

Abstract
Toll‐like receptor 4 (TLR4) recognizes bacterial lipopolysaccharide (LPS) and can also be activated by some Group 9/10 transition metals, which is believed to mediate immune hypersensitivity reactions. In this work, we test whether TLR4 can be activated by the Group 10 metal platinum and the platinum‐based chemotherapeutic cisplatin. Cisplatin is invaluable in childhood cancer treatment but its use is limited due to a permanent hearing loss (cisplatin‐induced ototoxicity, CIO) adverse effect. We demonstrate that platinum and cisplatin activate pathways downstream of TLR4 to a similar extent as the known TLR4 agonists LPS and nickel. We further show that TLR4 is required for cisplatin‐induced inflammatory, oxidative, and cell death responses in hair cells in vitro and for hair cell damage in vivo. Finally, we identify a TLR4 small molecule inhibitor able to curtail cisplatin toxicity in vitro. Thus, our findings indicate that TLR4 is a promising therapeutic target to mitigate CIO.
Keywords: chemotherapy, cisplatin, hearing loss, ototoxicity, TLR4
Subject Categories: Immunology, Chemical Biology, Signal Transduction
Cisplatin is invaluable in cancer treatment, but its use is limited due to cisplatin‐induced ototoxicity. TLR4 is activated by cisplatin, contributing to cisplatin‐induced hair cell death, but genetic and small molecule inhibition of TLR4 curtails ototoxicity.
Introduction
Toll‐like receptor 4 (TLR4) is a membrane‐bound pattern recognition receptor that is best characterized for its ability to initiate innate immune signaling upon detection of the Gram‐negative bacterial surface component lipopolysaccharide (LPS) (Kawasaki & Kawai, 2014). LPS detection requires the TLR4 co‐receptor MD‐2. Structural analyses have revealed that the LPS binding pocket is comprised of both MD‐2 and TLR4 on the external face of the membrane, with MD‐2 making a major contribution to agonist binding (Park et al, 2009; Park & Lee, 2013). LPS binding to the TLR4/MD‐2 complex induces TLR4 dimerization and signal propagation through adapter protein recruitment on the cytoplasmic face of the membrane (Park & Lee, 2013). Two canonical signaling pathways are activated through TLR4. The TLR4 adapter protein TIRAP engages the MyD88‐dependent signaling pathway that culminates in NF‐κB nuclear translocation and pro‐inflammatory cytokine production. TRAM, the alternate TLR4 adapter protein, engages TRIF resulting in IRF3 translocation to the nucleus and stimulation of type I interferon response (Kawasaki & Kawai, 2014).
It is also widely accepted that TLR4 is activated by other agonists including damage‐associated molecular patterns (DAMPs, e.g., HMGB1 and HSP70) and the fusion protein from respiratory syncytial virus (Kurt‐Jones et al, 2000; Lee & Seong, 2009; Rallabhandi et al, 2012; Gaikwad et al, 2017; Yuan et al, 2018). TLR4 was also found to mediate immune hypersensitivity reactions to the Group 9/10 transition metals nickel, cobalt, and palladium (Schmidt et al, 2010; Raghavan et al, 2012; Rachmawati et al, 2013). Mechanistically, metal binding to TLR4 induces receptor dimerization to activate downstream signaling (Raghavan et al, 2012). Platinum is a Group 10 transition metal that shares chemical properties with nickel and palladium but it is unknown whether it can activate TLR4.
Cisplatin is a platinum‐based, highly effective chemotherapeutic frequently used to treat solid tumors in children. In adults, it is used to treat ovarian, testicular, cervical, lung, head and neck, and bladder cancers (Dasari & Tchounwou, 2014). Cisplatin‐containing regimens contribute to a 5‐year survival rate that approaches 80% in childhood cancer patients and has become an asset to cancer therapy (American Childhood Cancer Organization, 2017). The anti‐tumor activity of cisplatin is based on its formation of intra‐strand and inter‐strand guanine crosslinks in DNA that prevent the strands from separating, or it alkylates DNA bases causing DNA miscoding (Siddik, 2003). This DNA modification activates multiple signal transduction pathways leading to cell‐cycle arrest and programmed cell death (Sorenson et al, 1990; Sarin et al, 2017; Maekawa et al, 2019).
Despite its effectiveness, cisplatin use is limited by the development of several toxicities that include nephrotoxicity, neurotoxicity, and ototoxicity. Although nephrotoxicity can be reversed by saline hydration and mannitol diuresis, there is no treatment for cisplatin‐induced neurotoxicity or ototoxicity (Rybak et al, 2009). The ototoxic effect of cisplatin leads to permanent bilateral hearing loss and is estimated to affect 26‐90% of children treated with cisplatin where age, treatment regimen, and concomitant factors also influence susceptibility (Skinner et al, 1990; Brock et al, 1991; Blakley & Myers, 1993; Li et al, 2004; Brock et al, 2012). Cisplatin‐induced ototoxicity (CIO) can have significant life‐long consequences in children by impairing speech and language development, impairing social‐emotional development, and increasing the risk of learning difficulties (Gurney et al, 2007; Gurney et al, 2009). Moreover, the likelihood of developing ototoxicity increases in a dose‐dependent manner, with nearly 100% of patients receiving high dosages of cisplatin (150–225 mg/m2) showing some degree of ototoxicity. This compromises anti‐cancer treatment, potentially impacting overall survival as cisplatin dose reduction or discontinuation is required to mitigate this ototoxicity (Kopelman et al, 1988; Chang & Chinosornvatana, 2010).
CIO is perhaps exacerbated because cisplatin accumulates preferentially in the cochlea of the inner ear (Breglio et al, 2017), and more particularly in the outer hair cells of the Organ of Corti, which are terminally differentiated mechanotransducers and the site of the first steps in sound perception (Lim, 1986). The cochlea is considered a closed system due to its isolated anatomical position and structure and, as such, is not able to rapidly flush out cisplatin and the metabolites generated in response (Lim, 1986). Apoptotic damage in the hair cells of the cochlea is the primary mechanism of cisplatin‐induced hearing loss (Rybak et al, 2007).
In the current study, we sought a mechanistic understanding of the signaling pathway activated by cisplatin to enable mitigation of its adverse long‐term effects. We found that cisplatin activates TLR4, independently of the MD‐2 co‐receptor. Further, deletion of Tlr4 in a murine inner ear cell line reduced cisplatin‐induced ototoxicity. Similarly, knockdown of Tlr4 homologs in zebrafish protected against cisplatin‐induced hair cell death. Moreover, we attenuated cisplatin ototoxic responses with the TLR4 chemical inhibitor, TAK‐242. These findings provide key insights into the etiology of cisplatin‐induced ototoxicity and are crucial to developing protective therapies against CIO, thereby improving the prognosis and long‐term health outcomes of cancer patients.
Results
Platinum and cisplatin activate TLR4 in vitro
Nickel, palladium, and cobalt (Group 9 and 10 transition metals) have been well characterized as TLR4 ligands that induce contact hypersensitivity (Rachmawati et al, 2013). Given that platinum is a Group 10 transition metal, we were interested in determining whether it also could serve as a TLR4 ligand. We investigated this using reporter cell lines that did (HEK‐hTLR4) or did not (HEK‐null2) stably express human TLR4 and its MD‐2/CD14 co‐receptors. These isogenic human embryonic kidney (HEK) cell lines also express a reporter of NF‐κB activation, where NF‐κB induces transcription of a secreted alkaline phosphatase (SEAP) reporter; these cells have been used previously to identify TLR4 ligands (Schmidt et al, 2010).
We treated HEK‐hTLR4 and HEK‐null2 cells with platinum (both platinum (II) and (IV) as chloride salts), or LPS or nickel as positive control TLR4 agonists, and monitored NF‐κB activation. As expected, we saw significant activation of NF‐κB in HEK‐hTLR4 cells treated with LPS or nickel compared to media‐only controls (Fig 1A). By contrast, HEK‐null2 cells showed no significant change in NF‐κB activation, demonstrating the effects were dependent on TLR4. HEK‐hTLR4 cells treated with platinum(II) or platinum(IV) also showed significant induction of SEAP activity compared to HEK‐null2 cells that was intermediate between nickel and LPS (Fig 1A). Separately, we assessed TLR4 activity by measuring its downstream induction of IL‐8 cytokine secretion in the same cells. IL‐8 secretion increased significantly in the HEK‐hTLR4 cells for LPS, nickel chloride, and platinum(IV) chloride (Fig 1B). Platinum(II) chloride induced IL‐8 secretion 10‐fold, though this was not statistically significant. Again, no IL‐8 secretion was observed in cells where hTLR4 was absent.
Figure 1. Platinum and cisplatin activate TLR4 in vitro .
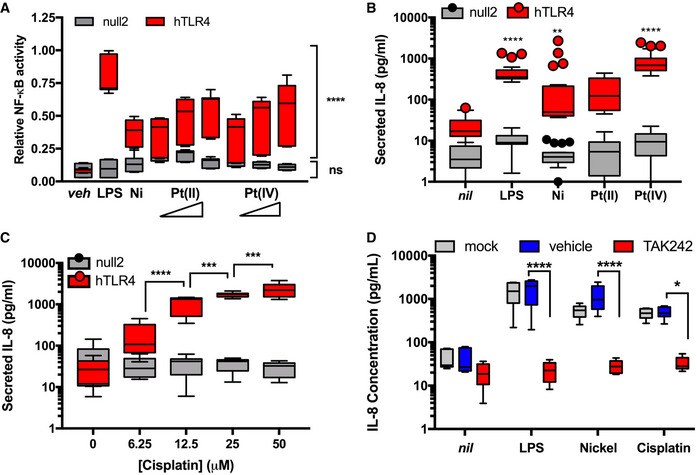
- Activity of an NF‐κB reporter relative to vehicle control in human embryonic kidney cells that express TLR4 (hTLR4) or an isogenic control cell line that does not express TLR4 (null2) and were stimulated with vehicle (veh), 1 ng/ml LPS, 400 µM nickel chloride, or 25, 50, or 100 µM platinum(II) chloride, and platinum(IV) chloride (n = 3 independent biological replicates).
- As per panel (A), secreted IL‐8 was monitored as a metric of TLR4 activation upon stimulation with 50 pg/ml LPS, 200 µM nickel chloride, 100 µM platinum(II) chloride, or 100 µM platinum(IV) chloride (n = 4 independent biological replicates).
- IL‐8 secretion in HEK‐hTLR4 and HEK‐null2 cells following treatment with cisplatin at the indicated concentrations (n = 3 independent biological replicates).
- IL‐8 secretion in HEK‐hTLR4 cells pre‐treated with 4 µ M TAK242 (TLR4 inhibitor) or vehicle, and subsequent treatment with 50 pg/ml LPS, 200 µM nickel chloride, or 25 µM cisplatin (n = 3 independent biological replicates). Mock cells were not subject to pre‐treatment prior to agonist addition.
Data Information: For all panels, actual individual data from each experiment are plotted as box (25th and 75th percentile borders; median central band) with Tukey whiskers. Statistical analyses were assessed by 2‐way ANOVA: in (A) hTLR4 compared to null2 cells; in (B) agonist treatment compared to non‐treated (nil); in (C) comparisons between successive concentrations; and in (D) comparisons between vehicle and TAK‐242 treatments. ns, not significant; *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001 (Dunnett’s test, A, B, D; Tukey test, C).
