

Abstract
SIRT7 is a NAD+‐dependent deacetylase that controls important aspects of metabolism, cancer, and bone formation. However, the molecular targets and functions of SIRT7 in the kidney are currently unknown. In silico analysis of kidney transcripts of the BXD murine genetic reference population revealed a positive correlation between Sirt7 and Slc12a7 mRNA expression, suggesting a link between the corresponding proteins that these transcripts encode, SIRT7, and the K‐Cl cotransporter KCC4, respectively. Here, we find that protein levels and activity of heterologously expressed KCC4 are significantly modulated depending on its acetylation status in Xenopus laevis oocytes. Moreover, SIRT7 interacts with KCC4 in a NAD+‐dependent manner and increases its stability and activity in HEK293 cells. Interestingly, metabolic acidosis increases SIRT7 expression in kidney, as occurs with KCC4. In contrast, total SIRT7‐deficient mice present lower KCC4 expression and an exacerbated metabolic acidosis than wild‐type mice during an ammonium chloride challenge. Altogether, our data suggest that SIRT7 interacts with, stabilizes and modulates KCC4 activity through deacetylation, and reveals a novel role for SIRT7 in renal physiology.
Keywords: kidney tubule, renal tubular acidosis, sirtuins
Subject Categories: Membrane & Intracellular Transport; Molecular Biology of Disease; Post-translational Modifications, Proteolysis & Proteomics
SIRT7 interacts with, stabilizes and modulates KCC4 activity through deacetylation, revealing a novel role for SIRT7 in renal physiology.
Introduction
Sirtuins (SIRT1‐7) are a conserved family of NAD+‐dependent deacetylases that differ in tissue expression, intracellular localization, enzymatic activity, and target proteins (Sauve et al, 2001; Houtkooper et al, 2012). Among the target proteins of the sirtuins are histones, several transcription factors, and a variety of enzymes involved in different cellular processes, such as proliferation, DNA repair, antioxidant activity, mitochondrial function, and metabolism (Baur et al, 2012). The role of sirtuins in renal physiology has begun to be evaluated (Hao & Haase, 2010; Hershberger et al 2017; Morigi et al, 2018). For instance, SIRT1 decreases sodium reabsorption and thus regulates sodium and water handling (Zhang et al, 2009). Moreover, an increase in SIRT1 activity, through a boost on NAD+ production, enhances mitochondrial function and protects the kidney from acute injury (Katsyuba et al, 2018). Similarly, SIRT3 and SIRT6 protects against acute kidney injury (Morigi et al, 2015) and kidney fibrosis (Cai et al, 2020), respectively. Although the kidney is one of the tissues that express the highest levels of Sirt7 mRNA and protein (Ford et al, 2006), no role of SIRT7 has been described in renal physiology. SIRT7 regulates transcription through activation of RNA polymerase I (Ford et al, 2006), deacetylates lysine 18 of histone H3 (Barber et al, 2012), and desuccinylates lysine 122 of histone H3 promoting chromatin condensation and DNA double‐strand breaks repair (Li et al, 2016). SIRT7‐deficient mice exhibit cardiac hypertrophy and inflammatory cardiomyopathy due to hyperacetylation of p53 (Vakhrusheva et al, 2008) and a multisystemic mitochondrial dysfunction (Ryu et al, 2014). Furthermore, recent studies have described a role for SIRT7 in adipogenesis (Cioffi et al, 2015; Fang et al, 2017), bone formation (Fukuda et al, 2018), and lipid metabolism (Shin et al, 2013; Ryu et al, 2014; Yoshizawa et al, 2014). In fact, SIRT7 modulates mitochondrial function through the deacetylation of the transcription factor GABPβ, a key regulator of the expression of mitochondrial proteins (Ryu et al, 2014).
Notably, SIRT7‐deficient mice display age‐dependent hearing loss, suggesting a malfunction in ion transport in the ear (Ryu et al, 2014). The hearing loss phenotype resembles that observed in mice lacking the K‐Cl cotransporter KCC4 (Boettger et al, 2002). KCC4 belongs to the SLC12 family of electroneutral cation‐chloride‐coupled cotransporters that are critical for several physiological processes, such as cell volume regulation, modulation of intracellular Cl− concentration, and transepithelial ion flux, which is critical for blood pressure and acid–base homeostasis (Arroyo et al, 2013). These transporters are regulated by phosphorylation/dephosphorylation (Pacheco‐Alvarez et al, 2006; Richardson et al, 2008; Glover et al, 2010) and ubiquitylation mechanisms (Ko et al, 2010; Arroyo et al, 2011; Gamba, 2012), but thus far, none have been shown to be regulated by acetylation/deacetylation processes. In the kidney, KCC4 is expressed in the proximal convoluted tubule, and the thick ascending limb of Henle’s loop (TAL), where it constitutes a pathway for cellular exit of K+ and Cl−, and in the α‐intercalated cells of the collecting duct (Melo et al, 2013a), where its activity is important for proton secretion to the tubular fluid (urine), thereby maintaining the acid–base balance (Gamba, 2005). Accordingly, KCC4 expression is increased during metabolic acidosis (Melo et al, 2013a), and mice deficient in KCC4 develop renal tubular acidosis (Boettger et al, 2002). Given the similar deafness phenotype observed in the SIRT7‐ and KCC4‐deficient mice, and the fact that both proteins are highly expressed in the kidney, we hypothesized that SIRT7 might be involved in KCC4 regulation. Here, using Xenopus laevis oocytes, HEK293 cells, a mouse model of metabolic acidosis, and total and renal tubular‐specific SIRT7‐deficient mice, we demonstrate that SIRT7 interacts with, stabilizes and modulates KCC4 activity through deacetylation, and therefore reveal a role for SIRT7 in renal physiology.
Results and Discussion
KCC4 acetylation decreases KCC4 protein abundance and activity in X. laevis oocytes
As a first approach, we performed an in silico analysis of the GeneNetwork database (www.genenetwork.org) in the kidneys of 41 recombinant inbred mouse strains of the BXD genetic reference population (Andreux et al, 2012), to evaluate whether Sirt7 mRNA abundance correlates with that of Slc12a7. We confirmed that the kidney is one of the tissues that exhibit the highest levels of Sirt7 mRNA expression (Fig 1A). Interestingly, Sirt7 expression significantly correlated (Pearson’s r = 0.597, P = 4.6E‐05) with Slc12a7 expression (Fig 1B), suggesting that the proteins encoded by these transcripts may have a functional interaction. Interestingly, KCC4 has been found to be acetylated at the putative acetylation site lysine 114 (K114) by high throughput proteomic analysis (Lundby et al, 2012), reinforcing the hypothesis that KCC4 activity could be modulated by acetylation. Thus, to evaluate whether acetylation is a post‐translational modification that affects KCC4, X. laevis oocytes injected with KCC4 were incubated with NAM, a deacetylase inhibitor, or NAD+, a sirtuin co‐substrate and co‐activator. KCC4 was acetylated after 4 h of incubation with NAM and remained deacetylated when incubated with NAD+ (Fig 1C). We then measured KCC4 protein levels after different incubation times with NAM and observed that KCC4 protein levels decreased more than 70% after 48 h of incubation with NAM (Fig 1D, upper panel). The decrease in KCC4 protein levels was paralleled by changes in KCC4 activity since incubation with NAM decreased KCC4 activity in a time‐dependent manner, reaching an 88% reduction after 48 h (Fig 1D, lower panel). The decrease in KCC4 protein levels was not attributed to a gradual decline in the expression system, as KCC4 expression increased in a time‐dependent manner in untreated oocytes (Fig 1E). Unfortunately, we were unable to confirm the acetylation site of KCC4 by mass spectrometry since the amount of immunoprecipitated KCC4 was not enough to identify a sufficient number of peptides. The difficulty to evaluate acetylation in membrane proteins has been previously reported (Griffin & Schnitzeer, 2011). However, the expression of a mutant KCC4 protein, in which the K114 lysine residue was replaced by glutamine (KCC4 K114Q) to mimic the charge of the hyperacetylated KCC4, was lower than wild‐type KCC4 and exhibited lower activity. In contrast, a second mutant, in which the K114 lysine residue was replaced by an arginine (KCC4 K114R) to mimic the hypoacetylated KCC4, exhibited higher expression and activity (Fig 1F). These results demonstrate that the activity of heterologously expressed KCC4 in X. laevis oocytes is modulated through acetylation and confirm that K114 is a key acetylation site.
Figure 1. KCC4 expression correlates with SIRT7 expression, and KCC4 activity is modulated by acetylation.
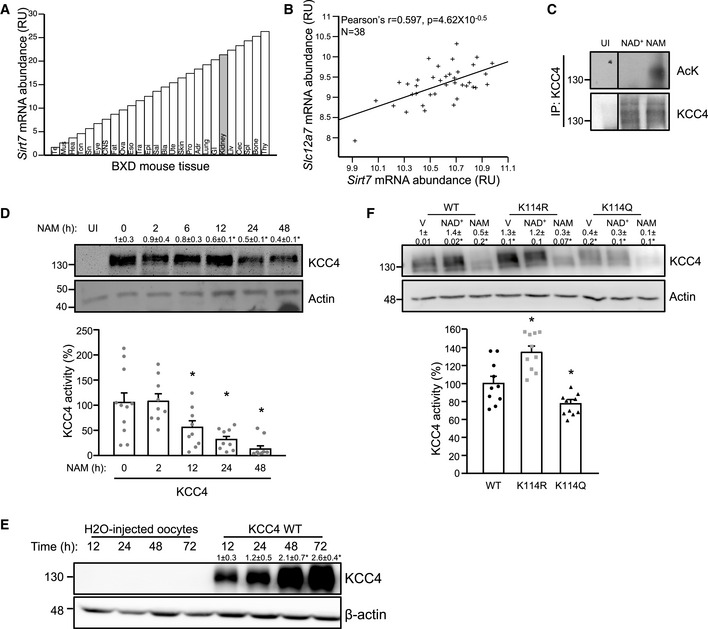
- In silico analysis of Sirt7 mRNA expression in different tissues of the BXD genetic reference population (www.genenetwork.org). Adr: Adrenal Gland mRNA, Bla: Bladder mRNA, Bone: Bone Tibia mRNA, CNS: CNS mRNA, Cec: Cecum mRNA, Epi: Epididymis mRNA, Eso: Esophagus mRNA, Eye: Eye mRNA, Fat: Peritoneal Fat mRNA, GI: GI Track mRNA, Hea: Heart mRNA, Kid: Kidney mRNA, Liv: Liver mRNA, Lung: Lung mRNA, Mus: Gastrocnemius Muscle mRNA, Ova: Ovary mRNA, Pro: Prostate mRNA, Sal: Salivary Gland mRNA, Skin: Skin Back mRNA, Sn: Sciatic Nerve mRNA, Spl: Spleen mRNA, Tes: Testis mRNA, Thy: Thymus mRNA, Ton: Tongue mRNA, Tra: Trachea mRNA, and Ute: Uterus mRNA.
