

Abstract
The final size and function of the adult central nervous system (CNS) are determined by neuronal lineages generated by neural stem cells (NSCs) in the developing brain. In Drosophila, NSCs called neuroblasts (NBs) reside within a specialised microenvironment called the glial niche. Here, we explore non‐autonomous glial regulation of NB proliferation. We show that lipid droplets (LDs) which reside within the glial niche are closely associated with the signalling molecule Hedgehog (Hh). Under physiological conditions, cortex glial Hh is autonomously required to sustain niche chamber formation. Upon FGF‐mediated cortex glial overgrowth, glial Hh non‐autonomously activates Hh signalling in the NBs, which in turn disrupts NB cell cycle progression and its ability to produce neurons. Glial Hh’s ability to signal to NB is further modulated by lipid storage regulator lipid storage droplet‐2 (Lsd‐2) and de novo lipogenesis gene fatty acid synthase 1 (Fasn1). Together, our data suggest that glial‐derived Hh modified by lipid metabolism mechanisms can affect the neighbouring NB’s ability to proliferate and produce neurons.
Keywords: Drosophila, glial niche, Hedgehog, lipid metabolism, neuroblast
Subject Categories: Metabolism, Neuroscience, Regenerative Medicine
The glial niche regulates the proliferation of neural stem cells in the Drosophila larval CNS. Glial Hh signaling autonomously facilitates cortex glial niche formation, and non‐autonomously regulates neuroblast proliferation dependent on the lipid regulators Fasn1 and Lsd2.
Introduction
Most stem cells reside within specialised groups of cells, collectively referred to as a niche, that provide the trophic, structural and nutritional microenvironment to sustain and protect the stem cells during development (Scadden, 2014). The niche relays developmental and physiological states of the animal to the stem cells and influences the stem cells’ ability to divide in accordance with the environmental state of the organism. Asymmetrically dividing and multipotent neural stem cells in both mammals and invertebrates are responsible for generating the adult nervous system (Homem & Knoblich, 2012).
In Drosophila, the vast majority of NBs are specified during embryogenesis, proliferate throughout larval development and terminate divisions during pupal stages. Type I NBs located within the ventral nerve cord (VNC) and the central brain (CB) are the predominant type of NBs, whilst type II NBs are eight NB lineages located on the dorsal surface of the CB (Homem & Knoblich, 2012). During each type I NB cell division, NB self‐renews and produces a smaller ganglion mother cell (GMC) that creates a limited number of neurons or glia (Fig 1A). The ability of NBs to divide and generate appropriate progeny number and cell diversity is determined by their ability to maintain asymmetric division, regulate the speed of their cell cycles and timely enter/exit the cell cycle at the beginning and end of neurogenesis (Homem & Knoblich, 2012). Cell intrinsic mechanisms such as the temporal regulation of NB identity via transcription factors that are expressed throughout the life time of the NBs impact on both the numbers and the types of neurons generated by the NB (Doe, 2017). However, more recently, attention has shifted towards understanding how cell extrinsic signals are interpreted by the NBs to alter their behaviour (Ramon‐Canellas et al, 2019).
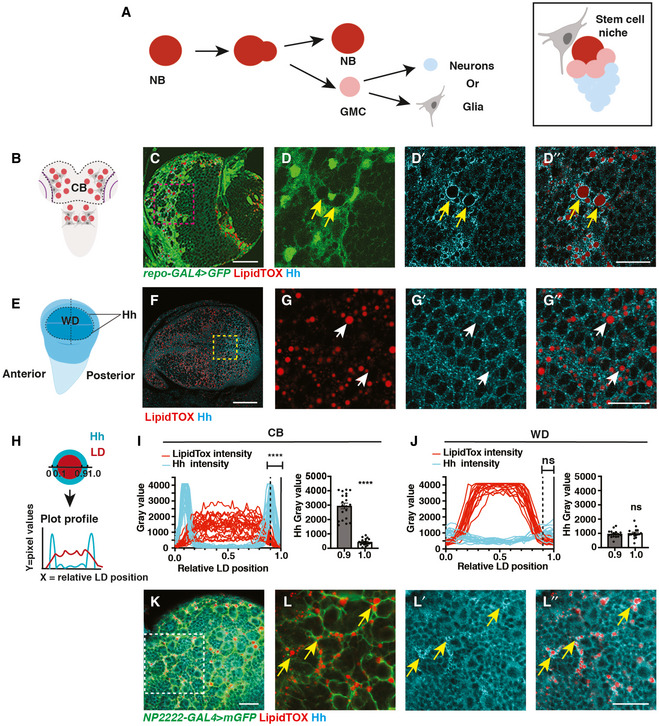
Figure 1. Hh is localised to the LDs within cortex glial cells.
-
ASchematic showing NBs that undergo asymmetric division to self‐renew and produce GMCs, which terminally differentiate to generate post‐mitotic neurons or glial cells (left). Each NB is surrounded by a microenvironment, composed of glial cells (right).
-
B–D”Representative images showing that Hh accumulates on the surface of LDs in glial cells of the CB (yellow arrows), quantified in (I) (n = 20 LDs). Glial cells are marked by repo‐GAL4 > GFP and CB is circled in (B).
-
E–G”In the posterior compartment of the developing wing disc (WD) pouch region where Hh is expressed, LDs and Hh are not tightly associated (white arrows), quantified in (J) (n = 17 LDs).
-
H–JHh‐LD association is quantified by plotting the pixel intensities of both Hh (cyan) and LDs (red) along a line across LDs. Y‐axis represents grey intensity values, and X‐axis represents relative LD position.
-
K, L”Hh‐LD associations are observed in the cortex glia (yellow arrows, NP2222‐GAL4 > mGFP).
Data information: Hh is detected with a Hh antibody and LDs are visualised with LipidTOX unless otherwise stated. (D‐D’’), (G‐G’’), (L‐L’’) are zoomed in images of (C, F, K), respectively. Scale bar = 50 μm in (C and F), scale bar = 20 μm in (D‐D’’, K‐L’’), scale bar = 10 μm in (G‐G’’). Error bar represents SEM. In (I): Welch’s t‐test, (****) P < 0.0001. In (J): unpaired t‐test, (ns) P = 0.7113.
Source data are available online for this figure.
Larval NBs and their progeny are surrounded by a scaffold of glial cell processes, which form the stem cell niche in the CNS (Fig 1A). Glial cells fall into three classes: (i) surface (perineural and subperineural) glia that enwrap the CNS to form the blood brain barrier (BBB); (ii) cortex glia that encapsulate neuronal soma and NBs; and (iii) neuropil glia that are located at the cortex–neuropil interface and form a sheath around the neuropil compartments (Freeman, 2015).
The intimate relationship between glial cells and NBs has been extensively studied in the context of NB entry into the cell cycle at the beginning of post‐embryonic neurogenesis shortly after larval hatch (Ding et al, 2020). Feeding has been shown to trigger insulin production by surface glial cells, which in turn activates the insulin/insulin‐like growth factor pathway in neighbouring NBs and stimulates their growth and proliferation via activation of the phosphoinositide 3‐kinase (PI3K) signalling pathway (Chell & Brand, 2010; Sousa‐Nunes et al, 2011). Once NBs enter into the cell cycle, glial cells continue to play active roles in promoting NB proliferation. These reactivated NBs are found in close association with cortex glia (Hoyle, 1986; Hoyle et al, 1986; Pereanu et al, 2005), and this contact is maintained through adhesion via E‐cadherin. Disruption of NB‐cortex glia contact affects the NB’s ability to undergo mitosis (Dumstrei et al, 2003; Doyle et al, 2017), and the failure to expand the glial membrane also affects both neuronal survival as well as NB cell cycle progression (Speder & Brand, 2018; Yuan et al, 2020). Diffusible molecules that pass from glial cells to influence NB behaviour include Dally‐like (Dlp) in the perineural glia, (Kanai et al, 2018) and Jellybelly (Jeb) in the cortex glia (Cheng et al, 2011). Furthermore, organelles such as lipid droplets (LDs) in the glial niche have been shown to buffer NBs proliferation from peroxidation chain reactions induced by oxidative stress (Bailey et al, 2015), suggesting that glial niche and the signalling molecules produced by these cells are important mediators of non‐autonomous regulation of NB behaviour during developmental and environmental stress.
In this study, we investigate how the stem cell niche and its dysfunction influence stem cell behaviour and the consequences on the brain as a whole. We found that the signalling molecule Hedgehog (Hh), involved in numerous developmental processes, resides within the cortex glial membrane that surrounds NBs. Hh is autonomously required to promote glial niche growth as well as acts non‐cell autonomously to activate the Hh signalling in the NB, triggering its delay in S phase progression. Maintaining cortex glial size is important, as overgrowth induced by FGF activation, a mutation implicated in glioblastoma (Morrison et al, 1994; Yamada et al, 1999; Dienstmann et al, 2014; Jimenez‐Pascual & Siebzehnrubl, 2019), phenocopied the effects of glial Hh activation on NBs. Indeed, inhibiting Hh rescued NB proliferation defects. Furthermore, we demonstrated that downstream of glial FGF signalling, Hh activity and its ability to signal to NBs are modulated by two lipid storage regulators Lsd‐2 and Fasn1. Together, our data show that a dysfunctional niche can non‐autonomously affect NB’s ability to produce the correct number of neurons that make up the adult CNS. This process mechanistically involves the Hh signalling pathway and its modulation by lipid metabolism.
Results
Hh is localised to the LDs within cortex glial cells
To identify potential morphogens that facilitate glia‐NB communication in the CB, we assayed for secreted molecules which are known to act in a paracrine fashion. We found that Hh is expressed at high levels at 96 h After Larval Hatching (96ALH) in glial cells labelled using Repo‐GAL4 > GFP (Fig 1B–D’). Hh is a morphogen that was first identified to regulate embryo segmentation and wing imaginal disc development (Nusslein‐Volhard & Wieschaus, 1980; Heemskerk & DiNardo, 1994). In the wing disc, it is expressed in the posterior compartment and is distributed in a gradient to regulate target gene expression in the anterior compartment (Fig 1E and F) (Chen et al, 2017). In the glial niche, however, we found Hh staining was mostly distributed in ring‐like structures in the glial cytoplasm (yellow arrows, Fig 1C and D’). We then assessed whether Hh is associated with specific organelles. LDs are round‐shaped organelles, consisting of a hydrophobic core for the storage of neutral lipids and a phospholipid monolayer containing LD surface proteins. As LDs have previously been reported to be enriched in the glial niche (Bailey et al, 2015; Kis et al, 2015), we therefore tested whether Hh ligands are associated with LDs. Using a neutral lipid stain, lipidTOX to visualise LDs and either an antibody, or a BAC encoded Hh:GFP to detect Hh (Chen et al, 2017), we found that Hh is localised to the surface of the LDs in the glial niche (yellow arrows, Figs 1C and D’’, I, and EV1A and B’’), but not in the wing disc (white arrows, Figs 1F and G’’, J, and Fig EV1, EV2, EV3, EV4, EV5’’) nor at earlier stages of development in the CB (48 ALH, Fig EV2A and B’’). Together, our data suggest that Hh is localised to the surface of LDs in the glial niche.
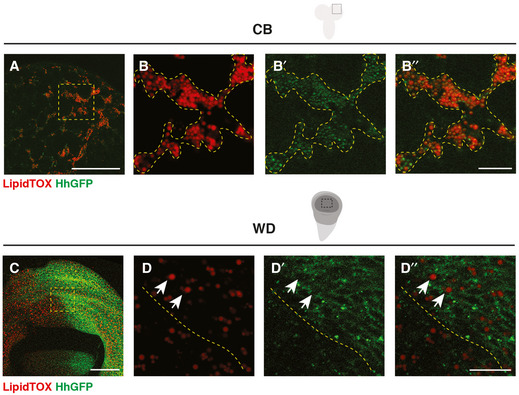
Figure EV1. Hh forms complexes with LDs in the CB but not the wing discs (related Fig 1).
-
A–B’’HhGFP and LDs are associated in the CB glial cells (outlined with yellow dashed lines).
-
C–D’’HhGFP is not associated with LDs in the posterior wing disc (white arrows, the posterior compartment is separated from the anterior with yellow dashed lines).
Data information: (B‐B’’, D‐D’’) are zoomed in images of (A and C), respectively. Scale bar = 50 μm in (A and C). Scale bar = 10 μm in (B‐B’’ and D‐D’’).
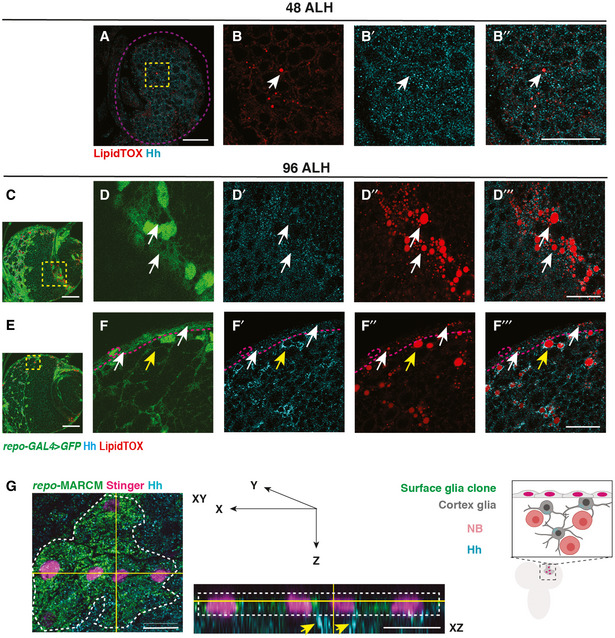
Figure EV2. Hh‐LD associations are specifically observed in cortex glial cells in the CB during late larval stages (related to Fig 1).
