known as Rahman syndrome (Tatton‐Brown et al, 2017; Takenouchi et al, 2018) which exhibits a specific DNA hypomethylation signature (Ciolfi et al, 2020). Future studies are required to determine whether the aberrant CpG methylation profiles associated with these disorders are directly caused by altered histone methylation or reflect patterns of gene expression. Nevertheless, the unique DNA methylation signatures can be reliable molecular biomarkers to classify genetic variants of uncertain significance (Aref‐Eshghi et al, 2017, 2019; Choufani et al, 2020) and facilitate accurate diagnosis of clinically overlapping disorders caused by mutations in distinct chromatin enzymes (Aref‐Eshghi et al, 2018).
Cancers
Dysregulation of DNA and histone methylation has been widely implicated in various types of human cancers. Whereas the mechanistic details are being actively investigated, several lines of evidence support an intimate interaction between DNA and histone methylation in driving cancer initiation and progression. First, like OGID syndromes, mutations in DNA‐ and histone‐modifying enzymes are found in closely related cancer types, particularly within hematological malignancies. In myeloid neoplasms, DNMT3A and TET2 are frequently mutated in acute myeloid leukemias (AML), myelodysplastic syndromes (MDSs), and myeloproliferative neoplasms (MPNs) (Delhommeau et al, 2009; Ley et al, 2010; Stegelmann et al, 2011; Haferlach et al, 2014). Inactivating mutations in EZH2 are also frequently found in MDS and MPN (Ernst et al, 2010), whereas loss‐of‐function mutations in SETD2 and chromosomal translocations of NSD1 and NSD3 were recurrently identified in acute leukemias (Jaju et al, 2001; Rosati et al, 2002; Zhu et al, 2014). In lymphoid malignancies, DNMT3A and TET2 mutations have been linked to adult acute lymphoblastic leukemias (ALL) (Grossmann et al, 2013) and T‐cell lymphomas (TCL), in particular angioimmunoblastic T‐cell lymphomas and peripheral T‐cell lymphomas, not otherwise specified (PTCL, NOS) (Couronné et al, 2012; Lemonnier et al, 2012; Sakata‐Yanagimoto et al, 2014). Gain‐of‐function mutations in NSD2 are found in 14% of ETV6/RUNX1‐fusion pediatric ALL (Jaffe et al, 2013), and modifiers of histone methylation and acetylation (KMT2D, SETD2, KMT2A, KDM6A, EP300, CREBBP) are collectively mutated in up to 36% of patients with PTCL, NOS (Ji et al, 2018; Watatani et al, 2019). On the other hand, mutations in KMT2C, KMT2D, and EZH2 are common events in diffuse large B‐cell lymphomas (DLBCLs) (Morin et al, 2010, 2011), where DNMT3A and TET2 mutations have also been reported albeit at lower frequencies (Asmar et al, 2013; Reddy et al, 2017). Furthermore, neomorphic mutations in the genes encoding metabolic enzymes IDH1 and IDH2, which produce an oncometabolite 2‐hydroxyglutarate that competitive inhibits both histone and DNA demethylation (Figueroa et al, 2010; Lu et al, 2012), are frequently found in AML and TCL (Mardis et al, 2009; Cairns et al, 2012). The widespread mutations affecting DNA/histone methylation machinery genes are consistent with the findings that genetic perturbations of these enzymes result in abnormal hematopoietic self‐renewal and differentiation (Challen et al, 2011; Mochizuki‐Kashio et al, 2011; Zhang et al, 2018b; Leonards et al, 2020), and epigenetic drugs, such as inhibitors of DNA methyltransferase, histone deacetylases, and IDH1/2, are approved to treat leukemias and lymphomas (Bates, 2020). Future investigations are required to address whether, and how, alterations in histone and DNA methylation pathways act redundantly or cooperatively to drive hematopoietic malignancies.
Second, although somatic mutations affecting DNMTs and TETs are rare in solid tumors, mutations in histone‐modifying enzymes and histones themselves can have a direct effect on reprogramming DNA methylome. As described above, NSD1 mutations and deletions in squamous cell carcinomas result in global DNA hypomethylation as a consequence of defective DNMT3A recruitment by H3K36me2 (Lee & Wiemels, 2016; Brennan et al, 2017; Papillon‐Cavanagh et al, 2017; Bui et al, 2018; Weinberg et al, 2019; Farhangdoost et al, 2021). Histone H3 lysine 36 to methionine (H3K36M) mutations deplete H3K36 methylation by inhibiting the catalytic activities of H3K36 methyltransferases, and H3K36M mutations are mutually exclusive with NSD1 loss‐of‐function mutations in head and neck squamous cell carcinomas (Lu et al, 2016; Papillon‐Cavanagh et al, 2017). Consistently, H3K36M mutation also leads to global decreases in DNA methylation (Rajagopalan et al, 2021). In contrast, SETD2 mutations in renal cell carcinoma and other cancer types are associated with global DNA hypermethylation (Tiedemann et al, 2016). This unexpected finding could be recapitulated in cellular models and is hypothesized to result from ectopic gain of H3K36me3 at large intergenic regions upon SETD2 inactivation. While counterintuitive, similar observation has been made in female germline following Setd2 deletion (Xu et al, 2019). Genetic inactivation of PRC2 complex is associated with malignant peripheral nerve sheath tumors (MPNST), and Wojcik et al (2019) demonstrated that PRC2‐null MPNST cells exhibit genome‐wide hypermethylation (Wojcik et al, 2019). This is likely due to a decreased ratio of H3K27me3/H3K36me2, as knockdown of NSD2 could reverse the changes in gene expression by PRC2 inactivation. Interestingly, oncohistone H3 lysine 27 to methionine (H3K27M) mutation—found in diffuse intrinsic pontine gliomas (DIPGs)—dominantly inhibits PRC2 activity and reduces global H3K27me3 yet results in DNA hypomethylation (Bender et al, 2013; Lewis et al, 2013). Furthermore, EZHIP/CXORF67, a germline‐specific gene, encodes a protein that binds to and blocks PRC2 spreading from H3K27me3‐occupied CpG islands through an H3K27M‐like mechanism (Jain et al, 2019; Ragazzini et al, 2019). EZHIP is silenced through promoter methylation in adult tissues, and its de‐repression upon DNA hypomethylation results in PRC2 inhibition (Bayliss et al, 2016; Piunti et al, 2019). These findings again highlight the complex interplay between H3K27me3 and DNA methylation. Lastly, a recent study points to a role of H3K4me1 in predisposing CpG island methylation during cancer progression (Skvortsova et al, 2019). In breast and prostate cancers, the invasion of methylation at the edges of CpG island is linked to enrichment of H3K4me1. Inactivation of KMT2D leads to a concurrent loss of H3K4me1 and CpG methylation. It would be interesting to determine the impact of DNA methylation reprogramming on gene expression and tumor development driven by mutations in HMTs and, consequently, whether these tumors display altered sensitivity to DNA hypomethylating therapies.
Third, altered DNA methylation is a well‐established pan‐cancer molecular hallmark. DNA hypomethylation across intergenic regions and DNA hypermethylation at promoter CpG islands have been described in many cancer contexts, independently of specific genetic mutations and tissue types (Baylin & Jones, 2016). Interestingly, both features can be predicted from pre‐existing histone methylation marks in corresponding normal cells (Fig 4). Promoter CpG islands that gain DNA methylation in cancers are marked by H3K27me3 in embryonic or tissue stem/progenitor cells (Ohm et al, 2007; Schlesinger et al, 2007; Widschwendter et al, 2007; McGarvey et al, 2008). More recently, it was shown that the ratio of H3K27me3 to H3K4me3 at bivalent promoters can predict the likelihood of cancer‐associated DNA hypermethylation (Dunican et al, 2020). On the other hand, the loss of DNA methylation in cancers is found to mainly occur at partially methylated domains (PMDs) (Zhou et al, 2018). PMDs are associated with nuclear lamina and late‐replicating regions (Berman et al, 2012). Recent evidence suggests that DNA hypomethylation at PMDs is coupled to mitotic cell division (possibly due to ineffective maintenance methylation) and gain of heterochromatic histone marks such as H3K9me3 (DEEP Consortium et al, 2018; Zhou et al, 2018). In contrast, regions of H3K36me3 where de novo DNMTs are targeted are protected from progressive hypomethylation (Zhou et al, 2018). Collectively, these correlative studies raise an intriguing possibility that by comparing DNA methylomes of cancer and histone methylomes of normal cells, one may be able to delineate the potential tissues and cells of origins for certain difficult‐to‐diagnose tumors.
Figure 4. Reprogramming of DNA methylation during cancer progression.
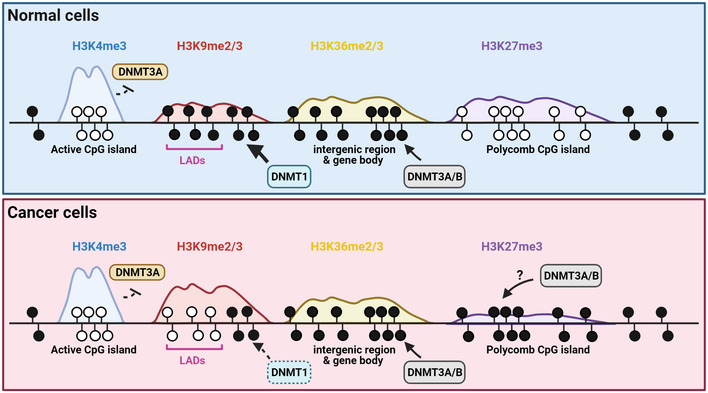
The transition from normal to cancerous state is associated with changes in genome‐wide patterns of DNA methylation. While promoter CpG islands of active genes marked by H3K4me3 remain free of DNA methylation, polycomb‐regulated promoter CpG islands become hypermethylated, possibly due to aberrant targeting of DNMT3A/B through an unknown mechanism. The gene‐poor, H3K9 methylation‐rich, late‐replicating lamina‐associated domains undergo progressive loss of maintenance methylation during cancer cell replication. Gene body and intergenic regions marked by H3K36 methylation are protected from such mitotically linked DNA hypomethylation, presumably due to the preferential targeting and activity of de novo methyltransferases DNMT3A/B.
Therapeutic implication
The close interplay between DNA and histone methylation enzymes in development and human diseases represents an opportunity for designing new therapeutic approaches. One strategy is to develop inhibitors of the “reader” domains that are key for enzyme interaction and activation. For example, two studies screened for inhibitors that prevent the binding between UHRF1’s TTD‐PHD finger and H3K9me3 (Houliston et al, 2017; Senisterra et al, 2018). These compounds can be used as powerful chemical probes and have the potential to become a new class of DNA hypomethylating agents with distinct mechanisms of action from that of catalytic inhibitors of DNMT1 for cancer therapy. Chemical probes for the NSD2‐PWWP and NSD3‐PWWP domains were recently reported (Böttcher et al, 2019), and similar approaches could be adopted to identify inhibitors of the DNMT3A/B PWWP domains to modulate de novo DNA methylation. Compared to inhibitors targeting the catalytic domains, reader domain inhibitors are expected to specifically correct disease‐associated mislocalization of methyltransferases while leaving their normal function unaffected, therefore providing a higher therapeutic index.