- Correlation plot for kidney mRNA expression of Sirt7 and Slc12a7 that codifies KCC4 in the mouse BxD genetic reference population (www.genenetwork.org).
- Oocytes injected with cRNA encoding KCC4 were incubated with NAD+ 200 μM and NAM 10 mM for 4 h. Lysates were immunoprecipitated with an anti‐KCC4 antibody and then analyzed by SDS–PAGE/immunoblotting using an anti‐acetylated–lysine (AcK) or anti‐KCC4 antibody. Non‐injected oocytes (UI) were used as a control.
- Oocytes expressing KCC4 were incubated with NAM for the indicated times, and KCC4 protein levels (upper panel), and activity (lower panel) were analyzed by SDS–PAGE/immunoblotting or Cl−‐dependent 86Rb+ uptake under hypotonic conditions.
- Oocytes injected with H20 or KCC4 were incubated for the indicated times, and KCC4 protein levels were analyzed by SDS–PAGE/immunoblotting.
- Oocytes were injected with either WT, K114R, or K114Q KCC4 and incubated with vehicle (V), NAD+ or NAM for 48 h, and KCC4 protein levels (upper pannel), and activity (lower pannel) were analyzed as in (D).
Data information: Data are presented as mean ± SEM. RU: relative units. Depicted blots are representative of three independent biological replicates and values shown are the mean of the three experiments. To evaluate KCC4 activity, we performed three independent biological experiments with oocytes obtained from different frogs and each experiment included at least 4 replicates. * Indicates a significant difference versus time 0, at P < 0.05 by Student’s t‐test.
Source data are available online for this figure.
Several studies using in vitro and in vivo models have demonstrated that the activity of electroneutral cation‐chloride cotransporters from the SLC12 family is regulated by post‐translational modifications such as phosphorylation and ubiquitylation (Gamba, 2012). On the one hand, phosphorylation mediated by with‐no‐lysine (WNK) serine/threonine kinases family in conjunction with the downstream Ste20‐related proline/alanine‐rich kinase (SPAK) or oxidative stress response 1 (OSR1) kinase plays a key role in the regulation of these cotransporters. Specifically, WNKs/SPAK phosphorylate and activate the Na+:Cl−‐coupled (NCC) members of the SLC12 family, NCC and NKCC1‐2 (Kahle et al, 2005; Rinehart et al, 2005; Chavez‐Canales et al, 2014), and inhibit the K+:Cl−‐coupled members, KCC1‐4 (de Los Heros et al, 2006; Garzon‐Muvdi et al, 2007; Rinehart et al, 2011). On the other hand, NCC and NKCC2 have been shown to be modulated by ubiquitylation through the homologous to E6‐AP C‐terminal (HECT)‐ligase neural precursor cell‐expressed developmentally downregulated gene 4 (NEDD4)‐2 (Arroyo et al, 2011; Ronzaud et al, 2013; Wu et al, 2013). To our knowledge, this is the first description of acetylation as a post‐translational modification that modulates the activity of an SLC12 cotransporter member, and our data suggest that SIRT7 may modulate this process.
SIRT7 interacts with and increases KCC4 activity and protein levels in X. laevis oocytes and HEK293 cells
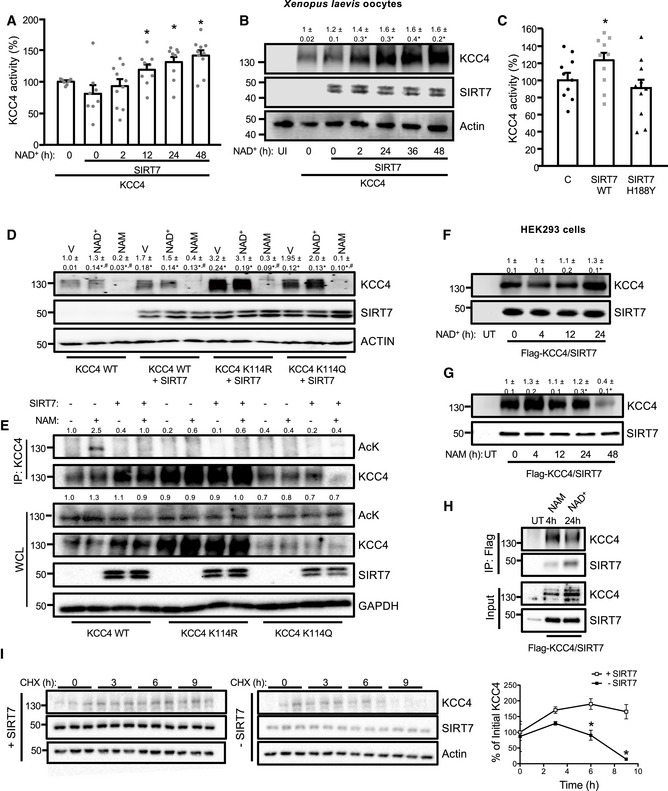
To evaluate whether SIRT7 modulates KCC4 activity, Xenopus oocytes were coinjected with KCC4 and SIRT7 cRNA and incubated with NAD+. KCC4 activity increased in a time‐dependent manner when the oocytes were incubated with NAD+ in the presence of SIRT7, reaching a 30% increase in activity at 48 h (Fig 2A). The change in KCC4 activity was associated with an increase in KCC4 protein levels (Fig 2B), which suggests that SIRT7 may increase KCC4 stability or synthesis. Notably, a SIRT7 mutant, in which the catalytic histidine H188 was replaced by tyrosine (SIRT7 H188Y), had no effect on KCC4 activity (Fig 2C). Neither 24‐h incubation with NAD+ modified the expression of KCC4 K114R or K114Q mutants (Fig 2D). We then tested the capability of SIRT7 to deacetylate the K114 lysine residue of KCC4 by injecting oocytes with the cRNA of KCC4 WT, KCC4 K114R, or KCC4 K114Q mutants, with or without SIRT7, and incubating with NAM for 4 h after 48 h of KCC4 cRNA injection. Interestingly, SIRT7 reduced global acetylation of oocytes incubated with NAM (Fig EV1), and most importantly, when we evaluated the acetylation status of KCC4 by immunoprecipitation, KCC4 WT was remarkably acetylated after incubation with NAM and remained deacetylated in the presence of SIRT7 (Fig 2E). Furthermore, the KCC4 K114R and KCC4 K114Q mutants were barely acetylated, and neither their acetylation status nor their expression was modified by the presence of SIRT7. These results confirm that K114 is the key acetylation site of KCC4 and that SIRT7 is able to deacetylate this site. To evaluate whether the effect of SIRT7 on KCC4 is conserved in mammalian cells, we co‐transfected SIRT7 and FLAG‐KCC4 in HEK293 cells and evaluated KCC4 protein levels. KCC4 protein levels increased by 50% after 24 h of incubation with NAD+ (Fig 2F), and decreased 80% after incubation with NAM (Fig 2G). Immunoprecipitation analysis using a FLAG antibody to immunoprecipitate KCC4 revealed that SIRT7 and KCC4 interact with each other after 24 h of incubation with NAD+, and the interaction was reduced after 4 h of incubation with NAM (Fig 2H). We assayed KCC4 stability to understand how SIRT7 increases KCC4 protein levels. For this purpose, we transfected KCC4 plasmids in the presence or absence of SIRT7 in HEK293 cells and blocked protein synthesis with cycloheximide 24 h after transfection. In the presence of SIRT7, KCC4 levels remained constant when cells were incubated with cycloheximide. However, when we used a shRNA against SIRT7, KCC4 stability was significantly reduced in the presence of cycloheximide when compared with cells transfected with a scramble shRNA (Fig 2I). Altogether, these observations suggest that SIRT7 interacts with and stabilizes KCC4 in a NAD+‐dependent fashion, increasing KCC4 protein levels and thus activity. Notably, this is the first report implicating a sirtuin in the regulation of a renal cotransporter activity, which may have implications in the modulation of acid–base homeostasis. Previously, it was demonstrated that SIRT1 might modulate Na+ balance by transcriptional repression of the epithelial Na+ channel α‐subunit (α‐ENaC) in the apical membrane of collecting duct principal cells (Zhang et al, 2009). However, the inhibition of α‐ENaC by SIRT1 is independent of its deacetylase activity (Zhang et al, 2009), which implies a different mechanism that the one exerted by SIRT7. Our results suggest that the mechanism by which SIRT7 increases KCC4 protein levels is through enhanced KCC4 stability. Proteins are degraded in the cell by two processes: a general process that involves lysosomal proteolysis (Sakamoto, 2002) and a selective process that eliminates specific proteins in an ubiquitin‐dependent manner by the proteasome (De Strooper et al, 2010). Although further research is required to evaluate whether the SIRT7‐mediated deacetylation of KCC4 directly modifies its ubiquitination, a recent report shows that SIRT7 modulates the ubiquitin‐proteasome pathway by binding the ubiquitin ligase complex, thereby inhibiting the ubiquitination and degradation of other proteins, such as the testicular orphan nuclear receptor, TR4 (Yoshizawa et al, 2014). Consistent with our observations of the role of protein acetylation and protein stability in KCC4 regulation, a previous report also linked the deacetylation of the SAM pointed domain‐containing ETS transcription factor (SPDEF) by SIRT1, with an increase in SPDEF stability (Lo Sasso et al, 2014).
Figure 2. SIRT7 interacts with, stabilizes, and increases KCC4 activity and protein levels.
-
A, BKCC4 activity (A), and protein levels (B) were evaluated in Xenopus oocytes expressing SIRT7 and KCC4 that were incubated for the indicated times with NAD+. Non‐injected oocytes (UI) were used as a control.
-
CKCC4 activity was evaluated in Xenopus oocytes injected with cRNA encoding KCC4 and coinjected with either wild‐type (SIRT7 WT) or the catalytically inactive SIRT7 (SIRT7 H188Y) for 48 h.
-
DOocytes were injected with either KCC4 WT, K114R, or K114Q for 48 h and then incubated with vehicle (V), NAD+ or NAM for 24 h, and KCC4 and SIRT7 protein levels were analyzed by SDS–PAGE/immunoblotting.
-
EOocytes were injected with either KCC4 WT, K114R, or K114Q with and without SIRT7 for 48 h and then incubated with vehicle (V) or NAM for 4 h. After lysis, KCC4 was immunoprecipitated using an anti‐KCC4 antibody, and the acetylation status of KCC4 was determined by SDS–PAGE/Western blotting using and anti‐acetyl lysine antibody.
-
F, GFLAG‐KCC4 and SIRT7 were transfected into HEK293 cells and treated for the indicated times with NAD+ (F) or NAM (G) and then analyzed by SDS–PAGE/immunoblotting using an anti‐KCC4 or anti‐SIRT7 antibody. Non‐transfected cells (UT) were used as a control.
-
HFLAG‐KCC4 and SIRT7 were transfected into HEK293 cells and treated for the indicated times with NAD+ and NAM. After lysis, KCC4 immunoprecipitation using an anti‐FLAG antibody was performed, and the interaction with SIRT7 was determined by SDS–PAGE/Western blotting.
