

The emergence of azole-resistant fungal pathogens has posed a great threat to public health worldwide. Although the molecular mechanism of azole resistance has been extensively investigated, the potential regulators of azole resistance remain largely unexplored.
KEYWORDS: Aspergillus fumigatus, SltA, azole resistance, ergosterol, drug efflux pump, Erg11A, Mdr1
ABSTRACT
The emergence of azole-resistant fungal pathogens has posed a great threat to public health worldwide. Although the molecular mechanism of azole resistance has been extensively investigated, the potential regulators of azole resistance remain largely unexplored. In this study, we identified a new function of the fungal specific C2H2 zinc finger transcription factor SltA (involved in the salt tolerance pathway) in the regulation of azole resistance of the human fungal pathogen Aspergillus fumigatus. A lack of SltA results in an itraconazole hypersusceptibility phenotype. Transcriptional profiling combined with LacZ reporter analysis and electrophoretic mobility shift assays (EMSA) demonstrated that SltA is involved in its own transcriptional regulation and also regulates the expression of genes related to ergosterol biosynthesis (erg11A, erg13A, and erg24A) and drug efflux pumps (mdr1, mfsC, and abcE) by directly binding to the conserved 5′-AGGCA-3′ motif in their promoter regions, and this binding is dependent on the conserved cysteine and histidine within the C2H2 DNA binding domain of SltA. Moreover, overexpression of erg11A or mdr1 rescues sltA deletion defects under itraconazole conditions, suggesting that erg11A and mdr1 are related to sltA-mediated itraconazole resistance. Most importantly, deletion of SltA in laboratory-derived and clinical azole-resistant isolates significantly attenuates drug resistance. Collectively, we have identified a new function of the transcription factor SltA in regulating azole resistance by coordinately mediating the key azole target Erg11A and the drug efflux pump Mdr1, and targeting SltA may provide a potential strategy for intervention of clinical azole-resistant isolates to improve the efficiency of currently approved antifungal drugs.
INTRODUCTION
Aspergillus fumigatus is a widespread opportunistic pathogen that causes invasive aspergillosis (IA) with high mortality in immunocompromised patients (1–4). Triazole drugs, such as fluconazole, itraconazole (ITZ), voriconazole (VRC), and posaconazole, are the primary therapeutic agents for the first-line treatment of IA (5–7). Notably, with the continuous application of azole antifungal drugs in clinical treatment, azole resistance of clinical A. fumigatus isolates has rapidly emerged over the past 2 decades (8–11). Several reports worldwide have demonstrated that patients infected with azole-resistant A. fumigatus isolates have a higher risk for therapy failure (12). Therefore, a better understanding of the molecular mechanisms underlying azole resistance will be useful for exploiting novel antifungal targets and facilitating the development of new antifungal drugs.
The mode of action of azole drugs involves in the inhibition of the lanosterol 14-α demethylation enzyme Erg11 (also referred to as Cyp51), resulting in depleted ergosterol biosynthesis and accumulation of toxic intermediates in cells, ultimately causing a change in membrane permeability, exudation of cellular contents, and cell death (13, 14). Mutations in Erg11 or its promoter are the most common mechanism of azole resistance in A. fumigatus (12). For example, mutations in the hot spots G54, G138, M220, and G448, which are positioned near the ligand access channels or close to the active site, block azole drug binding (15, 16). Tandem repeats in the promoter region (TR34/L98H and TR46/Y121F/T289A) cause increased transcription of Erg11 (17, 18). In addition to modifications of the Erg11 enzyme, the activation of the drug efflux pumps, such as the ABC transporter Cdr1B (19) and the major facilitator transporter Mdr1, have been reported to be responsible for azole resistance that resulted in reduced accumulation of the drug within fungal cells (20, 21). Several lines of evidence have shown that transcription factors play a critical role in the regulation of azole resistance in fungi. For instance, the basic helix-loop-helix (bHLH) transcriptional factor SrbA binds at the 34/46-mer duplicated in the promoter of Erg11A in azole-resistant isolates (18, 22–24). The CCAAT binding complex (CBC; a heterotrimer including HapB, HapC, and HapE) with the interaction partner HapX, a transcription factor involved in adaptation to iron deprivation, synergistically act as a negative regulator of sterol biosynthesis. CBC complex and SrbA bind competitively to the CGAAT motif within the azole resistance-associated region in the promoter of erg11A. Hence, deletion of CBC complex could enhance binding of SrbA to the promoter of erg11A, resulting in the upregulation of erg11A and drug resistance (24, 25). Furthermore, AtrR, a Zn2-Cys6 transcription factor, is also involved in azole resistance through its coregulation of the expression of erg11A and the drug exporter Cdr1B (20, 26). Recently, researchers have identified that the negative cofactor 2 (NCT) complex (NctA and NctB) acts as a key regulator of azole resistance by repressing srbA, atrR, erg11A, and cdr1B and activating hapC. Loss of NctA or NctB leads to drug resistance caused by increasing the ergosterol contents and decreasing the intracellular drug retention (27).
Recent studies on the mechanisms of clinical drug-resistant isolates revealed a highly complex cross-regulatory drug resistance transcriptional network that involved multiple regulators and multiple target genes (27). Thus, we speculate that apart from the identified regulators, other potential transcription factors may be involved in the regulation of drug resistance. For this purpose, we performed transcriptome sequencing (RNA-seq) on the A. fumigatus wild-type (WT) strain with or without itraconazole treatment and found that 16 transcription factors were significantly induced under itraconazole conditions. Among them, only deletion of AFUB_041100 showed hypersensitivity to itraconazole. Previous studies have reported that the homologue of AFUB_041100 in Aspergillus nidulans is SltA (involved in the salt tolerance pathway), a C2H2 zinc finger transcription factor that is involved in cation homeostasis by positively regulating the expression of enaA, a gene encoding a sodium pump ATPase (28, 29). In addition, SltA homologues such as Ace1 in an industrial strain of Hypocrea jecorina and SltA in the plant-pathogenic filamentous fungus Colletotrichum gloeosporioides (CgSltA) have also been characterized. Ace1 is a negative regulator of cellulase and xylanase, while CgSltA is a positive regulator of pmk1 and cat1, genes encoding a mitogen-activated protein (MAP) kinase and a carnitine acetyltransferase, respectively, which are required for appressorium formation (30, 31). In this study, we found that a previously identified transcription factor, SltA, has an unexplored function in azole resistance via directly regulating the expression of the drug target Erg11A and the drug pump Mdr1 in the human fungal pathogen A. fumigatus. A lack of SltA resulted in multidrug sensitivity, including to azoles, terbinafine, and simvastatin. Notably, deletion of SltA in the laboratory-derived and clinical azole-resistant isolates significantly attenuated drug resistance. Our findings suggest that fungus-specific SltA may be a promising drug target and may even possibly reverse resistance of clinical azole-resistant isolates, thereby improving the efficiency of currently approved antifungal drugs.
RESULTS
Screening and identification of sltA in A. fumigatus.
To identify previously unidentified transcription factors involved in azole resistance in A. fumigatus, we carried out RNA sequencing to compare the whole-genome transcriptomes of the wild-type strain grown in the presence and absence of 2 h of itraconazole treatment. We found that 16 candidate transcription factors were induced under itraconazole conditions (P < 0.05, log2 fold change [FC] ≥ 1 [Fig. 1A]). Reverse transcription-quantitative PCR (qRT-PCR) analyses of samples cultured under the same conditions were performed to validate the accuracy of the transcriptomes. As shown in Fig. 1A, the expression levels of the 16 genes were relatively high and correlated with the RNA-seq data (Pearson correlation was 0.689; P = 0.003). To further verify whether these genes were involved in the response to itraconazole treatment, we screened the phenotypes of all the deletion mutants on yeast extract, agar and glucose (YAG) medium supplemented with itraconazole; 12 of these deletion mutants were obtained from the A. fumigatus transcription factor deletion library (27) and the rest, which were absent from the library and included AFUB_063290, AFUB_051190, AFUB_003250 and AFUB_086150, were generated by homologous recombination and further confirmed by diagnostic PCR analyses (see Fig. S1C to G in the supplemental material). As shown in Fig. 1B, only the AFUB_041100 null mutant showed hypersensitivity to the antifungal itraconazole compared to the parental wild-type strain. When the concentration of itraconazole was 0.1 μg ml−1, the growth of the AFUB_041100 null mutant was totally inhibited (Fig. 1B), suggesting that this transcription factor, AFUB_041100, is required for A. fumigatus to respond to itraconazole. Bioinformatic analysis showed that the AFUB_041100 homologue in A. nidulans is sltA (AnsltA, AN2919; E value, 1.0e−130; identity ,41%), which encodes a C2H2 zinc finger transcription factor involved in cation homeostasis (32, 33). Here, we also refer to AFUB_041100 in A. fumigatus as sltA (AfsltA). Taken together, these data suggest that SltA is important for A. fumigatus in the response to azole treatment.
FIG 1.

Screening and identification of novel transcription factors that govern azole resistance in A. fumigatus. (A) Heat map for the expression levels, determined by RNA-seq and qRT-PCR, of 16 selected transcription factors induced under 2 h of itraconazole (ITZ) treatment in the parental wild-type strain. These genes are listed in descending order according to log2 fold change of mRNA level. The gene annotations were downloaded from AspGD (http://www.aspergillusgenome.org/) and Ensembl Fungi (http://fungi.ensembl.org/index.html). (B) Colony morphology of the indicated strains grown on YAG medium in the presence or absence of 0.05 or 0.1 μg ml−1 of itraconazole at 37°C for 36 h.
The C2H2-type transcription factor SltA confers resistance to itraconazole, and this function is highly conserved in Aspergillus species.