As another therapeutic strategy, combined inhibition of histone and DNA methylation has demonstrated synergy in cancer therapy. In gastric cancer, inhibition of EZH2 following DNA demethylation fully activates the expression of tumor suppressor RUNX3, suggesting a redundancy between H3K27me3 and CpG methylation in silencing certain genetic loci (Kodach et al, 2010). Indeed, in mESCs exposed to DNA hypomethylating culture condition, the initial de‐repression of retrotransposons is quickly silenced by H3K27me3 (Walter et al, 2016). Similarly, in breast cancer cells, global DNA hypomethylation seems to be compensated by gains in H3K9me3 and H3K27me3 with a remarkable allelic‐specific mutual exclusivity between these epigenetic marks (Hon et al, 2012). Accordingly, in colorectal cancer cells or chemotherapy‐resistant breast cancer lines, genetic or pharmacological inhibition of H3K27 or H3K9 methyltransferases significantly augments the effects of DNA hypomethylation on de‐repressing retrotransposons (Ohtani et al, 2018; Deblois et al, 2020). Since the expression of these transposable elements induces a “viral mimicry” that increases tumor infiltration (Chiappinelli et al, 2016), combined treatment of repressive HMTs and DNMTs may represent an effective strategy to sensitize poorly immune‐infiltrated tumors to immune checkpoint inhibitors.
Concluding remarks
It is increasingly evident that DNA and histone methylation marks cooperate to maintain patterns of epigenome in a stable and sometimes mitotically heritable manner. The molecular mechanisms, in large part, seem to involve the recognition (or the lack thereof) of chromatin modifications by the “reader” or “writer” domains of DNA‐ and histone‐modifying enzymes. As exciting advances in chromatin biochemistry and epigenomics continue to surface, researchers are equipped with powerful tools to gain a more comprehensive and deeper appreciation of how these epigenetic marks communicate (see also Box 1).
Box 1. In need of answers.
Are there additional functional domains in DNMTs and TET family enzymes that interact with modified histone to guide the genomic targeting of these DNA modifiers? Conversely, which histone‐modifying complexes are sensitive to CpG methylation and how?
How do multiple functional domains of DNMT cooperate to recognize histone modifications in the nucleosome context? What is the underlying biochemical and structural basis?
How does the imbalance between NSD1/H3K36me2, PRC2/H3K27me3, and DNMT3A/CpG methylation contribute to dysregulated gene expression during development and OGID syndromes?
Does aberrant histone–DNA methylation crosstalk contribute to cancer‐associated chromatin abnormalities? If yes, how?
What is the therapeutic potential for inhibitors of “reader” domains of DNA and histone methylation? How do we rationally combine DNA and histone methyltransferase inhibitors as more effective treatment strategy for human cancer and developmental diseases?
First, the majority of biochemical and structural analysis thus far involves isolated domains of chromatin enzymes and histone tail peptides. However, as illustrated by studies of UHRF1, extensive inter‐domain remodeling occurs during enzyme–histone engagement. Furthermore, since nucleosomes are the physiological substrates for chromatin‐modifying enzymes, the use of histone peptides may lead to incomplete or misguided mechanistic insights (Vaughan et al, 2018). Recent innovations in synthesizing chemically modified “designer” nucleosomes and nucleosome arrays provide ways to overcome this limitation (Holt & Muir, 2015). In addition, the revolution of cryogenic electron microscopy (cryo‐EM) has offered unprecedented opportunities to visualize the structure of fully assembled large chromatin complexes bound to nucleosomes (Jang & Song, 2019), such as the recently reported structure of DNMT3A2‐DNMT3B3‐nucleosome (Xu et al, 2020). We anticipate that these technologies will help reveal critical new insights into the molecular basis of DNA‐histone methylation crosstalk and uncover new functional domains, such as the recently described nuclear localization signal within the N‐terminal domain of DNMT3A1 (Zeng et al, 2020).
Second, our study of the histone and DNA methylation interplay is heavily based on bulk tissue cultures which offer limited physiological relevance. However, the recent explosion of new epigenome‐profiling technologies, such as CUT&RUN and CUT&Tag, enables analysis of genome‐wide histone modifications with extremely low cell numbers or even single cell (Ai et al, 2019; Carter et al, 2019; Hainer et al, 2019; Kaya‐Okur et al, 2019). Single‐cell CpG methylome analysis is also possible (Karemaker & Vermeulen, 2018). With these tools, we are now able to profile the epigenome in rare cell populations such as early embryos and adult tissue stem cells. Furthermore, we expect the rapid development of technology for simultaneous single‐cell analysis of histone and CpG methylation, which will allow interrogation of their interplay at unprecedented scale and resolution.
Finally, we look forward to the discovery and optimization of highly specific inhibitors that block the binding of chromatin “reader” domains to DNA and histone methylation. These inhibitors will serve as powerful probes to study the kinetics of chromatin interplay at high temporal resolution. Furthermore, compared to inhibitors of catalytic domains, “reader” domain inhibitors may be more effective in correcting mis‐targeting of chromatin enzyme activities, thus serving as potent and less toxic drug candidates for human developmental disorders and cancers driven by epigenetic abnormality.
Conflict of interest
The authors declare that they have no conflict of interest.
Acknowledgements
We apologize to colleagues whose work could not be cited because of space limitations. We thank the Lu laboratory for critical reading of the manuscript. CL is supported by funding from the National Institutes of Health (NIH) Grant R35GM138181 and the Pew‐Stewart Scholars for Cancer Research Award.
EMBO reports (2021) 22: e51803.
See the Glossary for abbreviations used in this article.
References
- Abdel‐Wahab O, Mullally A, Hedvat C, Garcia‐Manero G, Patel J, Wadleigh M, Malinge S, Yao J, Kilpivaara O, Bhat R et al (2009) Genetic characterization of TET1, TET2, and TET3 alterations in myeloid malignancies. Blood 114: 144–147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ai S, Xiong H, Li CC, Luo Y, Shi Q, Liu Y, Yu X, Li C, He A (2019) Profiling chromatin states using single‐cell itChIP‐seq. Nat Cell Biol 21: 1164–1172 [DOI] [PubMed] [Google Scholar]
- Aref‐Eshghi E, Bourque DK, Kerkhof J, Carere DA, Ainsworth P, Sadikovic B, Armour CM, Lin H (2019) Genome‐wide DNA methylation and RNA analyses enable reclassification of two variants of uncertain significance in a patient with clinical Kabuki syndrome. Hum Mutat 40: 1684–1689 [DOI] [PubMed] [Google Scholar]
- Aref‐Eshghi E, Rodenhiser DI, Schenkel LC, Lin H, Skinner C, Ainsworth P, Paré G, Hood RL, Bulman DE, Kernohan KD et al (2018) Genomic DNA methylation signatures enable concurrent diagnosis and clinical genetic variant classification in neurodevelopmental syndromes. Am J Hum Genet 102: 156–174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aref‐Eshghi E, Schenkel LC, Lin H, Skinner C, Ainsworth P, Paré G, Rodenhiser D, Schwartz C, Sadikovic B (2017) The defining DNA methylation signature of Kabuki syndrome enables functional assessment of genetic variants of unknown clinical significance. Epigenetics 12: 923–933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arita K, Isogai S, Oda T, Unoki M, Sugita K, Sekiyama N, Kuwata K, Hamamoto R, Tochio H, Sato M et al (2012) Recognition of modification status on a histone H3 tail by linked histone reader modules of the epigenetic regulator UHRF1. Proc Natl Acad Sci USA 109: 12950–12955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asmar F, Punj V, Christensen J, Pedersen MT, Pedersen A, Nielsen AB, Hother C, Ralfkiaer U, Brown P, Ralfkiaer E et al (2013) Genome‐wide profiling identifies a DNA methylation signature that associates with TET2 mutations in diffuse large B‐cell lymphoma. Haematologica 98: 1912–1920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auclair G, Borgel J, Sanz LA, Vallet J, Guibert S, Dumas M, Cavelier P, Girardot M, Forné T, Feil R et al (2016) EHMT2 directs DNA methylation for efficient gene silencing in mouse embryos. Genome Res 26: 192–202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballaré C, Lange M, Lapinaite A, Martin GM, Morey L, Pascual G, Liefke R, Simon B, Shi Y, Gozani O et al (2012) Phf19 links methylated Lys36 of histone H3 to regulation of Polycomb activity. Nat Struct Mol Biol 19: 1257–1265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartke T, Vermeulen M, Xhemalce B, Robson SC, Mann M, Kouzarides T (2010) Nucleosome‐interacting proteins regulated by DNA and histone methylation. Cell 143: 470–484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bates SE (2020) Epigenetic therapies for cancer. N Engl J Med 383: 650–663 [DOI] [PubMed] [Google Scholar]
- Baubec T, Colombo DF, Wirbelauer C, Schmidt J, Burger L, Krebs AR, Akalin A, Schübeler D (2015) Genomic profiling of DNA methyltransferases reveals a role for DNMT3B in genic methylation. Nature 520: 243–247 [DOI] [PubMed] [Google Scholar]
- Baylin SB, Jones PA (2016) Epigenetic determinants of cancer. Cold Spring Harb Perspect Biol 8: a019505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayliss J, Mukherjee P, Lu C, Jain SU, Chung C, Martinez D, Sabari B, Margol AS, Panwalkar P, Parolia A et al (2016) Lowered H3K27me3 and DNA hypomethylation define poorly prognostic pediatric posterior fossa ependymomas. Sci Transl Med 8: 366ra161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck DB, Petracovici A, He C, Moore HW, Louie RJ, Ansar M, Douzgou S, Sithambaram S, Cottrell T, Santos‐Cortez RLP et al (2020) Delineation of a human mendelian disorder of the DNA demethylation machinery: TET3 deficiency. Am J Hum Genet 106: 234–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender S, Tang Y, Lindroth AM, Hovestadt V, Jones DTW, Kool M, Zapatka M, Northcott PA, Sturm D, Wang W et al (2013) Reduced H3K27me3 and DNA hypomethylation are major drivers of gene expression in K27M mutant pediatric high‐grade gliomas. Cancer Cell 24: 660–672 [DOI] [PubMed] [Google Scholar]
