

A decade of research has shown that the molecule c-di-GMP functions as a central second messenger in many bacteria. A high level of c-di-GMP is associated with biofilm formation, whereas a low level of c-di-GMP is associated with a planktonic single-cell bacterial lifestyle. c-di-GMP is formed by diguanylate cyclases and is degraded by specific phosphodiesterases.
KEYWORDS: biofilm dispersal, c-di-GMP, phosphodiesterase, Pseudomonas aeruginosa
ABSTRACT
A decade of research has shown that the molecule c-di-GMP functions as a central second messenger in many bacteria. A high level of c-di-GMP is associated with biofilm formation, whereas a low level of c-di-GMP is associated with a planktonic single-cell bacterial lifestyle. c-di-GMP is formed by diguanylate cyclases and is degraded by specific phosphodiesterases. We previously presented evidence that the ectopic expression of the Escherichia coli phosphodiesterase YhjH in Pseudomonas aeruginosa results in biofilm dispersal. More recently, however, evidence has been presented that the induction of native c-di-GMP phosphodiesterases does not lead to a dispersal of P. aeruginosa biofilms. The latter result may discourage attempts to use c-di-GMP signaling as a target for the development of antibiofilm drugs. However, here, we demonstrate that the induction of the P. aeruginosa c-di-GMP phosphodiesterases PA2133 and BifA indeed results in the dispersal of P. aeruginosa biofilms in both a microtiter tray biofilm assay and a flow cell biofilm system.
INTRODUCTION
In recent years, it has become clear that bacterial biofilms constitute a significant part of antibiotic-resistant infections (1–3). In biofilms, bacteria are located in densely packed, slow-growing microcolonies concealed in a self-produced matrix of biopolymers (4). In this life mode, the bacteria attain the highest levels of resistance to our present assortment of antibiotics and our immune system (1, 2). Accordingly, biofilms are involved in a number of persistent infections, e.g., in the lungs of cystic fibrosis patients, in chronic wounds, in the urinary tract, and in association with various catheters and a variety of implanted medical devices. Because the use of conventional antimicrobial compounds in many cases cannot eradicate biofilms, there is an urgent need to develop alternative measures to combat biofilm-based infections.
Current research has shown that bacteria employ c-di-GMP signaling to regulate whether they form a biofilm or assume a planktonic lifestyle (5, 6). Diguanylate cyclase (DGC) enzymes catalyze the formation of the molecule c-di-GMP, whereas c-di-GMP phosphodiesterase (PDE) enzymes catalyze the degradation of c-di-GMP in bacteria. In addition to their catalytic GGDEF and EAL/HD-GYP domains, these enzymes have regulatory domains and are thought to regulate the lifestyle (planktonic versus biofilm) of bacteria in response to environmental cues. Bacteria in biofilms generally display an elevated cellular level of c-di-GMP, whereas planktonic bacteria generally display a low level of c-di-GMP. Genomic analyses have shown that the GGDEF and EAL domains are the most abundant motifs among bacteria, and c-di-GMP has been found to be the central biofilm regulator in all Gram-negative bacteria investigated to date (5). On the contrary, c-di-GMP is not produced by humans or other mammals, suggesting that c-di-GMP signaling could be an interesting antibiofilm drug target.
Reduction of the c-di-GMP level in bacteria has been suggested as a means of eradicating infections caused by biofilms (7, 8). In support of this strategy, we have previously provided evidence that the induction of the Escherichia coli PDE YhjH in Pseudomonas aeruginosa and Pseudomonas putida leads to biofilm dispersal (7, 9, 10). It has been anticipated that when forced away from the protective biofilm, planktonic bacteria become susceptible to the antimicrobial action of the host immune system and conventional antimicrobials. Accordingly, a recent study provided evidence that P. aeruginosa bacteria dispersed from biofilms become susceptible to antibiotics after a few hours (11). However, in that study, evidence was also provided that the induction of native PDEs in P. aeruginosa does not lead to biofilm dispersal. The E. coli PDE YhjH (also designated PdeH) that was employed in previous biofilm dispersal studies (7, 9, 10) has been shown to be a strongly expressed master PDE that eradicates the global effects of several DGCs by draining the global c-di-GMP pool (12). It is possible, therefore, that biofilm dispersal mediated by the induction of YhjH is a special case and that the induction of native PDEs in P. aeruginosa is not sufficient to induce biofilm dispersal. Therefore, in the present study, we have investigated if the induction of the P. aeruginosa PDEs BifA and PA2133 leads to the dispersal of P. aeruginosa biofilms.
RESULTS
Construction and characterization of P. aeruginosa strains with inducible c-di-GMP phosphodiesterases.
We chose to use the P. aeruginosa PDEs PA2133 and BifA for our studies. PA2133 and BifA are both characterized bona fide c-di-GMP PDEs (11, 13), and PA2133 was employed by Chambers et al. (11) in their study that suggested that the induction of a native c-di-GMP PDE is not sufficient to disperse P. aeruginosa biofilms. We PCR amplified the PA2133 and bifA genes from P. aeruginosa DNA and fused the genes to the arabinose-inducible PBAD promoter by cloning them into the vector pJN105 (14). Subsequently, the PBAD-PDE gene fusions were PCR amplified, moved to a mini-CTX vector, and inserted in the ϕCTX attB site of P. aeruginosa PAO1, resulting in the P. aeruginosa PBAD-PA2133 and P. aeruginosa PBAD-bifA strains. As expected, arabinose-mediated induction of the PDEs in these strains resulted in strongly diminished biofilm formation as determined using a microtiter tray assay where arabinose was added at the start (Fig. 1A). We noted that even without arabinose induction, the P. aeruginosa PBAD-PA2133 strain formed very little biofilm, whereas the P. aeruginosa PBAD-bifA strain showed somewhat reduced biofilm formation in comparison to the wild type (Fig. 1A). These results are probably due to the well-known leakiness of the PBAD promoter, and they indicate that PA2133 has higher PDE activity than BifA. Due to the low level of biofilm formation of the P. aeruginosa PBAD-PA2133 strain, we also inserted the PBAD-PA2133 and PBAD-bifA fusions into the ϕCTX attB site of a P. aeruginosa wspF mutant strain. The wspF mutant has an elevated level of c-di-GMP due to a hyperactive WspR DGC (15), and accordingly, the P. aeruginosa wspF PBAD-PA2133 strain formed a large amount of biofilm in the absence of arabinose induction, although it was smaller than the amount of biofilm formed by the P. aeruginosa wspF PBAD-bifA strain and the P. aeruginosa wspF strain (Fig. 1B [note the difference in the ordinate scales for Fig. 1A and B]). Again, the presence of an arabinose inducer resulted in reduced biofilm formation for the P. aeruginosa wspF PBAD-bifA and P. aeruginosa wspF PBAD-PA2133 strains (Fig. 1B). In addition to facilitating our biofilm dispersal studies, the use of the P. aeruginosa wspF mutant is of clinical relevance since such mutants are frequently isolated from cystic fibrosis patients (16).
FIG 1.

