

Metallo-β-lactamases (MBLs) are a growing clinical threat because they inactivate nearly all β-lactam-containing antibiotics, and there are no clinically available inhibitors. A significant number of variants have already emerged for each MBL subfamily.
KEYWORDS: IMP, S262G, V67F, beta-lactamase, carbapenemase, carbapenems, imipenemase
ABSTRACT
Metallo-β-lactamases (MBLs) are a growing clinical threat because they inactivate nearly all β-lactam-containing antibiotics, and there are no clinically available inhibitors. A significant number of variants have already emerged for each MBL subfamily. To understand the evolution of imipenemase (IMP) genes (blaIMP) and their clinical impact, 20 clinically derived IMP-1 like variants were obtained using site-directed mutagenesis and expressed in a uniform genetic background in Escherichia coli strain DH10B. Strains of IMP-1-like variants harboring S262G or V67F substitutions exhibited increased resistance toward carbapenems and decreased resistance toward ampicillin. Strains expressing IMP-78 (S262G/V67F) exhibited the largest changes in MIC values compared to IMP-1. In order to understand the molecular mechanisms of increased resistance, biochemical, biophysical, and molecular modeling studies were conducted to compare IMP-1, IMP-6 (S262G), IMP-10 (V67F), and IMP-78 (S262G/V67F). Finally, unlike most New Delhi metallo-β-lactamase (NDM) and Verona integron-encoded metallo-β-lactamase (VIM) variants, the IMP-1-like variants do not confer any additional survival advantage if zinc availability is limited. Therefore, the evolution of MBL subfamilies (i.e., IMP-6, -10, and -78) appears to be driven by different selective pressures.
INTRODUCTION
Antibiotic resistance is a continuing clinical concern (1). Based on a report from the CDC, there are 2.8 million infections from antibiotic-resistant bacteria in the United States each year, with about 35,000 deaths from these infections (2). The most abundant class of clinical antibiotics is the β-lactam-containing compounds, which include penicillins, cephalosporins, and last-resort drugs carbapenems (3). Production of β-lactamases is the most common bacterial mechanism to counteract β-lactams, which hydrolyze the β-lactam ring, rendering these drugs inactive (4). Currently, there are more than 2,800 distinct β-lactamases identified (5). The five β-lactamases currently of primary public health concern are Klebsiella pneumoniae carbapenemase (KPC), New Delhi metallo-β-lactamase (NDM), Verona integron-encoded metallo-β-lactamase (VIM), imipenemase (IMP), and oxacillinase-48-like carbapenemase (OXA-48) (6). NDM, VIM, and IMP belong to the B1 subclass of β-lactamases, and the enzymes belonging to the class B of β-lactamases are called metallo-β-lactamases (MBLs). These enzymes use Zn(II) at the active site for catalysis (7). To date, inhibitors for the MBLs that can be used clinically have not been reported (8).
Selective pressures are driving the evolution of resistance conferred by MBL genes: there are 29 clinical variants of NDM, 69 clinical variants of VIM, and 85 clinical variants of IMP as of May 2020 (9). While the increased use of β-lactam-based drugs is obviously a leading reason for the proliferation and diversification of β-lactamases, other selective pressures which may be driving the evolution of these enzymes are not fully understood. In the past, extensive studies on NDM variants (10–13) and VIM variants (14, 15) have reported specific insights enhancing our understanding of amino acid and substrate interactions in MBLs. Somewhat surprisingly, many NDM variants and at least one VIM variant have not evolved to process additional types of β-lactam substrates but rather to overcome zinc deprivation imposed by host defenses, indicating that zinc scarcity rather than substrate diversity is acting as the major selective pressure for NDM and VIM. For example, NDM variants that harbor M154L mutations have higher zinc binding affinities (10, 11, 13), and the H229R mutation in VIM-20 imparts increased thermostability at low Zn(II) concentrations (14). Understanding the selective pressures driving the evolution of B1 MBLs will inform drug design efforts. Toward that end, we investigate herein the properties of IMP variants, the third B1 MBL of major clinical importance.
IMP-1 was originally isolated from Serratia marcescens in Japan in 1991, and IMP-1 has been shown to hydrolyze all existing β-lactams except monobactams (16, 17). Since then, IMP-1 has spread widely and been found in Pseudomonas aeruginosa, Klebsiella pneumoniae, Acinetobacter baumannii, and Alcaligenes xylosoxidans (18–21). IMP-1 is encoded in both plasmids and integrons, and these highly mobile genetic elements are believed to be responsible for the rapid spread of IMP-1 in clinical isolates (22). Full-length IMP-type enzymes usually consist of 246 amino acid residues, while the mature periplasmic IMP-1 protein contains 228 residues and has a molecular mass of ca. 25 kDa (22). IMP-1 has been characterized through steady-state kinetics, stopped-flow kinetic studies, spectroscopic studies, and X-ray crystallography (23, 24). A phylogenetic tree based on the amino acid sequences of 85 IMP variants (Fig. S1) shows significant diversity among the IMP-type MBLs. The largest IMP-type subfamily is the IMP-1 cluster, which contains 20 members, and this subfamily appears distinct, suggesting different phylogenetic origins (22). At least five clinical variants from the IMP-1 cluster (IMP-6 [25], IMP-10 [26], IMP-25 [27], IMP-30 [28], and IMP-46 [29]) have been characterized, but extensive studies done in parallel with the entire IMP-1 cluster of variants have not been previously reported.
In this analysis, we provide an extensive comparison of variants within the IMP-1 cluster, completed in parallel. First, antibiotic susceptibility studies were conducted under standard conditions and under conditions with low zinc availability to better mimic infection sites, with each strain challenged by a panel of β-lactam drugs with various scaffolds. These tests reveal the overall impact of amino acid differences among these variants on resistance (Fig. 1). Several variants were shown to exhibit increased MIC values specifically toward the carbapenems (meropenem and ertapenem). Among these variants, the biochemical and biophysical properties of purified IMP-6, IMP-10, and IMP-78 were further compared to IMP-1 using native mass spectrometry (MS) and steady-state kinetics along with molecular modeling to understand the molecular mechanisms of increased resistance. Unlike previous findings with NDM variants and VIM-20, the IMP-1 cluster does not appear to be evolving in response to the selective pressure of zinc scarcity but rather in response to the structural differences introduced by β-lactam drug variants, most specifically meropenem and ertapenem. Despite sharing similar enzymatic mechanisms and encountering similar host defenses, the evolution of MBL subfamilies appears to be driven by different selective pressures.
FIG 1.
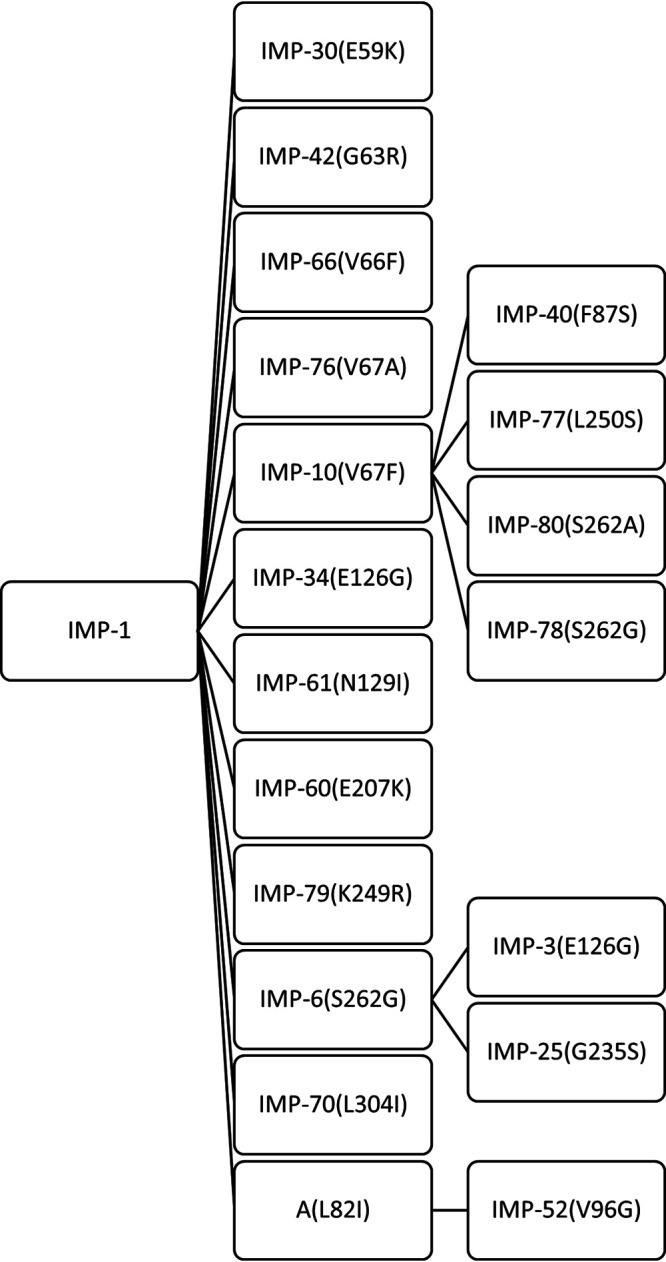
Mutated residues in clinically derived IMP-1 gene subfamily. In total, 20 members of IMP-1-like variants were selected from IMP-1 to IMP-80. The amino acid changes acquired with respect to the sequence of the ancestor are indicated in the branches (the standard numbering scheme for class B β-lactamases were used in this study [72]). IMP-A, which is not a clinical variant, was generated in order to construct double mutant IMP-52. This figure does not describe ancestral relationships but rather illustrates the construction method for the expression plasmids used here.
RESULTS
MIC studies under standard conditions.
E. coli DH10B strains harboring bla genes encoding IMP-1-like variants (Fig. 1) were used to assess the impact of substitutions on phenotype in antibiotic susceptibility studies. IMP-55 was excluded from our analysis because it contains more than 2 amino acid substitutions. Initially, we determined MIC values of these variants using standard conditions against a panel of three structural classes of β-lactams, including a penicillin (ampicillin), two cephalosporins (ceftazidime and cephalothin), and three carbapenems (imipenem, meropenem, and ertapenem) (Fig. 2). Not surprisingly, all strains containing IMP-1-like variants exhibited increased resistance to all tested antibiotics compared to that of strains harboring the empty vector (Table S1).
FIG 2.

Heat maps of fold changes in MIC values against a panel of antibiotics representing three structural classes of β-lactams. Heat map intensities for each family represent the fold change in MIC (see Tables S1 and S2) compared to that of the parent family member in standard or zinc-limited conditions.
To test whether different expression levels of clinical variants could have affected the MICs, immunoblots of the variants were obtained using an IMP-1-specific antibody (30). The amount of protein detected by the antibody using whole-cell extracts showed slight variability in expression but did not correlate with ampicillin, ceftazidime, cephalothin, imipenem, meropenem, or ertapenem MICs under standard or zinc-limited conditions. However, two interesting features were observed: (i) the IMP-60 variant was expressed at a lower level than the others, and (ii) IMP-30 migrated slightly faster than the other clinical variants in the SDS-PAGE gel (Fig. 3A and B). Each of these variants contains a Glu to Lys substitution (at different sites) and provides less resistance, but due to observed size or intensity differences, we did not interpret the direct cause to necessarily result from a change in enzyme activity caused by the substitution. Detection of IMP-60 was below the sensitivity of the antibody, and detection was only possible using increasing amounts of whole-cell culture (Fig. S4). qRT-PCR confirmed low-level RNA expression of IMP-60 (Fig. S5).