- Berman BP, Weisenberger DJ, Aman JF, Hinoue T, Ramjan Z, Liu Y, Noushmehr H, Lange CPE, van Dijk CM, Tollenaar RAEM et al (2012) Regions of focal DNA hypermethylation and long‐range hypomethylation in colorectal cancer coincide with nuclear lamina–associated domains. Nat Genet 44: 40–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackledge NP, Zhou JC, Tolstorukov MY, Farcas AM, Park PJ, Klose RJ (2010) CpG islands recruit a histone H3 lysine 36 demethylase. Mol Cell 38: 179–190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bostick M, Kim JK, Estève P‐O, Clark A, Pradhan S, Jacobsen SE (2007) UHRF1 plays a role in maintaining DNA methylation in mammalian cells. Science 317: 1760–1764 [DOI] [PubMed] [Google Scholar]
- Böttcher J, Dilworth D, Reiser U, Neumüller RA, Schleicher M, Petronczki M, Zeeb M, Mischerikow N, Allali‐Hassani A, Szewczyk MM et al (2019) Fragment‐based discovery of a chemical probe for the PWWP1 domain of NSD3. Nat Chem Biol 15: 822–829 [DOI] [PubMed] [Google Scholar]
- Bourc'his D (2001) Dnmt3L and the establishment of maternal genomic imprints. Science 294: 2536–2539 [DOI] [PubMed] [Google Scholar]
- Bowman RL, Levine RL (2017) TET2 in normal and malignant hematopoiesis. Cold Spring Harb Perspect Med 7: a026518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan K, Shin JH, Tay JK, Prunello M, Gentles AJ, Sunwoo JB, Gevaert O (2017) NSD1 inactivation defines an immune cold, DNA hypomethylated subtype in squamous cell carcinoma. Sci Rep 7: 17064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brien GL, Gambero G, O’Connell DJ, Jerman E, Turner SA, Egan CM, Dunne EJ, Jurgens MC, Wynne K, Piao L et al (2012) Polycomb PHF19 binds H3K36me3 and recruits PRC2 and demethylase NO66 to embryonic stem cell genes during differentiation. Nat Struct Mol Biol 19: 1273–1281 [DOI] [PubMed] [Google Scholar]
- Brinkman AB, Gu H, Bartels SJJ, Zhang Y, Matarese F, Simmer F, Marks H, Bock C, Gnirke A, Meissner A et al (2012) Sequential ChIP‐bisulfite sequencing enables direct genome‐scale investigation of chromatin and DNA methylation cross‐talk. Genome Res 22: 1128–1138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bui N, Huang JK, Bojorquez‐Gomez A, Licon K, Sanchez KS, Tang SN, Beckett AN, Wang T, Zhang W, Shen JP et al (2018) Disruption of NSD1 in head and neck cancer promotes favorable chemotherapeutic responses linked to hypomethylation. Mol Cancer Ther 17: 1585–1594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai L, Rothbart SB, Lu R, Xu B, Chen W‐Y, Tripathy A, Rockowitz S, Zheng D, Patel DJ, Allis CD et al (2013) An H3K36 methylation‐engaging Tudor motif of polycomb‐like proteins mediates PRC2 complex targeting. Mol Cell 49: 571–582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cairns RA, Iqbal J, Lemonnier F, Kucuk C, de Leval L, Jais J‐P, Parrens M, Martin A, Xerri L, Brousset P et al (2012) IDH2 mutations are frequent in angioimmunoblastic T‐cell lymphoma. Blood 119: 1901–1903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrozza MJ, Li B, Florens L, Suganuma T, Swanson SK, Lee KK, Shia W‐J, Anderson S, Yates J, Washburn MP et al (2005) Histone H3 methylation by Set2 directs deacetylation of coding regions by Rpd3S to suppress spurious intragenic transcription. Cell 123: 581–592 [DOI] [PubMed] [Google Scholar]
- Carter B, Ku WL, Kang JY, Hu G, Perrie J, Tang Q, Zhao K (2019) Mapping histone modifications in low cell number and single cells using antibody‐guided chromatin tagmentation (ACT‐seq). Nat Commun 10: 3747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Challen GA, Sun D, Jeong M, Luo M, Jelinek J, Berg JS, Bock C, Vasanthakumar A, Gu H, Xi Y et al (2011) Dnmt3a is essential for hematopoietic stem cell differentiation. Nat Genet 44: 23–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang Y, Sun L, Kokura K, Horton JR, Fukuda M, Espejo A, Izumi V, Koomen JM, Bedford MT, Zhang X et al (2011) MPP8 mediates the interactions between DNA methyltransferase Dnmt3a and H3K9 methyltransferase GLP/G9a. Nat Commun 2: 533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen T, Tsujimoto N, Li E (2004) The PWWP domain of Dnmt3a and Dnmt3b is required for directing DNA methylation to the major satellite repeats at pericentric heterochromatin. Mol Cell Biol 24: 9048–9058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Yin Q, Inoue A, Zhang C, Zhang Y (2019) Allelic H3K27me3 to allelic DNA methylation switch maintains noncanonical imprinting in extraembryonic cells. Science Adv 5: eaay7246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng J, Yang Y, Fang J, Xiao J, Zhu T, Chen F, Wang P, Li Z, Yang H, Xu Y (2013) Structural insight into coordinated recognition of trimethylated histone H3 lysine 9 (H3K9me3) by the plant homeodomain (PHD) and tandem tudor domain (TTD) of UHRF1 (ubiquitin‐like, containing PHD and RING finger domains, 1) protein. J Biol Chem 288: 1329–1339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiappinelli KB, Zahnow CA, Ahuja N, Baylin SB (2016) Combining epigenetic and immunotherapy to combat cancer. Cancer Res 76: 1683–1689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choufani S, Cytrynbaum C, Chung BHY, Turinsky AL, Grafodatskaya D, Chen YA, Cohen ASA, Dupuis L, Butcher DT, Siu MT et al (2015) NSD1 mutations generate a genome‐wide DNA methylation signature. Nat Commun 6: 10207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choufani S, Gibson WT, Turinsky AL, Chung BHY, Wang T, Garg K, Vitriolo A, Cohen ASA, Cyrus S, Goodman S et al (2020) DNA methylation signature for EZH2 functionally classifies sequence variants in three PRC2 complex genes. Am J Hum Genet 106: 596–610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciccone DN, Su H, Hevi S, Gay F, Lei H, Bajko J, Xu G, Li E, Chen T (2009) KDM1B is a histone H3K4 demethylase required to establish maternal genomic imprints. Nature 461: 415–418 [DOI] [PubMed] [Google Scholar]
- Ciolfi A, Aref‐Eshghi E, Pizzi S, Pedace L, Miele E, Kerkhof J, Flex E, Martinelli S, Radio FC, Ruivenkamp CAL et al (2020) Frameshift mutations at the C‐terminus of HIST1H1E result in a specific DNA hypomethylation signature. Clin Epigenet 12: 7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen AS, Gibson WT (2016) EED‐associated overgrowth in a second male patient. J Hum Genet 61: 831–834 [DOI] [PubMed] [Google Scholar]
- Cohen ASA, Yap DB, Lewis MES, Chijiwa C, Ramos‐Arroyo MA, Tkachenko N, Milano V, Fradin M, McKinnon ML, Townsend KN et al (2016) Weaver syndrome‐associated EZH2 protein variants show impaired histone methyltransferase function in vitro . Hum Mutat 37: 301–307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comet I, Riising EM, Leblanc B, Helin K (2016) Maintaining cell identity: PRC2‐mediated regulation of transcription and cancer. Nat Rev Cancer 16: 803–810 [DOI] [PubMed] [Google Scholar]
- Couronné L, Bastard C, Bernard OA (2012) TET2 and DNMT3A mutations in human T‐cell lymphoma. N Engl J Med 366: 95–96 [DOI] [PubMed] [Google Scholar]
- Cyrus SS, Cohen ASA, Agbahovbe R, Avela K, Yeung KS, Chung BHY, Luk H‐M, Tkachenko N, Choufani S, Weksberg R et al (2019) Rare SUZ12 variants commonly cause an overgrowth phenotype. Am J Med Genet C Semin Med Genet 181: 532–547 [DOI] [PubMed] [Google Scholar]
- DaRosa PA, Harrison JS, Zelter A, Davis TN, Brzovic P, Kuhlman B, Klevit RE (2018) A bifunctional role for the UHRF1 UBL domain in the control of hemi‐methylated DNA‐dependent histone ubiquitylation. Mol Cell 72: 753–765.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deblois G, Tonekaboni SAM, Grillo G, Martinez C, Kao YI, Tai F, Ettayebi I, Fortier A‐M, Savage P, Fedor AN et al (2020) Epigenetic switch‐induced viral mimicry evasion in chemotherapy‐resistant breast cancer. Cancer Discov 10: 1312–1329 [DOI] [PubMed] [Google Scholar]
- DEEP Consortium , Salhab A, Nordström K, Gasparoni G, Kattler K, Ebert P, Ramirez F, Arrigoni L, Müller F, Polansky JK et al (2018) A comprehensive analysis of 195 DNA methylomes reveals shared and cell‐specific features of partially methylated domains. Genome Biol 19: 150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deevy O, Bracken AP (2019) PRC2 functions in development and congenital disorders. Development 146: dev181354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delhommeau F, Dupont S, Valle VD, James C, Trannoy S, Massé A, Kosmider O, Le Couedic J‐P, Robert F, Alberdi A et al (2009) Mutation in TET2 in myeloid cancers. N Engl J Med 360: 2289–2301 [DOI] [PubMed] [Google Scholar]
- Dhayalan A, Rajavelu A, Rathert P, Tamas R, Jurkowska RZ, Ragozin S, Jeltsch A (2010) The Dnmt3a PWWP domain reads histone 3 lysine 36 trimethylation and guides DNA methylation. J Biol Chem 285: 26114–26120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dikow N, Maas B, Gaspar H, Kreiss‐Nachtsheim M, Engels H, Kuechler A, Garbes L, Netzer C, Neuhann TM, Koehler U et al (2013) The phenotypic spectrum of duplication 5q35.2‐q35.3 encompassing NSD1: is it really a reversed sotos syndrome? Am J Med Genet A 161: 2158–2166 [DOI] [PubMed] [Google Scholar]
- Dong KB, Maksakova IA, Mohn F, Leung D, Appanah R, Lee S, Yang HW, Lam LL, Mager DL, Schübeler D et al (2008) DNA methylation in ES cells requires the lysine methyltransferase G9a but not its catalytic activity. EMBO J 27: 2691–2701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douglas J, Hanks S, Temple IK, Davies S, Murray A, Upadhyaya M, Tomkins S, Hughes HE, Cole TRP, Rahman N (2003) NSD1 mutations are the major cause of Sotos syndrome and occur in some cases of Weaver syndrome but are rare in other overgrowth phenotypes. Am J Hum Genet 72: 132–143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dukatz M, Adam S, Biswal M, Song J, Bashtrykov P, Jeltsch A (2020) Complex DNA sequence readout mechanisms of the DNMT3B DNA methyltransferase. Nucleic Acids Res 48: 11495–11509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dukatz M, Holzer K, Choudalakis M, Emperle M, Lungu C, Bashtrykov P, Jeltsch A (2019) H3K36me2/3 binding and DNA binding of the DNA methyltransferase DNMT3A PWWP domain both contribute to its chromatin interaction. J Mol Biol 431: 5063–5074 [DOI] [PubMed] [Google Scholar]
