

Abstract
Fibroblasts are a major component of the microenvironment of most solid tumours. Recent research elucidated a large heterogeneity and plasticity of activated fibroblasts, indicating that their role in cancer initiation, growth and metastasis is complex and context‐dependent. Here, we performed genome‐wide expression analysis comparing fibroblasts in normal, inflammatory and tumour‐associated skin. Cancer‐associated fibroblasts (CAFs) exhibit a fibrotic gene signature in wound‐induced tumours, demonstrating persistent extracellular matrix (ECM) remodelling within these tumours. A top upregulated gene in mouse CAFs encodes for PRSS35, a protease capable of collagen remodelling. In human skin, we observed PRSS35 expression uniquely in the stroma of high‐grade squamous cell carcinomas. Ablation of PRSS35 in mouse models of wound‐ or chemically‐induced tumorigenesis resulted in aberrant collagen composition in the ECM and increased tumour incidence. Our results indicate that fibrotic enzymes expressed by CAFs can regulate squamous tumour initiation by remodelling the ECM.
Keywords: cancer‐associated fibroblasts, fibrosis, PRSS35, skin cancer, wound healing
Subject Categories: Cancer; Cell Adhesion, Polarity & Cytoskeleton
This genome‐wide expression study demonstrates that CAFs in skin tumors exhibit a fibrotic signature. Genetic deletion of PRSS35, a top upregulated gene in these CAFs, promotes wound induced tumorigenesis and collagen deposition in the ECM.
Introduction
Effective wound healing mechanisms in the skin are crucial for mammalian survival. Wound healing and tumorigenesis involve fibroblast infiltration and activation, extracellular matrix (ECM) remodelling, neovascularization and immune cell infiltration. Due to the many histological and molecular similarities between the processes that mediate wound healing and tumour formation, tumours are often referred to as “wounds that do not heal” (Flier et al, 1986; Schäfer & Werner, 2008). Chronic wounds and associated inflammation constitute well‐known risk factors for tumour initiation (Arwert et al, 2012). In skin, the association between wound healing and the development of squamous cell carcinoma (SCC) is remarkably clear in patients suffering from recessive dystrophic epidermolysis bullosa (RDEB), a rare genetic skin blistering disorder. Almost all RDEB patients will develop a malignant SCC in early adulthood at sites of previous injury (Fine et al, 2009). Cutaneous SCC represents the second most common skin cancer type in Caucasians. Also, in a transgenic mouse model in which constitutive expression of MAP kinase kinase‐1 (MEK1) in suprabasal keratinocytes drives epidermal hyperproliferation, skin is known to be predisposed to tumour formation at sites of injury (Hobbs et al, 2004). However, the molecular mechanisms that tip the balance between skin regeneration and tumour initiation are still largely unknown.
In both wound healing and tumorigenesis, a pivotal role of the tissue microenvironment in disease progression and outcome has been demonstrated (Foster et al, 2018). A crucial component of the wound and tumour microenvironment is the fibroblast, the major cell‐type responsible for ECM production. Interest in the tumour microenvironment and the role of cancer‐associated fibroblasts (CAFs) has been surging over the past two decades, as CAFs represent interesting drug targets. However, no unique markers that define CAFs have been identified so far, indicating the complexity and heterogeneity of this cell‐type (Sahai et al, 2020). Emerging data point to a role for fibroblasts in suppressing epithelial proliferation and neoplastic conversion under homeostatic conditions (Kalluri & Zeisberg, 2006; Kalluri, 2016). However, following injury or tumour initiation, fibroblasts become activated and are able to switch molecular signalling towards epithelial growth in order to sustain tissue regeneration or tumour growth (Sahai et al, 2020). The molecular mechanisms by which fibroblasts exert these functions are still largely unknown. Although extensive heterogeneity of fibroblasts has been reported, only recent studies were able to reveal different lineages of dermal fibroblasts with distinct morphological and functional characteristics (Driskell et al, 2013; Rinkevich et al, 2015). However, it is still largely unclear how embryonic lineage commitment controls the development of fibroblast subsets important for tumorigenesis.
An important function of fibroblasts is the production of collagen and ECM in connective tissue. The ECM is a dynamic structure that is continuously remodelled by specific enzymes. Dysregulation of ECM remodelling can result in various pathological conditions, such as in defective wound healing responses, fibrosis and the invasion of cancer cells in connective tissue (Bonnans et al, 2015). Fibroblasts are largely responsible for remodelling of the ECM to promote or maintain tumour development by synthesizing ECM components, promoting ECM proteolysis and regulating post‐translational modification of ECM (Egeblad et al, 2010; Kalluri, 2016). Cutaneous wound healing represents a fibrotic response, as tissue regeneration is accompanied by an influx of fibroblasts and an aberrant deposition of collagen‐rich connective tissue in the wound microenvironment. Transforming growth factor‐beta (TGF‐β) and the cytokines IL (interleukin)‐4 and IL‐13 are key immunological drivers of fibrotic tissue responses (Eming et al, 2017). These alterations that occur in fibroblasts are transient in normal wound healing but can persist in chronic wounds and developing tumours. Myofibroblasts are activated fibroblasts that express alpha‐smooth muscle actin (α‐SMA) and are present both in wounds and tumours, where they represent a major population of CAFs. Finally, fibrotic tissue represents a highly permissive microenvironment for the initiation of tumorigenic responses, and the accumulation of ECM during tumorigenesis has been shown to promote metastasis (Kaplan et al, 2005).
This study focusses on the molecular switches that tip the balance between the epithelial growth‐inhibitory and growth‐promoting roles of skin fibroblasts during inflammation and oncogenic transformation. In skin, CAF profiling of fibroblasts in dysplastic skin of a squamous skin carcinogenesis model revealed a pro‐inflammatory gene signature (Erez et al, 2010). Here, we show that in a mouse model of wound‐induced skin carcinogenesis CAFs display a fibrotic phenotype, with the serine protease PRSS35 as a top upregulated gene in CAFs relative to normal and inflammatory fibroblasts. PRSS35 is a protease that has been shown to cleave collagen‐I and to be implicated in fibrotic responses (Lebleu et al, 2013; Ichii et al, 2017). Interestingly, ablation of PRSS35 in mice results in enhanced sensitivity to wound‐ and chemically induced skin tumour formation, indicating an important role for PRSS35‐mediated fibrotic responses in the control of skin tumour formation.
Results
Dermal fibroblasts present a fibrotic phenotype in wound‐induced tumours
Fibroblasts are highly heterogeneous and can exhibit distinct phenotypes depending on their activation state (Schäfer & Werner, 2008; LeBleu & Kalluri, 2018; Sahai et al, 2020). To evaluate the specific phenotype of fibroblasts involved in inflammation versus wound‐induced skin tumorigenesis, a genome‐wide expression study was performed comparing gene expression profiles of “normal”, “inflammatory” and “papilloma‐associated” fibroblasts (Fig 1A). Inflammatory and papilloma‐associated fibroblasts were isolated from skin of transgenic mice expressing a constitutively active MEK1 selectively in differentiating keratinocytes (InvEE mice) (Hobbs et al, 2004). InvEE mice exhibit an inflammatory response in the skin and develop tumours at sites of full‐thickness skin wounding with an incidence depending on the extent of wounding (Arwert et al, 2010; Hoste et al, 2015). Fibroblasts were isolated from age‐ and gender‐matched wild‐type, InvEE and InvEE tumour‐associated back skin of PDGFRα‐H2B‐eGFP reporter mice (Hamilton et al, 2003) by flow cytometric sorting of GFP‐positive cells, as PDGFRα was previously shown to represent a robust marker for dermal fibroblasts (Erez et al, 2010). RNA was isolated from sorted cells, and expression profiling was performed by Affymetrix mouse genome arrays in order to identify a tumour‐specific fibroblast profile and to enable comparative analyses with previously generated expression profiles of CAFs in squamous carcinomas (Erez et al, 2010). Multiple genes encoding for collagens and other ECM molecules, as well as the enzymes modelling them, could be found among the top upregulated genes in fibroblasts isolated from wound‐induced papillomas (Pap) relative to fibroblasts isolated from inflamed (InvEE) or normal (WT) skin (Fig 1B and C). This fibrotic gene signature was also reflected in the top upregulated gene ontology (GO) terms in InvEE tumour‐associated fibroblasts, including fibroblast proliferation, response to wounding, collagen metabolic processing and ECM organization (Fig 1D). Interestingly, correlation analysis using Pearson correlation (significance level 0.05) of differentially expressed genes (DEGs) revealed that CAFs isolated from dysplastic Keratin‐14 Human papilloma virus 16 (K14‐HPV16) skin that were shown to exhibit a pro‐inflammatory gene signature (Erez et al, 2010), clustered together with our “inflamed” InvEE fibroblasts, but not with the wound‐induced tumour‐associated fibroblast population (Pap; Fig EV1A). Principal component analysis of both datasets confirmed that CAFs isolated from dysplastic skin in K14‐HPV16 mice clustered closely together with the inflamed fibroblasts isolated from non‐tumour bearing InvEE mice, while fibroblasts isolated from wound‐induced papillomas clearly represent a separate subset (Fig EV1B). Most genes characteristic for fibrosis, such as genes encoding for different collagens, Tenascin‐C, ADAM12, MMP16, α‐SMA, CCL12 and others, were more highly expressed in wound‐induced tumour‐associated fibroblasts (Pap) than in “inflamed” (InvEE) fibroblasts (Figs 1C and EV1A). The pro‐inflammatory gene signature that was reported to characterize CAFs in K14‐HPV16 mice was partially present in InvEE skin and wound‐induced papillomas, but these DEGs are not among the top upregulated “Pap” genes (Fig EV1C).