FIG 3.
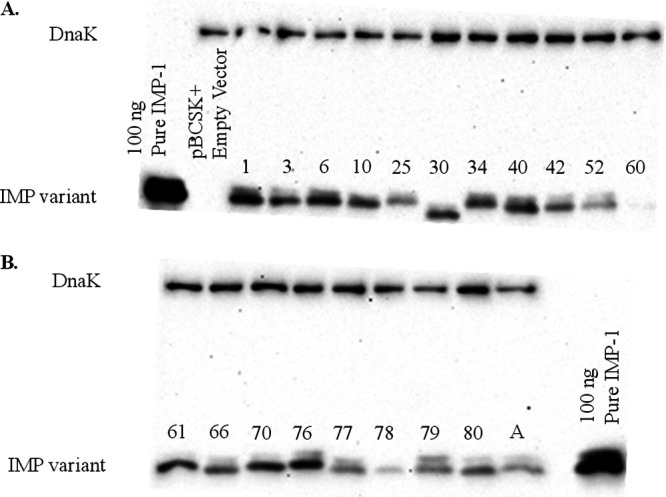
(A) Immunoblot of IMP variants investigated (IMP-1, -3, -6, -10, -25, -30, -34, -40, -42, -52, and -60). (B) Immunoblot of IMP variants investigated (IMP-61, -66, -70, -76, -77, -78, -79, -80, and -A). Both blots were prepared and exposed at the same time under the same conditions. DnaK was used as a constitutively expressed control. The IMP-60 variant was expressed at a lower level than the others. Low level expression of IMP-60 was further verified by RT-PCR (Fig. S5).
To facilitate visualization of the MIC data, a heat map was generated (Fig. 2). In this map, the fold changes in MIC values for the strains containing IMP-1 like variants were determined by normalizing to the MIC values of strains containing the parent IMP-1 gene. With ampicillin, strains with IMP-40 (V67F, F87S) and IMP-78 (V67F, S262G) exhibited 5-fold decreases in resistance. Strains with IMP-3 (S262G, E126G), IMP-6 (S262G), IMP-25 (S262G, G235S), IMP-76 (V67A), and IMP-80 (V67F, S262A) exhibited 4-fold decreases in resistance. Strains with IMP-10 (V67F) and IMP-77 (V67F, L250S) exhibited 3-fold decreases in resistance. The strain with IMP-66 (V66F) exhibited a 2-fold decrease in resistance. With cephalosporins, there are small differences in MIC values of the strains containing the variants compared to those of the strains containing IMP-1, with the exception of strains expressing IMP-3 (S262G, E126G) and IMP-6 (S262G), which exhibited 2-fold reduced resistance toward ceftazidime compared to that of the strain with the parent IMP-1 MBL. With imipenem, strains with IMP-3 (S262G, E126G) and IMP-6 (S262G), IMP-25 (S262G, G235S), and IMP-78 (V67F, S262G) exhibited 3-fold decrease in resistance. The strain with IMP-76 (V67A) exhibited 2-fold decreases in resistance.
With meropenem, the strain with IMP-78 (V67F, S262G) exhibited a 4-fold increase in resistance. Strains with IMP-3 (S262G, E126G), IMP-10 (V67F), IMP-25 (S262G, G198S), IMP-40 (V67F, F87S), IMP-66 (V66F), IMP-77 (V67F, L250S), and IMP-80 (V67F, S262A) exhibited 2-fold increases in resistance. With ertapenem, the strain with IMP-78 (V67F, S262G) exhibited a 4-fold increase in resistance. Strains with IMP-3 (S262G, E126G), IMP-6 (S262G), IMP-10 (V67F), IMP-25 (S262G, S235S), IMP-40 (V67F, F87S), and IMP-77 (V67F, L250S) exhibited 3-fold increases in resistance. Strains with IMP-61 (N129I), IMP-76 (V67A), and IMP-80 (V67F, S262A) exhibited 2-fold increases in resistance. In general, all of the strains exhibited decreased resistance toward ampicillin. Strains of IMP-1-like variants harboring S262G or V67F substitutions exhibited increased resistance toward meropenem and ertapenem compared to that of the strain containing the IMP-1 parent. Significantly, strains expressing IMP-78 exhibited the largest MIC changes compared to IMP-1. This result strongly suggests that strains containing IMP-78 (S262G/V67F) have evolved to better overcome treatment with carbapenem antibiotics.
MIC studies under Zn(II)-limiting conditions.
Previously, we found clinical variants of NDM and VIM that specifically increase antibiotic resistance when zinc availability is limited (10, 11, 13, 14). Therefore, we tested for a similar effect by determining MIC values for strains expressing IMP-1-like variants in the presence of EDTA against the same panel of antibiotics (Table S2). Unlike strains expressing NDM-1 and VIM-2 variants, the MIC values of strains harboring the IMP-1-like variants determined in the presence of EDTA were not substantially lower than those determined using standard conditions (Table 1). For example, the strain with IMP-1 exhibited a decrease by at least one serial dilution in resistance with ampicillin under Zn(II)-limiting conditions compared to standard conditions. Comparatively, strains with NDM-1 or VIM-2 exhibit decreases by seven or six serial dilutions, respectively. Similar results were also observed when using other substrates.
TABLE 1.

Abbreviations: AMP, ampicillin; EDTA, ethylenediaminetetraacetic acid; CAZ, ceftazidime; CF, cephalothin; IMI, imipenem; MER, meropenem; ERT, ertapenem; N.D., not determined.
Values are color coded to more easily visualize changes according to the following scheme: no coloration for little change; light, medium, and dark coloration for decreasing by approximately 2, 4, and ≥6 serial dilutions.
A heat map depicting the fold change for MIC values was generated from raw data as described above for the MIC values determined under standard conditions (Fig. 2). In general, the fold changes of strains with IMP-1-like variants were similar in standard and Zn(II)-limited conditions. Unlike most NDM variants and VIM-20, the IMP-1 like variants do not confer any additional survival advantage that is only observed when zinc availability is limited.
Overexpression, purification, and characterization of IMP-1-like variants.
Antibiotic susceptibility studies of the entire IMP-1 like cluster revealed a number of variants notable for their increased resistance toward carbapenems under standard conditions. In particular, IMP-6 (S262G) and IMP-10 (V67F), which have single mutations that are found together in IMP-78 (S262G/V67F), exhibited increased resistance. The S262G substitution is also found in IMP-3 and IMP-25, and the V67F substitution is also found in IMP-40, IMP-77, and IMP-80, but the combination of both S262G and V67F in IMP-78 achieved the greatest difference in resistance with meropenem and ertapenem. To better understand the origin of increased resistance, IMP-1, IMP-6, IMP-10, and IMP-78 were overexpressed and purified. The yields for tag-free IMP-1, IMP-6, IMP-10, and IMP-78 were 20.9, 11.0, 10.9, and 6.7 mg of protein/liter of LB medium, respectively. The homogeneity of the protein preparations was shown to be over 95% by SDS-PAGE (Fig. S2).
The predicted molecular weights for tag-free IMP-1, IMP-6, IMP-10, and IMP-78 are 25,438.09 Da, 25,408.07 Da, 25,486.14 Da, and 25,456.11 Da, as determined by the ExPASy-Comupte pI/MW tool (31). Native mass spectrometry gave the following molecular weights for IMP-1, IMP-6, IMP-10, and IMP-78: 25,565.45 Da, 25,538.18 Da, 25,617.27 Da, and 25,605.45 Da, respectively. The molecular weights determined via native mass spectrometry gave values similar to the theoretical values of IMP with two Zn(II) ions bound (Fig. S3).
Steady-state kinetics.
The effect of amino acid substitutions on the steady-state kinetics for substrate hydrolysis was determined using purified enzymes and common reaction conditions for four variants related to the most resistant variant (IMP-78): IMP-1 (reference sequence), IMP-6 (S262G), IMP-10 (V67F), and IMP-78 (V67F, S262G). Changes in the values with respect to IMP-1 were color coded to show improvement (defined as increased kcat, decreased Km) or worsening (decreased kcat, increased Km) of these values (Table 2). The S262G substitution increased kcat values for cephem substrates (ceftazidime and cephalothin) and also for carbapenems (imipenem, meropenem, and ertapenem), although to a lesser degree. The Km values were increased for cephems and also for one of the carbapenems (imipenem). These changes often offset each other so that the kcat/Km ratio was intermediate between increased, decreased, and not-changed results, depending on the individual substrate. The V67F substitution (IMP-10) showed similar effects, with increases in kcat values for cephem and carbapenem substrates and increases in Km values for most substrates. These combined to result in a more uniform increase in kcat/Km values for most cephem and carbapenem substrates (except imipenem). Although not an additive effect, similar impacts on kcat, Km, and kcat/Km values were observed for cephem and carbapenem substrates when the two substitutions were combined in IMP-78 (V67F, S262G). The largest single improvements in catalysis were observed for kcat of ceftazidime by IMP-78 (∼20-fold) and for kcat/Km of meropenem by IMP-6 (∼4-fold). Unexpectedly, catalysis of ampicillin hydrolysis was significantly worse by all of the variants. The S262G substitution (IMP-6) showed ∼10-fold decrease in kcat, although Km was maintained. The V67F substitution did not exhibit saturation kinetics, so the kcat/Km value was calculated directly and showed an ∼25-fold decrease. The effects were similar (although not additive) when combined in IMP-78, which has the largest decrease in kcat/Km (∼40-fold).
TABLE 2.
Kinetic parameters for antibiotic hydrolysis by IMP variantsa

Values are color coded to more easily visualize changes according to the following scheme: no coloration for little change; light, medium and dark coloration for approximately 2-, 5-, and ≥10-fold changes, with red hues for an improvement in values (increased kcat, decreased Km) and blue hues for the opposite trend.
Inhibition experiments with EDTA.
As a tool to assess zinc affinity of the variants, the half-maximal inhibitory concentration (IC50) values for EDTA inhibition of purified NDM-1, VIM-2, and IMP-1 were determined as 14 ± 1 μM, 200 ± 30 μM, and 170 ± 20 μM, respectively, all using the same experimental procedures and time constraints. These IC50 values are not interpreted as dissociation constants but rather as a gauge for ranking the relative affinities of the zinc ions required for catalysis by each of the three enzymes, clearly indicating that NDM-1 is much more susceptible to metal chelation than are IMP-1 or VIM-2.
Equilibrium dialysis.
To further determine if the differences in MIC and IC50 values with NDM-1, VIM-2, and IMP-1 were caused by different zinc affinities, equilibrium dialysis studies were conducted (Fig. 4). NDM-1 activity is very susceptible to EDTA. Metal analyses of the NDM-1 showed that the enzyme bound 1.7 equivalents (eq) of Zn(II) before EDTA is introduced. The zinc content decreased to 0.32 equivalents in the presence of 256 μM EDTA. Metal analyses indicate that VIM-2 has a third Zn(II) binding site in addition to the two Zn(II) ions at the active site, consistent with observations reported previously by us and other groups for VIM isoforms (14, 32, 33). The zinc content dropped from 2.60 eq to 0.99 eq when 256 μM EDTA was present. In the case of IMP-1, the zinc content dropped from 1.75 eq (in the absence of EDTA) to 1.31 eq when 256 μM EDTA was present. These results indicate that IMP-1 binds Zn(II) most tightly, followed by VIM-2, and then NDM-1, which has the weakest affinity.
