

Intra-abdominal candidiasis (IAC) is one of the most common yet underappreciated forms of invasive candidiasis. IAC is difficult to treat, and therapeutic failure and drug-resistant breakthrough infections are common in some institutions despite the use of echinocandins as first-line agents.
KEYWORDS: manogepix, MGX, APX001A, fosmanogepix, FMGX, APX001, intra-abdominal candidiasis, lesion, drug penetration, matrix-assisted desorption ionization–mass spectrometry imaging (MALDI-MSI), laser capture microdissection (LCM), fosmanogepix (FMGX), manogepix (MGX)
ABSTRACT
Intra-abdominal candidiasis (IAC) is one of the most common yet underappreciated forms of invasive candidiasis. IAC is difficult to treat, and therapeutic failure and drug-resistant breakthrough infections are common in some institutions despite the use of echinocandins as first-line agents. Fosmanogepix (FMGX, formerly APX001) is a first-in-class antifungal prodrug that can be administered both intravenously and orally. FMGX is currently in phase 2 clinical development for the treatment of life-threatening invasive fungal infections. To explore the pharmacological properties and therapeutic potential of FMGX for IAC, we evaluated both drug penetration and efficacy of the active moiety manogepix (MGX, formerly APX001A) in liver tissues in a clinically relevant IAC mouse model infected with Candida albicans. Matrix-assisted laser desorption ionization–mass spectrometry imaging (MALDI-MSI) and laser capture microdissection (LCM)-directed absolute drug quantitation were employed to evaluate drug penetration into liver abscess lesions both spatially and quantitatively. The partitioning of MGX into lesions occurred slowly after a single dose; however, robust accumulation in the lesion was achieved after 3 days of repeated dosing. Associated with this drug penetration pattern, reduction in fungal burden and clearance in the liver were observed in mice receiving the multiday FMGX regimen. In comparison, administration of micafungin resulted in marginal reduction in fungal burden at the end of 4 days of treatment. These results suggest that FMGX is a promising candidate for the treatment of IAC.
TEXT
Invasive candidiasis (IC), encompassing bloodstream Candida infections (candidemia), intra-abdominal candidiasis (IAC), and other deep-seated candidiasis, is the most common fungal disease in U.S. hospitals (1, 2). The global incidence of IC has been estimated at 750,000 cases annually (3), with an associated crude mortality rate in excess of 40% and an associated expenditure of ∼$46,000 per case (4). Despite candidemia being the focus in most studies involving IC, IAC poses huge threats to both immunocompetent and immunocompromised patient populations (5, 6). Studies have shown that factors associated with successful outcomes in IAC patients are prompt source control and appropriate antifungal therapy (7), in which the echinocandin antifungals are the recommended first-line agents (8). However, the clinical effectiveness of echinocandins in treating IAC is highly variable (9–12), and in some institutions, treatment failures and drug-resistant breakthrough infections can occur in half of the cases investigated (10). This may be due in part to the poor penetration of approved echinocandin drugs into the lesion tissue. Undoubtedly, there is an urgent need to develop new therapeutic strategies to meet this clinical challenge.
Fosmanogepix (FMGX, formerly APX001) is a first-in-class antifungal prodrug that is currently in phase 2 clinical development for the treatment of life-threatening invasive fungal infections caused by Candida spp. (ClinicalTrials registration no. NCT03604705 and NCT04148287), as well as Aspergillus spp. and rare molds (ClinicalTrials registration no. NCT04240886). FMGX is the N-phosphonooxymethyl prodrug of the active moiety manogepix (MGX, formerly APX001A). After intravenous (i.v.) or oral administration, FMGX is rapidly converted to MGX by systemic phosphatases. MGX has a novel mechanism of action and exerts antifungal activity against a wide spectrum of clinically relevant fungal pathogens. MGX inhibits the conserved fungal enzyme inositol acyltransferase Gwt1 in the glycosylphosphatidylinositol (GPI) anchor biosynthesis pathway, thereby disrupting the trafficking and anchoring of mannoproteins to the fungal cell wall (13, 14). Previous studies have demonstrated broad and potent in vitro activity of MGX against most Candida spp., with the exception of Candida krusei (14–18). Animal studies have also shown that FMGX is effective in vivo in treating disseminated candidiasis caused by major Candida pathogens, including Candida albicans, Candida glabrata, Candida auris, and Candida tropicalis (16, 19–23), but its effectiveness in the treatment of IAC is unknown. Of note, the major histopathological presentation of intra-abdominal abscesses with IAC is different from the homogeneous tissue invasion of disseminated candidiasis stemming from candidemia, making it difficult to predict the efficacy of FMGX in IAC from the candidemia animal model.
The objective of this study is to investigate the efficacy and the pharmacological properties of FMGX for the treatment of IAC, which may be instructional to the clinical application of this novel agent. The main histopathological manifestation of IAC is intra-abdominal abscesses. Therefore, we evaluated both drug penetration into liver abscesses and the standard assessment of fungal burden within the infected liver tissues in a clinically relevant IAC mouse model due to C. albicans. Matrix-assisted laser desorption ionization−mass spectrometry imaging (MALDI-MSI) and laser capture microdissection (LCM)-directed high-pressure liquid chromatography-tandem mass spectrometry (LC-MS/MS) were employed to evaluate drug penetration into liver abscess lesions both spatially and quantitatively. Liver burden reduction was measured as an efficacy index at multiple time points during the FMGX course of therapy and compared with the representative standard of care micafungin (MCF).
RESULTS
In vitro susceptibility.
C. albicans strain SC5314 was used to establish the intra-abdominal infections in our study. The MIC against this strain was 0.016 μg/ml for both manogepix and micafungin.
Plasma pharmacokinetics (PK) in IAC mice.
Given that MGX is metabolized considerably faster in mice than in humans (21, 24, 25), we employed an established mouse FMGX-dosing regimen involving administration of the nonspecific cytochrome P450 (CYP) inhibitor, 1-aminobenzotriazole (ABT) 2 h prior to FMGX dosing. ABT has no antifungal effect against multiple species either in vitro or in vivo and is not synergistic with antifungal agents (23, 26). This regimen has been proven to be nontoxic and effective at improving systemic exposure of MGX in several mouse models (23, 26–29). We examined the plasma drug concentrations of MGX in IAC mice following a single oral dose of FMGX at 78 mg/kg in conjunction with the pretreatment of ABT orally administered at 50 mg/kg on day 3 postinfection (day 1 dosing). This dosing regimen was previously shown in mice to achieve clinically relevant human drug exposures (24, 25). As shown in Fig. 1, the peak plasma drug concentration (Cmax) appeared at 3 h postdose, with a mean value of 16.15 μg/ml. The area under the concentration-time curve from time zero to 24 h postdose (AUC0−24) was measured as 265.1 μg · h/ml, and the elimination half-life was 15.08 h at the given dosage. This exposure in mice is similar to the safe and well-tolerated exposures achieved in healthy human volunteers in phase 1 clinical trials (24, 25).