Figure 1. Dermal CAFs from wound‐induced tumours exhibit a fibrotic gene signature.
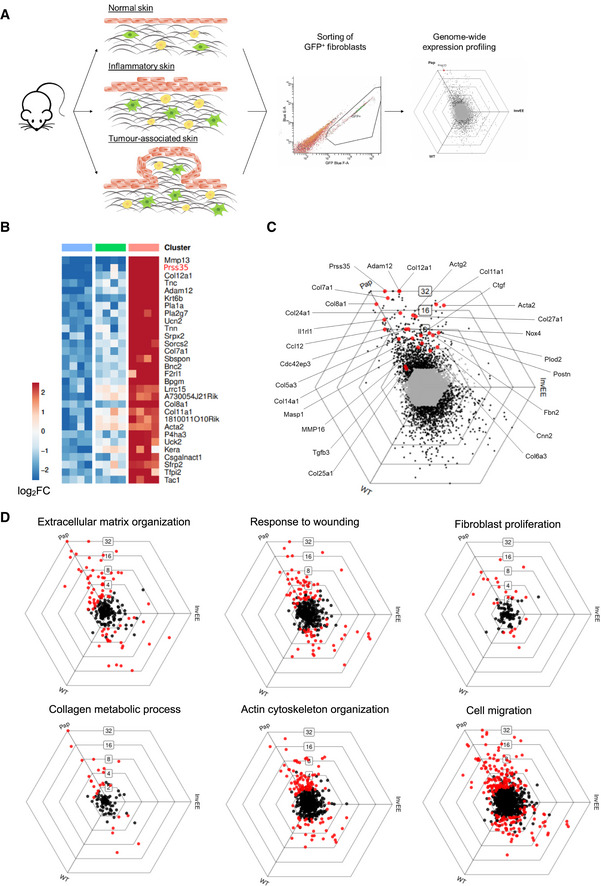
- Schematic representation of experimental set‐up for genome‐wide fibroblast expression profiling.
- Heat map depicting mean expression of the top 20 upregulated genes of fibroblast isolated from wound‐induced tumours (Pap) compared to fibroblasts from wild‐type (WT) and inflamed (InvEE) back skin (n = 3 per condition). PRSS35 is indicated in red as a top upregulated gene in CAFs.
- To visualize DEGs between subsets of fibroblasts sorted from homeostatic (WT), inflamed (InvEE) or tumour‐associated (Pap) skin, each gene was plotted in a hexagonal triwise diagram in which the position of a point represents the relative increased expression in one or two populations, whereas the distance from the origin represents the magnitude of expression. Genes that are 32‐fold or more upregulated are plotted on the outer grid line. Grey dots in the centre of the triwise plot represent genes that are not differentially expressed. Genes associated with a fibrotic response are depicted in red.
- Hexagonal triwise plots of genes belonging to the respective gene ontology (GO) term, which were among the highest upregulated GO terms. Dots in red represent genes that are differentially expressed between the three conditions.
Figure EV1. Comparison of the InvEE CAF signature with the pro‐inflammatory CAF signature characterized by Erez et al(2010).
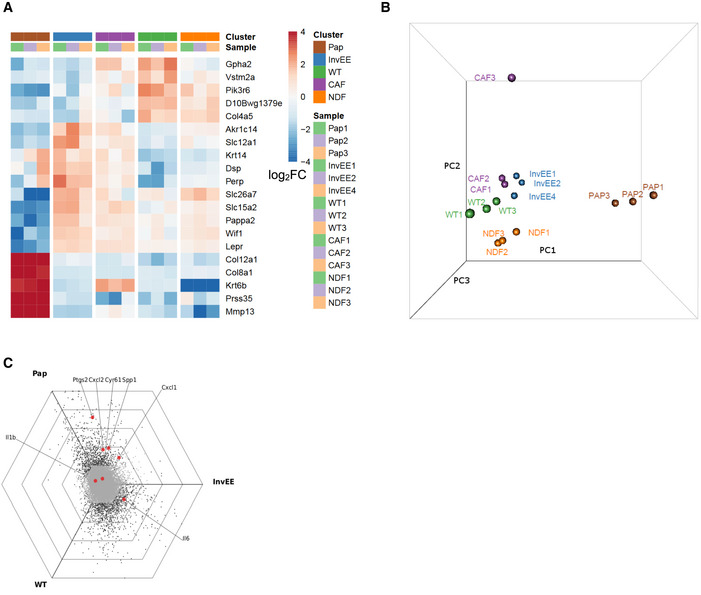
- Combined clustering of our dataset (WT: normal skin, InvEE: inflammatory skin and Pap: wound‐induced papillomas) and the dataset obtained by Erez et al (NDF: normal dermal fibroblasts and CAF: cancer‐associated fibroblasts) based on global differential gene expression.
- Principle component analysis of our dataset and the dataset from Erez et al purple: CAF, orange: NDF, blue: InvEE, green: WT and brown: Pap).
- Hexagonal triwise plot showing the pro‐inflammatory gene signature, determined by Erez et al and mapped on our gene expression analysis. (Ptgs2 = Cox2, SPP1 = Osteopontin, Cyr61 = CCN1).
Fibrosis is characterized by an increase in fibroblast proliferation and/or activation, which was reflected by one of the top upregulated GO terms “fibroblast proliferation” in papilloma‐associated fibroblasts (Fig 1D). The enhanced fibroblast proliferation could be verified by quantifying the number of fibroblasts present in wound‐induced InvEE‐PDGFRα‐eGFP tumours relative to inflamed or homeostatic skin by confocal microscopy analysis (Fig 2A and B). We observed a significant increase in the number of fibroblasts present in wounded skin relative to unwounded inflamed (InvEE) or homeostatic (WT) skin. In wound‐induced skin tumours, fibroblast numbers are even higher than in wounded skin and further increase during papilloma growth (Fig 2B). To investigate dermal ECM remodelling in the different conditions, we performed Herovici stainings. In normal and inflamed skin, the dermis contained mainly mature collagen, as visualized by purple Herovici staining, while scars and wound‐induced tumours showed highly active deposition of immature collagen as visualized by blue Herovici staining, indeed indicating persistent ECM remodelling in wound‐induced tumours (Fig 2C). Interestingly, the deposition of new collagen was present at the tumour base and in the tumour’s finger‐like stromal protrusions, demonstrating the occurrence of active ECM remodelling within the tumour. Another hallmark of fibrotic responses is increased serine protease activity, which we assessed by measuring the degradation of the chromogenic serine protease substrate Nα‐benzoyl‐DL‐arginine‐p‐nitroaniline (BAPNA). Serine protease activity was significantly increased in wound‐induced tumour lysates (Pap) relative to lysates from WT and InvEE skin (Fig 2D). Together, these data demonstrate that fibroblasts present in wound‐induced tumours exhibit a fibrotic, rather than an inflammatory, signature and exhibit persistent ECM remodelling.
Figure 2. Wound‐induced tumour formation comprises a fibrotic response.
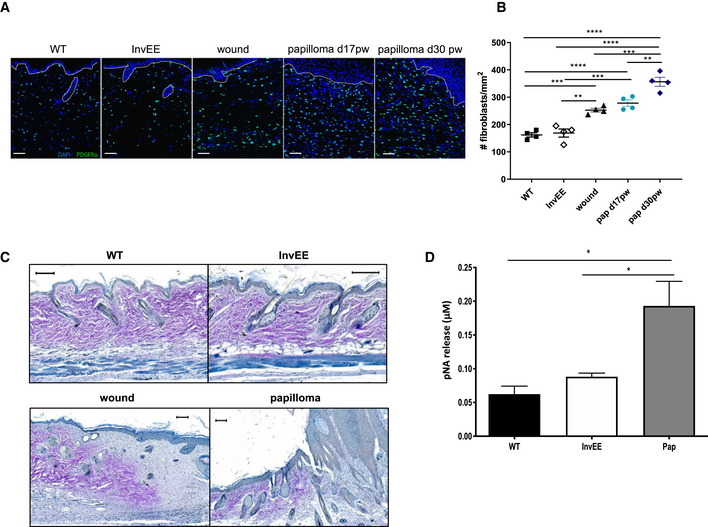
- Immunofluorescent images of skin sections from PDGFRα‐eGFP reporter mice with normal (WT, n = 4), inflamed (InvEE, n = 4), wounded (d14 post‐wounding; wound, n = 4) or tumour‐associated (Papilloma, Pap d17 and d30 post‐wounding, n = 4 per condition) skin. Nuclei were stained with Dapi (4’, 6‐diamidino‐2‐phenylindole; blue). Dotted line represents epidermal–dermal boundary. Scale bars: 50 µm.
- Quantification of the number of fibroblasts present in different skin conditions (n = 4 mice per condition; **P < 0.01; ***P < 0.001; ****P < 0.0001; One‐way ANOVA testing). Data represent means of multiple microscopic fields ± SEM.
- Herovici staining of sections of normal (WT), inflamed (InvEE), wound or tumour‐associated (Papilloma) skin. Immature collagen is stained blue and mature collagen is stained purple. Scale bars: 100 µm.
- pNA (p‐nitroaniline) release assessed by spectrophotometric analysis as a measure of serine protease activity in normal (WT, n = 5), inflamed (InvEE, n = 5) and tumour‐associated (Pap, n = 7) skin. (*P < 0.05; one‐way ANOVA testing). Data represent means ± SEM.