Phylogenetic analysis and homology searches of fungal genome databases revealed that SltA homologues exist only in filamentous fungi belonging to the Pezizomycotina subphylum. In contrast, there was no identifiable homologue in the Saccharomycotina, Taphrinomycotina, or Basidiomycota (Fig. 2A), implying that SltA is a filamentous fungus-specific protein. Moreover, all SltA homologues harbor the three C2H2 domain repeats (Fig. 2A), suggesting that SltA is a C2H2-type zinc finger transcription factor. To further verify the function of SltA in drug response, we constructed an sltA knockout strain by replacing the coding sequence with the Neurospora crassa pyr4 marker in the parental A1160 strain. A correct ΔsltA mutant was confirmed by diagnostic PCR (Fig. S1B). As shown in Fig. 2B, the ΔsltA mutant exhibited enhanced susceptibility to itraconazole compared to the parental wild-type strain when grown under itraconazole conditions. To confirm that the defective phenotype was specifically caused by the sltA deletion, we constructed sltA complemented strains by reintroducing the native A. fumigatus sltA or sltA orthologues from A. nidulans or Aspergillus flavus. Both native and orthologous sltA could rescue the defects of the ΔsltA mutant under itraconazole conditions (Fig. 2B), suggesting their highly conserved functions from SltA homologues among Aspergillus species in the response to itraconazole. Moreover, to further verify the relationship between azole susceptibility and the expression level of sltA, a strain overexpressing sltA (mRNA level increased approximately 4-fold [Fig. S2E]) was constructed and showed increased resistance to itraconazole compared to that of the parental wild-type strain (Fig. 2C and Table 1), implying that the expression of sltA contributes to the A. fumigatus response to itraconazole. We next questioned whether the expression of sltA would be affected by itraconazole. To test this, qRT-PCR was carried out with the A. fumigatus wild-type strain (A1160C′) (34) under itraconazole treatment. The results showed that after 2 h of itraconazole treatment, the sltA mRNA level increased approximately 5-fold compared to that of the untreated control (Fig. S2A), demonstrating that itraconazole treatment can increase the expression of SltA. Taken together, these data suggest that the conserved function of SltA is required for regulating resistance to itraconazole.
FIG 2.

SltA confers resistance to itraconazole, and this function is highly conserved in Aspergillus species. (A) Bioinformatic analysis for the homologues of C2H2-type transcription factor SltA in the Pezizomycotina subphylum. The phylogenetic tree was constructed with bootstrap values shown via MEGA 6 using the neighbor-joining method. The conserved C2H2 domains are highlighted in red. (B) Colony morphology of the indicated strains grown on YAG medium in the presence or absence of 0.025 or 0.05 μg ml−1 of itraconazole at 37°C for 48 h. (C) Equal series numbers of conidia (2 × 104, 2 × 103, and 2 × 102) from the indicated strains grown on YAG medium in the presence or absence of 0.05, 0.1, or 0.2 μg ml−1 of itraconazole at 37°C for 48 h.
TABLE 1.
MICs of itraconazole, voriconazole, bifonazole, fluconazole, terbinafine, and amphotericin B for sltA mutantsa
| Strain | MIC (μg ml−1) |
|||||
|---|---|---|---|---|---|---|
| ITZ | VRC | Bifo | Flu | Terb | AMB | |
| WT | 1 | 0.5 | 4 | 200 | 8 | 8 |
| ΔsltA | 0.5 | 0.25 | 2 | 100 | 4 | 8 |
| sltAC | 1 | 0.5 | 4 | 200 | 8 | 8 |
| C502S | 0.5 | 0.25 | 2 | 100 | 4 | 8 |
| H518A | 0.5 | 0.25 | 2 | 100 | 4 | 8 |
| SltAΔC | 0.5 | 0.25 | 2 | 100 | 4 | 8 |
| OE::sltA | 2 | 1 | 6 | 250 | 16 | 8 |
| R243Qb | 4 | 2 | 8 | NT | >16 | 8 |
| R243Q ΔsltA | 2 | 0.5 | 4 | NT | 8 | 8 |
| Shjt40c | 4 | 2 | 16 | NT | 16 | 8 |
| Shjt40 ΔsltA | 2 | 1 | 8 | NT | 8 | 8 |
| Shjt42bc | >8 | 2 | >16 | NT | >16 | 8 |
| Shjt42b ΔsltA | 4 | 1 | 8 | NT | 16 | 8 |
SltA is involved in antifungal drug resistance to azoles, terbinafine, and simvastatin but not amphotericin B or caspofungin.
To verify whether the sltA mutant could respond to other antifungal drugs, we performed drug susceptibility assays using five different classes of antifungal drugs, including azoles (itraconazole, voriconazole, fluconazole, and bifonazole), allylamines (terbinafine), statins (simvastatin), polyenes (amphotericin B), and echinocandins (caspofungin). Equal series numbers of conidia (2 × 104, 2 × 103, and 2 × 102) from the parental wild-type, ΔsltA, and sltA complemented strains were spotted onto YAG medium under the treatment of different antifungal drugs. As shown in Fig. 3A, the growth of the sltA mutant was inhibited by treatment with antifungal drugs, including azoles (itraconazole, voriconazole, fluconazole, and bifonazole), allylamines (terbinafine), and statins (simvastatin), which specifically inhibit the lanosterol 14-α demethylation enzyme Erg11, the squalene epoxidase Erg1, and the 3-hydroxy-3-methyl glutaryl coenzyme A (HMG-CoA) reductase Hmg1/2, respectively (14). In contrast, the ΔsltA mutant showed no detectable phenotype in response to amphotericin B or caspofungin (Fig. 3A). These results suggest that deletion of sltA results in multidrug sensitivity to drugs that impact the ergosterol biosynthetic pathway (azoles, terbinafine, and simvastatin) but not agents acting by other mechanisms (amphotericin B and caspofungin). Moreover, the MICs obtained from Etest strips and the Clinical and Laboratory Standards Institute (CLSI) method (35) showed that the ΔsltA mutant exhibited significantly reduced MIC values to itraconazole, voriconazole, and fluconazole compared to those of the parental wild-type and the sltA complemented strains (Fig. 3B and Table 1). These results imply that SltA may play an important role in regulating the ergosterol biosynthetic pathway.
FIG 3.
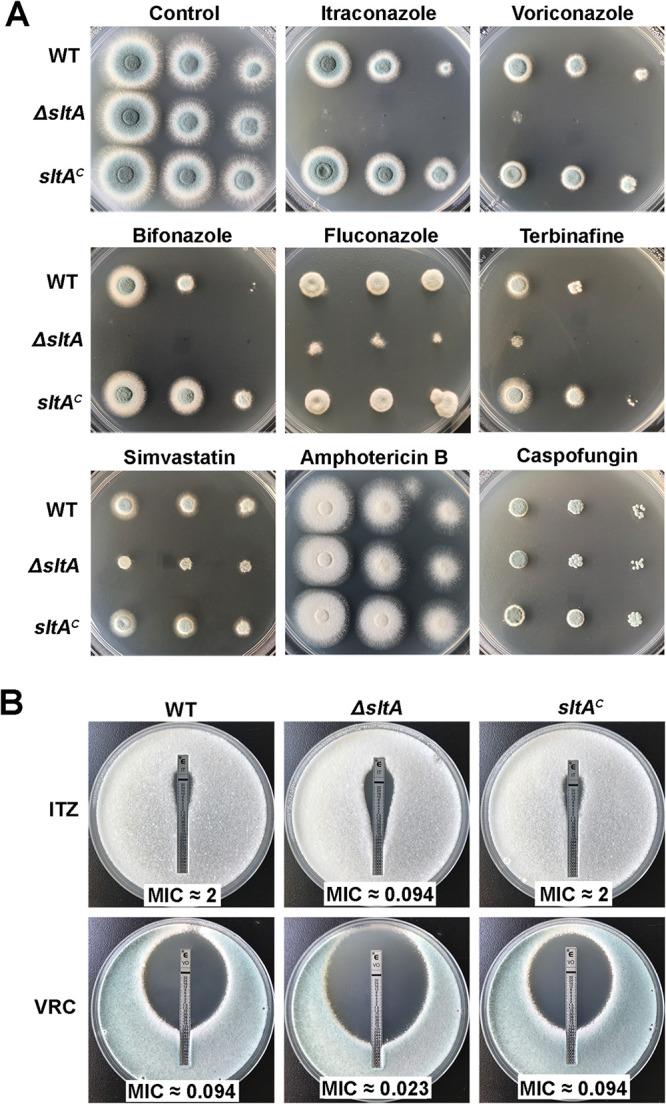
Deletion of sltA causes multidrug sensitivity to azoles, terbinafine, and simvastatin but not amphotericin B or caspofungin. (A) Equal series numbers of conidia (2 × 104, 2 × 103, and 2 × 102) from indicated strains inoculated on YAG medium without drug at 37°C for 1.5 days or with drug at 37°C for 2 days. The following antifungal drugs were used: 0.05 μg ml−1 of itraconazole, 0.1 μg ml−1 of voriconazole, 0.6 μg ml−1 of bifonazole, 200 μg ml−1 of fluconazole, 24 μg ml−1 of simvastatin, 0.2 μg ml−1 of terbinafine, 8 μg ml−1 of amphotericin B, and 0.6 μg ml−1 of caspofungin. (B) MIC Etest strips impregnated with a gradient of itraconazole (ITZ) or voriconazole (VRC) were placed onto YAG agar plates containing a lawn of conidia cultured for 36 h (the upper) or 48 h (the bottom) at 37°C before observation.
Conserved cysteine and histidine within the C2H2 DNA binding domain and the C terminus of SltA are required for itraconazole resistance.