FIG 1.

Plasma pharmacokinetics after a single oral dose of 78 mg/kg FMGX plus ABT.
Tissue PK and lesion penetration of MGX.
In the IAC model, lesion formation is observed most consistently in the liver among all of the intra-abdominal organs. Therefore, liver was selected as the target tissue for the pharmacokinetic and fungal tissue burden analyses. Tissue distribution and lesion penetration of MGX were analyzed in both single-dose and multidose studies. In the single-dose study, infected livers were collected at 1, 3, 6, and 24 h after dosing. Lesion (aggregate of necrotic core and inflammatory rim) and nonlesion (uninvolved healthy liver) tissues were dissected by LCM, then subjected to LC-MS/MS for MGX level quantitation. A rapid and significant partitioning of MGX to the liver was observed after a single dose of FMGX. The drug distribution kinetics (Fig. 2A) in the nonlesion liver tissues were similar to the plasma PK, but exceeded plasma drug concentrations at all four time points within the 24 h postdose course (mean MGX values of 18.9, 24.8, 23.1, and 9.6 μg/g for 1, 3, 6, and 24 h, respectively). In comparison, partitioning of MGX into the liver lesions occurred at a much slower rate, with the mean concentrations measured sequentially at 1.3, 4.6, 7.4, and 5.8 μg/g in lesions from 1 h to 24 h. The AUC0−24 was 143.8 μg · h/ml and 419.7 μg · h/ml in the lesion and nonlesion areas of the liver, respectively. To enable the comparison of MGX penetration into the lesion/nonlesion areas of the liver, lesion MGX levels were normalized to those in matched nonlesion liver tissues for each individual animal. The lesion/nonlesion MGX ratios demonstrated a significant (P = 0.003) time-dependent drug penetration of MGX into the abscess lesions (Fig. 2B).
FIG 2.
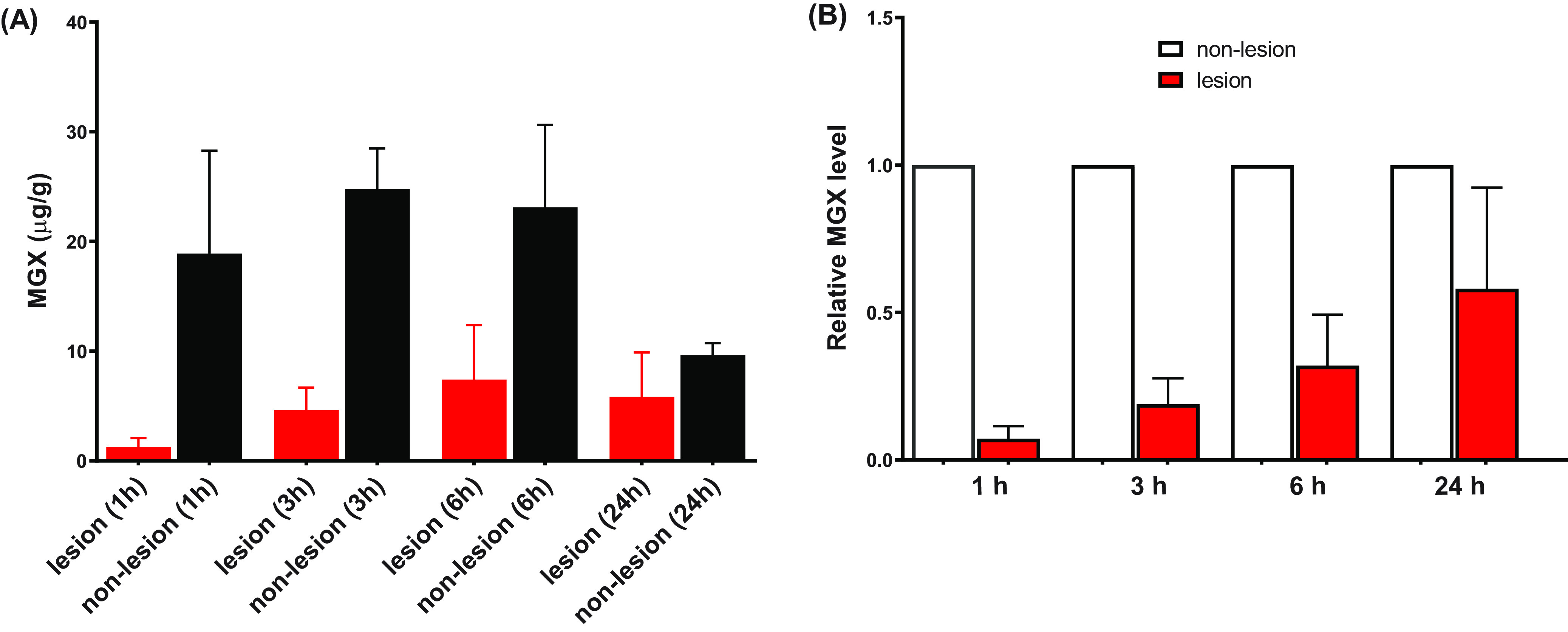
MGX penetration into infected livers is shown as (A) absolute quantitation of MGX in lesion and nonlesion compartments and (B) lesion/nonlesion MGX ratios, at selected time points after a single dose of FMGX. Error bars show the standard deviation between 4 liver samples.
Given the observed slow lesion partitioning of MGX after a single dose, we next asked whether a multidose regimen would further increase MGX lesion penetration. In the multidose study, once daily (QD) 78 mg/kg FMGX plus ABT treatment was administered to IAC mice starting on day 3 postinfection and continuing through days 4 and 5. At 24 h after the 2nd or 3rd dose (days 5 and 6), livers were collected and analyzed for drug exposure within and outside lesions, analogously to the single-dose study. As shown in Fig. 3, the mean values of MGX concentration in nonlesion liver tissues versus those in lesions were 5.5 versus 2.5 μg/g and 1.8 versus 2.9 μg/g after 2- and 3-day dosing of FMGX, respectively. When normalized to nonlesion tissues, lesion penetration of MGX after 2 doses was similar to that observed at 24 h after the single dose. However, a significant increase in MGX partitioning in lesions with a mean lesion/nonlesion MGX ratio of 1.92 (P = 0.0011) was observed in mice that received 3 doses of FMGX, suggesting accumulation.