PRSS35 is selectively upregulated in dermal wound‐induced CAFs
Our genome‐wide expression analysis identified PRSS35 as one of the top upregulated genes in wound‐induced papillomas (Figs 1B and 3A and B). PRSS35 is a serine protease with functional relevance in kidney fibrosis that can cleave collagen I (Lebleu et al, 2013; Ichii et al, 2017). The increased expression of PRSS35 in wound‐induced tumours was confirmed by quantitative RT–PCR (Q–PCR) of tissue lysates obtained from normal skin, InvEE skin, wounds (d14pw) and wound‐induced papillomas. We did not observe PRSS35 expression in normal skin, which is in agreement with previous reports (Miyakoshi et al, 2006). However, PRSS35 expression was induced in wounds, inflamed and tumour‐associated skin, but was strongest upregulated in wound‐induced papillomas (Fig 3C).
Figure 3. PRSS35 is selectively upregulated in wound‐induced tumours.
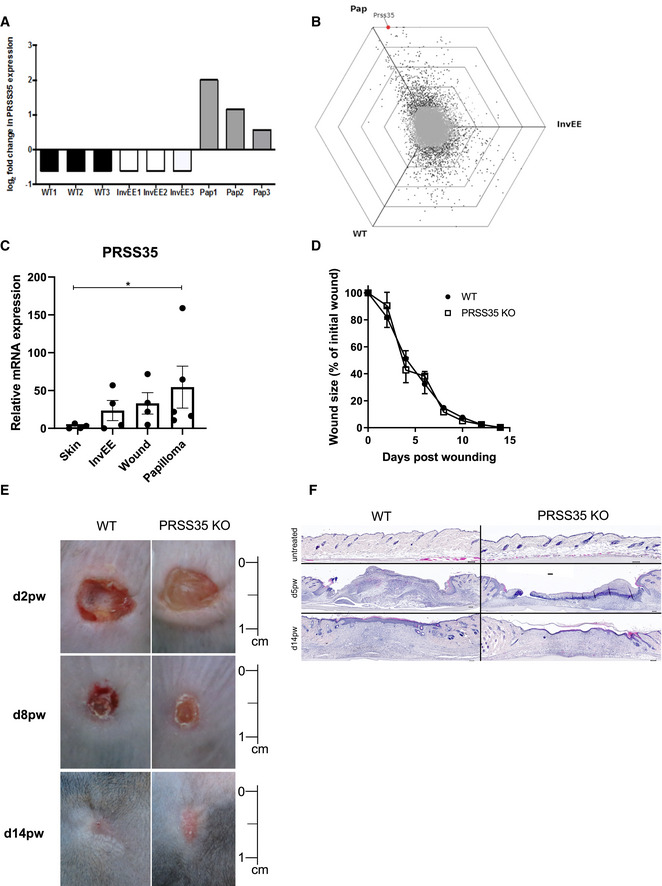
- Log2 fold change of probe intensities of PRSS35 gene expression in different subsets of dermal fibroblasts observed in Affymetrix GeneChip® Mouse 430 2.0 array (n = 3 per condition).
- Hexagonal triwise plot depicting the PRSS35 gene expression level.
- Relative mRNA levels of PRSS35 in lysates from normal skin (WT, n = 4 mice), inflamed skin (InvEE, n = 4 mice), wounds (d14pw, n = 4 mice) and wound‐induced papillomas (Pap, n = 5 mice). (*P = 0.0159; Mann–Whitney test). Data represent means of three technical replicates ± SEM.
- Wound healing dynamics of wild‐type (WT, n = 10) and PRSS35−/− (n = 11) mice after full‐thickness wounding with an 8‐mm punch biopsy. Wound size expressed as percentage of initial wound size (ns; Wilcoxon matched‐pairs signed rank test). Data represent means ± SEM.
- Representative pictures of back skin of WT and PRSS35−/− mice at different days post‐wounding (dpw). Scale bar: 1 cm.
- H&E‐stained skin sections of WT and PRSS35−/− wounds at different days post‐wounding. Scale bars: 100 µm.
In order to understand the in vivo function of PRSS35 in skin tumorigenesis, PRSS35‐deficient mice (B6J‐Prss35em1Irc; PRSS35−/− mice) were generated using CRISPR‐Cas9 gene‐editing technology (Fig EV2A and B). The genotype of the mice was confirmed by PCR on tail lysates and by Q–PCR on skin lysates (Fig EV2C and D). PRSS35‐deficient mice were born at the expected Mendelian ratio, were fertile and did not show any overt phenotype. To analyse the putative role of PRSS35 in wound healing, PRSS35‐deficient (KO) and control wild‐type (WT) mice were subjected to full‐thickness skin wounding. Wound healing rates were indistinguishable between PRSS35 KO mice and control littermates (Fig 3D and E), and histological analysis of cutaneous wounds at different stages of wound healing did not show marked morphological differences between PRSS35 WT and KO mice (Fig 3F).
Figure EV2. Generation of PRSS35 knockout (B6J‐Prss35em1Irc) mice.
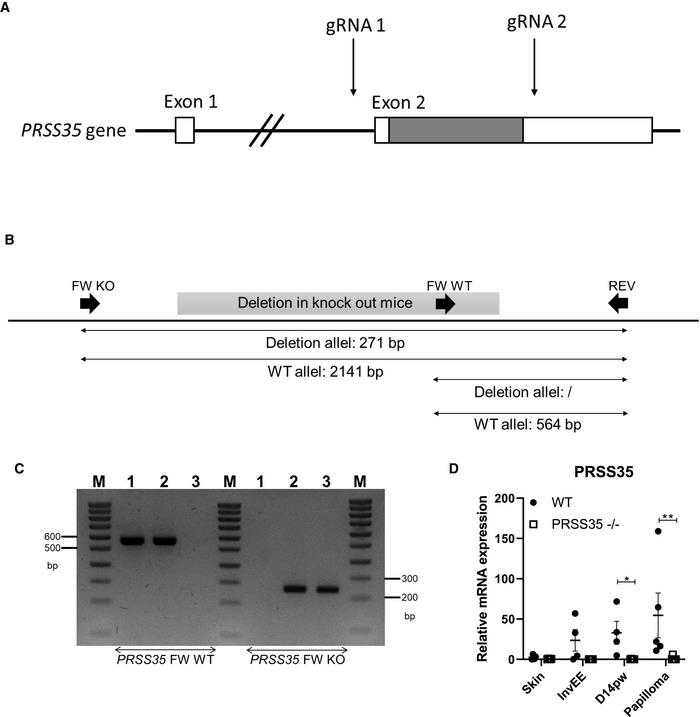
- Positioning of CRISPR‐Cas gRNAs on the PRSS35 gene. The sequence between the two gRNAs was deleted upon cleavage by CRISPR‐Cas9 at the site of the gRNAs. The coding region is shaded.
- Genotyping strategy for detection of the presence or absence of PRSS35. Schematic representation of the primer bindings sites used.
- An agarose gel image for PRSS35 genotyping. M: Ladder, 1‐3: genomic DNA extracted from tails from a WT, PRSS35 heterozygous and PRSS35 KO mouse.
- Relative mRNA expression levels of PRSS35 in tissue isolated from WT and PRSS35−/− mice (n ≥ 4 per condition; *P < 0.05, **P < 0.01; Mann–Whitney test).
PRSS35 mediates wound‐ and chemically‐induced skin tumorigenesis
To unravel the function of PRSS35 in cutaneous tumour initiation, we crossed PRSS35 KO mice with InvEE mice, and compound transgenic mice were wounded. Interestingly, InvEE‐PRSS35 KO mice exhibited an increased incidence in wound‐induced tumorigenesis and developed tumours earlier than their WT counterparts, indicating that PRSS35 can protect the skin from tumour initiation in the context of wounding (Fig 4A). Wound‐induced tumours from InvEE‐PRSS35 WT and KO mice did not exhibit clear morphological differences (Fig 4B).
Figure 4. Mice lacking PRSS35 are more sensitive to wound‐ and chemically‐induced carcinogenesis.
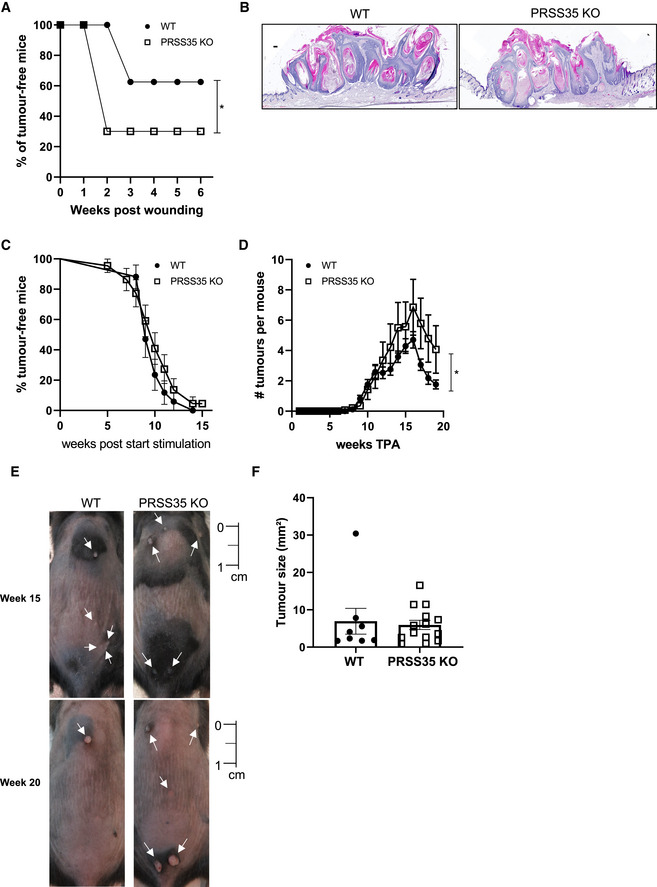
-
AIncidence of wound‐induced papilloma formation in InvEE PRSS35+/+ (WT; n = 8) and InvEE PRSS35−/− (PRSS35 KO; n = 10) mice at site of wounding. (*P = 0.0218; Gehan–Breslow–Wilcoxon test).