-
IHEK293 cells expressing KCC4 were incubated with NAD+ 200 μM and cycloheximide (CHX) for 0, 3, 6, and 9 h. KCC4 protein levels are shown at the indicated times in cells transfected with a scramble shRNA (+SIRT7) or a shRNA against SIRT7 (−SIRT7).
Data information: Data are presented as mean ± SEM. Depicted blots are representative of three independent biological replicates and values shown are the mean of the three experiments. To evaluate KCC4 activity, we performed three independent biological experiments with oocytes obtained from different frogs and each experiment included at least 4 replicates. *Indicates a significant difference versus time 0 or versus KCC4 WT or versus + SIRT7 at P < 0.05 by Student’s t‐test. # P < 0.05 when compared to Vehicle by Student’s t‐test.
Source data are available online for this figure.
Figure EV1. SIRT7 reduced global acetylation of oocytes incubated with NAM.
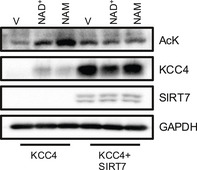
Oocytes were injected with KCC4 WT with and without SIRT7 for 48 h and then incubated with vehicle (V) or NAM for 4 h. KCC4 protein levels and acetylated proteins were analyzed by SDS–PAGE/immunoblotting using an anti‐acetyl lysine antibody.
SIRT7 regulates KCC4 activity during a decrease in external pH in X. laevis oocytes and HEK293 cells
We have previously shown that KCC4 activity is increased when the extracellular pH decreases or in response to acidosis (Bergeron et al, 2003; Melo et al, 2013a). To evaluate whether the increase of KCC4 activity by pH modification is related to changes in its acetylation status, X. laevis oocytes or HEK293 expressing KCC4 were incubated in media with different pH values, and KCC4 acetylation was evaluated. Interestingly, KCC4 acetylation levels were reduced when the extracellular pH decreased in both X. laevis oocytes (Appendix Fig S1) and HEK293 cells (Fig 3A). To confirm that SIRT7 regulates KCC4 activity during a decrease in external pH, we transfected HEK293 cells with shRNA against SIRT7 to abolish SIRT7 expression and evaluated KCC4 activity. Accordingly, increased KCC4 activity was observed when the extracellular pH was decreased and when SIRT7 was overexpressed. However, the increased KCC4 activity was not observed when shRNA against SIRT7 was co‐transfected (Fig 3B). Because SIRT7 has been described as a nucleolar protein (Michishita et al, 2005; Ford et al, 2006), the mechanism for its effect on a plasma membrane cotransporter, such as KCC4, is surprising. However, SIRT7 localization was recently demonstrated to be not only restricted to the nucleoli, but it was also shown to be a highly dynamic nuclear protein (Ryu et al, 2014); in addition, evidence suggests that there may be a cytoplasmic isoform of SIRT7 (Kiran et al, 2013). Interestingly, SIRT7 translocated from the nucleus to the cytoplasmic fraction when the extracellular pH was decreased (Fig 3C). These results suggest that SIRT7 modulates KCC4 protein levels and activity during variations in extracellular pH, and this may occur as SIRT7 translocates to the cytosol and gains access to KCC4.
Figure 3. SIRT7 regulates KCC4 activity during a decrease in external pH.
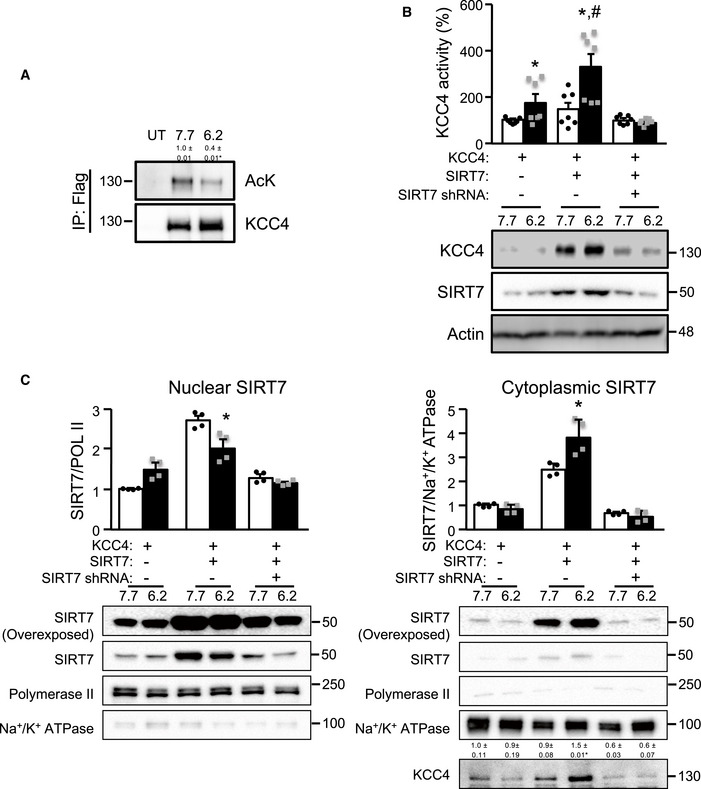
- KCC4 acetylation at the indicated pH in HEK293 transfected with KCC4. After lysis, KCC4 was immunoprecipitated using an anti‐FLAG antibody and then analyzed by SDS–PAGE/Western blotting using an anti‐acetylated–lysine (AcK) or anti‐KCC4 antibody. Non‐transfected cells (UT) were used as a control.
- HEK293 cells were transfected with KCC4 together with SIRT7 with a scramble shRNA (−) or a shRNA against SIRT7 (+), and KCC4 activity and protein level were determined at the indicated pH. SIRT7 expression in these conditions is presented in (C).
- Nuclear and cytoplasmic SIRT7 levels at different pH values in HEK293 cells. At a lower pH, SIRT7 appears to translocate to the cytoplasm.
Data information: Data are presented as mean ± SEM. Depicted blots are representative of three independent biological experiments and values shown are the mean of the three experiments. To evaluate KCC4 activity, we performed three independent biological experiments and each experiment included at least 3 replicates. * Indicates a significant difference versus pH = 7.7 at P < 0.05 by Student’s t‐test. # Indicates a significant difference versus KCC4 at pH = 6.2 at P < 0.05 by Student’s t‐test.
Source data are available online for this figure.
SIRT7 distribution along the nephron and SIRT7 expression increases during metabolic acidosis in the kidney of C57BL6 male mice
In the kidney, no information is available about the cellular distribution of SIRT7 along the nephron. Cortex and medulla presented similar levels of SIRT7 expression (Appendix Fig S2). In microdissected tubules, Sirt7 mRNA was detected in the proximal convoluted tubule (PCT), the proximal straight tubule (PST), the distal convoluted tubule (DCT), the connecting tube (CNT), the cortical collecting duct (CCD), and the outer medullary collecting duct (OMCD) and to a lesser extent in both the medullary and the cortical thick ascending limb (mTAL and cTAL) (Fig 4A). Our expression data are supported by recent single cell transcriptomic analysis of the mouse kidney showing Sirt7 expression in the different identified cell types (Park et al, 2018), and in a comparable level than Slc12a7 expression (Appendix Fig S3). Western blot analysis of microdissected tubules, which were previously validated (Spirli et al, 2019), showed that SIRT7 protein was present at highest level in the proximal tubules (PT), followed by DCT‐CNT, TAL, and CCD (Fig 4B). We then performed immunofluorescence to further characterize the cellular localization of SIRT7 in the nephron. With this technique, SIRT7 was mainly detected in the glomerulus (Fig 4C). The observed signal was specific since it was not observed in SIRT7‐deficient mice (Appendix Fig S4). Failure to observe expression in other nephron segments (as shown by Western blot) may be due to a lower sensitivity of the immunofluorescence technique. Notably, during alterations of acid–base homeostasis, such as in metabolic acidosis, we observed that both KCC4 and SIRT7 expression increased in the kidneys of mice with metabolic acidosis (Fig 4D), without changes in Sirt7 or Slc12a7 mRNA (Appendix Fig S5).
Figure 4. Characterization of renal SIRT7 expression in C57BL6 mice.
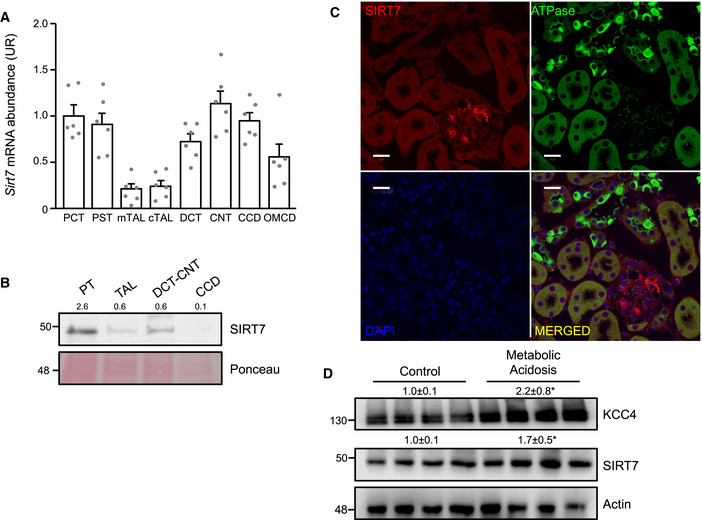
-
A, BSirt7 mRNA (A) and protein (B) abundance in microdissected renal tubules normalized to Rpl26 or Ponceau, respectively. Enrichment for each segment was previously validated by immunoblotting against segment‐specific markers as indicated in material and methods (Spirli et al, 2019).
-
CImmunolocalization of SIRT7 and ATPase in the kidneys of C57BL6 mice with metabolic acidosis (SIRT7, red; ATPase, green; DAPI, Blue). Scale bar: 20 μm. Controls of the specificity of antibodies using kidney of SIRT7‐deficient mice are provided in Appendix Fig S4.
-
DKCC4 and SIRT7 protein levels in kidneys from control mice or mice with metabolic acidosis. Values are the mean ± SEM.
Data information: Data are presented as mean ± SEM. For each segment, tubules isolated from different animals were pooled (PT n = 3, TAL n = 5, DCT/CNT n = 5, and CCD n = 5). The Western blot was performed with the pooled samples and repeated to assure reproducibility. Each lane of the Western blot depicted in D is a biological replicate as each lane represent one mouse. The blot was repeated with three other mice, and the quantification showed is the average of the seven mice. * Indicates a significant difference versus control at P < 0.05 by Student’s t‐test.
Source data are available online for this figure.