- Dunican DS, Mjoseng HK, Duthie L, Flyamer IM, Bickmore WA, Meehan RR (2020) Bivalent promoter hypermethylation in cancer is linked to the H327me3/H3K4me3 ratio in embryonic stem cells. BMC Biol 18: 25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duymich CE, Charlet J, Yang X, Jones PA, Liang G (2016) DNMT3B isoforms without catalytic activity stimulate gene body methylation as accessory proteins in somatic cells. Nat Commun 7: 11453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrlich M, Gama‐Sosa MA, Huang LH, Midgett RM, Kuo KC, McCune RA, Gehrke C (1982) Amount and distribution of 5‐methylcytosine in human DNA from different types of tissues of cells. Nucleic Acids Res 10: 2709–2721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epsztejn‐Litman S, Feldman N, Abu‐Remaileh M, Shufaro Y, Gerson A, Ueda J, Deplus R, Fuks F, Shinkai Y, Cedar H et al (2008) De novo DNA methylation promoted by G9a prevents reprogramming of embryonically silenced genes. Nat Struct Mol Biol 15: 1176–1183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst T, Chase AJ, Score J, Hidalgo‐Curtis CE, Bryant C, Jones AV, Waghorn K, Zoi K, Ross FM, Reiter A et al (2010) Inactivating mutations of the histone methyltransferase gene EZH2 in myeloid disorders. Nat Genet 42: 722–726 [DOI] [PubMed] [Google Scholar]
- Fang J, Cheng J, Wang J, Zhang Q, Liu M, Gong R, Wang P, Zhang X, Feng Y, Lan W et al (2016) Hemi‐methylated DNA opens a closed conformation of UHRF1 to facilitate its histone recognition. Nat Commun 7: 11197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farhangdoost N, Horth C, Hu B, Bareke E, Chen X, Li Y, Coradin M, Garcia BA, Lu C, Majewski J (2021) Chromatin dysregulation associated with NSD1 mutation in head and neck squamous cell carcinoma. Cell Rep 34: 108769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faust C, Schumacher A, Holdener B, Magnuson T (1995) The eed mutation disrupts anterior mesoderm production in mice. Development 121: 273–285 [DOI] [PubMed] [Google Scholar]
- Ferry L, Fournier A, Tsusaka T, Adelmant G, Shimazu T, Matano S, Kirsh O, Amouroux R, Dohmae N, Suzuki T et al (2017) Methylation of DNA ligase 1 by G9a/GLP recruits UHRF1 to replicating DNA and regulates DNA methylation. Mol Cell 67: 550–565.e5 [DOI] [PubMed] [Google Scholar]
- Figueroa ME, Abdel‐Wahab O, Lu C, Ward PS, Patel J, Shih A, Li Y, Bhagwat N, Vasanthakumar A, Fernandez HF et al (2010) Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 18: 553–567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischle W, Tseng BS, Dormann HL, Ueberheide BM, Garcia BA, Shabanowitz J, Hunt DF, Funabiki H, Allis CD (2005) Regulation of HP1‐chromatin binding by histone H3 methylation and phosphorylation. Nature 438: 1116–1122 [DOI] [PubMed] [Google Scholar]
- Fischle W, Wang Y, Jacobs SA, Kim Y, Allis CD, Khorasanizadeh S (2003) Molecular basis for the discrimination of repressive methyl‐lysine marks in histone H3 by Polycomb and HP1 chromodomains. Genes Dev 17: 1870–1881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster BM, Stolz P, Mulholland CB, Montoya A, Kramer H, Bultmann S, Bartke T (2018) Critical role of the UBL domain in stimulating the E3 ubiquitin ligase activity of UHRF1 toward chromatin. Mol Cell 72: 739–752.e9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu K, Bonora G, Pellegrini M (2020) Interactions between core histone marks and DNA methyltransferases predict DNA methylation patterns observed in human cells and tissues. Epigenetics 15: 272–282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuks F (2003) The DNA methyltransferases associate with HP1 and the SUV39H1 histone methyltransferase. Nucleic Acids Res 31: 2305–2312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao L, Emperle M, Guo Y, Grimm SA, Ren W, Adam S, Uryu H, Zhang Z‐M, Chen D, Yin J et al (2020) Comprehensive structure‐function characterization of DNMT3B and DNMT3A reveals distinctive de novo DNA methylation mechanisms. Nat Commun 11: 3355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatto S, Gagliardi M, Franzese M, Leppert S, Papa M, Cammisa M, Grillo G, Velasco G, Francastel C, Toubiana S et al (2017) ICF‐specific DNMT3B dysfunction interferes with intragenic regulation of mRNA transcription and alternative splicing. Nucleic Acids Res 45: 5739–5756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson WT, Hood RL, Zhan SH, Bulman DE, Fejes AP, Moore R, Mungall AJ, Eydoux P, Babul‐Hirji R, An J et al (2012) Mutations in EZH2 cause weaver syndrome. Am J Hum Genet 90: 110–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goll MG, Bestor TH (2005) Eukaryotic cytosine methyltransferases. Annu Rev Biochem 74: 481–514 [DOI] [PubMed] [Google Scholar]
- Grafodatskaya D, Chung BHY, Butcher DT, Turinsky AL, Goodman SJ, Choufani S, Chen Y‐A, Lou Y, Zhao C, Rajendram R et al (2013) Multilocus loss of DNA methylation in individuals with mutations in the histone H3 lysine 4 demethylase KDM5C. BMC Med Genomics 6: 1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossmann V, Haferlach C, Weissmann S, Roller A, Schindela S, Poetzinger F, Stadler K, Bellos F, Kern W, Haferlach T et al (2013) The molecular profile of adult T‐cell acute lymphoblastic leukemia: mutations in RUNX1 and DNMT3A are associated with poor prognosis in T‐ALL. Genes Chromosomes Cancer 52: 410–422 [DOI] [PubMed] [Google Scholar]
- Haferlach T, Nagata Y, Grossmann V, Okuno Y, Bacher U, Nagae G, Schnittger S, Sanada M, Kon A, Alpermann T et al (2014) Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia 28: 241–247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hainer SJ, Bošković A, McCannell KN, Rando OJ, Fazzio TG (2019) Profiling of pluripotency factors in single cells and early embryos. Cell 177: 1319–1329.e11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handa V, Jeltsch A (2005) Profound flanking sequence preference of Dnmt3a and Dnmt3b mammalian DNA methyltransferases shape the human epigenome. J Mol Biol 348: 1103–1112 [DOI] [PubMed] [Google Scholar]
- Hansen RS, Wijmenga C, Luo P, Stanek AM, Canfield TK, Weemaes CM, Gartler SM (1999) The DNMT3B DNA methyltransferase gene is mutated in the ICF immunodeficiency syndrome. Proc Natl Acad Sci USA 96: 14412–14417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison JS, Cornett EM, Goldfarb D, DaRosa PA, Li ZM, Yan F, Dickson BM, Guo AH, Cantu DV, Kaustov L et al (2016) Hemi‐methylated DNA regulates DNA methylation inheritance through allosteric activation of H3 ubiquitylation by UHRF1. eLife 5: e17101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y, Ecker JR (2015) Non‐CG methylation in the human genome. Annu Rev Genomics Hum Genet 16: 55–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y, Hariharan M, Gorkin DU, Dickel DE, Luo C, Castanon RG, Nery JR, Lee AY, Zhao Y, Huang H et al (2020) Spatiotemporal DNA methylome dynamics of the developing mouse fetus. Nature 583: 752–759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heyn P, Logan CV, Fluteau A, Challis RC, Auchynnikava T, Martin C‐A, Marsh JA, Taglini F, Kilanowski F, Parry DA et al (2019) Gain‐of‐function DNMT3A mutations cause microcephalic dwarfism and hypermethylation of Polycomb‐regulated regions. Nat Genet 51: 96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holoch D, Margueron R (2017) Mechanisms regulating PRC2 recruitment and enzymatic activity. Trends Biochem Sci 42: 531–542 [DOI] [PubMed] [Google Scholar]
- Holt M, Muir T (2015) Application of the protein semisynthesis strategy to the generation of modified chromatin. Annu Rev Biochem 84: 265–290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hon GC, Hawkins RD, Caballero OL, Lo C, Lister R, Pelizzola M, Valsesia A, Ye Z, Kuan S, Edsall LE et al (2012) Global DNA hypomethylation coupled to repressive chromatin domain formation and gene silencing in breast cancer. Genome Res 22: 246–258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houliston RS, Lemak A, Iqbal A, Ivanochko D, Duan S, Kaustov L, Ong MS, Fan L, Senisterra G, Brown PJ et al (2017) Conformational dynamics of the TTD–PHD histone reader module of the UHRF1 epigenetic regulator reveals multiple histone‐binding states, allosteric regulation, and druggability. J Biol Chem 292: 20947–20959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J‐L, Zhou BO, Zhang R‐R, Zhang K‐L, Zhou J‐Q, Xu G‐L (2009) The N‐terminus of histone H3 is required for de novo DNA methylation in chromatin. Proc Natl Acad Sci USA 106: 22187–22192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu L, Li Z, Wang P, Lin Y, Xu Y (2011) Crystal structure of PHD domain of UHRF1 and insights into recognition of unmodified histone H3 arginine residue 2. Cell Res 21: 1374–1378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunkapiller J, Shen Y, Diaz A, Cagney G, McCleary D, Ramalho‐Santos M, Krogan N, Ren B, Song JS, Reiter JF (2012) Polycomb‐like 3 promotes polycomb repressive complex 2 binding to CpG islands and embryonic stem cell self‐renewal. PLoS Genet 8: e1002576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imagawa E, Higashimoto K, Sakai Y, Numakura C, Okamoto N, Matsunaga S, Ryo A, Sato Y, Sanefuji M, Ihara K et al (2017) Mutations in genes encoding polycomb repressive complex 2 subunits cause Weaver syndrome. Hum Mutat 38: 637–648 [DOI] [PubMed] [Google Scholar]
- Ishiyama S, Nishiyama A, Saeki Y, Moritsugu K, Morimoto D, Yamaguchi L, Arai N, Matsumura R, Kawakami T, Mishima Y et al (2017) Structure of the Dnmt1 reader module complexed with a unique two‐mono‐ubiquitin mark on histone H3 reveals the basis for DNA methylation maintenance. Mol Cell 68: 350–360.e7 [DOI] [PubMed] [Google Scholar]
- Iwase S, Lan F, Bayliss P, de la Torre‐Ubieta L, Huarte M, Qi HH, Whetstine JR, Bonni A, Roberts TM, Shi Y (2007) The X‐linked mental retardation gene SMCX/JARID1C defines a family of histone H3 lysine 4 demethylases. Cell 128: 1077–1088 [DOI] [PubMed] [Google Scholar]
- Jackson JP, Lindroth AM, Cao X, Jacobsen SE (2002) Control of CpNpG DNA methylation by the KRYPTONITE histone H3 methyltransferase. Nature 416: 556–560 [DOI] [PubMed] [Google Scholar]
- Jaffe JD, Wang Y, Chan HM, Zhang J, Huether R, Kryukov GV, Bhang HC, Taylor JE, Hu M, Englund NP et al (2013) Global chromatin profiling reveals NSD2 mutations in pediatric acute lymphoblastic leukemia. Nat Genet 45: 1386–1391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain SU, Do TJ, Lund PJ, Rashoff AQ, Diehl KL, Cieslik M, Bajic A, Juretic N, Deshmukh S, Venneti S et al (2019) PFA ependymoma‐associated protein EZHIP inhibits PRC2 activity through a H3 K27M‐like mechanism. Nat Commun 10: 2146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaju RJ, Fidler C, Haas OA, Strickson AJ, Watkins F, Clark K, Cross NC, Cheng JF, Aplan PD, Kearney L et al (2001) A novel gene, NSD1, is fused to NUP98 in the t(5;11)(q35;p15.5) in de novo childhood acute myeloid leukemia. Blood 98: 1264–1267 [DOI] [PubMed] [Google Scholar]
- Jang S, Song J‐J (2019) The big picture of chromatin biology by cryo‐EM. Curr Opin Struct Biol 58: 76–87 [DOI] [PubMed] [Google Scholar]