FIG 3.

Liver lesion penetration of MGX at 24 h after repeated dosing of FMGX for 2 or 3 days. The significant lesion/nonlesion MGX concentration ratio difference is noted as ** (P < 0.01).
Spatial distribution of MGX in infected liver.
One limitation of the aforementioned lesion dissection by LCM was that the very small and shallow presentation of most lesions precluded us from separating two distinct subcompartments (necrotic core and inflammatory rim) within lesions, as doing so we would fail to procure enough material required for drug quantification. To gain a better understanding of the spatial distribution of MGX with details at higher resolution, MGX-specific ion maps (Fig. 4) were constructed for representative liver tissue sections using MALDI-MSI. For both the MGX and the deuterated MGX (d4-MGX) internal standard, the [M+H]+ ion of the reduced open ring structure of the isoxazole moiety in the MGX molecule was detected with greater sensitivity than the protonated MGX ion. For this reason, the reduced ion at m/z 361.166 was used to display the spatial distribution of MGX within infected tissue sections. For the single-dose imaging series, MGX distribution in nonlesion livers was generally homogeneous, and ion intensities in 3-h and 6-h samples were markedly higher relative to those in 1-h and 24-h tissues. Within lesions, MGX signals appeared at or after 3 h postdose, with varied intensity and spatial distribution. Close-up examination of the individual lesions and overlays of ion maps and hematoxylin and eosin (H&E)-stained images (Fig. 5) clearly demonstrated a gradual process of drug penetration and accumulation. MGX signals in lesions changed from barely visible at 1 h to slowly migrating to the outer rim of the abscess and increasing intensity from 3 to 6 h, reaching a therapeutic amount of drug and approaching the necrotic center by 24 h. Although MGX penetration into healthy liver tissue was faster than that into liver abscesses, the 24-h ion map showed that MGX was highly concentrated in the necrotic core, in which the main body of the fungal network (displayed by Grocott methenamine silver [GMS] staining in Fig. 5) resides. Moreover, the high intensity of the ion map from the liver lesion collected 24 h after 3 doses of FMGX reflects enhanced MGX drug penetration into the liver lesion over time (Fig. 6).
FIG 4.

Spatial distribution of MGX in infected livers after single FMGX dose. Matrix-assisted laser desorption ionization–mass spectrometry (MALDI MS) ion maps of MGX in representative liver tissues collected 1, 3, 6, and 24 h after a single oral dose of FMGX are displayed in the top panel. The hematoxylin and eosin (H&E) stains of the sections utilized for mass spectrometry imaging (MSI) are shown in the middle panel and Grocott methenamine silver (GMS) stains of adjacent sections are displayed in the bottom panel. Bar, 6 mm.
FIG 5.

Enlarged view of individual lesions at 1, 3, 6, and 24 h after single dosing of FMGX. Yellow outlines highlight the lesion area on each tissue section. MALDI MS ion maps of MGX are displayed in the top panel. The H&E stains of the sections utilized for MSI are shown in the second panel. A merged image of the H&E and MSI ion map of MGX is shown in the third panel. GMS staining of adjacent sections are displayed in the bottom panel. Bar, 2 mm.
FIG 6.
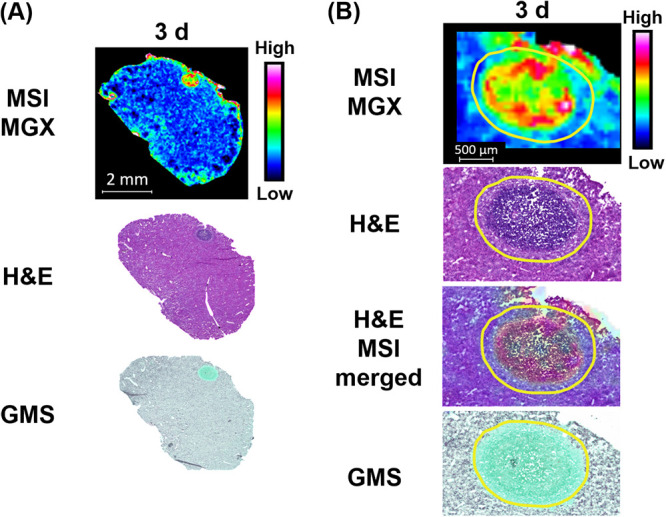
(A) Drug distribution in infected liver tissue after 3 doses of FMGX. (B) Higher-magnification images of the lesion. Outlines highlight the lesion area.
In vivo efficacy.
To test the therapeutic potential of MGX for IAC, we carried out an in vivo efficacy study in which mice infected with C. albicans were treated for 4 consecutive days with either the once-daily FMGX (78 mg/kg QD) plus ABT regimen, MCF at a dose in mice which results in exposures similar to what is clinically observed (5 mg/kg QD), or vehicle control. Liver fungal burdens were evaluated and compared among all dosing groups at predose and 24 h after each dosing (Fig. 7). Although a single dose of FMGX was not potent enough to decrease liver fungal burden, additional dosing significantly enhanced FMGX antifungal efficacy. Compared to mice given vehicle control, mice treated with FMGX for 2 days or 3 days had mean liver fungal burden reductions of 1.33 log10 CFU/g (P = 0.0034) and 1.53 log10 CFU/g (P = 0.0016), respectively. Fungal clearance (defined by repeated zero-CFU count in undiluted liver homogenate) was observed in 2 of 6 mice (33.3%) in the 3-day FMGX treatment arm. Most strikingly, fungal clearance was observed in all mice in the 4-day FMGX treatment arm. In comparison, liver burdens relative to vehicle control were not significantly reduced by MCF after 1 day of dosing (0.45 log10 CFU/g after a single dose; P = 0.4989); however, a 1.35 log10 CFU/g reduction (P = 0.0137) was observed after 2 days of MCF treatment. Continued administration of MCF beyond 2 days of dosing did not result in further statistically significant reductions in fungal burden, although liver burdens in these mice (4.28 and 4.84 log10 CFU/g for 3-day and 4-day MCF arms, respectively) were still numerically lower than that in vehicle control mice (4.78 and 5.08 log10 CFU/g for 3- and 4-day vehicle groups, respectively) and 1 mouse in the 4-day MCF group did clear the liver burden.
FIG 7.