KCC4 protein levels are reduced in the kidneys of total and inducible renal tubule‐specific SIRT7‐deficient mice, and this reduction is associated with an exacerbated metabolic acidosis in the total SIRT7‐deficient mice
We then evaluated KCC4 expression in the kidneys of SIRT7‐deficient mice to determine whether SIRT7 regulation of KCC4 protein levels was maintained in vivo. Consistent with our in vitro results obtained in SIRT7 loss‐of‐function cell models, KCC4 protein levels were also significantly reduced in the kidneys of SIRT7‐deficient mice (Fig 5A). This was not associated with a reduction in Slc12a7 mRNA abundance (Appendix Fig S6), supporting the idea that SIRT7 regulates KCC4 through a post‐transcriptional mechanism. Moreover, the increase in KCC4 expression detected in kidneys from wild‐type mice during ammonium chloride challenge was not observed in SIRT7‐deficient mice (Fig 5B). Notably, the decrease in KCC4 expression was accompanied by an increase in KCC4 acetylation in the SIRT7‐deficient mice (Fig 5B). We then performed an immunofluorescent staining of kidney sections from SIRT7+/+ and SIRT7−/− mice during control or acidosis conditions to evaluate whether KCC4 localization was affected by the absence of SIRT7. Interestingly, KCC4 was widely localized in the basolateral membrane of tubules in the kidney cortex and also in the basolateral membrane of intercalated cells (identified by H‐ATPase staining). KCC4 subcellular localization was not affected by SIRT7 deficiency. Nevertheless, we observed that the percentage of KCC4+ cells among the H‐ATPase+ cells increased in wild‐type mice treated with ammonium chloride (Fig 5C). However, this increase was not observed in SIRT7‐deficient mice, supporting the idea that SIRT7 may be important to modulate KCC4 acetylation level and stability in response to an acid load. We next assessed the acid–base status of the SIRT7‐deficient mice by measuring urinary pH to explore the physiological relevance of SIRT7 modulation of KCC4. Unexpectedly, the urinary pH and electrolytes were not modified in SIRT7‐deficient mice at normal conditions, nor even during ammonium chloride challenge, except for Na+ that was lower in SIRT7‐deficient mice (Table 1). Furthermore, we performed a blood chemistry analysis to evaluate systemic acid–base parameters beyond urinary pH in SIRT7‐deficient mice that were exposed to ammonium chloride challenge for 8 days. Interestingly, SIRT7‐deficient mice had lower pH, , and tCO2 than the wild‐type controls, indicating that an acid challenge is enough to unmask a metabolic acidosis‐prone phenotype in SIRT7‐deficient mice (Table 1). To further study the lack of renal tubular acidosis during basal conditions, we evaluated the expression of other members of the SLC12 family that are known to be involved in acid–base metabolism. Interestingly, the distal tubule NaCl and the ascending loop of Henle Na‐K‐2Cl cotransporters, NCC and NKCC2, respectively, were also reduced in the kidney of SIRT7‐deficient mice (Fig 5D). Moreover, we generated inducible renal tubule‐specific SIRT7‐deficient mice using a combination of the inducible Tet‐On and Cre‐loxP systems that would lack adaptation during basal conditions because the loss‐of‐function mutation would be induced for only a few weeks, as previously described (Arroyo et al, 2011; Ronzaud et al, 2013). Briefly, double‐transgenic Sirt7 flox/flox/Pax8‐rTA/LC1 (Sirt7Pax8/LC1) and control mice were treated with doxycycline to induce renal tubule‐specific Cre‐mediated recombination that led to SIRT7 deletion. Both the Sirt7Pax8/LC1 and control mice were treated with doxycycline to exclude any confounding effects produced by doxycycline on mitochondrial function (Moullan et al, 2015). Real‐time PCR and Western blot analysis 2 days after induction revealed a decrease in SIRT7 transcript and protein levels in both kidney and liver (Appendix Fig S7A). However, SIRT7 expression was regained in the liver 4 weeks after doxycycline induction (Appendix Fig S7B), while SIRT7 expression remained suppressed in the kidney (Fig EV2A). Notably, inducible renal tubule‐specific SIRT7‐deficient mice had a reduction in KCC4 protein levels (Fig EV2A), and a mild increase in urinary pH (Fig EV2B). Thus, these results indicate that SIRT7 modulates renal KCC4 expression in vivo.
Figure 5. KCC4 protein levels in the kidneys and metabolic acidosis status of SIRT7‐deficient mice and renal tubule‐specific SIRT7‐deficient mice.
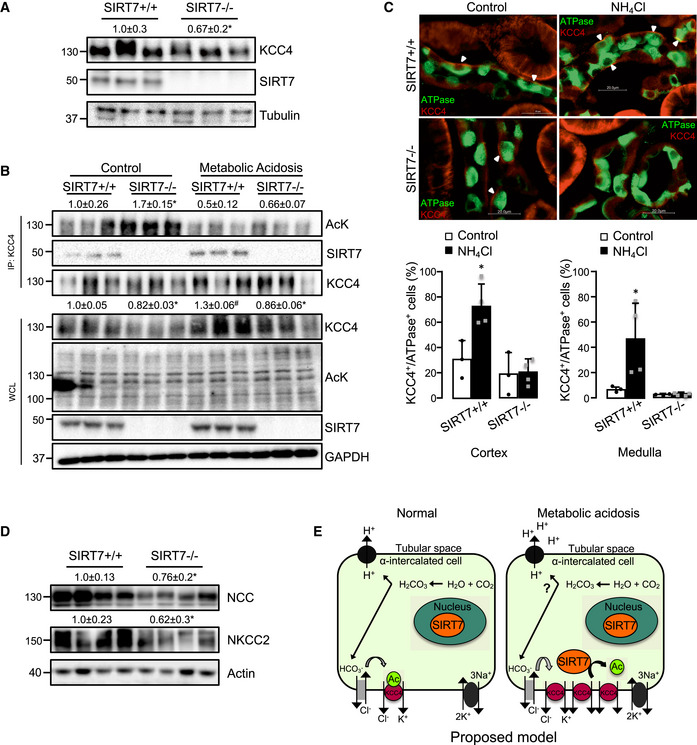
- KCC4 protein levels were determined in the kidneys of wild‐type (SIRT7+/+) and SIRT7‐deficient mice (SIRT7−/−). SIRT7 protein expression are absent in the kidneys of SIRT7‐deficient mice.
- KCC4 protein levels, acetylation status of KCC4, and interaction with SIRT7 in kidneys of SIRT7+/+ and SIRT7−/− mice under normal conditions (Control) or with metabolic acidosis.
- Immunolocalization of KCC4 and H‐ATPase in the kidneys of SIRT7+/+ and SIRT7−/− mice under normal conditions (Control) or with metabolic acidosis (NH4Cl). (KCC4, red; H‐ATPase, green). Scale bar: 20 μm. Cell count was performed automatically using the Gen5 software to report the percentage of KCC4 + cells among H‐ATPase + cells (lower panel).
- NCC and NKCC2 protein levels in the kidney of SIRT7+/+ and SIRT7−/− mice under basal conditions.
- Scheme illustrating the proposed mechanism by which SIRT7 regulates KCC4 in the kidney. Briefly, KCC4 maintains a low intracellular Cl− concentration to ensure that basolateral Cl−/ exchange remains active to allow extrusion of from the cell, specifically in α‐intercalated cells of the collecting duct, and therefore warranting a constant production of H+ that are secreted through the apical membrane to be eliminated in the urine. During stress, such as metabolic acidosis, SIRT7 levels increase in the cytoplasm, promoting the interaction and stability of KCC4. This interaction induces an increase in KCC4 protein levels and activity and therefore contributes to the increase of basolateral Cl− efflux, which decreases the intracellular Cl− concentration, thus maintaining the gradient for the Cl−/ exchanger to eliminate bicarbonate from the cells and increase the secretion of H+ to reestablish acid–base homeostasis.
Data information: Data are presented as mean ± SEM. Depicted blots are representative of two different Western blot analysis performed with six mice in total. The values presented are the mean of the seven mice. Depending on availability actin, tubulin or GAPDH were used as control. * Indicates a significant difference versus SIRT7+/+ at P < 0.05 by Student’s t‐test.
Source data are available online for this figure.
Table 1.
Blood gasometry and urine pH and electrolytes of mice exposed to 280 mM NH4Cl solution for 8 days.
| Control | NH4Cl | |||
|---|---|---|---|---|
| SIRT7+/+ | SIRT7−/− | SIRT7+/+ | SIRT7−/− | |
| Blood | ||||
| pH | 7.37 ± 0.03a | 7.37 ± 0.05a | 7.29 ± 0.04* ,a,b | 7.23 ± 0.05* ,#,b |
| (mM) | 17.5 ± 3.16a,b | 18.1 ± 1.78a | 15.3 ± 2.83a,b | 12.7 ± 1.21 # ,b |
| PCO2 | 30.5 ± 5.29 | 31.7 ± 4.24 | 31.2 ± 3.10 | 31.9 ± 3.13 |
| tCO2 (mM) | 18.2 ± 3.35a,b | 19.0 ± 1.79a | 16.2 ± 2.86a,b | 13.8 ± 1.47 # ,b |
| K+ (mM) | 3.78 ± 0.44 | 4.03 ± 0.16 | 3.74 ± 0.27 | 3.88 ± 0.35 |
| Na+ (mM) | 148 ± 1.79a,b | 147 ± 1.86b | 151 ± 2.05* ,a | 151 ± 1.17* ,a |
| Urine | ||||
| pH | 6.06 ± 0.21b | 6.05 ± 0.19b | 5.33 ± 0.29* ,a | 5.42 ± 0.16* ,a |
| K+/creatinine | 19.1 ± 6.28a,b | 21.4 ± 3.68a | 12.3 ± 3.14* ,b | 11.9 ± 3.36* ,b |
| Na+/creatinine | 17.7 ± 11.4a | 16.9 ± 6.34a | 9.90 ± 2.62a,b | 5.79 ± 3.60* ,#,b |
| Cl−/creatinine | 17.3 ± 12.2b | 17.4 ± 5.34b | 43.2 ± 5.85* ,a | 38.6 ± 2.76* ,a |
Values are the mean ± SEM (n = 5).
Letters, where a > b > c, indicate a statistically significant difference at P < 0.05 when analyzed with one‐way ANOVA followed by Tukey’s post hoc test.
P < 0.05 when compared to control by Student’s t‐test.
P < 0.05 when compared to SIRT7+/+ by Student’s t‐test.
Figure EV2. Renal KCC4 expression was reduced and urinary pH increased in renal tubule‐specific SIRT7‐deficient mice.
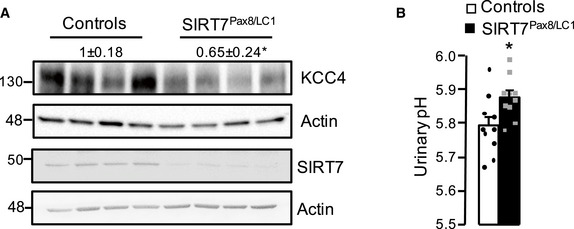
- SIRT7 and KCC4 protein levels in the kidney of control and renal tubule‐specific SIRT7‐deficient mice (SIRT7Pax8/LC1) 4 weeks after doxycycline treatment. Depicted blots are representative of two different Western blot analysis performed. *Indicates significant difference versus control mice at P < 0.05 by Student’s t‐test.