- Jani KS, Jain SU, Ge EJ, Diehl KL, Lundgren SM, Müller MM, Lewis PW, Muir TW (2019) Histone H3 tail binds a unique sensing pocket in EZH2 to activate the PRC2 methyltransferase. Proc Natl Acad Sci USA 116: 8295–8300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeanpierre M, Turleau C, Aurias A, Prieur M, Ledeist F, Fischer A, Viegas‐Pequignot E (1993) An embryonic‐like methylation pattern of classical satellite DNA is observed in ICF syndrome. Hum Mol Genet 2: 731–735 [DOI] [PubMed] [Google Scholar]
- Jeffries AR, Maroofian R, Salter CG, Chioza BA, Cross HE, Patton MA, Dempster E, Temple IK, Mackay DJG, Rezwan FI et al (2019) Growth disrupting mutations in epigenetic regulatory molecules are associated with abnormalities of epigenetic aging. Genome Res 29: 1057–1066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong M, Sun D, Luo M, Huang Y, Challen GA, Rodriguez B, Zhang X, Chavez L, Wang H, Hannah R et al (2014) Large conserved domains of low DNA methylation maintained by Dnmt3a. Nat Genet 46: 17–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jermann P, Hoerner L, Burger L, Schubeler D (2014) Short sequences can efficiently recruit histone H3 lysine 27 trimethylation in the absence of enhancer activity and DNA methylation. Proc Natl Acad Sci USA 111: E3415–E3421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji M‐M, Huang Y‐H, Huang J‐Y, Wang Z‐F, Fu D, Liu H, Liu F, Leboeuf C, Wang L, Ye J et al (2018) Histone modifier gene mutations in peripheral T‐cell lymphoma not otherwise specified. Haematologica 103: 679–687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones PA (2012) Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet 13: 484–492 [DOI] [PubMed] [Google Scholar]
- Jones PA, Liang G (2009) Rethinking how DNA methylation patterns are maintained. Nat Rev Genet 10: 805–811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurkowska RZ, Jeltsch A (2016) Enzymology of mammalian DNA methyltransferases. Adv Exp Med Biol 945: 87–122 [DOI] [PubMed] [Google Scholar]
- Karagianni P, Amazit L, Qin J, Wong J (2008) ICBP90, a novel Methyl K9 H3 binding protein linking protein ubiquitination with heterochromatin formation. Mol Cell Biol 28: 705–717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karemaker ID, Vermeulen M (2018) Single‐cell DNA methylation profiling: technologies and biological applications. Trends Biotechnol 36: 952–965 [DOI] [PubMed] [Google Scholar]
- Kaya‐Okur HS, Wu SJ, Codomo CA, Pledger ES, Bryson TD, Henikoff JG, Ahmad K, Henikoff S (2019) CUT&Tag for efficient epigenomic profiling of small samples and single cells. Nat Commun 10: 1930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- King AD, Huang K, Rubbi L, Liu S, Wang C‐Y, Wang Y, Pellegrini M, Fan G (2016) Reversible regulation of promoter and enhancer histone landscape by DNA methylation in mouse embryonic stem cells. Cell Rep 17: 289–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein CJ, Botuyan M‐V, Wu Y, Ward CJ, Nicholson GA, Hammans S, Hojo K, Yamanishi H, Karpf AR, Wallace DC et al (2011) Mutations in DNMT1 cause hereditary sensory neuropathy with dementia and hearing loss. Nat Genet 43: 595–600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kodach LL, Jacobs RJ, Heijmans J, van Noesel CJM, Langers AMJ, Verspaget HW, Hommes DW, Offerhaus GJA, van den Brink GR, Hardwick JCH (2010) The role of EZH2 and DNA methylation in the silencing of the tumour suppressor RUNX3 in colorectal cancer. Carcinogenesis 31: 1567–1575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo Y, Shen L, Cheng AS, Ahmed S, Boumber Y, Charo C, Yamochi T, Urano T, Furukawa K, Kwabi‐Addo B et al (2008) Gene silencing in cancer by histone H3 lysine 27 trimethylation independent of promoter DNA methylation. Nat Genet 40: 741–750 [DOI] [PubMed] [Google Scholar]
- Kong X, Chen J, Xie W, Brown SM, Cai Y, Wu K, Fan D, Nie Y, Yegnasubramanian S, Tiedemann RL et al (2019) Defining UHRF1 domains that support maintenance of human colon cancer DNA methylation and oncogenic properties. Cancer Cell 35: 633–648.e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo AJ, Cheung P, Chen K, Zee BM, Kioi M, Lauring J, Xi Y, Park BH, Shi X, Garcia BA et al (2011) NSD2 links dimethylation of histone H3 at lysine 36 to oncogenic programming. Mol Cell 44: 609–620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurotaki N, Imaizumi K, Harada N, Masuno M, Kondoh T, Nagai T, Ohashi H, Naritomi K, Tsukahara M, Makita Y et al (2002) Haploinsufficiency of NSD1 causes Sotos syndrome. Nat Genet 30: 365–366 [DOI] [PubMed] [Google Scholar]
- Lederer D, Grisart B, Digilio MC, Benoit V, Crespin M, Ghariani SC, Maystadt I, Dallapiccola B, Verellen‐Dumoulin C (2012) Deletion of KDM6A, a histone demethylase interacting with MLL2, in three patients with Kabuki syndrome. Am J Hum Genet 90: 119–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S‐T, Wiemels JL (2016) Genome‐wide CpG island methylation and intergenic demethylation propensities vary among different tumor sites. Nucleic Acids Res 44: 1105–1117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehnertz B, Ueda Y, Derijck AAHA, Braunschweig U, Perez‐Burgos L, Kubicek S, Chen T, Li E, Jenuwein T, Peters AHFM (2003) Suv39h‐mediated histone H3 lysine 9 methylation directs DNA methylation to major satellite repeats at pericentric heterochromatin. Curr Biol 13: 1192–1200 [DOI] [PubMed] [Google Scholar]
- Lemonnier F, Couronné L, Parrens M, Jaïs J‐P, Travert M, Lamant L, Tournillac O, Rousset T, Fabiani B, Cairns RA et al (2012) Recurrent TET2 mutations in peripheral T‐cell lymphomas correlate with TFH‐like features and adverse clinical parameters. Blood 120: 1466–1469 [DOI] [PubMed] [Google Scholar]
- Leonards K, Almosailleakh M, Tauchmann S, Bagger FO, Thirant C, Juge S, Bock T, Méreau H, Bezerra MF, Tzankov A et al (2020) Nuclear interacting SET domain protein 1 inactivation impairs GATA1‐regulated erythroid differentiation and causes erythroleukemia. Nat Commun 11: 2807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung DC, Dong KB, Maksakova IA, Goyal P, Appanah R, Lee S, Tachibana M, Shinkai Y, Lehnertz B, Mager DL et al (2011) Lysine methyltransferase G9a is required for de novo DNA methylation and the establishment, but not the maintenance, of proviral silencing. Proc Natl Acad Sci USA 108: 5718–5723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung D, Du T, Wagner U, Xie W, Lee AY, Goyal P, Li Y, Szulwach KE, Jin P, Lorincz MC et al (2014) Regulation of DNA methylation turnover at LTR retrotransposons and imprinted loci by the histone methyltransferase Setdb1. Proc Natl Acad Sci USA 111: 6690–6695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis PW, Müller MM, Koletsky MS, Cordero F, Lin S, Banaszynski LA, Garcia BA, Muir TW, Becher OJ, Allis CD (2013) Inhibition of PRC2 activity by a gain‐of‐function H3 mutation found in pediatric glioblastoma. Science 340: 857–861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ley TJ, Ding L, Walter MJ, McLellan MD, Lamprecht T, Larson DE, Kandoth C, Payton JE, Baty J, Welch J et al (2010) DNMT3A mutations in acute myeloid leukemia. N Engl J Med 363: 2424–2433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lhoumaud P, Badri S, Rodriguez‐Hernaez J, Sakellaropoulos T, Sethia G, Kloetgen A, Cornwell M, Bhattacharyya S, Ay F, Bonneau R et al (2019) NSD2 overexpression drives clustered chromatin and transcriptional changes in a subset of insulated domains. Nat Commun 10: 1–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B‐Z, Huang Z, Cui Q‐Y, Song X‐H, Du L, Jeltsch A, Chen P, Li G, Li E, Xu G‐L (2011) Histone tails regulate DNA methylation by allosterically activating de novo methyltransferase. Cell Res 21: 1172–1181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li E, Bestor TH, Jaenisch R (1992) Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell 69: 915–926 [DOI] [PubMed] [Google Scholar]
- Li H, Rauch T, Chen Z‐X, Szabó PE, Riggs AD, Pfeifer GP (2006) The histone methyltransferase SETDB1 and the DNA methyltransferase DNMT3A interact directly and localize to promoters silenced in cancer cells. J Biol Chem 281: 19489–19500 [DOI] [PubMed] [Google Scholar]
- Li T, Wang L, Du Y, Xie S, Yang X, Lian F, Zhou Z, Qian C (2018a) Structural and mechanistic insights into UHRF1‐mediated DNMT1 activation in the maintenance DNA methylation. Nucleic Acids Res 46: 3218–3231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Zhang Z, Chen J, Liu W, Lai W, Liu B, Li X, Liu L, Xu S, Dong Q et al (2018b) Stella safeguards the oocyte methylome by preventing de novo methylation mediated by DNMT1. Nature 564: 136–140 [DOI] [PubMed] [Google Scholar]
- Li Y, Zheng H, Wang Q, Zhou C, Wei L, Liu X, Zhang W, Zhang Y, Du Z, Wang X et al (2018c) Genome‐wide analyses reveal a role of Polycomb in promoting hypomethylation of DNA methylation valleys. Genome Biol 19: 18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lister R, Pelizzola M, Dowen RH, Hawkins RD, Hon G, Tonti‐Filippini J, Nery JR, Lee L, Ye Z, Ngo Q‐M et al (2009) Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 462: 315–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu C, Jain SU, Hoelper D, Bechet D, Molden RC, Ran L, Murphy D, Venneti S, Hameed M, Pawel BR et al (2016) Histone H3K36 mutations promote sarcomagenesis through altered histone methylation landscape. Science 352: 844–849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu C, Ward PS, Kapoor GS, Rohle D, Turcan S, Abdel‐Wahab O, Edwards CR, Khanin R, Figueroa ME, Melnick A et al (2012) IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 483: 474–478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luscan A, Laurendeau I, Malan V, Francannet C, Odent S, Giuliano F, Lacombe D, Touraine R, Vidaud M, Pasmant E et al (2014) Mutations in SETD2 cause a novel overgrowth condition. J Med Genet 51: 512–517 [DOI] [PubMed] [Google Scholar]