FIG 4.
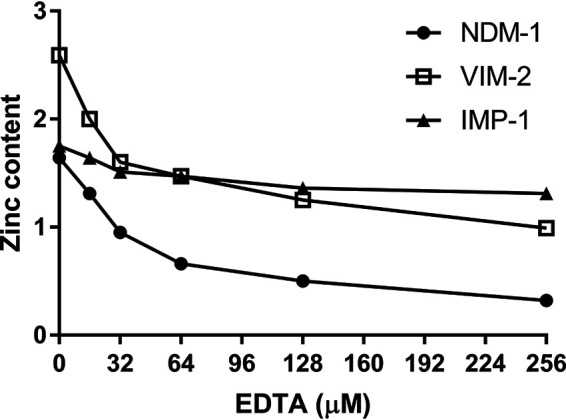
Metal content of NDM-1, VIM-2, and IMP-1 (8 μM) after dialysis with various concentrations of EDTA in 50 mM HEPES (pH 7.5) buffer.
Variant ΔΔG calculations.
To assess possible effects of IMP variant mutations on protein stability, we utilized Rosetta ddg via the ddg_monomer application (34) within the Rosetta software suite. As reported in Table S4, many variants exhibited reductions in stability compared to that of IMP-1, as measured in Rosetta energy units (REU). Rosetta employs multiple energy functions resulting in potentials that are different from conventional kJ mol−1 or kcal mol−1 units determined in physical experiments. As a result, the Rosetta suite utilizes REUs which are energies on an arbitrary scale. A study by Kellogg et al. 2010 (34) provides a thorough treatment of the relationship between REU values and actual energy values of kJ mol−1. Interestingly, while the V67F mutation found in IMP-10 resulted in a destabilization of 17.15 REU, adding in the S262G mutation to the V67F background stabilizes the protein by nearly 10 REU. Although still less stable than IMP-1, the S262G mutation may allow for additional flexibility near the active site, which could result in compensatory changes that aid in accommodating the disruptive V67F mutation. In comparison to IMP-1, IMP-6 harbors only the S262G mutation and exhibits a minor degree of stabilization by 0.63 REU. The lack of a corresponding substantive increase in stability, as seen with S262G in the V67F background, suggests that S262G is influential as a compensatory mutation but does not offer significant advantages in isolation.
Energy minimization of IMP variants with docked ligands.
A structure of IMP-1 (35) with bound zinc but no ligands bound at the active site was used as the basis for generating an IMP-1 model in SWISS-MODEL so as to generate a model with optimized geometry and limited clashes. Remaining clashes in the model were resolved using steepest descent (SD) and adopted basis Newton-Raphson (ABNR) minimization. Zn ions within the IMP-1 model were maintained within the active site using harmonic restraints with a sufficiently large force constant of 50 kcal/mol. The resulting IMP-1 model was used for making amino acid substitutions to produce models of the variants IMP-6, IMP-10, and IMP-78. Models of IMP-1, IMP-6, IMP-10, and IMP-78 with ampicillin or meropenem bound were generated by superimposing crystal structures of IMP-1:ampicillin (36) or IMP-1:meropenem (36) complexes onto the energy-minimized models, placing the ligands into the models, and minimizing the complexes with SD and ABNR, holding both the Zn(II) ions and ligand in place using harmonic restraints, to allow adjustment of the protein due to the presence of the ligand. The resulting IMP variant structures exhibit variations in position of the ligands within the active site, along with displacements of the β-hairpin loop (Fig. 5 and Table S5). The displacement of the β-hairpin loop and subsequent changes of protein-antibiotic/protein-zinc interactions were correlated with increasing activity against meropenem or ampicillin. Positioning of the β-hairpin loop is more greatly displaced in complexes with meropenem, which appears necessary to accommodate the larger substituent of meropenem. In comparison to the variation of β-hairpin loop positioning, which appears to influence the ability of Trp64 to serve as an active site cap, the positions of Val/Phe67 and Ser/Gly262 are nearly identical within each of the meropenem- or ampicillin-bound families of models, although differences in position of Val/Phe67 but not of Ser/Gly262 are observed across models from both families of complexes. Structural changes in the β-hairpin loop alter the position of the ligand in the active site. Meropenem binding to the IMP variants results in stronger ligand-protein interactions in comparison to IMP-1. In contrast, ampicillin binding displays simultaneously increased ligand-protein interactions and decreased ligand-zinc interactions.
FIG 5.
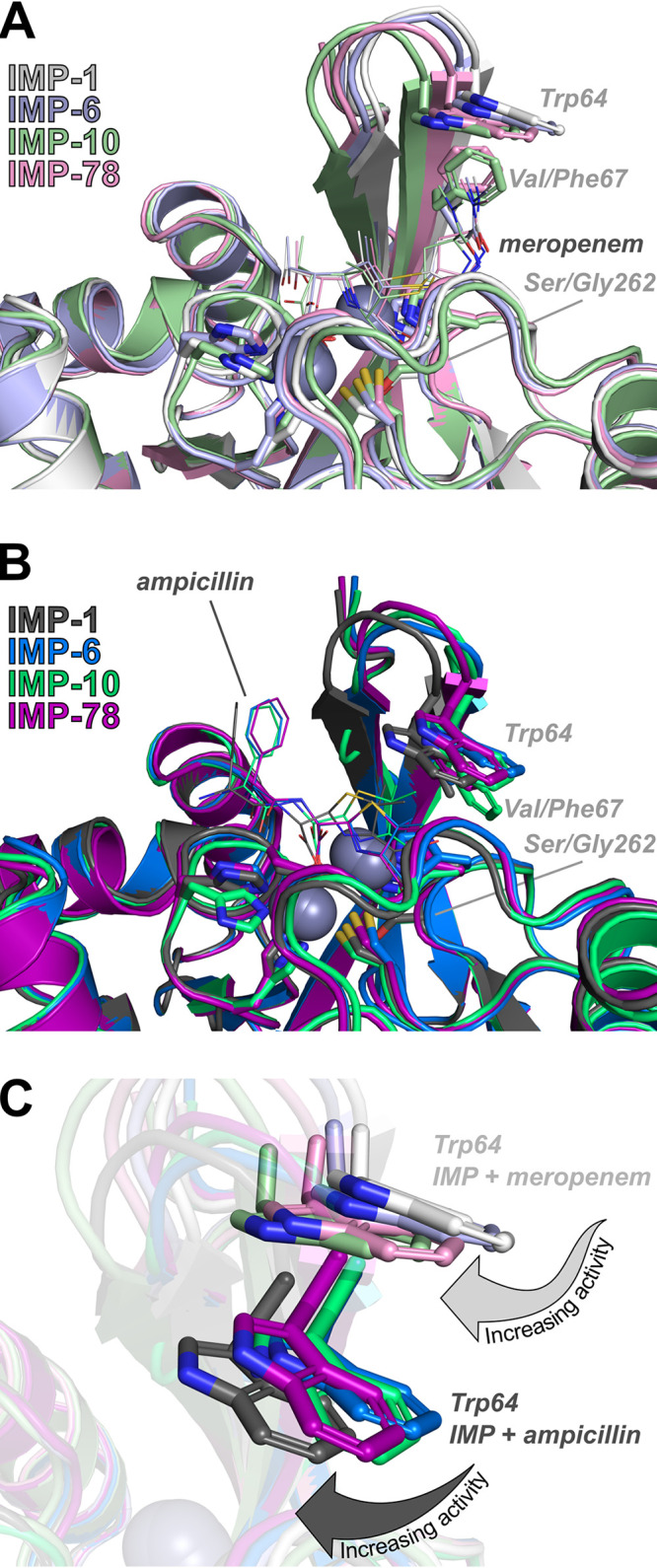
Role of IMP-78 S262G and V67F mutations. Energy-minimized models of complexes with hydrolyzed meropenem (A, lines) and hydrolyzed ampicillin (B, lines) for IMP-1, IMP-6, IMP-10, and IMP-78. Active site residues (sticks) coordinating zinc ions (spheres) are shown along with Trp64, Val/Phe67, and Ser/Gly262 (sticks). An overlay of energy-minimized models for all complexes with meropenem or ampicillin highlights changes in position and side chain rotamers for Trp64 (C).
DISCUSSION
A phylogenetic reconstruction using all available blaIMP protein sequences in GenBank illustrates significant sequence diversity within the IMP family of metallo-β-lactamases (Fig. S1). Previously, we used comparative analysis of clinical variants within other metallo-β-lactamase families to help identify which selective pressures are driving the evolution of these zinc-dependent enzymes. Somewhat surprisingly, we (and others) found that many NDM variants did not show significant differences from one another when resistance to a panel of structurally diverse β-lactams was measured under standard conditions (10, 11, 13). However, when we limited zinc availability to better mimic conditions found at infection sites, many NDM variants showed increased resistance, suggesting that these metalloenzymes evolved to overcome the dual selective pressures of β-lactam drugs and zinc scarcity imposed by the innate immune response. While not as widespread in the VIM family of metallo-β-lactamases, the variant VIM-20 also shows increased resistance in comparison with that in other variants when zinc availability is limited, suggesting that zinc scarcity may be a selective pressure driving the evolution of many metallo-β-lactamases (14). Here, we focused on the IMP family of metallo-β-lactamases and analyzed the largest phylogenetic cluster, which contains 20 clinical IMP variants, in parallel. This cluster contains IMP-1, which we use as the reference protein sequence, 11 variants encoding a single amino acid difference, and 7 variants containing two amino acid differences. IMP-55 is excluded from this study because it has five amino acid differences and will likely require a more complex analysis.
Determination of MIC values for structurally diverse β-lactam drugs containing penam (ampicillin), cephem (ceftazidime, cephalothin), and carbapenem (imipenem, meropenem, ertapenem) core structures under standard assay conditions revealed significant differences in resistance conferred by this set of variants (Fig. 2). Two variants (IMP-30, -60) showed less resistance against most β-lactams than did the others. However, these two variants showed either greatly decreased protein levels (IMP-60) or a smaller than predicted size (IMP-30) as gauged by SDS-PAGE and immunoblotting (Fig. 3A and B). The expression and maintenance of IMP-30 may be better accommodated in different bacterial strains and so were not considered further in our analyses. Despite low level expression of IMP-60, as revealed by immunoblotting and qRT-PCR (Fig. S4 and S5), we observe significant levels of resistance to ampicillin, ceftazidime, and cephalothin. We suspect that this variant, with an E207K substitution, may hydrolyze these substrates differently than carbapenems and may proceed through a different catalytic pathway. The IMP-42 variant showed slightly decreased resistance in comparison to that of IMP-1 for several antibiotics and a slight increase in resistance to ceftazidime, suggesting that this variant either may be evolving increased substrate selectivity or may instead represent a possible ancestor for the improved resistance found in IMP-1. Several other variants (IMP-34, -61, -52, -70, -79) showed only minor differences in MIC values for 2 or fewer β-lactam substrates, displaying resistance patterns very similar to those of IMP-1. The largest subset, consisting of the remaining variants (IMP-3, -6, -10, -25, -40, -66, -76, -77, -78, -80), shows a surprisingly similar pattern of changes in resistance with respect to those of IMP-1 despite the diversity of amino acid substitutions and locations. With respect to IMP-1, all of these variants contain either the V67F or S262G substitution, and all of these variants show improved resistance against the carbapenems meropenem and ertapenem and a marked decrease in resistance against the penam ampicillin. Changes in resistance to cephems (ceftazidime and cephalothin) as well as to imipenem (see below) are less uniform among the variants. From this set of results, it is clear that most IMP variants in this cluster appear to be evolving to counter the selective pressure provided by the carbapenem drugs meropenem and ertapenem. Particularly, the V67F and S262G substitutions appear to be key drivers of increased resistance in this IMP cluster. This conclusion is quite different from that reached for the NDM family of metallo-β-lactamase variants, which do not show any obvious differences in resistance patterns under standard conditions.