To determine whether the conserved cysteine and histidine within the DNA binding domain of SltA are required for sltA-mediated itraconazole resistance, we mutated the Cys residue at position 502 and the His residue at position 518 to a Ser and an Ala, respectively (Fig. S2B). qRT-PCR and Western blot analyses showed that mutated genes were expressed normally at transcript and protein levels (Fig. 4B and Fig. S2C), indicating that these mutants were successfully constructed. As shown in Fig. 4C, the sltAC502S and sltAH518A strains displayed itraconazole hypersusceptibility phenotypes similar to that of the full-length sltA deletion strain, suggesting that the conserved cysteine and histidine within the C2H2 DNA binding domain are indispensable for sltA-mediated itraconazole resistance. Next, to examine whether regions of SltA apart from the DNA binding domain were required for its function, a series of truncations were performed. As shown in Fig. 4A, we generated a mutant with a truncation of the N-terminal 485 residues (sltAΔN) and a mutant with a truncation of the C-terminal 292 residues (sltAΔC) and further analyzed their phenotypes. Surprisingly, the sltAΔN truncated gene could not be transcribed under the control of the native endogenous promoter (Fig. 4B), suggesting that the N-terminal region is required for the transcription of the sltA gene. To overcome this problem, we constructed a conditional tet-sltAΔN strain driven by the tet-on promoter, which could be induced in the presence of doxycycline (36). The hyphal growth and drug susceptibility of the parental wild-type strain and the ΔsltA mutant were not affected by the presence of doxycycline (Fig. 4C). With doxycycline induction, the tet-sltAΔN strain had mRNA expression levels comparable to that of the parental wild-type strain. In contrast, the tet-sltAΔN strain had no detectable sltA mRNA expression when doxycycline was absent (Fig. 4B), indicating that the Tet-SltA on/off system was functional in A. fumigatus. As shown in Fig. 4C, with doxycycline induction, the tet-sltAΔN strain exhibited a morphology similar to that of the parental wild-type strain in the presence of itraconazole. In comparison, the sltAΔC strain expressed the truncated version of SltA with a predicted molecular mass and had an itraconazole hypersusceptibility phenotype similar to that of the full-length sltA deletion (Fig. 4C and Fig. S2C). These results indicated that the C terminus, but not the N terminus, is required for sltA-mediated itraconazole resistance. The MICs of the sltA-related mutants for itraconazole, voriconazole, bifonazole, fluconazole, terbinafine, and amphotericin B were in agreement with the colony growth test under drug conditions (Table 1). Collectively, these results suggest that the C2H2 DNA binding domain and the C terminus of SltA are essential for its functions in itraconazole resistance.
FIG 4.

C2H2 DNA binding domain and C-terminal region of SltA are required for itraconazole resistance. (A) Schematic view of SltA protein domains (C2H2 DNA binding domain shown in blue) showing introduced site-directed mutations and N- and C-terminal truncations. (B) qRT-PCR analysis was performed after growth of cultures in MM for 18 h at 37°C. The tubA gene was used as an internal control. **, P < 0.01; ****, P < 0.0001; ns, P > 0.05 compared to the parental wild-type strain according to unpaired t test with Welch’s correction. tet-sltAΔN was induced (“on” state) in the presence of 5 mg liter−1 of doxycycline. (C) Colony morphology of the indicated strains grown on YAG medium in the presence or absence of 0.025 or 0.05 μg ml−1 of itraconazole at 37°C for 36 h. +Dox, strains were grown on medium with 5 mg liter−1 of doxycycline.
Increased expression of erg11A and mdr1 is responsible for sltA-mediated drug resistance.
To further dissect the mechanism underlying sltA-mediated itraconazole resistance, we performed RNA-seq analyses comparing the A. fumigatus wild type and the ΔsltA mutant cultured in liquid minimal medium (MM) at 37°C for 18 h, followed by 2 h with or without 0.5 μg ml−1 of itraconazole. Gene Ontology (GO) classification analysis showed that the differentially expressed genes were involved in drug transport, transmembrane transport, ion transport, and metabolic processes, including organic acid, carbohydrate, lipid, and cellular nitrogen metabolism (P < 0.05; |log2 FC| ≥ 1 [Fig. S3A]). Interestingly, the heat map based on the RNA-seq data combined with the KEGG analyses showed that SltA globally influenced the expression of genes related to the ergosterol biosynthetic pathway and drug transporters, which were previously reported to be involved in drug resistance (14) (Fig. 5A and Fig. S3B). Ergosterol biosynthetic genes, including erg11A, erg13A, and erg24A, and drug transporter genes, including abcE and especially mfsC and mdr1, were all downregulated in the ΔsltA mutant compared to the parental wild-type strain regardless of whether they were treated with itraconazole (Fig. 5A and B and Fig. S2D). qRT-PCR analyses of the expression of these six genes were highly correlated with the RNA-seq data (Pearson correlation from 0.685 to 0.917; P < 0.05 [Fig. 5B]). Collectively, these data suggest that the expression of these genes related to the ergosterol biosynthetic pathway (erg11A, erg13A, and erg24A) and drug transporters (mdr1, abcE, and mfsC) is dependent on SltA under both itraconazole and nonitraconazole conditions.
FIG 5.

Increased expression of erg11A and mdr1 contributes to sltA-mediated drug resistance. (A) Selected genes which were downregulated in the sltA mutant versus the parental wild-type strain without itraconazole (ΔsltA/WT, 0) or with 2 h of itraconazole treatment after 18 h of growth in liquid MM (ΔsltA/WT, +ITZ) are indicated. The majority of these genes were upregulated in the parental wild-type strain after itraconazole treatment (P value < 0.05; |log2 FC| > 1). These genes are listed in descending order from the fold change of each gene between the parental wild-type strain and the sltA mutant (ΔsltA/WT, 0). Genes which probably contributed to sltA-mediated itraconazole resistance are highlighted in red. The heat map was constructed in MeV (http://mev.tm4.org/) based on the RNA-seq data. Log2 fold change was scaled between −10 and 7. (B) Heat map for the expression levels of six selected genes involved in ergosterol biosynthesis and drug transport from panel A by qRT-PCR, which was performed after growth of cultures in liquid MM for 18 h at 37°C and subsequently for 2 h with or without 0.5 μg ml−1 of itraconazole. The tubA gene was used as an internal control. Samples were assessed in biological triplicates. Log2 fold change was scaled between −3.3 and 4.1. (C) β-Galactosidase activity driven by the indicated gene promoters in the parental wild-type and ΔsltA mutant strains. β-Galactosidase activities were measured from cultures of the related strains for 20 h at 37°C in liquid MM with a subsequent 3 h of growth under the same conditions or a subsequent 3-h shift into MM with 0.5 μg ml−1 of itraconazole. (D) Equal series numbers of conidia (2 × 104, 2 × 103, and 2 × 102) from indicated strains inoculated on YAG medium with or without 0.025 μg ml−1 of itraconazole at 37°C for 48 h.
Since the sltAC502S and sltAH518A strains exhibited azole-sensitive phenotypes similar to that of the sltA deletion mutant, we wondered whether the sltAC502S and sltAH518A point mutations affected the transcript expression of genes related to the ergosterol biosynthetic pathway and drug transporters. To verify this, we performed qRT-PCR to compare the mRNA levels of erg11A, erg13A, erg24A, mdr1, mfsC, and abcE between the wild-type and site-directed mutant strains. As predicted, all six genes were downregulated in the sltAC502S and sltAH518A strains (Fig. S2D), suggesting that the conserved cysteine and histidine within the SltA DNA binding domain are required for the transcriptional regulation process.
To further confirm whether the transcript levels of drug transporter genes (mdr1, mfsC, and abcE) and ergosterol biosynthetic genes (erg11A, erg13A, and erg24A) were truly dependent on SltA in vivo, we fused the promoters of these genes with a bacterial lacZ reporter gene and transformed them into the parental wild-type and ΔsltA mutant strains. Phenotypic analysis showed that the growth and drug susceptibility of the parental wild-type and ΔsltA mutant strains were not affected by transformation (Fig. S4A), suggesting that the lacZ reporter system was unable to affect colony growth phenotypes. In Fig. 5C, the x axis represents the incubation time for a β-galactosidase activity assay in vitro. Prolonged incubation time is needed for detecting the β-galactosidase activity of a strain with a low-expression gene like abcE. Therefore, instead of a uniform x/y axis scale, the corresponding x/y axis scales were set depending on the expression abundances of genes. Consequently, the β-galactosidase activity driven by erg11A(p)::lacZ increased significantly in the wild-type background strain upon itraconazole treatment. In contrast, the ΔsltAerg11A(p)::lacZ strain exhibited lower β-galactosidase activity than that of the wild-type background strain without itraconazole treatment, and there was no significant difference in the β-galactosidase activity in response to itraconazole. Similar results were also obtained for the mdr1(p)::lacZ and mfsC(p)::lacZ strains, suggesting that SltA was required for normal expression of these three genes. In comparison, the β-galactosidase activities driven by abcE(p)::lacZ and erg24A(p)::lacZ were induced in the presence of itraconazole, irrespective of the presence of sltA; however, the change in β-galactosidase activity was less pronounced in the sltA null background, suggesting that the expression of abcE and erg24A but not the response to ITZ was dependent on SltA. This may be due to a compensatory pathway associated with response to ITZ in the absence of SltA. In contrast, the β-galactosidase activity of the erg13A(p)::lacZ strain did not change in any conditions. These data indicate that the itraconazole-inducible genes mdr1, mfsC, abcE, erg11A, and erg24A are transcriptionally regulated in an SltA-dependent manner. Moreover, a previous study suggested that SltA binds to its own promoter region to maintain its own transcription (28). To confirm this hypothesis, we measured the β-galactosidase activities driven by sltA(p)::lacZ in the parental wild-type and ΔsltA strains. As shown in Fig. 5C, the activity driven by sltA(p)::lacZ in the ΔsltA mutant was significantly lower than that in the parental wild-type strain, suggesting that SltA is truly involved in its own transcriptional regulation. In addition, the β-galactosidase activity driven by sltA(p)::lacZ was induced in the presence of itraconazole, irrespective of the presence of sltA (Fig. 5C). These results suggest that there may be a compensatory pathway in response to ITZ in the absence of SltA.
To examine whether overexpression of the aforementioned downregulated genes could suppress the defects of the ΔsltA mutant under azole treatment conditions, we generated mutants harboring the corresponding overexpressed genes in the ΔsltA mutant, which were verified by qRT-PCR analysis (Fig. S2E). As shown in Fig. 5D, overexpression of mfsC, abcE, and erg24A failed to complement itraconazole resistance in the ΔsltA mutant, implying that these genes may be not involved in sltA-mediated itraconazole resistance. In comparison, overexpression of the azole target gene erg11A or the drug pump gene mdr1, both of which were itraconazole-inducible genes and significantly downregulated in ΔsltA (Fig. 5B and C and Fig. S2D), almost completely restored ΔsltA to WT-like growth under itraconazole conditions (Fig. 5D), suggesting that the sltA-mediated itraconazole resistance is mainly dependent on the expression of erg11A or mdr1. Collectively, these data suggest that erg11A and mdr1 might be related to sltA-mediated itraconazole resistance.