- Urinary pH in control and SIRT7Pax8/LC1 mice. Data are presented as mean ± SEM (n = 10). *Indicates significant difference versus control mice at P < 0.05 by Student’s t‐test.
Interestingly, SIRT7‐deficient mice show an age‐related hearing loss (Ryu et al, 2014), which is consistent with the phenotype observed in KCC4‐deficient mice, which develop sensorineural deafness (Boettger et al, 2002). Thus, the loss of KCC4 activity could, at least in part, contribute to the deafness in SIRT7‐deficient mice by altering K+ recycling in hair and Deiters’ cells. Of note, the KCC4‐deficient mice deafness is present since birth, suggesting that a complete lack of KCC4 has more severe consequences that a reduction in KCC4 expression. In this regard, SIRT7‐deficient mice did not exhibit renal tubular acidosis as was observed in the KCC4‐deficient mice. The absence of renal tubular acidosis in SIRT7‐deficient mice could be explained by at least two mechanisms. The SIRT7‐deficient mice exhibited reduced expression, but not a complete absence of KCC4. Hence, it is possible that, as might occurred with deafness, although lower, the expression level of KCC4 in SIRT7‐deficient mice is sufficient to prevent renal tubular acidosis. Second, the elimination of SIRT7 during development could lead to compensatory adaptations by activating other pathways to avoid renal tubular acidosis. This hypothesis is supported by the fact that inducible renal tubule‐specific SIRT7‐deficient (Sirt7Pax8/LC1) mice displayed mild renal tubular acidosis, as they have insufficient time to compensate for the loss of SIRT7. We have previously observed a similar compensatory mechanism in the regulation of NCC by NEDD4‐2 since the total NEDD4‐2‐deficient mice show no increase in NCC expression, whereas the inducible nephron‐specific NEDD4‐2‐deficient mice have an increase in NCC protein levels leading to the development of hypertension (Arroyo et al, 2011; Ronzaud et al, 2013). In this regard, we observed a reduced expression of NCC and NKCC2 in the kidney of global SIRT7‐deficient mice. Reduction in NCC and NKCC2 is known to produce metabolic alkalosis (Simon et al, 1996; Castañeda‐Bueno et al, 2014), which could counteract the renal tubular acidosis produced by the reduction of KCC4 in basal conditions. Although at this point, we cannot determine whether the reduction in NCC and NKCC2 is a result of a direct regulation of SIRT7, it helps to explain the lack of renal tubular acidosis in SIRT7‐deficient mice during basal conditions. Another limitation of our study is that at this point we cannot exclude that the metabolic acidosis observed in the SIRT7‐deficient mice is of non‐renal origin caused by the dysfunctional mitochondria in liver and heart (Ryu et al, 2014).
Our proposed model of KCC4 function (Melo et al, 2013a) underscores possible implications of SIRT7 in acid–base regulation (Fig 5E). In the basolateral membrane of α‐intercalated cells of the collecting duct, KCC4 maintains a low intracellular Cl− concentration to assure that the basolateral Cl−/ exchanger remains active. This low intracellular Cl− concentration allows the extrusion of from the cell, warranting a constant production of H+ ions that are secreted through the apical membrane to be eliminated in the urine, which typically has lower pH than plasma, thus maintaining the acid–base homeostasis in the organism. Supporting this hypothesis, loss of KCC4 function in mice is associated with the development of renal tubular acidosis (Boettger et al, 2002), and metabolic acidosis is associated with an increase in KCC4 expression in the basolateral membrane of the α‐intercalated cells of the collecting duct (Melo et al, 2013a). Our in vitro data suggest that this process requires SIRT7‐mediated deacetylation of KCC4. For instance, during stress, such as a decrease in external pH, SIRT7 levels increase in the cytoplasm, promoting its interaction with KCC4. The SIRT7/KCC4 interaction induces an increase in KCC4 protein stability that leads to enhanced KCC4 protein levels and activity and therefore could contributes to an increase in the basolateral Cl− efflux, decreasing the intracellular Cl− concentration, thus maintaining the gradient for the Cl−/ exchanger to eliminate bicarbonate from the cell. However, our in vivo data do not show a marked defect in acid–balance in the SIRT7‐deficient mice, which could be due to fact that KCC4 activity is not completely impaired or to the altered activity of other renal transporters that may produce a phenotype with a more complex etiology, suggesting that SIRT7 may have a more complex role in renal physiology.
In conclusion, we demonstrate that SIRT7 interacts with, deacetylates, and stabilizes the K‐Cl cotransporter KCC4 and promotes its activity. Through these actions, SIRT7 impacts the regulation of a renal transporter, and hence, we reveal a renal physiological process that is controlled by SIRT7. In addition, our results demonstrate that acetylation is a post‐translational modification that modulates the function of KCC4 and provide a model that may be a paradigm for the regulation of other transport proteins (e.g., of the many SLC type transporters). Thus, targeting protein acetylation could provide a potential new therapeutic approach to manage problems of acid–base homeostasis.
Materials and Methods
Functional expression of KCC4 and SIRT7 in Xenopus laevis oocytes
We assessed the effect of SIRT7 upon the activity of wild‐type KCC4 using the heterologous expression system of Xenopus laevis oocytes as previously described (Mount et al, 1999; Mercado et al, 2006; Cruz‐Rangel et al, 2011; Melo et al, 2013a; Melo et al, 2013b). In brief, stage V to VI oocytes were injected with wild‐type KCC4 cRNA at 5 ng/oocyte alone or coinjected with wild‐type SIRT7 cRNA at 10 ng/oocyte. 48 h later, the activity of the cotransporter was determined by assessing the Cl−‐dependent 86Rb+ uptake in hypotonic conditions. Oocytes were pre‐incubated in a Na+‐free, Cl−‐free medium (in mM: 50 N‐methyl‐D‐glucamine‐gluconate, 10 K+‐gluconate, 4.6 Ca2+‐gluconate, 1 Mg+‐gluconate, and 5 HEPES/Tris, pH 7.4) with 1 mM ouabain for 30 min at 32°C and then transferred to a KCl‐containing Na+‐free medium (in mM: 40 N‐methyl‐D‐glucamine‐Cl−, 10 KCl, 1.8 CaCl2, 1 MgCl2, and 5 HEPES/Tris, pH 7.4) with 1 mM ouabain and 1 μCi/ml of 86Rb+ and uptake continued for 60 min at 32°C. Chloride was substituted by gluconate for uptakes assays in the absence of this anion. At the end of the uptake period, cells were washed five times in the respective ice‐cold Na+‐free medium without isotope to remove extracellular fluid tracer. 86Rb+ uptake tracer activity was determined using a Beckman β‐scintillation counter. Ouabain prevents the activation of the Na+:K+‐ATPase, while the absence of extracellular Na+ and the hypotonicity prevents the 86Rb+ uptake via the endogenous Na+:K+:2Cl− cotransporter NKCC1 that is present in oocytes. For those assays in the presence of NAM or NAD+, the injected oocytes were incubated either with 10 mM NAM or 200 μM NAD+ right after the injection and the activity of the cotransporter was assessed 2, 4, 12, 24, or 48 h later. In another set of experiments, oocytes were injected with either the wild‐type KCC4 or with a K114R or K114Q mutant, and KCC4 activity was assessed as described above. The response of KCC4 to the medium acidification was determined under hypotonic conditions using similar preuptake and uptake solutions that were pH titrated from 6.0 to 8.0. cRNAs for injection were transcribed in vitro from WT, K114R or K114Q KCC4, and SIRT7 cDNAs linearized at the 3′ using the T7 RNA polymerase mMESSAGE kit (Ambion). At the end of the uptake assay, oocytes were lysed in 300 µl of ice‐cold lysis buffer [in mM: 50 Tris–HCl (pH 7.5), 1 EGTA, 1 EDTA, 50 sodium fluoride, 5 sodium pyrophosphate, 1 sodium orthovanadate, 1% (w/v) Nonidet P40, 0.27 sucrose, 0.1% 2‐mercaptoethanol, 5 NAM, 1 sodium butyrate, and protease inhibitors (1 tablet per 50 ml, Sigma). All experimental data are based on a minimum of 3 different experiments. The SIRT7 commercial vector (MC206249, Origene) was subcloned to the pGEMHE vector to obtain cRNA.
Construction of mutant cDNAs
We used the QuikChange mutagenesis system (Agilent Technologies) to construct the mutant cDNAs of KCC4 and SIRT7 used in this work. All of the cDNA constructs were confirmed by sequencing and subcloned in the Xenopus laevis expression vector pGEMHE (Mercado et al, 2006; Melo et al, 2013b). The full‐length mouse Sirt7 sequence in mammalian vector pcDNA3 was subcloned into pGEMHE using enzymes KpnI‐NotI. Then, its catalytic histidine H188 was replaced by tyrosine (SIRT7 H188Y) using the sense primer 5′‐ACCGCCATCTCAGAGCTCTATGGGAATATGTATATTGA‐3′ and the antisense primer 5′‐TCAATATACATATTCCCATAGAGCTCTGAGATGGCGGT‐3′. The lysine 114 from KCC4 was replaced by arginine (K114R) to mimic a hypoacetylated KCC4 using the sense primer 5′‐GGCGAGAGGTCAGAGCCCCACGC‐3′ and the antisense primer 5′‐GCGTGGGGCTCTGACCTCTCGCC‐3′. In addition, the lysine 114 from KCC4 was replaced by glutamine (K114Q) to mimic a hyperacetylated KCC4 using the sense primer 5′‐GGCGAGAGGTCCAGGCCCCACGC‐3′ and the antisense primer 5′‐GCGTGGGGCCTGGACCTCTCGCC‐3′.
HEK293 cell culture and treatments
The full‐length mouse KCC4 tagged with FLAG (Melo et al, 2013b) and SIRT7 cDNAs were subcloned into the mammalian expression vector pCMV5. For detection of SIRT7, a commercial primary antibody was used (sc‐365344, SCBT). To generate the transient cell lines, HEK‐293 cells underwent passages every 2–3 days to keep the cells in their exponential growth phase. Two hours before transfection, the cell suspension was centrifuged (300 gfor 5 min), resuspended in fresh medium at 5 × 106 cell/ml, and seeded in T25 flasks (25 cm2 and 80% confluence). Then cells were transfected at 37°C with 800 ng of each full‐length KCC4 and SIRT7 cDNA or empty vector using lipofectamine‐2000 (Invitrogen) following the manufacture’s recommendations. After 30 min at room temperature, the mixture was incubated with the cells for 6 h at 37°C, washed twice with 1 × phosphate buffered saline (PBS), and placed in Dulbecco’s modified Eagle medium (DMEM) supplemented with 50 U/ml penicillin, 50 µg/ml streptomycin, 10% FBS (Gibco), and 1 mg/ml G418 (Calbiochem, Merck Millipore) in a humidified atmosphere containing 95% air and 5% CO2 at 37 °C (Cruz‐Rangel et al, 2012; Melo et al, 2013b). Two days after transfection, cells were exposed to either isotonic or hypotonic Na+‐free medium (in mM: 100 N‐methyl‐D‐glucamine‐Cl−, 10 KCl, 1.8 CaCl2, 1 MgCl2, 5 sucrose, and 5 HEPES/Tris, pH 7.4) for 30 min with or without NAD+ 200 μM or NAM 10 mM for 0, 4, 12, 24, or 48h or with medium at pH of 6.2, 6.8, 7.2, or 7.7 for 16 h. The extracellular Na+ removal prevents the activation of the endogenous NKCC1 cotransporter. Following the treatment, to collect cell homogenates, cells were harvested in ice‐cold lysis buffer (in mM: 20 Tris–HCl pH 7.4, 1 EDTA, 50 NaCl, 1 EGTA, 0.5 Na3VO4, 1–2 β‐glycerophosphate, and 1% Triton X‐100), incubated for 10 min and then scraped. Cells homogenates were centrifuged (20 min at 11,000 g), and finally the supernatants were collected. Nuclear and cytosolic fractions were prepared using the NER‐PER Nuclear and Cytoplasmic Extraction reagents (78833, Thermo Scientific) following the manufacturer’s recommendations. For stability assay, HEK293 cells were transfected with 50 ng of KCC4, 100 ng of SIRT7, and 200 ng of a scramble shRNA or shRNA against SIRT7 (sc‐63031‐sh, Santa Cruz Biotechnology). After 24 h, cells were incubated with NAD+ 200 μM and 75 μg/ml of cycloheximide (C7698, Sigma‐Aldrich), and cell homogenates were obtained at 0, 3, 6 y 9 h as described above.