Source data are available online for this figure.
We next tested whether cisplatin, a platinum‐based chemotherapeutic, could also activate TLR4 considering its highly similar composition to platinum chloride. We found that cisplatin also induced IL‐8 secretion in HEK‐hTLR4 cells more than 100‐fold, but not in HEK‐null2 cells. Furthermore, cisplatin activation of TLR4 was dose‐dependent up to 50 μM, with pronounced toxicity limiting assessments at higher concentrations (Fig 1C). HEK‐null2 cells remained largely unresponsive at higher cisplatin concentrations.
To further examine cisplatin activation of TLR4, we studied MyD88‐dependent and MyD88‐independent signaling events downstream of TLR4 in response to cisplatin treatment. To this end, we monitored phospho‐signaling events and reporter assays in the mouse inner ear Organ of Corti cell line HEI‐OC1, which provides a popular in vitro model of drug‐induced hearing loss (Kalinec et al, 2016). We observed rapid phosphorylation of the MyD88‐dependent signaling components NF‐κB and p42/44 within 30 min of treatment (Fig EV1A). By contrast, we did not observe phosphorylation of IRF3 over the same time course. However, using a luciferase‐based IRF3 reporter we observed significant induction of this TRAM‐dependent signaling pathway 24 h after cisplatin treatment (Fig EV1B). The kinetics of signaling that we observed are consistent with those reported in the literature for TLR4 activation and for cisplatin treatment of HEI‐OC1 cells (Kawai & Akira, 2006; So et al, 2007, 2008; Chung et al, 2008).
Figure EV1. Cisplatin induces rapid MyD88‐dependent signaling and slower MyD88‐independent signaling downstream of Tlr4.
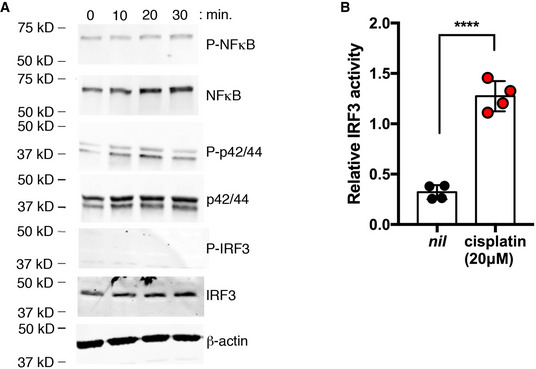
- Phospho‐signaling in HEI‐OC1 cells following treatment with 20 µM cisplatin or 10 ng/ml LPS at the indicated time points (representative of 3 independent biological replicates).
- IRF3 reporter activity in HEI‐OC1 cells treated with and without 20 µM cisplatin after 24 h (n = 4 independent biological replicates).
Data information: In (B), data are shown as mean and standard deviation. ****P < 0.0001 (unpaired Student’s t‐test).
Source data are available online for this figure.
To independently assess the requirement for TLR4 in cisplatin‐induced IL‐8 secretion in HEK‐hTLR4 cells, we repeated our treatments in the presence of a small molecule TLR4 inhibitor (TAK‐242) that binds to the intracellular domain of TLR4, disrupting its interactions with cytosolic adaptor proteins (Matsunaga et al, 2011). Chemical inhibition with TAK‐242 mitigated the effect of cisplatin on TLR4 activation similarly to nickel and LPS in HEK‐hTLR4 cells (Fig 1D). Taken together, these data demonstrate that platinum and cisplatin behave similarly to nickel and LPS with respect to their ability to activate TLR4.
Cisplatin activation of TLR4 does not require its co‐receptor, MD‐2
We next sought to appreciate the mechanisms of TLR4 activation upon cisplatin activation, as this would influence potential therapeutic design. Canonical TLR4 signaling after binding LPS requires the TLR4 co‐receptor MD‐2, whereas their requirements for metal‐based activation of TLR4 are less well defined (Nagai et al, 2002; Schmidt et al, 2010; Raghavan et al, 2012). To examine the role of the MD‐2 co‐receptor in TLR4 activation, we used an HEK cell background that stably expresses TLR4 but not the MD‐2 co‐receptor (HEK‐isoTLR4). HEK‐isoTLR4 cells were transfected with an empty vector control, or with a human MD‐2 expressing plasmid, and assayed for IL‐8 secretion upon treatment with TLR4 agonists. As expected, treatment with LPS did not yield a significant increase in secreted IL‐8 unless HEK‐isoTLR4 cells were transfected with MD‐2 (Fig 2A). This result confirmed that TLR4 was active in these cells and that they were MD‐2‐deficient. We obtained similar findings with nickel chloride, which is consistent with reports that nickel activation of TLR4 is MD‐2‐dependent (Raghavan et al, 2012; Oblak et al, 2015). By contrast, we observed significant secretion of IL‐8 in response to platinum(II) chloride, platinum(IV) chloride, and cisplatin in HEK‐isoTLR4 cells transfected with the empty vector. Although transfection of MD‐2 further enhanced IL‐8 secretion in response to these agonists, these findings indicate that unlike LPS and nickel, the platinum‐containing agonists are not strictly MD‐2 dependent. We also tested the DAMP, HMGB1 in this assay and observed that significant increases in IL‐8 secretion elicited by this TLR4 agonist also required MD‐2. These data suggest that cisplatin activation of TLR4 can occur independently of this DAMP, potentially mediated by its platinum constituent (Fig 2A).
Figure 2. Cisplatin activation of TLR4 is independent of the TLR4 co‐receptor, MD‐2.
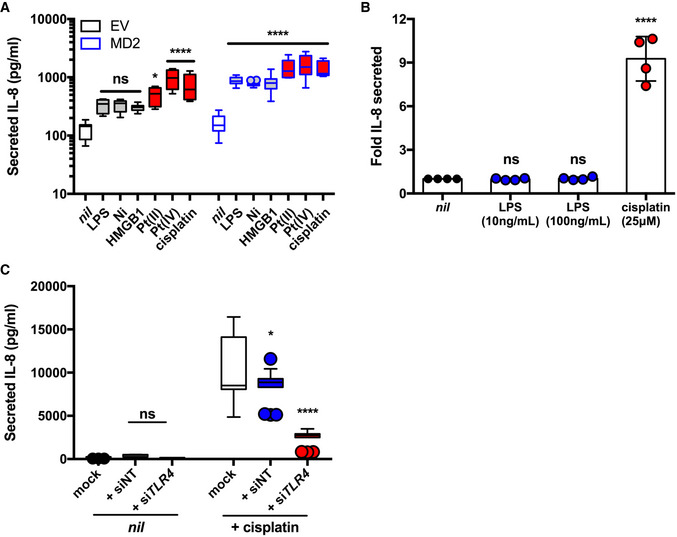
- IL‐8 secretion in HEK cells stably expressing hTLR4 but not MD‐2 (HEK‐isoTLR4), transfected with empty vector (EV) or MD‐2 and left untreated (nil) or treated with 1 ng/ml LPS, 200 µM nickel chloride, 2 µg/ml HMGB1, 100 µM platinum(II) chloride, 100 µM platinum(IV) chloride, or 25 µM cisplatin (n = 3 or 4 independent biological replicates).
- Fold IL‐8 secreted (relative to nil treatment) in MD‐2‐deficient HeLa cells treated with 10 or 100 ng/ml LPS or 25 µM cisplatin (n = 4 independent biological replicates).
- IL‐8 secretion in HeLa cells transfected with non‐targeting (siNT) or TLR4‐targeting (siTLR4) siRNA and left untreated (nil), or treated with 30 µM cisplatin (n = 3 independent biological replicates). Mock cells were not subject to siRNA treatment prior.
Data Information: Actual individual data are plotted as box (25th and 75th percentile borders; median central band) with Tukey whiskers (A, C) or mean and standard deviation (B). Statistical analyses were determined in comparison to nil treatments using 2‐way (A, C) or one‐way (B) ANOVA. ns, not significant; *P < 0.05; ****P < 0.0001 (Dunnett’s test).
Source data are available online for this figure.
To further investigate the requirement of the MD‐2 co‐receptor for cisplatin activation of TLR4, we used HeLa cells that have been reported to lack MD‐2 expression (Wyllie et al, 2000). We treated HeLa cells with cisplatin and LPS and observed that cisplatin, but not LPS, induced significant IL‐8 secretion (Fig 2B). To confirm that IL‐8 secretion elicited by cisplatin in HeLa cells was dependent on TLR4, we repeated cisplatin treatments in HeLa cells transfected with non‐targeting or TLR4‐targeting siRNA. We determined that siRNA treatment reduced TLR4 expression by > 75% (Fig EV2). Following TLR4 knockdown, we observed 70% lower cisplatin‐induced IL‐8 secretion indicating that secretion of this cytokine is mediated by TLR4 (Fig 2C). Taken together, these data indicate that TLR4 co‐receptors are dispensable for cisplatin activation of TLR4.
Figure EV2. TLR4 expression in HeLa cells is significantly reduced by transient silencing.
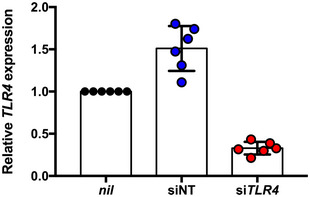
TLR4 expression levels (relative to nil treatment) in HeLa cells transfected with non‐targeting (siNT) or TLR4‐targeting (siTLR4) siRNA molecules and treated with 30 µM cisplatin.
Data information: Actual individual data from 2 independent experiments are plotted with mean and standard deviation indicated.
Source data are available online for this figure.
Tlr4 deletion mitigates cisplatin ototoxic responses in a murine inner ear cell line
Having shown that cisplatin can act as an agonist of TLR4 to induce a pro‐inflammatory response in vitro, we next asked whether TLR4 plays a role in mediating the molecular events that contribute to cisplatin‐induced ototoxicity. Cisplatin treatment induces the generation of reactive oxygen species (ROS) in the cochlea, which appear to be critical mediators of CIO (Clerici et al, 1996; van Ruijven et al, 2004). Hallmarks of in vitro cisplatin ototoxic responses, as modeled by Organ of Corti cell lines, include increased pro‐inflammatory IL‐6 signaling, which can upregulate ROS generation that in turn influence morphological and functional alterations leading to apoptotic cell death (Ravi et al, 1995; So et al, 2007; Kim et al, 2010).
We mutated Tlr4 in the mouse inner ear hair cell line HEI‐OC1 by CRISPR/Cas9. We established single‐cell clones of Tlr4‐edited cells, along with non‐targeting guide RNA‐edited control cells and conducted a primary screen to identify clones with diminished LPS responses. Sanger sequencing at the Tlr4 locus identified a clone with frame‐shift mutations in exon 1 (one adenine insertion or a four nucleotide deletion; Fig EV3A). Compared to control cells, the deletion clone exhibited decreased Tlr4 protein abundance (Fig EV3B), significantly reduced binding/internalization of a fluorescent LPS analog (Fig EV3C), and significantly reduced LPS‐induced cytokine secretion (Fig EV3D). Importantly, LPS‐induced IL‐6 secretion was enhanced 4‐fold upon complementation with ectopically expressed Tlr4 in the deletion cells, compared to less than 2‐fold in control cells (Fig EV3D, inset). Taken together with the genetic data, these results confirm a Tlr4 deletion in CRISPR‐targeted HEI‐OC1 cells.