SltA binds to the AGGCA motif in the promoters of ergosterol biosynthesis and drug pump genes and affects related gene expression and function.
Previous studies have shown that the SltA orthologue in A. nidulans regulates the expression of downstream target proteins such as SltB (AN6132), a chymotrypsin-like serine protease, through binding to the 5′-AGGCA-3′ sequence within its promoter region (28, 30, 33, 37). To identify the mechanism of SltA in the regulation of ergosterol biosynthesis and drug pump gene expression in A. fumigatus, we searched the promoters of mdr1, mfsC, abcE, erg11A, erg13A, and erg24A using multiple expectation maximizations (EM) for motif elicitation (MEME) analysis and found that these genes shared the consensus SltA-binding sites (AGGCA) in their promoters (Fig. 6A). In addition, MEME analysis showed that the AGGCA motif is conserved in the promoters of erg11A and mdr1 homologues from Aspergillus species (Table S1). To further investigate whether SltA can directly bind to the predicted AGGCA motif, we expressed and purified the DNA binding domain of SltA, of which the molecular weight is approximately 23 kDa (Fig. S4B), and performed electrophoretic mobility shift assays (EMSA). Interestingly, SltA is able to interact with the promoters of sltA, sltB, erg11A, erg13A, erg24A, mdr1, abcE, and mfsC (Fig. 6B). Adding unlabeled DNA probe (cold probe) could block SltA binding to the Cy5-labeled promoter fragments of these genes (Fig. 6B), highlighting the specificity of the protein-DNA interaction. To further confirm the specific binding of SltA to the AGGCA motif, we constructed the point mutation forms of the sltB, erg11A, and mdr1 probes, which mutated AGGCA into CAAAC. EMSA analysis showed no binding to the mutant probe for SltA protein (Fig. 6C), suggesting that SltA binds specifically to the AGGCA motif. To verify whether the conserved cysteine and histidine are required for the SltA binding ability, we used the mutated proteins SltAC502S and SltAH518A to performed EMSA. Our data showed that the SltAC502S and SltAH518A proteins could not bind to the Cy5-labeled promoter fragments of any of the tested genes (Fig. 6D), indicating that the conserved cysteine and histidine within the SltA DNA binding domain are indispensable for SltA binding to the promoters of these genes in vitro.
FIG 6.

SltA binds to the AGGCA motif in the promoters of ergosterol biosynthesis and drug pump genes and affects related gene expression and function. (A) The promoters of the indicated genes contain a conserved AGGCA motif. The “Start” column shows the number of base pairs upstream of ATG. (B) EMSA showed in vitro binding of the indicated amounts of purified SltA protein to Cy5-labeled promoter fragments (20 ng) of indicated genes. We used 100× nonlabeled DNA fragments (cold probe, 2 μg) as competition assays. (C) An EMSA with a mutated binding motif. The consensus binding motif AGGCA from the promoters of the sltB, erg11A, and mdr1 genes was mutated into CAAAC and used for EMSA analyses. (D) EMSA showed in vitro binding of the purified SltA, SltAC502S, and SltAH518A proteins (10 μg) to Cy5-labeled promoter fragments (20 ng) of the indicated genes. (E) Western blot analysis for the expression of Erg11A and Erg11B in the parental wild-type and ΔsltA mutant strains with or without 0.01 μg ml−1 of itraconazole. The protein actin was used as a loading control. Each protein was loaded in two replicates. The value of integrated option density (IOD; IODtarget protein/IODactin) measured by Image-Pro Plus (IPP) showed the relative protein quantity. (F) The same number of spores (2 × 107) from the indicated strains grown for 24 h at 37°C in MM with 0.5 mM calcium and 0.015 μg ml−1 of itraconazole for subsequent ergosterol measurement by HPLC. The ergosterol content of each mutant was normalized to that of the parental wild-type strain and is shown as relative fold change. Samples were assessed in biological triplicates. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ns, P > 0.05 according to Student’s t test with Welch’s correction. +, strains were grown on medium with 5 mg liter−1 of doxycycline. tet-sltAΔN was induced (“on” state) in the presence of doxycycline.
In addition, we performed Western blot assays to verify whether SltA affects the protein expression of the azole target Erg11. As shown in Fig. 6E, without itraconazole treatment, Erg11A and Erg11B protein levels were very low in the wild-type strain and the ΔsltA mutant. Upon itraconazole treatment, the expression of Erg11A and Erg11B was clearly induced in the wild-type strain and the ΔsltA mutant; however, the ΔsltA mutant exhibited much lower Erg11A and Erg11B expression than that of the wild-type strain (Fig. 6E). These data suggest that the expression of the azole targets Erg11A and Erg11B is partly dependent on SltA. Due to the downregulation of Erg11 expression in the ΔsltA mutant, we wondered whether the deletion of sltA affects the ergosterol content. Since the ΔsltA mutant grew poorly in liquid MM, to ensure that the wild-type and ΔsltA mutant strains maintained similar levels of growth, 0.5 mM calcium was added to the medium, as it was previously reported that calcium could rescue the growth defects of the ΔsltA mutant (Fig. S5C) (29). As shown in Fig. S5A and B, there was no difference in ergosterol content between the parental wild-type strain and the ΔsltA mutant with or without 0.5 mM calcium, suggesting that calcium does not change the ergosterol content and the lack of SltA does not influence the ergosterol content under normal conditions. Next, we wondered whether the ergosterol content in the ΔsltA mutant would change under itraconazole stress. As predicted, the ergosterol content of the ΔsltA mutant decreased by approximately 30% compared to that of the parental wild-type strain under itraconazole treatment conditions (Fig. 6F), suggesting that SltA plays a role in the biosynthesis of ergosterol under itraconazole treatment conditions in A. fumigatus. Moreover, we also measured the ergosterol contents of the other sltA-related mutation strains. The sltAC502S, sltAH518A, sltAΔC, and ΔsltA OE::erg24A strains all displayed significantly decreased ergosterol content compared to that of the parental wild-type strain. In contrast, the ΔsltA OE::erg11A and tet-sltAΔN strains had ergosterol contents that were comparable to that of the parental wild-type strain (Fig. 6F), which explained the phenotypes of these strains under itraconazole stress (Fig. 4C and Fig. 5D), indicating that decreased ergosterol content may contribute to the itraconazole hypersusceptibility phenotype of the ΔsltA mutant. Taken together, these data suggest that SltA binds specifically to the AGGCA motif in the promoters of ergosterol biosynthesis and drug pump genes and regulates related gene expression as well as ultimately regulating ergosterol production.
Deleting sltA in laboratory-derived and clinical azole-resistant strains significantly attenuates the degree of azole resistance.
Since SltA plays a critical role in azole resistance, we wondered whether disruption of the sltA gene in Cyp51A and non-Cyp51A-type drug-resistant mutants could reverse their drug resistance. Therefore, we deleted the sltA gene in the laboratory-derived azole-resistant strain Cox10R243Q (non-Cyp51A-type drug-resistant mutant), which has a mitochondrial dysfunction that triggers calcium signaling-dependent fungal multidrug resistance (21, 38), and the clinical azole-resistant strains Shjt40 and Shjt42b (Cyp51A mutants), both of which contain mutations in the promoter region of erg11A (TR34/L98H) (39). MIC assays showed that the azole-resistant strains (Cox10R243Q, Shjt40, and Shjt42b) displayed hyperresistance to azoles and terbinafine (Table 1). Notably, the MICs of the ΔsltA mutants (R243Q ΔsltA, Shjt40 ΔsltA, and Shjt42b ΔsltA) for azoles and terbinafine significantly decreased compared to that of the azole-resistant strains, suggesting that deletion of sltA in the background of these azole-resistant strains showed decreased resistance to azoles and terbinafine, which was consistent with the colony growth test under itraconazole conditions (Fig. 7A). Moreover, to further identify whether the expression of erg11A and mdr1 contributes to the changes in drug resistance in these azole-resistant strains with sltA deleted, qRT-PCR analysis was performed and demonstrated that similar to the downregulated expression of erg11A and mdr1 observed in the sltA deletion mutant in the A1160 background, the ΔsltA mutants (R243Q ΔsltA, Shjt40 ΔsltA, and Shjt42b ΔsltA) exhibited significantly decreased expression of erg11A and mdr1 compared to that of these parental azole-resistant strains (Fig. 7B). Collectively, our data demonstrated that deleting sltA in laboratory-derived and clinical azole-resistant strains significantly attenuates the degree of azole resistance by downregulating the expression levels of erg11A and mdr1.
FIG 7.

sltA gene deletion in laboratory-derived and clinical azole-resistant strains attenuates the degree of azole resistance. (A) Colony morphologies of the indicated strains (2 × 104, 2 × 103, and 2 × 102 conidia) grown on YAG medium at 37°C for 36 h with or without 0.2 μg ml−1 of itraconazole. (B) qRT-PCR analysis was performed after growth of cultures in MM for 18 h at 37°C and subsequently for 2 h of treatment with 0.5 μg ml−1 of itraconazole. The tubA gene was used as an internal control. Samples were assessed in biological triplicates. P values were calculated using unpaired t test with Welch’s correction: *, P < 0.05; **, P < 0.01; ***, P < 0.001.
DISCUSSION
Azole-resistant A. fumigatus isolates arising from increased azole usage in clinical and agricultural settings cause an increasing threat to human health due to the paucity of appropriate antifungal therapeutics for azole-resistant isolates (11, 40). Although previous studies have highlighted the important role of transcription factors, such as SrbA, AtrR, the CBC complex, and the NCT complex, in the regulation of azole resistance (20, 24, 27), the understanding of the transcription factors that regulate azole resistance remains incomplete. In this study, we demonstrated that the C2H2 zinc finger transcription factor SltA, which has been previously recognized as the regulator of cation homeostasis (31, 33), has an uncharacterized function in azole resistance via coregulating the expression of the drug target Erg11A and the drug efflux pump Mdr1 in A. fumigatus.