86Rb+ Uptake assay in HEK‐293 cells transfected with wild‐type Flag‐mKCC4 and mSIRT7
K+:Cl− cotransport was assessed on HEK‐293 cells 80% confluent by measuring Cl− ‐dependent 86Rb+ uptake (PerkinElmer) under isotonic or hypotonic conditions (300 or 250 mOsm, respectively). In brief, cells were transfected with 800 ng of each wild‐type FLAG‐mKCC4 and mSIRT7, and the uptake assay was performed 48 h post‐transfection. Cells were pre‐incubated in a Na+‐free, Cl−‐free medium (in mM: 100 N‐methyl‐D‐glucamine‐gluconate, 10 K+‐gluconate, 4.6 Ca2+‐gluconate, 1 Mg+‐gluconate, 5 sucrose, and 5 HEPES/Tris, pH 7.4) containing 1 mM ouabain for 30 min at 37°C. Cells were then transferred to a Na+‐free medium (in mM: 100 N‐methyl‐D‐glucamine‐Cl−, 10 KCl, 1.8 CaCl2, 1 MgCl2, 5 sucrose, and 5 HEPES/Tris, pH 7.4) containing 1 mM ouabain and 1 μCi/ml of 86Rb+ and uptake continued for 10 min at 37°C. At the end of the uptake period, cells were washed three times in the respective ice‐cold Na+‐free medium without isotope to remove extracellular fluid tracer and then lysed in 300 µl of ice‐cold lysis buffer (in mM: 50 Tris–HCl (pH 7.5), 1 EGTA, 1 EDTA, 50 sodium fluoride, 5 sodium pyrophosphate, 1 sodium orthovanadate, 1% (w/v) Nonidet P40, 0.27 sucrose, 0.1% 2‐mercaptoethanol and protease inhibitors (1 tablet per 50 ml, Sigma‐Aldrich). 86Rb+ uptake tracer activity was determined by Beckman β‐scintillation counter. To reach the isotonic conditions for HEK‐293 cells (~300 mOsm), mediums were supplemented with 1.7 g/100 ml sucrose. Removal of extracellular Na+ prevents 86Rb+ uptake by the endogenous NKCC1 cotransporter and the addition of ouabain prevents the activation of the Na+:K+‐ATPase. The Cl−‐dependent 86Rb+ uptake due to KCC4 was obtained by subtraction of uptake in Cl−‐free medium.
Metabolic acidosis model, and total and renal tubule‐specific SIRT7‐deficient mice
All animal procedures were performed in accordance with the Mexican Federal Regulation for Animal Experimentation and Care (NOM‐062‐ZOO‐2001) and approved by the Animal Ethics Research Committee of the Instituto Nacional de Ciencias Médicas y Nutrición Salvador Zubirán (Approval number 1560) or by the Swiss Federal Veterinary Office (Approval number VD3013b) and carried out in accordance with the local animal welfare act.
For the metabolic acidosis model, male C57B6 mice, weighing 20–25 g, were provided by the animal facility of the Instituto Nacional de Ciencias Médicas y Nutrición Salvador Zubirán. Mice were housed in regular polypropylene cages, with four‐five animals per cage, with light/dark cycles of 12/12 h at constant temperature and humidity of 20°C and 65%, respectively, during 2 days before the experimental period. To determine renal function mice were placed in metabolic cages the first and last day of the experiment to collect daily urine. Body mass and water intake were also determined. To induce metabolic acidosis, mice were fed with regular chow and were given free access to a solution with 280 mM ammonium chloride (NH4Cl) for 8 days. The control animals consisted of mice that were simultaneously carried through all procedures and fed the same diet and had free access to tap water. After the treatment period, animals were anesthetized with isoflurane, a blood sample was taken through cardiac puncture to perform a blood chemistry analysis. Blood gases were determined using i‐STAT ECG6 + cartridges and an i‐STAT1 Handheld Analyzer (Abaxis). Cortex and medulla from the kidney were rapidly removed and frozen in liquid nitrogen for further analysis. After the sacrifice, serum electrolytes, creatinine, and osmolarity were also determined.
Total SIRT7 deficient mice has been previously described (Ryu et al, 2014) and were bred on a pure C57BL/6J background. Mice of 9 weeks of age were placed in metabolic cages to collect 24‐h urine. Urine pH analysis was performed as previously described (Ronzaud et al, 2013). Kidney was dissected for mRNA and protein analysis. In another experiment, wild‐type and SIRT7‐deficient mice were fed with regular chow and were given free access to a solution with 280 mM ammonium chloride (NH4Cl) for 8 days to generate metabolic acidosis.
The inducible renal tubule‐specific SIRT7‐deficient mice were generated using a doxycycline (a tetracycline analog)‐inducible renal tubule‐specific Cre‐mediated recombination as previously described (Arroyo et al, 2013; Ronzaud et al, 2013). The Sirt7‐floxed mice used have been previously described and were bred on a pure C57BL/6J background (Ryu et al, 2014). Briefly, double‐transgenic Pax8‐rtTA/TRE‐LC1 mice (Pax8/LC1), which allow doxycycline (a tetracycline analog)‐inducible renal tubule‐specific Cre‐mediated recombination, were bred with mice homozygous for the Sirt7 floxed allele to obtain Sirt7flox/flox/Pax8‐rTA/LC1 inducible knockout mice (Sirt7 Pax8/LC1) and maintained in a mixed background. To induce the deletion of SIRT7, three‐ to 4‐week‐old Sirt7 wt/wt. Pax8/LC1, Sirt7wt/fl. Pax8/LC1 (controls) and Sirt7 flox/flox Pax8/LC1 animals were treated with doxycycline (0.5 mg/ml in drinking water with 2% sucrose) for 12 days. For further experiments, Sirt7 Pax8/LC1 and single transgenic Sirt7 Pax8 or Sirt7 LC1 animals (controls) were treated with doxycycline and the experiments started 4–6 weeks after the induction.
Total and renal tubule‐specific SIRT7‐deficient mice were adapted for 3 days under standard diet in metabolic cages to collect urine for 24 h. Urine pH analysis was performed as previously described (Ronzaud et al, 2013). After urine collection, kidneys were dissected for mRNA, and protein analysis and immunofluorescence as described below.
Immunofluorescence
Mice kidneys from wild‐type and SIRT7‐deficient mice with metabolic acidosis were perfused with 40 ml PBS followed by 50 ml 4% paraformaldehyde in PBS. After perfusion, tissues were incubated in 4% formaldehyde in PBS for at least 3 h and then incubated in 30% sucrose in PBS overnight at 4°C. The kidneys were mounted in OCT (Tissue‐Tek), and 5‐μm frozen sections were prepared. Antigen retrieval was performed in 100 mM citrate buffer, pH 6. Sections were blocked for 1 h in TBS‐tween 0.1% with 10% BSA and then incubated overnight at 4°C with primary antibodies diluted in 1% BSA in TBS‐tween against the B1 subunit of the H‐ATPase (Santa Cruz Biotechnologies sc‐21206, 1:100), SIRT7 (Santa Cruz Biotechnologies sc‐365344, 1:50) or KCC4 (Melo et al, 2013b, 1:200). For KCC4 immunofluorescence, antigen retrieval was performed by heating 2 min at 90°C in 100 mM citrate buffer. After incubation, sections were washed, incubated with secondary antibodies at room temperature for 1 h, then washed again. Secondary antibodies used were Alexa Fluor 488, donkey anti‐goat (1:400); Alexa Fluor 594, donkey anti‐mouse (1:400); Alexa Fluor 594, donkey anti‐rabbit (1:400) from Life Technologies. Tissue specimens were mounted on coverslips with 20 μl of Vectashield (Vector Labs). Slides were examined, and images were obtained with a 40× oil‐immersion objective mounted on a confocal inverted microscope (LSM 710 Duo, Carl Zeiss) or Cytation 1 (Biotek). Cell count was performed automatically using the Gen5 software. We first identified and counted H‐ATPase+ cells (green fluorescence) based on its size and fluorescence intensity, which was above a stabilized threshold determined by the background of the image. Then, we performed a subpopulation analysis by counting the cells that were positive for the red fluorescence (KCC4+ cells).
Western blot and coimmunoprecipitation analysis
Whole kidney, cortex, or medulla fractions were homogenized in lysis buffer (Tris–HCl 50 mM, NaCl 150 mM, EDTA 1 mM, NP‐40 1%) supplemented with protease inhibitors (Complete mini, Roche), phosphatase inhibitors (NaF 50 mM, NaP2O7 5 mM, Na3VO4 1 mM, or PhosSTOP (Roche)), and deacetylase inhibitors (NAM 5 mM, Sodium butyrate 1 mM). Homogenates were centrifuged 10,000 g, 10 min at 4°C, and supernatants were used in further analysis. Protein extracts from oocytes, HEK293 cells, and kidney were quantified by Bradford. Western blot was performed using 10–20 μg of total protein. Proteins were separated using a 7.5, 10, or 12% SDS–PAGE and transferred to nitrocellulose or PVDF membrane. KCC4 was identified using a previously characterized antibody (Karadsheh et al, 2004). To evaluate KCC4 acetylation, 500 μg of total protein of oocytes or HEK293 or kidney homogenates were immunoprecipitated using the KCC4 antibody. Western blot was performed using an antibody against acetylated lysines (#9441, Cell signaling), KCC4 or SIRT7. Detection was performed using ultra‐sensitive horseradish peroxidase chemiluminescence (Pierce). To determine the interaction between SIRT7 and KCC4, 500 μg of total protein of HEK293 or kidney homogenates were immunoprecipitated using anti‐SIRT7 antibody (Santa Cruz Biotechnology) or anti‐KCC4 antibody using magnetic beads coupled to protein G/A (Pierce), followed by a Western blot to identify both, SIRT7 and KCC4. Depending on availability, actin (Sigma), GAPDH (Abcam), or tubulin (Sigma) antibodies were used for normalization.