Figure EV3. Tlr4 −/− HEI‐OC1 cells show diminished LPS‐responsiveness unless complemented with Tlr4 .
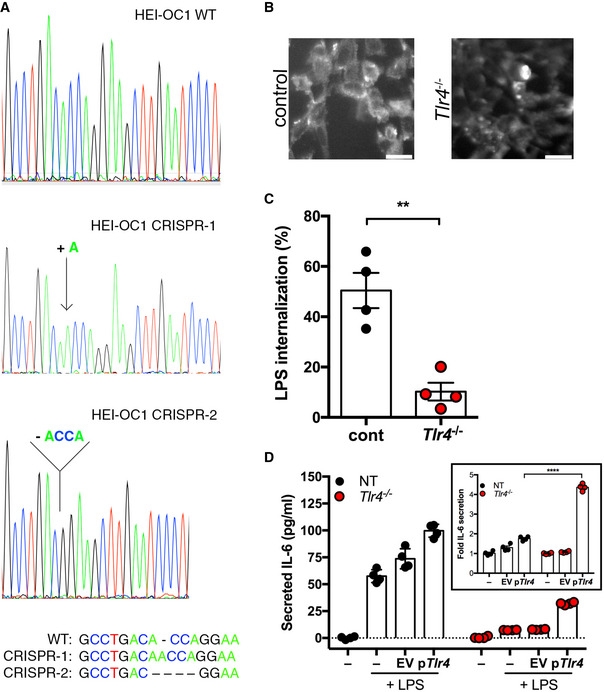
- Comparison of genomic DNA at the Tlr4 locus from Tlr4 −/− HEI‐OC1 and wild‐type cells. Sequences from the Tlr4 −/− cell line contained a single nucleotide insertion or four nucleotide deletion and summarized below.
- Anti‐TLR4 staining in Tlr4 −/− and control HEI‐OC1 cells. Bars (lower right) are 50 µm.
- Flow cytometric analysis of conjugated LPS internalization in Tlr4 −/− and control HEI‐OC1 cells (n = 4 independent biological replicates).
- IL‐6 secretion in Tlr4 −/− and control HEI‐OC1 cells transfected with empty vector (EV), Tlr4 (pTlr4), or left untransfected (−) and subsequently treated with 100 ng/ml LPS (n = 4 independent biological replicates). Inset, fold induction of IL‐6 secretion was determined relative to the untransfected cells treated with LPS.
Data information: In (C and D), data are presented as mean and standard deviation. Statistical comparisons were assessed by 2‐way ANOVA (D). **P < 0.01; ****P < 0.0001 (unpaired Student’s t‐test, C; Bonferroni test, D).
Source data are available online for this figure.
To examine the impact of the Tlr4 deletion on cisplatin ototoxic responses, we treated Tlr4 deletion and control HEI‐OC1 cells with cisplatin for 24 h to measure apoptosis, pro‐inflammatory cytokine secretion, and intracellular ROS generation. With increasing cisplatin concentrations, Tlr4‐deleted HEI‐OC1 cells had a higher level of viability than control, with a concomitant decrease in AnxV+/PI‐ cells suggesting that the cells were dying apoptotically (Fig 3A and B). Similarly, we observed a significant decrease in cisplatin‐induced ROS formation in Tlr4 deletion cells (Fig 3C). Moreover, Tlr4‐deleted cells had reduced IL‐6 secretion in response to cisplatin treatment compared to control cells (Fig 3D). Taken together, these data indicate that TLR4 is an important mediator of cisplatin ototoxic responses in inner ear hair cells.
Figure 3. Deletion of Tlr4 in a murine ear outer hair cell line (HEI‐OC1) reduces cisplatin‐induced ototoxic responses.
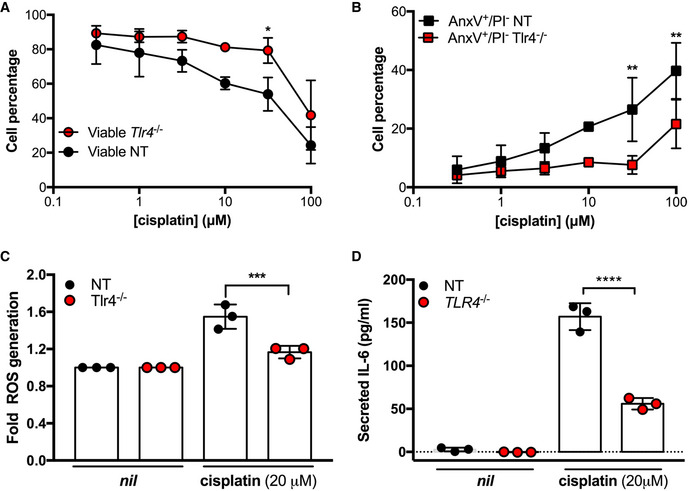
-
A–DHEI‐OC1 cells containing a Tlr4 deletion (Tlr4 −/−) were compared to HEI‐OC1 non‐targeting (NT) control cells and assessed for cell viability (A), Annexin V/propidium iodide staining (B), ROS generation (C), and IL‐6 secretion (D) following cisplatin treatment at the indicated concentrations (n = 3 independent biological replicates).
Data Information: In all panels, data are presented as mean and standard deviation. Statistical comparisons to NT at the same cisplatin concentration were assessed by 2‐way (A, B) or one‐way (C, D) ANOVA. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001 (Bonferroni test).
Source data are available online for this figure.
Cisplatin‐induced toxicity is consistent with its primary activation of TLR4
It has been previously reported that cisplatin induces the expression of Tlr4 leading to subsequent activation by LPS to potentiate cisplatin ototoxicity (Oh et al, 2011). This model describes a secondary effect of cisplatin on TLR4 activation. While our data demonstrating that cisplatin toxicity responses depend, at least in part, on Tlr4 could be consistent with this model, our observations in the HEK‐hTLR4 system suggest that cisplatin has a primary effect on TLR4 activation (e.g., co‐receptor‐independent TLR4 activation). To further characterize the effect of cisplatin in an ear outer hair cell line, we conducted kinetic analyses of Tlr4 activation to distinguish between primary (early) and secondary (later) effects. We examined IL‐6 secretion over time in HEI‐OC1 cells stimulated by the TLR4 agonists, cisplatin, and LPS. We observed that cisplatin‐ and LPS‐induced IL‐6 secretion followed similar kinetics for 4 h (Fig 4A). We confirmed that IL‐6 secretion induced by cisplatin at early time points was dependent on Tlr4 by performing similar experiments in our Tlr4‐deleted HEI‐OC1 cells, complemented with either Tlr4 or an empty vector. Here we observed no significant IL‐6 secretion over 8 h in this cell line unless complemented with Tlr4 (Fig 4B). Together, these data suggest that cisplatin is activating TLR4 in a primary manner.
Figure 4. Cisplatin has a primary role in Tlr4 activation in HEI‐OC1 cells.
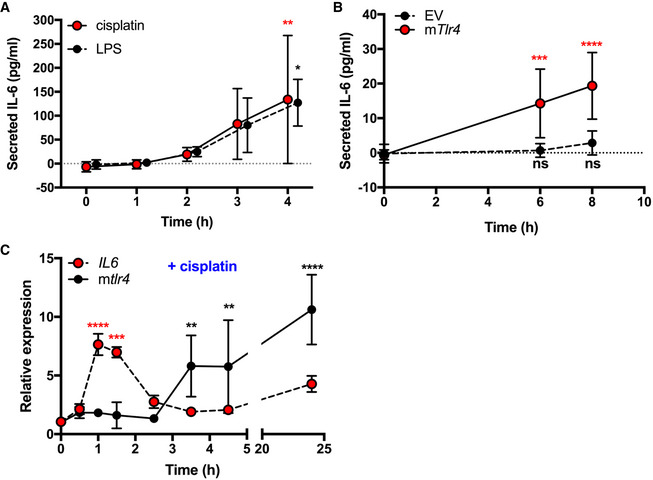
- IL‐6 secretion in HEI‐OC1 cells treated with 100 pg/ml LPS or 20 µM cisplatin (n = 3 or 4 independent biological replicates).
- IL‐6 secretion in TLR4−/− cells transfected with empty vector (EV) or mouse Tlr4 (mTlr4) following treatment with 20 µM cisplatin (n = 6 independent biological replicates).
- Il6 and Tlr4 transcript levels in HEI‐OC1 cells following treatment with 20 µM cisplatin for the indicated times (n = 3 independent biological replicates).
Data information: In all panels, data are presented as mean and standard deviation. Statistical comparisons to 0 h time point were assessed by 2‐way ANOVA. ns, not significant; *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001 (Dunnett’s test).
Source data are available online for this figure.
To further investigate TLR4 activation by cisplatin, we treated HEI‐OC1 cells with cisplatin and total RNA was extracted. We quantified the relative expression of both Il6 and Tlr4 over time in response to cisplatin treatment. Cisplatin treatment showed Il6 expression peaking after 1 h. Interestingly, there was a notable disparity in Il6 and Tlr4 expression patterns following cisplatin treatment, where Tlr4 expression remained relatively stable until sharply rising after 3.5 h (Fig 4C). The kinetics of cisplatin‐induced cytokine secretion in our experiments, coupled with our observations that cytokine gene expression preceded Tlr4 expression in response to cisplatin treatment, supports a model where cisplatin has a primary effect on Tlr4 activation.
Zebrafish homologs of TLR4 are required for cisplatin‐induced ototoxicity
Having shown that TLR4 played a critical role mediating cisplatin ototoxicity responses in vitro, we sought to examine the role of TLR4 in an in vivo CIO model. We chose to use zebrafish because it is a robust and widely accepted model of ototoxicity (Ton & Parng, 2005; Kari et al, 2007; Ou et al, 2007, 2010; Choi et al, 2013; Thomas et al, 2013; Uribe et al, 2013). Using established assays, we scored the health of neuromasts, which are mechanotransducing hair cells that bear structural, cellular, and physiological similarities to Organ of Corti outer hair cells (Coffin & Ramcharitar, 2016). Neuromast health can be visualized using the fluorescent dye DASPEI, which accumulates and stains viable hair cells. We used a dose–response format to establish a dose of cisplatin that robustly reduced hair cell viability in our hands (Fig 5A). A concentration of 15 µM was chosen for subsequent experiments because the dose–response curve indicated that this concentration yielded significant, but not total, loss of neuromast cell viability as determined through DASPEI staining. This concentration also yielded consistent results in morpholino experiments.
Figure 5. Zebrafish Tlr4 mediates cisplatin‐induced ototoxicity in vivo .
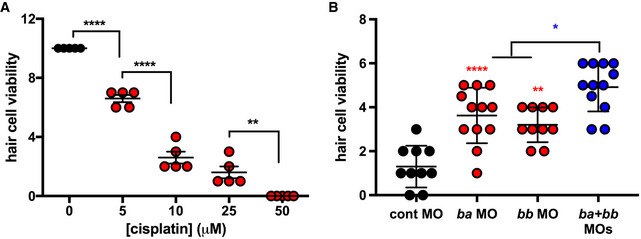
- Hair cell viability in larval zebrafish (assessed by DASPEI staining) following cisplatin treatment at the indicated concentrations.