-
A–B’’Hh and LDs are present at low levels at 48 ALH, and do not form specific association (white arrows, brain lobes are circled with purple dashed lines).
-
C–D’’’Hh and LDs are not associated in the optic lobe glial cells (white arrows).
-
E–F’’’Hh and LDs associate only in the cortex glial cells (yellow arrows) but not the surface glial cells (white arrows, surface glia are separated from cortex with magenta dashed lines).
-
GLeft and middle panel, representative image showing a surface glial clone (circled with white dashed lines, repo‐MARCM, glial nucleus marked by Stinger in pink). Hh is localised to the cortex glial cells (yellow arrows) underneath the clone marked in green. Right panel, a schematic depicting XZ cross‐section of CB glial cells and their relative position.
Data information: Glial cells are visualised with repo‐GAL4 > GFP in (C‐F’’’). (B‐B’’, D‐D’’’, F‐F’’’) are zoomed in images of (A, C, E), respectively. (C) is the same image as Fig 1C, with the optic lobe region highlighted in a yellow dashed square. Scale bar = 50 μm in (C, E) and XY section in (G). Scale bar = 20 μm in (A‐B’’, D‐D’’’, F‐F’’’), Scale bar = 10 μm for XZ section in (G).
Figure EV3. Effects of Hh overexpression and knockdown on Hh level, NB number and EdU index (related to Fig 2).
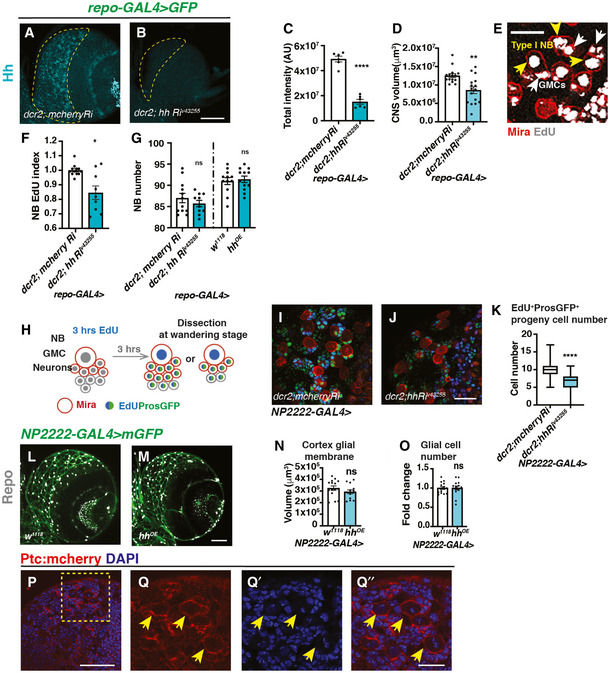
-
A–DRepresentative images showing pan‐glial Hh knockdown (repo‐GAL4 > GFP with UAS‐dcr2) efficiently reduces Hh staining in the CB (outlined with yellow dashed lines) and brain lobe size, quantified in (C) (n = 6, 6 brain lobes) and (D) (n = 15, 16 brain lobes), respectively.
-
ERepresentative image from EdU incorporation assays used throughout the manuscript. During a 15 min EdU pulse, type I NB (yellow arrow) and its GMC (white arrow) both incorporate EdU. EdU index quantifications include only EdU+ type I NBs.
-
FHh knockdown (repo‐GAL4 > GFP with UAS‐dcr2) significantly reduces NB EdU index (n = 12, 9 brain lobes).
-
GHh knockdown or overexpression in glial cells (repo‐Gal4>) does not significantly alter the number of CB NBs (n = 12, 10; 12, 12 brain lobes).
-
HSchematic depicting EdU pulse‐chase experiment. Larvae are fed with EdU‐containing food for 3 h and then chased with EdU‐free food for 3 h before CNS dissection at wandering stages. NBs and newly generated GMCs are marked with Mira; GMCs and newly generated neurons are marked with ProsGFP.
-
I–KRepresentative images showing that Hh knockdown in cortex glial cells (NP2222‐GAL4) significantly reduced the number of EdU+ cells that are marked by ProsGFP+, quantified in (K) (Box plot, the boxes extend from the 25th to 75th percentiles; the median is marked by a central band inside the box; and the whiskers go down to the minimum value and up to the maximum value. n = 94, 104 NB lineages imaged from 8, 8 brain lobes, respectively).
-
L–ORepresentative images showing that Hh overexpression in cortex glial cells (NP2222‐GAL4 > mGFP) does not alter cortex glial membrane size and total Repo+ glial cell numbers, quantified in (N) (n = 15, 14 brain lobes) and (O) (n = 15, 14 brain lobes), respectively.
-
P–Q’’Representative images showing Ptc:mcherry is expressed in NBs (yellow arrows). (Q‐Q’’) are zoomed in images of (P).
Data information: Scale bar = 50 μm in (A, B, E, L, M). Scale bar = 20 μm in (I, J, P, Q‐Q’’). Error bar represents SEM. In (C): unpaired t‐test, (****) P < 0.0001. In (D): Mann–Whitney test, (**) P = 0.0017. In (F): Welch’s t‐test, (*) P = 0.0101. In (G): unpaired t‐test, (ns) P = 0.3645; Mann–Whitney test, (ns) P = 0.7621. In (K): Welch’s t‐test, (****) P < 0.0001. In (N): unpaired t‐test, (ns) P = 0.5151. In (O): unpaired test, (ns) P = 0.9690.
Figure EV4. Glial htlACT overexpression affects NB asymmetric division, size and cell cycle exit (related to Fig 5).
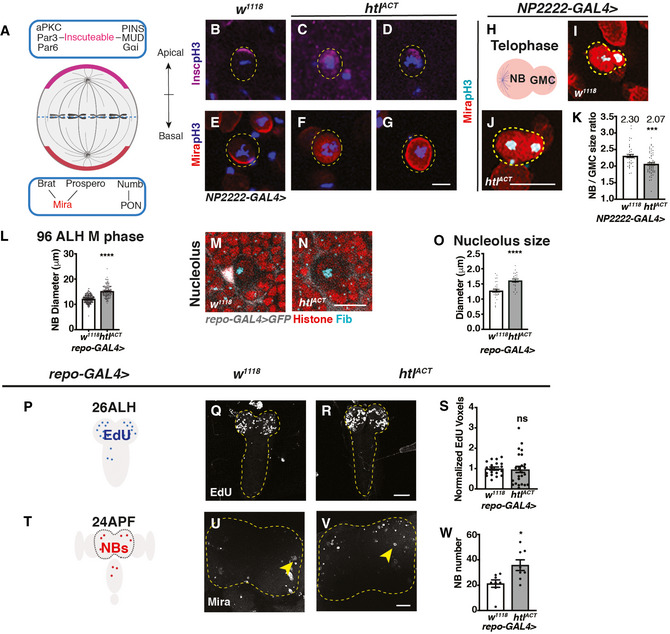
-
ASchematic depicting the distribution of polarity proteins in M phase NBs. Apical polarity proteins include the Par complex (aPKC/Par3/Par6), the PINS/MUD/Gαi complex and the adaptor protein, inscuteable (magenta); basal polarity complex comprises the cell fate determinants Brat/ Pros/ Numb and their adaptor proteins Mira (red) and PON.
-
B–GRepresentative images showing that in pH3+ NBs, Insc and Mira mislocalise to the cytoplasm or cortex upon FGF activation in cortex glia (NP2222‐GAL4 > htlACT).
-
HSchematic depicting a NB undergoing telophase.
-
I–KRepresentative images showing that NBs in telophase (Mira+; pH3+) give rise to more size‐symmetric daughter cells upon cortex glial (NP2222‐GAL4>) htlACT overexpression, quantified in (K) (n = 43, 63 NBs from 10, 9 brain lobes, respectively).
-
LGlial (repo‐GAL4>) htlACT overexpression causes an increase in M phase NB diameter (n = 70, 53 NBs from 12, 7 brain lobes, respectively).
-
M–ORepresentative images showing that NB nucleoli are significantly enlarged upon glial (repo‐GAL4>) htlACT overexpression, quantified in (O) (n = 33, 23 NBs from 9, 7 brain lobes). NBs are marked by Histone (red), surrounded by glial cells (grey, repo‐GAL4 > GFP), nucleoli are marked by Fib (Cyan).
-
P–SRepresentative images showing that the timing of NB cell cycle entry (visualised by EdU incorporation at 26ALH) is not significantly altered by pan‐glial (repo‐GAL4>) htlACT overexpression, quantified in (S), where EdU voxels are normalised to control (n = 19, 25 brains). The region of interest is outlined in yellow.
-
T–WRepresentative images showing that the number of CB NBs (Mira+) at 24APF is significantly increased with pan‐glial (repo‐GAL4>) htlACT overexpression, quantified in (W) (n = 8, 8 brains). The region of interest is outlined by yellow dashed lines and NBs are marked with yellow arrows.
Data information: NBs are outlined with yellow dashed lines in (B‐G, I and J). Scale bar = 50 μm in (Q, R, U, V). Scale bar = 10 μm in (B‐G); Scale bar = 20 μm in (I, J, M and N). Error bar represents SEM. In (K): Mann–Whitney test, (***) P = 0.0002. In (L): Welch’s t‐test, (****) P < 0.0001. In (O): unpaired t‐test, (****) P < 0.0001. In (S): Welch’s t‐test, (ns) P = 0.8152. In (W): unpaired t‐test, (*) P = 0.0134.
Figure EV5. Characterisation of the effects of glial hh, fasn1 and lsd2 RNAis on glial size, LDs and NB proliferation (related to Fig 6).
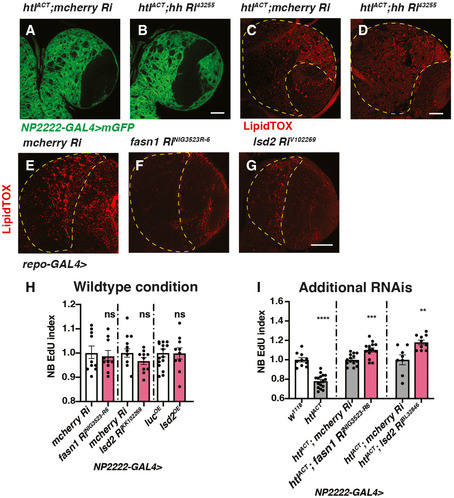
-
A–DRepresentative images showing that induction of hh RNAi in cortex glial cells with htlACT overexpression do not alter the size of cortex glial membrane (NP2222‐GAL4 > mGFP) nor the number of LDs in CB (outlined by yellow dashed lines).
-
E–GRepresentative images showing that glial (repo‐GAL4>) induction of RNAis against fasn1 and lsd2 efficiently reduce the number of LDs in CB (outlined by yellow dashed lines).
-
HKnockdown of lipogenesis genes fasn1 and lsd2 or overexpression of lsd2 using a cortex glial driver (NP2222‐GAL4>) do not significantly affect NB EdU index (n = 10, 10; 15, 10; 10, 10 brain lobes).
-
IThe NB EdU incorporation defects due to cortex glial (NP2222‐GAL4) overexpression of htlACT is rescued by overexpression of additional RNAis lines against fasn1 and lsd2 (related to Fig 6J; n = 10, 16; 14, 14; 8, 10 brain lobes). The NP2222‐GAL4 > w1118 versus htlACT columns depict the same data as those in Fig 5E.
Data information: Scale bar = 50 μm in (A–G). Error bar represents SEM. In (H): Mann–Whitney test, (ns) P = 0.9555; unpaired t‐test, (ns) P = 0.1799; unpaired t‐test, (ns) P = 0.9574. In (I): unpaired t‐test, (****) P < 0.0001; unpaired t‐test, (***) P = 0.0008; unpaired t‐test, (**) P = 0.0032.
We next explored whether Hh‐LD associations are specifically localised to a glial subtype. Hh‐LD associations were largely absent from both surface glial cells that forms the blood brain barrier (BBB) of the CNS (white arrows, Fig EV2E and F’’’), as well as optic lobe glial cells (white arrows, Fig EV2C and D’’’). In fact, Hh‐LD associations were enriched in the cortex glial cells, underneath the sheath‐like surface glial clone generated via repo‐MARCM (yellow arrows, Fig EV2G). Using a cortex‐specific driver, NP2222‐GAL4 (Hayashi et al, 2002; Awasaki et al, 2008), we confirmed that the Hh‐LD associations were localised to the cortex glia (Fig 1K and L’’).
Hh autonomously regulates cortex gliogenesis and non‐autonomously regulates NB proliferation
In the mouse brain, Hh has been shown to promote astrocyte proliferation (Takezaki et al, 2011; Chandra et al, 2015; Ugbode et al, 2017). In glioblastoma (GBM), the Hh/Gli1 signalling pathway acts to accelerate cell proliferation (Chandra et al, 2015). To investigate the role of Hh in Drosophila cortex glial cells, where Hh is most abundant, we used pan‐glial driver repo‐GAL4 to express hh RNAi together with UAS‐Dcr2 (Dicer‐2) which sufficiently depleted Hh expression and reduced overall CNS size (Fig EV3, EV4, EV5). The reduction in CNS volume was accounted for by a significant decrease in Repo+ glial cell number and membrane size (labelled using NP2222‐GAL4 > GFP, Fig 2A–D). Using a pan‐glial driver repo‐GAL4, we found cortex glial chambers were significantly disrupted upon Hh knockdown (Speder & Brand, 2018; Yuan et al, 2020), leading to clustering of NBs (compare Fig 2G to F). However, overexpression of Hh did not affect glial cell number nor membrane size (Fig EV3, EV4, EV5). This suggests Hh is necessary but not sufficient for glial expansion during CNS development.