- Lynch MD, Smith AJH, De Gobbi M, Flenley M, Hughes JR, Vernimmen D, Ayyub H, Sharpe JA, Sloane‐Stanley JA, Sutherland L et al (2012) An interspecies analysis reveals a key role for unmethylated CpG dinucleotides in vertebrate Polycomb complex recruitment: an interspecies analysis of chromatin bivalency. EMBO J 31: 317–329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallona I, Ilie IM, Karemaker ID, Butz S, Manzo M, Caflisch A, Baubec T (2021) Flanking sequence preference modulates de novo DNA methylation in the mouse genome. Nucleic Acids Res 49: 145–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manzo M, Wirz J, Ambrosi C, Villaseñor R, Roschitzki B, Baubec T (2017) Isoform‐specific localization of DNMT3A regulates DNA methylation fidelity at bivalent CpG islands. EMBO J 36: 3421–3434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mardis ER, Ding L, Dooling DJ, Larson DE, McLellan MD, Chen K, Koboldt DC, Fulton RS, Delehaunty KD, McGrath SD et al (2009) Recurring mutations found by sequencing an acute myeloid leukemia genome. N Engl J Med 361: 1058–1066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez‐Garcia E, Popovic R, Min D‐J, Sweet SMM, Thomas PM, Zamdborg L, Heffner A, Will C, Lamy L, Staudt LM et al (2011) The MMSET histone methyl transferase switches global histone methylation and alters gene expression in t(4;14) multiple myeloma cells. Blood 117: 211–220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin‐Herranz DE, Aref‐Eshghi E, Bonder MJ, Stubbs TM, Choufani S, Weksberg R, Stegle O, Sadikovic B, Reik W, Thornton JM (2019) Screening for genes that accelerate the epigenetic aging clock in humans reveals a role for the H3K36 methyltransferase NSD1. Genome Biol 20: 146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGarvey KM, Van Neste L, Cope L, Ohm JE, Herman JG, Van Criekinge W, Schuebel KE, Baylin SB (2008) Defining a chromatin pattern that characterizes DNA‐hypermethylated genes in colon cancer cells. Cancer Res 68: 5753–5759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin K, Flyamer IM, Thomson JP, Mjoseng HK, Shukla R, Williamson I, Grimes GR, Illingworth RS, Adams IR, Pennings S et al (2019) DNA methylation directs polycomb‐dependent 3D genome re‐organization in naive pluripotency. Cell Rep 29: 1974–1985.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meilinger D, Fellinger K, Bultmann S, Rothbauer U, Bonapace IM, Klinkert WEF, Spada F, Leonhardt H (2009) Np95 interacts with de novo DNA methyltransferases, Dnmt3a and Dnmt3b, and mediates epigenetic silencing of the viral CMV promoter in embryonic stem cells. EMBO Rep 10: 1259–1264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meissner A, Mikkelsen TS, Gu H, Wernig M, Hanna J, Sivachenko A, Zhang X, Bernstein BE, Nusbaum C, Jaffe DB et al (2008) Genome‐scale DNA methylation maps of pluripotent and differentiated cells. Nature 454: 766–770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Mierlo G, Dirks RAM, De Clerck L, Brinkman AB, Huth M, Kloet SL, Saksouk N, Kroeze LI, Willems S, Farlik M et al (2019) Integrative proteomic profiling reveals PRC2‐dependent epigenetic crosstalk maintains ground‐state pluripotency. Cell Stem Cell 24: 123–137.e8 [DOI] [PubMed] [Google Scholar]
- Ming X, Zhang Z, Zou Z, Lv C, Dong Q, He Q, Yi Y, Li Y, Wang H, Zhu B (2020) Kinetics and mechanisms of mitotic inheritance of DNA methylation and their roles in aging‐associated methylome deterioration. Cell Res 30: 980–996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mochizuki‐Kashio M, Mishima Y, Miyagi S, Negishi M, Saraya A, Konuma T, Shinga J, Koseki H, Iwama A (2011) Dependency on the polycomb gene Ezh2 distinguishes fetal from adult hematopoietic stem cells. Blood 118: 6553–6561 [DOI] [PubMed] [Google Scholar]
- Mohn F, Weber M, Rebhan M, Roloff TC, Richter J, Stadler MB, Bibel M, Schübeler D (2008) Lineage‐specific polycomb targets and de novo DNA methylation define restriction and potential of neuronal progenitors. Mol Cell 30: 755–766 [DOI] [PubMed] [Google Scholar]
- Morin RD, Johnson NA, Severson TM, Mungall AJ, An J, Goya R, Paul JE, Boyle M, Woolcock BW, Kuchenbauer F et al (2010) Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B‐cell lymphomas of germinal‐center origin. Nat Genet 42: 181–185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morin RD, Mendez‐Lago M, Mungall AJ, Goya R, Mungall KL, Corbett RD, Johnson NA, Severson TM, Chiu R, Field M et al (2011) Frequent mutation of histone‐modifying genes in non‐Hodgkin lymphoma. Nature 476: 298–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morselli M, Pastor WA, Montanini B, Nee K, Ferrari R, Fu K, Bonora G, Rubbi L, Clark AT, Ottonello S et al (2015) In vivo targeting of de novo DNA methylation by histone modifications in yeast and mouse. eLife 4: e06205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musselman CA, Avvakumov N, Watanabe R, Abraham CG, Lalonde M‐E, Hong Z, Allen C, Roy S, Nuñez JK, Nickoloff J et al (2012) Molecular basis for H3K36me3 recognition by the Tudor domain of PHF1. Nat Struct Mol Biol 19: 1266–1272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myant K, Termanis A, Sundaram AYM, Boe T, Li C, Merusi C, Burrage J, Heras JIL, Stancheva I (2011) LSH and G9a/GLP complex are required for developmentally programmed DNA methylation. Genome Res 21: 83–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nady N, Lemak A, Walker JR, Avvakumov GV, Kareta MS, Achour M, Xue S, Duan S, Allali‐Hassani A, Zuo X et al (2011) Recognition of multivalent histone states associated with heterochromatin by UHRF1 protein. J Biol Chem 286: 24300–24311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nanty L, Carbajosa G, Heap GA, Ratnieks F, van Heel DA, Down TA, Rakyan VK (2011) Comparative methylomics reveals gene‐body H3K36me3 in Drosophila predicts DNA methylation and CpG landscapes in other invertebrates. Genome Res 21: 1841–1850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neri F, Incarnato D, Krepelova A, Rapelli S, Pagnani A, Zecchina R, Parlato C, Oliviero S (2013a) Genome‐wide analysis identifies a functional association of Tet1 and Polycomb repressive complex 2 in mouse embryonic stem cells. Genome Biol 14: R91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neri F, Krepelova A, Incarnato D, Maldotti M, Parlato C, Galvagni F, Matarese F, Stunnenberg HG, Oliviero S (2013b) Dnmt3L antagonizes DNA methylation at bivalent promoters and favors DNA methylation at gene bodies in ESCs. Cell 155: 121–134 [DOI] [PubMed] [Google Scholar]
- Neri F, Rapelli S, Krepelova A, Incarnato D, Parlato C, Basile G, Maldotti M, Anselmi F, Oliviero S (2017) Intragenic DNA methylation prevents spurious transcription initiation. Nature 543: 72–77 [DOI] [PubMed] [Google Scholar]
- Ng SB, Bigham AW, Buckingham KJ, Hannibal MC, McMillin MJ, Gildersleeve HI, Beck AE, Tabor HK, Cooper GM, Mefford HC et al (2010) Exome sequencing identifies MLL2 mutations as a cause of Kabuki syndrome. Nat Genet 42: 790–793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishiyama A, Yamaguchi L, Sharif J, Johmura Y, Kawamura T, Nakanishi K, Shimamura S, Arita K, Kodama T, Ishikawa F et al (2013) Uhrf1‐dependent H3K23 ubiquitylation couples maintenance DNA methylation and replication. Nature 502: 249–253 [DOI] [PubMed] [Google Scholar]
- Noh K‐M, Wang H, Kim HR, Wenderski W, Fang F, Li CH, Dewell S, Hughes SH, Melnick AM, Patel DJ et al (2015) Engineering of a histone‐recognition domain in Dnmt3a alters the epigenetic landscape and phenotypic features of mouse ESCs. Mol Cell 59: 89–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Carroll D, Erhardt S, Pagani M, Barton SC, Surani MA, Jenuwein T (2001) The polycomb‐group GeneEzh2 is required for early mouse development. Mol Cell Biol 21: 4330–4336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohm JE, McGarvey KM, Yu X, Cheng L, Schuebel KE, Cope L, Mohammad HP, Chen W, Daniel VC, Yu W et al (2007) A stem cell–like chromatin pattern may predispose tumor suppressor genes to DNA hypermethylation and heritable silencing. Nat Genet 39: 237–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohtani H, Liu M, Zhou W, Liang G, Jones PA (2018) Switching roles for DNA and histone methylation depend on evolutionary ages of human endogenous retroviruses. Genome Res 28: 1147–1157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okano M, Bell DW, Haber DA, Li E (1999) DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 99: 247–257 [DOI] [PubMed] [Google Scholar]
- Ooi SKT, Qiu C, Bernstein E, Li K, Jia D, Yang Z, Erdjument‐Bromage H, Tempst P, Lin S‐P, Allis CD et al (2007) DNMT3L connects unmethylated lysine 4 of histone H3 to de novo methylation of DNA. Nature 448: 714–717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otani J, Nankumo T, Arita K, Inamoto S, Ariyoshi M, Shirakawa M (2009) Structural basis for recognition of H3K4 methylation status by the DNA methyltransferase 3A ATRX–DNMT3–DNMT3L domain. EMBO Rep 10: 1235–1241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papillon‐Cavanagh S, Lu C, Gayden T, Mikael LG, Bechet D, Karamboulas C, Ailles L, Karamchandani J, Marchione DM, Garcia BA et al (2017) Impaired H3K36 methylation defines a subset of head and neck squamous cell carcinomas. Nat Genet 49: 180–185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasini D, Bracken AP, Jensen MR, Denchi EL, Helin K (2004) Suz12 is essential for mouse development and for EZH2 histone methyltransferase activity. EMBO J 23: 4061–4071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perino M, Mierlo G, Karemaker ID, Genesen S, Vermeulen M, Marks H, Heeringen SJ, Veenstra GJC (2018) MTF2 recruits Polycomb Repressive Complex 2 by helical‐shape‐selective DNA binding. Nat Genet 50: 1002–1010 [DOI] [PubMed] [Google Scholar]
