

Abstract
Despite significant steps in our understanding of Alzheimer’s disease (AD), many of the molecular processes underlying its pathogenesis remain largely unknown. Here, we focus on the role of non‐coding RNAs produced by small interspersed nuclear elements (SINEs). RNAs from SINE B2 repeats in mouse and SINE Alu repeats in humans, long regarded as “junk” DNA, control gene expression by binding RNA polymerase II and suppressing transcription. They also possess self‐cleaving activity that is accelerated through their interaction with certain proteins disabling this suppression. Here, we show that similar to mouse SINE RNAs, human Alu RNAs, are processed, and the processing rate is increased in brains of AD patients. This increased processing correlates with the activation of genes up‐regulated in AD patients, while increased intact Alu RNA levels correlate with down‐regulated gene expression in AD. In vitro assays show that processing of Alu RNAs is accelerated by HSF1. Overall, our data show that RNAs from SINE elements in the human brain show a similar pattern of deregulation during amyloid beta pathology as in mouse.
Keywords: Alu RNAs, Alzheimer’s disease, small interspersed nuclear element, transcriptome
Subject Categories: Chromatin, Epigenetics, Genomics & Functional Genomics; Molecular Biology of Disease; Neuroscience
SINE Alu RNAs in the human brain, similar to their counterpart SINE B2 RNAs in mouse, show increased processing in amyloid beta pathology. Increased Alu processing further correlates with AD‐linked gene expression changes.
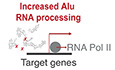
Introduction
As the human life expectancy continues to expand, the number of people with aging‐associated diseases, including late‐onset Alzheimer’s disease (AD), is expected to increase. This will generate a wave of new cases in the upcoming years, putting a strain on societies worldwide (Cornutiu, 2015; Montgomery et al, 2018). Amyloid pathology is a required feature of AD, where amyloid beta peptides and their aggregated forms have been associated with AD pathogenesis in brains of Alzheimer’s disease patients as well as neural cell toxicity in mouse models of amyloid pathology (Ittner et al, 2010; Bloom, 2014; Gonzalez et al, 2018; Mehla et al, 2019). However, despite the significant steps made in understanding AD pathogenesis during the last decades, diagnostic tests and clinical trials for potential therapeutic factors in AD remain inconclusive, as AD remains a highly complex disease, with extreme heterogeneity and many of the molecular processes underlying its pathogenesis still elusive (Bertram & Tanzi, 2008; Korczyn, 2012; Toledo et al, 2013; Hane et al, 2017). Among such molecular mechanisms that have attracted only a little attention until now are transcriptome changes in a class of RNAs produced by retrotransposons called SINE RNAs.
Retrotransposons are the dominant repetitive transposable elements in the mammalian genome (Natt & Thorsell, 2016). SINE RNAs are non‐coding RNAs generated by a class of non‐autonomous retrotransposons called small interspersed nuclear elements (SINEs), which consist of 100–500 base pairs (Tatosyan & Kramerov, 2016). The most successful SINE retroelement in the human genome, Alu SINE, has an amplification rate of one new insertion per 20 human births leading to the current one million transcripts in the genome (Walters et al, 2009; Deininger, 2011). While most of retrotransposons are inactive through epigenetic regulatory mechanisms, Alu, together with some other retroelements such as L1 (a LINE element) can be active (Mu et al, 2016; Platt et al, 2018). For a long time, most SINEs have been regarded as part of the so called “junk DNA”, a part of the non‐coding genome with no apparent function (Karijolich et al, 2017). More recently, it was shown that SINE genomic elements contribute to genomic diversity through their integration into multiple genomic sites, by providing novel splicing sites, promoter elements, enhancers and transcription factor binding sites or disrupting existing ones (Kaaij et al, 2019; Zhang et al, 2019).
However, little is known about the role of the non‐coding RNAs transcribed by SINE genomic elements themselves. Two of the most frequent subclasses of SINEs, the B2 and Alu retrotransposons, are present in millions of copies in mouse and humans, respectively, and are either transcribed by RNA polymerase III independently or by RNA polymerase II as parts of naïve transcripts in which they are embedded. RNAs from these elements have been known for years to be up‐regulated during response to various types of cellular stress. In particular, levels of RNAs transcribed from SINEs such as B1 and B2 (mouse), Alu (humans), PRE‐1 (swine), Bm 1 (silkworm), and C (rabbit) have been reported to increase during heat shock (Tatosyan & Kramerov, 2016; Funkhouser et al, 2017). However, their exact role in this process remained unknown until a number of studies have revealed that levels of SINE B2 and Alu RNAs are elevated during response to stress to suppress transcription through their binding of RNA polymerase II (Espinoza et al, 2007; Mariner et al, 2008; Yakovchuk et al, 2009; Ponicsan et al, 2010, 2015). This binding contributes to the genome‐wide transcriptional repression observed during cellular stress by suppressing housekeeping genes, potentially facilitating redistribution of energy resources of the cell to support survival (Mariner et al, 2008). Subsequently, we showed that SINE RNAs can also mediate cellular response to stress in a different way. In particular, we showed that SINE B2 RNAs in mouse show a protein‐accelerated self‐cleavage activity (Zovoilis et al, 2016; Cheng et al, 2020; Hernandez et al, 2020), leading to their fragmentation and thus interfering with their capability of binding to and suppressing RNA polymerase II. As a result, SINE RNAs in mouse act as transcriptional switches during response to stress stimuli by binding RNA polymerase II at several stress response genes in the pro‐cellular stress state and suppressing their transcription. Upon application of a stress stimulus, B2 RNAs are processed, leading to the release of the delayed or stalled RNA polymerase at stress response genes and, thus, their transcription (Zovoilis et al, 2016). Thus, SINE RNAs, based on their processing status, play an important role in stress response through either suppressing (as full‐length RNAs) or activating (during processing) gene expression (Appendix Fig S1 provides a model combining what is known for this mode of regulation of gene expression in mouse).
Interestingly, in a recent study we have shown that this processing and destabilization of B2 RNAs is connected with a pathologic process in the cell, namely amyloid pathology in mouse neural cells (Cheng et al, 2020). In particular, increased amyloid beta load during the active neurodegenerative phase acts as a continuous stimulus that causes an increase in a key stress response factor, called Hsf1. Increased Hsf1 levels accelerate B2 RNA processing and lead the Hsf1/B2 RNA/stress response genes axis to “lock” into an activated mode with accompanying high levels of pro cell death genes, such as p53 (Cheng et al, 2020).
The above findings have revealed a role for SINE RNA processing in amyloid beta pathology in mouse brain and raised the intriguing possibility that a similar mode of deregulation of SINE RNA processing may also exist in human brain. In humans, SINE Alu RNAs are also able to bind and inhibit RNA Pol II alike to B2 RNAs (Mariner et al, 2008). We have found that, in vitro, much alike B2 RNAs, Alu RNAs are also self‐cleaving RNAs and can become destabilized (Hernandez et al, 2020). Thus, in vivo, SINE Alu RNAs in human brains could also be subject to a similar RNA processing as SINE B2 RNAs in mouse brains, assigning a broader role to SINE RNA processing in brain molecular physiology and AD pathogenesis. Here, we investigate this hypothesis by examining whether Alu RNAs are processed in human brains, whether their processing ratio is deregulated in brains of AD patients compared to healthy aging individuals, and the potential mechanisms underlying such a deregulation.
Results
Short RNA sequencing identifies processing at the right arm of Alu RNAs in human hippocampus
Much alike SINE B2 RNAs, SINE Alu RNAs can bind RNA polymerase II and suppress transcription. In the absence of an intact Alu RNA region binding Pol II, for example, after fragmentation of the Alu RNA, this binding and suppression of Pol II cannot take place (Fig 1A; Yakovchuk et al, 2009; Ponicsan et al, 2010). This is of particular interest, given that we have recently shown that Alu RNAs, as in the case of B2 RNAs, are self‐cleaving RNAs (Hernandez et al, 2020). In mouse, we have shown that processing of SINE RNAs in both NIH/3T3 cells and mouse hippocampus takes place at a position within the RNA region that binds and suppresses RNA polymerase II (Cheng et al, 2020). Interestingly, previous studies have identified a similar Pol II binding and suppression region also in Alu RNA. In particular, Alu RNA consists of two parts (arms) and the Alu RNA’s 3′ part (right arm), including the nucleotide bridge connecting the two arms, has been reported as the part of the Alu RNAs that is necessary for binding and suppression of RNA polymerase II (Mariner et al, 2008). Thus, we questioned whether Alu RNAs are processed within their Pol II binding and repression region also in human brain.
Figure 1. Processing areas of Alu RNAs in the human hippocampus as revealed by short RNA‐seq.
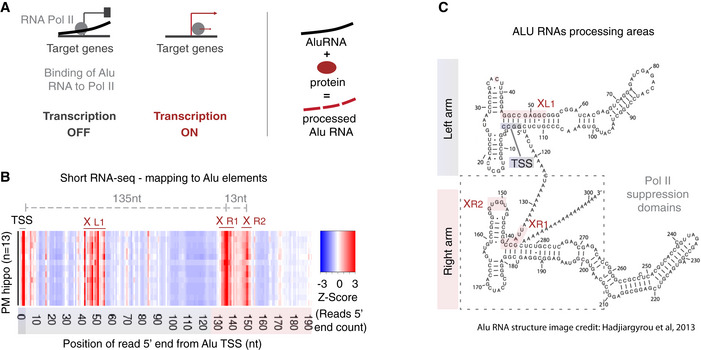
- Mode of regulation of transcription by Alu RNAs through suppression of RNA polymerase II (left panel) and protein‐accelerated self‐cleaving processing properties of Alu RNAs (right panel) as described in previous studies (Mariner et al, 2008; Yakovchuk et al, 2009; Ponicsan et al, 2010; Hernandez et al, 2020).
- Plotting of the position of the 5′ end of Alu RNA fragments across the Alu metagene to depict potential processing areas of Alu RNAs in all post‐mortem hippocampal tissues (PM hippo) from patients from the CBB. Each row in the heatmap depicts the distribution of counts of the 5′ ends of reads mapped across the Alu metagene for each patient. The x‐axis represents a metagene combining all unique Alu RNA sequences (ALUome) aligned at the start site of their consensus sequence with numbers representing the distance from the transcriptional start site (TSS) area. Heatmap density corresponds to normalized counts of the 5′ end of the reads with red corresponding to higher density of these 5′ ends at a specific position. XL1, XR1, and XR2 denote the Alu processing areas defined by the high‐density areas in the heatmap at specific positions of the Alu metagene, with the middle letter corresponding to the arm of the Alu RNA (see C, L = left, R = right), in which the area is located.
- Processing areas of Alu RNAs on the secondary Alu RNA structure. Secondary structure of Alu RNA adapted from Hadjiargyrou and colleagues (Hadjiargyrou & Delihas, 2013). As in our previous studies (Zovoilis et al, 2016; Cheng et al, 2020), we depict the SINE RNA processing areas (highlighted in pink) based on short RNA‐seq data and mapping of the 5′ ends of Alu RNA fragments. X mark the cleavage sites of Alu RNA that correspond to enriched processing areas (the high densities of 5′ end fragments distribution) at the heatmap of (B). The rectangle depicts the critical region that binds and suppresses RNA Pol II based on (Mariner et al, 2008) that may be affected due to processing points.
To test this, we extracted RNA from post‐mortem hippocampal tissues from 13 individuals with no clinical signs of Alzheimer’s disease from the Calgary Brain Bank (CBB). We then employed an RNA sequencing approach that has been customized for the sequencing of SINE RNAs and their fragments (short RNA‐seq). This library construction approach reduces any potential bias introduced through RNA fragmentation, that is used in standard long RNA‐seq protocols, which is optimal for the identification of short SINE Alu RNA fragments (< 200 nt) that may be produced by Alu RNA processing. The version of short RNA sequencing we have used for Alu RNAs is similar to the version used in our previous work for SINE B2 RNAs (Zovoilis et al, 2016), but modified to include also the full‐length Alu RNAs that are approximately 300 nt long (see Materials and Methods).
In contrast to B2 elements studied before in inbred mouse models and cell lines, Alu elements show increased genomic sequence variability when tested at the population level, for example, in humans. To account for this, after short RNA‐seq, we performed mapping of the sequenced reads not against the human reference genome but against a generated list of all unique Alu sequences that are available in the UCSC Genome Browser (genome.ucsc.edu) Repeat Masker track (repeatmasker.org). Sequenced reads were mapped against the generated unique “ALUome” with an aligner normally used for genomic mapping (no splicing or soft‐clipping). Despite being computationally more intensive, this approach also helps to eliminate potential biases due to truncated Alu forms, Alu copy number variations, insertions, and deletions that may exist in the genome of tested individuals as well as sequences that are generated through splicing and could intervene with the mapping process.