Liver burden reduction and clearance comparison at predose, 24 h after 1-, 2-, 3-, and 4-day treatments of FMGX, MCF, and vehicle control. The dotted line is the limit of detection (LOD) at 22.7 CFU/g. Symbols on the x axis represent mice with no recoverable liver burden (clearance). The percentages of mice with liver burden clearance are plotted as bars on the right y axis underneath burdens of each corresponding group. The significant burden differences are labeled as * (P < 0.05) and ** (P < 0.01).
Histopathological examination of livers harvested from mice who received a 4-day treatment with FMGX showed that lesions had not detectable fungi (Fig. 8A) or had small numbers of rounded fungal cells or largely distorted hyphal elements in necrosis (Fig. 8B). The viability of these morphologically damaged fungal cells was unknown. In contrast, a dense and highly entangled fungal network composed of long branching filaments in the center of the lesion was observed in mice treated with 4 days of MCF (Fig. 8C). These findings are consistent with the fungal burden assessment results.
FIG 8.

GMS-stained liver samples collected at 24 h after 4 days of treatment with FMGX or MCF. FMGX-treated liver had either (A) lesion with no detectable fungal cells or (B) lesion containing a small amount of morphologically disrupted fungal cells in necrosis. (C) A dense and well-organized fungal network residing in the lesion core was observed for the MCF-treated sample. Yellow arrows in the enlarged views (right; magnification, ×40) of black boxed areas (left; magnification, ×5) indicate fungal cells.
DISCUSSION
The antifungal pipeline has progressed at a very slow pace for the past 2 decades, with very few new chemical entities discovered and no new drug classes added to the antifungal armamentarium. Currently, only four classes of drugs are approved to treat systemic fungal infections (30, 31). For certain diseases, current antifungal therapies are suboptimal, and thus therapeutic failure and resistance development are increasingly encountered in clinical settings (32). This has been problematic for the management of IAC patients utilizing echinocandins as first-line agents, and some clinicians have raised concerns about whether echinocandins penetrate sufficiently into the infected tissue sites (10). Our previous studies found that even though MCF penetrated into intra-abdominal abscesses after multiple doses, it failed to reach the pharmacodynamic target (PD)-associated drug exposure within this infected tissue compartment (33, 34). These results provide a rationale for the limited efficacy of MCF in IAC mouse studies (33, 34), as well as for the clinical observations in patients (10). Clearly, sufficient lesion penetration is a premise of successful antifungal effect in the treatment of IAC. Therefore, in the present study, we investigated the efficacy and pharmacological properties of FMGX for the treatment of IAC, which may inform the clinical development of this novel agent.
Compared to currently available antifungal drugs, FMGX is both structurally and mechanistically unique. Despite the lack of in vitro activity against C. krusei, MGX has demonstrated favorable antifungal profiles, both in vitro and in vivo, against diverse yeast and mold pathogens, including emerging multidrug-resistant and difficult-to-treat fungal species (14–23, 26, 28, 35, 36). Of note, all previous in vivo studies involving Candida species were carried out in disseminated candidiasis models, and the efficacy of FMGX to treat IAC had not been investigated until the present study. Given the clinical significance of IAC and the imperfect scenario of using the first-line echinocandins to treat IAC patients, it is important to explore different therapeutic strategies with clinical drug candidates such as FMGX. It is worth noting that, after FMGX dosing, MGX is metabolized rapidly in mice, with a half-life of ∼1.4 to 2.75 h (21), whereas the half-life in humans (∼2 to 2.5 days) is much longer (24, 25). To counter this rapid metabolism in mice, previous studies utilized frequent dosing, including twice a day (BID), three times a day (TID), or once every 6 h (q6h), to achieve exposures similar to those observed in humans (16, 21, 22). Recently, we found that pretreatment of mice with ABT, a nonselective CYP inhibitor, 2 h prior to once-daily FMGX dosing can effectively slow down the metabolic process of MGX in mice, resulting in plasma exposures comparable to those in humans (23). Subsequently, several mouse efficacy studies have used once-daily 78 mg/kg FMGX plus ABT as the standard regimen, since this dosing regimen results in a MGX exposure similar to that observed in humans (24, 25). Improved efficacy as a result of enhanced PK was consistently observed (26–28, 37). Therefore, we have adopted the same strategy in our current study, and the 78 mg/kg of FMGX plus ABT dose (29) was used for all experiments.
MGX was found to be rapidly and extensively distributed to nonlesion liver tissues, with drug levels mirroring day one plasma PK, albeit with higher liver drug concentrations across the measured time points. This observation was consistent with a previous report about tissue distribution of MGX in rats and monkeys using whole-body autoradiography (QWBA) (38), in which the highest-radioactivity Cmax values in rats were found in bile and fat tissues, followed by liver. However, this rapid partitioning of MGX was not observed in the lesion compartment. The average lesion/nonlesion drug ratios were only about 0.07 at 1 h after a single dose, and gradually increased over time to about 0.58 at 24 h postdose, suggesting that MGX accumulation into the abscess occurs over time. This is likely due to the fact that abscess core is nonvascularized, and hence the drug can only diffuse passively through this matrix. Passive drug diffusion into a necrotic lesion can be a slow process, and the diffusion efficiency inversely correlates with hydrophobicity and tissue protein binding of the compound, as well as with the size of the necrotic core (39). Nonetheless, enhanced lesion penetration was observed with continued dosing. The drug concentration gradient between lesion and nonlesion compartments was reversed after 3 doses of FMGX, and drug levels in lesions were 1.92-fold higher than those in nonlesion liver tissues. It was noticed that absolute MGX concentrations in nonlesion compartments were lower in mice that received 3 doses of FMGX compared to concentrations in those that received 2 doses, and the mean systemic drug exposure was also lower in mice treated for 3 days (5.5 μg/ml at 24 h after 3 days versus 3.6 μg/ml at 24 h after 2 days of FMGX treatment). This finding was unexpected, and it may be a stochastic result of the small group sample size and relatively large individual variations. It is also possible that repeated ABT dosing may have inversely affected the CYP inhibition effect, reflected by the decreased systemic MGX level in multiday-treated mice compared to single-dosed mice. However, whether this projection stands true, and the underlying mechanism, warrant further investigation.
To increase the spatial resolution of our analysis and to gain a holistic view of drug distribution at the infected tissue site, we captured MSI snapshots of representative liver sections from both single-dose and 3-day treatment courses. Interestingly, highly heterogenous intralesion MGX distribution was visualized across the single-dosed tissue sections. MGX ion intensity at 6 h after single dosing was observed to be high in the inflammatory rim, where immune cells (neutrophils, lymphocytes, and macrophages) aggregate, but low in the necrotic core. However, the 24-h snapshot showed that the necrotic core was the hot spot of MGX deposition, whereas MGX signal in the inflammatory rim was much less abundant. This changing pattern is a function of the kinetics of drug binding and passive diffusion of the free fraction of MGX. Acquisition of ion maps on tissue sections collected after 3 days dosing further provided convincing evidence for sustained accumulation of MGX into necrotic core, consistent with the LCM data.