- Hair cell viability in larval zebrafish pre‐treated with control‐, tlr4ba‐, and/or tlr4bb‐targeting morpholino oligonucleotides (MO) and subsequently treated with 15 µM cisplatin.
Data information: In both panels, each data point represents a score of hair cell integrity in an individual animal (taken from multiple samples per animal) with lines representing mean and standard deviation. Statistical comparisons between cisplatin concentrations (A) or to control morpholino (B, except as indicated in blue) were assessed by one‐way ANOVA. *P < 0.05; **P < 0.01; ****P < 0.0001 (Tukey test).
Source data are available online for this figure.
Zebrafish have two tlr4 genes, designated tlr4ba and tlr4bb that are orphan receptors. They are not activated by LPS but chimeric experiments show that they are linked to the NF‐κB signaling pathway (Sepulcre et al, 2009). Prior to bath application of cisplatin, we knocked down the tlr4ba homolog, the tlr4bb homolog, or both tlr4ba and tlr4bb homologs, using morpholinos that were previously validated thoroughly for specificity and efficacy (Sepulcre et al, 2009; He et al, 2015; Chang et al, 2016). Knockdown of either tlr4ba or tlr4bb was significantly protective against CIO (Fig 5B). Moreover, a protective effect against cisplatin‐induced neuromast toxicity was observed with two independent tlr4bb‐targeting morpholinos that disrupt gene splicing or translation (Fig EV4). Notably, we observed that protection from CIO could be further enhanced by combinatorial knockdown of both tlr4ba and tlr4bb, further supporting the role of zebrafish tlr4 in CIO (Fig 5B).
Figure EV4. Cisplatin‐induced neuromast toxicity is reduced in independent tlr4bb knockdowns.
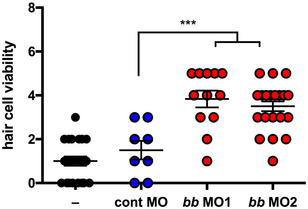
Hair cell viability in larval zebrafish pre‐treated with control‐, splice‐targeting tlr4bb (MO1) or translation‐targeting tlr4bb (MO2) morpholinos prior to treatment with 15 µM cisplatin.
Data information: Each data point represents a score of hair cell integrity in an individual animal (taken from multiple samples per animal) with lines representing mean and standard deviation. Statistical comparisons to control morpholino were assessed by one‐way ANOVA. ***P < 0.001 (Tukey test).
Source data are available online for this figure.
Chemical inhibition of TLR4 decreases cisplatin ototoxicity responses in vitro
Our data suggest TLR4 may be a druggable therapeutic target to mitigate CIO. We next sought to examine the effect of a TLR4 chemical inhibitor on cisplatin toxicity in HEI‐OC1 cells. We blocked the TLR4 signaling pathway in these cells by pre‐treating them with TAK‐242 or vehicle control prior to treatment with LPS or cisplatin and concurrent TAK‐242/vehicle treatment. We chose this inhibitor, rather than one that targets TLR4/MD‐2 interactions, because our findings indicated that TLR4 activation by cisplatin is independent of MD‐2. Analysis of secreted IL‐6 levels indicated that cells treated with TAK‐242 released significantly less IL‐6 protein in comparison to the vehicle control in response to both cisplatin and LPS agonists (Fig 6A). Similarly, ROS generation was significantly reduced in cells treated with cisplatin and TAK‐242 compared to cisplatin and vehicle treatments (Fig 6B). Overall, these results indicate that cisplatin ototoxic responses that underlie hearing loss can be mitigated using a chemical inhibitor of TLR4 in an ear outer hair cell line.
Figure 6. Small molecule inhibition of Tlr4 mitigates cisplatin‐induced ototoxic responses in HEI‐OC1 cells.
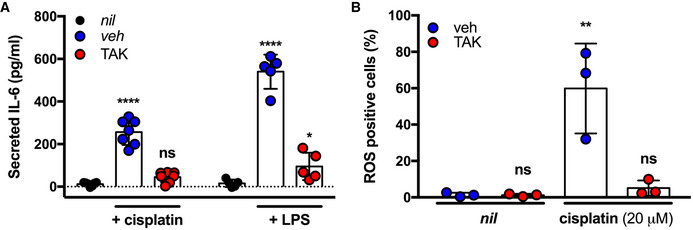
- IL‐6 secretion in HEI‐OC1 cells pre‐treated with DMF vehicle (veh), 4 µM TAK242 (TAK), or left untreated (nil) following treatment with 10 ng/ml LPS or 20 µM cisplatin (n = 5–7 independent biological replicates).
- ROS generation in HEI‐OC1 cells pre‐treated with DMF vehicle (veh), 4 µM TAK242 (TAK), or left untreated (nil) following treatment with 20 µM cisplatin (n = 3 independent biological replicates).
Data Information: In all panels, data are presented as mean and standard deviation. Statistical comparisons to nil treatment were assessed by 2‐way (A) or one‐way (B) ANOVA. *P < 0.05; **P < 0.01; ****P < 0.0001(Dunnett’s test).
Source data are available online for this figure.
Discussion
In this work, we have shown that TLR4 is a critical mediator of cisplatin‐induced ototoxic responses in an ear outer hair cell line and in zebrafish. This is the result of cisplatin activating TLR4 based on its structural similarity to platinum chloride. Moreover, we show for the first time that a small molecule inhibitor of TLR4 can mitigate cisplatin ototoxic responses in ear outer hair cells. Taken together, this work sets the stage to focus on TLR4 as a therapeutic target for the mitigation of CIO and establishes appropriate model systems for these preclinical efforts.
ROS generation and apoptosis induction in outer hair cells are considered the basis of how cisplatin kills these critical mechanotransducing cells; however, it is poorly understood how these responses are elicited. Previous studies have identified key roles for the TNFα and NF‐κB inflammatory signaling pathways in transducing cisplatin ototoxic responses but the upstream processes were less defined (So et al, 2007; Chung et al, 2008). Our work now identifies TLR4 as a bridge between cisplatin and these signaling pathways since TLR4 activation can induce NF‐κB signaling and TNFα secretion (Smolinska et al, 2011; Kawasaki & Kawai, 2014). Interestingly, some reports have demonstrated the activation of TLR4 as one of the main pathways causing ototoxicity by aminoglycoside treatment and cochlear inflammation after acoustic injury (Hirose et al, 2014; Koo et al, 2015; Vethanayagam et al, 2016). The specific ligands that activate TLR4 in these conditions are not defined; however, in general, each of these types of damage increases Tlr4 expression levels in the cochlea within hours (Hirose et al, 2014; Vethanayagam et al, 2016).
It has been reported that a Tlr4 deletion in C3H/HeJ mice partially mitigated cisplatin‐induced nephrotoxicity (Cenedeze et al, 2007). This work was interpreted to suggest, but did not confirm, that damage‐associated molecular patterns activate TLR4 in C3H/HeJ mice and cause cisplatin‐induced renal toxicity (Cenedeze et al, 2007). Others have reported that LPS acts coordinately with cisplatin to increase inflammatory responses and cellular damage in renal and cochlear cells (Ramesh et al, 2007; Oh et al, 2011). Specifically, Oh et al suggested that cisplatin plays a secondary role in TLR4 activation by upregulating the expression of Tlr4 for subsequent activation by LPS (Oh et al, 2011). Our study contributes to a new understanding of the association of cisplatin and TLR4 in the induction of CIO. Our data clearly indicate that cisplatin can activate TLR4 in vitro and does so in a manner that is mechanistically disparate from LPS. We observed that TLR4 activation by cisplatin in an ear outer hair cell line (assessed by cytokine secretion) occurred with similar kinetics to TLR4 activation by LPS. Furthermore, we noted that cisplatin treatment induced Tlr4 expression at later time points than LPS. Taken together, our data argue that cisplatin has a primary effect on TLR4 activation, similar to the TLR4 agonists LPS and nickel.
While highly structurally distinct from LPS, metal contact allergens have been shown to signal through direct TLR4 interactions (Schmidt et al, 2010; Rachmawati et al, 2013). Our results suggest that platinum chloride and cisplatin are also capable of activating TLR4. Given that platinum is also a Group 10 transition metal, we speculated that platinum and cisplatin may activate TLR4 in a manner analogous to nickel, rather than LPS. Our in vitro analyses showed that LPS and HMGB1 required MD‐2 for significant TLR4 activation, which is consistent with the literature (Kawai & Akira, 2006; Yang et al, 2015). By contrast, cisplatin, platinum(II), and platinum(IV) were able to activate TLR4 signaling in the absence of the TLR4 co‐receptor, MD‐2. This is notable because it has been reported that this co‐receptor is required for effective TLR4 activation by nickel, suggesting possible functional differences within the Group 10 metals in their capacity to activate TLR4 (Raghavan et al, 2012; Oblak et al, 2015). Nickel is proposed to form critical interactions with TLR4 histidine residues (456 and 458) on the ectodomain of human TLR4 that facilitate the dimerization of TLR4 complexes and subsequent signaling (Schmidt et al, 2010; Raghavan et al, 2012). The role of these residues in cisplatin activation of TLR4 remains to be studied and could help glean information on whether cisplatin behaves as a direct ligand of TLR4 by analogy to nickel. While our kinetic analyses of cisplatin responses in HEI‐OC1 cells strongly suggest that cisplatin has a primary effect on TLR4 activation, it does not confirm a direct interaction between cisplatin and TLR4 and further study is required to establish this.
Zebrafish have proved to be a robust model system for studying cisplatin‐mediated hair cell death by monitoring neuromast viability (Ou et al, 2007; Eimon & Rubinstein, 2009; Chowdhury et al, 2018). Moreover, this model has been used to investigate potential otoprotective therapies (Ton & Parng, 2005; Kim et al, 2008; Coffin et al, 2010; Choi et al, 2013; Hong et al, 2013; Lee et al, 2015; Thomas et al, 2015; Pang et al, 2018; Rocha‐Sanchez et al, 2018). It is, however, well recognized that zebrafish tlr4ba/bb are distinct from mammalian TLR4. TLR4 homologs appear to have been lost from the genomes of many fish species, suggesting a very disparate role for TLR4 compared to its centrality in mammalian responses to LPS endotoxin. Indeed, zebrafish TLR4 homologs appear to be unresponsive to LPS, which has been attributed to a lack of an MD‐2 ortholog in zebrafish (Sepulcre et al, 2009). Nevertheless, chimeric mammalian TLR4 and zebrafish tlr4ba/bb constructs studied in vitro showed that the intracellular domains of the zebrafish proteins could interact with downstream signaling components in the TLR4 pathway (Sepulcre et al, 2009). Our finding that zebrafish TLR4 homologs are required for CIO is consistent with their expression in zebrafish hair cells (Barta et al, 2018; Lush et al, 2019) and is in line with the in vitro and murine inner ear cell studies presented in this work. This further supports the identification of TLR4 as a key mediator of cisplatin‐induced ototoxicity. These results suggest that zebrafish can be an important model for screening TLR4 antagonists as putative otoprotectants against CIO. They also raise the intriguing possibility that zebrafish tlr4ba/bb, heretofore orphan receptors with no known agonist, could be sensors of Group 10 transition metals.