Subsequently, for every patient, an Alu metagene model was constructed, plotting cumulatively the distribution of the 5′ ends of all sequenced RNAs aligning to the ALUome with regard to their distance from the Alu elements’ start. These 5′ end read distribution models for each patient across the Alu metagene are presented one above the other in the form of a heatmap in Fig 1B. Increased color density in the heatmap represents increased number of 5′ ends of sequenced reads mapping to specific positions in the Alu metagene, thus enabling the study of both full‐length Alu RNAs (mapping within the first 15 nt to account for the variability in sequence among Alus) and their fragments (mapping either at the transcription start site—TSS or beyond) (Fig 1B). This approach enabled the determination in our short RNA sequencing data of three processing areas at Alu RNAs. These processing areas are highlighted in the previously described structure of the Alu consensus sequence (Hadjiargyrou & Delihas, 2013) depicted in Fig 1C and annotated as X L(eft arm)1, X R(ight arm)1 and XR2. The processing areas include two positions (XR1 and XR2) located within the right arm and the Pol II binding region (depicted as a rectangle in Fig 1C). Due to the very high prevalence of small insertions/deletions in the ALUome with regard to Alu consensus sequence, in the current study we opted for the definition of processing areas rather than processing points to present more accurately this level of sequence variability. The distances presented in Fig 1B correspond to the distance between the peaks of distributions around each high‐density area, with the peak in the TSS region marking position 0 in the distance from TSS.
These data show that, in human hippocampus, SINE Alu RNAs are processed within their Pol II binding and suppression region, suggesting that the mode of regulation of gene expression though SINE RNA processing previously described in mouse may extend also to human brain.
Alu RNA processing is accelerated in hippocampi of AD patients
We have previously shown that in the hippocampi of a mouse model of amyloid beta pathology, SINE B2 RNA processing is accelerated. We questioned whether a similar mode of deregulation is present in humans. To this end, we employed post‐mortem hippocampal tissue from a group of patients from the Calgary Brain Bank (CBB) clinically diagnosed with AD that was compared to the non‐AD individuals tested above. Post‐mortem delay was comparable between the two groups. De‐identified clinical and pathological data for the CBB patients used in the current study are deposited as metadata in the controlled access next‐generation sequencing repository (see Materials and Methods).
In total, we performed short RNA‐seq in RNA from hippocampi from 24 patients, 13 with no clinical signs of AD and 11 diagnosed with AD (labeled as no AD and AD in Fig 2, respectively). RNAs were mapped against the ALUome as described above and 5′ end counts of Alu RNA fragments mapping to the Pol II binding region of Alu RNA (right arm/positions > 130nt from Alu start) were calculated. Subsequently, we identified Alu RNAs that are differentially processed in this region between AD and no AD patients (Fig 2A). These Alu RNAs are listed in Dataset EV1, while processing levels for these Alu RNAs for the two patient groups are presented as a heatmap in Fig 2B. As shown in Fig 2B, more than 80% of identified Alu RNAs are highly processed in hippocampi of AD patients, with these results being consistent within each group despite the expected variability and heterogeneity among individuals.
Figure 2. Alu RNA processing ratio is increased in the hippocampi of AD patients in CBB cohort.
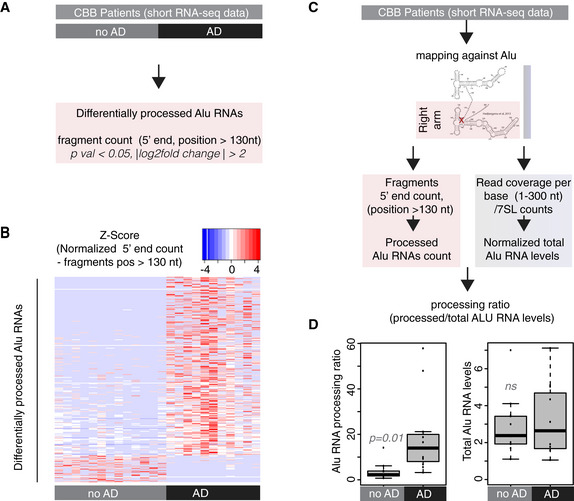
- Analysis design for the identification of differentially processed Alu RNAs between no AD (n = 13) and AD (n = 11) patients of the CBB cohort using the DESeq2 R package. Statistical significance was estimated using the Wald significance tests within the DESeq2 package.
- Normalized counts of processed fragments mapping to the right arm of Alu RNAs that are differentially processed (rows) between AD and no AD for each CBB patient (columns) (Dataset EV1). Red corresponds to higher normalized counts of processed Alu RNA fragments.
- Analysis design for calculation of the Alu RNA processing ratio in CBB patients short RNA‐seq data.
- Boxplots depict differences in hippocampi of AD and no AD patients regarding SINE Alu RNA processing ratio (left panel) (a P value of 0.05 was considered as threshold for statistical significance with P = 0.01, n = 24, unpaired non‐directional t‐test) and in total Alu RNA levels (right panel) (ns = not significant). In the boxplots, the central band (the line that divides the box into 2 parts) represents the median of the data, the ends of the box show the upper (Q3) and lower (Q1) quartiles, the extreme line shows Q3 + 1.5 × IQR to Q1 − 1.5 × IQR (the highest and lowest value excluding potential outliers), while dots beyond the extreme line show potential outliers.
These data revealed an increased number of Alu RNA fragments in AD patients, suggesting that the processing of Alu RNAs may be higher in these patients. In order to factor in any differences in the number of fragments due to changes in total Alu RNA levels, counts of Alu RNA processing fragments were normalized to total read coverage per base across the full‐length Alu RNA sequence (from start to end) (Fig 2C). As in case of mouse short RNA‐seq in our previous studies (Zovoilis et al, 2016; Cheng et al, 2020), in order to factor in any changes in these levels due to any SINE specific degradation of Alu RNAs, we normalized our data with a housekeeping Pol III non‐coding RNA transcript (7SL RNA) the levels of which do not vary across AD and no AD patients (see Materials and Methods). This approach enabled us to estimate the Alu RNA processing ratio in AD and no AD patients, confirming substantially higher levels of Alu RNA processing in AD patients compared to no AD (Fig 2D, left panel) (P = 0.01, n = 11 (AD)/13 (no AD)), but not a change in total Alu RNA levels (Fig 2D, right panel).
Thus, hippocampi of AD patients are characterized by higher destabilization and processing ratios of Alu RNAs in the right arm including the RNA Pol II binding domain, consistent with the similar increased processing of B2 SINE RNAs previously described in the hippocampi of mouse models.
Standard RNA sequencing is able to detect the processing area in the right arm of Alu RNAs
Our strategy until now involved as a proof of principle the employment of short RNA sequencing as the method of choice for studying Alu RNA processing given its proven ability to detect SINE RNA fragments in mouse in the past. Our strategy also involved human hippocampi as the tissue of choice in order to maintain consistency with our previous SINE RNA studies data in mouse (Cheng et al, 2020; Fig 3A). However, when it comes to large‐scale transcriptome studies in AD, this approach has a number of limitations. Firstly, short RNA‐seq is not the first choice for study of mRNAs. In case of large transcriptome projects involving hundreds of patients compared to our small patient cohort, this approach would create the need for additional sequencing per sample, significantly increasing the cost. Moreover, a number of large‐scale RNA‐seq projects in AD patients have been completed using standard RNA‐seq with a focus on protein coding RNAs. These studies have already generated an immense amount of publicly available data not only in hippocampus but also in other brain regions that are affected during AD, such as cortex. Thus, despite the increased transcript noise that RNA fragmentation in standard RNA‐seq may cause, we were compelled to test whether available large‐scale standard RNA‐seq projects could enable identification of SINE Alu RNA processing at least at the Pol II binding region of the Alu RNA right arm.
Figure 3. Processing areas of Alu RNAs in the human cortex as revealed by RNA‐seq.
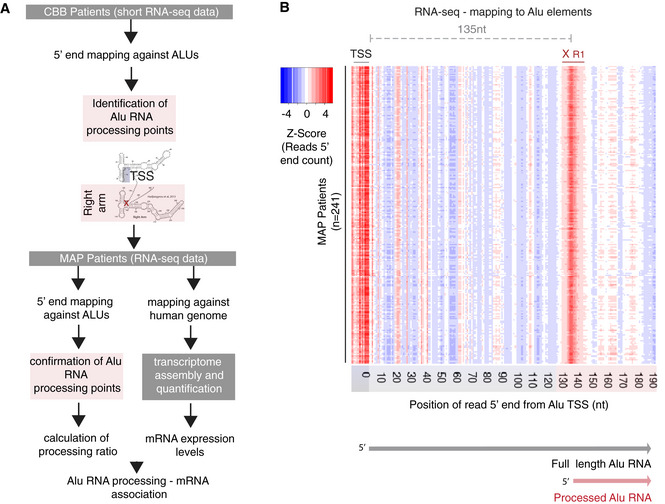
- Two‐tier analysis strategy and design for the confirmation of processing areas in Alu RNAs in human brain combining different sequencing approaches and patient cohorts (CBB cohort, n = 24; MAP cohort, n = 241).
- Plotting of the position of the 5′ end of Alu RNA fragments across the Alu metagene to depict potential processing areas of Alu RNAs in all post‐mortem cortex tissues from patients from the MAP cohort. As in Fig 1B, each row represents the distribution of normalized counts of the 5′ ends of reads mapped across the Alu metagene for each patient. As in Fig 1B, x‐axis numbers represent the distance from the transcriptional start site (TSS) area and heatmap density corresponds to normalized counts of the 5′ end of the reads with red for higher density of these 5′ ends at a specific position. XR1 as in Fig 1B.
In particular, we questioned whether publicly available poly‐A selection‐based RNA sequencing approaches in AD patients would allow the identification of Alu RNA processing in the Alu right arm. This could be possible in humans compared to mouse for a number of reasons. Firstly, Alu RNAs in humans are much longer than B2 RNAs (300 nt vs. 180 nt). With a processing area at 135 nt downstream of TSS, the resulting Alu RNA fragment size would fall within the range of most standard RNA‐seq protocols in contrast to shorter SINE B2 RNA fragments in mouse. Secondly, despite being non‐coding, SINE RNAs contain long 3′ end adenosine stretches. This would enable their sequencing in most standard RNA‐seq projects that use poly(A) adapters or selection strategies. In addition, due to the poly(A) tail selection, this approach would enable filtering out processed transcripts at TSS and counting only of full‐length Alu RNAs at this position in contrast to short RNA‐seq which at TSS could include both types of transcripts (see Materials and Methods).
To test this, we employed RNA‐seq data from one of the largest transcriptome projects in AD completed until now, the Memory and Aging Project (MAP) study that is part of the ROSMAP study. MAP is a longitudinal, epidemiologic clinical‐pathologic cohort study with an emphasis on decline in cognitive and motor function and risk of Alzheimer’s disease that began in 1997 and is run from Rush University (Bennett et al, 2012). The Broad Institute’s Genomics Platform performed the RNA‐seq (Mostafavi et al, 2018). The study employed poly(A)‐based RNA sequencing for the study of mRNAs and includes RNA sequencing data from the gray matter of the dorsolateral prefrontal cortex, accompanied with various clinical and demographic metadata including clinical diagnosis, Braak staging, ApoE genotype, and age. Sequence data from this project were obtained from the AMP‐AD Knowledge Portal (https://adknowledgeportal.synapse.org), and FASTQ files were reconstructed and then re‐analyzed with the exact same pipelines used in our CBB patient cohort. In addition, given that these data offer the possibility to study also mRNA expression, we applied in parallel a typical mRNA analysis pipeline, involving mapping against the genome (with splicing), transcriptome assembly, and quantification, in order to be able to associate our findings in SINE Alu RNAs with gene expression at a later stage (Fig 3A). Based on our preliminary quality control, we identified potential batch effects in samples sequenced within a specific time period (year 2013), and these samples were excluded from further analysis. In total, data from 241 MAP patients were analyzed.
As shown in Fig 3B, mapping of the 5′ ends of reads aligning to the ALUome across the Alu metagene confirmed that Alu RNAs can indeed be poly(A) selected and sequenced through this approach generating clear strong read distribution density at the TSS region at the beginning of the Alu elements. Most importantly, it revealed the exact same processing area (XR1—135nt downstream of TSS) we have been able to identify through short RNA‐seq above, suggesting that generation of this fragment is one of the first steps of processing of the initial poly(A) full‐length Alu RNA. In contrast, both XL1 and XR2 areas identified in short RNA‐seq were not prominent in standard RNA‐seq, suggesting that these are processing fragments without the 3′ poly(A) end, that can be recognized only with methods such as short RNA‐seq that detect both poly(A) and non‐poly(A) fragments. The level of transcriptional noise expected due to library RNA fragmentation was low and amenable, with none of the other density areas competing the density at XR1. Moreover, identification of the XR1 processing area was consistent across all MAP patients tested (Fig 3B).
These data show that study of Alu RNA processing is possible also when using standard RNA‐seq techniques, expanding significantly our potential to investigate SINE RNAs in already completed large transcriptome studies in human brain.
Alu RNA processing is accelerated in the cortex of AD patients
As in the case of hippocampus, cortex regions are among the primary targets of amyloid pathology in AD. To this end, we investigated whether Alu RNA processing ratio is increased in the dorsolateral prefrontal cortex of AD MAP patients similarly to CBB hippocampal tissues. MAP includes patients with a wide range of clinical diagnosis, including no cognitive impairment (NCI), mild cognitive impairment (MCI) and AD, as well as of patients classified in all Braak stages (from 0 to VI). Similar to our CBB cohort, patients from the MAP cohort were separated into two categories, no AD and AD, based on clinical diagnosis and Braak staging (see Materials and Methods).