Microdissection of mouse renal tubule segments and analysis
For RNA extraction, glomeruli and nephron segments were microdissected from male 8–10‐week‐old C57BL6 mice (Charles River Breeding Laboratories). RNA extraction and real‐time PCR and validation of the micro dissected nephron segments are described here (Cheval et al, 2011). The following primers were used for real‐time PCR analysis: Sirt7 FW 5′‐AGAAGGCAGCCACAGTAGGA‐3′, Sirt7 RV 5′‐TTTCTTCCTTTTTGCACGCT‐3′; Rpl26 FW 5′‐GCTAATGGCACAACCGTC‐3′, Rpl26 RV 5′‐TCTCGATCGTTTCTTCCTTGTAT‐3′. For protein extraction, kidneys from 10‐to 12‐week‐old male C57BL6 mice (Charles River Breeding Laboratories) were perfused, and nephron segments were microdissected as described previously (Christensen et al, 2011) and resuspended directly in loading buffer. For each segment, tubules isolated from different animals were pooled (PT n = 3, TAL n = 5, DCT/CNT n = 5 and CCD n = 5) and used for Western blotting. Enrichment for each segment was validated by immunoblotting against segment‐specific markers as previously reported (Spirli et al, 2019). The proteins were transferred to nitrocellulose membrane and incubated with the SIRT7 antibody (sc‐365344, SCBT).
RNA extraction and Real‐Time PCR
For KCC4 mRNA expression, total RNA was prepared from homogenized kidneys using TRIzol (Invitrogen). cDNAs were prepared by reverse transcription of 1,000 ng of total RNA using the MMLV enzyme (Invitrogen). KCC4 cDNA was amplified in 96‐well plates using a SYBR green PCR kit (Qiagen) in a LC480 cycler (Roche) using the following primers: fwd 5′‐CACTGCATCCCATACCACAG‐3′ and rvs 5′‐GAACTACGCCGTTGAGCTTC‐3′. Data were normalized to β2‐microglobulin and 36B4 using the Pfaffl method. For Sirt7 mRNA expression in inducible renal tubule‐specific SIRT7‐deficient mice, total RNA was extracted using TRIzol (Invitrogen) and RNasy kit (Qiagen). 1 ug of RNA was reverse transcribed using Superscipt II (Invitrogen) and random hexamers. Real‐Time PCR analysis was performed in 7500 Fast Real‐Time PCR System (Applied Biosystems), using TaqMan gene expression assay (Applied Biosystems) for Sirt7 and Gapdh. The following TaqMan probes were used: Mm01248607_m1‐Sirt7 and Mm99999915_g1‐Gapdh. The data were normalized to Gapdh and relative mRNA expression is shown.
Statistical analyses
All data are presented as mean ± SEM. The results were considered statistically significant when P‐values were < 0.05 using an unpaired two‐tailed Student’s t‐test when comparing two groups, or a one‐way ANOVA followed by Bonferroni multiple comparison test when comparing more than two groups. Data were analyzed using GraphPad Prism (GraphPad Software, version 7.0).
Author contributions
Design of research studies: LGN, ZM, RDR, AM, ART, JA, OS, and GG; Experiments and acquiring and analyzing data: ZM, RDR, AM, LAV‐V, MC‐B, YRL, DR, LR‐V, GM‐A, AML‐B, MSH, ADe, ADo, LC, and LGN; Original idea and writing manuscript: LGN; Manuscript revision: ZM, RDR, ART, AM, DR, JA, OS, GG, and LGN; Funding and different resources: ART, NT, JA, OS, GG, and LGN. Final approval of the manuscript: All authors.
Conflict of interest
J.A. is a SAB member of Astellas.
Supporting information
Appendix
EVFigs
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Acknowledgements
This work was supported by grants 23 and A1‐S‐8290 from the Mexican Council of Science and Technology (CONACYT) to GG, 154939 to ART, 155949 to LGN, and 302510 to NT, the EPFL, Systems X (SySX.ch 2013/153), and the Swiss National Science Foundation Grants 31003A‐140780 to JA, 310030_189171 to OS, the Leducq Foundation to OS, the National Centre of Competence in Research “NCCR‐Kidney.ch” to OS, networking support by the COST Action ADMIRE BM1301 to OS. We thank the members of the Auwerx laboratory for discussion.
EMBO reports (2021) 22: e50766.
Contributor Information
Lilia G Noriega, Email: lgnoriegal@gmail.com.
Gerardo Gamba, Email: gamba@biomedicas.unam.mx.
Data availability
No data were deposited in a public database.
References
- Andreux PA, Williams EG, Koutnikova H, Houtkooper RH, Champy MF, Henry H, Schoonjans K, Williams RW, Auwerx J (2012) Systems genetics of metabolism: the use of the BXD murine reference panel for multiscalar integration of traits. Cell 150: 1287–1299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arroyo JP, Kahle KT, Gamba G (2013) The SLC12 family of electroneutral cation‐coupled chloride cotransporters. Mol Aspects Med 34: 288–298 [DOI] [PubMed] [Google Scholar]
- Arroyo JP, Lagnaz D, Ronzaud C, Vazquez N, Ko BS, Moddes L, Ruffieux‐Daidie D, Hausel P, Koesters R, Yang B et al (2011) Nedd4‐2 modulates renal Na+‐Cl‐ cotransporter via the aldosterone‐SGK1‐Nedd4‐2 pathway. J Am Soc Nephrol 22: 1707–1719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barber MF, Michishita‐Kioi E, Xi Y, Tasselli L, Kioi M, Moqtaderi Z, Tennen RI, Paredes S, Young NL, Chen K et al (2012) SIRT7 links H3K18 deacetylation to maintenance of oncogenic transformation. Nature 487: 114–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baur JA, Ungvari Z, Minor RK, Le Couteur DG, de Cabo R (2012) Are sirtuins viable targets for improving healthspan and lifespan? Nat Rev Drug Discov 11: 443–461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergeron MJ, Gagnon E, Wallendorff B, Lapointe JY, Isenring P (2003) Ammonium transport and pH regulation by K(+)‐Cl(‐) cotransporters. Am J Physiol Renal Physiol 285: F68–F78 [DOI] [PubMed] [Google Scholar]
- Boettger T, Hubner CA, Maier H, Rust MB, Beck FX, Jentsch TJ (2002) Deafness and renal tubular acidosis in mice lacking the K‐Cl co‐transporter KCC4. Nature 416: 874–878 [DOI] [PubMed] [Google Scholar]
- Cai J, Liu Z, Huang X, Shu S, Hu X, Zheng M, Tang C, Liu Y, Chen G, Sun L et al (2020) The deacetylase sirtuin 6 protects against kidney fibrosis by epigenetically blocking b‐catenin target gene expression. Kidney Int 97: 106–118 [DOI] [PubMed] [Google Scholar]
- Castañeda‐Bueno M, Cervantes‐Perez LG, Rojas‐Vega L, Arroyo‐Garza I, Vazquez N, Moreno E, Gamba G (2014) Modulation of NCC activity by low and high K(+) intake: insights into the signaling pathways involved. Am J Physiol Renal Physiol 306: F1507–F1519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavez‐Canales M, Zhang C, Soukaseum C, Moreno E, Pacheco‐Alvarez D, Vidal‐Petiot E, Castaneda‐Bueno M, Vazquez N, Rojas‐Vega L, Meermeier NP et al (2014) WNK‐SPAK‐NCC cascade revisited: WNK1 stimulates the activity of the Na‐Cl cotransporter via SPAK, an effect antagonized by WNK4. Hypertension 64: 1047–1053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheval L, Pierrat F, Dossat C, Genete M, Imbert‐Teboul M, Doung Van Houyen JP, Poulain J, Wincker P, Weissenbach J, Piquelman D et al (2011) Atlas of gene expression in the mouse kidney: new features of glomerular parietal cells. Physiol Genomics 43: 161–173 [DOI] [PubMed] [Google Scholar]
- Christensen BM, Zuber AM, Loffing J, Stehle JC, Deen PM, Rossier BC, Hummler E (2011) alphaENaC‐mediated lithium absorption promotes nephrogenic diabetes insipidus. J Am Soc Nephrol 22: 253–261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cioffi M, Vallespinos‐Serrano M, Trabulo SM, Fernandez‐Marcos PJ, Firment AN, Vazquez BN, Vieira CR, Mulero F, Camara JA, Cronin UP et al (2015) MiR‐93 controls adiposity via inhibition of Sirt7 and Tbx3. Cell Rep 12: 1594–1605 [DOI] [PubMed] [Google Scholar]
- Cruz‐Rangel S, Gamba G, Ramos‐Mandujano G, Pasantes‐Morales H (2012) Influence of WNK3 on intracellular chloride concentration and volume regulation in HEK293 cells. Pflugers Arch 464: 317–330 [DOI] [PubMed] [Google Scholar]
- Cruz‐Rangel S, Melo Z, Vazquez N, Meade P, Bobadilla NA, Pasantes‐Morales H, Gamba G, Mercado A (2011) Similar effects of all WNK3 variants on SLC12 cotransporters. Am J Physiol Cell Physiol 301: C601–608 [DOI] [PubMed] [Google Scholar]
- de los Heros P, Kahle KT, Rinehart J, Bobadilla NA, Vazquez N, San Cristobal P, Mount DB, Lifton RP, Hebert SC, Gamba G (2006) WNK3 bypasses the tonicity requirement for K‐Cl cotransporter activation via a phosphatase‐dependent pathway. Proc Natl Acad Sci USA 103: 1976–1981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Strooper B (2010) Proteases and proteolysis in Alzheimer disease: a multifactorial view on the disease process. Physiol Rev 90: 465–494 [DOI] [PubMed] [Google Scholar]
- Fang J, Ianni A, Smolka C, Vakhrusheva O, Nolte H, Kruger M, Wietelmann A, Simonet NG, Adrian‐Segarra JM, Vaquero A et al (2017) Sirt7 promotes adipogenesis in the mouse by inhibiting autocatalytic activation of Sirt1. Proc Natl Acad Sci USA 114: E8352–E8361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford E, Voit R, Liszt G, Magin C, Grummt I, Guarente L (2006) Mammalian Sir2 homolog SIRT7 is an activator of RNA polymerase I transcription. Genes Dev 20: 1075–1080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda M, Yoshizawa T, Karim MF, Sobuz SU, Korogi W, Kobayasi D, Okanishi H, Tasaki M, Ono K, Sawa T et al (2018) SIRT7 has a critical role in bone formation by regulating lysine acylation of SP7/Osterix. Nat Commun 9: 2833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamba G (2005) Molecular physiology and pathophysiology of electroneutral cation‐chloride cotransporters. Physiol Rev 85: 423–493 [DOI] [PubMed] [Google Scholar]
- Gamba G (2012) Regulation of the renal Na+‐Cl‐ cotransporter by phosphorylation and ubiquitylation. Am J Physiol Renal Physiol 303: F1573–F1583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garzon‐Muvdi T, Pacheco‐Alvarez D, Gagnon KB, Vazquez N, Ponce‐Coria J, Moreno E, Delpire E, Gamba G (2007) WNK4 kinase is a negative regulator of K+‐Cl‐ cotransporters. Am J Physiol Renal Physiol 292: F1197–F1207 [DOI] [PubMed] [Google Scholar]