-
BH&E‐stained skin sections of wound‐induced papillomas of InvEE PRSS35+/+ and InvEE PRSS35−/− mice. Scale bars: 100 µm.
-
C, DIncidence of papilloma formation (ns: non‐significant; Gehan–Breslow–Wilcoxon test) (C) and number of tumours per mouse (D) in wild‐type (WT, n = 17) and PRSS35 KO (n = 22) mice treated with DMBA‐TPA (*P = 0.0171; Wilcoxon matched‐paired rank test). Data represent means ± SEM.
-
ERepresentative pictures of the back skin of WT and PRSS35 KO mice undergoing the DMBA‐TPA treatment at week 15 and week 20 after the start of TPA treatment. White arrows indicate tumours. Scale bar: 1 cm.
-
FTumour size at week 20 after the start of TPA treatment. The biggest tumour of each mouse was measured (WT; n = 8; KO; n = 14; ns: Mann–Whitney test). Data represent means ± SEM.
To validate these findings in a second, independent mouse model of skin carcinogenesis, we induced tumours in PRSS35 WT and KO mice via the well‐established two‐stage DMBA‐TPA (7,12‐dimethylbenz(a)anthracene/12‐O‐tetradecanoylphorbol‐13‐acetate) protocol for chemical induction of skin tumours (Abel et al, 2009). Mice were first exposed to a single dose of DMBA, inducing oncogenic mutations in H‐Ras (initiation), followed by repeated topical application with TPA, to promote tumorigenesis, 3 times a week for 15 weeks, which allowed mutagenized cells to expand and form papillomas. While tumour incidence (the amount of mice that developed a tumour) was comparable between PRSS35 KO and control mice (Fig 4C), PRSS35 KO mice developed significantly more tumours relative to WT mice (Fig 4D and E), indicating that the tumour microenvironment enables the formation of more tumours in the absence of PRSS35. No difference in tumour size was observed between WT and KO tumours at the time of sacrifice (Fig 4F). These data demonstrate that the presence of PRSS35 impacts on skin tumour formation in both wound‐ and chemically‐induced carcinogenesis.
PRSS35 levels are downregulated by stimulation with TGF‐β1, and in vivo ablation of PRSS35 alters collagen deposition in the ECM of wounds and tumours
To understand the cell‐autonomous effect of PRSS35 in fibroblasts and to investigate the molecular mechanisms by which PRSS35 could mediate skin tumour formation, we isolated primary dermal fibroblasts from WT and PRSS35 KO skin. Assessment of the proliferation rate of these fibroblasts did not show differences between both genotypes (Fig EV3A). In agreement with our in vivo findings, no significantly altered in vitro wound healing responses were observed between WT and KO fibroblasts in scratch wound assays (Fig EV3B). We next stimulated primary dermal fibroblast cultures with TGF‐β1, a known activator of fibrotic responses (Caja et al, 2018). PRSS35 is expressed in cultured primary mouse fibroblasts, and we observed a significant decrease of PRSS35 expression upon treatment of the cells with TGF‐β1 (Fig 5A). TGF‐β1 stimulation of primary fibroblasts induced enhanced expression levels of growth factors and ECM components associated with fibrosis (Fig 5C and D) (Castor et al, 1979; Kalluri, 2016), however, no differences in expression levels of these genes could be observed between PRSS35‐proficient and PRSS35‐deficient fibroblasts.
Figure EV3. PRSS35 does not alter proliferation or inflammatory responses in fibroblasts.
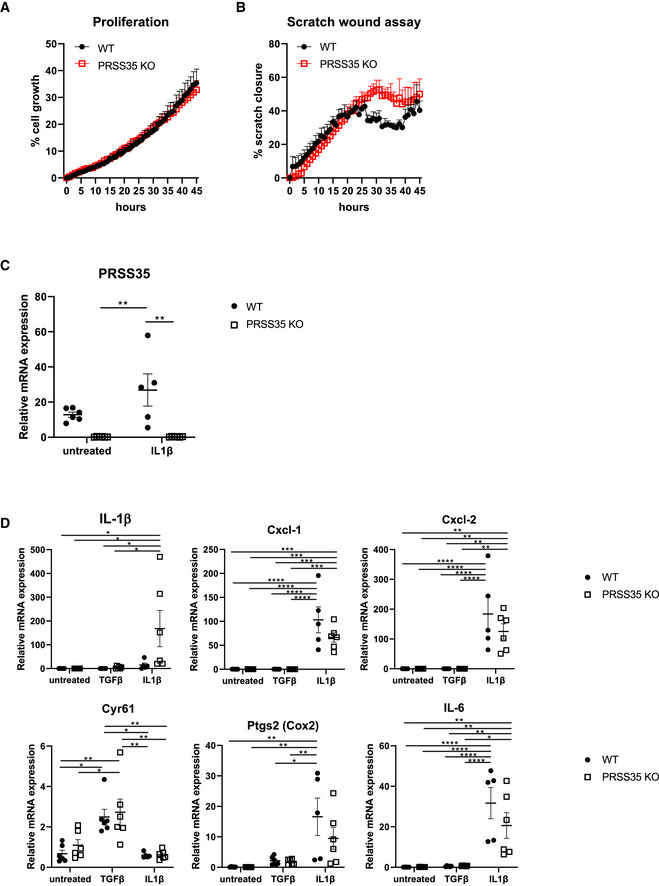
-
ACell proliferation assay on PRSS35 WT and PRSS35 KO primary fibroblasts, as measured through live cell imaging (n = 3 biological replicates per condition, ns, Wilcoxon matched‐paired rank test). Data represent means ± SEM
-
BScratch wound assay of PRSS35 WT and PRSS35 KO primary fibroblasts. Scratch width was measured over time with live cell imaging (n = 3 per condition, ns, Wilcoxon matched‐paired rank test). Data represent means ± SEM
-
C, DRelative RNA expression of the indicated genes in primary fibroblast isolated from WT and PRSS35 KO mice, untreated or after 24 h stimulation with 10 ng/ml TGF‐β (n = 6 per genotype) or 20 ng/ml IL‐1β (WT, n = 5; PRSS35 KO, n = 6) (*P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001; two‐way ANOVA with multiple comparisons). Data represent means ± SEM.
Figure 5. PRSS35 expression by primary fibroblasts is inhibited by TGF‐β stimulation, and in vivo ablation of PRSS35 leads to an altered collagen deposition in skin wounds and tumours.
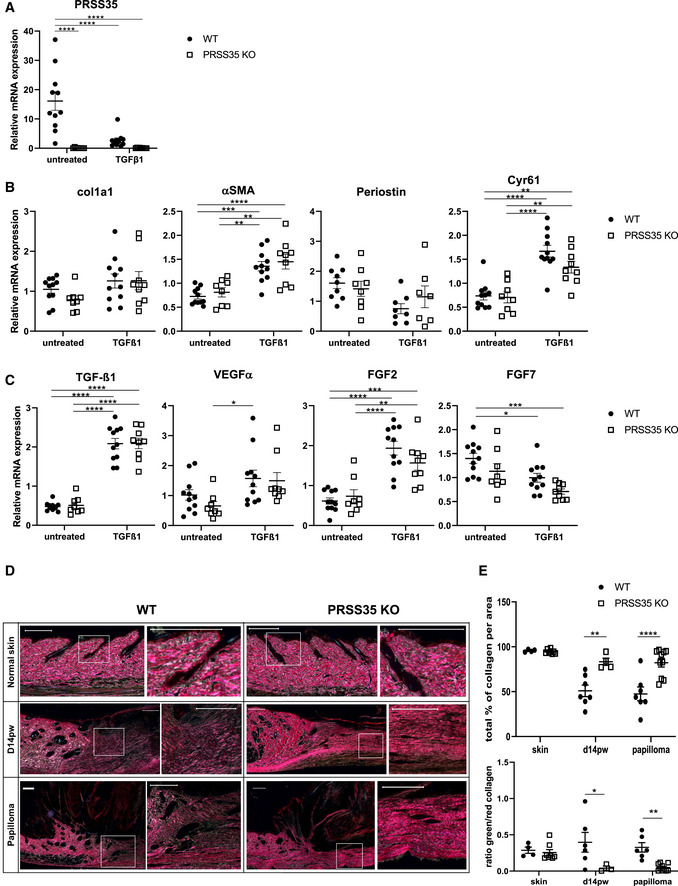
- Relative mRNA expression levels of PRSS35 in primary fibroblast cultures isolated from WT and PRSS35 KO mice, untreated or after 24 h stimulation with 10 ng/ml TGF‐β1 (n > 8 biological replicates per condition) (****P < 0.0001; two‐way ANOVA with multiple comparisons). Data represent means of three technical replicates ± SEM.