As in the case of hippocampus, based on RNAs mapped against the ALUome, we calculated 5′ end counts of Alu RNA fragments mapping to the Pol II binding region of Alu RNA (right arm/position > 130nt) of MAP patients. Subsequently, we identified Alu RNAs that are differentially processed in this region between AD and no AD patients (Fig 4A). Resembling our observations in the hippocampus, the vast majority of differentially processed Alus (> 80%) depict increased processing levels in the dorsolateral prefrontal cortex of the AD patient group. These Alu RNAs are listed in Dataset EV2 and are presented for all MAP AD and no AD patients in the heatmap of Fig 4B. In contrast to short RNA‐seq, in standard RNA‐seq, due to poly(A) selection, 5′ end counts at TSS correspond only to full‐length Alu RNAs. Thus, we have been able to calculate the Alu processing ratio factoring in the full‐length Alu RNA levels as described in Fig 4C. As in case of hippocampal samples, the Alu RNA processing ratio is significantly elevated in the dorsolateral prefrontal cortices of AD patients (Fig 4D left panel) (P < 0.001, n = 67 (AD)/51 (no AD)). Full‐length Alu RNA levels were also slightly increased, but not to the extent of the processing ratio (Fig 4D, right panel, P = 0.005).
Figure 4. Alu RNA processing ratio is increased in the cortex of AD patients in the MAP cohort.
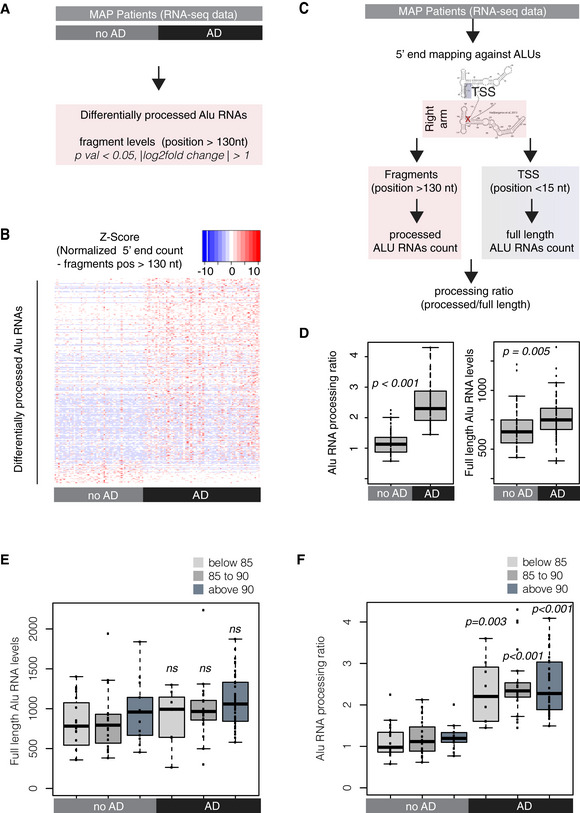
- Analysis design for the identification of differentially processed Alu RNAs between AD (n = 67) and no AD patients (n = 51) of the MAP cohort using the DESeq2 R package. Statistical significance was estimated using the Wald significance tests within the DESeq2 package.
- Analysis design for calculation of the Alu RNA processing ratio in MAP patients’ RNA‐seq data.
- Boxplots depict differences in cortex of AD and no AD patients regarding SINE Alu RNA processing ratio (left panel) (a P value of 0.05 was considered as threshold for statistical significance with P < 0.001, n = 118, unpaired non‐directional t‐test) and in full‐length Alu RNAs (P = 0.005). Boxplot representation as in subfigure 2D.
- Boxplots depict full‐length Alu RNA levels in cortex of AD and no AD patients separated into three age groups. No significant difference observed between the different age groups of either AD or no AD patients or for the comparisons between no AD and AD of each age group (unpaired non‐directional t‐test, with P < 0.05 considered the threshold for statistical significance, n for no AD = 19 (below 85), 19 (between 85–90), 13 (above 90); n for AD = 8 (below 85), 21 (between 85–90), 38 (above 90)). Boxplot representation as in subfigure 2D.
- Boxplots depict differences in SINE Alu RNA processing ratio in cortex of AD and no AD patients separated into three age groups. No significant difference observed between the different age groups of either AD or no AD patients. A P value 0.05 was considered as threshold for statistical significance for the comparisons between no AD and AD of each age group (unpaired non‐directional t‐test, with P = 0.003 for the comparison below 85, and P < 0.001 for the other two comparisons and n numbers as in (E)). Boxplot representation as in subfigure 2D.
We then questioned whether our results may be influenced by the patient’s age. This is of particular interest given that aging constitutes one of the main risk factors for AD. As shown in Fig 4E and F, increased Alu RNA processing ratio is a distinct characteristic of AD patients independently of their age, while no difference was observed regarding full‐length Alu RNA levels.
We then decided to investigate the association of Alu RNA processing ratio with additional clinical and molecular indicators of AD included in the dorsolateral prefrontal cortices of patients of the MAP study. In particular, the MAP patient cohort includes also a number of patients with clinical diagnosis or pathology data in‐between the two categories of no AD and AD patients tested above (for example, patients with MCI, and/or intermediate Braak stage). Thus, we questioned whether changes in the Alu RNA processing ratio can mirror differences in the clinical diagnosis and the Braak staging not only of AD and no AD patient categories but of all MAP patients, including those in the middle of the assessed clinical range. Indeed, as shown in Fig 5A and B, processing ratios are gradually being elevated as we move from low to higher severity cases both regarding clinical diagnosis and Braak staging. Moreover, the MAP study includes information about additional molecular genetics indicators, such as APOE genotypes. APOE gene is associated with the risk of developing late‐onset Alzheimer’s disease, with the variant APOE e4 (E4) conferring a higher risk (Green et al, 2009; Genin et al, 2011). Interestingly, as shown in Appendix Fig S2A, when all MAP patients are sorted based on their Alu RNA processing ratio (upper panel of Appendix Fig S2A), and the presence of the E4 variant in the genotype of each of them is plotted (lower panel of Appendix Fig S2A), we observed higher Alu RNA processing ratios in most patients with this variant.
Figure 5. Alu RNA processing ratio is associated with clinical, pathology, genetic, and molecular markers of AD in MAP patients.
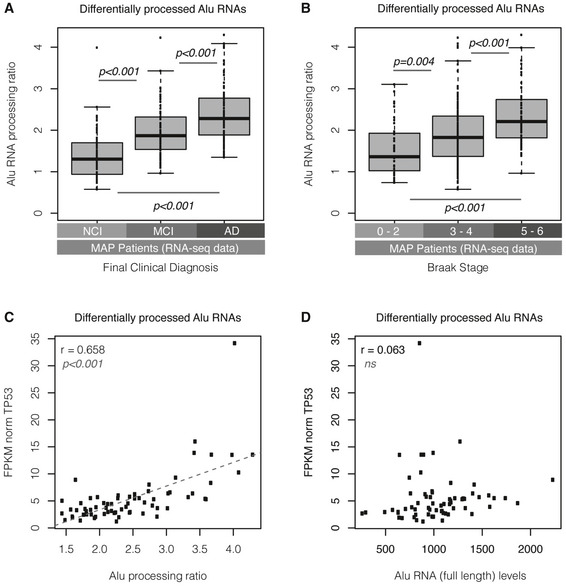
- Boxplots depict differences in SINE Alu RNA processing ratio in all MAP patients with regard to final clinical diagnosis (P < 0.01 for the comparison between NCI and MCI, between NCI and AD, and between MCI and AD, unpaired, non‐directional t‐test). NCI = no cognitive impairment (n = 71), MCI = mild cognitive impairment (n = 67), AD = Alzheimer’s disease (n = 87). Boxplot representation as in subfigure 2D.
- Boxplots depict differences in SINE Alu RNA processing ratio in all MAP patients (n = 241) with regard to Braak stage classified into three categories (P = 0.004 for the comparison between 0–2 and 3–4, and P < 0.001 for the comparisons between 3–4 and 5–6 and between 0–2 and 5–6, unpaired, non‐directional t‐test, n = 39 [0–2], n = 148 [3–4] and n = 54 [5–6]). Boxplot representation as in subfigure 2D.
- Scatterplot depicting the positive correlation between TP53 expression values and Alu RNA processing ratio in AD patients (r = 0.65, P < 0.001). Statistical test is based on Pearson’s product moment correlation coefficient and follows a t distribution using the cor.test function in R package stats.
- Scatterplot depicting no correlation between TP53 expression values and full‐length Alu RNA levels in AD patients (r = 0.06, no correlation, ns = non‐significant). Statistical test as in subfigure 5C.
The above data suggest that Alu RNA processing is increased also in the cortex of AD patients and it is associated with major clinical, genetic, and molecular pathology markers of AD.
Changes in Alu RNA processing ratio are associated with changes in P53 levels
Neural cell death leading to brain atrophy is a hallmark of AD disease. P53 (TP53) constitutes one of the major markers of cell death, and P53 deregulation has been connected with AD molecular pathology, soluble amyloid beta oligomers, and tau hyperphosphorylation (reviewed at Jazvinscak Jembrek et al, 2018; Chang et al, 2012). In our previous study in the mouse (Cheng et al, 2020), we found that SINE B2 RNA processing is associated with high P53 levels, likely as a result of consistently high levels of stress response genes upstream of P53 that are activated by increased B2 RNA processing. Since the MAP patient data included levels of mRNAs in these tissues, we questioned whether a similar association between P53 (TP53) levels and Alu RNA processing ratio exists also in humans. The scatterplot of Fig 5C depicts this relationship, revealing a moderately strong correlation between P53 levels and Alu RNA processing ratio (r = 0.65, P < 0.001) in AD patients. This strong correlation was only observed with Alu RNA processing ratio (that is indicative of increased gene activation) and not with levels of full‐length Alu RNAs (Fig 5D) (that are indicative of suppression of gene expression). Similar results were observed, as shown in Appendix Fig S2B, when we investigated the relationship between P53 and Alu RNA processing ratio in all MAP patients, which confirmed the association between the expression levels of this gene and Alu RNA processing (r = 0.58, P < 0.001) but no association with the full‐length Alu RNA levels (Appendix Fig S2C).
These data raised the question whether gene expression of members of pathways that are upstream of P53 is also associated with Alu RNA processing ratio.
Changes in Alu RNA expression and processing are associated with changes in gene expression
As shown above, changes in the Alu RNA processing ratio correlate with P53 levels in MAP patients, which is consistent with our previous studies in mouse fibroblasts and neural cells that connected the activation of gene expression in pro‐apoptotic/pro‐survival pathways with SINE RNA processing (Zovoilis et al, 2016; Cheng et al, 2020). We decided to investigate the global gene expression in MAP AD patients and search for possible associations between SINE RNA processing ratio and transcriptome changes as it has been previously shown for mouse. To this end, we first analyzed the MAP mRNA data and identified genes that are differentially expressed between AD and no AD patients. These genes are divided into two lists including genes that are up‐regulated in AD patients (up‐regulated genes, Dataset EV3) and genes that are down‐regulated in AD patients (down‐regulated genes, Dataset EV4).
Should the mode of regulation for activated genes in humans be the same as the one described in mouse (Appendix Fig S1), we would expect a strong positive correlation between high Alu processing ratio and gene expression of up‐regulated genes (Fig 6A, left panel). In the same way, should the mode of regulation for suppressed genes be the same as the one previously described in mouse, we would expect a negative correlation between high full‐length (un‐processed) Alu RNA levels and gene expression of down‐regulated genes (Fig 6A, right panel). To test these two hypotheses, we sorted the patients based either on their Alu RNA processing ratio (Fig 6B, upper panel) or their full‐length Alu RNA levels (Fig 6C, upper panel), respectively, while in the lower panels, we depicted gene expression levels of up‐ and down‐regulated genes, for each of these patients sorted in the same order as the sorted processing ratio or full‐length levels (Fig 6B and C lower panels, respectively).
Figure 6. Alu RNA expression and processing levels are associated with transcriptome changes in the cortex of MAP patients.
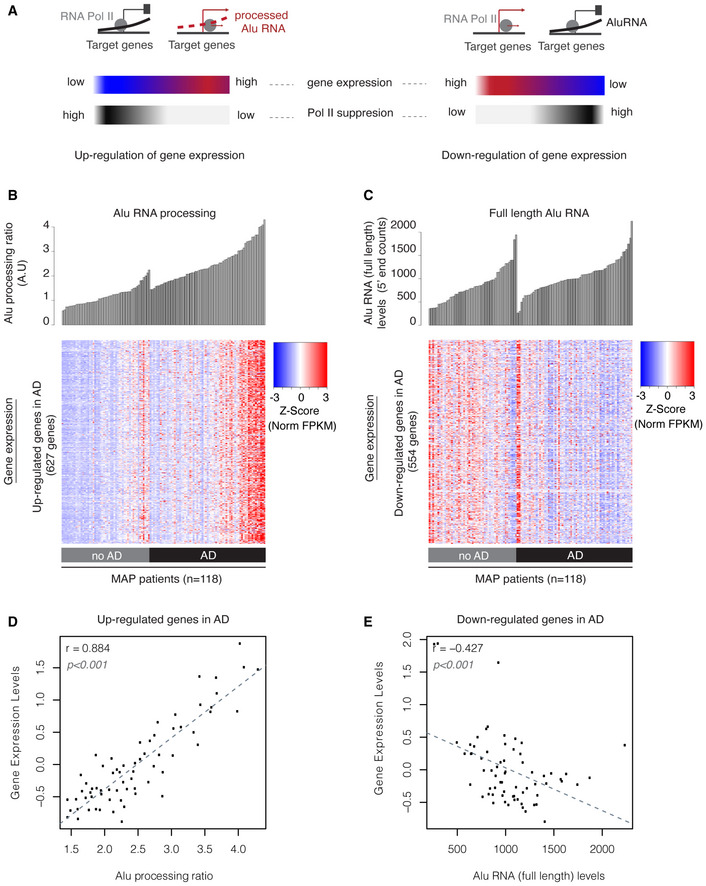
- Graphical representation of the expected transcriptome changes with regard to Alu RNA expression and processing levels based on previous findings in mouse (Appendix Fig S1) and prior reports on the ability of Alu RNAs to control transcription (Mariner et al, 2008; Yakovchuk et al, 2009; Ponicsan et al, 2010) and become self‐cleaved (Hernandez et al, 2020).