Transcriptome analysis of the A. fumigatus parental wild-type strain grown in the presence and absence of the azole drug itraconazole revealed that the expression of the transcription factor SltA was induced under azole stress, implying a potential role for SltA in azole resistance. Phenotypic analysis further demonstrated that a lack of sltA caused sensitivity to ergosterol biosynthesis pathway inhibitors, including azoles, terbinafine, and simvastatin, but not to the ergosterol molecule-targeting antifungal amphotericin B or the cell wall component β-1,3-glucan inhibitor caspofungin (41), implying that SltA may have an impact on the ergosterol biosynthetic pathway.
In the industrial fungus Trichoderma reesei, the SltA homologue, named Ace1, regulates the expression of cellulase cellobiohydrolase (Cbh1) by binding to the AGGCA motif within the promoter of cbh1 (30). MEME analysis revealed that the conserved AGGCA motif exists in the promoter regions of genes related to ergosterol biosynthesis (erg11A, erg13A, and erg24A) and drug pumps (mdr1, mfsC, and abcE) in A. fumigatus. EMSAs confirmed that SltA can directly bind to these target genes in vitro. Notably, the cysteine and histidine residues within the C2H2 DNA binding domain were indispensable for the binding ability of SltA. Moreover, the sltAC502S and sltAH518A point mutation strains showed azole susceptibility phenotypes similar to that of the ΔsltA mutant due to the loss of the ability to bind to the promoters of related genes (erg11A, erg13A, erg24A, mdr1, mfsC, and abcE), implying that SltA may function to regulate the expression of genes encoding ergosterol biosynthesis enzymes and drug pumps. In line with this notion, qRT-PCR and lacZ reporter assays showed that the expression of these genes, most notably erg11A, mdr1, and mfsC, was downregulated in the ΔsltA mutant compared to the parental wild-type strain, suggesting that these genes may contribute to sltA-mediated itraconazole resistance. Interestingly, only overexpression of erg11A and mdr1 and not mfsC could completely restore the azole resistance of the ΔsltA mutant, indicating the dominant role of Erg11A and Mdr1 in response to azole treatment. Moreover, Western blot experiments along with ergosterol content analyses showed that the protein expression of the azole targets Erg11A and Erg11B decreased in the ΔsltA mutant, resulting in decreased ergosterol contents in the ΔsltA mutant under azole stress compared to that of the parental wild-type strain. These results suggested that the decreased ergosterol contents may contribute to the azole susceptibility phenotype of the ΔsltA mutant. Notably, the mRNA level of mdr1 and erg11A decreased by approximately 50% in the ΔsltA mutant, implying that other transcription factors may participate in controlling the expression of mdr1 and erg11A. Indeed, we previously demonstrated that the transcription factor CrzA regulates mdr1 expression by binding the calcium-dependent serine-threonine phosphatase-dependent response element (CDRE) motif in the promoter of mdr1 in vitro, and other researchers demonstrated that the expression of erg11A is controlled by the transcription factors SrbA and AtrR, and the NCT complex (21, 22, 26). Taken together, our results strongly suggested that the C2H2 transcription factor SltA contributes to azole resistance by coregulating erg11A and mdr1 in A. fumigatus.
Most importantly, deleting sltA in lab-derived and clinical drug-resistant strains could significantly attenuate their multidrug resistance by downregulating the expression of erg11A and mdr1. We demonstrated that azole-resistant A. fumigatus isolates with a non-erg11A mutation (Cox10R243Q) and with erg11A mutations (Shjt40 and Shjt42b) all displayed remarkably decreased MIC values to azoles and terbinafine when sltA was deleted. The key roles of the azole drug target Erg11A and the ABC transporter Mdr1 in the regulation of azole resistance have been documented not only for Aspergillus but also for other pathogenic fungi, such as Cryptococcus neoformans, Candida albicans, Magnaporthe grisea, and Fusarium graminearum (42–50). Therefore, our results raise the possibility that targeting SltA and its orthologues in other fungal pathogens could be a potential therapeutic strategy for intervention of clinical azole-resistant isolates to improve the efficacy of antifungal drugs. In summary, we identified a novel function of the transcription factor SltA in regulating azole resistance by coordinately mediating the azole target Erg11A and the drug efflux pump Mdr1. A possible working model of sltA-mediated azole resistance in A. fumigatus is presented in Fig. 8. Our findings shed new light on how filamentous fungi adapt to azole stress by coregulating the expression of ergosterol biosynthesis and drug pump genes.
FIG 8.

A putative working model for sltA-mediated itraconazole resistance. SltA directly binds to the conserved AGGCA motif to contribute to azole resistance via regulating the expression of the drug target Erg11A and the drug transporter Mdr1 in A. fumigatus, and this binding is dependent on the conserved cysteine and histidine within the C2H2 DNA binding domain. The cysteine and histidine were mutated to Ser and Ala, respectively, which are marked with stars. Under itraconazole conditions, sltA is induced at the mRNA level. SltA transcriptionally regulates drug transporters (Mdr1, AbcE, and MfsC) to extrude excess intracellular itraconazole. On the other hand, SltA transcriptionally regulates ergosterol biosynthetic enzymes (Erg11A, Erg13A, and Erg24A) to withstand itraconazole and is involved in the biosynthesis of ergosterol under itraconazole conditions.
MATERIALS AND METHODS
Strains, oligonucleotides, media, and transformation.
The genotype of each strain is listed in Table S2 in the supplemental material. The oligonucleotides used in this study are displayed in Table S3. A. fumigatus strain A1160 was purchased from the Fungal Genetics Stock Center (FGSC). For all experiments, A1160C′ was used as the parental wild-type strain by reintroducing pyrG into A. fumigatus strain A1160 (51). For RNA-seq, qRT-PCR, Western blotting, ergosterol measurement, and LacZ reporter assays, A. fumigatus strains were grown on liquid minimal medium (MM) containing 1% glucose, trace elements, and 50 ml liter−1 of 20× salt (pH 6.5) plus 0.5 mM calcium to ensure that the wild type and the ΔsltA mutant maintained similar levels of growth. For the colony growth tests, A. fumigatus strains were grown on YAG (yeast extract, agar, and glucose), containing 2% glucose, trace elements, 0.5% yeast extract and 2% agar. Transformation of A. fumigatus was performed according to the previous study (52).
Construction of deletion mutants and complemented strains.
The sltA gene was replaced with the pyr4 selectable marker by homologous recombination in A. fumigatus strain A1160. The pyr4 gene was amplified from plasmid pAL5 (purchased from FGSC) with the primer pair Pyr4-F/Pyr4-R. The 5′ and 3′ flanking regions of the sltA gene were amplified from A. fumigatus A1160 genomic DNA with the primer pairs sltA-P1/sltA-P3 and sltA-P4/sltA-P6, respectively, and then the flanking regions of sltA and the pyr4 selectable marker were fused together with the nested primer pair sltA-P2/sltA-P5. The fusion product was transformed into the parental A. fumigatus A1160 strain to generate DWA01 (ΔsltA). A similar strategy was used to construct the strains DWA35 (ΔAFUB_063290), DWA36 (ΔAFUB_051190), DWA37 (ΔAFUB_003250), and DWA38 (ΔAFUB_086150). The hph gene was amplified from plasmid pAN7-1 with the primer pair Hph-F/Hph-R. The 5′ and 3′ flanking regions of the sltA gene were amplified from A. fumigatus A1160 genomic DNA with the primer pairs sltA-P1/sltA-P3hph and sltA-P4hph/sltA-P6, respectively, and then the flanking regions of the sltA and hph selectable marker were fused together with the nested primer pair sltA-P2/sltA-P5. The fusion product was transformed into strains Shjt40, Shjt42b, and Cox10R243Q to generate strains DWA32 (Shjt40 ΔsltA), DWA33 (Shjt42b ΔsltA), and DWA34 (Cox10R243Q ΔsltA), respectively. The sltA-complemented fragments were amplified with the primer pair, Com-sltA-F/Com-sltA-R and then cloned into plasmid pAN7-1 with the ClonExpress MultiS one-step cloning kit (Vazyme Biotech Co., Ltd.; C113-02). The complementary plasmid pWL01 was transformed into DWA01 to generate DWA02 (sltAC). Phanta Max master mix (Vazyme Biotech Co., Ltd.; P515) was used to amplify and fuse PCR products according to the manufacturer’s manual.
Construction of sltA mutation and homologue complementation strains.
To construct an AnSltA open reading frame (ORF) driven by the AfsltA native promoter, fragments containing regions flanking the 5′ and 3′ AfsltA ORF were amplified from A. fumigatus A1160 with primer pairs Com-sltA-F/sltA-P3free and sltA-P4free/Com-sltA-R. The coding sequence of the AnSltA ORF was amplified with the primer pair ComAnsltA-F/ComAnsltA-R from A. nidulans genomic DNA. The three PCR products were cloned into plasmid pAN7-1 to generate the plasmid pWL02, which was subsequently transformed into DWA01 to generate DWA03 (ΔsltAAnSltA). A similar strategy was used to construct strain DWA04 (ΔsltAAflSltA) with plasmid pWL03.
To generate strain DWA06 (sltAC502S), the fragments were cloned with primer pairs Com-sltA-F/C502S-R and C502S-F/Com-sltA-R, and then the two fragments were cloned into plasmid pAN7-1 to generate plasmid pWL04, which was transformed into DWA01. A similar strategy was used to construct strain DWA07 (sltAH518A) with plasmid pWL05.
To generate the truncation of sltA, the fragments were cloned with primer pairs Com-sltA-F/sltA-P3free and ΔN-F/Com-sltA-R. The two fragments were cloned into plasmid pAN7-1 to generate plasmid pWL06, which was transformed into DWA01 to generate strain DWA08 (sltAΔN). A similar strategy was used to construct strain DWA09 (sltAΔC) and DWA10 (tet-sltAΔN) with plasmids pWL07 and pWL08, respectively.
Construction of strains overexpressing related genes.