Next, we considered changes to resistance patterns when zinc availability was limited to better match conditions at infection sites. Compared to that of NDM-1 and VIM-2, the resistance afforded by IMP-1 is much less affected by zinc scarcity, showing only minor weakening of resistance upon zinc sequestration (Table 1). When the IMP variants in this cluster are compared to each other under these conditions (Fig. 2), they show an even more uniform pattern of resistance, with the only occurrences of higher resistance restricted to the carbapenems meropenem and ertapenem and a more widespread loss of resistance to ampicillin and the cephems. In contrast with our previous studies of NDM variants, no new instances of enhanced resistance are revealed by using conditions of limited zinc availability that were not already observed under standard assay conditions. This set of experiments indicates that the evolution of the largest set of clinical IMP variants is likely not driven by zinc scarcity but rather by selective pressure enforced by structurally different β-lactam drugs, predominantly the carbapenems meropenem and ertapenem for which the majority of variants provide increased resistance.
To better understand the mechanisms behind increased resistance in this IMP cluster, we chose to study a purified variant that has the largest increase in resistance to meropenem and ertapenem and combines the two most predominant mutations associated with this phenotype: IMP-78 (V67F, S262G). For comparison, we also used the reference enzyme IMP-1 and the variants representing the single mutants that comprise IMP-78: IMP-6 (S262G) and IMP-10 (V67F). The residue at position 67 is located within the active site and provides a conserved hydrophobic patch at the base of a substrate-binding loop. The residue at position 262 is slightly more distant, and although residue 262 buttresses the active site, it does not make direct contact with substrates.
Kinetic parameters and molecular mechanisms have been studied previously in the single mutants IMP-6 (25, 37–40) and IMP-10 (26, 30), and this study is the first analysis of their combination in IMP-78. We found that the four variants all expressed to similar levels and were each purified containing approximately two equivalents of bound zinc ions (Fig. S3). Except for ampicillin, the kcat values for all substrates were greater in IMP-6, -10, and -78 than in IMP-1, in most cases with IMP-78 displaying the highest value. Notably, kcat,IMP-78 for ceftazidime was ∼20-fold higher. Except for in a few cases, the higher kcat values for these substrates were almost always offset by an accompanying increase in Km values, with the most notable being a 10-fold higher Km,IMP-78 for imipenem. When kcat/Km is considered, these values then reflect a mixed impact of the mutants on this panel of substrates, in general resulting in modest increases in activity for cephems and carbapenems, with imipenem as an exception (see below). Due to differential drug accumulation and high enzyme concentrations in the periplasm, it is difficult to predict a priori which steady-state parameters will best predict resistance in MIC studies. Here, we see that neither kcat nor kcat/Km accurately predicts each difference observed in MIC values, but kcat/Km is the value that best reflects the pattern of the most significant changes in the variants’ resistance for penams and carbapenems in microbial culture. Notably, while kcat/Km,IMP-78 values for cephems and carbapenems increased, the kcat/Km,IMP-78 for imipenem decreased slightly, and that for ampicillin decreased by 97%. Similar differences for penam and carbapenem substrates were reflected in the MIC data (Fig. 2), suggesting that changes in catalytic efficiency of these variants may be the main factor driving changes in resistance and that the corresponding loss of ampicillin activity might be mechanistically linked.
Comparison of the kinetic parameters for IMP-1, IMP-6 (S262G), and IMP-10 (V67F) with previously published data does show some differences from those measured in different laboratories, but most of the general trends are quite similar, notably the worsening of kinetic parameters for ampicillin along with improvements for the carbapenems meropenem and ertapenem (25, 26, 30, 37–40). The reproducibility of the observed trends among different investigators and our observation of similar trends in MICs for most of the variants within this cluster adds confidence to our interpretation that selective pressure provided by the carbapenems such as meropenem and ertapenem are driving the evolution of these variants and that ampicillin is not.
Market data from Japan, where most of these IMP variants were first isolated (Table S3), indicate that sales of both meropenem (2-fold) and ampicillin (2.2-fold) increased from 2004 to 2016 (41). This raises the question of why blaIMP may be evolving in response to meropenem but not to ampicillin. One possibility is that other resistance determinants may coexist in complex carbapenem-resistant infections. For example, blaIMP genes often coexist with blaOXA and blaCTX-M genes (42, 43). OXA-type carbapenemases and CTX-M are quite diverse but generally hydrolyze ampicillin, with lower activities against imipenem and meropenem (44, 45). Coexpression of OXA-type carbapenemases would be expected to reduce these selective pressures on blaIMP evolution enabling IMP to evolve into a “specialist enzyme” that is more selective for meropenem and ertapenem. In the same market study, meropenem was the only carbapenem listed in the top 10 parenteral agents in 2016 (8% total antimicrobial consumption) (41), consistent with this antibiotic providing a driving selective pressure.
Another question that emerges from our analysis of this IMP cluster is why ampicillin activity (and often imipenem activity) is lost in concert with gains in activity for meropenem and ertapenem. Are these changes mechanistically linked? Lowered ampicillin kcat/Km values stem from both lowered kcat and raised Km values, suggesting that both catalysis and binding may be impaired in the IMP-6, -10, and -78 variants, and the loss in imipenem kcat/Km appears to not be driven by kcat but rather by raised Km, possibly again indicating impaired binding. A simple hypothesis is that weakening the interactions between ligand and enzyme can help to alleviate hurdles in reaction coordinate, such as a slowly released product or an intermediate in the reaction that has a rate-limiting decay. In the case of low Km substrates (meropenem and ertapenem), this weakening of interactions may help to speed product release or intermediate decay. However, for high Km substrates (ampicillin), a further loss in affinity would more adversely affect catalysis. LaCuran et al. propose a compelling explanation for IMP-10 (V67F) in which mutation of this active site residue better accommodates the side chains of meropenem, bound in its neutral form, but introduces interactions that disfavor binding of ampicillin or imipenem (in its cationic form, due to the higher pKa of its formamidine substituent) (30). This mechanism is consistent with our observation that throughout the IMP-1 cluster of variants, gains in resistance (and kcat/Km values) toward meropenem are most often linked to decreased activity toward ampicillin and imipenem. Saturation mutagenesis at this site also reflects this sensitivity, where only Val and Ile were tolerated when libraries were selected using ampicillin (46). As noted previously for IMP-6 (46), IMP-78 and similar enzymes in this cluster appear to be evolving as specialist enzymes, with specialization perhaps enabled by coexistence of other resistance determinants.
The more distant S262G substitution (IMP-6) appears to have a very similar effect on steady-state kinetic parameters, resulting in increased kcat/Km for meropenem and ertapenem but notable decreases for ampicillin. Oelschlaeger et al. provide a “domino effect” model whereby the distant change at position 262 is transferred to changes in flexibility at the active site, destabilizing the Zn2 ligand His250 (38). Changes in activity are predicted to be substrate-dependent due to differences in interactions between β-lactam substituents and the binding site and the differential ability of Zn2 to stabilize the anionic reaction intermediate proposed for this enzyme. Although these mutations are found on different sides of the protein’s αββα sandwich domain, we find here that the functional result of combining S262G with V67F is less than quantitatively additive, indicating that the impact on the active site is either not entirely complementary or that there is a ceiling for improvements through these mechanisms.
The activity assays results are interesting when interpreted in light of the estimated stabilities generated by Rosetta ddG. While IMP-6 is predicted to be slightly more stable than IMP-1 and significantly more stable than IMP-10 and IMP-78, it exhibits the smallest increase in activity against meropenem, suggesting that predicted stability of the enzyme is not necessarily directly linked to activity. In comparison to the results with meropenem, differences in predicted stabilities relative to IMP-1 of IMP-6, IMP-10, and IMP-78 do not yield a consistent trend. Together, these results suggest that while the V67F and V67F/S262G mutations found in IMP-10 and IMP-78 may perturb the structure of the variant, relative to IMP-1, these perturbations are not so severe as to substantively reduce the effective amount of enzyme and decrease activity across the board. Interestingly, consistent with the Oleschlaeger et al. domino effect, changes in flexibility for IMP-6, IMP-10, and IMP-78, which could be manifested as potential reductions in predicted stability, may allow for conformations that are optimized for catalyzing the hydrolysis of meropenem at the expense of ampicillin hydrolysis.
Further definition of the mechanism enabling enhanced meropenem hydrolysis at the expense of ampicillin hydrolysis is enabled by examining energy-minimized models for complexes of IMP-1, IMP-6, IMP-10, and IMP-78 with meropenem (Fig. 5A) or ampicillin (Fig. 5B). Energy-minimized model complexes with ampicillin or meropenem appear to identify a closing of the β-hairpin loop over the active site correlated with IMP variants that exhibit increased kcat values. Although variations in positioning of active site residues coordinating the zinc ions are observed across the entire family of models, movement of the β-hairpin to cap the active site is the sole structural change correlated with changes in kcat values. Interestingly, movement of the β-hairpin closer to the bound antibiotic also places Trp64 closer to the active site (Fig. 5C), potentially serving as a lid that constrains the position of the substrate for hydrolysis. In contrast, shifting Trp64 further from the active site could allow for greater movement of the substrate, possibly accommodating conformations that are nonproductive or less productive for catalysis. For example, IMP-10 exhibits the poorest performance against ampicillin, and Trp64 is rotated away, serving as a poorer cap to the active site. In contrast, IMP-1 exhibits the highest activity against ampicillin, and the Trp64 side chain is rotated toward ampicillin, while the β-hairpin pushes the entire residue closer. For meropenem IMP-10 and IMP-78 β-hairpins are closest to the meropenem, while the Trp64 side chains are rotated to close off the top of the active site. In comparison, shifting of the β-hairpins for IMP-6 and IMP-1 progressively pull Trp64 further from the meropenem, while the active site becomes more open due to rotation of the Trp64 side chain. Although changes in the position and orientation of Trp64 are due, in part, to changes in position of the β-hairpin backbone, changes in position and orientation for Trp64 are also aided by rotations around the χ1 side chain rotamer for complexes with ampicillin and changes in both χ1 and χ2 side chain rotamers for complexes with meropenem. As the β-hairpin loop closes and moves closer to the antibiotic in the active site, the position of the antibiotic within the active site is also shifted. This shift is more pronounced with ampicillin than with meropenem, as indicated by the larger root mean square division (RMSD) values of the ligand relative to IMP-1 (Table S5). The changes in position of the antibiotic can also be quantified by interactions within the active site. Using both ampicillin and meropenem, the interaction energies of the antibiotic with the protein and the antibiotic with the zinc ions were computed for IMP-1, IMP-6, IMP-10, and IMP-78 using the energy-minimized models. In the case of ampicillin binding, IMP-6 and IMP-78 interact over twice as strongly with ampicillin relative to IMP-1 at the expense of a 2-fold decrease in interaction with the zinc ions, while IMP-10 displays ampicillin interactions similar to those of IMP-1. In contrast, meropenem binding causes smaller overall changes in RMSD of the ligand in the active site than does IMP-1 (Table S5); however, these changes result in a stronger protein-meropenem interaction for all three mutants, while the meropenem-Zn(II) interactions are on the same order of magnitude as IMP-1. Interestingly, these results correlate well with the trends of the kinetic parameters and MICs that show improvements for meropenem and decreased activity toward ampicillin and may serve as an explanation of how the structural changes of the β-hairpin loop propagate to the active site to alter protein activity and resistance.