Figure 2. Hh autonomously regulates cortex gliogenesis and non‐autonomously regulates NB proliferation.
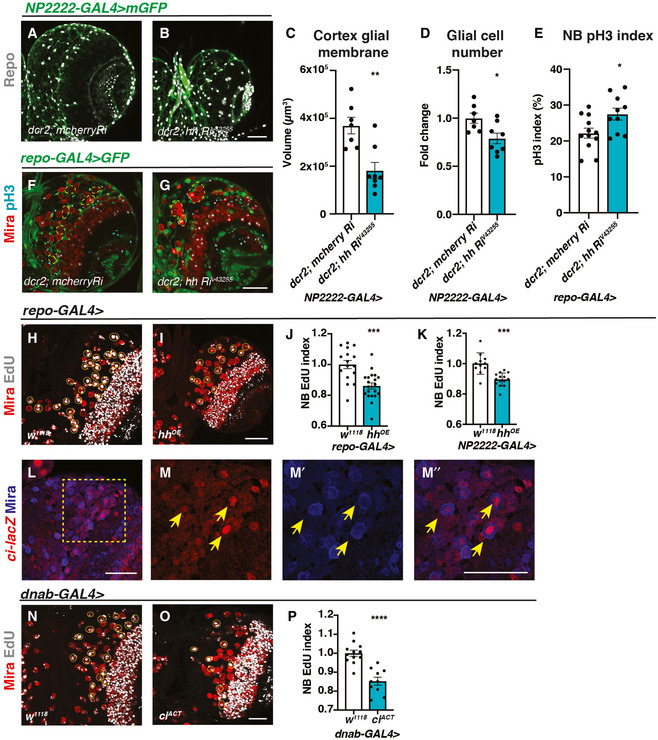
-
A–DRepresentative images showing that upon knockdown of Hh in cortex glial cells (NP2222‐GAL4 > mGFP with UAS‐dcr2), cortex glial membrane and overall Repo + glial cell number are significantly reduced, quantified in (C) (n = 7, 8 brain lobes) and (D) (n = 7, 8 brain lobes), respectively.
-
E–GHh knockdown in glia (repo‐GAL4 > GFP) results in niche disruption and clustering of NBs (circled with yellow dashed line), as well as an increase in the percentage of NBs in M phase (pH3+), quantified in (E) (n = 12, 10 brain lobes).
-
H–KRepresentative images showing that Hh overexpression using pan‐glial (repo‐GAL4) and cortex glial (NP2222‐GAL4) drivers both result in a decrease in NB EdU index, quantified in (J) (n = 16, 20 brain lobes) and (K) (n = 11, 14 brain lobes), respectively.
-
L–M”Representative images showing ci‐lacZ is expressed in NBs (yellow arrows). (M‐M’’) are zoomed in images of (L).
-
N–POverexpression of ciACT in NBs (dnab‐GAL4) reduces EdU index, quantified in (P) (n = 12, 10 brain lobes).
Data information: NBs are marked with Mira and EdU+ NBs are circled by yellow dashed line. Scale bar = 50 μm. Error bar represents SEM. In (C): Mann–Whitney test, (**) P = 0.0059. In (D): unpaired t‐test, (*) P = 0.0176. In (E): unpaired t‐test, (*) P = 0.0215. In (J): unpaired t‐test, (***) P = 0.0002. In (K): unpaired t‐test, (***) P = 0.0001. In (P): unpaired t‐test, (****) P < 0.0001.
Source data are available online for this figure.
Given the role of Hh as a secreted ligand that can act over short range within the NB lineage (Chai et al, 2013) and that it is highly expressed in the cortex glial niche surrounding NBs, it is plausible that glial Hh non‐autonomously affects NB proliferation. We next explored the potential impact of glial Hh on the activities of type I NBs. As Hh is required to maintain the glial niche (Fig 2A–D, F and G) and niche impairment has been shown to induce NB elimination (Read, 2018), we first assessed for alterations in NB number. We found that pan‐glial Hh knockdown (repo‐GAL4) did not significantly alter NB number (Fig EV3G), suggesting that NB survival is unaffected. We then investigated the effects of glial Hh knockdown on NB proliferation. Glial Hh knockdown using repo‐GAL4 induced a small increase in the percentage of NBs in M phase (pH3 index; Fig 2E). To assess NB S phase progression, we examined EdU (5‐ethynyl‐2′‐deoxyuridine) incorporation during a 15‐min time window (EdU index, Fig EV3E, yellow arrows). Here, we found EdU incorporation was significantly reduced upon glial Hh knockdown, suggesting that fewer NBs entered into the S phase of the cell cycle (Fig EV3F). Interestingly, a similar alteration of NB pH3 and EdU index was observed upon glial niche impairment caused by PI3K signalling inhibition (Speder & Brand, 2018). Therefore, it is likely that glial Hh depletion indirectly causes NBs to stall at M phase via inhibition of cortex glial chamber formation. To investigate the effect of glial Hh depletion on NB progeny production, we conducted EdU pulse‐chase assay, where larvae were fed food supplemented with EdU for 3 h and chased for 3 h in EdU‐free food (Fig EV3H). Using pros:GFP which marks individual NB lineages, we found the number of GFP+ and EdU+ cells per lineage was significantly reduced upon cortex glial Hh knockdown (Fig EV3, EV4, EV5K). Together, these results suggest that NB proliferation is inhibited upon cortex glial niche impairment caused by Hh knockdown.
The subperineural glial Dlp and the cortex glial Jeb promote NB proliferation during development; however, overexpression of these signalling molecules in the glial niche was not sufficient to increase NB cell cycle rate (Cheng et al, 2011; Kanai et al, 2018). We next assessed the effect of glial Hh overexpression on NB behaviour. Pan‐glial induction of Hh did not significantly alter NB number (Fig EV3G). However, pan‐glial and cortex glial‐specific Hh overexpression caused a reduction in NB EdU incorporation (Fig 2H–K). Using reporter lines of Hh activity ci ‐lacZ (Schwartz et al, 1995) and Ptc:mCherry (Varjosalo & Taipale, 2008; Chen et al, 2017), we found that Hh signalling is highly active in the NBs (Figs 2L and M’’ and EV3P and Q’’), consistent with a previous report by (Chai et al, 2013). Furthermore, activation of Hh transcriptional activator cubitus interruptus (ciNc5m5m or ciACT) with a NB‐specific driver dnab‐GAL4 (Maurange et al, 2008) significantly reduced NB EdU index (Fig 2N–P), phenocopying the effects of glial Hh overexpression. Together, our data suggest that high levels of glial Hh expression restrict NB cell cycle progression.
Hh activity is modulated by Lsd‐2
Lipid storage droplet‐2 (Lsd‐2) is the Drosophila orthologue of the mammalian perilipin2 and a widely used marker of LDs (Teixeira et al, 2003) (Fig 3A and B’’). Using a GFP reporter against Lsd‐2 together with Hh antibody, we observed that Lsd‐2 and Hh colocalise to the surface of LDs in the cortex glia (yellow arrows, Fig 3C and D’’). To explore whether Lsd‐2 affects Hh activity, we knocked down Lsd‐2 whilst overexpressing Hh in cortex glial cells (NP2222‐GAL4>). Lsd‐2 is known to block the access of lipases, thus promoting lipid storage (Teixeira et al, 2003). As expected, knockdown of Lsd‐2 caused a significant reduction in LDs (Fig 3E–G). Furthermore, this also significantly rescued the NB EdU incorporation defects caused by hh overexpression (Fig 3H–K), suggesting Lsd‐2 modulates Hh’s ability to signal to NBs.
Figure 3. Hh activity is modulated by Lsd‐2.
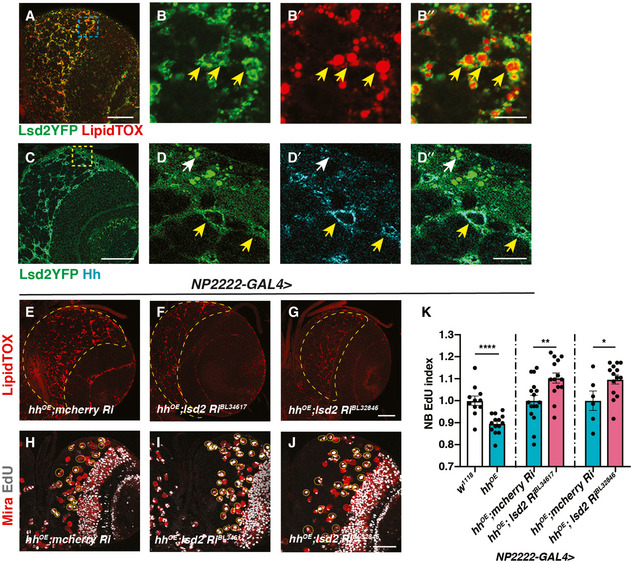
-
A–B”Representative images showing that Lsd2YFP is localised to the surface of LDs (yellow arrows).
-
C–D”Lsd2YFP co‐localises with Hh antibody staining in the cortex glia (yellow arrows) but not surface glial cells (white arrow).
-
E–GLsd‐2 knockdown in cortex glial cells (NP2222‐GAL4) where hh is overexpressed effectively reduces LD number in CB (outlined in yellow dashed lines).
-
H–KRepresentative images showing that NB EdU index is rescued upon Lsd‐2 knockdown in cortex glial cells (NP2222‐GAL4) where hh is overexpressed, quantified in (K) (n = 11, 14; 16, 14; 6, 14 brain lobes). The NP2222‐GAL4 > w1118 versus hhOE columns depict the same data as Fig 2K. EdU+ NBs are circled with yellow, dashed lines.
Data information: (B‐B’’, D‐D’’) are zoomed in images of (A and C). Scale bar = 50 μm in (A, C, E‐J). Scale bar = 10 μm in (B‐B’’ and D‐D’’). Error bar represents SEM. In (K): unpaired t‐test, (***) P = 0.0001; unpaired t‐test, (**) P = 0.0053; unpaired t‐test, (*) P = 0.0349.
Source data are available online for this figure.
FGFR, but not EGFR or InR, activation induces cortex glial overgrowth
Given that we found Hh activity is modulated by a lipid modulator Lsd‐2, it is possible that Hh might act in disease contexts such as glioblastoma, where glial cells undergo increased proliferation and lipid metabolism alterations (Geng & Guo, 2017). As Hh/LD associations mostly localised to the cortex glia that enwrap NBs, we explored the role for cortex glial overgrowth, using previously characterised glioma models, involving the activation of FGF, InR and EGFR (Read et al, 2009; Witte et al, 2009; Reddy & Irvine, 2011; Avet‐Rochex et al, 2012). Firstly, we characterised the effect of overexpression of a wild‐type form of InR, constitutively activated form of the FGF receptor Heartless (Htl) and EGFR. Consistent with the observation of (Avet‐Rochex et al, 2012), we found htlACT but not InRwt overexpression caused an expansion of the cortex glial niche which enwraps NBs (Fig 4A–C, A’–C’). In contrast, EgfrACT overexpression, which acts through Ras signalling, did not affect cortex glial niche size (Fig 4A and A’, D and D’). Using cortex glial‐specific NP2222‐GAL4 and wrapper‐GAL4 (Coutinho‐Budd et al, 2017; Richier et al, 2017), we found that cortex glial overexpression of htlACT, but not InRwt or EgfrACT, led to an increase in total glial cell numbers (Fig 4E–G, I–K). Finally, htlACT overexpression significantly increased the size of the cortex glial membrane (Fig 4E and F, H). Together, our data suggest the activation of FGF, but not InR or EGFR, induced cortex glial niche overgrowth.
Figure 4. Activation of htlACT but not InRwt and EgfrACT induces cortex glial overgrowth.
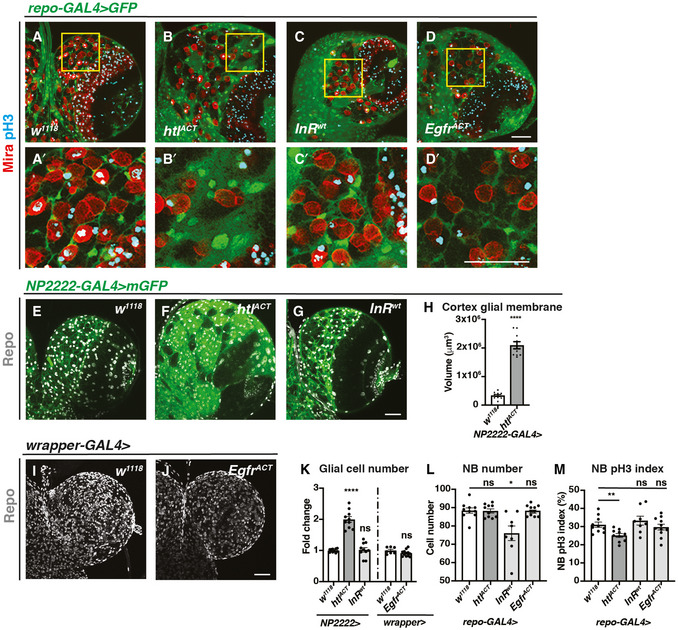
-
A–D’Representative images showing that pan‐glial overexpression of htlACT, but not InRwt or EgfrACT causes an expansion of cortex glia that enwraps NBs. Glial cells are marked with repo‐GAL4 > GFP, and NBs are marked with Mira. (A’, B’, C’ and D’) are zoomed in images of (A, B, C and D), respectively.