Recently, Breglio et al reported that sensory hair cells could be protected from aminoglycoside‐induced ototoxicity through exosome‐mediated activation of TLR4 by the DAMP, HSP70 (Breglio et al, 2020). This work, and our data, positions TLR4 as a critical mediator of ototoxicity. Here we show that TLR4 contributes to cisplatin‐induced ototoxicity, while Breglio et al studied aminoglycoside‐induced ototoxicity. It is notable that in contrast to cisplatin, as we report here, aminoglycosides are not known to activate TLR4 as a primary event. Moreover, Breglio et al reported that the TLR4‐activating DAMP, HSP70, required an exosomal context to mediate otoprotection through TLR4, which raises the possibility that other factors in the exosome may modulate TLR4 responses. In their report, Bregio et al did not characterize the signaling event downstream of TLR4 that correspond to HSP70‐mediated protection from aminoglycoside‐induced ototoxicity. However, it is known that TLR4 can activate both pro‐inflammatory signaling (NF‐κb, MAPK) and anti‐inflammatory signaling (PI3K, AKT) (Siegemund & Sauer, 2012).
In summary, our data argue that cisplatin plays a primary role in activating TLR4, independently of LPS, suggesting that TLR4 is a critical mediator of CIO. This is reinforced by our observation that genetic or chemical inhibition of Tlr4 in an ear outer hair cell line or in zebrafish significantly reduced cisplatin toxicity. To our knowledge, our TAK242 data represent the first demonstration that a small molecule inhibitor of TLR4 can mitigate cisplatin toxicity. Given that cisplatin activation of TLR4 is distinct from LPS, this affords an opportunity to develop tailored therapies that specifically target cisplatin activation of TLR4. The findings in this study bring us closer to understanding the mechanisms involved in CIO, with TLR4 as a primary target for the rational design of otoprotectants, and bettering health outcomes for cancer patients while conserving the success of cisplatin chemotherapy.
Materials and Methods
Cell culture and treatments
The murine inner ear cell line HEI‐OC1 cells (a kind gift from Dr. Federico Kalinec, UCLA) were grown in DMEM supplemented with 10% FBS (Gibco, 123483‐020) and 5% penicillin–streptomycin (1 unit penicillin/ml and 0.1 mg streptomycin/ml, Sigma, P4333). HEI‐OC1 cells were grown at 33°C in the presence of 10% CO2. HEK (human embryonic kidney)‐hTLR4 and HEK‐null2 cell lines (cat# hkb‐htlr4 and hkb‐null2, Invivogen) are isogenic reporter cell lines stably transfected with a secreted alkaline phosphatase reporter under the control of five tandem NF‐κB response elements. HEK‐hTLR4 cells also stably express human TLR4 and MD‐2. HEK‐isoTLR4 cells stably express TLR4 but not its co‐receptor MD‐2 (cat# 293‐htlr4a; Invivogen). These cells were grown in DMEM supplemented with 10% FBS, 5% penicillin–streptomycin, and 100 µg/ml Normocin (Invivogen) at 37°C and 5% CO2. Cells were routinely seeded in 96‐well plates (5 × 103 cells/well), 24‐well plates (7.0 × 104 cells/well), 12‐well plates (1.1 × 105 cells/well), or 6‐well plates (1.5–2.5 × 105 cells/well). HeLa cells (ATCC CCL‐2) were grown in DMEM supplemented with 10% FBS (Gibco, 123483‐020) and 5% penicillin–streptomycin (1 unit penicillin/ml and 0.1 mg streptomycin/ml, Sigma, P4333) and routinely grown at 37°C and 5% CO2. Cisplatin (Teva, 02402188), LPS (Invitrogen, L23351), nickel chloride hexahydrate (Sigma, 654507), platinum (II) chloride (Sigma, 520632), or platinum (IV) chloride (Sigma, 379840) was added to cells 48 h after seeding in fresh media. Vehicle (DMF; Fisher Scientific, D1331) or TAK242 (Cayman, 243984‐11‐4) was added to cell culture in fresh media 1 h prior to treatments. Following 1 h pre‐treatment, media was aspirated and vehicle or TAK242 were added to cells in combination with cisplatin, LPS, or nickel treatments for 24 or 48 h. All reagents were assessed for endotoxin contamination ˃ 0.125 ԐU using Pyrotell Gel Clot Formulation kit for bacterial endotoxin testing (Pyrotell, GS125‐5). LPS and low endotoxin water (˂ 0.005 ԐU; HyClone, SH30529.02) were used as positive and negative controls, respectively. All TLR4 agonists (except LPS) tested negative for endotoxin.
CRISPR‐Cas9 mediated Tlr4 knock out
Mouse Tlr4 was targeted for mutation in HEI‐OC1 cells using TrueCutTM Cas9 Protein V2 (Invitrogen, A36498), TrueGuideTM tracrRNA (Invitrogen, A35507), and TrueGuideTM Syn crRNA (Invitrogen, A35509‐CRISPR511653) and gRNA (Target: GATTCAAGCTTCCTGGTGTC). TrueGuide™ Syn crRNA, Negative Control, (Invitrogen, A35519) was used as a non‐targeting crRNA in this assay. Gene editing efficiency was verified using the GeneArtTM. Genomic Cleavage Detection kit (Invitrogen, A24372) in a pooled cell population. Procedures were carried out based on the manufacturer’s protocols. Single‐cell clones were then isolated for further validation using limited dilution in 96‐well plates. Tlr4 deletion clones were then screened for loss of LPS‐induced IL‐6 cytokine secretion. Genomic DNA from selected clones was amplified at the Tlr4 locus and analyzed by Sanger Sequencing using primer pair: 5′‐CCTCCAGTCGGTCAGCAAAC‐3′ and 5′‐CTAAGCAGAGCACACACAGGG‐3′.
Immunocytochemistry
Immunofluorescence staining was used to examine levels of TLR4 protein from control and Tlr4‐deleted HEI‐OC1 cells. Cells were seeded in a 96‐well plate and fixed after 24 h in 4% paraformaldehyde for 20 min at room temperature. After washing 3X with PBS, blocking was performed using 3% BSA for 1 h. Cells were then stained using mouse anti‐TLR4 primary antibody (Invitrogen, 13‐9041‐80) and Alexa Fluor 488 Goat anti‐mouse IgG secondary antibody (Jackson ImmunoResearch; 115‐545‐146). Antibodies were used at 1:200 dilution. Finally, secondary antibody was removed by washing with PBS 3X and PBS was added to the wells. Images were acquired using an Evos FL Auto microscope and manufacturer’s software.
Cell viability assays
MTT reagent (ACROS, 158990010) was added to 1 mg/ml to seeded cells, 24 or 48 h post‐treatment. When required, aliquots of the supernatant were collected for ELISAs before the addition of MTT. Plates were incubated at 33°C at 10% CO2 (HEI‐OC1) or 37°C at 5% CO2 (HEK) for 4 h in the dark. Next, supernatants were replaced with DMSO (Sigma, D109) and incubated with shaking at room temperature for 20 min. Absorbance at 590 nm was collected in a plate reader (SpectraMax i3x, Molecular Devices). For the purposes of cell viability dose–response curves, the mean absorbance for a no‐treatment control was considered 100% cell viability so that cell viability (%) of treatment = (absorbancetreatment/absorbancecontrol) × 100.
NF‐κB activation assays
To measure TLR4 activation in the HEK‐null2 and HEK‐hTLR4 cells, an integrated NF‐κB reporter system was used. The reporter is a secreted alkaline phosphatase enzyme that is transcriptionally regulated by NF‐κB. The secreted alkaline phosphatase assay was performed according to the manufacturer’s instructions with slight modifications. Cells were cultured in DMEM containing 10% FBS, 1% penicillin/streptomycin, and 0.1 mg/ml Normocin (ant‐nr‐2, Invivogen). HEK‐Blue selection (hb‐sel Invivogen) and zeocin (ant‐zn‐05 Invivogen) were applied to HEK‐hTLR4 and HEK‐null2 cells, respectively, every 5 passages to maintain stable cell transfection. Cells were seeded in a 96‐well dish at 1.4 × 105 cells/ml (HEK‐hTLR4) and 2.8 × 105 cells/ml (HEK‐null2) in HEK detection media (Cat# hb‐det3 Invivogen) as per manufacturer’s protocol. Cells were treated upon seeding with media, DMF (vehicle for platinum (II) chloride) or TLR4 agonists. Alkaline phosphatase activity was measured by reading absorbance at 620 nm after 36 h of stimulation (SpectraMax i3x reader, Molecular Devices).
ELISAs
As an alternate method of assessing TLR4 activation, IL‐6 secretion was quantified in HEI‐OC1 because this cytokine is a key mediator of cisplatin toxicity in HEI‐OC1 cells (So et al, 2007). IL‐8 secretion was previously reported as marker of TLR4 activation in HEK‐hTLR4 cells and was chosen for our experiments using related cell lines (Schmidt et al, 2010). In addition, both IL‐6 and IL‐8 have been reported to be upregulated by cisplatin in human cells (Kiss et al, 2020), while mice do not contain a true gene ortholog for IL8 precluding its direct characterization. Colorimetric protein assays were conducted using commercial human IL‐8 ELISA and mouse IL‐6 ELISA kits (Invitrogen; 88‐8086, 88‐7064) according to the manufacturer’s protocol. Supernatants were collected from 12‐well plates or 24‐well plates 0, 0.25, 0.5, 1, 2, 3, 24, or 48 h post‐treatment (leaving half the volume in the well for subsequent MTT assays). Protein secretion was normalized to the number of viable cells to account for agonist toxicity.
For experiments in HeLa cells, 4 × 105 cells were seeded in each well of a 6‐well dish and grown for 2 days prior to agonist treatment. Supernatants were collected 48 h post‐treatment and analyzed. For experiments involving siRNA knockdown of TLR4, cells were seeded at 3 × 105 cells per well in 6‐well dishes and treated with siRNA after 24 h of growth. Transfected cells were treated with agonists after 24 h and supernatants collected 72 h post‐agonist treatment.
Western Blot Analysis
HEI‐OC1 cells were treated with 20 µM cisplatin for 10, 20, or 30 min or 24 h and washed with ice‐cold PBS. 10 ng/ml LPS treatment for 10 min was used as a control, and cells were processed identically. Total cell proteins were extracted using NP40 Cell Lysis Buffer supplemented with EDTA‐free protease inhibitor and sodium orthovanadate. 20 µg of protein lysate was separated using a 10% Bio‐Rad TGX Gel System (Cat #4561034), transferred to nitrocellulose membranes (Bio‐Rad), and blocked with LI‐COR Intercept (PBS) Blocking Buffer for 30 min at room temperature. Subsequently, membranes were incubated with anti‐IRF3 (4302S), anti‐P‐IRF3 (4947S), anti‐NF‐ĸB P65 (8242S), anti‐P‐NF‐ĸB P65 (3033L), anti‐p42/44 (9102S), anti‐P‐p42/44 (9101S), or anti‐beta‐Actin (3700S) antibodies from Cell Signaling Technology overnight at 4◦C. After washing with TBST, membranes were incubated with IRDye 680RD Goat anti‐Rabbit IgG secondary antibody (LI‐COR Bioscience, 92568071) or IRDye 680RD Goat anti‐mouse IgG secondary antibody (LI‐COR Bioscience, 92668020) for 1 h at room temperature. Membranes were washed with TBST and then scanned with an Odyssey Infrared Imaging System (LI‐COR Bioscience).