In the in vivo efficacy evaluation experiments, an increased antifungal effect was observed over time in mice receiving FMGX therapy. With each additional daily dose of FMGX, a greater reduction in liver fungal burden and/or tissue sterilization was observed (Fig. 7). This temporal pattern of antifungal efficacy is likely associated with the accumulation over time of MGX within the liver lesion, which is the location for the main body of the fungal population (33). Within the local environment of the liver lesion, drug pressure may be low and tolerable for the large fungal mass in the beginning of the FMGX therapy, explaining the lack of fungal burden reduction on day 1. However, after multiple doses and the accumulation of MGX within the lesions, the antifungal efficacy was clearly observed. In the current study, 4 days of FMGX treatment eliminated any measurable fungal burden in the infected livers (Fig. 7). In contrast, the standard-of-care representative MCF therapy was only initially effective after therapy started, but the activity was not sufficient in overcoming persistent fungal growth when the therapy course was extended to 4 days. Histopathological evaluation of two extra mice after the 4-day treatment further confirmed the in vivo antifungal efficacy difference between these two drugs (Fig. 8).
Previous PK/PD studies have defined MGX as an AUC/MIC-driven drug, and the 24-h total drug AUC (tAUC)/MIC values resulting in kidney burden stasis ranged from 6,678 to 12,952 for three C. albicans strains with MGX MICs of 0.002 to 0.008 μg/ml (21). In our study, the tAUC/MIC was 8,988 in lesion tissue, 26,228 in nonlesion tissue, and 19,162 in plasma, using parameters obtained after a single dose of FMGX. There are a few important differences between the previous study and ours which must be noted, including different animal models (neutropenic IC versus immunocompetent IAC), different C. albicans strains, different dosing regimens (FMGX q6h versus FMGX+ABT QD), different drug exposure measurement (plasma versus plasma, lesion, and nonlesion tissue), and different efficacy endpoints (kidney burden versus liver burden). In our study, the lesion tAUC/MIC fell within the range of the previously defined preclinical PK/PD target and was associated with a high degree of in vivo fungicidal activity, as evident by marked liver burden clearance in all mice after 4 days of treatment.
In summary, we have investigated the therapeutic potential of FMGX for the treatment of IAC. After dosing FMGX in conjunction with ABT, the resulting MGX exposures in mice were similar to clinically seen exposures. At these exposures, MGX demonstrated marked in vivo potency in clearing liver burden after multiple doses. In comparison, the first-line echinocandin drug MCF was initially effective, but liver burdens were only marginally reduced at the end of a 4-day treatment. Importantly, we have also characterized drug penetration of MGX into intra-abdominal abscess lesions. MGX demonstrated time-dependent accumulation in the liver lesion compartment upon repeat dosing, in line with the observed in vivo efficacy. Overall, our results suggest that FMGX is a promising therapeutic option for the treatment of IAC patients.
MATERIALS AND METHODS
Antifungal agents.
Fosmanogepix, manogepix, manogepix-d4 (Amplyx Pharmaceuticals, Inc., San Diego, CA), and micafungin (Astellas Pharma Inc., Tokyo, Japan) were obtained as standard powders from their manufacturers. Antifungal drug formulation was performed according to the manufacturer’s instructions as previously described (23). ABT was purchased from Sigma and was dissolved in sterile water at 10 mg/ml prior to use.
Strain.
Candida albicans strain SC5314 was grown in yeast extract-peptone-dextrose (YPD) broth at 37°C with shaking overnight. Cells were washed, counted, and prepared in a sterile stool matrix containing a final concentration of 1 × 108 CFU/ml for infection as previously described (33). MICs for MGX and MCF were determined using the broth microdilution method following Clinical and Laboratory Standards Institute (CLSI) protocol M27-A3 (40).
Animals.
Female 20- to 22-g CD1 mice (Charles River, Wilmington, MA) were housed in the animal biosafety level 2 Research Animal Facility at Center for Discovery and Innovation, Hackensack Meridian Health (HMH). All experimental procedures were performed in accordance with National Research Council guidelines and approved by the HMH Institutional Animal Care and Use Committee (IACUC).
Mouse model of intra-abdominal candidiasis, PK, and efficacy evaluation.
A well-established, immunocompetent IAC mouse model was used for this study (41). Mice were infected intraperitoneally (i.p.) with 100 μl cell suspension prepared in sterile stool matrix containing of 1 × 107 CFU of C. albicans SC5314, as previously described (33). All drug treatment was initiated on day 3 postinfection. For the single-dose PK study, mice received a single oral (p.o.) treatment of 78 mg/kg FMGX and sacrificed at 1, 3, 6, and 24 h posttreatment (4 mice per time point). ABT was administered at 50 mg/kg via oral gavage 2 h prior to FMGX dosing. Livers were explored for abscesses, dissected, placed on a cryohistology tray, snap-frozen in liquid nitrogen, and stored at −80°C for MALDI-MSI and LCM-directed drug quantification. Blood was also collected at 0, 0.5, 1, 3, 6, and 24 h posttreatment for plasma drug concentration measurement. For multidose PK, mice received 2 or 3 doses of once-daily treatment of oral FMGX at 78 mg/kg. ABT at 50 mg/kg was orally administered 2 h prior to each FMGX dose. Livers and blood were collected for drug exposure evaluation (5 mice per time point) at 24 h after completion of each regimen. The 4-day efficacy study included three treatment arms, FMGX (78 mg/kg p.o. QD) (plus ABT), MCF (5 mg/kg i.p. QD), or phosphate-buffered saline (PBS) (p.o. QD). Liver fungal burden was assessed predose (n = 3), 24 h after single dose, and at 2 days, 3 days, and 4 days treatment (n = 6 mice per regimen per time point).
Tissue sectioning.
Using an CM 1860 UV cryostat (Leica, Buffalo Grove, IL), 10-μm thick tissue sections were cut and mounted onto positively charged slides for MALDI-MSI analysis and GMS and H&E staining. Adjacent 25-μm thick tissue sections were mounted onto a thin polymer membrane slide (polyethylene terephthalate [PET]) for laser capture microdissection. All slides were air dried for 10 min, sealed in a small airtight sealable bag, and transferred to −80°C storage until analysis.
Laser capture microdissection.