-
E–KRepresentative images showing that cortex glial overexpression of htlACT but not InRwt or EgfrACT causes an increase in glial cell (Repo+) numbers and cortex glial membrane size, quantified in (K) (n = 10, 10, 10; 7, 12 brain lobes) and (H) (n = 10, 10 brain lobes), respectively. NP2222‐GAL4 > mGFP is used to mark cortex glial membrane in (E‐G) and wrapper‐GAL4 > is used in (I, J).
-
LGlial (repo‐GAL4>) overexpression of InRwt but not htlACT or EgfrACT significantly reduces the number of CB NBs (n = 10, 10, 8, 11 brain lobes).
-
MGlial (repo‐GAL4>) overexpression of htlACT but not InRwt or EgfrACT significantly reduces the pH3 index of CB NBs (marked with Mira and pH3 in (A‐D’)) (n = 10, 10, 8, 11 brain lobes).
Data information: Scale bar = 50 μm. Error bar represents SEM. In (H): Welch’s t‐test, (****) P < 0.0001. In (K): Welch’s t‐test, (****) P < 0.0001; Welch’s t‐test, (ns) P = 0.7905; unpaired t‐test, (ns) P = 0.0941. In (L): unpaired t‐test, (ns) P = 0.9140; Welch’s t‐test, (*) P = 0.0170; unpaired t‐test, (ns) P = 0.9825. In (M): unpaired t‐test, (**) P = 0.0038; unpaired t‐test, (ns) P = 0.4172; unpaired t‐test, (ns) P = 0.5523.
Source data are available online for this figure.
We next explored the impact of glial niche overgrowth on NBs. Using the pan‐glial driver repo‐GAL4, we measured NB cell number and its impact on the cell cycle assessed with pH3 index. We found that InRwt, but not htlACT and EgfrACT overexpression, caused a reduction in NB number, reminiscent of the NB elimination phenotype reported for glial PvrACT overexpression (Read, 2018) (Fig 4L). In addition, only overexpression of htlACT, which induced cortex glial overgrowth, caused a reduction of NB pH3 index, whilst InRwt and EgfrACT overexpression did not significantly alter NB cell cycle progression (Fig 4M). Together, these data indicate that cortex glial niche is the key glial subset that mediates glia‐NB crosstalk, and its expansion driven by FGF activation affected NB cell cycle progression.
Cortex glial overgrowth mediated by FGF activation slows down NB cell cycle progression
To further investigate the effect of glial FGF activation on NB cell cycle, we assessed S phase progression via EdU incorporation. FGF activation using pan‐glial (repo‐GAL4) as well as cortex glial (NP2222‐GAL4) drivers caused a significant reduction in NB EdU index (Fig 5A–E). Consistent with the reduction in pH3 index (Fig 4M), these experiments confirm that the cortex glia is responsible for NB cell cycle delay induced by FGF activation.
Figure 5. Cortex glial overgrowth mediated by htlACT overexpression triggers NB cell cycle delay.
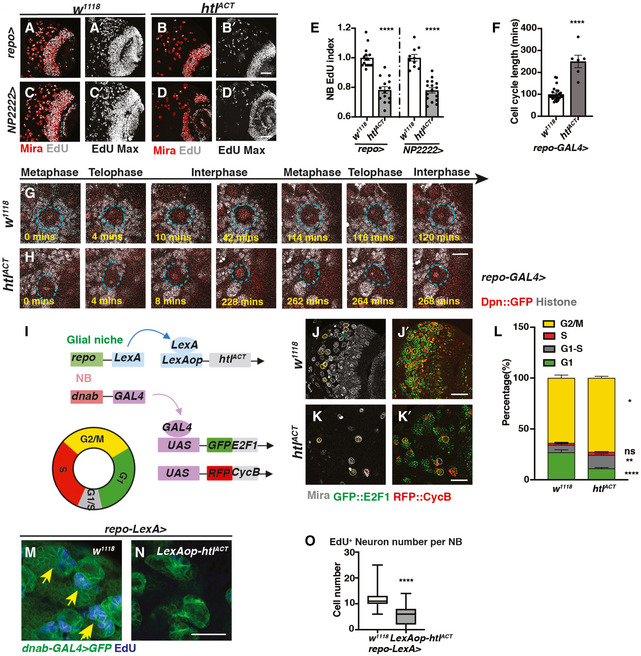
-
A–ERepresentative images showing that both pan‐glial (repo‐GAL4>) and cortex glial (NP2222‐GAL4>) htlACT overexpression significantly reduce NB EdU index, quantified in (E) (n = 15, 14; 10, 16 brain lobes). (A, B, C, D) are single sections with Mira and EdU staining, and (A’, B’ C’, D’) are Z‐projection of the EdU staining.
-
F–HRepresentative still images from ex vivo CNS live imaging at 72ALH showing that pan‐glial (repo‐GAL4) htlACT overexpression lengthens NB cell cycle, quantified in (F) (n = 25, 6 NBs imaged from three brains per genotype). The cell cycle length is measured as the length between consecutive divisions. NBs (Dpn::GFP, red; Histone RFP, grey) are circled with blue dashed lines.
-
ISchematic depicting concurrent glial FGF activation (repo‐LexA > LexAop‐htlACT), and NB overexpression of fly‐FUCCI (dnab‐GAL4 > UAS‐GFP::E2F1, UAS‐RFP::CycB). The fly‐FUCCI system utilises the fusion protein GFP::E2F1 (a marker for cells in G2, M and G1 phase) and RFP::CycB ( a marker for cells in S, G2, M phase) to monitor cell cycle progression. Cells in G1 phase are GFP+ RFP‐ (green), cells in G2/ M phase are GFP+RFP+ (yellow), and cells in S phase are GFP‐RFP+ (red), whereas cells in G1‐S transition are weakly labelled by both GFP and RFP (grey).
-
J–LRepresentative images showing that the percentage of NBs in G1‐S transition and G2/M phase are both significantly increased with significantly less cells remaining in G1 phase, quantified in (L) (n = 9, 10 brain lobes). NBs in G1 phase (Mira+, GFP+) are circled by green dashed lines; NBs in G1‐S transition (Mira+, GFP‐RFP‐) are circled by grey dashed lines; NBs in S phase (Mira+, RFP+) are circled by red dashed lines, and NBs in G2/M phase (Mira+, GFP+RFP+) are circled by yellow dashed lines.
-
M–ORepresentative images showing that the number of EdU+ neurons generated per NB is significantly reduced upon pan‐glial overexpression of FGF (repo‐LexA > LexAop‐htlACT; yellow arrows), quantified in (O) (Box plot, the boxes extend from the 25th to 75th percentiles; the median is marked by a central band inside the box; and the whiskers go down to the minimum value and up to the maximum value. n = 94, 127 NB lineages imaged from five and seven brain lobes, respectively). NB lineages are marked with dnab‐gal4 > GFP.
Data information: Scale bar = 50 μm in (A–D’, J–K’); Scale bar = 10 μm in (G, H); Scale bar = 20 μm in (M, N). Error bar represents SEM unless otherwise stated. In (E): unpaired t‐test, (****) P < 0.0001; unpaired t‐test, (****) P < 0.0001. In (F): Mann–Whitney test, (****) P < 0.0001. In (L): G1: Mann–Whitney test, (****) P < 0.0001; G1‐S: unpaired t‐test, (**) P = 0.0064; S: unpaired t‐test, (ns) P = 0.1757; G2/M: unpaired t‐test, (*) P = 0.0171. In (O): Mann–Whitney test, (****) P < 0.0001.
Source data are available online for this figure.
We next examined the cell cycle length of NBs using live cell imaging on explanted brains, where three brains containing multiple NBs labelled with Dpn::GFP and His::RFP (Fig 5G and H) were imaged. We found FGF activation with repo‐GAL4 induced a severe slowing down of the cell cycle at 96ALH, such that we could not capture any entire NB cell cycles within an 8‐h time window. Previously, it was reported that NBs cycle faster during earlier developmental stages (Maurange et al, 2008; Chai et al, 2013), and therefore, we imaged NB divisions at 72 ALH. We observed that glial FGF activation lengthened NB cell cycle from 100.2 ± 6.0 min to 250.0 ± 28.4 min (Fig 5F, Movies EV1 and EV2).
To decipher which phase of the cell cycle was affected, we generated flies expressing htlACT downstream of a LexA operator (LexAop) (Lai & Lee, 2006). Overexpressing htlACT using repo‐LexA/LexAop system enabled us to concurrently induce NB‐specific expression of Fly‐Fucci using dnab‐GAL4 (as depicted in Fig 5I schematic). With Fly‐Fucci, cell cycle phases can be identified using combinations of two fluorescent fusion proteins (Zielke et al, 2014). Surprisingly, we found glial FGF activation significantly increased the percentage of NBs in G2/M and G1‐S transition at the expense of cells in G1 phase (Fig 5J–L). As the percentage of NBs in M phase (reflected by pH3 index) was reduced upon glial FGF activation, we conclude that these NBs are potentially stalled at G2/ G2‐M and G1‐S transitions of the cell cycle. As a result, NBs cannot efficiently enter S phase or M phase, as indicated by reduced EdU and pH3 indices (Fig 5A–E, and 4M), and, therefore, undergo a dramatically lengthened cell cycle (Fig 5F).
We expect that slowing down of the NB cell cycle upon glial FGF activation would consequentially affect the number of neurons generated by NBs. To test this hypothesis, we conducted EdU pulse‐chase assay (3‐h feeding followed by a 4‐h chase) and counted the number of EdU+ neurons per lineage, which are marked by dnab‐GAL4::UAS‐GFP. We found that overexpression of htlACT with repo‐LexA/LexAop significantly reduced the number of EdU+ neurons generated per NB (Fig 5M–O). Together, our data indicate that FGF activation in cortex glial cells prevents NB cell cycle progression and its ability to produce the correct number of neurons.
Glial FGF activation affects NB asymmetric division, size and cell cycle exit
Given that cell polarity that contributes to NB asymmetric division is established in the G2/M phase, we then assessed whether NB asymmetric division is affected upon glial FGF activation. In the wild type, inscuteable, an adaptor protein that connects the aPKC/Par3/Par6 complex to the PINS/MUD/Gαi complex, forms a crescent at the apical side during NB mitosis (Doe, 1996); these apical complexes further direct the localisation of cell fate determinants (Brat/Pros/Numb) and their adaptor proteins Mira (Shen et al, 1997) and PON to the basal cortex (as depicted in Fig EV4A). The correct distribution of polarity proteins ensures the generation of a larger daughter NB and a smaller GMC upon asymmetric division (Fig EV4H). We found mitotic NBs displayed cytoplasmic or cortical localisation of Mira and Insc upon glial FGF activation (Fig EV4, EV5). Furthermore, telophase NBs and GMCs were also found to be more similar in size (evaluated as NB /GMC size ratio in Fig EV4, EV5K). Intriguingly, both NB and its daughter cells were larger than their wild‐type counterparts, and consistent with this, the average size of mitotic NBs was also significantly larger upon glial htlACT overexpression (Fig EV4L, from 12.44 ± 0.16μm to 15.55 ± 0.34μm). This increase in cell size was coupled with elevated cellular growth, as indicated by an increase in nucleoli size (Fig EV4, EV5, from 1.28 ± 0.05μm to 1.61 ± 0.05μm). It is therefore likely that the delay between consecutive cell cycles allowed these NBs to grow larger.
To investigate when glial FGF starts to impact on NB proliferation, we assessed NB EdU index at 26ALH, a time point when most NBs reactivate from quiescence, and commence post‐embryonic neurogenesis (Fig EV4P). We found glial htlACT overexpression did not significantly affect NB EdU incorporation during a 1‐h EdU pulse (Fig EV4, EV5), suggesting that glial FGF does not affect NB reactivation at the beginning of neurogenesis. We then examined whether NB termination at 24 h after pupal formation (24APF) is altered (Fig EV4T). Glial htlACT overexpression resulted in the presence of increased number of NBs at 24APF (Fig EV4, EV5), suggesting NB cell cycle exit is possibly delayed. However, it is not clear whether NBs that persist are capable of dividing. Together, we conclude glial FGF mostly exerts its effects on reducing NB cell proliferation during late larval neurogenesis, coinciding with the time when Hh‐LD associations become highly enriched in the cortex glia. Taken together, our results revealed that NB activities including its proliferation, asymmetric division and termination are affected by cortex glial niche overgrowth driven by FGF activation.