We used the primer pair OE::sltA-F/OE::sltA-R to amplify a fragment of the sltA ORF from A. fumigatus A1160 genomic DNA. Then the fragment was cloned into the reconstituted plasmid vector pBARGPE1, containing the Aspergillus nidulans glyceraldehyde-3-phosphate dehydrogenase (AngpdA) promoter, the trpC terminator, and the hygromycin resistance gene, to construct plasmid pWL09, which was transformed into the WT to generate strain DWA05 (OE::sltA). Similarly, the ORFs of mdr1, mfsC, abcE, erg11A, and erg24A were amplified with primer pairs OE::mdr1-F/OE::mdr1-R, OE::mfsC-F/OE::mfsC-R, OE::abcE-F/OE::abcE-R, OE::erg11A-F/OE::erg11A-R, and OE::erg24A-F/OE::erg24A-R, respectively, which were cloned into pBARGPE1 to generate plasmids pWL10 to pWL14. Then plasmids pWL10 to pWL14 were transformed into strain DWA01 (ΔsltA) to generate strains DWA11 to DWA15, respectively.
Construction of strains for the LacZ reporter assay.
The lacZ DNA fragment was amplified from Escherichia coli with the primer pair LacZ-S/LacZ-TrpC-A. The promoter of mdr1 was amplified from A. fumigatus A1160 genomic DNA with the primer pair Pmdr1-F/Pmdr1-R-lacZ. Then the two fragments were cloned into plasmid pAN7-1 to generate plasmid pWL15, which was transformed into the WT and DWA01 (ΔsltA) to generate DWA16 (WTmdr1(p)::lacZ) and DWA19 (ΔsltAmdr1(p)::lacZ), respectively. A similar strategy was used to construct strains DWA17 (WTmfsC(p)::lacZ), DWA20 (ΔsltAmfsC(p)::lacZ), DWA18 (WTabcE(p)::lacZ), DWA21 (ΔsltAabcE(p)::lacZ), DWA22 (WTerg11A(p)::lacZ), DWA25 (ΔsltAerg11A(p)::lacZ), DWA23 (WTerg13A(p)::lacZ), DWA26 (ΔsltAerg13A(p)::lacZ), DWA24 (WTerg24A(p)::lacZ), DWA27 (ΔsltAerg24A(p)::lacZ), DWA39 (WTsltA(p)::lacZ), and DWA40 (ΔsltAsltA(p)::lacZ) with plasmids pWL16 to pWL21.
Construction of green fluorescent protein (GFP)-labeled strains.
We transformed the plasmid containing the Erg11A::GFP cassette (53) into the WT and DWA01 (ΔsltA) to generate DWA28 (WTErg11A::GFP) and DWA29 (ΔsltAErg11A::GFP), respectively. Similarly, We transformed the plasmid containing the Erg11B::GFP cassette (53) into the WT and DWA01 (ΔsltA) to generate DWA30 (WTErg11B::GFP) and DWA31 (ΔsltAErg11B::GFP), respectively.
Construction of FLAG-labeled strains.
To construct the in situ SltA-FLAG-tagged strain (DWA41), fragments containing regions flanking the 5′ and 3′ AfsltA termination codon (TAA) were amplified from A. fumigatus A1160 with primer pairs SltA-FLAG-P1/SltA-FLAG-P3 and SltA-FLAG-P4/SltA-FLAG-P6, respectively. The coding sequence of FLAG::pyr4 was amplified with the primer pair FLAG-F/Pyr4-R. Then the three fragments were fused together with the nested primer pair SltA-FLAG-P2/SltA-FLAG-P5. The fusion PCR products were then transformed into the parental A. fumigatus A1160 strain.
To generate strain DWA42 (SltAC502S-FLAG), the fragments were cloned with primer pairs Com-sltA-F/C502S-R and C502S-F/FLAG-R-PAN7-1 from DWA41 genomic DNA, and then the two fragments were cloned into plasmid pAN7-1 to generate plasmid pWL22, which was transformed into DWA01. A similar strategy was used to construct strain DWA43 (SltAH518A-FLAG) with plasmid pWL23.
To generate strain DWA44 (SltAΔC-FLAG), the fragments were cloned with primer pairs Com-sltA-F/ΔC-R-FLAG and FLAG-F/Hph-R. The two fragments were cloned into plasmid Blunt-Zero (Vazyme Biotech Co., Ltd.; C601-01) to generate plasmid pWL24, which was transformed into DWA01.
Antifungal susceptibility testing.
Testing for susceptibility to antifungal drugs was performed by the broth-based microdilution method according to CLSI document M38-A2 and the previously described method (21, 35). Briefly, the same numbers of spores (1 × 105 conidia) from related mutants were diluted into RPMI 1640 medium containing gradient diluent antifungal drugs, transferred into 96-well plates, and cultured at 35°C for 48 h.
Construction of a phylogenetic tree.
The amino acid sequences of SltA homologues were downloaded from the FungiDB (http://fungidb.org/fungidb) and NCBI (https://www.ncbi.nlm.nih.gov) websites. The DNA binding domain was obtained based on SMART-predicted analysis (http://smart.embl-heidelberg.de). The phylogenetic tree was constructed with bootstrap values shown via MEGA 6 using the neighbor-joining method.
Total sterol extraction and analysis.
The total sterol extraction was performed as previously described (53), with a few modifications for sample treatment. In brief, the related strains were grown in MM, MM plus 0.5 mM calcium, and MM with 0.5 mM calcium and 0.015 μg ml−1 of ITZ for 24 h. Then the mycelia were harvested for sterol extraction and analysis. Briefly, the ergosterol of 200 mg of mycelium powder was extracted in 3 ml of 25% KOH (volmethanol:volethanol = 3:2) and vortexed for 1 min, subsequently subjected to water bath heating to 85°C for 1 h and extraction with 1 ml of distilled water and 3 ml of hexane, and vortexed for 3 min; the supernatant was transferred into new tubes and dried. Finally, the samples were resolved in 1 ml of methanol and were measured with a high-performance liquid chromatograph (HPLC; Agilent Technologies) at 280 nm on an AQ-C18 column (250 mm by 4.6 mm, 5 μm).
qRT-PCR and RNA-seq analysis.
A. fumigatus conidia were inoculated into liquid MM with 0.5 mM calcium and shaken on a rotary shaker at 220 rpm and 37°C for 18 h with a subsequent 2-h growth under the same conditions or a subsequent 2-h shift to a 0.5-μg ml−1 itraconazole condition. RNA isolation and qRT-PCR analysis were performed as previously described (34). For RNA-seq analysis, all samples were prepared to perform digital transcriptome analyses through the RNA-seq approach (Shanghai Sangon Biotech Co., Ltd., China). For qRT-PCR analysis, total RNA samples were extracted and purified as described in the UNlQ-10 column TRIzol total RNA isolation kit (Sangon Biotech; B511361). The synthesis of cDNA was performed according to the protocol described in the HiScript II Q RT SuperMix for qPCR kit (Vazyme; R223-01).
β-Galactosidase assays.
A. fumigatus conidia were inoculated into MM with 0.5 mM calcium, shaken on a rotary shaker at 220 rpm and 37°C for 20 h, and then grown for a further 3 h under the same conditions or shifted to 3 h of growth with 0.5 μg ml−1 of itraconazole. The mycelia were harvested for β-galactosidase assays as previously described (34). Briefly, the protein was extracted in PEB buffer (60 mM Na2HPO4·7H2O, 40 mM NaH2PO4·H2O, 10 mM KCl, 1 mM MgSO4·7H2O, 1 mM EDTA [pH 7.0]) and incubated on ice for 15 min and vortexed. The tubes were spun at 13,000 × g for 15 min at 4°C, and the supernatant was transferred to a new tube. The reaction was started by adding 100 μl of protein in PEB buffer, 900 μl of Z buffer (60 mM Na2HPO4·7H2O, 40 mM NaH2PO4·H2O, 10 mM KCl, 1 mM MgSO4·7H2O [pH 7.0]), and 200 μl of o-nitrophenyl-β-d-galactopyranoside (ONPG), diluted to 4 mg ml−1 in Z buffer, at 37°C and stopped by adding 500 μl of 1 M Na2CO3. The values of β-galactosidase activity were calculated as previously described (34).
Western blotting.
A. fumigatus conidia were inoculated into MM with 0.5 mM calcium and shaken on a rotary shaker at 220 rpm at 37°C for 24 h with or without 0.01 μg ml−1 of itraconazole, and then the mycelia were harvested for Western blotting as previously described (54). Briefly, for Erg11A and Erg11B GFP-tagged strains, the total protein of A. fumigatus mycelium powder was extracted in lysis buffer (50 mM HEPES [pH 7.4], 137 mM KCl, 10% glycerol, 1 mM EDTA, 1 μg ml−1 of pepstatin A, 1 μg ml−1 of leupeptin, 1 mM phenylmethylsulfonyl fluoride [PMSF]). For SltA FLAG-tagged strains, the mycelia were ground with liquid nitrogen and alkaline lysis buffer (0.2 M NaOH and 0.2% β-mercaptoethanol), and a mediated protein isolation strategy was followed as previously described (54). Equal amounts of protein measured by a Bio-Rad (Hercules, CA) protein assay kit were loaded in duplicate onto 10% SDS-PAGE gels and subsequently transferred to a polyvinylidene difluoride (PVDF) membrane (Immobilon-P; Millipore) in 384 mM glycine, 50 mM Tris (pH 8.4), and 20% methanol at 250 mA for 2 h. The membrane was blocked in phosphate-buffered saline containing 5% milk and 0.1% Tween 20 for 2 h at room temperature. Then the membrane was probed sequentially with 1:3,000 dilutions of anti-GFP antibody (Roche; catalogue no. 11 814 460 001) or anti-actin antibody (ICN Biomedicals Inc.; clone C4) and goat anti-mouse IgG-horseradish peroxidase and was developed by chemiluminescence (ECL; Amersham).
Recombinant SltA protein purification and EMSA.