- Glover M, Mercier ZA, Figg N, O'Shaughnessy KM (2010) The activity of the thiazide‐sensitive Na+‐Cl cotransporter is regulated by protein phosphatase PP4. Can J Physiol Pharmacol 88: 986–995 [DOI] [PubMed] [Google Scholar]
- Griffin NM, Schnitzeer JE (2011) Overcoming key technological challenges in using mass spectrometry for mapping cell surfaces in tissues. Mol Cell Proteomics 10: 1–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao CM, Haase VH (2010) Sirtuins and their relevance to the kidney. J Am Soc Nephrol 21: 1620–1627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hershberger KA, Martin AS, Hirschey MD (2017) Role of NAD(+) and mitochondrial sirtuins in cardiac and renal diseases. Nat Rev Nephrol 13: 213–225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houtkooper RH, Pirinen E, Auwerx J (2012) Sirtuins as regulators of metabolism and healthspan. Nat Rev Mol Cell Biol 13: 225–238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahle KT, Rinehart J, de Los HP, Louvi A, Meade P, Vazquez N, Hebert SC, Gamba G, Gimenez I, Lifton RP (2005) WNK3 modulates transport of Cl‐ in and out of cells: implications for control of cell volume and neuronal excitability. Proc Natl Acad Sci USA 102: 16783–16788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karadsheh MF, Byun N, Mount DB, Delpire E (2004) Localization of the KCC4 potassium‐chloride cotransporter in the nervous system. Neuroscience 123: 381–391 [DOI] [PubMed] [Google Scholar]
- Katsyuba E, Mottis A, Zietak M, De Franco F, van der Velpen V, Gariani K, Ryu D, Cialabrini L, Matilainen O, Liscio P et al (2018) De novo NAD(+) synthesis enhances mitochondrial function and improves health. Nature 563: 354–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiran S, Chatterjee N, Singh S, Kaul SC, Wadhwa R, Ramakrishna G (2013) Intracellular distribution of human SIRT7 and mapping of the nuclear/nucleolar localization signal. FEBS J 280: 3451–3466 [DOI] [PubMed] [Google Scholar]
- Ko B, Kamsteeg EJ, Cooke LL, Moddes LN, Deen PM, Hoover RS (2010) RasGRP1 stimulation enhances ubiquitination and endocytosis of the sodium‐chloride cotransporter. Am J Physiol Renal Physiol 299: F300–F309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Shi L, Yang S, Yan R, Zhang D, Yang J, He L, Li W, Yi X, Sun L et al (2016) SIRT7 is a histone desuccinylase that functionally links to chromatin compaction and genome stability. Nat Commun 7: 12235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo Sasso G, Ryu D, Mouchiroud L, Fernando SC, Anderson CL, Katsyuba E, Piersigilli A, Hottiger MO, Schoonjans K, Auwerx J (2014) Loss of Sirt1 function improves intestinal anti‐bacterial defense and protects from colitis‐induced colorectal cancer. PLoS One 9: e102495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundby A, Lage K, Weinert BT, Bekker‐Jensen DB, Secher A, Skovgaard T, Kelstrup CD, Dmytriyev A, Choudhary C, Lundby C et al (2012) Proteomic analysis of lysine acetylation sites in rat tissues reveals organ specificity and subcellular patterns. Cell Rep 2: 419–431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melo Z, Cruz‐Rangel S, Bautista R, Vazquez N, Castaneda‐Bueno M, Mount DB, Pasantes‐Morales H, Mercado A, Gamba G (2013a) Molecular evidence for a role for K(+)‐Cl(‐) cotransporters in the kidney. Am J Physiol Renal Physiol 305: F1402–F1411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melo Z, de los Heros P, Cruz‐Rangel S, Vazquez N, Bobadilla NA, Pasantes‐Morales H, Alessi DR, Mercado A, Gamba G (2013b) N‐terminal serine dephosphorylation is required for KCC3 cotransporter full activation by cell swelling. J Biol Chem 288: 31468–31476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercado A, Broumand V, Zandi‐Nejad K, Enck AH, Mount DB (2006) A C‐terminal domain in KCC2 confers constitutive K+‐Cl‐ cotransport. J Biol Chem 281: 1016–1026 [DOI] [PubMed] [Google Scholar]
- Michishita E, Park JY, Burneskis JM, Barrett JC, Horikawa I (2005) Evolutionarily conserved and nonconserved cellular localizations and functions of human SIRT proteins. Mol Biol Cell 16: 4623–4635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morigi M, Perico L, Rota C, Longaretti L, Conti S, Rottoli D, Novelli R, Remuzzi G, Benigni A (2015) Sirtuin 3‐dependent mitochondrial dynamic improvements protect against acute kidney injury. J Clin Invest 125: 715–726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morigi M, Perico L, Benigni A (2018) Sirtuins in renal health and disease. J Am Soc Nephrol 29: 1799–1809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moullan N, Mouchiroud L, Wang X, Ryu D, Williams EG, Mottis A, Jovaisaite V, Frochaux MV, Quiros PM, Deplancke B et al (2015) Tetracyclines disturb mitochondrial function across eukaryotic models: a call for caution in biomedical research. Cell Rep 10: 1681–1691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mount DB, Mercado A, Song L, Xu J, George AL, Delpire E, Gamba G (1999) Cloning and characterization of KCC3 and KCC4, new members of the cation‐chloride cotransporter gene family. J Biol Chem 274: 16355–16362 [DOI] [PubMed] [Google Scholar]
- Pacheco‐Alvarez D, San Cristobal P, Meade P, Moreno E, Vazquez N, Munoz E, Diaz A, Juarez ME, Gimenez I, Gamba G (2006) The Na‐Cl cotransporter is activated and phosphorylated at the amino terminal domain upon intracellular chloride depletion. J Biol Chem 281: 28755–28763 [DOI] [PubMed] [Google Scholar]
- Park J, Shrestha R, Qiu C, Kondo A, Huang S, Werth M, Li M, Barasch J, Suszták K (2018) Single‐cell transcriptomics of the mouse kidney reveals potential cellular targets of kidney disease. Science 360: 758–763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson C, Rafiqi FH, Karlsson HK, Moleleki N, Vandewalle A, Campbell DG, Morrice NA, Alessi DR (2008) Activation of the thiazide‐sensitive Na+‐Cl− cotransporter by the WNK‐regulated kinases SPAK and OSR1. J Cell Sci 121: 675–684 [DOI] [PubMed] [Google Scholar]
- Rinehart J, Kahle KT, de Los HP, Vazquez N, Meade P, Wilson FH, Hebert SC, Gimenez I, Gamba G, Lifton RP (2005) WNK3 kinase is a positive regulator of NKCC2 and NCC, renal cation‐Cl‐ cotransporters required for normal blood pressure homeostasis. Proc Natl Acad Sci USA 102: 16777–16782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinehart J, Vazquez N, Kahle KT, Hodson CA, Ring AM, Gulcicek EE, Louvi A, Bobadilla NA, Gamba G, Lifton RP (2011) WNK2 kinase is a novel regulator of essential neuronal cation‐chloride cotransporters. J Biol Chem 286: 30171–30180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronzaud C, Loffing‐Cueni D, Hausel P, Debonneville A, Malsure SR, Fowler‐Jaeger N, Boase NA, Perrier R, Maillard M, Yang B et al (2013) Renal tubular NEDD4‐2 deficiency causes NCC‐mediated salt‐dependent hypertension. J Clin Invest 123: 657–665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu D, Jo YS, Lo Sasso G, Stein S, Zhang H, Perino A, Lee JU, Zeviani M, Romand R, Hottiger MO et al (2014) A SIRT7‐Dependent acetylation switch of GABPbeta1 controls mitochondrial function. Cell Metab 20: 856–869 [DOI] [PubMed] [Google Scholar]
- Sakamoto KM (2002) Ubiquitin‐dependent proteolysis: its role in human diseases and the design of therapeutic strategies. Mol Genet Metab 77: 44–56 [DOI] [PubMed] [Google Scholar]
- Sauve AA, Celic I, Avalos J, Deng H, Boeke JD, Schramm VL (2001) Chemistry of gene silencing: the mechanism of NAD+‐dependent deacetylation reactions. Biochemistry 40: 15456–15463 [DOI] [PubMed] [Google Scholar]
- Shin J, He M, Liu Y, Paredes S, Villanova L, Brown K, Qiu X, Nabavi N, Mohrin M, Wojnoonski K et al (2013) SIRT7 represses Myc activity to suppress ER stress and prevent fatty liver disease. Cell Rep 5: 654–665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon DB, Karet FE, Hamdan JM, DiPietro A, Sanjad SA, Lifton RP (1996) Bartter's syndrome, hypokalaemic alkalosis with hypercalciuria, is caused by mutations in the Na‐K‐2Cl cotransporter NKCC2. Nat Genet 13: 183–188 [DOI] [PubMed] [Google Scholar]
- Spirli A, Cheval L, Debonneville A, Penton D, Ronzaud C, Maillard M, Doucet A, Loffing J, Staub O (2019) The serine‐threonine kinase PIM3 is an aldosterone‐regulated protein in the distal nephron. Physiol Rep 7: e14177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vakhrusheva O, Smolka C, Gajawada P, Kostin S, Boettger T, Kubin T, Braun T, Bober E (2008) Sirt7 increases stress resistance of cardiomyocytes and prevents apoptosis and inflammatory cardiomyopathy in mice. Circ Res 102: 703–710 [DOI] [PubMed] [Google Scholar]
- Wu J, Liu X, Lai G, Yang X, Wang L, Zhao Y (2013) Synergistical effect of 20‐HETE and high salt on NKCC2 protein and blood pressure via ubiquitin‐proteasome pathway. Hum Genet 132: 179–187 [DOI] [PubMed] [Google Scholar]
- Yoshizawa T, Karim MF, Sato Y, Senokuchi T, Miyata K, Fukuda T, Go C, Tasaki M, Uchimura K, Kadomatsu T et al (2014) SIRT7 controls hepatic lipid metabolism by regulating the ubiquitin‐proteasome pathway. Cell Metab 19: 712–721 [DOI] [PubMed] [Google Scholar]
- Zhang D, Li S, Cruz P, Kone BC (2009) Sirtuin 1 functionally and physically interacts with disruptor of telomeric silencing‐1 to regulate alpha‐ENaC transcription in collecting duct. J Biol Chem 284: 20917–20926 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
EVFigs
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Data Availability Statement
No data were deposited in a public database.