Luciferase reporter assay
HEI‐OC1 cells (1 × 105) were seeded in a 24‐well plate. Cell density was about 70% on the day of the cell transfection with the IRF3 luciferase reporter construct, p55‐CIB‐Luc. The IRF3 reporter was co‐transfected along with a Renilla luciferase construct (kindly provided by Tom Hobman, University of Alberta) as a constitutive expression reporter. Cells were then stimulated with 20 µM cisplatin for 24 h. Next, cells were washed with PBS and lysed with 250 µl Luciferase Lysis Buffer per well (10% glycerol, 25 mM glycylglycine, 4 mM EGTA, 15 mM MgSO4, 0.1% Triton X‐100, and freshly added 1 µM dithiothreitol (DTT)). Cell lysates were stored at −20°C up to a week. On the day of the experiment, samples were thawed and 50 µl of cell lysates were aliquoted in duplicate in a white 96‐well plate for Renilla and firefly luciferase assays. We used D‐luciferin (Gold Biotechnology, USA, at the final concentration of 70 µM) and the coelenterazine (Gold Biotechnology, USA, at the final concentration of 1.4 µM) as the firefly and Renilla luciferase substrates, respectively, prepared according to the manufacturer’s specifications. 100 µl of substrates were added sequentially to cell lysates in each well and incubated 5 min. in the absence of light. Luciferase activity was measured on a multimodal plate reader.
LPS internalization assay
We assessed the extent of LPS internalization to characterize our HEI‐OC1 Tlr4 deletion cell line compared to the non‐targeting control cells. Tlr4‐deletion and control HEI‐OC1 cells were grown up to 90% confluence in 6‐well plates in complete DMEM. Cells were then treated with 5 μg/ml ultrapure Alexa Fluor™ 488 LPS (Life Technologies, L23351) or with non‐fluorescent LPS (eBioscience™, 00‐4976‐93) as a control for 4 h. Cellular LPS internalization was assayed via flow cytometry after quenching with 1 mg/ml Trypan blue. 50,000 cells were read and gated by forward and side scatter for selecting live cells and then single cells. Cellular uptake of Alexa Fluor™ 488 LPS was shown based on the median fluorescence intensity (MFI) at 488 nm excitation wavelength and 525 nm emission wavelength. Samples treated with non‐fluorescent LPS were used to subtract background auto‐fluorescence. Data analysis was performed using FlowJo_V10 software.
Apoptosis assay
We monitored apoptosis induction as a hallmark response of cisplatin treatment in vitro. HEI‐OC1 cells were treated with increasing concentrations of cisplatin for 24 h. After 24 h, cells were harvested and washed once with PBS and once with 1× Annexin V Binding Buffer (Thermo Fisher Scientific, V13246). Following washes, cells were re‐suspended in 100 μl Annexin V Binding Buffer. Cells were then stained following the manufacturers’ protocol. In brief, 5 μl of FITC‐Annexin V and 1 μl of diluted propidium iodide solution (100 μg/ml working solution) were added to each sample. Samples were then incubated at room temperature in the dark for 15 min. Samples were diluted with 400 μl Annexin V Binding Buffer and acquired on an Attune NxT flow cytometer (Thermo Fisher Scientific). A minimum of 10,000 events were acquired in each sample. Following the acquisition, samples were analyzed using FlowJo (BD Biosciences).
ROS detection assay
We monitored ROS generation as a hallmark response of cisplatin treatment in vitro using the Total ROS‐ID detection kit (Enzo Life Sciences, ENZ‐51011). 5 × 103 HEI‐OC1, Tlr4‐deleted HEI‐OC1, or control cells were seeded in 96‐well plates. Cell density was approximately 70–80% on the day of the cisplatin (20 µM) treatment. Cells were then stained following the manufacturers’ protocol. In brief, supernatants were removed and cells were washed with 1X ROS Wash Buffer. Cells were then stained with 100 µl/well of ROS Detection Solution for 60 min at 37°C in the dark. No washing was required prior to sample analysis using the SpectraMax i3x fluorescence plate reader. Excitation and emission were monitored at 488 and 520 nm, respectively.
For total ROS measurements of HEI‐OC1 cells in response to TAK242 treatment, cells were pre‐treated with 4 µM TAK242 or DMF for 1 h before cisplatin stimulation for 24 h. One million cells were trypsinized and then washed with 1× ROS Wash Buffer. Cells were then stained following the manufacturers’ protocol. In brief, cells were re‐suspended in 500 µl of the ROS Detection Solution for 30 min. No washing was required prior to sample analysis using an Attune NxT flow cytometer (Thermo Fisher Scientific). A minimum of 10,000 events were acquired for each sample. Following acquisition, samples were analyzed using FlowJo (BD Biosciences).
Cell transfections
HEK‐isoTLR4 cells were transfected with a human MD‐2 expression clone (OriGene; RC204686) to assess cytokine secretion in response to TLR4 agonist treatments. HEI‐OC1 cells were transfected with a mouse Tlr4 expression clone (OriGene; MR210887) to test for complementation of the Tlr4 deletion strain. Transfections of HEK cells were carried out with Lipofectamine 3000 (Invitrogen, L30000 IS) and of HEI‐OC1 cells with jetPRIME (Polyplus, CA89129‐924) reagents in 24‐well plates, with 0.5 μg of DNA according to the manufacturer’s specification. Cells were treated 24 h post‐transfection with cisplatin, LPS, or nickel.
qPCR assay
HEI‐OC1 cells were seeded in 6‐well plates with 250,000 cells/well. Cells were allowed to grow for 48 h to achieve a 70% confluency prior to treatment with cisplatin. Cells were all treated simultaneously. RNA extraction (Bio‐Rad Aurum Total RNA Mini Kit) was performed at the appropriate time points after cisplatin addition (0, 1, 2, 3, 4, and 24 h post‐treatment). cDNA synthesis was performed using the iScript gDNA‐Clear cDNA Synthesis kit (Bio‐Rad, 1725034). Final qPCR was performed using TLR4 (qMmuCIP0035732) and HPRT (qMmuCEP0054164) primer‐probe assays obtained from Bio‐Rad and the associated SSO Advanced Universal Probe Super Mix kit (Bio‐Rad 1725281).
siRNA gene knockdown
Cells were seeded in 6‐well plates with 150,000 cells/well. Cells were allowed to grow for 24 h to achieve 50–60% confluency and were transfected with 5 nM of non‐targeting/negative control siRNA or the appropriate TLR4 siRNA (hs. Ri.TLR4.13) using a dsiRNA TriFECTa Kit from Integrated DNA Technologies in conjunction with RNAiMAX transfection reagent (Thermo Fisher). Cells were allowed to grow for another 24 h following transfection prior to treatment with cisplatin. Exposure to cisplatin continued for 72 h prior to the collection of supernatant detection of secreted IL‐8 by ELISA and the completion of MTT cell viability assays for normalization as previously indicated.
Animal ethics and zebrafish husbandry
Zebrafish were kept at the University of Alberta following a 14:10 light/dark cycle at 28°C cycle as previously described (Westerfield, 2000). They were raised, bred, and maintained following an institutional Animal Care and Use Committee approved protocol AUP00000077, operating under guidelines set by the Canadian Council of Animal Care.
Assessing CIO in larval zebrafish
Wild‐type (AB strain) zebrafish were grown to 5 days post‐fertilization (dpf) in standard E3 embryo media (Westerfield, 2000) and were bath‐treated with either 0, 5, 10, 25, or 50 μM of cisplatin in 6‐well plates, with 10–15 zebrafish larvae per well. After a 20‐h incubation with cisplatin at 28°C, wells were washed with embryo media before the fish were incubated in media containing 0.01% 2‐[4‐(dimethylamino) styryl]‐1‐ethylpyridinium iodide (DASPEI, Sigma‐Aldrich) to stain for neuromast mitochondrial activity for 20 min. Wells were washed again in embryo media and zebrafish larvae anesthetized with 4% tricaine. Neuromasts were imaged under a Leica M165 FC dissecting microscope equipped with a fluorescent filter. A standard scoring method for zebrafish hair cell viability was used (Chowdhury et al, 2018): Five posterior lateral line (PLL) neuromasts for each fish were assigned a score representing cell viability based on DASPEI fluorescent intensity (2 for no noticeable decline, 1.5 for minor decline, 1 for moderate decline, 0.5 for severe decline, and 0 for complete loss of fluorescent intensity). These five scores were summed for each individual (10 = all hair cells appear normal and viable; 0 = intense ototoxicity).
Morpholino knockdown of TLR4 homologs
Previously validated anti‐sense knockdown reagents (Morpholinos (Sepulcre et al, 2009; He et al, 2015; Chang et al, 2016)) against tlr4ba and tlr4bb (Gene Tools, LLC; Philomath, OR) were delivered to developing zebrafish. Two tlr4bb morpholinos were used, the first translation blocking: trl4bb‐MO1 (5′‐AATCATCCGTTCCCCATTTGACATG‐3′) and the second splice blocking: tlr4bb‐MO2 (5′‐CTATGTAATGTTCTTACCTCGGTAC‐3′). A splice blocking tlr4ba‐MO2 (5′‐GTAATGGCATTACTTACCTTGACAG‐3′) was also used. All gene‐specific morpholinos have been previously described and thoroughly vetted for efficacy and specificity to the gene target (Sepulcre et al, 2009; He et al, 2015; Chang et al, 2016). A standard control morpholino (5′‐CCTCTTACCTCAGTTACAATTTATA‐3′) was used as a negative control. Injection solution for morpholinos consisted of 0.1 M KCl, 0.25% dextran red, either the standard control or gene‐specific morpholinos to effective dose and nuclease‐free water. One‐cell stage newly fertilized embryos were positioned on an agarose plate and injected with 5 ng of morpholino. At 2dpf, gene‐specific morpholino injected fish, control morpholino injected fish, and un‐injected fish were added to separate wells of a 6‐well plate with 10–15 fish per well. Fish were incubated with 15 μM cisplatin for 20‐h before being washed, DASPEI stained, imaged, and analyzed as described above.
Statistical analyses
TLR4 activation across multiple cell lines was analyzed by 2‐way ANOVA with Bonferroni multiple comparison test between samples and Dunnett’s multiple comparison test against a control sample (nil or vehicle). TLR4 activation in a single cell line was analyzed by one‐way ANOVA using Dunnett’s multiple comparison test to a control sample (nil or siNT). Cisplatin responses tested in HEI‐OC1 were analyzed by 2‐way ANOVA at multiple concentrations, or one‐way ANOVA at a single concentration of cisplatin, using Bonferroni multiple comparison test. Time course experiments in HEI‐OC1 cells were analyzed by 2‐way ANOVA with Dunnett’s multiple comparison test. Neuromast scores were analyzed via one‐way ANOVA with Tukey’s multiple comparison test. All statistical analyses were performed using Prism 7.2.