- Association between SINE Alu RNA ratio (upper panel) and gene expression levels (lower panel) of genes up‐regulated in AD (Dataset EV3, |log2FoldChange| > 0.5). Every column in both panels corresponds to the same MAP patient of either the no AD or AD group. Patients in each group are sorted from left to right in an ascending order with regard to Alu RNA processing ratio. Every row in the heatmap corresponds to one gene, with color density representing normalized FPKM values from RNA‐seq for this gene for each patient (columns) (red represents high and blue low expression levels).
- Association between full‐length Alu RNA levels (upper panel) and gene expression levels (lower panel) of genes down‐regulated in AD (Dataset EV4, |log2FoldChange| > 0.5). Every column in both panels corresponds to the same MAP patient of either the no AD or AD group. Patients in each group are sorted from left to right in an ascending order with regard to full‐length Alu RNA levels. Every row in the heatmap corresponds to one gene, with color density representing normalized FPKM values from RNA‐seq for this gene for each patient (columns) (red represents high and blue low expression levels).
- Scatterplot depicting the positive correlation between average gene expression values of up‐regulated genes in Fig 6B and Alu RNA processing ratio in AD patients (r = 0.88, P < 0.001). Statistical test is based on Pearson’s product moment correlation coefficient and follows a t distribution using the cor.test function in R package stats.
- Scatterplot depicting the negative correlation between average gene expression values of down‐regulated genes in Fig 6C and full‐length Alu RNA levels in AD patients (r = −0.42, P < 0.001). Statistical test as in subfigure 6D.
As shown in Fig 6B, consistent with our hypothesis, increase of Alu RNA processing ratio correlates with gradual increase in the expression levels of up‐regulated genes. In fact, gene expression levels of up‐regulated genes are strongly correlated with Alu RNA processing ratio in AD patients (Fig 6D r = 0.88, P < 0.001). No such correlation is observed between up‐regulated genes and full‐length Alu RNA levels (Appendix Fig S3), in which sorting based on full‐length Alu RNA levels generates a rather random distribution of gene expression densities across patients. In contrast, as shown in Fig 6C and E, full‐length Alu RNA levels correlate negatively with gene expression levels of down‐regulated genes (r = −0.42, P < 0.001), in accordance with our hypothesis, while as shown in Appendix Fig S4, there is a much weaker correlation when the relationship of these genes with Alu RNA processing ratio is tested (r = −0.26).
The changes in expression levels in AD relevant genes reported by RNA‐seq correspond to final mRNA levels in the cell and not to the initial levels transcribed by Pol II. These initially transcribed levels may have been subject to a cascade of potential post‐transcriptional modifications and processing that may have affected stability and half‐life of the mRNAs. Thus, final mRNA levels reported by RNA‐seq may not correspond completely to the elongation activity of RNA polymerase II at the chromatin level, for which we are mostly interested in the current study. To this end, we have also employed ChIP‐seq data for histone 3 lysine 9 acetylation (H3K9ac) that are available for the same patients. H3K9ac is a chromatin mark for the switch of RNA Pol II from transcription initiation to elongation (Gates et al, 2017). Thus, testing this chromatin mark corresponds very well to RNA Pol II elongation activity. As shown in Appendix Fig S5, H3K9ac occupancy downstream of the transcription start site is significantly increased in AD patients for AD up‐regulated genes compared to (i) down‐regulated genes (Appendix Fig S5A) or (ii) a random set of non‐AD differentiating genes (Appendix Fig S5B). This finding supports that there is an increase in Pol II elongation in AD up‐regulated genes.
These data show that transcriptome changes in SINE RNA are associated with transcriptome and chromatin‐wide changes observed in brains of AD patients.
Targeting of Alu RNAs leads to activation of those AD up‐regulated genes that are strongly associated with Alu RNA processing
We then questioned to what extend the observed transcriptome‐wide changes in AD patients are connected with Alu RNAs ability to suppress transcription, which would justify the increase of expression of potential target genes in AD due to increased Alu RNA processing and alleviation of such suppression.
Despite the overall positive correlation between gene expression and Alu RNA processing ratio in AD patients described in Fig 6, it is unlikely that this correlation applies to all 2,860 genes that were found to be up‐regulated in AD (Dataset EV3). To clarify this point, we estimated the correlation coefficient (r) between gene expression and Alu RNA processing of each of these 2,860 genes as well as for a random set of genes that are not differentially expressed in AD as reference. Subsequently, we classified tested genes in two main categories: one including results either non‐significant (P val < 0.05) or with weak/no correlation (r ≤ 0.5) and one including results that are both statistically significant and depict strong or very strong correlation (r more than 0.5). As shown in Fig 7A, in accordance with our findings in Fig 6, the vast majority of AD up‐regulated genes (88%) were classified to the significant and strong correlation group compared to only 26% in the control set of genes.
Figure 7. Alu RNA destabilization leads to increase in expression of Alu RNA processing correlated genes.
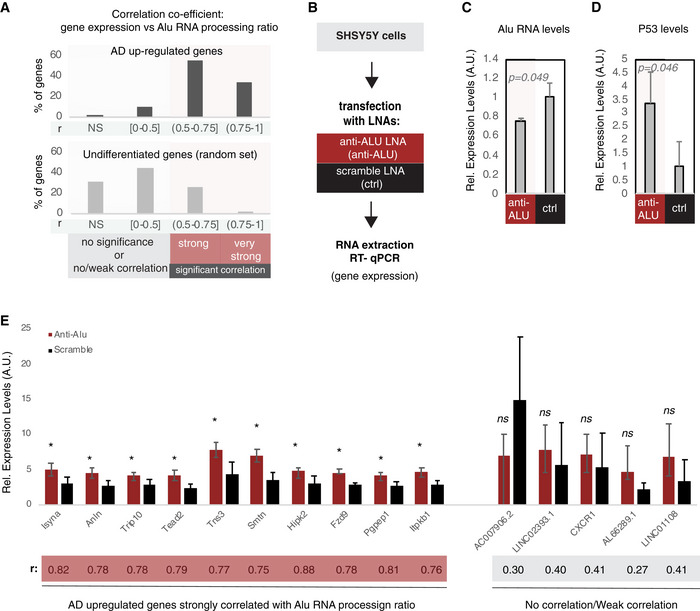
- Correlation between gene expression and Alu RNA processing ratio for AD up‐regulated genes (Dataset EV3) (upper panel) and a random set of non‐differentially expressed genes (Dataset EV5) (lower panel). For every gene, a correlation coefficient was calculated (Pearson correlation) together with the respective P‐value. Statistical test is based on Pearson’s product moment correlation coefficient and follows a t distribution using the cor.test function in R package stats. Based on the r and P value, genes were classified into four categories: (i) NS, non‐significant for P value > 0.05, (ii) no correlation or weak correlation (r ≤ 0.05), (iii) significant (P < 0.05) and strong correlation (0.5 < r ≤ 0.75), and (iv) significant (P < 0.05) and very strong correlation (0.75 < r). The bar graphs represent the percentage of each category for each set of genes. The exact r and P values of each gene are listed in Dataset EV6 for the upper panel and Dataset EV7 for the lower panel.
- Experimental design for targeting Alu RNAs in a cell culture assay employing SHSY5Y neural cells.
- Expression levels of full‐length Alu RNA (RT–qPCR) in the Alu RNA KD experiment. Statistical significance (P value threshold 0.05) for the comparison between anti‐Alu LNA‐treated samples (anti‐Alu) and samples treated with a scramble control LNA (ctrl) with P = 0.049 and n = 3, unpaired non‐directional t‐test. Error bars represent standard deviation from the mean.
- Expression levels of P53 (RT–qPCR) in the Alu RNA KD experiment. Statistical significance (P value threshold 0.05) for levels in anti‐Alu LNA‐treated samples (anti‐Alu) greater than samples treated with a scramble control LNA (ctrl) with P = 0.046 and n = 3, unpaired directional t‐test. Error bars represent standard deviation from the mean.
- Expression levels (RT–qPCR)_of selected genes from panel A that are either strongly correlated with Alu RNA processing (left, r > 0.5) or only weakly/not correlated (right, r < 0.5). Statistical significance (P value threshold 0.05) for anti‐Alu greater than control (depicted as asterisk, n = 3/group, unpaired directional t‐test, error bars represent standard deviation from the mean). Pearson correlation coefficient for each gene is depicted below the name of each gene.
Gene ontology and pathway analysis of the AD genes identified above to be strongly correlated with Alu RNA processing revealed a statistically significant enrichment (P adj < 0.05) for terms directly related with response to cellular stress, cell proliferation, cell death, P53 function, and regulation of transcription and RNA polymerase activity (Dataset EV8). In particular, KEGG pathway analysis of these genes (Dataset EV8, middle) revealed enrichment of terms for a number of cell signaling pathways upstream of P53, originally described in cancer, but subsequently attributed also to regulation of learning and memory, chromatin and synaptic plasticity and AD pathogenesis in brain. Among these pathways are MAPK and PI3K‐AKT signaling pathways (Appendix Fig S6) as well as the RAS and HIPPO signaling pathways (Appendix Fig S7). These results show that not only P53 but also members of upstream regulatory pathways of P53 are strongly associated with Alu RNA processing. Moreover, Gene Ontology term enrichment analysis of Alu RNA processing correlated genes (Dataset EV8, right) revealed among the top enrichment terms, multiple terms related with regulation of transcription from RNA polymerase II.
We then questioned whether by inducing an artificial degradation of Alu RNAs, we would be able to induce expression of these Alu RNA processing correlated genes in a neural cell culture model frequently used in AD molecular pathology experiments (SHSY5Y cells; Jazvinscak Jembrek et al, 2018). To achieve this, we employed a similar approach that we used in our previous study in mouse (LNAs against SINE RNAs). Application of a set of LNAs against Alu RNAs was able to reduce levels of Alu RNAs compared to the control LNA (Fig 7B and C). Similarly to mouse neural cells and SINE B2 RNAs, targeting of the Alu RNAs in SHSY5Y cells resulted in the increase of the expression levels of selected AD up‐regulated genes that are strongly correlated with Alu RNA processing ratio and also of P53 levels (Fig 7D and E). The increase in gene expression occurred in the absence of any stress stimulus, suggesting that these genes are under the suppressive control of Alu RNAs. At the same time, Alu RNA destabilization did not affect expression of 5 AD up‐regulated genes with a weak or no correlation to Alu RNA processing that were used as negative controls.
HSF1 can accelerate Alu RNA processing in vitro
We have shown before that SINE B2 and SINE Alu RNA processing can be attributed to an endogenous self‐cleaving ability of these RNAs that is accelerated in the presence of a protein called Ezh2 in a riboswitch manner (Hernandez et al, 2020). Subsequently, we expanded the list of B2 RNA processing accelerators to include a protein that is key in cellular response to stress called heat shock factor (Hsf1) and we showed that increased SINE RNA processing during amyloid beta toxicity in neural cells in mouse can be at least partially attributed to increased levels of Hsf1 (Cheng et al, 2020).
Thus, we questioned whether HSF1 in humans is also a potential accelerator of SINE Alu RNA processing as it is for SINE B2 RNAs in mouse. To test this, we incubated the Alu RNA in the presence of the HSF1 protein in vitro under the same conditions used in our previous studies. Then, we measured levels of the remaining full‐length Alu RNA over time in the presence and the absence of HSF1. As shown in Fig 8A, incubation of the full‐length Alu RNA in vitro led to processing and destabilization of the Alu RNA in the presence of HSF1. We then tested the impact on acceleration of Alu RNA processing by Hsf1 in the case of heat denaturation of Hsf1. As shown in Fig 8B, heat denaturation of Hsf1 resulted in abrogation of its ability to accelerate Alu RNA processing (Fig 8B). Similar results were obtained with the incubation of Alu RNA with another RNA binding protein of similar size to Hsf1, poly‐A polymerase (Hsf1 ~ 60 kDa, poly‐A polymerase ~ 56 kDa) (Fig 8B). In order to take into account any RNA destabilization due to non‐specific degradation, hydrolysis or Alu RNA endogenous self‐cleavage, we also incubated Alu RNA in the absence of any HSF1 but in the same buffer and for the same time as the sample with the HSF1 protein, and we also used as controls other RNAs in parallel reactions. In the presence of HSF1, none of these RNAs depicted the destabilization observed in Alu RNAs (Fig 8C).