Hh mediates the effects of glial FGF signalling on NB proliferation
Given that glial Hh overexpression or activation of the Hh signalling cascade in NBs similarly inhibits NB proliferation and induces polarity protein delocalisation (Chai et al, 2013); it is therefore plausible that Hh lies downstream of glial FGF to regulate NB activity. To test this hypothesis, we first assessed whether niche‐derived signals including Hh and its modulators such as LD synthesis enzymes were altered. By RT‐qPCR, we found hh mRNA was upregulated by 8‐fold (normalised to rpl32) upon glial FGF activation (repo‐GAL4, Fig 6A). Amongst genes that promote LD storage (Fig 6B), fatty acid synthase‐1 (fasn1) (Smith et al, 2003) and lsd2 (an antagonist of lipases which we have shown to regulate Hh activity, Fig 3) were also significantly upregulated (Fig 6A). Furthermore, Hh staining was found to be more widely distributed in the glial cytoplasm, rather than restricted to the ring‐like structures surrounding LDs upon FGF activation (Fig 6C–G, evaluated by the ratio of glial cytoplasm that contains Hh staining).
Figure 6. Hh and lipid metabolism regulators mediate the effects of glial htlACT on NB proliferation.
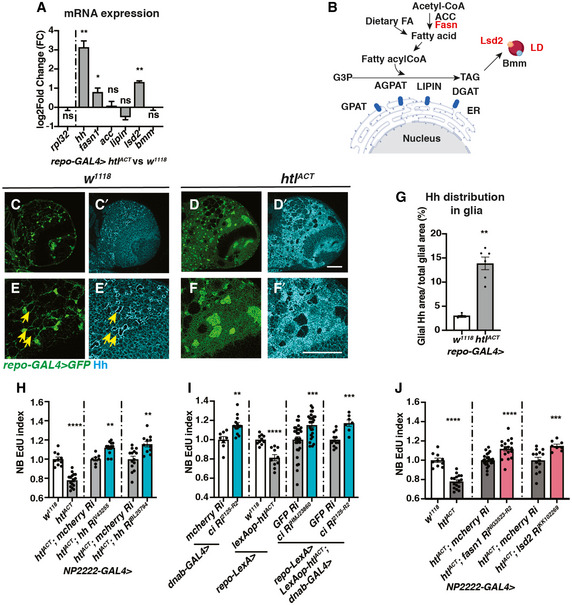
-
APan‐glial (repo‐GAL4) htlACT overexpression causes upregulation of hh, fasn1 and lsd2 transcripts (n = 3 biological replicates pooled from 20 brains for each genotype; for each biological replicate, we ran three technical replicates for each PCR reaction). The lipogenesis genes (acc, lipin), and lipolysis gene bmm transcripts are not significantly altered. We utilised rpl32 as a reference gene in these experiments, as it is not altered by htlACT overexpression. The data are represented by log2‐fold change relative to the control (repo‐GAL4 > w1118).
-
BSchematic depicting lipogenesis and lipolysis. Lipogenesis begins with de novo synthesis of fatty acids by carboxylation of cytosolic acetyl‐CoA via acetyl‐CoA carboxylase (ACC) and elongation of fatty chain via fatty acid synthase (Fasn, red). Dietary‐derived and de novo‐generated fatty acids are converted into fatty acylCoA, which re‐localises to ER and participates in triglyceride (TAG) synthesis with glycerol‐3 phosphate (G3P). This process is mediated by a series of enzymes: glycerol‐3‐phosphate acyltransferase (GPAT), acylCoA acylglycerol‐3‐phasphte acyltransferases (AGPAT), Lipin (a phosphatidate phosphatase) and diacylglycerol acyltransferase (DGAT, red). TAG is translocated from the ER to the core of the intracellular organelles called LDs. On the surface of LDs, a triglyceride lipase called Brummer (Bmm), and its inhibitor Lsd‐2, antagonistically control TAG storage.
-
C–GRepresentative images showing that Hh staining normally localised to a ring‐like structure (yellow arrows), becomes delocalises to the glial cytoplasm upon htlACT overexpression, quantified in (E) (n = 4, 6 brain lobes). Glial cells are marked with repo‐GAL4 > GFP. (E‐F’) are zoomed in images of (C‐D’).
-
HCortex glial (NP2222‐GAL4>) overexpression of two independent hh RNAis significantly rescue EdU incorporation defects caused by htlACT overexpression (n = 10, 16; 7, 12; 14, 11 brain lobes). The NP2222‐GAL4 > w1118 versus htlACT columns depict the same data as those in Fig 5E.
-
IKnockdown of NB Hh signalling pathway (dnab‐GAL4 > UAS‐ciRNAi) rescues NB EdU incorporation defects induced by glial htlACT overexpression (repo‐LexA > LexAop‐htlACT). Induction of ciRNAi in NBs alone increases NB EdU incorporation (n = 8, 13; 10, 10; 30, 26; 12, 7 brain lobes).
-
JThe NB EdU incorporation defects due to cortex glial (NP2222‐GAL4>) overexpression of htlACT is significantly rescued by overexpression of RNAis against fasn1 and lsd2, compared to corresponding control RNAis (n = 10, 16; 25, 17; 14, 8 brain lobes). The NP2222‐GAL4 > w1118 versus htlACT columns depict the same data as those in Fig 5E. The control column for htlACT; lsd2 RiKK102269 depicts the same data as the control column for htlACT; hh RiBL25794 in Fig 6H.
Data information: Scale bar = 50 μm. Error bar represents SEM. In (A), t‐test was conducted to compare log transformed fold change: rpl32: Welch’s t‐test, (ns) P = 0.9355; hh: unpaired t‐test, (**) P = 0.0061; fasn1: unpaired t‐test, (*) P = 0.0242; acc: unpaired t‐test, (ns) P = 0.7570; lipin: unpaired t‐test, (ns) P = 0.1222; lsd2: unpaired t‐test, (**) P = 0.0074; bmm: unpaired t‐test, (ns) P = 0.9356. In (G): Mann–Whitney test, (**) P = 0.0095. In (H): unpaired t‐test, (****) P < 0.0001; Mann–Whitney test, (**) P = 0.0031; unpaired t‐test, (**) P = 0.0012. In (I): unpaired t‐test, (**) P = 0.0019; unpaired t‐test, (****) P < 0.0001; Mann–Whitney test, (***) P = 0.0006; unpaired t‐test, (***) P = 0.0003. In (J): unpaired t‐test, (****) P < 0.0001; unpaired t‐test, (****) P < 0.0001; Welch’s t‐test, (***) P = 0.0004.
Source data are available online for this figure.
We next assessed the role of Hh signalling downstream of glial FGF activation. Hh knockdown (RNAi efficiency tested in (Tian et al, 2015)) rescued NB S phase delay (Fig 6H) without affecting glial niche size (Fig EV5A and B). In addition, we used a LexA/LexAop binary expression system in conjunction with the GAL4/UAS system, to simultaneously activate glial FGF and inhibit NB Hh signalling. Induction of RNAi against ci in the NB caused a significant increase in EdU incorporation (Fig 6I, dnab‐GAL4 > ci RNAi). Together with glial FGF activation, Ci knockdown also significantly rescued NB EdU index (Fig 6I). Together, our results suggest that Hh mediates glia‐NB crosstalk downstream of glial FGF activation.
Lipid metabolism genes lie downstream of glial FGF‐NB crosstalk
As we previously showed that Lsd‐2 modulates Hh activity (Fig 3), we hypothesise that lipid metabolism enzymes function upstream of Hh to regulate NB behaviour. Consistent with this, we found induction of hh RNAi upon cortex glial FGF activation did not alter the number of LDs (Fig EV5C and D). We next explored whether lipid metabolism genes mediate the effects of glial FGF activation on NB proliferation. Glial expression of RNAis targeting lipogenesis genes fasn1 and lsd2 efficiently reduced LDs (Fig EV5E–G), but were not required for wild‐type NB S phase progression (Fig EV5H). However, knockdown of Fasn1 and Lsd2 significantly rescued NB S phase progression defects caused by htlACT overexpression (Figs 6J and Fig EV5I), suggesting lipid metabolism genes function downstream of glial FGF‐NB crosstalk.
Fasn1 and Rasp affect glial Hh palmitoylation to regulate NB cell cycle
Hh is known to be synthesised as a precursor protein (HhN) which undergoes a series of post‐translational modifications within the secretory pathway (Fig 7A) (Mann & Beachy, 2004). The N‐ and C‐termini of Hh proteins are covalently modified with palmitate and cholesterol, respectively (Porter et al, 1996; Pepinsky et al, 1998). Palmitate is a 16‐carbon saturated fatty acid, which can either be diet‐derived or synthesised via de novo fatty acid synthesis, mediated by enzymes such as Fasn and ACC (Schiller & Bensch, 1971; Slakey et al, 1979; Innis, 2016). Given Fasn1, an enzyme involved in de novo synthesis of palmitate, is also involved in glial FGF‐NB crosstalk, we assessed whether Hh palmitoylation is required downstream of glial FGF signalling. Palmitoylation of the N‐terminal of HhN is mediated by a dedicated acyltransferase in the ER called Rasp in Drosophila (Fig 7A). In the embryo, Rasp has been shown to be required for Hh diffusion (Chamoun et al, 2001; Lee & Treisman, 2001; Micchelli et al, 2002). We found cortex glial knockdown of Rasp whilst not required for NB S phase progression (Fig 7B) was sufficient to rescue NB EdU incorporation defects caused by FGF activation (Fig 7B, NP2222‐GAL4 > htlACT).
Figure 7. Fasn1 affects glial Hh palmitoylation to regulate its signalling to NBs.
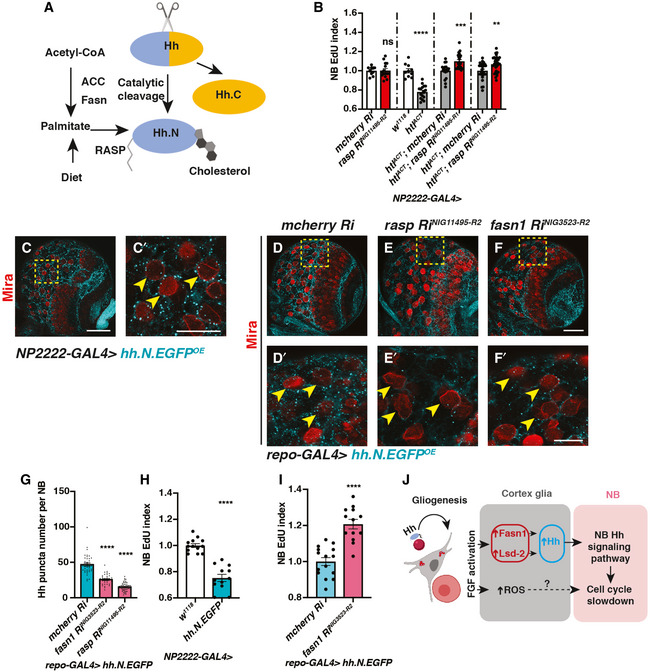
-
ASchematic depicting Hh auto‐processing, which starts with the cleavage of the protein into a C‐terminal part (Hh.C, yellow) and a N‐terminal part (Hh.N, blue), with simultaneous covalent addition of cholesterol. Palmitate, from either diet or de novo lipogenesis (via ACC and Fasn), is added onto Hh‐N, in a reaction catalysed by an acyltransferase, encoded by rasp.
-
BInhibition of palmitoylation (via two independent rasp RNAis) rescues NB EdU incorporation defects induced by cortex glial (NP2222‐GAL4>) htlACT overexpression, whilst knockdown of Rasp in cortex glial cells alone does not alter NB EdU index (n = 10, 15; 10, 16; 18, 21; 26, 29 brain lobes). The NP2222‐GAL4 > w1118 versus htlACT columns depict the same data as those in Fig 5E.
-
C–C’Representative images showing that Hh.N.EGFP (which cannot undergo cholesterol modification) are found as puncta on the surface of NBs (yellow arrows) when overexpressed in neighbouring cortex glial cells (NP2222‐GAL4>).
-
D–GRepresentative images showing that knockdown of Fasn1 in glial cells (repo‐GAL4>), where Hh.N.EGFP is overexpressed, significantly reduces the number of Hh.N.EGFP puncta on the surface of NBs (yellow arrows), phenocopying the effect of Rasp knockdown, quantified in (G) (n = 38, 38, 57 NBs from 4, 4, 8 brain lobes, respectively).
-
HCortex glial (NP2222‐GAL4>) overexpression of Hh.N.EGFP significantly reduces NB EdU incorporation (n = 13,12 brain lobes).
-
IKnockdown of Fasn1 in glial cells rescues NB EdU incorporation defects, caused by glial Hh.N.EGFP overexpression, quantified in (I) (n = 15, 13 brain lobes).
-
JSchematic depicting our working model. During development, Hh tethered to LDs are localised to cortex glial cells, to activate gliogenesis. Non‐autonomously, excessive glial Hh inhibits NB cell cycle progression. Upon cortex glial‐specific FGF activation, increased Hh modified by Fasn1 and Lsd‐2 together with increased ROS prevent NB proliferation.
Data information: (C’, D’, E’ and F’) are zoomed in images of (C, D, E and F), respectively; NBs are marked with Mira. Scale bar = 50 μm in (C, D, E, F). Scale bar = 20 μm in (C’, D’, E’ and F’). Error bar represents SEM. In (B): unpaired t‐test, (ns) P = 0.9403; unpaired t‐test, (****) P < 0.0001; Mann–Whitney test, (***) P = 0.0004; Mann–Whitney test, (**) P = 0.0047. In (G): Welch’s t‐test, (****) P < 0.0001; Welch’s t‐test, (****) P < 0.0001. In (H): unpaired t‐test, (****) P < 0.0001. In (I): unpaired t‐test, (****) P < 0.0001.
Source data are available online for this figure.