- Relative mRNA expression levels of the indicated ECM genes in primary fibroblast cultures (col1a1: collagen type 1 alpha 1; αSMA, alpha‐smooth muscle actin; Cyr61: Cysteine‐rich angiogenic inducer 61 (=CCN1)) in primary fibroblast cultures isolated from WT and PRSS35 KO mice, untreated or after 24 h stimulation with 10 ng/ml TGFβ1 (n ≥ 7 biological replicates per condition) (**P < 0.01, ***P < 0.001, ****P < 0.0001; two‐way ANOVA with multiple comparisons). Data represent means of three technical replicates± SEM.
- Relative mRNA expression levels of the indicated cytokines/growth factors in primary fibroblast cultures (n ≥ 8 per condition) (VEGF‐α, vascular endothelial growth factor‐alpha; FGF2: basic fibroblast growth factor; FGF7: keratinocyte growth factor) (*P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001; two‐way ANOVA with multiple comparisons). Data represent means of three technical replicates ± SEM.
- Picrosirius red staining of normal skin, day 14 post‐wounding (d14pw) skin or wound‐induced papillomas from WT and PRSS35 KO mice. Red colouring indicates thick fibres, green colouring indicates thin fibres. Scale bars: 200 µm.
- Quantification of picrosirius red staining as total collagen density (upper panel) and ratio of red versus green picrosirius staining (lower panel). Using QuPath Bioimage analysis software pixels colouring red or green were determined per area. (n ≥ 4 technical replicates per condition) (*P < 0.05, **P < 0.01, ****P < 0.0001; two‐way ANOVA with multiple comparisons). Data represent means ± SEM.
A previous study described a pro‐inflammatory gene signature in CAFs isolated from mouse squamous carcinomas, which could be partially reproduced by stimulation of dermal fibroblasts with the inflammatory cytokine IL‐1β (Erez et al, 2010). However, treatment of PRSS35 WT and KO primary skin fibroblasts with the inflammatory cytokine IL‐1β did not result in significant changes in the expression profile of inflammatory signature genes between both genotypes (Fig EV3C). PRSS35 expression levels were not significantly increased in WT dermal fibroblasts that were treated with IL‐1β (Fig EV3D), indicating that PRSS35 expression is not upregulated in inflammatory conditions.
Since PRSS35 did not significantly alter fibrotic responses in cultured dermal fibroblasts, but has previously been implicated in collagen remodelling (Lebleu et al, 2013; Ichii et al, 2017), we next analysed the collagen content in wounds and tumours of PRSS35‐proficient versus ‐deficient skin stained with Picrosirius red and imaged under polarized light (Junqueira et al, 1979; Rich & Whittaker, 2005). The collagen composition of WT and PRSS35 KO skin exhibited a comparable ratio of red (thick fibres) and green (thin fibres) coloured collagen fibres in homeostatic conditions. However, in re‐epithelialized wounds and papillomas, PRSS35 KO skin clearly showed a more dense collagen matrix characterized by red‐stained collagen, while in WT wounds and tumours the collagen fibres were more loosely organized, with thin green‐stained fibres (Fig 5D and E). Quantification of this staining confirmed that there is significantly more collagen per area in scars and papillomas of PRSS35 KO compared to WT mice and that there are significantly less thin collagen fibres compared to thick collagen fibres in scars and papillomas of PRSS35 KO skin (Fig 5E). These data indicate that collagen deposition by fibroblasts during wound healing and tumorigenesis is altered in PRSS35 KO skin and more directed towards the formation of thick collagen fibres.
PRSS35 is selectively upregulated in high‐grade human SCCs
So far, no markers have been identified that allow discrimination of different stages of squamous cell carcinomas in human patients. As we observed PRSS35 expression in wound‐induced carcinomas and an enhanced tumour progression in PRSS35‐deficient mice subjected to skin carcinogenesis protocols, we probed PRSS35 expression in human keloids, which are defined as hypertrophic scars that are considered benign fibrotic lesions (Seifert & Mrowietz, 2009), and in differently staged SCCs by confocal microscopy. No PRSS35 expression could be observed in keloids or in low‐grade SCCs (n = 5 per condition). However, high‐grade, undifferentiated SCCs exhibited marked PRSS35 expression, which was most apparent in the tumour stroma at the dermal–epidermal interface, indicating that PRSS35 is selectively upregulated in high‐grade human SCCs (Fig 6A and B; n = 5).
Figure 6. PRSS35 is selectively upregulated in high‐grade human squamous cell carcinomas.
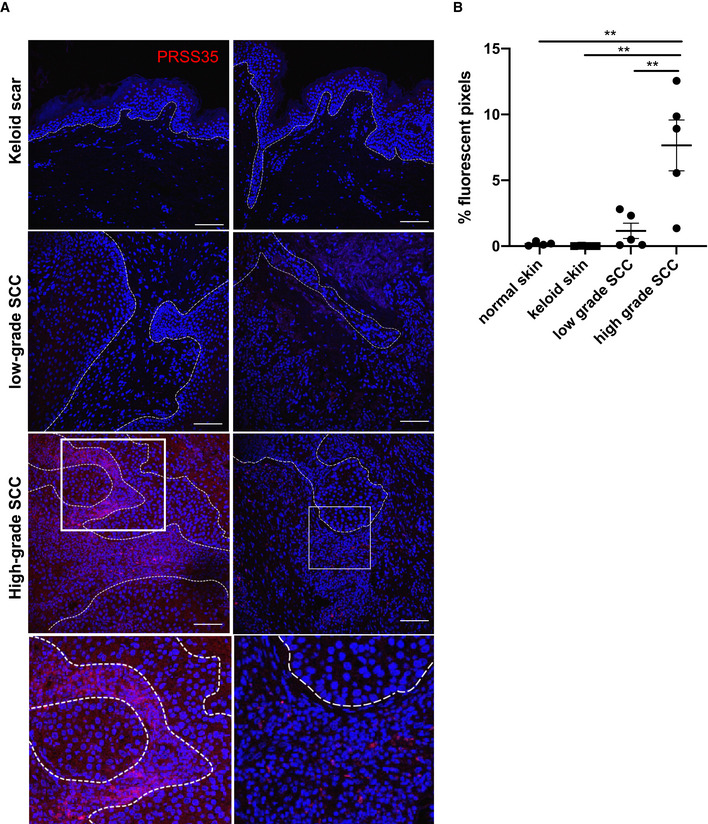
- Human skin sections were stained with a PRSS35‐specific antibody (red) and counterstained with Dapi (n = 5 per condition). Boxed areas are magnifications as shown in the lower panel, and dotted line represents the epidermal–dermal boundary. Scale bars: 100 µm.
- Quantification of PRSS35‐positive signal by Volocity analysis (n ≥ 4 per condition; **P < 0.01; one‐way ANOVA testing). Data represent means of multiple microscopic fields ± SEM.
In conclusion, we have demonstrated that cancer‐associated fibroblasts exhibit a persistent fibrotic phenotype in wound‐induced skin tumours. We showed that fibrotic proteases secreted by these CAFs can have a major influence on tumour development and should be considered as potential therapeutic targets in clinical cancer treatment. We also provided evidence for PRSS35 as a potential biomarker that enables discrimination between high‐grade malignant SCCs and well‐differentiated low‐grade SCCs in human patients.
Discussion
The tumour microenvironment critically impacts on cancer initiation and progression. Here, we showed that cancer‐associated fibroblasts in wound‐induced papillomas exhibit a fibrotic phenotype, enabling ECM remodelling during cancer growth and progression. Our genome‐wide expression analysis of wound‐induced CAFs markedly differs from the findings reported by Erez et al(2010), who demonstrated a predominantly pro‐inflammatory gene signature in CAFs isolated from squamous carcinomas. Our data showed that, while some inflammatory mediators are indeed upregulated in wound‐induced CAFs, the main DEGs are linked to fibrotic responses. Indeed, clustering of both datasets demonstrated that the CAF population defined by Erez et alresembles our “inflamed” fibroblast condition but differs significantly from fibroblasts isolated from wound‐induced papillomas. This discrepancy could be explained by the fact that wound‐induced papillomas in InvEE mice represent an exophytic tumour type, enabling precise surgical isolation of tumours with minimal contamination of surrounding inflamed stroma, while the K14‐HPV16 mouse model used by Erez et alnvolves hyperplasia of the epidermis, followed by dysplastic lesion development that progresses into squamous cell carcinomas (Erez et al, 2010). In their study, the authors stated that they profiled fibroblasts from dysplastic skin regions that are early neoplastic lesions, which could represent a more “inflammatory” and earlier stage in the process of carcinogenesis progression than the CAFs in our InvEE model. Indeed, single‐cell sequencing data of oesophageal cancer at different stages of tumour development showed that distinct fibroblast clusters are dominating different stages of cancer development, with a surge of fibroblasts expressing a strong inflammatory gene signature at the early inflammatory stage, which is then replaced by a fibroblast cluster with a predominant TGF‐β signalling signature during dysplasia and carcinoma in situ stages (Yao et al, 2020). Another possible explanation is the fact that a different subpopulation of activated fibroblasts could be involved in the two different animal models. CAFs have previously been shown to either be “myoCAFs” that exhibit a matrix‐remodelling and contractile phenotype, or “iCAFs”, presenting an immunomodulatory phenotype (Sahai et al, 2020). GO analysis of DEGs in wound‐induced CAFs point to persistent ECM and cytoskeletal remodelling, while K14‐HPV16 CAFs express various cytokines that are potent inducers of inflammatory responses. Indeed, different activating triggers induce different CAF subpopulations (Bordignon et al, 2019). This would suggest that wound‐induced InvEE tumours largely depend on myoCAFs, while K14‐HPV16 squamous tumours contain mainly iCAFs. Co‐existence of two distinct CAF subpopulations, that largely correspond to myoCAFs and iCAFs, within a tumour has been documented in pancreatic cancer where these subpopulations are spatially separated, with myoCAFs residing close to the neoplastic cells, while iCAFs resided further away from the cancer‐initiating cells (Öhlund et al, 2017). In human head and neck squamous cell carcinomas (HNSCCs), single‐cell analysis also revealed a high representation of myofibroblasts within the tumour stroma, and TGF‐β, alongside other fibrotic genes, was identified as highly upregulated within these tumours (Puram et al, 2017).