Through the parallel analysis of the structures and biochemical activities of clinical MBL variants, insights can be gained about the selective pressures driving the evolution of these resistance determinants. While NDM variants appear to be evolving to overcome limited zinc availability rather than changes in substrate structure, this cluster of IMP-1-like variants appears to be driven by the selective pressure of meropenem drug treatment. NDM appears to have a fairly uniform substrate selectivity among its variants, but the majority of the IMP-1-like variants appear to be evolving to be more specialist enzymes for particularly potent carbapenem drugs. The differences in each group’s response to zinc scarcity likely reflect attributes of the two enzymes rather than vastly different clinical environments. Soluble constructs of NDM-1 have a markedly weak affinity for Zn2 (dissociation constant [Kd] = 2 μM) (47) that is improved approximately 10-fold in clinical variants (10), but IMP-1 has a stronger affinity for zinc binding across the board (Kd,Zn1 < 60 nM and Kd,Zn2 = 0.3 μM) (48). Here, we find that both resistance (Table 1) and zinc binding (Fig. 4) are much less affected in IMP-1 by treatment with a strong chelator (EDTA) but NDM-1 resistance, activity (10), and zinc binding are severely compromised. Poor zinc affinity is not an inherent weakness found in IMP-1 and is therefore not reflected in the evolution of its variants. Regarding evolution as a specialist enzyme, it is not clear why this phenotype has emerged within IMP-1 cluster and not the more newly identified NDM family (49–52), but these results suggest that further monitoring of MBL clinical variants and study of the biochemical and biophysical bases of resistance determinants is warranted.
MATERIALS AND METHODS
Cloning of blaIMP-1-like variants for cell viability assays.
Coding sequences for IMP-1-like variants were generated by site-directed mutagenesis using nonoverlapping primers and the previously described vector pBC SK(+)-blaIMP-1 as the template (40). After PCR, the amplified genes were added directly to a kinase-ligase-DpnI enzyme mix (NEB) for rapid, room-temperature circularization and template removal. A 2-μl aliquot of the ligation mixture was used to transform 30 μl of Escherichia coli DH5α chemically competent cells (Lucigen), and the transformation mixtures were spread on lysogeny broth (LB) plates containing 50 μg/ml chloramphenicol. Plasmid DNA, purified from a recultured single colony, was used to sequence and confirm the insert DNA sequences (Eurofins Genomics). E. coli DH10B were transformed with plasmids encoding IMP-1-like variants for subsequent MIC measurements. Although IMP-55 is within the IMP-1 cluster, it is excluded from this analysis because it contains more than 2 amino acid substitutions (see below).
Minimum inhibitor concentration (MIC) measurements.
MIC measurements were performed in triplicate using the Mueller-Hinton (MH) agar dilution method according to the Clinical Laboratory and Standards Institute (CLSI) protocol (53). All cell viability assays were conducted in E. coli DH10B in order to provide a uniform genetic background in which to evaluate the variants. Briefly, bacterial cultures containing blaIMP-1 variants cloned into a uniform expression vector pBC SK(+) were grown overnight at 37°C in cation-adjusted Mueller-Hinton broth (CAMHB). The cultures were diluted, and a Steers replicator was used to deliver 10 μl of a diluted overnight culture containing approximately 104 CFU. MICs were determined for the following: meropenem (Fresenius Kabi), imipenem, ertapenem (Merck), ampicillin, cephalothin, and ceftazidime (Sigma-Aldrich). MICs were also determined (as described above) with the addition of 50 μM EDTA (Promega Corp.) to the Mueller-Hinton agar in order to evaluate antibiotic susceptibility under conditions where Zn(II) availability was low (10, 14). MIC values are reported in Tables S1 and S2. To facilitate interpretation of the MIC data, we generated a heat map. Heat map intensities were determined by determining the fold change of MIC for each IMP-1 variant compared to that of the MIC of the parent variant.
Immunoblotting.
Immunoblotting was used to evaluate the expression levels of wild-type and clinical variants of IMP-1. These experiments were done under conditions comparable to those in the susceptibility assays as previously described (30). Five-milliliter cultures of E. coli DH10B cells containing pBC SK(+) phagemids harboring the various blaIMP genes in Mueller-Hinton II broth containing 20 μg/ml chloramphenicol were grown at 37°C to an optical density at 600 nm (OD600) of 0.8. Fifty-microliter aliquots of whole cells from these cultures were pelleted and frozen overnight. Pellets were resuspended in 20 μl of loading buffer. Whole cells were then separated via SDS-PAGE and subsequently transferred to a polyvinylidene difluoride (PVDF) membrane (Novex, Life Technologies, Carlsbad, CA) by electroblotting. After blocking for 1 h with 5% nonfat dry milk, the presence of IMP was detected by incubation in 5% nonfat dry milk with anti-IMP-1 polyclonal antibodies (1 μg/ml) and anti-DnaK (Enzo Life Science, Farmingdale, New York) (1/10,000 dilution) overnight at 4°C. The membrane was washed four times, 15 min each, in Tris-buffered saline (pH 7.4) containing 0.1% Tween 20 and subsequently incubated in 5% nonfat dry milk with a 1/10,000 dilution of horseradish peroxidase (HRP)-protein G conjugate (Bio-Rad, Hercules, California) and 1/10,000 dilution of goat anti-mouse IgG-HRP (Santa Cruz Biotechnology, Santa Cruz, California). After four additional washes, the membrane was processed for exposure using the ECL kit (GE Healthcare, Chicago, Illinois) and FOTO/AnalystVR FX (Fotodyne, Hartland, Wisconsin). Additional immunoblotting was done with 50-μl, 100-μl, 200-μl, and 500-μl aliquots of whole cells expressing IMP-60.
qRT-PCR of IMP-60 in pBC SK(+).
RNA from mid-log-phase IMP-1 and IMP-60 clones was isolated with the Qiagen RNeasy kit (Qiagen, Venlo, Netherlands). RNA was Dnase-treated and confirmed to be DNA-free via normal PCR. Serial dilutions of RNA (500 ng to 7.8 ng) were used as the template in each reaction including a no-template control. Internal IMP primers and pBCSK(+) CAT primers were used to normalize expression. qRT-PCR was performed using the Bio-Rad Universal Sybr green kit and the CFX real-time PCR instrument (Bio-Rad, Hercules, California) following the manufacturer’s recommendations.
Cloning, overexpression, and purification of IMP-1-like variants.
An IMP-1-encoding DNA sequence, codon optimized for E. coli and corresponding to residues 19 to 246, was synthesized and inserted into a pUC57 vector by Genscript Biotech Corp. The insert was subsequently subcloned into a pET-28a(+)-TEV plasmid between NdeI/XhoI restriction sites to generate the overexpression plasmid pET-28a (+)-TEV-IMP-1. The expression plasmids for IMP-6 and IMP-10, which harbors the S262G and V67F mutations, respectively, were generated by site-directed mutagenesis using nonoverlapping primers and the pET-28a (+)-TEV-IMP-1 plasmid as the template. After the PCR, the amplified sequences were added directly to a kinase-ligase-DpnI enzyme mix (NEB) for rapid, room temperature circularization and template removal. A 2-μl aliquot of the ligation mixture was used to transform 30 μl of E. coli DH5α chemically competent cells (Lucigen), and the transformation mixture was spread onto a lysogeny broth (LB) plate containing 50 μg/ml kanamycin. Plasmid DNA, purified from a single colony, was used to sequence and confirm the inserted sequence (Eurofins Genomics). Plasmid pET-28a (+)-TEV-IMP-10 was subsequently used as the template to introduce the second mutation (S262G) to obtain plasmid pET-28a (+)-TEV-IMP-78.
Identical overexpression and purification procedures were utilized for IMP-1, IMP-6, IMP-10, and IMP-78 constructs. Each overexpression plasmid was transformed into E. coli express BL21(DE3) chemically competent cells (Lucigen), and the transformation mixture was plated on an LB agar plate containing 50 μg/ml kanamycin. A single colony was transferred into 50 ml of LB broth, containing 50 μg/ml kanamycin, and the culture was shaken overnight at 30°C. The overnight culture (10 ml) was transferred into 2 flasks with each containing 1 liter of LB with 50 μg/ml kanamycin. The resulting culture was grown at 37°C with a shaking speed of 220 rpm until an OD600 of 0.6 was reached. Protein production was induced by adding isopropyl-β-d-thiogalactopyranoside (IPTG; final concentration of 0.5 mM) to each culture while providing ZnCl2 (final concentration of 100 μM). The cell cultures were shaken overnight at 18°C, and the cells were harvested by centrifugation for 10 min at 8,000 × g. The resulting pellets were resuspended in 40 ml of 50 mM HEPES (pH 7.5) containing 500 mM NaCl. Cells were lysed by passing the mixture two times through a French press at a pressure between 15,000 to 20,000 lb/in2. The insoluble components were removed after pelleting in a centrifuge for 1 h at 32,000 × g. The resulting solution was mixed with imidazole (final concentration is 50 mM) and loaded onto a HisTrap HP (5 ml, GE) column. The column was washed with 10 column volumes of 50 mM HEPES (pH 7.5) containing 500 mM NaCl. Bound proteins were eluted using 50 mM HEPES, pH 7.5, containing 500 mM NaCl and 500 mM imidazole. The imidazole in the eluted protein mixture was removed by dialysis against 2 liters of 50 mM HEPES (pH 7.5) containing 500 mM NaCl. Tobacco Etch Virus (TEV) protease was added at a protease to target protein ratio of 1:50 (wt/wt), and the mixture was incubated overnight. The resulting truncated IMP-1-like variants were then repurified by passage through a HisTrap column, which removed both the His6-TEV enzyme and the His6 tag peptide cleaved from the N terminus of IMP constructs.
Purified recombinant IMP-1 variants (before and after TEV digestion) were analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) using Fisher EZ-Gel solution (12.5%), a Mini-PROTEAN System (Bio-Rad Laboratories), and Coomassie blue stain. Protein concentrations were determined by UV-Vis absorbance at 280 nm using the calculated molar absorptivities (ε280nm = 46,410 M−1cm−1 for IMP-1 variants containing the poly-His tag and ε280nm = 44,920 M−1cm−1 for the IMP variants without the poly-His tag).
Nano-ESI-MS-based analysis.