Distinct subcompartments of infected liver tissue (e.g., abscesses and surrounding uninvolved tissue) totaling 1 million to 3 million μm2 were carefully dissected from between 4 and 6 serial liver tissue sections using an LMD 6 (CC7000) system (Leica) (42). Lesion areas were identified optically from the bright field image scan and by comparison to the adjacent sectioned GMS- and H&E-stained tissue. The dissected tissues were collected into 0.25-ml standard PCR tubes and immediately stored in the −80°C freezer unless analyzed immediately.
Drug quantitation by LC-MS/MS.
Prior to analysis, the PCR tubes containing LCM samples were thawed at room temperature. A 50-μl of extraction solution (1:1 acetonitrile-methanol [ACN-MeOH] containing 10 ng/ml verapamil internal standard), 10 μl of 1:1 ACN-H2O solution, and 2 μl of PBS buffer were added, then tubes were sonicated for 10 min and centrifuged at 4,000 rpm for 5 min at room temperature. Standard curve and quality control tubes were created by combining 50 μl of extraction solution, 10 μl of serial diluted manogepix or MCF in ACN-H2O, and 2 μl of liver homogenate from untreated animals (42), followed by sonication and centrifugation as described above. Aliquots (50 μl) of supernatant were transferred from both study samples and standards for LC-MS/MS analysis and diluted with an additional 50 μl of deionized water into a 96-well deep-well plate.
LC-MS analysis was performed on an AB Sciex Qtrap 6500+ instrument (ON, Canada) coupled to a Shimadzu high-performance liquid chromatography (HPLC) system. Chromatography was performed with an Agilent SB-C8 column (2.1 mm × 30 mm; particle size, 3.5 μm) using a reverse phase gradient. Mobile phase A was 0.1% formic acid in 100% water and mobile phase B was 0.1% formic acid in 100% ACN. Drug quantitation in tissue was conducted using the transition m/z 359.10/264.10 for MGX and 455.40/165.20 for verapamil, which was used as internal standard.
MALDI MSI sample preparation.
Upon analysis, slides were removed from the −80°C freezer and desiccated for 20 min prior to matrix deposition. DHB at 20 mg/ml (50% MeOH and 0.1% trifluoroacetic acid [TFA]) containing 5 pmol/μl d4-MGX (internal standard) was deposited onto the tissue sections using an M5 sprayer (HTX Imaging, Chapel Hill, NC). The spraying parameters were as follows: 60 μl/min flow rate, 1,200 mm/min nozzle velocity, 60°C nozzle temperature, 3-mm track spacing, 10 lb/in2 N2 gas, 30 spray cycles, and a crisscross spray pattern.
MALDI-MSI.
A solariX 7T MALDI Fourier-transform ion cyclotron resonance (FT-ICR) mass spectrometer (Bruker Daltonics, Billerica, MA) equipped with a SmartBeam II Nd:YAG (355-nm) laser was employed to perform the MALDI imaging experiments. The instrument was operated in the positive ion mode in the mass range of m/z 150 to 3,000, with a transient length of 0.7340 s, resulting in an estimated resolving power of 99,000 at m/z 400. The continuous accumulation of selected ions (CASI) function was applied with the quadrupole (Q1) mass of m/z = 350 and an isolation window of 200.0 m/z. The laser power was set at 17%, accumulating 300 laser shots/pixel. Data were acquired at a spatial resolution of 50 μm. After data acquisition, the slides were washed with 70% ethanol and stained with hematoxylin and eosin (H&E) following the manufacturer’s protocol. Slides were then scanned in using the Pannoramic DESK II DW scanner (3DHistech, Budapest, Hungary).
All MSI data were processed using the SCiLS Lab MVS 2020a Pro software (Bruker Daltonics). Single-dosed samples along with their stained sections were incorporated into one data set for signal intensity comparison across the time points postdose. MSI images were directly merged with their H&E sections to demonstrate MGX distribution within each section. Ion maps of MGX were visualized using the rainbow scale color scheme and the weak denoising function. The same data processing steps were applied to the repeatedly dosed data.
Statistical analysis.
Absolute drug concentrations were graphed and statistically analyzed in Prism 9.0.0 (GraphPad Software, Inc., San Diego, CA). Drug levels in different tissue compartments and liver burden counts at various time points were compared by one-way analysis of variance (ANOVA), and Dunn’s multiple comparison was used for the post hoc analyses. Statistical significance was defined as a P value of <0.05.
ACKNOWLEDGMENTS
This work was supported by Amplyx Pharmaceuticals, Inc., research contract to Y.Z. and D.S.P., by NIH grant R01AI109025 to D.S.P., and by NIH grants S10 OD023524-01 and S10 OD018072-01 to V.D.
We thank Enriko Dolgov for his great help with the animal experiments.
D.S.P. serves on advisory boards for Cidara, Amplyx, Scynexis, Astellas, Matinas, and N8 Medical. D.S.P. has an issued U.S. patent concerning echinocandin resistance. K.J.S. was an employee of Amplyx and is now an independent consultant at Hearts Consulting Group, LLC.