The persistent ECM remodelling response that we observed in wound‐induced CAFs has also been reported in human advanced high‐grade serous ovarian cancer (HGSOC) metastases and in 12 other human cancer types, suggesting a common ECM response in primary and metastatic cancer. This ECM‐associated signature could also predict disease severity, indicating the importance of the matrix response for cancer progression (Pearce et al, 2018). In triple‐negative breast cancer and pancreatic cancer three different CAF populations were identified, two of which, the inflammatory and the antigen‐presenting CAFs, were also found in normal tissue, while the myoCAF population was uniquely present in the tumour tissue, again implying that fibrotic responses are indeed a driving force in tumorigenesis (Sebastian et al, 2020).
Profiling of dermal fibroblasts in our study was based on PDGFRα expression, which represents a robust marker for fibroblasts present in murine and human skin (Driskell et al, 2013; Philippeos et al, 2018). However, cells expressing PDGFRα in the skin are known to encompass distinct subpopulations, which play different roles in tissue development and regeneration (Driskell et al, 2013; Rinkevich et al, 2015; Rognoni et al, 2016) and may not capture the total spectrum of lineage plasticity of dermal fibroblasts in tumorigenesis. Recent single‐cell RNA‐sequencing studies have indeed revealed highly plastic fibroblast populations in wounded skin (Guerrero‐Juarez et al, 2019; Phan et al, 2020). Hence, performing similar studies on CAFs at single‐cell level will give a broader view on the full spectrum of fibroblast plasticity in tumorigenesis.
One of the top upregulated genes in wound‐induced CAFs was PRSS35, a gene previously implicated in fibrotic kidney responses (Lebleu et al, 2013). The study by Lebleu et al demonstrated that PRSS35 expression is enhanced in mouse and human fibrotic kidneys, as is its inhibitor human epididymis protein 4 (HE4). Neutralizing HE4 prevented kidney fibrosis by accelerating collagen type 1 degradation, indicating that PRSS35 can protect from fibrosis (Lebleu et al, 2013). We observed that depletion of PRSS35 resulted in enhanced tumour formation in wound‐ and chemically‐induced skin carcinogenesis, suggesting that upregulation of PRSS35 aims to reduce the fibrotic reaction caused by the activated fibroblasts and TGF‐β secretion in wound‐induced tumorigenesis. This hypothesis was strengthened by the observation that the collagen matrix in wounds and papillomas of mouse skin lacking PRSS35 is more densely packed than in wild‐type skin. This could be due to a different deposition of collagen by PRSS35‐deficient fibroblasts, which is less probable as we showed that cultured fibroblasts lacking PRSS35 express collagen1a1 to a similar extent as normal fibroblasts. More likely, PRSS35 is able to cleave the collagen that is deposited during wound healing and neoplastic responses, thereby altering the structure of collagen fibres in the matrix. In our study, we made use of mice lacking PRSS35 in all cell types. Although a functional role for PRSS35 has so far only been described in fibroblasts, we can, however, not exclude that other cell‐types can also secrete PRSS35 and thereby influence ECM remodelling.
So far, little research has been done on PRSS35 in the context of cancer. However, a possible role for this protease in cancer progression has been described in metastatic lung cancer (Schliekelman et al, 2011) and in melanoma progression (Yan et al, 2013). Interestingly, when we probed the RNA‐sequencing data published in the Pearce et al(2018) study for PRSS35 expression levels, we observed an increased PRSS35 expression in several HGSOC biopsy samples with a high disease score, indicating that PRSS35 might be overexpressed in a range of human tumour types. Reporter studies in mice have demonstrated active TGF‐β signalling in chemically induced skin tumours that are transitioning into malignant SCCs (Oshimori et al, 2015). Our observations that PRSS35, a protease present in fibrotic conditions, is expressed in the tumour stroma of malignant human SCCs, confirm these data. Also, in human HNSCCs, PRSS35 was identified as a highly upregulated gene in one of two CAF subpopulations that were present in lymph node metastases (Puram et al, 2017).
In conclusion, our data have shown that CAFs isolated from wound‐induced tumours display a fibrotic gene signature with high expression of the collagen‐remodelling enzyme PRSS35, and that depletion of PRSS35 results in a more densely packed collagen matrix, which may contribute to the enhanced squamous tumour formation in PRSS35 knockout mice. Moreover, we have shown that PRSS35 expression is uniquely present in high‐grade SCCs, thereby laying the grounds for its use as a biomarker in staging these malignant skin tumours in patients.
Materials and Methods
Mice
The following mouse lines were used: PRSS35 KO (B6J‐Prss35em1Irc), InvEE (Hobbs et al, 2004) and PDGFRα‐GFP (Hamilton et al, 2003). InvEE mice were maintained on an F1 genetic background (CBA × C57Bl/6) and kept heterogeneous for the MEK1 transgene. Transgene negative littermates were used as wild‐type (WT) controls. All mice in the DMBA/TPA experiments were on a C57/Bl6J genetic background. Mice were housed in individually ventilated cages at the VIB Center for Inflammation Research, in a specific pathogen‐free animal facility. All experiments on mice were conducted according to institutional, national and European animal regulations. Animal protocols were approved by the ethics committee of Ghent University.
Generation of PRSS35 knockout mice
PRSS35 KO (B6J‐Prss35em1Irc) mice were generated by the Transgenic mouse Core Facility (TmCF) of the VIB‐UGent Center for Inflammation Research using the CRISPR/Cas9 system. Synthetic Alt‐R® CRISPR‐Cas9 crRNA (Integrated DNA Technologies, IDT) with protospacer sequences 5’‐TACTCTAGGAAGAACCTAGG‐3’ and 5’‐ACAGAGAGTGGGCTCCTGTT‐3’ were duplexed with synthetic Alt‐R® CRISPR‐Cas9 tracrRNA (IDT). cr/tracrRNA duplexes (100 ng/µl) were complexed with Alt‐R® S.p. Cas9 Nuclease V3 (500 ng/µl) (IDT). The resulting RNP complex was electroporated into C57BL/6J zygotes using a Nepa21 electroporator with electrode CUY501P1‐1.5. Electroporated embryos were incubated overnight in Embryomax KSOM medium (Merck, Millipore) in a CO2 incubator. The following day, 2‐cell embryos were transferred to pseudo‐pregnant B6CBAF1 foster mothers. The resulting pups were screened by PCR over the target region using primers 5’‐ATGCTTCCAAACTCTGAC‐3’ and 5’‐CTAAACCTCCTGTCTCCC‐3’. PCR bands were Sanger sequenced to identify the exact nature of the deletion. Mouse line B6J‐RNF213em1Irc contains an allele with a deletion of 1870 bp (chr9+: 86754911‐86756780) removing all coding sequences of the Prss35 gene.
Genome‐wide expression profiling
Single‐cell suspensions were prepared from skin as previously described (Collins et al, 2011). Briefly, dissected skin was scraped free of muscle and dermal fat and incubated overnight at 4°C in 0.25% trypsin solution. The interfollicular epidermis was scraped away, and dermis was minced and enzymatically dissociated for 45 min at 37°C using a mixture of 1.25 mg/ml collagenase type I (Invitrogen), 0.5 mg/ml collagenase type II (Worthington), 0.5 mg/ml collagenase type IV (Sigma), 0.1 mg/ml hyaluronidase IVS (Sigma) and 50 U/ml DNase I (Roche). Normal, inflammatory and papilloma‐associated fibroblasts were isolated by flow cytometric sorting on a BD FACSAria cell sorting system of GFP‐positive cells (purity ≥ 90%) from 7‐week‐old wild‐type, InvEE and InvEE tumour‐associated back skin of male mice containing a PDGFRα‐H2B‐eGFP allele. RNA was isolated from sorted cells using the Qiagen RNeasy kit and amplified once. Amplified RNA was hybridized to Affymetrix MG430.2A arrays. Samples were subsequently analysed using R/Bioconductor, the oligo package was used for RMA‐normalization, and limma was used to perform differential expression analysis with an adjustment of the P‐values for multiple testing via the Benjamin and Hochberg algorithm.
Gene ontology and clustering analysis
In order to compare our microarray data with the previously generated dataset (Erez et al, 2010), ComBat correction from the sva packages was applied to adjust for the batch effect. Consecutively, principal component analysis (PCA) was performed to visualize clustering of the samples. Gene ontology (GO) was analysed using org.Mm.egGO2ALLEGS from the org.Mm.eg.db package with translation of the identifiers to ENREZID and filtering to only Biological Process GO terms. Gene sets were tested for unidirectional enrichment and visualized using the Triwise package.
Quantitative real‐time PCR
Total RNA was isolated using TRIzol reagent (Invitrogen) and Aurum Total RNA Isolation Mini Kit (Bio‐Rad), according to manufacturer’s instructions. Synthesis of cDNA was performed using Sensifast cDNA Synthesis Kit (Bioline) according to the manufacturer’s instructions. cDNA was amplified on quantitative PCR in a total volume of 5 μl with SensiFAST SYBR® No‐ROX Kit (Bioline) and specific primers on a LightCycler 480 (Roche). The reactions were performed in triplicates. The sequences of the mouse‐specific primers are listed in Table 1.