Figure 8. HSF1 accelerates Alu RNA processing in vitro .
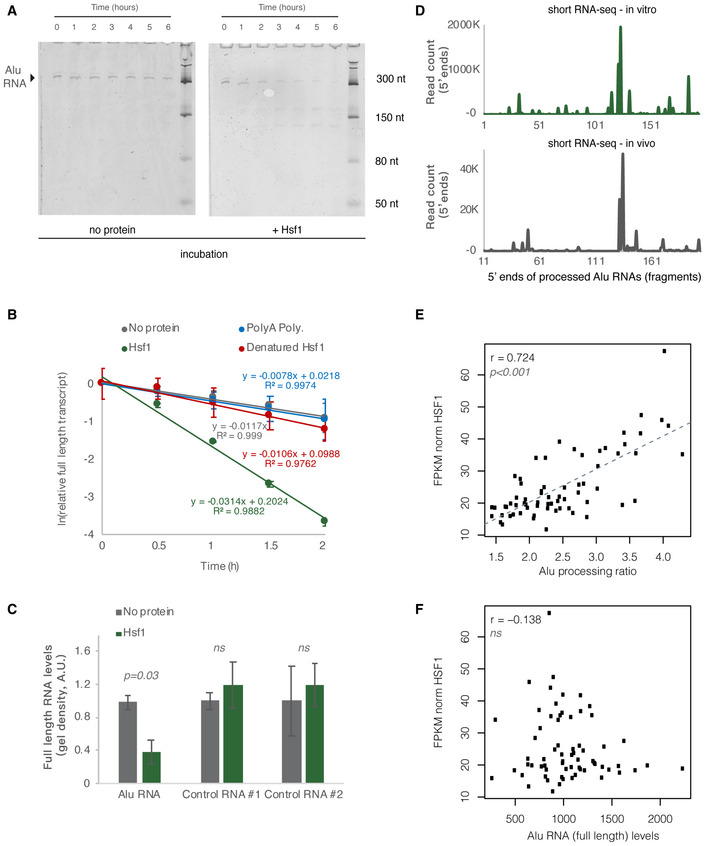
- In vitro incubation of one of the Alu RNA consensus sequences for different incubation periods. In vitro transcribed Alu RNA (67 nM) incubated at 37°C with 250 nM HSF1 in the course of 6 h with time intervals of 1 h.
- Comparison among Hsf1 (~ 60 kDa), denatured HSF1, poly‐A polymerase (~ 56 kDa), and no protein (just TAP buffer) with regard to Alu RNA processing (estimating relative full‐length RNA remaining) (two replicates). The full gels are available as Source Data for Fig 8. Relative full‐length RNA remaining was calculated using ImageJ area under the curve software over time. Error bars represent standard deviation from the mean.
- Comparison among Alu RNA (three replicates) and two control RNAs (two replicates) regarding the full‐length RNA levels remaining after in vitro incubation for 90 min at 37°C with HSF1. Sizes of control RNAs are control for RNA #1, 143nt and for control RNA #2, 432nt. Incubation in the absence of HSF1 but presence of the same buffer (TAP) was used as control to take into account any non‐HSF1‐specific RNA destabilization due to non‐specific degradation. Threshold for statistical significance was a P value of 0.05 with P = 0.03 (unpaired, non‐directional t‐test) for the comparison between HSF1 and no protein incubation (n = 3, for Alu RNA). The full gels are available as Source Data for Fig 8. Error bars represent standard deviation from the mean.
- Plotting of the position of the first base (5′ end) of Alu RNA fragments across the Alu consensus sequence produced by Alu RNA that has been processed in vitro for 90 min at 37°C in the presence of HSF1 (upper panel) and compared with one of the in vivo samples of Fig 1 (lower panel). The x‐axis represents an Alu RNA metagene aligned at the start site of the Alu consensus sequence, and the y‐axis shows the 5′ end count for Alu RNA fragments aligning to any position downstream of position +1. The in vivo sample x‐axis depicts a 11nt sift compared to the in vitro one.
- Scatterplot depicting the positive correlation between HSF1 mRNA expression values and Alu RNA processing ratio in MAP AD patients (r = 0.72, P < 0.001). Statistical test is based on Pearson’s product moment correlation coefficient and follows a t distribution using the cor.test function in R package stats.
- Scatterplot depicting lack of correlation between HSF1 mRNA expression values and full‐length Alu RNA levels in MAP AD patients (ns = non‐significant, with P value 0.05 as the significance threshold). Statistical test as in subfigure 8E.
Source data are available online for this figure.
Then, we purified the Alu RNA fragments derived in vitro from the incubation with HSF1 and subjected them to the same short RNA sequencing protocol we used for the post‐mortem samples in order to compare the Alu RNA fragmentation pattern in vitro with that in vivo. As shown in Fig 8D, we have been able to identify also in vitro the major cut at Alu RNA right arm we had observed in vivo. Interestingly, the position of the cut was slightly shifted for 11nt toward the 5′ end, across the poly‐A bridge. This finding implies that the initial cut in vivo may be subsequently subjected to a yet unclear form of maturation that trims the 5′ end of the generated fragment. Alternatively, given the great genomic sequence diversity of Alu RNA elements among humans, we cannot exclude that the single Alu consensus sequence we used for the in vitro synthesis of the RNA may not correspond completely to the most frequently represented Alu RNAs in the samples we tested from the patients that may include deletions compared to this consensus. However, the lack of whole‐genome sequencing data from these patients makes clarifying this point challenging.
These data show that B2 RNA and Alu RNA may share a common factor, HSF1, as a potential accelerator of their processing. To assess this potential in our context, we tested the association of Alu RNA processing ratio with regard to HSF1 mRNA levels in the brains of MAP AD patients. Indeed, as shown in Fig 8E a strong correlation was found between them in AD patients (r = 0.72, P < 0.001). In contrast, when an association between HSF1 levels and full‐length Alu RNA levels was tested, no correlation was found (Fig 8F).
These data suggest that similarities observed between mice and humans in vitro may also extend to SINE RNA processing in human brain.
Discussion
Elucidation of the molecular mechanisms underlying AD pathogenesis comprises an important part of efforts currently underway to understand better this debilitating disease. To this end, recent high‐throughput approaches and advances in our understanding of novel elements of human transcriptome’s architecture such as RNAs from SINE transposable elements may offer novel perspectives regarding molecular pathology of neurodegenerative diseases.
SINE Alu elements constitute a substantial part of the repetitive non‐coding genome in humans, being present in millions of copies in human DNA (Walters et al, 2009). Although SINE elements had been regarded for long as genetic parasites and part of “junk” DNA, numerous studies in recent years have started shedding light into their potential functions in increasing genomic regulatory capabilities through generation of new genomic regulatory sites (Deininger, 2011). However, little is known about the role in human health and disease of the non‐coding RNAs generated by these SINE repeats. The identification, some years ago, of the critical ability of SINE B2 and SINE Alu RNAs to regulate transcription through suppression of RNA polymerase II, both in humans and in mouse, opened a totally new avenue of possibilities regarding their potential role in cellular function as well as their potential involvement in molecular pathologies (Espinoza et al, 2007; Mariner et al, 2008; Walters et al, 2009; Yakovchuk et al, 2009; Ponicsan et al, 2010, 2015). Subsequently, our recent discovery of the SINE RNA ribozyme/riboswitch potential in response to cellular stress expanded further these possibilities (Zovoilis et al, 2016; Hernandez et al, 2020) and resulted in the identification of a connection between transcriptome deregulation observed in amyloid beta pathology in mouse brain and increased processing of SINE B2 RNAs accelerated by high Hsf1 levels (Cheng et al, 2020).
Here, we show that SINE Alu RNAs are processed also in human brains and reveal increased processing of SINE RNAs as a novel type of transcriptome deregulation observed in AD (Fig 9). These findings allow us to extend to human AD patients our previous conclusions in murine models of AD and deduce that SINE RNAs may be important components of the molecular mechanisms underlying this disease. To this end, this is the first human disease identified to be connected with the ribozyme‐based processing of SINE RNAs and also with a ribozyme‐based activity in humans in general.
Figure 9. Representation of the observed changes in Alu RNA processing in AD.
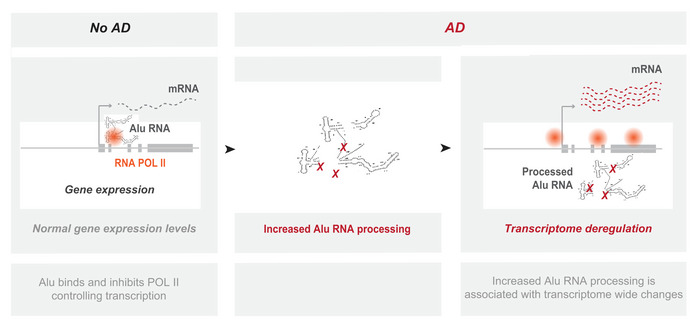
In no AD brains, basal Alu RNA processing levels maintain control of gene transcription through regulation of RNA Pol II. In contrast, in AD pathology, various, yet unknown, factors lead to an increase in Alu RNA processing associated with transcriptome‐wide deregulation of gene expression.
The hypothesis that transposable elements such as Alu elements may play a role in neurodegeneration is not new. Non‐coding RNAs and transposable elements in the context of neurodegenerative diseases have been shown to play interesting roles in how neural cells regulate pro‐survival and pro‐death pathways. For example, tau has been shown to activate transposable elements in Alzheimer’s disease, while pathogenic tau‐induced piRNA depletion has been shown to promote neuronal death through transposable element dysregulation in neurodegenerative tauopathies (Sun et al, 2018). Moreover, a described protein–RNA interaction between the engrailed homeoprotein b and the LINE‐1 retrotransposon has been shown to prevent neurodegeneration in adult dopaminergic neurons (Blaudin de The et al, 2018). A potential connection of Alu elements with neurodegeneration has been documented in a number of reports, and it is very well outlined in a review by Larsen and colleagues (Larsen et al, 2017) that points into the impact of insertions of Alu elements into genes such as TOMM40 that control mitochondrial function. In addition, more than 95% of Adenosine‐to‐Inosine RNA editing sites have been discovered within Alu regions (Huang et al, 2018) and it is now well established that the major targets for these modifications are Alu elements embedded in mRNAs, especially the retro‐transposition‐incompetent ones (Nakahama & Kawahara, 2020). In particular, RNA editing in protein coding genes has been shown to be connected with recoding (changes in the protein coding sequence) of protein coding RNAs associated with neurodegenerative diseases including Alzheimer’s disease (Singh, 2012; Khermesh et al, 2016; Annese et al, 2018; Krestel & Meier, 2018; Larsen et al, 2018; Kanata et al, 2019).
However, until now the role of Alu elements in neurodegeneration has been described mainly as a passive one, via disruption of genomic integrity though their integration into critical regions in the genome or as mediators of RNA editing of the mRNAs in which they are embedded. Here, we present evidence about a more active role of SINEs in regulation of gene expression in neurodegenerative diseases, through an independent ribozyme‐related function of the transcripts generated by these elements. Moreover, a pioneering study by the Ambati laboratory (Kaneko et al, 2011) revealed a critical role of Alu RNAs in pathogenesis of macular degeneration, through cellular toxicity effects of Alu RNAs. Thus, in addition to the potential direct impact that Alu RNA processing can have on gene transcription through abolition of Pol II suppression, it is likely that increased Alu RNA processing we observe in AD patients may impact cells also in other ways such as through the generation of toxic Alu RNA species.
In this study, we employed RNA‐seq data from two different sequencing approaches, one customized for these RNAs (short RNA‐seq) and a standard one used in one of the larger transcriptome studies in AD, in order to test whether existing standard RNA‐seq datasets can be used to study Alu RNA processing beyond the more specific short RNA‐seq approach. Our findings indeed indicate that these data can be used in case of humans for the study of Alu RNA processing. Being able to use standard RNA‐seq to study Alu RNA processing has significant advantages with regard to the more specific short RNA‐seq approach given that existing publicly available data have been generated using standard protocols. Moreover, the list of Alu RNA sequences that were found to be differentially processed in hippocampus of CBB patients was different from that in the prefrontal cortex of MAP patients. This difference could be attributed to either the different tissue source (different transcription levels in different tissues) or to different demographics between the two patient cohorts (genetic variability/CNVs, SNPs, insertions/deletions), or to a combination of both. Thus, despite the end result being the same (i.e., increased processing of Alu RNAs), the source of these RNAs remains a population of a quite diverse set of Alu genomic elements reflecting human genomic variability, which denotes the need for cost‐effective ways to study Alu RNA processing in order to include large patient cohorts that are representative of such variability.