For expressing the DNA binding domain of SltA in vitro, two parts of exons of the DNA binding domain of SltA were amplified with two pairs of primers, EX-sltA-P1NdeI/EX-sltA-P2 and EX-sltA-P3/EX-sltA-P4EcoRI, then cloned into pET-30a(+), and subsequently transformed into express competent BL21(DE3) cells (TransGen Biotech). The BL21(DE3) cells (optical density at 600 nm [OD600] up to 0.6 to 0.8) were grown in LB medium at 16°C for 12 h under 0.5 mM isopropyl-β-d-thiogalactopyranoside (IPTG) induction, and protein purification was performed as previously described using nickel-nitrilotriacetic acid (Ni-NTA) agarose (55). A similar strategy was used to express the DNA binding domain of SltAC502S and SltAH518A. EMSA was performed as previously described (54, 55). Each reaction mixture contained 20 ng of Cy5-labeled probe and 5 μg or 10 μg of purified protein. For nonspecific competitor or cold probe, 1 μg of salmon sperm DNA or a 100-fold nonlabeled DNA probe (2 μg) was added. To confirm the specific binding of SltA to the binding motif AGGCA, the AGGCA motif within the promoter of the sltB gene was mutated into CAAAC with the primer pairs EMSA-sltB-F/EMSA-MUsltB-R and EMSA-MUsltB-F/EMSA-sltB-R, and then the two fragments were fused with the primer Cy5 to generate the mutant probe for sltB. A similar strategy was used to generate the mutant probes for erg11A and mdr1. The Cy5-labeled probes containing conserved AGGCA motif were amplified as previously described (54) and were detected with an Odyssey machine.
Data availability.
The raw Illumina sequencing data were uploaded in the SRA with accession numbers SRR12338578 to SRR12338580. A processed format of the RNA-seq data sets is included in Data Set S1.
Supplementary Material
ACKNOWLEDGMENTS
This work was financially supported by the National Key R & D Program of China (2019YFA0904900) and the National Natural Science Foundation of China (31861133014 and 31770086), Program for Jiangsu Excellent Scientific and Technological Innovation team (17CXTD00014), the Priority Academic Program Development (PAPD) of Jiangsu Higher Education Institutions (L.L.), and National Natural Science Foundation of China (31900404) and Natural Science Foundation of the Jiangsu Higher Education Institutions of China (19KJB180017) to Y.Z. This work was also supported by the Wellcome Trust (208396/Z/17/Z) (M.J.B.).
REFERENCES
- 1.Pappy L, Lee S, Shah A, Bernstein M. 2019. Pulmonary aspergillosis leading to disseminated disease in an immunocompetent patient. Chest 156:A685. doi: 10.1016/j.chest.2019.08.663. [DOI] [Google Scholar]
- 2.Dagenais TRT, Keller NP. 2009. Pathogenesis of Aspergillus fumigatus in invasive aspergillosis. Clin Microbiol Rev 22:447–465. doi: 10.1128/CMR.00055-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cabanes L, Plaza P, Moragon EM. 2018. Chronic necrotizing pulmonary aspergillosis in an immunocompetent patient. Med Clin 150:451. doi: 10.1016/j.medcle.2017.10.047. [DOI] [PubMed] [Google Scholar]
- 4.Toor A, Sharma D, Krishnamurthy M. 2019. Lethal pulmonary aspergillosis in an immunocompetent patient. Chest 156:A656. doi: 10.1016/j.chest.2019.08.638. [DOI] [Google Scholar]
- 5.Karthaus M, Buchheidt D. 2013. Invasive aspergillosis: new insights into disease, diagnostic and treatment. Curr Pharm Des 19:3569–3594. doi: 10.2174/13816128113199990330. [DOI] [PubMed] [Google Scholar]
- 6.Roemer T, Krysan DJ. 2014. Antifungal drug development: challenges, unmet clinical needs, and new approaches. Cold Spring Harb Perspect Med 4:a019703. doi: 10.1101/cshperspect.a019703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Esquivel BD, Smith AR, Zavrel M, White TC. 2015. Azole drug import into the pathogenic fungus Aspergillus fumigatus. Antimicrob Agents Chemother 59:3390–3398. doi: 10.1128/AAC.05003-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Latge JP, Chamilos G. 2019. Aspergillus fumigatus and aspergillosis in 2019. Clin Microbiol Rev 33:e00140-18. doi: 10.1128/CMR.00140-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lestrade PPA, Meis JF, Melchers WJG, Verweij PE. 2019. Triazole resistance in Aspergillus fumigatus: recent insights and challenges for patient management. Clin Microbiol Infect 25:799–806. doi: 10.1016/j.cmi.2018.11.027. [DOI] [PubMed] [Google Scholar]
- 10.Garcia-Rubio R, Cuenca-Estrella M, Mellado E. 2017. Triazole resistance in Aspergillus species: an emerging problem. Drugs 77:599–613. doi: 10.1007/s40265-017-0714-4. [DOI] [PubMed] [Google Scholar]
- 11.Ren JB, Jin XX, Zhang Q, Zheng Y, Lin DL, Yu YL. 2017. Fungicides induced triazole-resistance in Aspergillus fumigatus associated with mutations of TR46/Y121F/T289A and its appearance in agricultural fields. J Hazard Mater 326:54–60. doi: 10.1016/j.jhazmat.2016.12.013. [DOI] [PubMed] [Google Scholar]
- 12.Rybak JM, Fortwendel JR, Rogers PD. 2019. Emerging threat of triazole-resistant Aspergillus fumigatus. J Antimicrob Chemother 74:835–842. doi: 10.1093/jac/dky517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Perez-Cantero A, Lopez-Fernandez L, Guarro J, Capilla J. 2020. Azole resistance mechanisms in Aspergillus: update and recent advances. Int J Antimicrob Agents 55:105807. doi: 10.1016/j.ijantimicag.2019.09.011. [DOI] [PubMed] [Google Scholar]
- 14.Ferreira MED, Colombo AL, Paulsen I, Ren Q, Wortman J, Huang J, Goldman MHS, Goldman GH. 2005. The ergosterol biosynthesis pathway, transporter genes, and azole resistance in Aspergillus fumigatus. Med Mycol 43:S313–S319. [DOI] [PubMed] [Google Scholar]
- 15.Verweij PE, Howard SJ, Melchers WJG, Denning DW. 2009. Azole-resistance in Aspergillus: proposed nomenclature and breakpoints. Drug Resist Updat 12:141–147. doi: 10.1016/j.drup.2009.09.002. [DOI] [PubMed] [Google Scholar]
- 16.Mohammadi F, Hashemi SJ, Seyedmousavi SM, Akbarzade D. 2018. Isolation and characterization of clinical triazole resistance Aspergillus fumigatus in Iran. Iran J Public Health 47:994–1000. [PMC free article] [PubMed] [Google Scholar]
- 17.Willger SD, Puttikamonkul S, Kim KH, Burritt JB, Grahl N, Metzler LJ, Barbuch R, Bard M, Lawrence CB, Cramer RA. 2008. A sterol-regulatory element binding protein is required for cell polarity, hypoxia adaptation, azole drug resistance, and virulence in Aspergillus fumigatus. PLoS Pathog 4:e1000200. doi: 10.1371/journal.ppat.1000200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Blatzer M, Barker BM, Willger SD, Beckmann N, Blosser SJ, Cornish EJ, Mazurie A, Grahl N, Haas H, Cramer RA. 2011. SREBP coordinates iron and ergosterol homeostasis to mediate triazole drug and hypoxia responses in the human fungal pathogen Aspergillus fumigatus. PLoS Genet 7:e1002374. doi: 10.1371/journal.pgen.1002374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fraczek MG, Bromley M, Buied A, Moore CB, Rajendran R, Rautemaa R, Ramage G, Denning DW, Bowyer P. 2013. The cdr1B efflux transporter is associated with non-cyp51a-mediated itraconazole resistance in Aspergillus fumigatus. J Antimicrob Chemother 68:1486–1496. doi: 10.1093/jac/dkt075. [DOI] [PubMed] [Google Scholar]
- 20.Paul S, Stamnes M, Thomas GH, Liu H, Hagiwara D, Gomi K, Filler SG, Moye-Rowley WS. 2019. AtrR is an essential determinant of azole resistance in Aspergillus fumigatus. mBio 10:e02563-18. doi: 10.1128/mBio.02563-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li YQ, Zhang YW, Zhang C, Wang HC, Wei XL, Chen PY, Lu L. 2020. Mitochondrial dysfunctions trigger the calcium signaling-dependent fungal multidrug resistance. Proc Natl Acad Sci U S A 117:1711–1721. doi: 10.1073/pnas.1911560116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Blosser SJ, Cramer RA. 2012. SREBP-dependent triazole susceptibility in Aspergillus fumigatus is mediated through direct transcriptional regulation of erg11A (cyp51A). Antimicrob Agents Chemother 56:248–257. doi: 10.1128/AAC.05027-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chung D, Barker BM, Carey CC, Merriman B, Werner ER, Lechner BE, Dhingra S, Cheng C, Xu W, Blosser SJ, Morohashi K, Mazurie A, Mitchell TK, Haas H, Mitchell AP, Cramer RA. 2014. ChIP-seq and in vivo transcriptome analyses of the Aspergillus fumigatus SREBP SrbA reveals a new regulator of the fungal hypoxia response and virulence. PLoS Pathog 10:e1004487. doi: 10.1371/journal.ppat.1004487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gsaller F, Hortschansky P, Furukawa T, Carr PD, Rash B, Capilla J, Muller C, Bracher F, Bowyer P, Haas H, Brakhage AA, Bromley MJ. 2016. Sterol biosynthesis and azole tolerance is governed by the opposing actions of SrbA and the CCAAT binding complex. PLoS Pathog 12:e1005775. doi: 10.1371/journal.ppat.1005775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Furukawa T, Scheven MT, Misslinger M, Zhao C, Hoefgen S, Gsaller F, Lau J, Jochl C, Donaldson I, Valiante V, Brakhage AA, Bromley MJ, Haas H, Hortschansky P. 2020. The fungal CCAAT-binding complex and HapX display highly variable but evolutionary conserved synergetic promoter-specific DNA recognition. Nucleic Acids Res 48:3567–3590. doi: 10.1093/nar/gkaa109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hagiwara D, Miura D, Shimizu K, Paul S, Ohba A, Gonoi T, Watanabe A, Kamei K, Shintani T, Moye-Rowley WS, Kawamoto S, Gomi K. 2017. A novel Zn-2-Cys(6) transcription factor AtrR plays a key role in an azole resistance mechanism of Aspergillus fumigatus by coregulating cyp51A and cdr1B expressions. PLoS Pathog 13:e1006096. doi: 10.1371/journal.ppat.1006096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Furukawa T, van Rhijn N, Fraczek M, Gsaller F, Davies E, Carr P, Gago S, Fortune-Grant R, Rahman S, Gilsenan JM, Houlder E, Kowalski CH, Raj S, Paul S, Cook P, Parker JE, Kelly S, Cramer RA, Latge JP, Moye-Rowley S, Bignell E, Bowyer P, Bromley MJ. 2020. The negative cofactor 2 complex is a key regulator of drug resistance in Aspergillus fumigatus. Nat Commun 11:427. doi: 10.1038/s41467-019-14191-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mellado L, Calcagno-Pizarelli AM, Lockington RA, Cortese MS, Kelly JM, Arst HN, Espeso EA. 2015. A second component of the SltA-dependent cation tolerance pathway in Aspergillus nidulans. Fungal Genet Biol 82:116–128. doi: 10.1016/j.fgb.2015.