To confirm the molecular mass and zinc content of IMP-1, IMP-6, IMP-10, and IMP-78, nanoelectrospray ionization-MS (nano-ESI-MS) analysis was used. Tris(2-carboxyethyl)phosphine hydrochloride (TCEP) (final concentration, 1 mM) and ZnCI2 (final concentration, 100 μM) were added to purified protein samples (50 μM), and the mixtures were incubated for 1 h. Adventitiously bound Zn(II) was removed by dialysis against 100 mM ammonium acetate (pH 7.5) overnight. A Thermo Scientific LTQ Orbitrap XL hybrid ion trap-orbitrap mass spectrometer equipped with a nanoelectrospray ionization (nano-ESI) probe (Thermo Fisher Scientific, San Jose, CA, USA) with positive-mode protein detection was used to analyze the samples. The major parameters were capillary temperature, 180°C; sheath gas, 0; auxiliary gas, 0; sweep gas, 0; spray voltage, 1.1 to 1.5 kV; tube-lens, 150 V; and capillary voltage, 35 V. A full scan ranging from m/z 1,000 to 4,000 was used. The resolution was set to 30,000. Automatic gain control was set at 3 × 104 in full scan, 1 × 104 in structured illumination microscopy (SIM), 1 × 104 in sequential mass spectrometry (MSn), and 3,000 in zoom for ion trap, 3 × 106 in full scan, 1 × 105 in SIM, and 1 × 105 in MSn for Fourier transform. The nano-ESI source was equipped with an offline nano electrospray ion source head unit (part number ES260) (Thermo Fisher Scientific), and the source was constructed based on published work, with modifications (54). Briefly, a platinum wire (0.25-mm diameter) was inserted in the center of the offline unit and in a pulled-glass capillary with a tip inner diameter (i.d.) of about 1 μm (produced in-house from a glass capillary; i.d. 0.8 mm, outer diameter [o.d.] 1.5 mm) using a micropipette puller (model P-87 Flaming/Brown micropipette puller; Sutter Instrument, Inc., USA). The sample solution (5 μl) was loaded into the pulled-glass capillary by an infusion syringe (Thermo Scientific, USA). The platinum wire was inserted in the center of the capillary. The position of capillary tip was then adjusted to approximately 3 mm away from the MS inlet.
Steady-state kinetic assay.
Steady-state kinetic parameters (kcat, Km, kcat/Km) were determined with ampicillin, ceftazidime, cephalothin, imipenem (from Goldbio, St. Louis, MO), meropenem (Millipore Sigma), and ertapenem (from Research Products International Corp., Mt Prospect, IL) in 50 mM HEPES (pH 7.5), 150 mM NaCl, supplemented with 10 μM ZnSO4. Quartz microcuvettes were used with a final assay volume of 0.3 ml. Briefly, 140 μl of 2× buffer (100 mM HEPES (pH 7.5), 300 mM NaCl, 20 μM ZnSO4) and the desired amount of substrate (stocks diluted in deionized water) were added into a quartz cuvette. After mixing, 20 μl of an enzyme stock solution in 1× buffer was added and mixed. Absorbance changes due to the disappearance of substrate were monitored for 0.3 min at 25°C using a UV-visible spectrophotometer (Varian Cary 20). In some cases, the final concentration of the enzyme was adjusted to keep the observed rates linear within this time frame, and then rates were normalized using a linear relationship between enzyme concentration and activity. The following difference extinction coefficients and wavelengths were used: ampicillin (Δε235 = −670 M−1 cm−1), cephalothin (Δε262 = −8,610 M−1 cm−1), ceftazidime (Δε260 = −10,500 M−1 cm−1), imipenem (Δε297 = −10,930 M−1 cm−1), meropenem (Δε298 = −7,200 M−1 cm−1), and ertapenem (Δε295 = −10,940 M−1 cm−1). All reactions were conducted in triplicate. The steady-state Km and kcat values for each substrate were derived by fitting the concentration dependence of initial rates directly by the Michaelis-Menten equation using KaleidaGraph 4.5 (Synergy Software, Reading, PA), with the fitting errors reported.
Inhibition experiments with EDTA.
The relative metal ion affinities were estimated for NDM-1, VIM-2, and IMP-1 by measuring IC50 values for EDTA using the conditions described below. The preparation of NDM-1 and VIM-2 is described previously (10, 14). The IC50 values should not be interpreted as dissociation constants but rather as a gauge for ranking relative zinc affinities of the three enzymes. Briefly, 40 μl of various EDTA stock solutions (diluted in deionized water) were added into wells of a 96-well polystyrene plate, followed by addition of 100 μl of stock solutions of each enzyme in 100 mM HEPES (pH 7.0) (for NDM-1 and IMP-1, this solution was supplemented with 4 mM CHAPS [3-[(3-cholamidopropyl)-dimethylammonio]-1-propanesulfonate]). EDTA and enzyme mixtures were preincubated for 20 min, and the reaction was initiated upon addition of 60 μl of a 50 μM chromacef stock solution (a chromogenic substrate) (55). The absorbance at 442 nm was monitored continuously for 0.3 min at 25°C. The EDTA concentrations in 200 μl final reaction volume were 0, 0.01, 0.1, 1, 25, 50, 100, 500, 1,000, 2,500, 5,000, and 10,000 μM. The final concentrations of NDM-1, IMP-1, and VIM-2 were 5, 0.25, and 1 nM, respectively. IC50 values were determined by direct fitting of the concentrations of EDTA and the percent activities using the following equation: activity (%) = [100/(1 + (IC50/[I])h)], where h is fitted as the Hill coefficient via KaleidaGraph (Synergy, Reading, PA), and the fitting errors were reported.
Equilibrium dialysis and metal analyses.
Samples of NDM-1, VIM-2, and IMP-1 (final concentrations of 8 μM) in 5 ml of 50 mM HEPES (pH 7.5) were mixed with EDTA at concentrations of 0 to 256 μM. After incubation for 60 min, the solutions were dialyzed versus 500 ml of metal-free, 50 mM HEPES (pH 7.5) overnight (dialysis tubing molecular weight cutoff [MWCO] 6,000 to 8,000, Fisherbrand). The Zn(II) content in the resulting protein samples was determined using Atomic Absorption Spectrometer (AA, PerkinElmer PinAAcle 500). The emission wavelength was set to 213.86 nm, as previously described (56). Calibration curves were generated using serial dilutions of Fisherbrand zinc metal standard.
Variant ΔΔG calculations.
The ddg_monomer (34) application within the Rosetta software suite was used to estimate the stabilizing or destabilizing effects of mutations observed in IMP variants. First, the structure of IMP-1 (5EV6) (57) was relaxed using relax within the Rosetta software suite. A library of 10 relaxed models were prepared to optimize packing. A custom constraints file was used to restrict distances between active site zinc ions and the atoms from IMP side chains that coordinate the zinc ions. The relaxed model with the lowest score, reported in Rosetta energy units (REU), was chosen as the starting point for ddg_monomer. All IMP variants and IMP-1 were simulated in ddg_monomer to produce and score a family of 50 models. The predicted ΔG value for each variant, provided in REUs, was averaged across all 50 models for each variant. The ΔΔG value for each variant was then calculated using the equation below as the difference between ΔG values for the variant and IMP-1.
Energy minimization of docked complexes.
To further understand the effects of the IMP-1 mutations on the binding modes of ampicillin and meropenem, ligand-bound models of IMP-1, IMP-6, IMP-10, and IMP-78 were created. Analysis of the deposited IMP-1 structures in the protein data bank in the absence of ligand revealed the flexibility of the β-hairpin in regard to positioning near the active site. The crystal structure of S. marcescens IMP-1 (5Y5B) (35) was selected since the position of the hairpin in this structure was relatively open to the active site, allowing ligand to be placed without severe steric hindrance. Next, a homology model of E. coli IMP-1 was generated by SWISS-MODEL (58–62). The root mean square deviation (RMSD) of the crystal structure and homology model is ∼0.25 Å. The quality of the model was further verified by PROCHECK (63) and MolProbity (64). The E. coli IMP-1 model was then minimized using the CHARMM program (65, 66) and the CHARMM general force field (cgenff) (67, 68) to relieve any bad contacts; 50 steps of steepest descent minimization (SD) and 100 steps of adopted basis Newton-Raphson (ABNR) minimization were performed while constraining the Zn in the active site using harmonic restraints and a force constant of 50 kcal/mol. The step size of the minimization algorithms was set to 0.001. We used a constant dielectric of 1.0 and atom-based cutoffs with a shifting function for electrostatics and a switching function for van der Waals interactions. The cutoff radius for generating the nonbonded list was 14 Å with the switching and shifting functions invoked between 10 Å and 12 Å. Next, the IMP-1 variants were constructed by making the appropriate substitution(s) in this IMP-1 structure, and further minimization was performed using an additional 50 steps of SD and 100 steps of ABNR to relieve any bad contacts due to the amino acid substitution(s). Ligand-bound conformations of IMP-1 and the IMP variants were created by superimposing the IMP models with hydrolyzed meropenem- (5N0H) (36) or ampicillin -bound(5O2F) (69) NDM-1 structures to minimize the RMSD, and the ligand coordinates were merged with the IMP models. The resulting structures were then minimized with 25 steps of SD and 1,000 steps of ABNR, holding both the zinc ions and ligand in the active site using harmonic restraints with a force constant of 50 kcal/mol to allow adjustment of the protein due to the presence of the ligand. Then, the restraints on the ligand were removed and another 5,000 steps of ABNR were performed. The system was set up and the force field parameters were obtained using the CHARMM-GUI PDB reader (70, 71).
Supplementary Material
ACKNOWLEDGMENTS
This research was supported in part by National Institutes of Health (grant GM111926 to W.F., M.W.C., R.A.B., D.L.T., and R.C.P.; grant R35 GM128595 to R.C.P.; and grant R15 GM134454 to M.W.C.), and by the Robert A. Welch Foundation (grant F-1572 to W.F.). Additionally, research reported in this publication was supported by the National Institute of Allergy and Infectious Diseases of the National Institutes of Health (NIH) to R.A.B. under award numbers R01AI100560, R01AI063517, and R01AI072219. This study was also supported in part by funds and/or facilities provided by the Cleveland Department of Veterans Affairs, award number 1I01BX001974 to R.A.B., from the Biomedical Laboratory Research & Development Service of the VA Office of Research and Development, and the Geriatric Research Education and Clinical Center VISN 10. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH or the Department of Veterans Affairs.
Footnotes
Supplemental material is available online only.
REFERENCES
- 1.Fair RJ, Tor Y. 2014. Antibiotics and bacterial resistance in the 21st century. Perspect Medicin Chem 6:25–64. doi: 10.4137/PMC.S14459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.CDC. 2019. More people in the United States dying from antibiotic-resistant infections than previously estimated. https://www.cdc.gov/media/releases/2019/p1113-antibiotic-resistant.html. Accessed March 19th, 2020.