REFERENCES
- 1.Clancy CJ, Nguyen MH. 2013. Finding the “missing 50%” of invasive candidiasis: how nonculture diagnostics will improve understanding of disease spectrum and transform patient care. Clin Infect Dis 56:1284–1292. doi: 10.1093/cid/cit006. [DOI] [PubMed] [Google Scholar]
- 2.Wisplinghoff H, Bischoff T, Tallent SM, Seifert H, Wenzel RP, Edmond MB. 2004. Nosocomial bloodstream infections in US hospitals: analysis of 24,179 cases from a prospective nationwide surveillance study. Clinical infectious diseases: an official publication of the Infectious Diseases Society of America 39:309–317. doi: 10.1086/421946. [DOI] [PubMed] [Google Scholar]
- 3.Bongomin F, Gago S, Oladele RO, Denning DW. 2017. Global and multi-national prevalence of fungal diseases—estimate precision. J Fungi (Basel, Switzerland) 3:57. doi: 10.3390/jof3040057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Strollo S, Lionakis MS, Adjemian J, Steiner CA, Prevots DR. 2016. Epidemiology of hospitalizations associated with invasive candidiasis, United States, 2002–2012(1). Emerg Infect Dis 23:7–13. doi: 10.3201/eid2301.161198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Montravers P, Leroy O, Eckmann C. 2015. Intra-abdominal candidiasis: it’s still a long way to get unquestionable data. Intensive Care Med 41:1682–1684. doi: 10.1007/s00134-015-3894-y. [DOI] [PubMed] [Google Scholar]
- 6.Montravers P, Dupont H, Eggimann P. 2013. Intra-abdominal candidiasis: the guidelines-forgotten non-candidemic invasive candidiasis. Intensive Care Med 39:2226–2230. doi: 10.1007/s00134-013-3134-2. [DOI] [PubMed] [Google Scholar]
- 7.Vergidis P, Clancy CJ, Shields RK, Park SY, Wildfeuer BN, Simmons RL, Nguyen MH. 2016. Intra-abdominal candidiasis: the importance of early source control and antifungal treatment. PLoS One 11:e0153247. doi: 10.1371/journal.pone.0153247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pappas PG, Kauffman CA, Andes DR, Clancy CJ, Marr KA, Ostrosky-Zeichner L, Reboli AC, Schuster MG, Vazquez JA, Walsh TJ, Zaoutis TE, Sobel JD. 2016. Clinical practice guideline for the management of candidiasis: 2016 update by the Infectious Diseases Society of America. Clin Infect Dis 62:e1–e50. doi: 10.1093/cid/civ933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Andes DR, Safdar N, Baddley JW, Playford G, Reboli AC, Rex JH, Sobel JD, Pappas PG, Kullberg BJ, Mycoses Study Group. 2012. Impact of treatment strategy on outcomes in patients with candidemia and other forms of invasive candidiasis: a patient-level quantitative review of randomized trials. Clin Infect Dis 54:1110–1122. doi: 10.1093/cid/cis021. [DOI] [PubMed] [Google Scholar]
- 10.Shields RK, Nguyen MH, Press EG, Clancy CJ. 2014. Abdominal candidiasis is a hidden reservoir of echinocandin resistance. Antimicrob Agents Chemother 58:7601–7605. doi: 10.1128/AAC.04134-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sganga G, Wang M, Capparella MR, Tawadrous M, Yan JL, Aram JA, Montravers P. 2019. Evaluation of anidulafungin in the treatment of intra-abdominal candidiasis: a pooled analysis of patient-level data from 5 prospective studies. Eur J Clin Microbiol Infect Dis 38:1849–1856. doi: 10.1007/s10096-019-03617-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pappas PG, Rotstein CM, Betts RF, Nucci M, Talwar D, De Waele JJ, Vazquez JA, Dupont BF, Horn DL, Ostrosky-Zeichner L, Reboli AC, Suh B, Digumarti R, Wu C, Kovanda LL, Arnold LJ, Buell DN. 2007. Micafungin versus caspofungin for treatment of candidemia and other forms of invasive candidiasis. Clin Infect Dis 45:883–893. doi: 10.1086/520980. [DOI] [PubMed] [Google Scholar]
- 13.Watanabe NA, Miyazaki M, Horii T, Sagane K, Tsukahara K, Hata K. 2012. E1210, a new broad-spectrum antifungal, suppresses Candida albicans hyphal growth through inhibition of glycosylphosphatidylinositol biosynthesis. Antimicrob Agents Chemother 56:960–971. doi: 10.1128/AAC.00731-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Miyazaki M, Horii T, Hata K, Watanabe NA, Nakamoto K, Tanaka K, Shirotori S, Murai N, Inoue S, Matsukura M, Abe S, Yoshimatsu K, Asada M. 2011. In vitro activity of E1210, a novel antifungal, against clinically important yeasts and molds. Antimicrob Agents Chemother 55:4652–4658. doi: 10.1128/AAC.00291-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bugli F, Posteraro B, Papi M, Torelli R, Maiorana A, Paroni Sterbini F, Posteraro P, Sanguinetti M, De Spirito M. 2013. In vitro interaction between alginate lyase and amphotericin B against Aspergillus fumigatus biofilm determined by different methods. Antimicrob Agents Chemother 57:1275–1282. doi: 10.1128/AAC.01875-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hager CL, Larkin EL, Long L, Abidi FZ, Shaw KJ, Ghannoum MA. 2018. In vitro and in vivo evaluation of the antifungal activity of APX001A/APX001 against Candida auris. Antimicrob Agents Chemother 62:e02319-17. doi: 10.1128/AAC.02319-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhu Y, Kilburn S, Kapoor M, Chaturvedi S, Shaw KJ, Chaturvedi V. 2020. In vitro activity of manogepix against multidrug-resistant and panresistant Candida auris from the New York outbreak. Antimicrob Agents Chemother 64:e01124-20. doi: 10.1128/AAC.01124-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Berkow EL, Lockhart SR. 2018. Activity of novel antifungal compound APX001A against a large collection of Candida auris. J Antimicrob Chemother 73:3060–3062. doi: 10.1093/jac/dky302. [DOI] [PubMed] [Google Scholar]
- 19.Hata K, Horii T, Miyazaki M, Watanabe NA, Okubo M, Sonoda J, Nakamoto K, Tanaka K, Shirotori S, Murai N, Inoue S, Matsukura M, Abe S, Yoshimatsu K, Asada M. 2011. Efficacy of oral E1210, a new broad-spectrum antifungal with a novel mechanism of action, in murine models of candidiasis, aspergillosis, and fusariosis. Antimicrob Agents Chemother 55:4543–4551. doi: 10.1128/AAC.00366-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wiederhold NP, Najvar LK, Fothergill AW, McCarthy DI, Bocanegra R, Olivo M, Kirkpatrick WR, Everson MP, Duncanson FP, Patterson TF. 2015. The investigational agent E1210 is effective in treatment of experimental invasive candidiasis caused by resistant Candida albicans. Antimicrob Agents Chemother 59:690–692. doi: 10.1128/AAC.03944-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhao M, Lepak AJ, VanScoy B, Bader JC, Marchillo K, Vanhecker J, Ambrose PG, Andes DR. 2018. In vivo pharmacokinetics and pharmacodynamics of APX001 against Candida spp. in a neutropenic disseminated candidiasis mouse model. Antimicrob