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Findon H, Calcagno-Pizarelli AM, Martinez JL, Spielvogel A, Markina-Inarrairaegui A, Indrakumar T, Ramos J, Penalva MA, Espeso EA, Arst HN. 2010. Analysis of a novel calcium auxotrophy in Aspergillus nidulans. Fungal Genet Biol 47:647–655. doi: 10.1016/j.fgb.2010.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Saloheimo A, Aro N, Ilmen M, Penttila M. 2000. Isolation of the ace1 gene encoding a Cys(2)-His(2) transcription factor involved in regulation of activity of the cellulase promoter cbh1 of Trichoderma reesei. J Biol Chem 275:5817–5825. doi: 10.1074/jbc.275.8.5817. [DOI] [PubMed] [Google Scholar]
- 31.Dubey AK, Barad S, Luria N, Kumar D, Espeso EA, Prusky DB. 2016. Cation-stress-responsive transcription factors SItA and CrzA regulate morphogenetic processes and pathogenicity of Colletotrichurn gloeosporioides. PLoS One 11:e0168561. doi: 10.1371/journal.pone.0168561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.O’Mahony RJ, Burns ATH, Millam S, Hooley P, Fincham DA. 2002. Isotropic growth of spores and salt tolerance in Aspergillus nidulans. Mycol Res 106:1480–1486. doi: 10.1017/S0953756202006949. [DOI] [Google Scholar]
- 33.Spielvogel A, Findon H, Arst HN, Araujo-Bazan L, Hernandez-Ortiz P, Stahl U, Meyer V, Espeso EA. 2008. Two zinc finger transcription factors, CrzA and SltA, are involved in cation homoeostasis and detoxification in Aspergillus nidulans. Biochem J 414:419–429. doi: 10.1042/BJ20080344. [DOI] [PubMed] [Google Scholar]
- 34.Cai ZD, Du WL, Zhang Z, Guan LY, Zeng QQ, Chai YF, Dai CC, Lu L. 2018. The Aspergillus fumigatus transcription factor AceA is involved not only in Cu but also in Zn detoxification through regulating transporters CrpA and ZrcA. Cell Microbiol 20:e12864. doi: 10.1111/cmi.12864. [DOI] [PubMed] [Google Scholar]
- 35.Espinel-Ingroff A, Cuenca-Estrella M, Fothergill A, Fuller J, Ghannoum M, Johnson E, Pelaez T, Pfaller MA, Turnidge J. 2011. Wild-type MIC distributions and epidemiological cutoff values for amphotericin B and Aspergillus spp. for the CLSI broth microdilution method (M38-A2 document). Antimicrob Agents Chemother 55:5150–5154. doi: 10.1128/AAC.00686-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Helmschrott C, Sasse A, Samantaray S, Krappmann S, Wagener J. 2013. Upgrading fungal gene expression on demand: improved systems for doxycycline-dependent silencing in Aspergillus fumigatus. Appl Environ Microbiol 79:1751–1754. doi: 10.1128/AEM.03626-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mellado L, Arst HN, Espeso EA. 2016. Proteolytic activation of both components of the cation stress-responsive Slt pathway in Aspergillus nidulans. Mol Biol Cell 27:2598–2612. doi: 10.1091/mbc.E16-01-0049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wei XL, Chen PY, Gao RS, Li YQ, Zhang AX, Liu FF, Lu L. 2017. Screening and characterization of a non-cyp51A mutation in an Aspergillus fumigatus cox10 strain conferring azole resistance. Antimicrob Agents Chemother 61:e02101-16. doi: 10.1128/AAC.02101-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu MS, Zeng R, Zhang LL, Li DM, Lv GX, Shen YN, Zheng HL, Zhang QQ, Zhao JJ, Zheng N, Liu WD. 2015. Multiple cyp51A-based mechanisms identified in azole-resistant isolates of Aspergillus fumigatus from China. Antimicrob Agents Chemother 59:4321–4325. doi: 10.1128/AAC.00003-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Abad A, Fernandez-Molina JV, Bikandi J, Ramirez A, Margareto J, Sendino J, Hernando FL, Ponton J, Garaizar J, Rementeria A. 2010. What makes Aspergillus fumigatus a successful pathogen? Genes and molecules involved in invasive aspergillosis. Rev Iberoam Micol 27:155–182. doi: 10.1016/j.riam.2010.10.003. [DOI] [PubMed] [Google Scholar]
- 41.Wiederhold NP. 2018. The antifungal arsenal: alternative drugs and future targets. Int J Antimicrob Agents 51:333–339. doi: 10.1016/j.ijantimicag.2017.09.002. [DOI] [PubMed] [Google Scholar]
- 42.Jin LY, Cao ZR, Wang Q, Wang YC, Wang XJ, Chen HB, Wang H. 2018. MDR1 overexpression combined with ERG11 mutations induce high-level fluconazole resistance in Candida tropicalis clinical isolates. BMC Infect Dis 18:162. doi: 10.1186/s12879-018-3082-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Feng WL, Yang J, Yang L, Li Q, Zhu X, Xi ZQ, Qiao ZS, Cen W. 2018. Research of Mrr1, Cap1 and MDR1 in Candida albicans resistant to azole medications. ExpTher Med 15:1217–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gołąbek K, Strzelczyk JK, Owczarek A, Cuber P, Ślemp-Migiel A, Wiczkowski A. 2015. Selected mechanisms of molecular resistance of Candida albicans to azole drugs. Acta Biochim Pol 62:247–251. doi: 10.18388/abp.2014_940. [DOI] [PubMed] [Google Scholar]
- 45.Basso LR, Gast CE, Bruzual I, Wong B. 2015. Identification and properties of plasma membrane azole efflux pumps from the pathogenic fungi Cryptococcus gattii and Cryptococcus neoformans. J Antimicrob Chemother 70:1396–1407. doi: 10.1093/jac/dku554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sionov E, Chang YC, Garraffo HM, Dolan MA, Ghannoum MA, Kwon-Chung KJ. 2012. Identification of a Cryptococcus neoformans cytochrome P450 lanosterol 14 alpha-demethylase (Erg11) residue critical for differential susceptibility between fluconazole/voriconazole and itraconazole/posaconazole. Antimicrob Agents Chemother 56:1162–1169. doi: 10.1128/AAC.05502-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gupta A, Chattoo BB. 2008. Functional analysis of a novel ABC transporter ABC4 from Magnaporthe grisea. FEMS Microbiol Lett 278:22–28. doi: 10.1111/j.1574-6968.2007.00937.x. [DOI] [PubMed] [Google Scholar]
- 48.Yang JY, Zhang QY, Liao MJ, Xiao M, Xiao WJ, Yang S, Wan N. 2009. Expression and homology modelling of sterol 14 alpha-demethylase of Magnaporthe grisea and its interaction with azoles. Pest Manag Sci 65:260–265. doi: 10.1002/ps.1680. [DOI] [PubMed] [Google Scholar]
- 49.Abou Ammar G, Tryono R, Doll K, Karlovsky P, Deising HB, Wirsel SGR. 2013. Identification of ABC transporter genes of Fusarium graminearum with roles in azole tolerance and/or virulence. PLoS One 8:e79042. doi: 10.1371/journal.pone.0079042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu X, Yu F, Schnabel G, Wu JB, Wang ZY, Ma ZH. 2011. Paralogous cyp51 genes in Fusarium graminearum mediate differential sensitivity to sterol demethylation inhibitors. Fungal Genet Biol 48:113–123. doi: 10.1016/j.fgb.2010.10.004. [DOI] [PubMed] [Google Scholar]
- 51.Jiang H, Shen Y, Liu W, Lu L. 2014. Deletion of the putative stretch-activated ion channel Mid1 is hypervirulent in Aspergillus fumigatus. Fungal Genet Biol 62:62–70. doi: 10.1016/j.fgb.2013.11.003. [DOI] [PubMed] [Google Scholar]
- 52.Zhai PF, Song JX, Gao L, Lu L. 2019. A sphingolipid synthesis-related protein OrmA in Aspergillus fumigatus is responsible for azole susceptibility and virulence. Cell Microbiol 21:e13092. doi: 10.1111/cmi.13092. [DOI] [PubMed] [Google Scholar]
- 53.Song JX, Zhai PF, Zhang YW, Zhang CY, Sang H, Han GZ, Keller NP, Lu L. 2016. The Aspergillus fumigatus damage resistance protein family coordinately regulates ergosterol biosynthesis and azole susceptibility. mBio 7:e01919-15. doi: 10.1128/mBio.01919-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Long N, Orasch T, Zhang S, Gao L, Xu X, Hortschansky P, Ye J, Zhang F, Xu K, Gsaller F, Straßburger M, Binder U, Heinekamp T, Brakhage AA, Haas H, Lu L. 2018. The Zn(2)Cys(6)-type transcription factor LeuB cross-links regulation of leucine biosynthesis and iron acquisition in Aspergillus fumigatus. PLoS Genet 14:e1007762. doi: 10.1371/journal.pgen.1007762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Huang W, Shang YF, Chen PL, Gao Q, Wang CS. 2015. MrpacC regulates sporulation, insect cuticle penetration and immune evasion in Metarhizium robertsii. Environ Microbiol 17:994–1008. doi: 10.1111/1462-2920.12451. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw Illumina sequencing data were uploaded in the SRA with accession numbers SRR12338578 to SRR12338580. A processed format of the RNA-seq data sets is included in Data Set S1.