- 3.Bush K, Bradford PA. 2016. β-Lactams and β-lactamase inhibitors: an overview. Cold Spring Harb Perspect Med 6:a025247. doi: 10.1101/cshperspect.a025247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tooke CL, Hinchliffe P, Bragginton EC, Colenso CK, Hirvonen VHA, Takebayashi Y, Spencer J. 2019. β-Lactamases and β-lactamase inhibitors in the 21st century. J Mol Biol 431:3472–3500. doi: 10.1016/j.jmb.2019.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bush K. 2018. Past and present perspectives on β-lactamases. Antimicrob Agents Chemother 62:e01076-18. doi: 10.1128/AAC.01076-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.van Duin D, Doi Y. 2017. The global epidemiology of carbapenemase-producing Enterobacteriaceae. Virulence 8:460–469. doi: 10.1080/21505594.2016.1222343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Palzkill T. 2013. Metallo-beta-lactamase structure and function. Ann N Y Acad Sci 1277:91–104. doi: 10.1111/j.1749-6632.2012.06796.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ju LC, Cheng Z, Fast W, Bonomo RA, Crowder MW. 2018. The continuing challenge of metallo-beta-lactamase inhibition: mechanism matters. Trends Pharmacol Sci 39:635–647. doi: 10.1016/j.tips.2018.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Naas T, Oueslati S, Bonnin RA, Dabos ML, Zavala A, Dortet L, Retailleau P, Iorga BI. 2017. Beta-lactamase database (BLDB) - structure and function. J Enzyme Inhib Med Chem 32:917–919. doi: 10.1080/14756366.2017.1344235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cheng Z, Thomas PW, Ju L, Bergstrom A, Mason K, Clayton D, Miller C, Bethel CR, VanPelt J, Tierney DL, Page RC, Bonomo RA, Fast W, Crowder MW. 2018. Evolution of New Delhi metallo-beta-lactamase (NDM) in the clinic: effects of NDM mutations on stability, zinc affinity, and mono-zinc activity. J Biol Chem 293:12606–12618. doi: 10.1074/jbc.RA118.003835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stewart AC, Bethel CR, VanPelt J, Bergstrom A, Cheng Z, Miller CG, Williams C, Poth R, Morris M, Lahey O, Nix JC, Tierney DL, Page RC, Crowder MW, Bonomo RA, Fast W. 2017. Clinical variants of New Delhi metallo-beta-lactamase are evolving to overcome zinc scarcity. ACS Infect Dis 3:927–940. doi: 10.1021/acsinfecdis.7b00128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Makena A, Brem J, Pfeffer I, Geffen RE, Wilkins SE, Tarhonskaya H, Flashman E, Phee LM, Wareham DW, Schofield CJ. 2015. Biochemical characterization of New Delhi metallo-β-lactamase variants reveals differences in protein stability. J Antimicrob Chemother 70:463–469. doi: 10.1093/jac/dku403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bahr G, Vitor-Horen L, Bethel CR, Bonomo RA, González LJ, Vila AJ. 2017. Clinical evolution of New Delhi metallo-β-lactamase (NDM) optimizes resistance under Zn(II) deprivation. Antimicrob Agents Chemother 62:e01849-17. doi: 10.1128/AAC.01849-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cheng Z, Shurina BA, Bethel CR, Thomas PW, Marshall SH, Thomas CA, Yang K, Kimble RL, Montgomery JS, Orischak MG, Miller CM, Tennenbaum JL, Nix JC, Tierney DL, Fast W, Bonomo RA, Page RC, Crowder MW. 2019. A single salt bridge in VIM-20 increases protein stability and antibiotic resistance under low-zinc conditions. mBio 10:e02412-19. doi: 10.1128/mBio.02412-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Makena A, Düzgün AÖ, Brem J, McDonough MA, Rydzik AM, Abboud MI, Saral A, Çiçek AÇ, Sandalli C, Schofield CJ. 2015. Comparison of Verona integron-borne metallo-β-lactamase (VIM) variants reveals differences in stability and inhibition profiles. Antimicrob Agents Chemother 60:1377–1384. doi: 10.1128/AAC.01768-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Osano E, Arakawa Y, Wacharotayankun R, Ohta M, Horii T, Ito H, Yoshimura F, Kato N. 1994. Molecular characterization of an enterobacterial metallo beta-lactamase found in a clinical isolate of Serratia marcescens that shows imipenem resistance. Antimicrob Agents Chemother 38:71–78. doi: 10.1128/aac.38.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bush K, Jacoby GA. 2010. Updated functional classification of β-lactamases. Antimicrob Agents Chemother 54:969–976. doi: 10.1128/AAC.01009-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Watanabe M, Iyobe S, Inoue M, Mitsuhashi S. 1991. Transferable imipenem resistance in Pseudomonas aeruginosa. Antimicrob Agents Chemother 35:147–151. doi: 10.1128/aac.35.1.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Koh TH, Babini GS, Woodford N, Sng LH, Hall LM, Livermore DM. 1999. Carbapenem-hydrolysing IMP-1 beta-lactamase in Klebsiella pneumoniae from Singapore. Lancet 353:2162. doi: 10.1016/s0140-6736(05)75604-x. [DOI] [PubMed] [Google Scholar]
- 20.Jones RN, Deshpande LM, Bell JM, Turnidge JD, Kohno S, Hirakata Y, Ono Y, Miyazawa Y, Kawakama S, Inoue M, Hirata Y, Toleman MA. 2004. Evaluation of the contemporary occurrence rates of metallo-beta-lactamases in multidrug-resistant Gram-negative bacilli in Japan: report from the SENTRY antimicrobial surveillance program (1998–2002). Diagn Microbiol Infect Dis 49:289–294. doi: 10.1016/j.diagmicrobio.2004.04.007. [DOI] [PubMed] [Google Scholar]
- 21.Shibata N, Doi Y, Yamane K, Yagi T, Kurokawa H, Shibayama K, Kato H, Kai K, Arakawa Y. 2003. PCR typing of genetic determinants for metallo-beta-lactamases and integrases carried by gram-negative bacteria isolated in Japan, with focus on the class 3 integron. J Clin Microbiol 41:5407–5413. doi: 10.1128/jcm.41.12.5407-5413.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bush K, Bradford PA. 2020. Epidemiology of β-lactamase-producing pathogens. Clin Microbiol Rev 33:e00047-19. doi: 10.1128/CMR.00047-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Concha NO, Janson CA, Rowling P, Pearson S, Cheever CA, Clarke BP, Lewis C, Galleni M, Frere JM, Payne DJ, Bateson JH, Abdel-Meguid SS. 2000. Crystal structure of the IMP-1 metallo beta-lactamase from Pseudomonas aeruginosa and its complex with a mercaptocarboxylate inhibitor: binding determinants of a potent, broad-spectrum inhibitor. Biochemistry 39:4288–4298. doi: 10.1021/bi992569m. [DOI] [PubMed] [Google Scholar]
- 24.Griffin DH, Richmond TK, Sanchez C, Moller AJ, Breece RM, Tierney DL, Bennett B, Crowder MW. 2011. Structural and kinetic studies on metallo-beta-lactamase IMP-1. Biochemistry 50:9125–9134. doi: 10.1021/bi200839h. [DOI] [PubMed] [Google Scholar]
- 25.Yano H, Kuga A, Okamoto R, Kitasato H, Kobayashi T, Inoue M. 2001. Plasmid-encoded metallo-β-lactamase (IMP-6) conferring resistance to carbapenems, especially meropenem. Antimicrob Agents Chemother 45:1343–1348. doi: 10.1128/AAC.45.5.1343-1348.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Iyobe S, Kusadokoro H, Takahashi A, Yomoda S, Okubo T, Nakamura A, O’Hara K. 2002. Detection of a variant metallo-beta-lactamase, IMP-10, from two unrelated strains of Pseudomonas aeruginosa and an alcaligenes xylosoxidans strain. Antimicrob Agents Chemother 46:2014–2016. doi: 10.1128/aac.46.6.2014-2016.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Oelschlaeger P, Aitha M, Yang H, Kang JS, Zhang AL, Liu EM, Buynak JD, Crowder MW. 2015. Meropenem and chromacef intermediates observed in IMP-25 metallo-beta-lactamase-catalyzed hydrolysis. Antimicrob Agents Chemother 59:4326–4330. doi: 10.1128/AAC.04409-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pegg KM, Liu EM, Lacuran AE, Oelschlaeger P. 2013. Biochemical characterization of IMP-30, a metallo-β-lactamase with enhanced activity toward ceftazidime. Antimicrob Agents Chemother 57:5122–5126. doi: 10.1128/AAC.02341-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang CJ, Faheem M, Dang P, Morris MN, Kumar P, Oelschlaeger P. 2019. Mutation S115T in IMP-type metallo-β-lactamases compensates for decreased expression levels caused by mutation S119G. Biomolecules 9:724. doi: 10.3390/biom9110724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.LaCuran AE, Pegg KM, Liu EM, Bethel CR, Ai N, Welsh WJ, Bonomo RA, Oelschlaeger P. 2015. Elucidating the role of residue 67 in IMP-type metallo-β-lactamase evolution. Antimicrob Agents Chemother 59:7299–7307. doi: 10.1128/AAC.01651-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wilkins MR, Gasteiger E, Bairoch A, Sanchez JC, Williams KL, Appel RD, Hochstrasser DF. 1999. Protein identification and analysis tools in the ExPASy server. Methods Mol Biol 112:531–552. doi: 10.1385/1-59259-584-7:531. [DOI] [PubMed] [Google Scholar]
- 32.Kupper MB, Herzog K, Bennink S, Schlömer P, Bogaerts P, Glupczynski Y, Fischer R, Bebrone C, Hoffmann KM. 2015. The three-dimensional structure of VIM-31–a metallo-β-lactamase from Enterobacter cloacae in its native and oxidized form. FEBS J 282:2352–2360. doi: 10.1111/febs.13283. [DOI] [PubMed] [Google Scholar]
- 33.Christopeit T, Yang KW, Yang SK, Leiros HKS. 2016. The structure of the metallo-β-lactamase VIM-2 in complex with a triazolylthioacetamide inhibitor. Acta Crystallogr F Struct Biol Commun 72:813–819. doi: 10.1107/S2053230X16016113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kellogg EH, Leaver-Fay A, Baker D. 2011. Role of conformational sampling in computing mutation-induced changes in protein structure and stability. Proteins 79:830–838. doi: 10.1002/prot.22921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wachino J-i, Kanechi R, Nishino E, Mochizuki M, Jin W, Kimura K, Kurosaki H, Arakawa Y. 2019. 4-Amino-2-sulfanylbenzoic acid as a potent subclass B3 metallo-β-lactamase-specific inhibitor applicable for distinguishing metallo-β-lactamase subclasses. Antimicrob Agents Chemother 63:e01197-19. doi: 10.1128/AAC.01197-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Raczynska JE, Shabalin IG, Minor W, Wlodawer A, Jaskolski M. 2018. A close look onto structural models and primary ligands of metallo-β-lactamases. Drug Resist Updat 40:1–12. doi: 10.1016/j.drup.2018.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Iyobe S, Kusadokoro H, Ozaki J, Matsumura N, Minami S, Haruta S, Sawai T, O’Hara K. 2000. Amino acid substitutions in a variant of IMP-1 metallo-beta-lactamase. Antimicrob Agents Chemother 44:2023–2027. doi: 10.1128/aac.44.8.2023-2027.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Oelschlaeger P, Schmid RD, Pleiss J. 2003. Modeling domino effects in enzymes: molecular basis of the substrate specificity of the bacterial metallo-β-lactamases IMP-1 and IMP-6. Biochemistry 42:8945–8956. doi: 10.1021/bi0300332. [DOI] [PubMed] [Google Scholar]