- Piunti A, Smith ER, Morgan MAJ, Ugarenko M, Khaltyan N, Helmin KA, Ryan CA, Murray DC, Rickels RA, Yilmaz BD et al (2019) CATACOMB: an endogenous inducible gene that antagonizes H3K27 methylation activity of Polycomb repressive complex 2 via an H3K27M‐like mechanism. Sci Adv 5: eaax2887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin W, Wolf P, Liu N, Link S, Smets M, La Mastra F, Forné I, Pichler G, Hörl D, Fellinger K et al (2015) DNA methylation requires a DNMT1 ubiquitin interacting motif (UIM) and histone ubiquitination. Cell Res 25: 911–929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ragazzini R, Pérez‐Palacios R, Baymaz IH, Diop S, Ancelin K, Zielinski D, Michaud A, Givelet M, Borsos M, Aflaki S et al (2019) EZHIP constrains Polycomb Repressive Complex 2 activity in germ cells. Nat Commun 10: 1–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajagopalan KN, Chen X, Weinberg DN, Chen H, Majewski J, Allis CD, Lu C (2021) Depletion of H3K36me2 recapitulates epigenomic and phenotypic changes induced by the H3.3K36M oncohistone mutation. Proc Natl Acad Sci USA 118: e2021795118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajakumara E, Wang Z, Ma H, Hu L, Chen H, Lin Y, Guo R, Wu F, Li H, Lan F et al (2011) PHD finger recognition of unmodified histone H3R2 links UHRF1 to regulation of euchromatic gene expression. Mol Cell 43: 275–284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao B, Shibata Y, Strahl BD, Lieb JD (2005) Dimethylation of histone H3 at lysine 36 demarcates regulatory and nonregulatory chromatin genome‐wide. Mol Cell Biol 25: 9447–9459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rayasam GV, Wendling O, Angrand P‐O, Mark M, Niederreither K, Song L, Lerouge T, Hager GL, Chambon P, Losson R (2003) NSD1 is essential for early post‐implantation development and has a catalytically active SET domain. EMBO J 22: 3153–3163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddington JP, Perricone SM, Nestor CE, Reichmann J, Youngson NA, Suzuki M, Reinhardt D, Dunican DS, Prendergast JG, Mjoseng H et al (2013) Redistribution of H3K27me3 upon DNA hypomethylation results in de‐repression of Polycomb target genes. Genome Biol 14: R25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy A, Zhang J, Davis NS, Moffitt AB, Love CL, Waldrop A, Leppa S, Pasanen A, Meriranta L, Karjalainen‐Lindsberg M‐L et al (2017) Genetic and functional drivers of diffuse large B cell lymphoma. Cell 171: 481–494.e15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren W, Fan H, Grimm SA, Guo Y, Kim JJ, Yin J, Li L, Petell CJ, Tan X‐F, Zhang Z‐M et al (2020) Direct readout of heterochromatic H3K9me3 regulates DNMT1‐mediated maintenance DNA methylation. Proc Natl Acad Sci USA 117: 18439–18447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren W, Gao L, Song J (2018) Structural basis of DNMT1 and DNMT3A‐mediated DNA methylation. Genes 9: 620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinaldi L, Datta D, Serrat J, Morey L, Solanas G, Avgustinova A, Blanco E, Pons JI, Matallanas D, Von Kriegsheim A et al (2016) Dnmt3a and Dnmt3b associate with enhancers to regulate human epidermal stem cell homeostasis. Cell Stem Cell 19: 491–501 [DOI] [PubMed] [Google Scholar]
- Rondelet G, Dal Maso T, Willems L, Wouters J (2016) Structural basis for recognition of histone H3K36me3 nucleosome by human de novo DNA methyltransferases 3A and 3B. J Struct Biol 194: 357–367 [DOI] [PubMed] [Google Scholar]
- Rosati R, La Starza R, Veronese A, Aventin A, Schwienbacher C, Vallespi T, Negrini M, Martelli MF, Mecucci C (2002) NUP98 is fused to the NSD3 gene in acute myeloid leukemia associated with t(8;11)(p11.2;p15). Blood 99: 3857–3860 [DOI] [PubMed] [Google Scholar]
- Rosenfeld JA, Kim KH, Angle B, Troxell R, Gorski JL, Westemeyer M, Frydman M, Senturias Y, Earl D, Torchia B et al (2013) Further evidence of contrasting phenotypes caused by reciprocal deletions and duplications: duplication of NSD1 causes growth retardation and microcephaly. Mol Syndromol 3: 247–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothbart SB, Dickson BM, Ong MS, Krajewski K, Houliston S, Kireev DB, Arrowsmith CH, Strahl BD (2013) Multivalent histone engagement by the linked tandem Tudor and PHD domains of UHRF1 is required for the epigenetic inheritance of DNA methylation. Genes Dev 27: 1288–1298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothbart SB, Krajewski K, Nady N, Tempel W, Xue S, Badeaux AI, Barsyte‐Lovejoy D, Martinez JY, Bedford MT, Fuchs SM et al (2012) Association of UHRF1 with methylated H3K9 directs the maintenance of DNA methylation. Nat Struct Mol Biol 19: 1155–1160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rottach A, Frauer C, Pichler G, Bonapace IM, Spada F, Leonhardt H (2010) The multi‐domain protein Np95 connects DNA methylation and histone modification. Nucleic Acids Res 38: 1796–1804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowe HM, Friedli M, Offner S, Verp S, Mesnard D, Marquis J, Aktas T, Trono D (2013) De novo DNA methylation of endogenous retroviruses is shaped by KRAB‐ZFPs/KAP1 and ESET. Development 140: 519–529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakata‐Yanagimoto M, Enami T, Yoshida K, Shiraishi Y, Ishii R, Miyake Y, Muto H, Tsuyama N, Sato‐Otsubo A, Okuno Y et al (2014) Somatic RHOA mutation in angioimmunoblastic T cell lymphoma. Nat Genet 46: 171–175 [DOI] [PubMed] [Google Scholar]
- Salhab A, Nordström K, Gasparoni G, Kattler K, Ebert P, Ramirez F, Arrigoni L, Müller F, Polansky JK, Cadenas C et al (2018) A comprehensive analysis of 195 DNA methylomes reveals shared and cell‐specific features of partially methylated domains. Genome Biol 19: 150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sankaran SM, Wilkinson AW, Gozani O (2016) A PWWP domain of histone‐lysine N‐methyltransferase NSD2 binds to dimethylated Lys36 of histone H3 and regulates NSD2 function at chromatin. J Biol Chem 291: 8465–8474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schenkel LC, Aref‐Eshghi E, Skinner C, Ainsworth P, Lin H, Paré G, Rodenhiser DI, Schwartz C, Sadikovic B (2018) Peripheral blood epi‐signature of Claes‐Jensen syndrome enables sensitive and specific identification of patients and healthy carriers with pathogenic mutations in KDM5C. Clin Epigenetics 10: 21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlesinger Y, Straussman R, Keshet I, Farkash S, Hecht M, Zimmerman J, Eden E, Yakhini Z, Ben‐Shushan E, Reubinoff BE et al (2007) Polycomb‐mediated methylation on Lys27 of histone H3 pre‐marks genes for de novo methylation in cancer. Nat Genet 39: 232–236 [DOI] [PubMed] [Google Scholar]
- Schmitges FW, Prusty AB, Faty M, Stützer A, Lingaraju GM, Aiwazian J, Sack R, Hess D, Li L, Zhou S et al (2011) Histone methylation by PRC2 is inhibited by active chromatin marks. Mol Cell 42: 330–341 [DOI] [PubMed] [Google Scholar]
- Sendžikaitė G, Hanna CW, Stewart‐Morgan KR, Ivanova E, Kelsey G (2019) A DNMT3A PWWP mutation leads to methylation of bivalent chromatin and growth retardation in mice. Nat Commun 10: 1884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senisterra G, Zhu HY, Luo X, Zhang H, Xun G, Lu C, Xiao W, Hajian T, Loppnau P, Chau I et al (2018) Discovery of small‐molecule antagonists of the H3K9me3 binding to UHRF1 tandem tudor domain. SLAS Discov 23: 930–940 [DOI] [PubMed] [Google Scholar]
- Sharif J, Muto M, Takebayashi S, Suetake I, Iwamatsu A, Endo TA, Shinga J, Mizutani‐Koseki Y, Toyoda T, Okamura K et al (2007) The SRA protein Np95 mediates epigenetic inheritance by recruiting Dnmt1 to methylated DNA. Nature 450: 908–912 [DOI] [PubMed] [Google Scholar]
- Shirane K, Miura F, Ito T, Lorincz MC (2020) NSD1‐deposited H3K36me2 directs de novo methylation in the mouse male germline and counteracts Polycomb‐associated silencing. Nat Genet 52: 1088–1098 [DOI] [PubMed] [Google Scholar]
- Shirohzu H, Kubota T, Kumazawa A, Sado T, Chijiwa T, Inagaki K, Suetake I, Tajima S, Wakui K, Miki Y et al (2002) Three novel DNMT3B mutations in Japanese patients with ICF syndrome. Am J Med Genet 112: 31–37 [DOI] [PubMed] [Google Scholar]
- Singh P, Li AX, Tran DA, Oates N, Kang E‐R, Wu X, Szabó PE (2013) De novo DNA methylation in the male germ line occurs by default but is excluded at sites of H3K4 methylation. Cell Rep 4: 205–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skvortsova K, Masle‐Farquhar E, Luu P‐L, Song JZ, Qu W, Zotenko E, Gould CM, Du Q, Peters TJ, Colino‐Sanguino Y et al (2019) DNA hypermethylation encroachment at CpG island borders in cancer is predisposed by H3K4 monomethylation patterns. Cancer Cell 35: 297–314.e8 [DOI] [PubMed] [Google Scholar]
- Sobreira N, Brucato M, Zhang L, Ladd‐Acosta C, Ongaco C, Romm J, Doheny KF, Mingroni‐Netto RC, Bertola D, Kim CA et al (2017) Patients with a Kabuki syndrome phenotype demonstrate DNA methylation abnormalities. Eur J Hum Genet 25: 1335–1344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Statham AL, Robinson MD, Song JZ, Coolen MW, Stirzaker C, Clark SJ (2012) Bisulfite sequencing of chromatin immunoprecipitated DNA (BisChIP‐seq) directly informs methylation status of histone‐modified DNA. Genome Res 22: 1120–1127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stegelmann F, Bullinger L, Schlenk RF, Paschka P, Griesshammer M, Blersch C, Kuhn S, Schauer S, Döhner H, Döhner K (2011) DNMT3A mutations in myeloproliferative neoplasms. Leukemia 25: 1217–1219 [DOI] [PubMed] [Google Scholar]
- Stewart KR, Veselovska L, Kim J, Huang J, Saadeh H, Tomizawa S, Smallwood SA, Chen T, Kelsey G (2015) Dynamic changes in histone modifications precede de novo DNA methylation in oocytes. Genes Dev 29: 2449–2462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streubel G, Watson A, Jammula SG, Scelfo A, Fitzpatrick DJ, Oliviero G, McCole R, Conway E, Glancy E, Negri GL et al (2018) The H3K36me2 methyltransferase Nsd1 demarcates PRC2‐mediated H3K27me2 and H3K27me3 domains in embryonic stem cells. Mol Cell 70: 371–379.e5 [DOI] [PubMed] [Google Scholar]
- Suzuki MM, Bird A (2008) DNA methylation landscapes: provocative insights from epigenomics. Nat Rev Genet 9: 465–476 [DOI] [PubMed] [Google Scholar]
- Tachibana M, Matsumura Y, Fukuda M, Kimura H, Shinkai Y (2008) G9a/GLP complexes independently mediate H3K9 and DNA methylation to silence transcription. EMBO J 27: 2681–2690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takenouchi T, Uehara T, Kosaki K, Mizuno S (2018) Growth pattern of Rahman syndrome. Am J Med Genet A 176: 712–714 [DOI] [PubMed] [Google Scholar]
- Tamaru H, Selker EU (2001) A histone H3 methyltransferase controls DNA methylation in Neurospora crassa . Nature 414: 277–283 [DOI] [PubMed] [Google Scholar]