Table 1.
The sequences of the mouse‐specific primers
| Fw | Rev | |
|---|---|---|
| PRSS35 | ACGGGAGGATACAGTAAGCATC | CTGTTTCCGACCCATCAGAGA |
| TGFβ1 | GCTGAACCAAGGAGACGGAATA | GAGTTTGTTATCTTTGCTGTCACAAGA |
| col1a1 | CCCAAGGAAAAGAAGCACGTC | ACATTAGGCGCAGGAAGGTCA |
| αSMA | CCAGAGCAAGAGAGGGATCCT | TGTCGTCCCAGTTGGTGATG |
| Periostin | CGGAGAGCCAGTCATTAAAA | TGCAAGGTCTCTCCTGTTTC |
| Cyr61 | GCACTCTGGGTTGTCATTGG | GCAATTGGAAAAGGCAGCTC |
| VEGFα | CTTGTTCAGAGCGGAGAAAGC | ACATCTGCAAGTACGTTCGTT |
| IL‐6 | GAGGATACCACTCCCAACAGACC | AAGTGCATCATCGTTGTTCATACA |
| FGF2 | CAAGAACGGCGGCTTCTTC | GGAAGAAACAGTATGGCCT |
| IL1β | TGG GCC TCA AAG GAA AGA | GGT GCT GAT GTA CCA GTT |
| Cox2 | GAAATATCAGGTCATTGGTGGAG | ATACATCATCAGACCAGGCA |
| Cxcl‐2 | ACAGAAGTCATAGCCACTCTC | TTAGCCTTGCCTTTGTTCAG |
| Cxcl‐1 | GAGCCTCTAACCAGTTCCAG | TGAGTGTGGCTATGACTTCG |
Histology
Skin tissue was dissected and fixed in 4% paraformaldehyde and embedded in paraffin. Sections were stained with haematoxylin/eosin (H&E) or with specific staining. The Herovici staining was performed as previously described (Herovici, 1963). Following dewaxing, slides were incubated with Weigert’s haematoxylin (Sigma) for 10 min to stain the nuclei. The sections were then stained for 3 min with the staining solution (50 ml Van Gieson staining kit (Sigma), 50 ml Methyl blue (0,05 % aq.), 10 ml glycerol, 0.5 ml Lithium carbonate) The slides were washed with 1% acetic acid for 2 min. The slides were dehydrated with ethanol and xylene and mounted with Entallan (Merck). Picrosirius red staining was carried out with the commercially available “Picro Sirius Red Stain Kit” (Abcam) according to the manufacturers’ instructions. Picrosirius red staining was quantified using QuPath Bioimage analysis software. The software was trained to classify a pixel as “red”, “green” or “empty”. The same classifier was used for all samples. The percentage of “red” or “green” pixels per selected area was determined.
Immunofluorescence staining
Frozen sections from PGDFRα‐H2B‐eGFP mice were fixed in 4% paraformaldehyde/PBS pH 7.4, incubated with DAPI to stain nuclei, washed in PBS and then mounted using the DAKO mounting reagent (DAKO). Paraffin‐embedded human skin sections were subjected to heat‐mediated antigen retrieval (citrate buffer, pH 6) and blocked with 2% BSA, 0,02% fish skin gelatin and 10% goat serum for 1 h in PBS at room temperature. Sections were incubated with PRSS35 antibody (Abcam, ab151970; 1:1,000) overnight at 4°C in antibody solution, followed by 1 h incubation at room temperature with secondary antibody (DyLight 555 goat‐anti‐rabbit) and DAPI. Fluorescent PRSS35 staining was quantified by setting a threshold based on intensity of fluorescence. The intensity interval used for thresholding was identical for all samples. The percentage of positive pixel counts in this intensity interval per total area of skin was quantified using Volocity software version 6.3 (Perkin Elmer).
Serine protease activity assay
The skin and tumour samples were lysed in 500 μl Tris–HCl (pH 8,5) using the precellys 24 tissue homogenizer (Bertin instruments). The supernatant was isolated, and protein concentration was determined. The reaction mix constituted 10 µl 1 mM Bapna (Nα‐Benzoyl‐L‐arginine 4‐nitroanilide hydrochloride) in DMSO, 40 µg protein, Tris–HCl (pH 8.5) to a total volume of 200 µl. The reaction was incubated for 25 h at 37°C at 100 rpm. The liberated p‐nitroaniline was measured at 405 nm.
Wounding
Full‐thickness wounds were made on shaved back skin by using 8‐mm punch biopsy needles (Stiefel Instruments) under analgesia and general anaesthesia in 8‐week‐old InvEE and control littermates. For wound healing experiments, assuming standard deviations of 10–15%, an experimental group of n = 10 is needed to obtain statistical power of 90% (significance level 0.05) and detect a difference between means of 20%, using two‐tailed paired T‐testing. No animals were excluded from any experiment.
DMBA/TPA two‐stage carcinogenesis
Chemical carcinogenesis experiments were performed, as previously described (Abel et al, 2009). In brief, back skin of 8‐week‐old mice was shaved and treated once with 100 nmol (25 μg) DMBA in 200 μl acetone. Seven days later, mice were treated with 6 nmol (3.7 μg) TPA three times weekly for 15 weeks. Mice were allocated to groups based on genotypes, but housed in a randomized manner. Tumour incidence and burden were assessed once a week by two independent researchers who were blinded to the group allocations.
Cell cultures
Primary fibroblasts were isolated from mice ears, as previously described (Moore & Allen, 2013). Shortly, ears were removed from mice, disinfected, minced into small pieces and incubated overnight at 37°C in DMEM medium with 4 mg/ml collagenase D (Roche) and 4 mg/ml dispase (Gibco). The next day, tissue was further dissociated by pipetting, passed through a cell strainer twice, centrifuged, resuspended and grown on a 150 mm2 plate. Cells were cultured in DMEM (Gibco) supplemented with 10% FCS and Penicillin/Streptomycin. Cells were left untreated or stimulated for 24 h either with TGFβ1 (10 ng/ml) or IL‐1β (20 ng/ml). Proliferation assay and scratch wound assay were performed with the IncuCyte® Live‐Cell Analysis Systems.
Statistics
Statistical analyses were performed using GraphPad Prism 8. Data are presented as the means ± SEM. Specific tests included Fisher’s exact tests, one or two‐way analysis of variance followed by Tukey’s multiple comparison test, unpaired t‐tests and Mann–Whitney tests. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Human samples
All patient specimens were obtained after informed, written consent, in accordance with Helsinki guidelines and with appropriate consent from the relevant institutional review board (Ghent University and Otto‐von‐Guericke University Magdeburg).
Author contributions
LvH performed experiments, analysed data and drafted the manuscript, KL and H‐KV performed experiments and analysed data, JR, NV, GD and YS analysed microarray data, SRQ, IH and KO provided human skin sections and clinical data. TH generated PRSS35‐deficient mice. FMW and GvL analysed data and revised the manuscript. EH designed and performed experiments, analysed data, drafted and revised the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Review Process File
Acknowledgements
We thank Laetitia Bellen, Dimitri Huygebaert and Dieter Vanhede for animal care, and all members of the van Loo laboratory for suggestions and discussions. We acknowledge the VIB Bioimaging core for their help with imaging and analysis. L.v.H is supported by an FWO FR fellowship, I.H. is supported by an FWO postdoctoral fellowship. G.D. is supported by AIRC MFAG 2018. E.H. is supported by an FWO postdoctoral fellowship and a CRIG Young‐Investigator grant. Research in the van Loo lab is financed by research grants from the FWO, Stichting tegen Kanker, Kom op Tegen Kanker and the “Concerted Research Actions” (GOA) of the Ghent University.
EMBO reports (2021) 22: e51573.
Contributor Information
Geert van Loo, Email: geert.vanloo@irc.vib-ugent.be.
Esther Hoste, Email: esther.hoste@irc.vib-ugent.be.
Data availability
The datasets produced in this study are available in the following database: Gene Expression Omnibus GSE156197 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE156197).