For our analysis, we have used RNA‐seq data from bulk tissue specimens. Despite their undisputable value, such type of data cannot directly distinguish between neuroglia and neurons. As shown in Appendix Fig S8, RNA‐seq data used in this study express both markers that are predominantly neuronal (such as SLC17A7) and markers that are mainly non‐neuronal (such as AQP4) in contrast to RNA‐seq dataset from exclusively non‐neuronal cells (Appendix Fig S8A–C). Application of a cellular deconvolution approach revealed that approx. 43% of the cells that contribute to the MAP RNA‐seq data are neuronal cells (Appendix Fig S8D and E). Moreover, both neuronal and neuroglia cells express Alu RNAs (Appendix Fig S8F). These data exclude the possibility that our findings could be attributed exclusively to either neuronal or non‐neuronal cells within this brain tissue, though the exact contribution of each cell type to our results remains unknown. The same applies to the observed correlation between P53 and Alu RNA processing ratio. It is currently unclear whether this is result of microgliosis, astrogliosis neuronal death, or combination of these, since our RNA‐seq is not single cell specific and all the identified pathways (Dataset EV8, Appendix Figs S6 and S7) are cellular stress response and signaling pathways that are universally expressed in all these cell types.
In our analysis, we found no correlation between HSF1 levels and full‐length Alu RNAs. We have included the HSF1 vs. full‐length plot in Fig 8 as a control to the HSF1 vs. processing ratio plot. In that way, we wanted to exclude the possibility that Alu RNAs may be under the direct transcriptional control of HSF1, which would confound our findings by causing an increase in the denominator of our estimated Alu RNA processing ratio. Our results show that HSF1 is unlikely to be an upstream direct regulator of Alu RNA transcription, as there is practically no correlation with Alu RNA full‐length levels, and that HSF1 rather exerts its action on Alu RNAs by increasing the proportion of fragmented Alu RNAs. As with many gene circuits and pathways involved in cellular response to stress, there are usually a number of compensatory cellular homeostasis mechanisms in place that through positive and negative feedbacks regulate RNA levels. In our case, it would be reasonable to expect that in vivo there are compensatory pathways that would respond to the reduction of Alu RNA levels by increased processing through an increase of Pol III Alu RNA transcription. This could even result in an increase of total Alu RNA levels after chronic exposure to such stimulus as observed in Fig 4D.
Due to the repetitive nature of Alu RNAs, it is currently difficult to make any direct conclusions regarding how many of the Alu sequences tested here originate from Pol III transcripts and how many from transcripts embedded into mRNAs (likely nascent ones). As shown in Appendix Fig S9, we tried to address this question indirectly by repeating our mapping against the genome, and separating the Alu elements, against which we map the RNA fragments, into two categories: (i) Alu elements that fall within gene regions, and (ii) Alu elements outside of gene regions. Despite Alu multiple mapping, a level of information regarding genomic positions that generate Alu RNAs is expected to be maintained through this approach. Therefore, if the mapped Alu RNAs originated exclusively from either only Pol III Alu elements or mRNA embedded Alu elements, we would expect at least some difference in the distribution of fragments between Alu elements of these two categories, as the genic ones overlaps with mRNAs. As shown in Appendix Fig S9, the fact that distribution models are very similar between the two categories implies that both types of Alu elements may contribute to Alu RNA processing. However, given the limitations posed by the repetitive nature of Alu RNAs mentioned above, it remains difficult to provide an exact number regarding the portion of B2 RNA fragments produced by each category.
In the current study, we have justified higher levels of Alu RNA fragments in AD patients as a result of higher processing and destabilization of Alu RNAs, using as reference the full‐length Alu RNA levels estimated through the poly(A) selection‐based RNA‐seq in MAP patients. However, it is important to note that acceleration of Alu self‐cleavage is likely only one of the ways through which the cell may control Alu RNA levels. Other pathways may also be at play including nucleases or protection through A‐I editing. In fact, the 11nt shift in the cutting point we observe in vivo compared to the in vitro processed RNA in Fig 8D implies an additional Alu RNA processing step in vivo beyond its self‐cleaving activity. We are at the moment unaware of any nuclease defects in AD patients that may be specific to Alu RNA, but such a possibility can also not be excluded.
Our study leaves also some additional open questions. This study is the first to confirm an association between SINE RNA transcriptome and genome‐wide transcriptome changes in a human tissue. We also showed that in the context of a neural cell line, targeting SINE Alu RNAs in humans elicits the up‐regulation of various genes in agreement with what was previously described in mouse. However, it remains unclear whether this applies also in vivo since performing such studies in vivo is not possible.
Moreover, our data tested in humans the potential role of HSF1 in SINE RNA processing previously observed in mouse only in vitro. Future studies should examine that impact also in cell systems that may resemble neural cells in human brain. Finally, it currently remains unknown which other factors may be implicated in the increased processing of Alu RNAs in AD beyond HSF1. Elucidating the factors that are upstream of this process may provide additional insight regarding the mechanisms underlying molecular pathology of AD and help advance further our understanding of these RNAs as potential novel therapeutic targets and not just transcriptional noise and “junk DNA” products.
Materials and Methods
Post‐mortem hippocampal tissues
Hippocampal tissues were acquired by the Calgary Brain Bank following the respective institutional approvals. De‐identified clinical metadata are available as a separate phenotype file accompanying the next‐generation sequencing data. Aggregate clinical data were as follows: AD, seven males and four females, with a. median age of 77; no AD, seven males, six females, with a median age of 64. Tissues from hippocampus (approx. 0.75 cm3 cubes) were homogenized in 5 ml TRIzol reagent: 15‐min incubation and subsequent grinding using a pestle until nothing but insoluble connective tissue remained. The homogenized mix was pipetted up and down and the solution was stored at −80°C. 1 ml of homogenized mixture was phase separate by the addition of 200 μl of chloroform (Sigma, C2432) and mixed by inversion, incubated for 3 min and centrifuged at 12,500 g for 15 min at 4°C. The top (aqueous) layer was transferred to a new tube and mixed with 500 μl of isopropanol (Fisher, 67‐63‐0), followed by a 1‐h incubation at −20°C and centrifugation at 12,500 g for 10 min at 4°C. The supernatant was removed and the pellet was washed and mixed with 1 ml of 75% ethanol, followed by a centrifugation at 7,600 g for 5 min at 4°C. The supernatant was removed and the pellet was allowed to dry for 1 min before eluting in 50 μl of nuclease free H2O. The eluted RNA was heated at 55°C for 15 min and subsequently incubated with 1 μl of DNaseI (NEB, M0303), 10 μl of 10× DNaseI buffer (NEB, B0303), and 39 μl of nuclease free H2O for 15 min at 37°C. The RNA was further cleaned using the Zymo Research RNA clean and concentrator kit ‐25 (R1017). The RNA was stored at −80°C. Estimation of RIN scores for the purified RNAs revealed no difference between the AD and no AD patients (Appendix Fig S10B).
RNA in vitro transcription and RNA‐protein incubations
Alu consensus template was ordered as IDT g‐blocks™ (lower case denotes the T7 promoter sequence): AluY: 5′‐taatacgactcactatagGCCGGGCGCGGTGGCTCACGCCTGTAATCCCAGCACTTTGGGAGGCCGAGGCGGGCGGATCACGAGGTCAGGAGATCGAGACCATCCTGGCTAACACGGTGAAACCCCGTCTCTACTAAAAATACAAAAAATTAGCCGGGCGTGGTGGCGGGCGCCTGTAGTCCCAGCTACTCGGGAGGCTGAGGCAGGAGAATGGCGTGAACCCGGGAGGCGGAGCTTGCAGTGAGCCGAGATCGCGCCACTGCACTCCAGCCTGGGCGACAGAGCGAGACTCCGTCTCA; templates were amplified by PCR using a T7 promoter sequence as the forward primer: 5′‐TAATACGACTCACTATAG and a reverse primer: 5′‐TGAGACGGAGTCTCGCTC. The amplified g‐blocks were then gel band excised and extracted using the BioBasic EZ‐10 gel extraction kit (BS353). The gel excised DNA was used as an additional PCR template and the amplified g‐block was in vitro transcribed by the Hiscribe T7 High Yield RNA Synthesis Kit (NEB, E2040S) for 2 h at 37°C. RNA was cleaned using the Qiagen RNeasy MinElute Kit. RNA controls used are Control#1(G‐44U): 5′GCCCCGUUGCAAUGGAAUGACAGCGGGUAUGUUAAACAACCCCAUCCGUCAUGGAGACAGGUGGACGUUAAAUAUAAACCUGAAGAUUAAACAUGACUGAAUCUUUUGCUACUAGAAUGGUGAGCAAGGGCGAGGAGCUGUUC 3′ and Control #2 (Zika 3′ scramble): CCGCAGGCGAGUGAUGCAGAUAAAAACAUACUCGUCCAAUCCGAAAAUGAUCGGCGGUCUUUCAGCGCACGCGAGCGAGCGACCGCCUUCGGCUGAUGCGCACGCCGCGCGCAGGGACAGGUACAUCUUCAGAGGGAGCGGUCAGAAAUUGCACACGAUAGAAGGAAAGACCCUAGAAUGCCCCACUCGGGGAUGUGCGCCACAAAGGACUCCGGCAGUGAAGUUGAAUCGAGCGCCGAGGAGAUAGCCCGCCAGCACGACCACGGCGGACAGGGUGCGGCGCGCCAUGGAUUUGUGGGACUUCCAAAUAAGGCGUCGCUGAAGGCUUACUGAAUCUCUUCACAGAUGUGCAGGGAACUCUACAAGCAGGAUUAGGGCCUCCUACACUUGCGCACACGCACCGGUAGAAAUCAACGGGACGAUCUACGAUCA. Both RNAs were generously donated by the Wieden and Patel laboratory respectively, from the University of Lethbridge.
In vitro experiments were done as described before (Zovoilis et al, 2016; Cheng et al, 2020; Hernandez et al, 2020) and included a refolding reaction for the RNA samples with 300 mM NaCl into nuclease free water. The incubation of 2 pmoles of Alu transcripts (0.33 pm for Fig 8A) occurred as follows: RNA was incubated for 1 min at 50°C and cooled at a rate of 1°C/10 s until 4°C. Samples were then incubated at 37°C with the addition of TAP buffer (final reaction concentrations: 5 nM Tris pH 7.9, 0.5 mM MgCl2, 0.02 mM EDTA, 0.01% NP40, 1% glycerol, 0.2 mM DTT) or protein samples diluted in TAP buffer and folding buffer (reaction concentration: 300 mM NaCl, 50 mM Hepes). Degradation of Alu RNA was analyzed on 8 M urea 10% PAGE gels, stained with SYBR II (Invitrogen, S7564). Samples were mixed with Novex™ 2× TBE‐Urea loading buffer (Invitrogen, LC6876) and denatured at 95°C for 5 min before loading. Electrophoresis occurred in 1× TBE buffer at 180 V. Gel analysis was performed using Amersham Typhoon instruments. Band absorbance was analyzed using ImageJ area under the curve software and normalization by the ratio of experimental over initial.
HSF1 protein incubations were performed with phosphorylated, recombinant, His‐tagged HSF1 (Enzo Life Sciences, ADI‐SPP‐902). HSF1 working concentrations were 250 nM unless otherwise specified, diluted in TAP buffer.
Short RNA‐seq
5 µg total RNA was PNK phosphorylated (85 µl RNA, 10 µl 10× PNK buffer, 5 µl T4 PNK [NEB, M0201], incubated at 37°C for 1 h). The RNA was then cleaned using the MinElute RNA Cleanup Kit (Qiagen, 74204) with a customized protocol for total RNA recovery: in brief, 100 µl sample mixed with 350 µl supplied RPE buffer and 525 µl 100% ethanol. The samples were centrifuged and washed as recommended. 3,000 ng of remaining RNA was 2× rRNA depleted (NEB, E6130). The library was then prepared using NEBNext® Small RNA Library Prep Set for Illumina (NEB, E7330) with 1:10 adapter dilutions. Final PCR used 18 cycles and kit optimized index primers. Sample clean ups were performed as recommended using Omega NGS Total Pure Mag Beads (Omega) and following amplification, samples were twice 1.2× bead cleaned. Quality control analysis was performed using the Agilent bioanalyzer 2100 RNA pico kit and DNA HS kit. Quantification of libraries was done by qPCR using the NEBNext library quant kit for Illumina and library sizes were analyzed using the Agilent bioanalyzer 2100 HS DNA kit. Equimolar amounts were prepared for sequencing. Libraries were sequenced on the Illumina HiSeq platform in pairwise fashion. Appendix Fig S10A shows a representative Bioanalyzer electropherogram of one of these libraries.
SH‐SY5Y cell culture
SH‐SY5Y neuroblastoma cell line was obtained from ATCC (CRL‐2266). Cells were cultured in 1% pen/strep treated EMEM (Sigma, M4655) and F12 Ham (Sigma, N6658) mixed 1:1, 1% l‐glutamine (200 mM; Gibco, 25030149), 1% Non‐essential amino acids (SAFC, M7145), and 10% FBS (Sigma, F4135). Before any assay, cells were thawed and plated in a T75 flask. During subculture, cell pellet was resuspended in 10 ml fresh medium and re‐plated. Cells were maintained in 5% CO2, 37°C. Cell passaging was done by incubating at 37°C with trypsin (Gibco, 12605028) for 5 min. Trypsin was then inhibited 1:1 with fresh medium, and cells are then centrifuged. The pellet was washed with 5 ml PBS, resuspended in fresh medium, and plated.