To test whether Hh palmitoylation is required for its ability to signal to NBs, we expressed a GFP tagged and non‐cholesterol modifiable form of HhN in cortex glial cells (Wendler et al, 2006; Hartman et al, 2013). HhN‐GFP was mostly localised to puncta in the glial cytoplasm, and some of these puncta made contact with NBs (Fig 7C‐C’, NP2222‐GAL4). The number of puncta making contact with NBs was significantly reduced upon knockdown of Rasp (Fig 7E‐E’ compared to D‐D’, G), suggesting that palmitoylation is required for HhN transport from glia to NBs. Similar to full‐length Hh (Fig 2J and K), glial HhN expression caused a reduction in NB EdU incorporation (Fig 7H), suggesting that palmitoylated Hh is sufficient for glia‐NB crosstalk.
Similar to Rasp knockdown, Fasn1 knockdown caused a reduction in the number of HhN‐GFP puncta that made contact with NBs (Fig 7D–G). To test whether Fasn1 is functionally required to mediate HhN’s ability to cause NB S phase progression defects, we knocked down Fasn1 whilst overexpressed HhN. In this setting, Fasn1 knockdown rescued EdU incorporation defects caused by HhN expression (Fig 7I). Together, our data suggest that palmitoylation via Rasp and Fasn1, in addition to regulation by Lsd‐2, are required for Hh function in the context of glia‐NB crosstalk.
Glial ROS acts in parallel to lipid‐Hh signalling to regulate NB proliferation in the FGF‐driven glioma model
Lipid metabolism alteration in glia has previously been linked to reactive oxygen species (ROS), where excessive ROS production causes LD accumulation, which in turn triggers neurodegeneration (Bailey et al, 2015; Liu et al, 2015). Furthermore, ROS upregulation is known to promote glioblastoma progression (Schieber & Chandel, 2014). We therefore tested whether lipid metabolism changes in the FGF‐driven glioma model are correlated with ROS levels. Using ROS‐inducible gstD promoter‐GFP reporter (Sykiotis & Bohmann, 2008), we detected a 5‐fold increase in gstD‐GFP upon FGF activation in glial cells (Appendix Fig S1A–C). To decipher the effect of ROS manipulation on lipid metabolism, we overexpressed a Catalase (Anderson et al, 2005) and a superoxide dismutase 1 (Martin et al, 2009) in cortex glial cells where FGF was activated to suppress ROS. And we found the number of LDs, an indicator of lipid metabolism, was not altered upon ROS reduction (Appendix Fig S1D’, E’, F’, and H). These manipulations were however effective in partially rescuing NB EdU incorporation defects caused by glial FGF activation (Appendix Fig S1I) without affecting glial overgrowth (Appendix Fig S1D–G). We conclude that the production of ROS upon FGF activation in the glial niche potentially acts in parallel with lipid‐Hh signalling to inhibit NB proliferation.
Collectively, our findings indicate that the expression of Hh in the cortex glia is required for the formation of glial chambers. Moreover, we demonstrate that expression of Hh in the cortex glial niche must be restrained to prevent ectopic Hh signalling in the NBs. We showed that the ability of glial Hh to signal to NBs is modulated by lipid metabolism enzymes Fasn1 and Lsd‐2. Upon overgrowth of the cortex glial niche induced by FGF activation, Hh, lipid metabolism regulators as well as ROS are upregulated in the niche, which in turn slow down the NB cell cycle and affect its ability to generate a full repertoire of neurons (Fig 7J).
Discussion
In the mammalian CNS, neuron and glia interactions are complex. Astrocytes that are structurally related to the cortex glia in Drosophila enwrap multiple neurons and NSCs, and are known to modulate adult neurogenesis through soluble signals such as morphogens and extracellular matrix proteins (Spampinato et al, 2019). Similarly, the Drosophila cortex glial niche, which form chambers that encapsulate NB and its progeny, creates microenvironments that are required for NB maintenance and neuronal maturation (Dumstrei et al, 2003; Pereanu et al, 2005; Coutinho‐Budd et al, 2017; Speder & Brand, 2018; Yuan et al, 2020). Furthermore, glial overgrowth observed in the context of glioblastoma has recently been shown to affect NBs and neuronal survival (Read, 2018; Portela et al, 2019). In this study, we have identified a new mechanism of glia‐NB crosstalk via Hh modulated by lipid regulators that affect NB cell cycle progression and lineage size.
During development, NB lineages have been shown to produce its own pool of Hh (Chai et al, 2013), which likely act redundantly with the glial Hh. However, glial Hh levels must be tightly regulated to ensure NB can progress through the cell cycle, and produce the appropriate number and types of neurons. Similar to pro‐proliferative roles of Hh in the astrocytes of the mammalian brain (Ugbode et al, 2017), Drosophila glial‐ Hh is autonomously required for the growth of cortex glial cells and the formation of the glia chambers which surround NBs. Hh knockdown caused NB clustering which in turn induced moderate NB cell cycle delay and reduced neuron production, reminiscent of phenotypes reported for glial PI3K inhibition which also caused disrupted glial chambers (Speder & Brand, 2018). This contrasts with Hh and Smo mutant NB phenotype, previously reported by Chai et al (2013), where they found the loss of NB Hh signalling caused a significant increase in NB clone size. Therefore, our data support the model that glial Hh is required for niche elaboration, which in turn affects NB proliferation.
Whilst moderate level of Hh is permissive for NB cell cycle progression, excess glial Hh through either its overexpression or via FGF activation also affects NB cell cycle progression and its ability to produce neurons. In this case, excess glial Hh non‐autonomously activated Hh signalling in the NBs, which is sensitive to this ligand. Different to other niche‐derived secreted molecules, such as Dlp (Kanai et al, 2018) and Jeb (Cheng et al, 2011), which are necessary but not sufficient to induce changes in the NB, Hh plays a physiologically relevant role in disease models of glial overgrowth caused by FGFR activation. TCGA data show that FGFR1–4 is expressed in different combinations in glioblastoma patient samples, and FGFR1 is an important contributor to poor outcome in glioblastoma (Jimenez‐Pascual & Siebzehnrubl, 2019).
In the Drosophila brain, cortex glia appears to the key glial subtype that is responsible for glia‐NB crosstalk. Only FGF but not glial InR and EGFR activation caused an expansion of cortex glia (Avet‐Rochex et al, 2012, 2014), which in turn increased Hh levels and reduced NB proliferation. In cortex glial cells, Hh closely associates with LDs which are analogous to lipoproteins that have been shown to transport Hh for long‐range signalling (Panakova et al, 2005; Palm et al, 2013). Different from lipoproteins, LDs mainly act as lipid storage organelles and are less mobile. Consistent with this, upon FGF activation, genes that promote lipid synthesis and storage were upregulated. So how does lipid metabolism regulate Hh function? We showed two mechanisms. The first mechanism involves the interaction between lipid storage regulator Lsd‐2 and Hh that colocalise on the surface of LDs and that Lsd‐2 modulates Hh’s ability to signal to NBs. It is not yet clear how Lsd‐2 directly regulates Hh activity, but we think it is possible that Lsd‐2 might physically interact with Hh and affect its secretion, or alternatively, Lsd‐2 competes with Hh for positions on the surface of LDs and pushes Hh into the cytoplasm. The second mechanism involves Fasn1and Rasp which regulates Hh‐post‐translational modification. Given that lipid synthesis and LDs have recently been reported to be upregulated in glioma, and are emerging as important biomarkers and metabolic targets (Guo et al, 2013; Geng & Guo, 2017), it would be interesting to further explore how lipid metabolism regulators affect signalling in this context. In addition to the Hh‐LD axis, ROS which is implicated in glioblastoma (Schieber & Chandel, 2014) also lies downstream of FGF‐mediated glia‐NB crosstalk. Knockdown of both Hh and ROS axes significantly rescued NB cell proliferation defects caused by FGF activation in the glial cells. The phenomenon we reported here together with other reports that glioblastoma affects the survival or proliferation rate of its neighbours (Read, 2018; Portela et al, 2019) poses an interesting but yet unexplored prospect that glioma outcompetes NBs within the CNS for limited energy and nutrient resources.
Materials and Methods
Reagents and Tools Table
| Reagent/Resource | Reference or Source |
|---|---|
| Oligonucleotides and other sequence‐based reagents | |
|
RT‐qPCR primers for rpl32 Forward primer: CCGCTTCAAGGGACAGTATCTG Reverse primer: ATCTCGCCGCAGTAAACGC |
Akkouche et al (2017) |
|
RT‐qPCR primers for fasn1 Forward primer: TAAGGAGGTCTGCACAAAGCC Reverse primer: CGGTGAGAGGGTGATGATCG |
PrimerBlast |
|
RT‐qPCR primers for acc Forward primer: CCTCATTAACCCGCGCTACA Reverse primer: TTTTCAGCGCAATGGTGGTC |
PrimerBlast |
|
RT‐qPCR primers for lipin Forward primer: AAACGAAGCTGAGACGGAGAA Reverse primer: GGTTTTGCTCTTGGACACCTC |
FlyPrimerBank |
|
RT‐qPCR primers for lsd2 Forward primer: ATTGCCCGTGGTAAATGCG Reverse primer: CGAAGACACGATTTTTGCCTTT |
FlyPrimerBank |
|
RT‐qPCR primers for bmm Forward primer: GTCTCCTCTGCGATTTGCCAT Reverse primer: CTGAAGGGACCCAGGGAGTA |
FlyPrimerBank |
|
RT‐qPCR primers for hh Forward primer: TGCTCCGTCAAGTCAGATTCG Reverse primer: GTTGGCGGTCATGCTCAAAA |
PrimerBlast |
Methods and Protocols
Fly husbandry and strains
Fly stocks were reared on standard Drosophila media at 25°C. Crosses for overexpression and knockdown experiment were set up at 25°C, and after a day, the progenies were moved to 29°C, unless otherwise stated.
The fly strains used were as follows: repo‐GAL4 (BDSC7415), NP2222‐GAL4 (KYOTO112830), wrapper‐GAL4 (Coutinho‐Budd et al, 2017; Richier et al, 2017), dnab‐GAL4 (From Alex Gould laboratory), repo‐LexA::GAD (BDSC67096), w1118,UAS‐htlACT (BDSC5367), UAS‐EgfrACT (BDSC59843), UAS‐InRwt (BDSC8262), LexAop‐htlACT (generated in this paper), UAS‐GFP, UAS‐mGFP, UAS‐dcr2, UAS‐FUCCI (BDSC55110), UAS‐lacZ, UAS‐luc (BDSC64774), UAS‐hh (from Thomas B. Kornberg lab), UAS‐hh.N.GFP (BDSC81023), UAS‐lsd2 (from Alex Gould lab), UAS‐ciNc5m5m(ACT) (from Yu Cai lab), UAS‐cat.A (BDSC24621), UAS‐Sod1.A (BDSC24754),UAS‐mcherryRNAi (BDSC35785), UAS‐GFPRNAi (BSDC9331), UAS‐fasn1RNAis (NIG3523R‐2, NIG3523R‐6), UAS‐lsd2RNAis (VDRC102269, BDSC34617 and BDSC32846), UAS‐hhRNAis (VDRC43255, BDSC25794), UAS‐ciRNAis (NIGHMJ23860, NIG2125R‐2), UAS‐raspRNAis (NIG11495R‐1, NIG11495R‐2), dpnGFP (BDSC59755), his2AV‐mRFP (Kieran Harvey laboratory), Hh:GFP BAC, Ptc:mcherry BAC, ci‐lacZ (all three lines are generated by Thomas B. Kornberg laboratory), lsd2YFP (KYOTO115301) and gstD‐GFP (From Tatsushi Igaki laboratory), prosGFP (VDRC318418). The repo‐MARCM stock was as follows: UAS‐RedStinger; repo‐flp, repo‐GAL4, UAS‐actinGFP; FRT82B, tub‐gal80 (from Joseph M. Bateman laboratory). w;;FRT82B was used to generate surface glial clones.
Immunostaining
Larval and pupal brains were dissected in PBS, fixed for 25 min in 4% formaldehyde (Sigma‐Aldrich, #F8775) in PBS and rinsed in 0.5% PBST (PBS + 0.5% Triton X‐100 (Sigma‐Aldrich, #T8787)). For immunostaining, brains were incubated with primary antibodies overnight at 4°C, followed by an overnight secondary antibody incubation at °C. Samples were mounted in 80% glycerol in PBS for image acquisition. The primary antibodies used were mouse anti‐Mira (1:50; gift from Alex Gould), rat anti‐Mira (1:100, Abcam, #ab197788), rabbit anti‐Mira (1:200, gift from Rita Sousa‐Nunes), rat anti‐pH3 (1:500; Abcam, #ab10543), chick anti‐GFP (1:2000; Abcam, #ab13970), rabbit anti‐RFP (1:100, Abcam, #ab62341), mouse anti‐Fibrillarin (1:200, Abcam, #ab4566), rabbit anti‐Hh (1:500, gift from Isabel Guerrero) and rabbit anti‐Insc (1:20, gift from William Chia). Secondary donkey antibodies conjugated to Alexa 555 and Alexa 647 and goat antibody conjugated to Alexa 405, 488, 555 and 647 (Molecular Probes) were used at 1:500. DAPI (Molecular Probes) was used at 1:10,000.