References
- Abel EL, Angel JM, Kiguchi K, DiGiovanni J (2009) Multi‐stage chemical carcinogenesis in mouse skin: Fundamentals and applications. Nat Protoc 4: 1350–1362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arwert EN, Hoste E, Watt FM (2012) Epithelial stem cells, wound healing and cancer. Nat Rev Cancer 12: 170–180 [DOI] [PubMed] [Google Scholar]
- Arwert EN, Lal R, Quist S, Rosewell I, van Rooijen N, Watt FM (2010) Tumor formation initiated by nondividing epidermal cells via an inflammatory infiltrate. Proc Natl Acad Sci 107: 19903–19908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnans C, Chou J, Werb Z (2015) Remodelling the extracellular matrix in development and disease. Nat Rev Mol Cell Biol 15: 786–801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bordignon P, Bottoni G, Xu X, Popescu AS, Truan Z, Guenova E, Kofler L, Jafari P, Ostano P, Röcken M et al(2019) Dualism of FGF and TGF‐β signaling in heterogeneous cancer‐associated fibroblast activation with ETV1 as a critical determinant. Cell Rep 28: 2358–2372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caja L, Dituri F, Mancarella S, Caballero‐Diaz D, Moustakas A, Giannelli G, Fabregat I (2018) TGF‐β and the tissue microenvironment: Relevance in fibrosis and cancer. Int J Mol Sci 19: 1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castor CW, Heiss PR, Gray RH, Seidman JC (1979) Connective tissue formation by lung fibroblasts in vitro . Am Rev Respir Dis 120: 107–119 [DOI] [PubMed] [Google Scholar]
- Collins CA, Kretzschmar K, Watt FM (2011) Reprogramming adult dermis to a neonatal state through epidermal activation of β‐catenin. Development 138: 5189–5199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driskell RR, Lichtenberger BM, Hoste E, Kretzschmar K, Simons BD, Charalambous M, Ferron SR, Herault Y, Pavlovic G, Ferguson‐Smith AC et al(2013) Distinct fibroblast lineages determine dermal architecture in skin development and repair. Nature 504: 277–281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egeblad M, Rasch MG, Weaver VM (2010) Dynamic interplay between the collagen scaffold and tumor evolution. Curr Opin Cell Biol 22: 697–706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eming SA, Wynn TA, Martin P (2017) Inflammation and metabolism in tissue repair and regeneration. Science 356: 1026–1030 [DOI] [PubMed] [Google Scholar]
- Erez N, Truitt M, Olson P, Hanahan D (2010) Cancer‐associated fibroblasts are activated in incipient neoplasia to orchestrate tumor‐promoting inflammation in an NF‐ k B‐dependent manner. Cancer Cell 17: 135–147 [DOI] [PubMed] [Google Scholar]
- Fine JD, Johnson LB, Weiner M, Li KP, Suchindran C (2009) Epidermolysis bullosa and the risk of life‐threatening cancers: The National EB Registry experience, 1986–2006. J Am Acad Dermatol 60: 203–211 [DOI] [PubMed] [Google Scholar]
- Flier JS, Underhill LH, Dvorak HF (1986) Tumors: wounds that do not heal. N Engl J Med 315: 1650–1659 [DOI] [PubMed] [Google Scholar]
- Foster DS, Longaker MT, Jeffrey A, Foster DS, Jones RE, Ransom RC, Longaker MT, Norton JA (2018) The evolving relationship of wound healing and tumor stroma. JCI Insight 3: e99911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerrero‐Juarez CF, Dedhia PH, Jin S, Ruiz‐Vega R, Ma D, Liu Y, Yamaga K, Shestova O, Gay DL, Yang Z et al(2019) Single‐cell analysis reveals fibroblast heterogeneity and myeloid‐derived adipocyte progenitors in murine skin wounds. Nat Commun 10: 650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton TG, Klinghoffer RA, Corrin PD, Soriano P (2003) Evolutionary divergence of platelet‐derived growth factor alpha receptor signaling mechanisms. Mol Cell Biol 23: 4013–4025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herovici C (1963) A polychrome stain for differentiating precollagen from collagen. Stain Technol 38: 204–206 [PubMed] [Google Scholar]
- Hobbs RM, Silva‐Vargas V, Groves R, Watt FM (2004) Expression of activated MEK1 in differentiating epidermal cells is sufficient to generate hyperproliferative and inflammatory skin lesions. J Invest Dermatol 123: 503–515 [DOI] [PubMed] [Google Scholar]
- Hoste E, Arwert EN, Lal R, South AP, Salas‐Alanis JC, Murrell DF, Donati G, Watt FM (2015) Innate sensing of microbial products promotes wound‐induced skin cancer. Nat Commun 6: 5932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichii O, Kimura J, Okamura T, Horino T, Nakamura T, Sasaki H, Hosni Ali Elewa Y, Kon Y (2017) IL‐36a regulates Tubulointerstitial inflammation in the Mouse Kidney. Front Immunol 8: 1346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Junqueira LCU, Bignolas G, Brentani RR (1979) Picrosirius staining plus polarization microscopy, a specific method for collagen detection in tissue sections. Histochem J 11: 447–455 [DOI] [PubMed] [Google Scholar]
- Kalluri R (2016) The biology and function of fibroblasts in cancer. Nat Rev Cancer 16: 582–598 [DOI] [PubMed] [Google Scholar]
- Kalluri R, Zeisberg M (2006) Fibroblasts in cancer. Nat Rev Cancer 6: 392–401 [DOI] [PubMed] [Google Scholar]
- Kaplan RN, Riba RD, Zacharoulis S, Bramley AH, Vincent L, Costa C, MacDonald DD, Jin DK, Shido K, Kerns SA et al(2005) VEGFR1‐positive haematopoietic bone marrow progenitors initiate the pre‐metastatic niche. Nature 438: 820–827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeBleu VS, Kalluri R (2018) A peek into cancer‐associated fibroblasts: Origins, functions and translational impact. Dis Model Mech 11: dmm029447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebleu VS, Teng Y, Connell JTO, Charytan D, Müller GA, Müller CA, Sugimoto H, Kalluri R (2013) Identification of human epididymis protein‐4 as a fibroblast‐derived mediator of fibrosis. Nat Med 19: 227–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyakoshi K, Murphy MJ, Yeoman RR, Mitra S, Dubay CJ, Hennebold JD (2006) The identification of novel ovarian proteases through the use of genomic and bioinformatic methodologies1. Biol Reprod 75: 823–835 [DOI] [PubMed] [Google Scholar]
- Moore CB, Allen IC (2013) Primary ear fibroblast derivation from mice. Methods Mol Biol 1031: 65–70 [DOI] [PubMed] [Google Scholar]
- Öhlund D, Handly‐Santana A, Biffi G, Elyada E, Almeida AS, Ponz‐Sarvise M, Corbo V, Oni TE, Hearn SA, Lee EJ et al(2017) Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J Exp Med 214: 579–596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oshimori N, Oristian D, Fuchs E (2015) TGF‐β promotes heterogeneity and drug resistance in squamous cell carcinoma. Cell 160: 963–976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearce OMT, Delaine‐Smith RM, Maniati E, Nichols S, Wang J, Böhm S, Rajeeve V, Ullah D, Chakravarty P, Jones RR et al(2018) Deconstruction of a metastatic tumor microenvironment reveals a common matrix response in human cancers. Cancer Discov 8: 304–319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phan QM, Fine GM, Salz L, Herrera GG, Wildman B, Driskell IM, Driskell RR (2020) Lef1 expression in fibroblasts maintains developmental potential in adult skin to regenerate wounds. Elife 9: 1–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philippeos C, Telerman SB, Oulès B, Pisco AO, Shaw TJ, Elgueta R, Lombardi G, Driskell RR, Soldin M, Lynch MD et al(2018) Spatial and single‐cell transcriptional profiling identifies functionally distinct human dermal fibroblast subpopulations. J Invest Dermatol 138: 811–825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puram SV, Tirosh I, Parikh AS, Lin DT, Regev A, Bernstein BE (2017) Single‐cell transcriptomic analysis of primary and metastatic tumor ecosystems in head and neck cancer. Cell 171: 1611.e1–1611.e24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rich L, Whittaker P (2005) Collagen and Picrosirius red staining: a polarized light assessment of fibrillar hue and spatial distribution. Braz J morphol Sci 22: 97–104 [Google Scholar]
- Rinkevich Y, Walmsley GG, Hu MS, Maan ZN, Newman AM, Drukker M, Januszyk M, Krampitz GW, Gurtner GC, Lorenz HP et al(2015) Identification and isolation of a dermal lineage with intrinsic fibrogenic potential. Science 348: aaa2151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rognoni E, Gomez C, Pisco AO, Rawlins EL, Simons BD, Watt FM, Driskell RR (2016) Inhibition of β‐catenin signalling in dermal fibroblasts enhances hair follicle regeneration during wound healing. Development 143: 2522–2535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahai E, Astsaturov I, Cukierman E, DeNardo DG, Egeblad M, Evans RM, Fearon D, Greten FR, Hingorani SR, Hunter T et al(2020) A framework for advancing our understanding of cancer‐associated fibroblasts. Nat Rev Cancer 20: 174–186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schäfer M, Werner S (2008) Cancer as an overhealing wound: an old hypothesis revisited. Nat Rev Mol Cell Biol 9: 628–638 [DOI] [PubMed] [Google Scholar]
- Schliekelman MJ, Gibbons DL, Faca VM, Creighton CJ, Rizvi ZH, Zhang Q, Wong C‐H, Wang H, Ungewiss C, Ahn Y‐H et al(2011) Tumor and stem cell biology targets of the tumor suppressor miR‐200 in regulation of the epithelial‐mesenchymal transition in cancer. Cancer Res 71: 7670–7682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebastian A, Hum NR, Martin KA, Gilmore SF, Peran I, Byers SW, Wheeler EK, Coleman MA, Loots GG (2020) Single‐cell transcriptomic analysis of tumor‐derived fibroblasts and normal tissue‐resident fibroblasts reveals fibroblast heterogeneity in breast cancer. Cancers 12: 1307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seifert O, Mrowietz U (2009) Keloid scarring: bench and bedside. Arch Dermatol Res 301: 259–272 [DOI] [PubMed] [Google Scholar]
- Yan G, Eller MS, Elm C, Larocca CA, Ryu B, Panova IP, Dancy BM, Bowers EM, Meyers D, Lareau L et al(2013) Selective inhibition of p300 HAT blocks cell cycle progression, induces cellular senescence, and inhibits the DNA damage response in melanoma cells. J Invest Dermatol 133: 2444–2452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao J, Cui Q, Fan W, Ma Y, Chen Y, Liu T, Zhang X, Xi Y, Wang C, Peng L et al(2020) Single‐cell transcriptomic analysis in a mouse model deciphers cell transition states in the multistep development of esophageal cancer. Nat Commun 11: 3715 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Review Process File
Data Availability Statement
The datasets produced in this study are available in the following database: Gene Expression Omnibus GSE156197 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE156197).