LNA transfections
500,000 cells were plated in a 6‐well plate well. 15 min after plating, 100 µM of scramble LNA (Qiagen, custom) and 12.5 µM of each anti‐Alu LNA (100 µM total pooled; IDT, custom) were prepared and applied dropwise in solution with 10.7% Hiperfect transfecting agent (Qiagen, 301704). Transfections occurred for 12 h before treating the cells with trypsin as described above to displace and pellet the cells. Cells were then added to a vial with 1 ml TRIzol and allowed at room temperature for 5 min. Pellets in TRIzol were then stored at −80°C. RNA extraction was performed as described with hippocampus tissue above. The anti‐Alu LNAs are as follows (* denotes a phosphorothioate bond and +denotes an LNA bond): 1.+C*+A*+C*+G*C*C*C*G*G*C*T*+A*+A*+T*+T 2.+C*+A*+C*+G*C*C*C*A*G*C*T*+A*+A*+T*+T 3.+C*+A*+T*+G*C*C*C*G*G*C*T*+A*+A*+T*+T 4.+T*+C*+C*+T*G*C*C*T*C*A*G*C*+C*+T*+C*+C 5.+G*+G*+A*+G*T*C*T*C*G*C*T*+C*+T*+G*+T 6.+G*+G*+A*+G*T*C*T*C*A*C*T*+C*+T*+G*+T 7.+A*+G*+A*+G*T*C*T*C*G*C*T*+C*+T*+G*+T 8.+T*+C*+G*+G*C*C*T*C*C*C*A*A*+A*+G*+T*+G. The scramble LNA used is as follows: +C*+C*+T*+C*A*A*T*T*T*T*A*+T*+C*+A*+C.
RT–qPCR
Reverse transcription was performed by the standard Thermo‐Fisher M‐MLV reverse transcription protocol using the M‐MLV reverse transcriptase (Invitrogen, 28025013) with 500 ng RNA. Quantitative PCR was performed using the Luna Universal qPCR mix (NEB, M3003). Reactions were set up so cDNA was diluted in water 1:20, constituting 20% of the overall reaction. Primers were diluted to 10 µM, constituting 5% of the overall reaction, and the master mix was added to 50% of the overall reaction. Total reaction volume was 10 µl. Standard dilution pools were set up in four standard dilutions from a pooled sample mixture, diluted 1:5, 1:10, 1:20, and 1:40. Thermocycler conditions were as follows: 3 min at 95°C (15 s at 95°C, 30 s at 54°C, 30 s at 66°C) × 40 cycles. qPCR was performed using the Bio‐Rad CFX384 Real‐time detection system and measurements were taken during extension steps. Primers used are included in Table EV1. Experiments were performed is three biological replicates and three technical replicates.
Bioinformatics analysis
For Calgary Brain Bank (CBB) short RNA‐seq data analysis, standard Illumina adaptor sequences were trimmed off using cutadapt‐1.18 (https://doi.org/10.14806/ej.17.1.200). For the construction of the “ALUome”, we retrieved Alu sequences for all identified Alu elements (within the range of 280–320 bp in order to exclude truncated forms) from UCSC hg38 Repeat Masker (August 2018) and subsequently collapsed sequences present in our list more than once into one unique representative sequence, resulting only in unique Alu sequences. Alu RNA sequences mapping within known exons, upstream/downstream 300 bp of exons, snoRNAs, microRNAs, and tRNAs were filtered out (using as annotation: Hg38, UCSC NCBI RefSeq, sno/miRNA, tRNA Genes). Reads for each sample were mapped to the ALUome using bwa‐0.7.17 (Li & Durbin, 2009) aln single end mode, with parameters: maximum edit distance: 0, maximum number of gap opens: 0, disallow a long deletion within bp toward the 3′ end: 100, disallow an indel within bp toward the ends: 100, maximum edit distance in the seed: 0, mismatch penalty: 4, gap open penalty: 22, gap extension penalty: 12. SAM format files generated from mapping were converted to BAM format files using samtools‐1.6 (Li et al, 2009) and to files in BED format with bamToBed utility from BEDTools‐2.26.0 (Quinlan & Hall, 2010). Then, start positions of reads mapping in the sense direction to each unique Alu of the ALUome were retrieved. For each sample, a matrix was generated with Alu sequences as rows and counts of read starts for each Alu position as columns, normalized to FASTQ file reads (per 100 million reads). Sum of counts from position above 130 were calculated for each Alu and patient, and then, the respective count matrices were generated combining each Alu (rows) with the counts in the patients (columns) for position above 130. R (version 3.4.3) (https://www.R‐project.org/) package DESeq2 (Love et al, 2014) was applied to counts above 130 between AD and no AD patients to get differentially expressed processed Alus. To calculate the Alu RNA processing ratio, Babraham NGS analysis suite Seqmonk 1.45.4 (https://www.bioinformatics.babraham.ac.uk/projects/seqmonk/) was used to obtain the number of reads overlapping with RNA 7SL as well as the read coverage per base along all Alu loci. Processing ratio for each sample was calculated by the processed Alu count obtained from mapping fragments to unique Alus normalized by the RNA 7SL counts and total Alu read coverage per base: [processed fragments (position above 130)/(Alu read coverage per base/RNA 7SL count)].
For ROSMAP study data, RNA‐seq data from the AMP‐AD Knowledge Portal (https://adknowledgeportal.synapse.org) for the MAP project (Synapse ID: syn3219045) (Bennett et al, 2012) were acquired and paired end FASTQ files of each sample were converted from original data in BAM format using utility bam2fq in samtools‐1.6. Then, sequencing data were mapped against the ALUome and analyzed generating Alu count matrices. For each sample, a matrix was generated with Alu sequences as rows and counts of read starts for each Alu position as columns, normalized to FASTQ file reads (per 100 million reads). Sum of counts from position 0 to 15 (full‐length Alu) and above 130 (processed Alus) were calculated for each Alu and patient, and then, the respective count matrices were generated combining each Alu (rows) with the counts in the patients (columns) for position 0 to 15 and above 130. To get differentially expressed processed Alus, the R package DESeq2 was applied to counts above 130 between no AD patients (with a final clinical consensus diagnosis of no cognitive impairment and a Braak stage: 0–III) and AD (final clinical consensus diagnosis: Alzheimer’s disease and no other cause of cognitive impairment, Braak stage: IV–VI). Processing ratio for each sample was calculated by dividing the processed Alu count obtained from mapping to unique Alus with the full‐length Alu level count mentioned above.
The decision for the selection of the appropriate method for estimating Alu RNA processing ratio in short RNA‐seq and RNA‐seq data was based on whether the applied RNA selection and library construction protocol maintains both fragments generated by the cut at XR1 (one overlapping with left arm and one overlapping right arm) or only the one at the right arm. As shown in Appendix Fig S10C, short RNA‐seq maintains both fragments as depicted by the existence of two major 3′ end densities, one around XR1 and one around the expected end of the full‐length Alu RNA. In contrast, RNA‐seq of poly‐A RNA filters out the fragment overlapping the left arm, as now increased density is observed around XR1 position. This is in accordance with the poly‐A selection step used, which would select only the full‐length fragment and the processed fragment at the right arm. Thus, position 0–15 is expected in the standard RNA‐seq protocol to map only full‐length Alu reads, which is why the count at this position is used for the estimation of the processing ratio at Fig 4. In contrast, for short RNA‐seq, the 0–15 position count number includes both full‐length and fragmented RNAs. Thus, for the estimation of the processing ratio in short RNA‐seq samples, we used the total Alu read coverage per base that is independent from the position of the mapping of the 5′ end of the reads. As in our previous studies in mouse, the SINE RNA levels were normalized to a Pol III transcript (7SL RNA) to account for non‐specific degradation by nucleases. As shown in Appendix Fig S10D, as expected, we observed no difference in 7SL levels between the two groups of patients, supporting the use for the per sample normalization.
Regarding gene expression analysis of MAP patients, reads for each sample were mapped to the reference genome acquired from ensemble (GRCh38/November 2018, primary assembly) using hisat2‐2.1.0, in single end mode, with the following parameters: Report alignments tailored for transcript assemblers including StringTie and searches for at most 1 distinct, primary alignments for each read (Kim et al, 2019). SAM format files generated from mapping were converted to BAM format files using samtools‐1.6 and to files in BED format with bamToBed utility from BEDTools‐2.26.0. FPKM (Fragments Per Kilobase of transcript per Million) for genes were generated using StringTie‐1.3.4d, with the following annotation: ensembl GRCh38 patch 94 .gff3 file, and parameters: limits the processing of read alignments to only estimate and output the assembled transcripts matching the reference transcripts given in annotation and excluding non‐regular chromosomes, then normalized gene FPKM with RN7SL2. R package DESeq2 was applied to gene counts to perform differential gene expression analysis between no AD and AD individuals (with P adj < 0.05 used as thresholds for reporting). For data visualization, statistics, and differential expression analysis, we employed R (version 3.4.3) with r in scatterplots corresponding to Pearson correlation coefficient (https://www.R‐project.org/). In the boxplots, the central band (the line that divides the box into two parts) represents the median of the data, the ends of the box shows the upper (Q3) and lower (Q1) quartiles, the extreme line shows Q3 + 1.5 × IQR to Q1 − 1.5 × IQR (the highest and lowest value excluding potential outliers), while dots beyond the extreme line show potential outliers.
For the cellular deconvolution approach, we used the R (version 3.6.1) package MuSiC (Wang et al, 2019). This was implemented to estimate cell type proportions from MAP project data based on the Allen Brain Map Anterior Cingulate Cortex (ACC) SMART‐seq dataset (https://portal.brain‐map.org/atlases‐and‐data/rnaseq/human‐v1‐acc‐smart‐seq). Markers that are enriched in non‐neuronal cells. For panel B image credit: Allen Institute. © [2015] Allen Institute for Brain Science. [Allen Mouse Brain Atlas]. Available from: [https://celltypes.brain‐map.org/rnaseq/human_ctx_smart‐seq] based on (Tasic et al, 2018; Hodge et al, 2019).
Gene expression analysis of microglia RNA‐seq data (Synapse ID: syn11468526) from ROSMAP study was also conducted with the same analysis pipeline as described for MAP patients above, and subsequently, gene expression levels of the two datasets were compared.
Patients with available H3K9ac ChIP‐seq data from ROSMAP study (Synapse ID: syn4896408) overlapping with tested MAP RNA‐seq patients were identified, and filtered bam files against the provided peak file (H3K9acDomains.bed) were obtained with the intersectBed utility from BEDTools‐2.26.0 (Quinlan & Hall, 2010). Then, metagene plots of the filtered bam files for the different gene lists were generated using Seqmonk.
Ethics approvals
CBB patient data analysis was performed under the following institutional approvals: University of Lethbridge Human Subject Research Committee (HSRC), protocol #2019‐059 and Calgary Brain Bank provisions under the University of Calgary Conjoint Health Research Ethics Board Study ID: REB17‐2289.
Author contributions
YC: Bioinformatics analysis, statistical analysis, establishment and testing of the analysis pipelines, data visualization and writing of the manuscript; LS: testing of the next‐generation‐sequencing and in vitro assay pipelines, library construction, RT–qPCRs, in vitro assays, data visualization and writing of the manuscript; BG: establishment and testing of the next‐generation‐sequencing pipelines, library construction; AAV: collection of tissues; MM: data interpretation; JTJ: pathology analysis, provision of material, study design, data interpretation; AZ: conception and design, establishment and testing of data generation and analysis pipelines, data interpretation, data visualization and writing of the manuscript, overall supervision.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Table EV1
Dataset EV1
Dataset EV2
Dataset EV3
Dataset EV4
Dataset EV5
Dataset EV6
Dataset EV7
Dataset EV8
Review Process File
Source Data for Figure 8
Acknowledgements
This work has been supported by an Explorations Grant # 201700011 to AZ and MM from Alberta Innovates and the Alberta Prion Research Institute, a Grant # 201900003 to AZ and MM from the Alzheimer Society of Alberta and Northwest Territories and the Alberta Prion Research Institute, a Discovery Grant # RGPIN‐2018‐05955 to AZ from NSERC, the BioNet Alberta grant to AZ from Genome Canada and a Compute Canada Resource Allocation Grant to AZ. AZ is supported by the Canada Research Chairs Program and the Canada Foundation for Innovation and is a former EMBO and DFG long‐term fellow. YC is supported by an Alberta Innovates (AITF) fellowship. We are grateful to Dr. Angeliki Pantazi for extensively reviewing, editing, and commenting on the manuscript. We are grateful to Dr. Trushar Patel and Dr. H.J. Wieden and their laboratories for providing us with the two control RNAs. The results published here are in part based on data obtained from the AMP‐AD Knowledge Portal (https://adknowledgeportal.synapse.org). Study data were provided by the Rush Alzheimer’s Disease Center, Rush University Medical Center, Chicago. Data collection was supported through funding by NIA grants P30AG10161 (ROS), R01AG15819 (ROSMAP; genomics and RNA‐seq), R01AG17917 (MAP), R01AG30146, R01AG36042 (5hC methylation, ATACseq), RC2AG036547 (H3K9Ac), R01AG36836 (RNA‐seq), R01AG48015 (monocyte RNA‐seq) RF1AG57473 (single nucleus RNA‐seq), U01AG32984 (genomic and whole‐exome sequencing), U01AG46152 (ROSMAP AMP‐AD, targeted proteomics), U01AG46161(TMT proteomics), U01AG61356 (whole‐genome sequencing, targeted proteomics, ROSMAP AMP‐AD), the Illinois Department of Public Health (ROSMAP), and the Translational Genomics Research Institute (genomic). Additional phenotypic data can be requested at www.radc.rush.edu.