EdU labelling and pulse‐chase
EdU in vitro labelling was used to identify actively dividing NBs, and larval brains were incubated in 10 μM EdU /PBS for 10–15 min (for 96ALH brains) or 1 h (Fig EV4, EV5), followed by fixation, and development using Click‐iT Plus EdU Cell Proliferation Kit for imaging, Alexa Fluor 647 dye (Invitrogen, #C10640), following the manufacturer’s instruction. The brain tissues were then stained with Mira to label NBs. Control and experimental brains were processed in the same tube to ensure they were exposed to the same incubation conditions.
EdU pulse‐chase was used to identify the progeny of dividing NBs. 3rd instar larvae were fed with instant fly food supplemented with 100 μg/ml EdU (Lee et al, 2006) for 3 h. They were then transferred to standard Drosophila media for a 3 or 4 h EdU chase. Wandering stage larvae were collected for brain dissection, followed by fixation, development and immunostaining as described above.
LD staining
For LD staining, larval brains were dissected in PBS, fixed and rinsed in PBS before incubation in HCS LipidTOX Red Neutral Lipid Stain (Invitrogen, #H34476, 1:1,000 in PBS) for 1 h. These samples were then rinsed and mounted in PBS and imaged directly to preserve LD morphology. For experiments that require LD staining together with immunostaining, the tissues were rinsed three times in PBS after immunostaining to remove all PBST and incubated with LD dyes as described above. The tissues were mounted in 80% glycerol in PBS for imaging.
Imaging and image processing
Images were collected on a Leica SP5 or Olympus FV3000 confocal microscopes and analysed using Fiji (https://imagej.net/Fiji). Z stacks of CBs were imaged, and the ventral side of the CB was shown as the representative image unless otherwise stated.
Live cell imaging
Dissected brains (72ALH) were cultured in Schneider’s culture medium supplemented with 10% inactivated FBS, 2% Penicillin‐Streptomycin solution (Sigma‐Aldrich, #P4458), 20 μM glutamine and Schneider’s culture medium (Gibco, #21720024) and dissected fat body from the same animals. The brains were imaged in a μ‐Slide 8 well (Ibidi, #80806) on an Olympus FV3000 microscope using resonance scanner in 16Bit mode, with a 40×/0.95 lens and 2× zoom. Z stacks with 2 μm intervals were captured every 2 min over a period of 3–8 h. Laser intensity was kept low to avoid cytotoxicity. AVI movies were generated using Fiji.
All the images were processed using Adobe Photoshop and compiled using Adobe Illustrator. For the purpose of better presentation, image brightness adjustments were applied equally to controls and experiments.
Quantitative reverse transcription PCR
For gene expression analysis, 20 dissected late 3rd‐instar larval brains were lysed in 300 μl TRI Reagent (ZYMO Research, #R2061) to form one biological replica. Three biological replicates were prepared for each genotype: repo‐GAL4 > w1118 and repo‐GAL4 > htlACT. Total RNA was extracted using a Direct‐zol RNA Microprep Kit (ZYMO Research, #R2061), and 1μg of total RNA from each sample was reverse transcribed into cDNA using ProtoScript II First Strand cDNA Synthesis Kit (NEB, #E6560S) according to the manufacturer’s instructions. The qPCR was performed using the stepOnePlus real‐time PCR system (Applied Biosystems) using Fast SYBR Green master mix reagent (Applied Biosystems, #4385612). Gene expression levels were normalised to rpl32 (in Fig 6A, one representative data set of rpl32 was plotted) and calculated using the 2‐ΔΔCt method. The primers were either designed using Primer‐BLAST (https://www.ncbi.nlm.nih.gov/tools/primer‐blast/) or obtained from FlyPrimerBank (https://www.flyrnai.org/flyprimerbank).
Listed in Reagents Table.
Molecular cloning
A constitutively active form of htl, comprising the dimerisation domain of the bacteriophage λ repressor (Michelson et al, 1998), was amplified from the genomic DNA of the fly stock: UAS‐htlACT (BDSC5367), using a forward primer, 5’‐CAACTGCAACTACTGAAATCTGCC‐3’, and a reverse primer 5’‐ CCCCCTCTAGATTAATAATTACACCACTTCTGC‐3’. The resulting PCR product was digested with NotI (Promega, #R6431) and XbaI (Promega, #R6181), which cut at the restriction sites as indicated in the reverse primer (underlined above). The plasmid, pJFRC19‐13XLexAop2‐IVS‐myr::GFP (Addgene, #26224), was identically digested to remove myr::GFP. The restriction fragment, NotI‐htlACT‐XbaI, was subsequently cloned into the digested LexAop vector (Pfeiffer et al, 2010). The reconstructed plasmid was sequenced and injected into flies carrying an attP2 docking site (BDSC25710).
Quantification and analysis
Hh and LipidTOX intensity profile
A flat line was drawn across each LDs as described in Fig 1H, and single pixel values of Hh and LipidTOX staining were generated along line using “Analyze‐Plot Profile” tool of Fiji. The intensity profiles of Hh and LipidTOX for each LDs were compiled using Prism‐GraphPad, with Y‐axis reflecting pixel values and X‐axis reflecting relative position on LDs. The relative LD position was generated by dividing the line position values to each line length. This normalisation was conducted because line length is different in each sample due to the variations in LD size. The Hh intensity at the position X_0.9 (Fig 1H, at the surface of LDs) was compared with that of the position X_1.0 (Fig 1H, just outside of LDs) to illustrate the Hh‐LD association described in Fig 1C–G’’.
Cortex glial membrane size, LD volume and glial number measurement
Cortex glial membrane volume (NP2222‐GAL4 > UAS‐mGFP) and LD volume were measured from three‐dimensional reconstruction of confocal Z stacks (2‐μm step‐size) with Volocity software (Improvision). Glial cell number was automatically counted with a Fiji plug‐in “DeadEasy Larval Glia” (Forero et al, 2012).
Cell cycle speed
The number of type I NBs in each brain lobe was manually counted using Fiji. type I NBs were distinguished from other Mira+ cells by size and morphology: Newly generated GMCs are smaller than type I NBs as shown in Fig EV3E (white arrows). Type II NBs are associated with more Mira+ progeny cells.
EdU index: For 26ALH larval brains, EdU voxels of the whole brain were measured with Volocity software (Improvision) to indicate NB re‐entry into cell cycle. Glial EdU voxels represents only a small amount of the total EdU voxels (Sousa‐Nunes et al, 2011). Normalised EdU voxels were calculated by dividing EdU voxels to the mean voxel counts of the control. For 96ALH larval brains, the number of EdU+ type I NBs was counted in the CB of each brain lobe. EdU index is calculated as number of EdU+ NBs normalised to control EdU+ NBs. The number of EdU+ NBs reflects the NBs that progress through S phase in a 10 or 15‐min time window relative to control. We have utilised GAL4 driver > w1118, UAS‐mcherryRNAi or UAS‐luc interchangeably as controls in our experiments, as we found EdU incorporation did not significantly alter between these controls (Appendix Fig S2). The total number of type I NBs is not altered in experiments where Edu index was used to determine NB cell cycle speed. For EdU pulse‐chase quantification, the number of progeny cells that inherit EdU from each dividing NBs was counted to indicate the speed by which NBs generate their progeny.
pH3 index: represented as the % of type I NBs in M phase (pH3+)/ the total number of type I NBs.
Cell cycle lengths in NBs were measured as the time between two consecutive cell divisions. The cell cycle lengths of NBs from at least three different ex vivo brains were plotted for each genotype.
CNS and cell size measurement
Brain lobes, NB, GMC and nucleoli diameters were estimated by averaging orthogonal measurements of diameter, with a single confocal section on Fiji. The volume of each brain lobe (CNS) was calculated by the formula: V = 4/3Π r3.
Localisation of asymmetric determinants was assessed in M phase NBs that display a condensed metaphase plate marked by pH3. Clear crescent localisation was counted as correct localisation, and cytoplasmic or cortical localisation was counted as mislocalisation.
The distribution of Hh in glia was measured on Fiji with the formula: The area of glia that contains Hh / Total glial area. The detailed procedure: Total glial area: Adjust Threshold (Default) > Analysis > measure (area); The area of glia that contains Hh: Create selection of glial channel by Adjusting Threshold (Default) > Restore selection in the Hh channel > clear outside > measure area.
Total intensity
The sum projection confocal image was used for the intensity measurement on Fiji using the formula: CTCF (corrected total cell fluorescence) = Integrated density – (Area of selected cell x mean fluorescence of background readings) (McCloy et al, 2014).
Statistical analysis
P‐values were calculated by two‐tailed, unpaired Student’s t‐test, with equal sample variance; Welch’s correction was applied in case of unequal variances. Kolmogorov–Smirnov test was used to test data normality. Mann–Whitney test was used when data deviated from a normal distribution. Data are presented as mean ± SEM in the main text.
Author contributions
QD, MZ, FF, TL and SG conducted the experiments; QD and LYC designed the experiments and wrote the paper.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Movie EV1
Movie EV2
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
Acknowledgements
We are grateful to Alex Gould, Thomas B. Kornberg, Isabel Guerrero, Yu Cai, William Chia, Tatsushi Igaki, Joseph M. Bateman, Kieran Harvey, Helena Richardson, Gary Hime and Philip Batterham for generous sharing of antibodies and fly stocks. We would like to thank Bloomington Drosophila Stock Center, Vienna Drosophila Resource Center, Fly Stocks of National Institute of Genetics, KYOTO Stock Center, Developmental Studies Hybridoma Bank and Addgene for fly stocks and plasmids. We would like to also thank OZDros for Drosophila quarantine, Peter MacCallum Cancer Institute Microscopy core and Biological Optical Microscopy platform at the University of Melbourne for technical assistance. We would like to thank Charles Robin and Mike Murray for sharing their microinjection facility with us. We are grateful to Kieran Harvey, Helena Richardson and Andrew Cox for critical reading of the manuscript. Schematic pictures in the figures are created with BioRender. QD is funded by a Melbourne Research Scholarship, LYC is funded by an ARC Future Fellowship, and LYC’s laboratory is supported by funding from the NHMRC, ARC and the Peter MacCallum Cancer Foundation.
EMBO reports (2021) 22: e52130.
Data availability
This study includes no data deposited in external repositories.