- 39.Oelschlaeger P, Pleiss J. 2007. Hydroxyl groups in the ββ sandwich of metallo-β-lactamases favor enzyme activity: Tyr218 and Ser262 pull down the lid. J Mol Biol 366:316–329. doi: 10.1016/j.jmb.2006.11.027. [DOI] [PubMed] [Google Scholar]
- 40.Liu EM, Pegg KM, Oelschlaeger P. 2012. The sequence-activity relationship between metallo-β-lactamases IMP-1, IMP-6, and IMP-25 suggests an evolutionary adaptation to meropenem exposure. Antimicrob Agents Chemother 56:6403–6406. doi: 10.1128/AAC.01440-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tsutsui A, Yahara K, Shibayama K. 2018. Trends and patterns of national antimicrobial consumption in Japan from 2004 to 2016. J Infect Chemother 24:414–421. doi: 10.1016/j.jiac.2018.01.003. [DOI] [PubMed] [Google Scholar]
- 42.Zhao WH, Hu ZQ. 2011. IMP-type metallo-β-lactamases in Gram-negative bacilli: distribution, phylogeny, and association with integrons. Crit Rev Microbiol 37:214–226. doi: 10.3109/1040841X.2011.559944. [DOI] [PubMed] [Google Scholar]
- 43.Yano H, Ogawa M, Endo S, Kakuta R, Kanamori H, Inomata S, Ishibashi N, Aoyagi T, Hatta M, Gu Y, Yamada M, Tokuda K, Kunishima H, Kitagawa M, Hirakata Y, Kaku M. 2012. High frequency of IMP-6 among clinical isolates of metallo-β-lactamase-producing Escherichia coli in Japan. Antimicrob Agents Chemother 56:4554–4555. doi: 10.1128/AAC.00617-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Walther-Rasmussen J, Høiby N. 2006. OXA-type carbapenemases. J Antimicrob Chemother 57:373–383. doi: 10.1093/jac/dki482. [DOI] [PubMed] [Google Scholar]
- 45.Poirel L, Ortiz de la Rosa J-M, Richard A, Aires-de-Sousa M, Nordmann P. 2019. CTX-M-33, a CTX-M-15 derivative conferring reduced susceptibility to carbapenems. Antimicrob Agents Chemother 63:e01515-19. doi: 10.1128/AAC.01515-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Materon IC, Beharry Z, Huang W, Perez C, Palzkill T. 2004. Analysis of the context dependent sequence requirements of active site residues in the metallo-beta-lactamase IMP-1. J Mol Biol 344:653–663. doi: 10.1016/j.jmb.2004.09.074. [DOI] [PubMed] [Google Scholar]
- 47.Thomas PW, Zheng M, Wu S, Guo H, Liu D, Xu D, Fast W. 2011. Characterization of purified New Delhi metallo-β-lactamase-1. Biochemistry 50:10102–10113. doi: 10.1021/bi201449r. [DOI] [PubMed] [Google Scholar]
- 48.Yamaguchi Y, Ding S, Murakami E, Imamura K, Fuchigami S, Hashiguchi R, Yutani K, Mori H, Suzuki S, Arakawa Y, Kurosaki H. 2011. A demetallation method for IMP-1 metallo-β-lactamase with restored enzymatic activity upon addition of metal ion(s). Chembiochem 12:1979–1983. doi: 10.1002/cbic.201100342. [DOI] [PubMed] [Google Scholar]
- 49.Liu Y, Zhang H, Zhang X, Jiang N, Zhang Z, Zhang J, Zhu B, Wang G, Zhao K, Zhou Y. 2019. Characterization of an NDM-19-producing Klebsiella pneumoniae strain harboring 2 resistance plasmids from China. Diagn Microbiol Infect Dis 93:355–361. doi: 10.1016/j.diagmicrobio.2018.11.007. [DOI] [PubMed] [Google Scholar]
- 50.Liu Z, Li J, Wang X, Liu D, Ke Y, Wang Y, Shen J. 2018. Novel variant of New Delhi metallo-β-lactamase, NDM-20, in Escherichia coli. Front Microbiol 9:248. doi: 10.3389/fmicb.2018.00248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liu L, Feng Y, McNally A, Zong Z. 2018. blaNDM-21, a new variant of blaNDM in an Escherichia coli clinical isolate carrying blaCTX-M-55 and rmtB. J Antimicrob Chemother 73:2336–2339. doi: 10.1093/jac/dky226. [DOI] [PubMed] [Google Scholar]
- 52.El-Badawy MF, El-Far SW, Althobaiti SS, Abou-Elazm FI, Shohayeb MM. 2020. The first Egyptian report showing the co-existence of bla (NDM-25), bla (OXA-23), bla (OXA-181), and bla (GES-1) among carbapenem-resistant K. pneumoniae clinical isolates genotyped by BOX-PCR. Infect Drug Resist 13:1237–1250. doi: 10.2147/IDR.S244064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.CLSI. 2020. Performance standards for antimicrobial susceptibility testing, thirtieth ed.
- 54.Harvey SR, Porrini M, Stachl C, MacMillan D, Zinzalla G, Barran PE. 2012. Small-molecule inhibition of c-MYC:MAX leucine zipper formation is revealed by ion mobility mass spectrometry. J Am Chem Soc 134:19384–19392. doi: 10.1021/ja306519h. [DOI] [PubMed] [Google Scholar]
- 55.Yu S, Vosbeek A, Corbella K, Severson J, Schesser J, Sutton LD. 2012. A chromogenic cephalosporin for β-lactamase inhibitor screening assays. Anal Biochem 428:96–98. doi: 10.1016/j.ab.2012.06.006. [DOI] [PubMed] [Google Scholar]
- 56.Clark D, Dagnall RM, West TS. 1973. The atomic absorption determination of zinc with a graphite furnace. Anal Chim Acta 63:11–18. doi: 10.1016/S0003-2670(01)82169-3. [DOI] [Google Scholar]
- 57.Hinchliffe P, González MM, Mojica MF, González JM, Castillo V, Saiz C, Kosmopoulou M, Tooke CL, Llarrull LI, Mahler G, Bonomo RA, Vila AJ, Spencer J. 2016. Cross-class metallo-β-lactamase inhibition by bisthiazolidines reveals multiple binding modes. Proc Natl Acad Sci U S A 113:E3745–E3754. doi: 10.1073/pnas.1601368113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Waterhouse A, Bertoni M, Bienert S, Studer G, Tauriello G, Gumienny R, Heer FT, de Beer TP, Rempfer C, Bordoli L, Lepore R, Schwede T. 2018. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res 46:W296–W303. doi: 10.1093/nar/gky427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bienert S, Waterhouse A, de Beer Tjaart AP, Tauriello G, Studer G, Bordoli L, Schwede T. 2017. The SWISS-MODEL repository—new features and functionality. Nucleic Acids Res 45:D313–D319. doi: 10.1093/nar/gkw1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Guex N, Peitsch MC, Schwede T. 2009. Automated comparative protein structure modeling with SWISS-MODEL and Swiss-PdbViewer: a historical perspective. Electrophoresis 30:S162–S173. doi: 10.1002/elps.200900140. [DOI] [PubMed] [Google Scholar]
- 61.Studer G, Rempfer C, Waterhouse AM, Gumienny R, Haas J, Schwede T. 2020. QMEANDisCo—distance constraints applied on model quality estimation. Bioinformatics 36:2647. doi: 10.1093/bioinformatics/btaa058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bertoni M, Kiefer F, Biasini M, Bordoli L, Schwede T. 2017. Modeling protein quaternary structure of homo- and hetero-oligomers beyond binary interactions by homology. Sci Rep 7:10480. doi: 10.1038/s41598-017-09654-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. 1993. PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Crystallogr 26:283–291. doi: 10.1107/S0021889892009944. [DOI] [Google Scholar]
- 64.Chen VB, Arendall WB, 3rd, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC. 2010. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr 66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Brooks BR, Brooks CL, III, Mackerell AD, Jr, Nilsson L, Petrella RJ, Roux B, Won Y, Archontis G, Bartels C, Boresch S, Caflisch A, Caves L, Cui Q, Dinner AR, Feig M, Fischer S, Gao J, Hodoscek M, Im W, Kuczera K, Lazaridis T, Ma J, Ovchinnikov V, Paci E, Pastor RW, Post CB, Pu JZ, Schaefer M, Tidor B, Venable RM, Woodcock HL, Wu X, Yang W, York DM, Karplus M. 2009. CHARMM: the biomolecular simulation program. J Comput Chem 30:1545–1614. doi: 10.1002/jcc.21287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.MacKerell AD, Jr, Brooks B, Brooks CL, III, Nilsson L, Roux B, Won Y, Karplus M. 2002. CHARMM: the energy function and its parameterization, encyclopedia of computational chemistry doi: 10.1002/0470845015.cfa007. [DOI]
- 67.Vanommeslaeghe K, Hatcher E, Acharya C, Kundu S, Zhong S, Shim J, Darian E, Guvench O, Lopes P, Vorobyov I, Mackerell AD, Jr. 2010. CHARMM general force field: a force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J Comput Chem 31:671–690. doi: 10.1002/jcc.21367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.MacKerell AD, Bashford D, Bellott M, Dunbrack RL, Evanseck JD, Field MJ, Fischer S, Gao J, Guo H, Ha S, Joseph-McCarthy D, Kuchnir L, Kuczera K, Lau FT, Mattos C, Michnick S, Ngo T, Nguyen DT, Prodhom B, Reiher WE, Roux B, Schlenkrich M, Smith JC, Stote R, Straub J, Watanabe M, Wiorkiewicz-Kuczera J, Yin D, Karplus M. 1998. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J Phys Chem B 102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 69.Diehl A, Roske Y, Ball L, Chowdhury A, Hiller M, Molière N, Kramer R, Stöppler D, Worth CL, Schlegel B, Leidert M, Cremer N, Erdmann N, Lopez D, Stephanowitz H, Krause E, van Rossum B-J, Schmieder P, Heinemann U, Turgay K, Akbey Ü, Oschkinat H. 2018. Structural changes of TasA in biofilm formation of Bacillus subtilis. Proc Natl Acad Sci U S A 115:3237–3242. doi: 10.1073/pnas.1718102115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Jo S, Kim T, Iyer VG, Im W. 2008. CHARMM-GUI: a web-based graphical user interface for CHARMM. J Comput Chem 29:1859–1865. doi: 10.1002/jcc.20945. [DOI] [PubMed] [Google Scholar]
- 71.Jo S, Cheng X, Islam SM, Huang L, Rui H, Zhu A, Lee HS, Qi Y, Han W, Vanommeslaeghe K, MacKerell AD, Roux B, Im W. 2014. Chapter eight - CHARMM-GUI PDB manipulator for advanced modeling and simulations of proteins containing nonstandard residues, p 235–265. In Karabencheva-Christova T (ed), Advances in Protein Chemistry and Structural Biology, vol 96. Academic Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Garau G, García-Sáez I, Bebrone C, Anne C, Mercuri P, Galleni M, Frère J-M, Dideberg O. 2004. Update of the standard numbering scheme for class B β-lactamases. Antimicrob Agents Chemother 48:2347–2349. doi: 10.1128/AAC.48.7.2347-2349.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.