Author contributions
Conceptualization: GB, AL, NMP, IKD, CD, AMR, WTA, APB; Investigation: GB, AL, NMP, IKD, CD, AMR; Formal Analysis: GB, AL, NMP, IKD, CD, AMR, WTA, APB; Funding acquisition and Supervision: WTA, APB; Writing – original draft: GB, AL, NMP, AMR, APB; Writing – review & editing: all authors.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Acknowledgements
This work was supported by operating grants to APB from the Canadian Institutes of Health Research (MY2‐155361) and Hair Massacure (FN 2489) and to WTA from the Natural Sciences and Engineering Research Council of Canada (RGPIN‐2019‐04825). This research has also been funded by the Li Ka Shing Institute of Virology (LKSIoV) (LKSIOVSF) and the generous support of the Stollery Children’s Hospital Foundation through the Women and Children’s Health Research Institute (WCHRI) (WCHRIRRG 1958). GB was supported by studentships from the Kids with Cancer Society and WCHRI (2631). AL was supported by studentships from the Alberta Cancer Foundation (27176) and WCHRI (2749). CD was supported by a studentship from the LKSIoV. APB holds a Canada Research Chair (Tier 2) in Functional Genomic Medicine and this research was undertaken, in part, thanks to funding from the Canada Research Chairs Program (231622). AMR is an ISAC SRL Emerging Leader. Some experiments were performed at the University of Alberta Faculty of Medicine & Dentistry Flow Cytometry Facility, which receives financial support from the Faculty of Medicine & Dentistry and Canada Foundation for Innovation (CFI) awards to contributing investigators. We thank Ray Ishida and Tom Hobman (University of Alberta) for technical assistance and reagents for the luciferase assays. We thank Maria Febbraio, Shayla Mosley, and Kristi Baker (University of Alberta) for technical assistance and reagents for the phospho‐Western blotting.
EMBO reports (2021) 22: e51280.
Data availability
This study includes no data deposited in external repositories.
References
- American Childhood Cancer Organization (2017) US Childhood Cancer Statistics
- Barta CL, Liu H, Chen L, Giffen KP, Li Y, Kramer KL, Beisel KW, He DZ (2018) RNA‐seq transcriptomic analysis of adult zebrafish inner ear hair cells. Sci Data 5: 180005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blakley BW, Myers SF (1993) Patterns of hearing loss resulting from cis‐platinum therapy. Otolaryngol Head Neck Surg 109: 385–391 [DOI] [PubMed] [Google Scholar]
- Breglio AM, May LA, Barzik M, Welsh NC, Francis SP, Costain TQ, Wang L, Anderson DE, Petralia RS, Wang YX et al (2020) Exosomes mediate sensory hair cell protection in the inner ear. J Clin Invest 130: 2657–2672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breglio AM, Rusheen AE, Shide ED, Fernandez KA, Spielbauer KK, McLachlin KM, Hall MD, Amable L, Cunningham LL (2017) Cisplatin is retained in the cochlea indefinitely following chemotherapy. Nat Commun 8: 1654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brock PR, Bellman SC, Yeomans EC, Pinkerton CR, Pritchard J (1991) Cisplatin ototoxicity in children: a practical grading system. Med Pediatr Oncol 19: 295–300 [DOI] [PubMed] [Google Scholar]
- Brock PR, Knight KR, Freyer DR, Campbell KC, Steyger PS, Blakley BW, Rassekh SR, Chang KW, Fligor BJ, Rajput K et al (2012) Platinum‐induced ototoxicity in children: a consensus review on mechanisms, predisposition, and protection, including a new International Society of Pediatric Oncology Boston ototoxicity scale. J Clin Oncol 30: 2408–2417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cenedeze MA, Goncalves GM, Feitoza CQ, Wang PM, Damiao MJ, Bertocchi AP, Pacheco‐Silva A, Camara NO (2007) The role of toll‐like receptor 4 in cisplatin‐induced renal injury. Transpl Proc 39: 409–411 [DOI] [PubMed] [Google Scholar]
- Chang KW, Chinosornvatana N (2010) Practical grading system for evaluating cisplatin ototoxicity in children. J Clin Oncol 28: 1788–1795 [DOI] [PubMed] [Google Scholar]
- Chang MY, Cheng YC, Hsu SH, Ma TL, Chou LF, Hsu HH, Tian YC, Chen YC, Sun YJ, Hung CC et al (2016) Leptospiral outer membrane protein LipL32 induces inflammation and kidney injury in zebrafish larvae. Sci Rep 6: 27838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi J, Im GJ, Chang J, Chae SW, Lee SH, Kwon SY, Chung AY, Park HC, Jung HH (2013) Protective effects of apocynin on cisplatin‐induced ototoxicity in an auditory cell line and in zebrafish. J Appl Toxicol 33: 125–133 [DOI] [PubMed] [Google Scholar]
- Chowdhury S, Owens KN, Herr RJ, Jiang Q, Chen X, Johnson G, Groppi VE, Raible DW, Rubel EW, Simon JA (2018) Phenotypic optimization of urea‐thiophene carboxamides to yield potent, well tolerated, and orally active protective agents against aminoglycoside‐induced hearing loss. J Med Chem 61: 84–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung WH, Boo SH, Chung MK, Lee HS, Cho YS, Hong SH (2008) Proapoptotic effects of NF‐kappaB on cisplatin‐induced cell death in auditory cell line. Acta Otolaryngol 128: 1063–1070 [DOI] [PubMed] [Google Scholar]
- Clerici WJ, Hensley K, DiMartino DL, Butterfield DA (1996) Direct detection of ototoxicant‐induced reactive oxygen species generation in cochlear explants. Hear Res 98: 116–124 [DOI] [PubMed] [Google Scholar]
- Coffin AB, Ou H, Owens KN, Santos F, Simon JA, Rubel EW, Raible DW (2010) Chemical screening for hair cell loss and protection in the zebrafish lateral line. Zebrafish 7: 3–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coffin AB, Ramcharitar J (2016) Chemical ototoxicity of the fish inner ear and lateral line. In: Fish hearing and bioacoustics: an anthology in Honor of Arthur N Popper and Richard R Fay, Sisneros JA (ed.) pp. 419–437. Cham: Springer International Publishing; [DOI] [PubMed] [Google Scholar]
- Dasari S, Tchounwou PB (2014) Cisplatin in cancer therapy: molecular mechanisms of action. Eur J Pharmacol 740: 364–378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eimon PM, Rubinstein AL (2009) The use of in vivo zebrafish assays in drug toxicity screening. Expert Opin Drug Metab Toxicol 5: 393–401 [DOI] [PubMed] [Google Scholar]
- Gaikwad S, Patel D, Agrawal‐Rajput R (2017) CD40 negatively regulates ATP‐TLR4‐activated inflammasome in microglia. Cell Mol Neurobiol 37: 351–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurney JG, Krull KR, Kadan‐Lottick N, Nicholson HS, Nathan PC, Zebrack B, Tersak JM, Ness KK (2009) Social outcomes in the Childhood Cancer Survivor Study cohort. J Clin Oncol 27: 2390–2395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurney JG, Tersak JM, Ness KK, Landier W, Matthay KK, Schmidt ML, Children's Oncology G (2007) Hearing loss, quality of life, and academic problems in long‐term neuroblastoma survivors: a report from the Children's Oncology Group. Pediatrics 120: e1229–1236 [DOI] [PubMed] [Google Scholar]
- He Q, Zhang C, Wang L, Zhang P, Ma D, Lv J, Liu F (2015) Inflammatory signaling regulates hematopoietic stem and progenitor cell emergence in vertebrates. Blood 125: 1098–1106 [DOI] [PubMed] [Google Scholar]
- Hirose K, Li S‐ZZ, Ohlemiller KK, Ransohoff RM (2014) Systemic lipopolysaccharide induces cochlear inflammation and exacerbates the synergistic ototoxicity of kanamycin and furosemide. J Assoc Res Otolaryngol 15: 555–570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong SJ, Im GJ, Chang J, Chae SW, Lee SH, Kwon SY, Jung HH, Chung AY, Park HC, Choi J (2013) Protective effects of edaravone against cisplatin‐induced hair cell damage in zebrafish. Int J Pediatr Otorhinolaryngol 77: 1025–1031 [DOI] [PubMed] [Google Scholar]
- Kalinec G, Thein P, Park C, Kalinec F (2016) HEI‐OC1 cells as a model for investigating drug cytotoxicity. Hear Res 335: 105–117 [DOI] [PubMed] [Google Scholar]
- Kari G, Rodeck U, Dicker AP (2007) Zebrafish: an emerging model system for human disease and drug discovery. Clin Pharmacol Ther 82: 70–80 [DOI] [PubMed] [Google Scholar]
- Kawai T, Akira S (2006) TLR signaling. Cell Death Differ 13: 816–825 [DOI] [PubMed] [Google Scholar]
- Kawasaki T, Kawai T (2014) Toll‐like receptor signaling pathways. Front Immunol 5: 461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim CH, Kang SU, Pyun J, Lee MH, Hwang HS, Lee H (2008) Epicatechin protects auditory cells against cisplatin‐induced death. Apoptosis 13: 1184–1194 [DOI] [PubMed] [Google Scholar]
- Kim H‐JJ, Lee J‐HH, Kim S‐JJ, Oh GS, Moon H‐DD, Kwon K‐BB, Park C, Park BH, Lee H‐KK, Chung S‐YY et al (2010) Roles of NADPH oxidases in cisplatin‐induced reactive oxygen species generation and ototoxicity. J Neurosci 30: 3933–3946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiss E, Abdelwahab E, Steib A, Papp E, Torok Z, Jakab L, Smuk G, Sarosi V, Pongracz JE (2020) Cisplatin treatment induced interleukin 6 and 8 production alters lung adenocarcinoma cell migration in an oncogenic mutation dependent manner. Respir Res 21: 120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koo JW, Quintanilla‐Dieck L, Jiang M, Liu J, Urdang ZD, Allensworth JJ, Cross CP, Li H, Steyger PS (2015) Endotoxemia‐mediated inflammation potentiates aminoglycoside‐induced ototoxicity. Sci Transl Med 7: 298ra118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopelman J, Budnick AS, Sessions RB, Kramer MB, Wong GY (1988) Ototoxicity of high‐dose cisplatin by bolus administration in patients with advanced cancers and normal hearing. Laryngoscope 98: 858–864 [DOI] [PubMed] [Google Scholar]
- Kurt‐Jones EA, Popova L, Kwinn L, Haynes LM, Jones LP, Tripp RA, Walsh EE, Freeman MW, Golenbock DT, Anderson LJ et al (2000) Pattern recognition receptors TLR4 and CD14 mediate response to respiratory syncytial virus. Nat Immunol 1: 398–401 [DOI] [PubMed] [Google Scholar]
- Lee KM, Seong SY (2009) Partial role of TLR4 as a receptor responding to damage‐associated molecular pattern. Immunol Lett 125: 31–39 [DOI] [PubMed] [Google Scholar]