References
- Akkouche A, Mugat B, Barckmann B, Varela‐Chavez C, Li B, Raffel R, Pelisson A, Chambeyron S (2017) Piwi Is Required during Drosophila embryogenesis to license dual‐strand piRNA clusters for transposon repression in adult ovaries. Mol Cell 66: 411–419 e414 [DOI] [PubMed] [Google Scholar]
- Anderson PR, Kirby K, Hilliker AJ, Phillips JP (2005) RNAi‐mediated suppression of the mitochondrial iron chaperone, frataxin, in Drosophila . Hum Mol Genet 14: 3397–3405 [DOI] [PubMed] [Google Scholar]
- Avet‐Rochex A, Kaul AK, Gatt AP, McNeill H, Bateman JM (2012) Concerted control of gliogenesis by InR/TOR and FGF signalling in the Drosophila post‐embryonic brain. Development 139: 2763–2772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avet‐Rochex A, Maierbrugger KT, Bateman JM (2014) Glial enriched gene expression profiling identifies novel factors regulating the proliferation of specific glial subtypes in the Drosophila brain. Gene Expr Patterns 16: 61–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Awasaki T, Lai SL, Ito K, Lee T (2008) Organization and postembryonic development of glial cells in the adult central brain of Drosophila . J Neurosci 28: 13742–13753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey AP, Koster G, Guillermier C, Hirst EM, MacRae JI, Lechene CP, Postle AD, Gould AP (2015) Antioxidant role for lipid droplets in a stem cell niche of Drosophila . Cell 163: 340–353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chai PC, Liu Z, Chia W, Cai Y (2013) Hedgehog signaling acts with the temporal cascade to promote neuroblast cell cycle exit. PLoS Biol 11: e1001494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamoun Z, Mann RK, Nellen D, von Kessler DP, Bellotto M, Beachy PA, Basler K (2001) Skinny Hedgehog, an acyltransferase required for palmitoylation and activity of the Hedgehog signal. Science 293: 2080–2084 [DOI] [PubMed] [Google Scholar]
- Chandra V, Das T, Gulati P, Biswas NK, Rote S, Chatterjee U, Ghosh SN, Deb S, Saha SK, Chowdhury AK et al (2015) Hedgehog signaling pathway is active in GBM with GLI1 mRNA expression showing a single continuous distribution rather than discrete high/low clusters. PLoS One 10: e0116390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chell JM, Brand AH (2010) Nutrition‐responsive glia control exit of neural stem cells from quiescence. Cell 143: 1161–1173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W, Huang H, Hatori R, Kornberg TB (2017) Essential basal cytonemes take up Hedgehog in the Drosophila wing imaginal disc. Development 144: 3134–3144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng LY, Bailey AP, Leevers SJ, Ragan TJ, Driscoll PC, Gould AP (2011) Anaplastic lymphoma kinase spares organ growth during nutrient restriction in Drosophila . Cell 146: 435–447 [DOI] [PubMed] [Google Scholar]
- Coutinho‐Budd JC, Sheehan AE, Freeman MR (2017) The secreted neurotrophin Spatzle 3 promotes glial morphogenesis and supports neuronal survival and function. Genes Dev 31: 2023–2038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dienstmann R, Rodon J, Prat A, Perez‐Garcia J, Adamo B, Felip E, Cortes J, Iafrate AJ, Nuciforo P, Tabernero J (2014) Genomic aberrations in the FGFR pathway: opportunities for targeted therapies in solid tumors. Ann Oncol 25: 552–563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding WY, Huang J, Wang H (2020) Waking up quiescent neural stem cells: molecular mechanisms and implications in neurodevelopmental disorders. PLoS Genet 16: e1008653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doe CQ (1996) Spindle orientation and asymmetric localization in Drosophila: both inscuteable? Cell 86: 695–697 [DOI] [PubMed] [Google Scholar]
- Doe CQ (2017) Temporal patterning in the Drosophila CNS. Annu Rev Cell Dev Biol 33: 219–240 [DOI] [PubMed] [Google Scholar]
- Doyle SE, Pahl MC, Siller KH, Ardiff L, Siegrist SE (2017) Neuroblast niche position is controlled by phosphoinositide 3‐kinase‐dependent DE‐cadherin adhesion. Development 144: 820–829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumstrei K, Wang F, Hartenstein V (2003) Role of DE‐cadherin in neuroblast proliferation, neural morphogenesis, and axon tract formation in Drosophila larval brain development. J Neurosci 23: 3325–3335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forero MG, Kato K, Hidalgo A (2012) Automatic cell counting in vivo in the larval nervous system of Drosophila . J Microsc 246: 202–212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman MR (2015) Drosophila central nervous system glia. Cold Spring Harb Perspect Biol 7: a020552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geng F, Guo D (2017) Lipid droplets, potential biomarker and metabolic target in glioblastoma. Intern Med Rev 3: 10.18103/imr.v3i5.443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo D, Bell EH, Chakravarti A (2013) Lipid metabolism emerges as a promising target for malignant glioma therapy. CNS Oncol 2: 289–299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartman TR, Strochlic TI, Ji Y, Zinshteyn D, O'Reilly AM (2013) Diet controls Drosophila follicle stem cell proliferation via Hedgehog sequestration and release. J Cell Biol 201: 741–757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi S, Ito K, Sado Y, Taniguchi M, Akimoto A, Takeuchi H, Aigaki T, Matsuzaki F, Nakagoshi H, Tanimura T et al (2002) GETDB, a database compiling expression patterns and molecular locations of a collection of Gal4 enhancer traps. Genesis 34: 58–61 [DOI] [PubMed] [Google Scholar]
- Heemskerk J, DiNardo S (1994) Drosophila Hedgehog acts as a morphogen in cellular patterning. Cell 76: 449–460 [DOI] [PubMed] [Google Scholar]
- Homem CC, Knoblich JA (2012) Drosophila neuroblasts: a model for stem cell biology. Development 139: 4297–4310 [DOI] [PubMed] [Google Scholar]
- Hoyle G (1986) Glial cells of an insect ganglion. J Comp Neurol 246: 85–103 [DOI] [PubMed] [Google Scholar]
- Hoyle G, Williams M, Phillips C (1986) Functional morphology of insect neuronal cell‐surface/glial contacts: the trophospongium. J Comp Neurol 246: 113–128 [DOI] [PubMed] [Google Scholar]
- Innis SM (2016) Palmitic acid in early human development. Crit Rev Food Sci Nutr 56: 1952–1959 [DOI] [PubMed] [Google Scholar]
- Jimenez‐Pascual A, Siebzehnrubl FA (2019) Fibroblast growth factor receptor functions in glioblastoma. Cells 8: 715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanai MI, Kim MJ, Akiyama T, Takemura M, Wharton K, O'Connor MB, Nakato H (2018) Regulation of neuroblast proliferation by surface glia in the Drosophila larval brain. Sci Rep 8: 3730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kis V, Barti B, Lippai M, Sass M (2015) Specialized cortex glial cells accumulate lipid droplets in Drosophila melanogaster . PLoS One 10: e0131250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai SL, Lee T (2006) Genetic mosaic with dual binary transcriptional systems in Drosophila . Nat Neurosci 9: 703–709 [DOI] [PubMed] [Google Scholar]
- Lee JD, Treisman JE (2001) Sightless has homology to transmembrane acyltransferases and is required to generate active Hedgehog protein. Curr Biol 11: 1147–1152 [DOI] [PubMed] [Google Scholar]
- Lee CY, Wilkinson BD, Siegrist SE, Wharton RP, Doe CQ (2006) Brat is a Miranda cargo protein that promotes neuronal differentiation and inhibits neuroblast self‐renewal. Dev Cell 10: 441–449 [DOI] [PubMed] [Google Scholar]
- Liu L, Zhang K, Sandoval H, Yamamoto S, Jaiswal M, Sanz E, Li Z, Hui J, Graham BH, Quintana A et al (2015) Glial lipid droplets and ROS induced by mitochondrial defects promote neurodegeneration. Cell 160: 177–190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann RK, Beachy PA (2004) Novel lipid modifications of secreted protein signals. Annu Rev Biochem 73: 891–923 [DOI] [PubMed] [Google Scholar]
- Martin I, Jones MA, Grotewiel M (2009) Manipulation of Sod1 expression ubiquitously, but not in the nervous system or muscle, impacts age‐related parameters in Drosophila . FEBS Lett 583: 2308–2314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurange C, Cheng L, Gould AP (2008) Temporal transcription factors and their targets schedule the end of neural proliferation in Drosophila . Cell 133: 891–902 [DOI] [PubMed] [Google Scholar]
- McCloy RA, Rogers S, Caldon CE, Lorca T, Castro A, Burgess A (2014) Partial inhibition of Cdk1 in G 2 phase overrides the SAC and decouples mitotic events. Cell Cycle 13: 1400–1412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micchelli CA, The I, Selva E, Mogila V, Perrimon N (2002) Rasp, a putative transmembrane acyltransferase, is required for Hedgehog signaling. Development 129: 843–851 [DOI] [PubMed] [Google Scholar]
- Michelson AM, Gisselbrecht S, Buff E, Skeath JB (1998) Heartbroken is a specific downstream mediator of FGF receptor signalling in Drosophila . Development 125: 4379–4389 [DOI] [PubMed] [Google Scholar]
- Morrison RS, Yamaguchi F, Saya H, Bruner JM, Yahanda AM, Donehower LA, Berger M (1994) Basic fibroblast growth factor and fibroblast growth factor receptor I are implicated in the growth of human astrocytomas. J Neurooncol 18: 207–216 [DOI] [PubMed] [Google Scholar]
- Nusslein‐Volhard C, Wieschaus E (1980) Mutations affecting segment number and polarity in Drosophila . Nature 287: 795–801 [DOI] [PubMed] [Google Scholar]
- Palm W, Swierczynska MM, Kumari V, Ehrhart‐Bornstein M, Bornstein SR, Eaton S (2013) Secretion and signaling activities of lipoprotein‐associated Hedgehog and non‐sterol‐modified Hedgehog in flies and mammals. PLoS Biol 11: e1001505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panakova D, Sprong H, Marois E, Thiele C, Eaton S (2005) Lipoprotein particles are required for Hedgehog and wingless signalling. Nature 435: 58–65 [DOI] [PubMed] [Google Scholar]
- Pepinsky RB, Zeng C, Wen D, Rayhorn P, Baker DP, Williams KP, Bixler SA, Ambrose CM, Garber EA, Miatkowski K et al (1998) Identification of a palmitic acid‐modified form of human sonic Hedgehog. J Biol Chem 273: 14037–14045 [DOI] [PubMed] [Google Scholar]
- Pereanu W, Shy D, Hartenstein V (2005) Morphogenesis and proliferation of the larval brain glia in Drosophila . Dev Biol 283: 191–203 [DOI] [PubMed] [Google Scholar]
- Pfeiffer BD, Ngo TT, Hibbard KL, Murphy C, Jenett A, Truman JW, Rubin GM (2010) Refinement of tools for targeted gene expression in Drosophila . Genetics 186: 735–755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portela M, Venkataramani V, Fahey‐Lozano N, Seco E, Losada‐Perez M, Winkler F, Casas‐Tinto S (2019) Glioblastoma cells vampirize WNT from neurons and trigger a JNK/MMP signaling loop that enhances glioblastoma progression and neurodegeneration. PLoS Biol 17: e3000545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter JA, Young KE, Beachy PA (1996) Cholesterol modification of Hedgehog signaling proteins in animal development. Science 274: 255–259 [DOI] [PubMed] [Google Scholar]
- Ramon‐Canellas P, Peterson HP, Morante J (2019) From early to late neurogenesis: neural progenitors and the glial niche from a fly's point of view. Neuroscience 399: 39–52 [DOI] [PubMed] [Google Scholar]
- Read RD, Cavenee WK, Furnari FB, Thomas JB (2009) A Drosophila model for EGFR‐Ras and PI3K‐dependent human glioma. PLoS Genet 5: e1000374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Read RD (2018) Pvr receptor tyrosine kinase signaling promotes post‐embryonic morphogenesis, and survival of glia and neural progenitor cells in Drosophila . Development 145: dev164285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy BV, Irvine KD (2011) Regulation of Drosophila glial cell proliferation by Merlin‐Hippo signaling. Development 138: 5201–5212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richier B, Vijandi CM, Mackensen S, Salecker I (2017) Lapsyn controls branch extension and positioning of astrocyte‐like glia in the Drosophila optic lobe. Nat Commun 8: 317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scadden DT (2014) Nice neighborhood: emerging concepts of the stem cell niche. Cell 157: 41–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schieber M, Chandel NS (2014) ROS function in redox signaling and oxidative stress. Curr Biol 24: R453–R462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiller H, Bensch K (1971) De novo fatty acid synthesis and elongation of fatty acids by subcellular fractions of lung. J Lipid Res 12: 248–255 [PubMed] [Google Scholar]
- Schwartz C, Locke J, Nishida C, Kornberg TB (1995) Analysis of cubitus interruptus regulation in Drosophila embryos and imaginal disks. Development 121: 1625–1635 [DOI] [PubMed] [Google Scholar]
- Shen CP, Jan LY, Jan YN (1997) Miranda is required for the asymmetric localization of Prospero during mitosis in Drosophila . Cell 90: 449–458 [DOI] [PubMed] [Google Scholar]
- Slakey LL, Ferrick TJ, Ness GC, Porter JW (1979) De novo fatty acid synthesis and fatty acid elongation catalyzed by subcellular fractions from hog and human aorta. Lipids 14: 451–457 [DOI] [PubMed] [Google Scholar]
- Smith S, Witkowski A, Joshi AK (2003) Structural and functional organization of the animal fatty acid synthase. Prog Lipid Res 42: 289–317 [DOI] [PubMed] [Google Scholar]
- Sousa‐Nunes R, Yee LL, Gould AP (2011) Fat cells reactivate quiescent neuroblasts via TOR and glial insulin relays in Drosophila . Nature 471: 508–512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spampinato SF, Bortolotto V, Canonico PL, Sortino MA, Grilli M (2019) Astrocyte‐derived paracrine signals: relevance for neurogenic niche regulation and blood‐brain barrier integrity. Front Pharmacol 10: 1346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speder P, Brand AH (2018) Systemic and local cues drive neural stem cell niche remodelling during neurogenesis in Drosophila . eLife 7: e30413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sykiotis GP, Bohmann D (2008) Keap1/Nrf2 signaling regulates oxidative stress tolerance and lifespan in Drosophila . Dev Cell 14: 76–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takezaki T, Hide T, Takanaga H, Nakamura H, Kuratsu J, Kondo T (2011) Essential role of the Hedgehog signaling pathway in human glioma‐initiating cells. Cancer Sci 102: 1306–1312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teixeira L, Rabouille C, Rorth P, Ephrussi A, Vanzo NF (2003) Drosophila Perilipin/ADRP homologue Lsd2 regulates lipid metabolism. Mech Dev 120: 1071–1081 [DOI] [PubMed] [Google Scholar]
- Tian A, Shi Q, Jiang A, Li S, Wang B, Jiang J (2015) Injury‐stimulated Hedgehog signaling promotes regenerative proliferation of Drosophila intestinal stem cells. J Cell Biol 208: 807–819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ugbode CI, Smith I, Whalley BJ, Hirst WD, Rattray M (2017) Sonic Hedgehog signalling mediates astrocyte crosstalk with neurons to confer neuroprotection. J Neurochem 142: 429–443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varjosalo M, Taipale J (2008) Hedgehog: functions and mechanisms. Genes Dev 22: 2454–2472 [DOI] [PubMed] [Google Scholar]
- Wendler F, Franch‐Marro X, Vincent JP (2006) How does cholesterol affect the way Hedgehog works? Development 133: 3055–3061 [DOI] [PubMed] [Google Scholar]
- Witte HT, Jeibmann A, Klambt C, Paulus W (2009) Modeling glioma growth and invasion in Drosophila melanogaster . Neoplasia 11: 882–888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada SM, Yamaguchi F, Brown R, Berger MS, Morrison RS (1999) Suppression of glioblastoma cell growth following antisense oligonucleotide‐mediated inhibition of fibroblast growth factor receptor expression. Glia 28: 66–76 [DOI] [PubMed] [Google Scholar]
- Yuan X, Sipe CW, Suzawa M, Bland ML, Siegrist SE (2020) Dilp‐2‐mediated PI3‐kinase activation coordinates reactivation of quiescent neuroblasts with growth of their glial stem cell niche. PLoS Biol 18: e3000721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zielke N, Korzelius J, van Straaten M, Bender K, Schuhknecht GFP, Dutta D, Xiang J, Edgar BA (2014) Fly‐FUCCI: a versatile tool for studying cell proliferation in complex tissues. Cell Rep 7: 588–598 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Movie EV1
Movie EV2
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
Data Availability Statement
This study includes no data deposited in external repositories.