EMBO reports (2021) 22: e52255.
Data availability
Short RNA‐seq raw fastq data from the CBB patients are available under controlled access through the European Genome‐Phenome Archive (EGA), https://ega‐archive.org/studies/EGAS00001004973, under the dataset accession number EGAD00001006886, and the study access number EGAS00001004973. MAP patient RNA‐seq data have been obtained from the AMP‐AD Knowledge Portal Synapse ID: syn3219045 (Bennett et al, 2012).
References
- Annese A, Manzari C, Lionetti C, Picardi E, Horner DS, Chiara M, Caratozzolo MF, Tullo A, Fosso B, Pesole G et al (2018) Whole transcriptome profiling of Late‐Onset Alzheimer’s Disease patients provides insights into the molecular changes involved in the disease. Sci Rep 8: 4282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett DA, Schneider JA, Arvanitakis Z, Wilson RS (2012) Overview and findings from the religious orders study. Curr Alzheimer Res 9: 628–645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertram L, Tanzi RE (2008) Thirty years of Alzheimer’s disease genetics: the implications of systematic meta‐analyses. Nat Rev Neurosci 9: 768–778 [DOI] [PubMed] [Google Scholar]
- Blaudin de The FX, Rekaik H, Peze‐Heidsieck E, Massiani‐Beaudoin O, Joshi RL, Fuchs J, Prochiantz A (2018) Engrailed homeoprotein blocks degeneration in adult dopaminergic neurons through LINE‐1 repression. EMBO J 37: e97374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloom GS (2014) Amyloid‐beta and tau: the trigger and bullet in Alzheimer disease pathogenesis. JAMA Neurol 71: 505–508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang JR, Ghafouri M, Mukerjee R, Bagashev A, Chabrashvili T, Sawaya BE (2012) Role of p53 in neurodegenerative diseases. Neurodegener Dis 9: 68–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Y, Saville L, Gollen B, Isaac C, Belay A, Mehla J, Patel K, Thakor N, Mohajerani MH, Zovoilis A (2020) Increased processing of SINE B2 ncRNAs unveils a novel type of transcriptome deregulation in amyloid beta neuropathology. Elife 9: e61265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornutiu G (2015) The epidemiological scale of Alzheimer’s disease. J Clin Med Res 7: 657–666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deininger P (2011) Alu elements: know the SINEs. Genome Biol 12: 236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espinoza CA, Goodrich JA, Kugel JF (2007) Characterization of the structure, function, and mechanism of B2 RNA, an ncRNA repressor of RNA polymerase II transcription. RNA 13: 583–596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funkhouser SA, Steibel JP, Bates RO, Raney NE, Schenk D, Ernst CW (2017) Evidence for transcriptome‐wide RNA editing among Sus scrofa PRE‐1 SINE elements. BMC Genom 18: 360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gates LA, Shi J, Rohira AD, Feng Q, Zhu B, Bedford MT, Sagum CA, Jung SY, Qin J, Tsai MJ et al (2017) Acetylation on histone H3 lysine 9 mediates a switch from transcription initiation to elongation. J Biol Chem 292: 14456–14472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genin E, Hannequin D, Wallon D, Sleegers K, Hiltunen M, Combarros O, Bullido MJ, Engelborghs S, De Deyn P, Berr C et al (2011) APOE and Alzheimer disease: a major gene with semi‐dominant inheritance. Mol Psychiatry 16: 903–907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez C, Armijo E, Bravo‐Alegria J, Becerra‐Calixto A, Mays CE, Soto C (2018) Modeling amyloid beta and tau pathology in human cerebral organoids. Mol Psychiatry 23: 2363–2374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green RC, Roberts JS, Cupples LA, Relkin NR, Whitehouse PJ, Brown T, Eckert SL, Butson M, Sadovnick AD, Quaid KA et al (2009) Disclosure of APOE genotype for risk of Alzheimer’s disease. N Engl J Med 361: 245–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadjiargyrou M, Delihas N (2013) The intertwining of transposable elements and non‐coding RNAs. Int J Mol Sci 14: 13307–13328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hane FT, Robinson M, Lee BY, Bai O, Leonenko Z, Albert MS (2017) Recent progress in Alzheimer’s disease research, part 3: diagnosis and treatment. J Alzheimers Dis 57: 645–665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez AJ, Zovoilis A, Cifuentes‐Rojas C, Han L, Bujisic B, Lee JT (2020) B2 and ALU retrotransposons are self‐cleaving ribozymes whose activity is enhanced by EZH2. Proc Natl Acad Sci USA 117: 415–425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodge RD, Bakken TE, Miller JA, Smith KA, Barkan ER, Graybuck LT, Close JL, Long B, Johansen N, Penn O et al (2019) Conserved cell types with divergent features in human versus mouse cortex. Nature 573: 61–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Cao Y, Li J, Liu Y, Zhong W, Li X, Chen C, Hao P (2018) A survey on cellular RNA editing activity in response to Candida albicans infections. BMC Genom 19: 43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ittner LM, Ke YD, Delerue F, Bi M, Gladbach A, van Eersel J, Wolfing H, Chieng BC, Christie MJ, Napier IA et al (2010) Dendritic function of tau mediates amyloid‐beta toxicity in Alzheimer’s disease mouse models. Cell 142: 387–397 [DOI] [PubMed] [Google Scholar]
- Jazvinscak Jembrek M, Slade N, Hof PR, Simic G (2018) The interactions of p53 with tau and Ass as potential therapeutic targets for Alzheimer’s disease. Prog Neurogibol 168: 104–127 [DOI] [PubMed] [Google Scholar]
- Kaaij LJT, Mohn F, van der Weide RH, de Wit E, Buhler M (2019) The ChAHP complex counteracts chromatin looping at CTCF sites that emerged from SINE expansions in mouse. Cell 178: 1437–1451 [DOI] [PubMed] [Google Scholar]
- Kanata E, Llorens F, Dafou D, Dimitriadis A, Thune K, Xanthopoulos K, Bekas N, Espinosa JC, Schmitz M, Marin‐Moreno A et al (2019) RNA editing alterations define manifestation of prion diseases. Proc Natl Acad Sci USA 116: 19727–19735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneko H, Dridi S, Tarallo V, Gelfand BD, Fowler BJ, Cho WG, Kleinman ME, Ponicsan SL, Hauswirth WW, Chiodo VA et al (2011) DICER1 deficit induces Alu RNA toxicity in age‐related macular degeneration. Nature 471: 325–330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karijolich J, Zhao Y, Alla R, Glaunsinger B (2017) Genome‐wide mapping of infection‐induced SINE RNAs reveals a role in selective mRNA export. Nucleic Acids Res 45: 6194–6208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khermesh K, D’Erchia AM, Barak M, Annese A, Wachtel C, Levanon EY, Picardi E, Eisenberg E (2016) Reduced levels of protein recoding by A‐to‐I RNA editing in Alzheimer’s disease. RNA 22: 290–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Paggi JM, Park C, Bennett C, Salzberg SL (2019) Graph‐based genome alignment and genotyping with HISAT2 and HISAT‐genotype. Nat Biotechnol 37: 907–915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korczyn AD (2012) Why have we failed to cure Alzheimer’s disease? J Alzheimers Dis 29: 275–282 [DOI] [PubMed] [Google Scholar]
- Krestel H, Meier JC (2018) RNA editing and retrotransposons in neurology. Front Mol Neurosci 11: 163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen PA, Lutz MW, Hunnicutt KE, Mihovilovic M, Saunders AM, Yoder AD, Roses AD (2017) The Alu neurodegeneration hypothesis: a primate‐specific mechanism for neuronal transcription noise, mitochondrial dysfunction, and manifestation of neurodegenerative disease. Alzheimers Dement 13: 828–838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen PA, Hunnicutt KE, Larsen RJ, Yoder AD, Saunders AM (2018) Warning SINEs: Alu elements, evolution of the human brain, and the spectrum of neurological disease. Chromosome Res 26: 93–111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R (2009) Fast and accurate short read alignment with Burrows‐Wheeler transform. Bioinformatics 25: 1754–1760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Genome Project Data Processing S (2009) The Sequence Alignment/Map format and SAMtools. Bioinformatics 25: 2078–2079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S (2014) Moderated estimation of fold change and dispersion for RNA‐seq data with DESeq2. Genome Biol 15: 550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariner PD, Walters RD, Espinoza CA, Drullinger LF, Wagner SD, Kugel JF, Goodrich JA (2008) Human Alu RNA is a modular transacting repressor of mRNA transcription during heat shock. Mol Cell 29: 499–509 [DOI] [PubMed] [Google Scholar]
- Mehla J, Lacoursiere SG, Lapointe V, McNaughton BL, Sutherland RJ, McDonald RJ, Mohajerani MH (2019) Age‐dependent behavioral and biochemical characterization of single APP knock‐in mouse (APP(NL‐G‐F/NL‐G‐F)) model of Alzheimer’s disease. Neurobiol Aging 75: 25–37 [DOI] [PubMed] [Google Scholar]
- Montgomery W, Ueda K, Jorgensen M, Stathis S, Cheng Y, Nakamura T (2018) Epidemiology, associated burden, and current clinical practice for the diagnosis and management of Alzheimer’s disease in Japan. Clinicoecon Outcomes Res 10: 13–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mostafavi S, Gaiteri C, Sullivan SE, White CC, Tasaki S, Xu J, Taga M, Klein HU, Patrick E, Komashko V et al (2018) A molecular network of the aging human brain provides insights into the pathology and cognitive decline of Alzheimer’s disease. Nat Neurosci 21: 811–819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mu X, Ahmad S, Hur S (2016) Endogenous retroelements and the host innate immune sensors. Adv Immunol 132: 47–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakahama T, Kawahara Y (2020) Adenosine‐to‐inosine RNA editing in the immune system: friend or foe? Cell Mol Life Sci 77: 2931–2948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natt D, Thorsell A (2016) Stress‐induced transposon reactivation: a mediator or an estimator of allostatic load? Environ Epigenet 2: dvw015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platt RN II, Vandewege MW, Ray DA (2018) Mammalian transposable elements and their impacts on genome evolution. Chromosome Res 26: 25–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponicsan SL, Kugel JF, Goodrich JA (2010) Genomic gems: SINE RNAs regulate mRNA production. Curr Opin Genet Dev 20: 149–155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponicsan SL, Kugel JF, Goodrich JA (2015) Repression of RNA polymerase II transcription by B2 RNA depends on a specific pattern of structural regions in the RNA. Noncoding RNA 1: 4–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, Hall IM (2010) BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26: 841–842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh M (2012) Dysregulated A to I RNA editing and non‐coding RNAs in neurodegeneration. Front Genet 3: 326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun W, Samimi H, Gamez M, Zare H, Frost B (2018) Pathogenic tau‐induced piRNA depletion promotes neuronal death through transposable element dysregulation in neurodegenerative tauopathies. Nat Neurosci 21: 1038–1048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasic B, Yao Z, Graybuck LT, Smith KA, Nguyen TN, Bertagnolli D, Goldy J, Garren E, Economo MN, Viswanathan S et al (2018) Shared and distinct transcriptomic cell types across neocortical areas. Nature 563: 72–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatosyan KA, Kramerov DA (2016) Heat shock increases lifetime of a small RNA and induces its accumulation in cells. Gene 587: 33–41 [DOI] [PubMed] [Google Scholar]
- Toledo JB, Shaw LM, Trojanowski JQ (2013) Plasma amyloid beta measurements – a desired but elusive Alzheimer’s disease biomarker. Alzheimers Res Ther 5: 8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walters RD, Kugel JF, Goodrich JA (2009) InvAluable junk: the cellular impact and function of Alu and B2 RNAs. IUBMB Life 61: 831–837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Park J, Susztak K, Zhang NR, Li M (2019) Bulk tissue cell type deconvolution with multi‐subject single‐cell expression reference. Nat Commun 10: 380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yakovchuk P, Goodrich JA, Kugel JF (2009) B2 RNA and Alu RNA repress transcription by disrupting contacts between RNA polymerase II and promoter DNA within assembled complexes. Proc Natl Acad Sci USA 106: 5569–5574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang XO, Gingeras TR, Weng Z (2019) Genome‐wide analysis of polymerase III‐transcribed Alu elements suggests cell‐type‐specific enhancer function. Genome Res 29: 1402–1414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zovoilis A, Cifuentes‐Rojas C, Chu HP, Hernandez AJ, Lee JT (2016) Destabilization of B2 RNA by EZH2 activates the stress response. Cell 167: 1788–1802 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Table EV1
Dataset EV1
Dataset EV2
Dataset EV3
Dataset EV4
Dataset EV5
Dataset EV6
Dataset EV7
Dataset EV8
Review Process File
Source Data for Figure 8
Data Availability Statement
Short RNA‐seq raw fastq data from the CBB patients are available under controlled access through the European Genome‐Phenome Archive (EGA), https://ega‐archive.org/studies/EGAS00001004973, under the dataset accession number EGAD00001006886, and the study access number EGAS00001004973. MAP patient RNA‐seq data have been obtained from the AMP‐AD Knowledge Portal Synapse ID: syn3219045 (Bennett et al, 2012).