- Lee SK, Oh KH, Chung AY, Park HC, Lee SH, Kwon SY, Choi J (2015) Protective role of quercetin against cisplatin‐induced hair cell damage in zebrafish embryos. Hum Exp Toxicol 34: 1043–1052 [DOI] [PubMed] [Google Scholar]
- Li Y, Womer RB, Silber JH (2004) Predicting cisplatin ototoxicity in children: the influence of age and the cumulative dose. Eur J Cancer 40: 2445–2451 [DOI] [PubMed] [Google Scholar]
- Lim DJ (1986) Functional structure of the organ of Corti: a review. Hear Res 22: 117–146 [DOI] [PubMed] [Google Scholar]
- Lush ME, Diaz DC, Koenecke N, Baek S, Boldt H, St Peter MK, Gaitan‐Escudero T, Romero‐Carvajal A, Busch‐Nentwich EM, Perera AG et al (2019) scRNA‐Seq reveals distinct stem cell populations that drive hair cell regeneration after loss of Fgf and Notch signaling. Elife 8: e44431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maekawa H, Inoue T, Ouchi H, Jao TM, Inoue R, Nishi H, Fujii R, Ishidate F, Tanaka T, Tanaka Y et al (2019) Mitochondrial damage causes inflammation via cGAS‐STING SIGNALING IN ACUTE KIDNEY INJURY. Cell Rep 29: 1261–1273 e1266 [DOI] [PubMed] [Google Scholar]
- Matsunaga N, Tsuchimori N, Matsumoto T, Ii M (2011) TAK‐242 (resatorvid), a small‐molecule inhibitor of Toll‐like receptor (TLR) 4 signaling, binds selectively to TLR4 and interferes with interactions between TLR4 and its adaptor molecules. Mol Pharmacol 79: 34–41 [DOI] [PubMed] [Google Scholar]
- Nagai Y, Akashi S, Nagafuku M, Ogata M, Iwakura Y, Akira S, Kitamura T, Kosugi A, Kimoto M, Miyake K (2002) Essential role of MD‐2 in LPS responsiveness and TLR4 distribution. Nature 3: 667–672 [DOI] [PubMed] [Google Scholar]
- Oblak A, Pohar J, Jerala R (2015) MD‐2 determinants of nickel and cobalt‐mediated activation of human TLR4. PLoS One 10: e0120583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh G‐SS, Kim H‐JJ, Choi J‐HH, Shen A, Kim C‐HH, Kim S‐JJ, Shin S‐RR, Hong S‐HH, Kim Y, Park C et al (2011) Activation of lipopolysaccharide‐TLR4 signaling accelerates the ototoxic potential of cisplatin in mice. J Immunol 186: 1140–1150 [DOI] [PubMed] [Google Scholar]
- Ou HC, Raible DW, Rubel EW (2007) Cisplatin‐induced hair cell loss in zebrafish (Danio rerio) lateral line. Hear Res 233: 46–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ou HC, Santos F, Raible DW, Simon JA, Rubel EW (2010) Drug screening for hearing loss: using the zebrafish lateral line to screen for drugs that prevent and cause hearing loss. Drug Discov Today 15: 265–271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang J, Xiong H, Zhan T, Cheng G, Jia H, Ye Y, Su Z, Chen H, Lin H, Lai L et al (2018) Sirtuin 1 and autophagy attenuate cisplatin‐induced hair cell death in the mouse cochlea and zebrafish lateral line. Front Cell Neurosci 12: 515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park BS, Lee J‐OO (2013) Recognition of lipopolysaccharide pattern by TLR4 complexes. Exp Mol Med 45: e66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park BS, Song DH, Kim HM, Choi BS, Lee H, Lee JO (2009) The structural basis of lipopolysaccharide recognition by the TLR4‐MD‐2 complex. Nature 458: 1191–1195 [DOI] [PubMed] [Google Scholar]
- Rachmawati D, Bontkes HJ, Verstege MI, Muris J, von Blomberg BM, Scheper RJ, van Hoogstraten IM (2013) Transition metal sensing by Toll‐like receptor‐4: next to nickel, cobalt and palladium are potent human dendritic cell stimulators. Contact Dermatitis 68: 331–338 [DOI] [PubMed] [Google Scholar]
- Raghavan B, Martin SF, Esser PR, Goebeler M, Schmidt M (2012) Metal allergens nickel and cobalt facilitate TLR4 homodimerization independently of MD2. EMBO Rep 13: 1109–1115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rallabhandi P, Phillips RL, Boukhvalova MS, Pletneva LM, Shirey KA, Gioannini TL, Weiss JP, Chow JC, Hawkins LD, Vogel SN et al (2012) Respiratory syncytial virus fusion protein‐induced toll‐like receptor 4 (TLR4) signaling is inhibited by the TLR4 antagonists Rhodobacter sphaeroides lipopolysaccharide and eritoran (E5564) and requires direct interaction with MD‐2. MBio 3: e00218‐12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramesh G, Zhang B, Uematsu S, Akira S, Reeves WB (2007) Endotoxin and cisplatin synergistically induce renal dysfunction and cytokine production in mice. Am J Physiol Renal Physiol 293: F325–332 [DOI] [PubMed] [Google Scholar]
- Ravi R, Somani SM, Rybak LP (1995) Mechanism of cisplatin ototoxicity: antioxidant system. Pharmacol Toxicol 76: 386–394 [DOI] [PubMed] [Google Scholar]
- Rocha‐Sanchez SM, Fuson O, Tarang S, Goodman L, Pyakurel U, Liu H, He DZ, Zallocchi M (2018) Quinoxaline protects zebrafish lateral line hair cells from cisplatin and aminoglycosides damage. Sci Rep 8: 15119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Ruijven MW, de Groot JC, Smoorenburg GF (2004) Time sequence of degeneration pattern in the guinea pig cochlea during cisplatin administration. A quantitative histological study. Hear Res 197: 44–54 [DOI] [PubMed] [Google Scholar]
- Rybak LP, Mukherjea D, Jajoo S, Ramkumar V (2009) Cisplatin ototoxicity and protection: clinical and experimental studies. Tohoku J Exp Med 219: 177–186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rybak LP, Whitworth CA, Mukherjea D, Ramkumar V (2007) Mechanisms of cisplatin‐induced ototoxicity and prevention. Hear Res 226: 157–167 [DOI] [PubMed] [Google Scholar]
- Sarin N, Engel F, Kalayda GV, Mannewitz M, Cinatl J Jr, Rothweiler F, Michaelis M, Saafan H, Ritter CA, Jaehde U et al (2017) Cisplatin resistance in non‐small cell lung cancer cells is associated with an abrogation of cisplatin‐induced G2/M cell cycle arrest. PLoS One 12: e0181081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt M, Raghavan B, Müller V, Vogl T, Fejer G, Tchaptchet S, Keck S, Kalis C, Nielsen PJ, Galanos C et al (2010) Crucial role for human Toll‐like receptor 4 in the development of contact allergy to nickel. Nat Immunol 11: 814–819 [DOI] [PubMed] [Google Scholar]
- Sepulcre MP, Alcaraz‐Perez F, Lopez‐Munoz A, Roca FJ, Meseguer J, Cayuela ML, Mulero V (2009) Evolution of lipopolysaccharide (LPS) recognition and signaling: fish TLR4 does not recognize LPS and negatively regulates NF‐kappaB activation. J Immunol 182: 1836–1845 [DOI] [PubMed] [Google Scholar]
- Siddik ZH (2003) Cisplatin: mode of cytotoxic action and molecular basis of resistance. Oncogene 22: 7265–7279 [DOI] [PubMed] [Google Scholar]
- Siegemund S, Sauer K (2012) Balancing pro‐ and anti‐inflammatory TLR4 signaling. Nat Immunol 13: 1031–1033 [DOI] [PubMed] [Google Scholar]
- Skinner R, Pearson AD, Amineddine HA, Mathias DB, Craft AW (1990) Ototoxicity of cisplatinum in children and adolescents. Br J Cancer 61: 927–931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smolinska MJ, Page TH, Urbaniak AM, Mutch BE, Horwood NJ (2011) Hck tyrosine kinase regulates TLR4‐induced TNF and IL‐6 production via AP‐1. J Immunol 187: 6043–6051 [DOI] [PubMed] [Google Scholar]
- So H, Kim H, Lee J‐HH, Park C, Kim Y, Kim E, Kim J‐KK, Yun K‐JJ, Lee K‐MM, Lee H‐YY et al (2007) Cisplatin cytotoxicity of auditory cells requires secretions of proinflammatory cytokines via activation of ERK and NF‐kappaB. J Assoc Res Otolaryngol 8: 338–355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- So H, Kim H, Kim Y, Kim E, Pae H‐OO, Chung H‐TT, Kim H‐JJ, Kwon K‐BB, Lee K‐MM, Lee H‐YY et al (2008) Evidence that cisplatin‐induced auditory damage is attenuated by downregulation of pro‐inflammatory cytokines via Nrf2/HO‐1. J Assoc Res Otolaryngol 9: 290–306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorenson CM, Barry MA, Eastman A (1990) Analysis of events associated with cell cycle arrest at G2 phase and cell death induced by cisplatin. J Natl Cancer Inst 82: 749–755 [DOI] [PubMed] [Google Scholar]
- Thomas AJ, Hailey DW, Stawicki TM, Wu P, Coffin AB, Rubel EW, Raible DW, Simon JA, Ou HC (2013) Functional mechanotransduction is required for cisplatin‐induced hair cell death in the zebrafish lateral line. J Neurosci 33: 4405–4414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas AJ, Wu P, Raible DW, Rubel EW, Simon JA, Ou HC (2015) Identification of small molecule inhibitors of cisplatin‐induced hair cell death: results of a 10,000 compound screen in the zebrafish lateral line. Otol Neurotol 36: 519–525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ton C, Parng C (2005) The use of zebrafish for assessing ototoxic and otoprotective agents. Hear Res 208: 79–88 [DOI] [PubMed] [Google Scholar]
- Uribe PM, Mueller MA, Gleichman JS, Kramer MD, Wang Q, Sibrian‐Vazquez M, Strongin RM, Steyger PS, Cotanche DA, Matsui JI (2013) Dimethyl sulfoxide (DMSO) exacerbates cisplatin‐induced sensory hair cell death in zebrafish (Danio rerio). PLoS One 8: e55359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vethanayagam RR, Yang W, Dong Y, Hu BH (2016) Toll‐like receptor 4 modulates the cochlear immune response to acoustic injury. Cell Death Dis 7: e2245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westerfield M (2000) The zebrafish book. A guide for the laboratory use of zebrafish (Danio rerio). Eugene, OR: University of Oregon Press; [Google Scholar]
- Wyllie DH, Kiss‐Toth E, Visintin A, Smith SC, Boussouf S, Segal DM, Duff GW, Dower SK (2000) Evidence for an accessory protein function for Toll‐like receptor 1 in anti‐bacterial responses. J Immunol 165: 7125–7132 [DOI] [PubMed] [Google Scholar]
- Yang H, Wang H, Ju Z, Ragab AA, Lundbäck P, Long W, Valdes‐Ferrer SI, He M, Pribis JP, Li J et al (2015) MD‐2 is required for disulfide HMGB1‐dependent TLR4 signaling. J Exp Med 212: 5–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan X, Hu T, He H, Qiu H, Wu X, Chen J, Wang M, Chen C, Huang S (2018) Respiratory syncytial virus prolifically infects N2a neuronal cells, leading to TLR4 and nucleolin protein modulations and RSV F protein co‐localization with TLR4 and nucleolin. J Biomed Sci 25: 13 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Data Availability Statement
This study includes no data deposited in external repositories.