Agents Chemother 62:e02542-17. doi: 10.1128/AAC.02542-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wiederhold NP, Najvar LK, Shaw KJ, Jaramillo R, Patterson H, Olivo M, Catano G, Patterson TF. 2019. Efficacy of delayed therapy with fosmanogepix (APX001) in a murine model of Candida auris invasive candidiasis. Antimicrob Agents Chemother 63:e01120-19. doi: 10.1128/AAC.01120-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhao Y, Lee MH, Paderu P, Lee A, Jimenez-Ortigosa C, Park S, Mansbach RS, Shaw KJ, Perlin DS. 2018. Significantly improved pharmacokinetics enhances in vivo efficacy of APX001 against echinocandin- and multidrug-resistant Candida isolates in a mouse model of invasive candidiasis. Antimicrob Agents Chemother 62:e00425-18. doi: 10.1128/AAC.00425-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hodges MR, Ople E, Shaw KJ, Mansbach R, Van Marle SJ, Van Hoogdalem E-J, Wedel P, Kramer W. 2017. First-in-human study to assess safety, tolerability and pharmacokinetics of APX001 administered by intravenous infusion to healthy subjects. Open Forum Infect Dis 4:S526–S526. doi: 10.1093/ofid/ofx163.1370. [DOI] [Google Scholar]
- 25.Hodges MR, Ople E, Shaw KJ, Mansbach R, Van Marle SP, Van Hoogdalem E-J, Kramer W, Wedel P. 2017. Phase 1 study to assess safety, tolerability and pharmacokinetics of single and multiple oral doses of APX001 and to investigate the effect of food on APX001 bioavailability. Open Forum Infect Dis 4:S534–S534. doi: 10.1093/ofid/ofx163.1390. [DOI] [Google Scholar]
- 26.Gebremariam T, Alkhazraji S, Alqarihi A, Jeon HH, Gu Y, Kapoor M, Shaw KJ, Ibrahim AS. 2018. APX001 is effective in the treatment of murine invasive pulmonary aspergillosis. Antimicrob Agents Chemother 63:e01713-18. doi: 10.1128/AAC.01713-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shaw KJ, Schell WA, Covel J, Duboc G, Giamberardino C, Kapoor M, Moloney M, Soltow QA, Tenor JL, Toffaletti DL, Trzoss M, Webb P, Perfect JR. 2018. In vitro and in vivo evaluation of APX001A/APX001 and other Gwt1 inhibitors against Cryptococcus. Antimicrob Agents Chemother 62:e00523-18. doi: 10.1128/AAC.00523-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Alkhazraji S, Gebremariam T, Alqarihi A, Gu Y, Mamouei Z, Singh S, Wiederhold NP, Shaw KJ, Ibrahim AS. 2019. Fosmanogepix (APX001) is effective in the treatment of immunocompromised mice infected with invasive pulmonary scedosporiosis or disseminated fusariosis. Antimicrob Agents Chemother 64:e01735-19. doi: 10.1128/AAC.01735-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shaw KJ, Ibrahim AS. 2020. Fosmanogepix: a review of the first-in-class broad spectrum agent for the treatment of invasive fungal infections. J Fungi (Basel, Switzerland) 6:239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wiederhold NP. 2018. The antifungal arsenal: alternative drugs and future targets. Int J Antimicrob Agents 51:333–339. doi: 10.1016/j.ijantimicag.2017.09.002. [DOI] [PubMed] [Google Scholar]
- 31.Li D, She X, Calderone R. 2020. The antifungal pipeline: the need is established. Are there new compounds? FEMS Yeast Res 20:foaa023. doi: 10.1093/femsyr/foaa023. [DOI] [PubMed] [Google Scholar]
- 32.Hope W, Drusano GL, Rex JH. 2016. Pharmacodynamics for antifungal drug development: an approach for acceleration, risk minimization and demonstration of causality. J Antimicrob Chemother 71:3008–3019. doi: 10.1093/jac/dkw298. [DOI] [PubMed] [Google Scholar]
- 33.Zhao Y, Prideaux B, Nagasaki Y, Lee MH, Chen PY, Blanc L, Ho H, Clancy CJ, Nguyen MH, Dartois V, Perlin DS. 2017. Unraveling drug penetration of echinocandin antifungals at the site of infection in an intra-abdominal abscess model. Antimicrob Agents Chemother 61:e01009-17. doi: 10.1128/AAC.01009-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee A, Prideaux B, Zimmerman M, Carter C, Barat S, Angulo D, Dartois V, Perlin DS, Zhao Y. 2019. Penetration of ibrexafungerp (formerly SCY-078) at the site of infection in an intra-abdominal candidiasis mouse model. Antimicrob Agents Chemother 64:e02268-19. doi: 10.1128/AAC.02268-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pfaller MA, Duncanson F, Messer SA, Moet GJ, Jones RN, Castanheira M. 2011. In vitro activity of a novel broad-spectrum antifungal, E1210, tested against Aspergillus spp. determined by CLSI and EUCAST broth microdilution methods. Antimicrob Agents Chemother 55:5155–5158. doi: 10.1128/AAC.00570-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Castanheira M, Duncanson FP, Diekema DJ, Guarro J, Jones RN, Pfaller MA. 2012. Activities of E1210 and comparator agents tested by CLSI and EUCAST broth microdilution methods against Fusarium and Scedosporium species identified using molecular methods. Antimicrob Agents Chemother 56:352–357. doi: 10.1128/AAC.05414-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Viriyakosol S, Kapoor M, Okamoto S, Covel J, Soltow QA, Trzoss M, Shaw KJ, Fierer J. 2018. APX001 and other Gwt1 inhibitor prodrugs are effective in experimental Coccidioides immitis pneumonia. Antimicrob Agents Chemother 63:e01715-18. doi: 10.1128/AAC.01715-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mansbach R, Shaw KJ, Hodges MR, Coleman S, Fitzsimmons ME. 2017. Absorption, distribution, and excretion of 14C-APX001 after single-dose administration to rats and monkeys. Open Forum Infect Dis 4:S472–S472. doi: 10.1093/ofid/ofx163.1209. [DOI] [Google Scholar]
- 39.Sarathy JP, Zuccotto F, Hsinpin H, Sandberg L, Via LE, Marriner GA, Masquelin T, Wyatt P, Ray P, Dartois V. 2016. Prediction of drug penetration in tuberculosis lesions. ACS Infect Dis 2:552–563. doi: 10.1021/acsinfecdis.6b00051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Clinical and Laboratory Standards Institute. 2008. Reference method for broth dilution antifungal susceptibility testing of yeasts, 3rd ed. CLSI document M27-A3. Clinical and Laboratory Standards Institute, Wayne, PA. [Google Scholar]
- 41.Cheng S, Clancy CJ, Xu W, Schneider F, Hao B, Mitchell AP, Nguyen MH. 2013. Profiling of Candida albicans gene expression during intra-abdominal candidiasis identifies biologic processes involved in pathogenesis. J Infect Dis 208:1529–1537. doi: 10.1093/infdis/jit335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zimmerman M, Blanc L, Chen PY, Dartois V, Prideaux B. 2018. Spatial quantification of drugs in pulmonary tuberculosis lesions by laser capture microdissection liquid chromatography mass spectrometry (LCM-LC/MS). J Vis Exp 134:57402. doi: 10.3791/57402. [DOI] [PMC free article] [PubMed] [Google Scholar]