- Tatton‐Brown K, Hanks S, Ruark E, Zachariou A, Duarte SDV, Ramsay E, Snape K, Murray A, Perdeaux ER, Seal S et al (2011) Germline mutations in the oncogene EZH2 cause Weaver syndrome and increased human height. Oncotarget 2: 1127–1133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatton‐Brown K, Loveday C, Yost S, Clarke M, Ramsay E, Zachariou A, Elliott A, Wylie H, Ardissone A, Rittinger O et al (2017) Mutations in epigenetic regulation genes are a major cause of overgrowth with intellectual disability. Am J Hum Genet 100: 725–736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatton‐Brown K, Seal S, Ruark E, Harmer J, Ramsay E, del Vecchio DS, Zachariou A, Hanks S, O’Brien E, Aksglaede L et al (2014) Mutations in the DNA methyltransferase gene DNMT3A cause an overgrowth syndrome with intellectual disability. Nat Genet 46: 385–388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tauber M, Kreuz S, Lemak A, Mandal P, Yerkesh Z, Veluchamy A, Al‐Gashgari B, Aljahani A, Cortés‐Medina LV, Azhibek D et al (2020) Alternative splicing and allosteric regulation modulate the chromatin binding of UHRF1. Nucleic Acids Res 48: 7728–7747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiedemann RL, Hlady RA, Hanavan PD, Lake DF, Tibes R, Lee J‐H, Choi J‐H, Ho TH, Robertson KD (2016) Dynamic reprogramming of DNA methylation in SETD2‐deregulated renal cell carcinoma. Oncotarget 7: 1927–1946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaughan RM, Dickson BM, Whelihan MF, Johnstone AL, Cornett EM, Cheek MA, Ausherman CA, Cowles MW, Sun Z‐W, Rothbart SB (2018) Chromatin structure and its chemical modifications regulate the ubiquitin ligase substrate selectivity of UHRF1. Proc Natl Acad Sci USA 115: 8775–8780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veland N, Hardikar S, Zhong Y, Gayatri S, Dan J, Strahl BD, Rothbart SB, Bedford MT, Chen T (2017) The arginine methyltransferase PRMT6 regulates DNA methylation and contributes to global DNA hypomethylation in cancer. Cell Rep 21: 3390–3397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velasco G, Grillo G, Touleimat N, Ferry L, Ivkovic I, Ribierre F, Deleuze J‐F, Chantalat S, Picard C, Francastel C (2018) Comparative methylome analysis of ICF patients identifies heterochromatin loci that require ZBTB24, CDCA7 and HELLS for their methylated state. Hum Mol Genet 27: 2409–2424 [DOI] [PubMed] [Google Scholar]
- Vezzoli A, Bonadies N, Allen MD, Freund SMV, Santiveri CM, Kvinlaug BT, Huntly BJP, Göttgens B, Bycroft M (2010) Molecular basis of histone H3K36me3 recognition by the PWWP domain of Brpf1. Nat Struct Mol Biol 17: 617–619 [DOI] [PubMed] [Google Scholar]
- Viré E, Brenner C, Deplus R, Blanchon L, Fraga M, Didelot C, Morey L, Eynde AV, Bernard D, Vanderwinden J‐M et al (2006) The Polycomb group protein EZH2 directly controls DNA methylation. Nature 439: 871–874 [DOI] [PubMed] [Google Scholar]
- Vlachogiannis G, Niederhuth CE, Tuna S, Stathopoulou A, Viiri K, de Rooij DG, Jenner RG, Schmitz RJ, Ooi SKT (2015) The Dnmt3L ADD domain controls cytosine methylation establishment during spermatogenesis. Cell Rep 10: 944–956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wachter E, Quante T, Merusi C, Arczewska A, Stewart F, Webb S, Bird A (2014) Synthetic CpG islands reveal DNA sequence determinants of chromatin structure. eLife 3: e03397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter M, Teissandier A, Pérez‐Palacios R, Bourc'his D (2016) An epigenetic switch ensures transposon repression upon dynamic loss of DNA methylation in embryonic stem cells. eLife 5: e11418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Shen J, Yang Z, Chen P, Zhao B, Hu W, Lan W, Tong X, Wu H, Li G et al (2011) Structural basis for site‐specific reading of unmodified R2 of histone H3 tail by UHRF1 PHD finger. Cell Res 21: 1379–1382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Hevi S, Kurash JK, Lei H, Gay F, Bajko J, Su H, Sun W, Chang H, Xu G et al (2009) The lysine demethylase LSD1 (KDM1) is required for maintenance of global DNA methylation. Nat Genet 41: 125–129 [DOI] [PubMed] [Google Scholar]
- Wasson JA, Simon AK, Myrick DA, Wolf G, Driscoll S, Pfaff SL, Macfarlan TS, Katz DJ (2016) Maternally provided LSD1/KDM1A enables the maternal‐to‐zygotic transition and prevents defects that manifest postnatally. eLife 5: e08848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watatani Y, Sato Y, Miyoshi H, Sakamoto K, Nishida K, Gion Y, Nagata Y, Shiraishi Y, Chiba K, Tanaka H et al (2019) Molecular heterogeneity in peripheral T‐cell lymphoma, not otherwise specified revealed by comprehensive genetic profiling. Leukemia 33: 2867–2883 [DOI] [PubMed] [Google Scholar]
- Weemaes CM, van Tol MJ, Wang J, van Ostaijen‐ten Dam MM, van Eggermond MC, Thijssen PE, Aytekin C, Brunetti‐Pierri N, van der Burg M, Graham Davies E et al (2013) Heterogeneous clinical presentation in ICF syndrome: correlation with underlying gene defects. Eur J Hum Genet 21: 1219–1225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberg DN, Papillon‐Cavanagh S, Chen H, Yue Y, Chen X, Rajagopalan KN, Horth C, McGuire JT, Xu X, Nikbakht H et al (2019) The histone mark H3K36me2 recruits DNMT3A and shapes the intergenic DNA methylation landscape. Nature 573: 281–286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisenberger DJ, Velicescu M, Cheng JC, Gonzales FA, Liang G, Jones PA (2004) Role of the DNA methyltransferase variant DNMT3b3 in DNA methylation. Mol Cancer Res 2: 62–72 [PubMed] [Google Scholar]
- Widschwendter M, Fiegl H, Egle D, Mueller‐Holzner E, Spizzo G, Marth C, Weisenberger DJ, Campan M, Young J, Jacobs I et al (2007) Epigenetic stem cell signature in cancer. Nat Genet 39: 157–158 [DOI] [PubMed] [Google Scholar]
- Winkelmann J, Lin L, Schormair B, Kornum BR, Faraco J, Plazzi G, Melberg A, Cornelio F, Urban AE, Pizza F et al (2012) Mutations in DNMT1 cause autosomal dominant cerebellar ataxia, deafness and narcolepsy. Hum Mol Genet 21: 2205–2210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojcik JB, Marchione DM, Sidoli S, Djedid A, Lisby A, Majewski J, Garcia BA (2019) Epigenomic reordering induced by polycomb loss drives oncogenesis but leads to therapeutic vulnerabilities in malignant peripheral nerve sheath tumors. Cancer Res 79: 3205–3219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, Coskun V, Tao J, Xie W, Ge W, Yoshikawa K, Li E, Zhang Y, Sun YE (2010) Dnmt3a‐dependent nonpromoter DNA methylation facilitates transcription of neurogenic genes. Science 329: 444–448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, Zhang Y (2014) Reversing DNA methylation: mechanisms, genomics, and biological functions. Cell 156: 45–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie S, Jakoncic J, Qian C (2012) UHRF1 double tudor domain and the adjacent PHD finger act together to recognize K9me3‐containing histone H3 tail. J Mol Biol 415: 318–328 [DOI] [PubMed] [Google Scholar]
- Xie W, Schultz MD, Lister R, Hou Z, Rajagopal N, Ray P, Whitaker JW, Tian S, Hawkins RD, Leung D et al (2013) Epigenomic analysis of multilineage differentiation of human embryonic stem cells. Cell 153: 1134–1148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xin Z, Tachibana M, Guggiari M, Heard E, Shinkai Y, Wagstaff J (2003) Role of histone methyltransferase G9a in CpG methylation of the Prader‐Willi syndrome imprinting center. J Biol Chem 278: 14996–15000 [DOI] [PubMed] [Google Scholar]
- Xu Q, Xiang Y, Wang Q, Wang L, Brind’Amour J, Bogutz AB, Zhang Y, Zhang B, Yu G, Xia W et al (2019) SETD2 regulates the maternal epigenome, genomic imprinting and embryonic development. Nat Genet 1: 844–856 [DOI] [PubMed] [Google Scholar]
- Xu T‐H, Liu M, Zhou XE, Liang G, Zhao G, Xu HE, Melcher K, Jones PA (2020a) Structure of nucleosome‐bound DNA methyltransferases DNMT3A and DNMT3B. Nature 586: 151–155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Li J, Rong B, Zhao B, Wang M, Dai R, Chen Q, Liu H, Gu Z, Liu S et al (2020b) DNMT3A reads and connects histone H3K36me2 to DNA methylation. Protein & Cell 11: 150–154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarychkivska O, Shahabuddin Z, Comfort N, Boulard M, Bestor TH (2018) BAH domains and a histone‐like motif in DNA methyltransferase 1 (DNMT1) regulate de novo and maintenance methylation in vivo . J Biol Chem 293: 19466–19475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying Q‐L, Wray J, Nichols J, Batlle‐Morera L, Doble B, Woodgett J, Cohen P, Smith A (2008) The ground state of embryonic stem cell self‐renewal. Nature 453: 519–523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J‐R, Lee C‐H, Oksuz O, Stafford JM, Reinberg D (2019) PRC2 is high maintenance. Genes Dev 33: 903–935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan W, Xu M, Huang C, Liu N, Chen S, Zhu B (2011) H3K36 methylation antagonizes PRC2‐mediated H3K27 methylation. J Biol Chem 286: 7983–7989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng Y, Chen T (2019) DNA methylation reprogramming during mammalian development. Genes 10: 257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng Y, Ren R, Kaur G, Hardikar S, Ying Z, Babcock L, Gupta E, Zhang X, Chen T, Cheng X (2020) The inactive Dnmt3b3 isoform preferentially enhances Dnmt3b‐mediated DNA methylation. Genes Dev 34: 1546–1558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Jeong M, Huang X, Wang XQ, Wang X, Zhou W, Shamim MS, Gore H, Himadewi P, Liu Y et al (2020) Large DNA methylation nadirs anchor chromatin loops maintaining hematopoietic stem cell identity. Mol Cell 78: 506–521.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Charlton J, Karnik R, Beerman I, Smith ZD, Gu H, Boyle P, Mi X, Clement K, Pop R et al (2018a) Targets and genomic constraints of ectopic Dnmt3b expression. eLife 7: e40757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Jurkowska R, Soeroes S, Rajavelu A, Dhayalan A, Bock I, Rathert P, Brandt O, Reinhardt R, Fischle W et al (2010) Chromatin methylation activity of Dnmt3a and Dnmt3a/3L is guided by interaction of the ADD domain with the histone H3 tail. Nucleic Acids Res 38: 4246–4253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y‐L, Sun J‐W, Xie Y‐Y, Zhou Y, Liu P, Song J‐C, Xu C‐H, Wang L, Liu D, Xu A‐N et al (2018b) Setd2 deficiency impairs hematopoietic stem cell self‐renewal and causes malignant transformation. Cell Res 28: 476–490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Q, Zhang J, Chen R, Wang L, Li B, Cheng H, Duan X, Zhu H, Wei W, Li J et al (2016a) Dissecting the precise role of H3K9 methylation in crosstalk with DNA maintenance methylation in mammals. Nat Commun 7: 12464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Sun H, Wang H (2016b) Long noncoding RNAs in DNA methylation: new players stepping into the old game. Cell Biosci 6: 45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou W, Dinh HQ, Ramjan Z, Weisenberger DJ, Nicolet CM, Shen H, Laird PW, Berman BP (2018) DNA methylation loss in late‐replicating domains is linked to mitotic cell division. Nat Genet 50: 591–602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu H, Geiman TM, Xi S, Jiang Q, Schmidtmann A, Chen T, Li E, Muegge K (2006) Lsh is involved in de novo methylation of DNA. EMBO J 25: 335–345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu H, Wang G, Qian J (2016) Transcription factors as readers and effectors of DNA methylation. Nat Rev Genet 17: 551–565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X, He F, Zeng H, Ling S, Chen A, Wang Y, Yan X, Wei W, Pang Y, Cheng H et al (2014) Identification of functional cooperative mutations of SETD2 in human acute leukemia. Nat Genet 46: 287–293 [DOI] [PMC free article] [PubMed] [Google Scholar]