Amount of biofilm formed in microtiter tray wells by P. aeruginosa, P. aeruginosa PBAD-bifA, and P. aeruginosa PBAD-PA2133 (A) and by P. aeruginosa wspF, P. aeruginosa wspF PBAD-bifA, and P. aeruginosa wspF PBAD-PA2133 (B) after 10 h of growth in the presence (+) or absence (−) of arabinose. Biofilm biomass was determined by the use of a crystal violet staining assay. Average amounts of biofilm biomass from three replicate cultures are shown. The bars indicate standard deviations, and the asterisks indicate significant differences (***, P < 0.001; ****, P < 0.0001). wt, wild type.
In order to assess the level of c-di-GMP in our strains, we used a plasmid-based cdrA-gfp fluorescent reporter, which has been shown to gauge the c-di-GMP level in P. aeruginosa (17). The cdrA-gfp reporter was transformed into the P. aeruginosa PBAD-PA2133 strain, the P. aeruginosa PBAD-bifA strain, as well as the P. aeruginosa wild type as a control. The P. aeruginosa wspF PBAD-PA2133 and P. aeruginosa wspF PBAD-bifA strains, however, form clumps in liquid culture due to the overexpression of c-di-GMP-regulated exopolysaccharide, which prevented proper fluorescence measurements. Therefore, to assess c-di-GMP levels in the wspF strains using the cdrA-gfp fluorescent reporter, we constructed the P. aeruginosa psl pel wspF PBAD-PA2133 and P. aeruginosa psl pel wspF PBAD-bifA strains, which do not form clumps since the Psl and Pel exopolysaccharide genes are deleted. Subsequently, the cdrA-gfp reporter plasmid was transformed into the constructed strains with deleted exopolysaccharide genes as well as a P. aeruginosa psl pel wspF strain as a control. As shown in Fig. 2, PDE induction with arabinose reduced the fluorescence readout from all the strains except the P. aeruginosa PBAD-PA2133 strain, which had a very low fluorescence readout even in the absence of induction (note the difference in the ordinate scales for Fig. 2A and B). In general, even in the absence of arabinose, the strains with the PBAD-PDE fusions showed a reduced fluorescence readout compared to the control strains without the PBAD-PDE fusions (Fig. 2). The results are in agreement with the biofilm quantifications shown in Fig. 1, and they support the suggestion that the PBAD-PA2133 and PBAD-bifA fusions to some extent are leaky and that PA2133 has higher PDE activity than BifA.
FIG 2.

c-di-GMP levels gauged by the use of a cdrA-gfp fluorescent reporter in cultures formed in microtiter tray wells by P. aeruginosa/pCdrA-gfp, P. aeruginosa PBAD-bifA/pCdrA-gfp, and P. aeruginosa PBAD-PA2133/pCdrA-gfp (A) and by P. aeruginosa wspF pel psl/pCdrA-gfp, P. aeruginosa wspF pel psl PBAD-bifA/pCdrA-gfp, and P. aeruginosa wspF pel psl PBAD-PA2133/pCdrA-gfp (B) after 10 h of growth in the presence (+) or absence (−) of arabinose. The average amount of GFP fluorescence/OD600 was calculated from 2 biological replicates, each with three technical replicates. The bars indicate standard deviations, and the asterisks indicate significant differences (*, P < 0.05; ****, P < 0.0001).
Induction of PBAD-PA2133 or PBAD-bifA leads to dispersal of P. aeruginosa biofilms grown in microtiter trays.
We employed a microtiter tray-based assay to investigate the effect of the induction of native PDEs on P. aeruginosa biofilms. In this assay, P. aeruginosa initially forms biofilm and subsequently enters an intrinsic dispersal phase, probably due to substrate limitation (18). We employed an assay that reveals the phase (biofilm formation versus intrinsic dispersal) of the biofilms (19). This was achieved by inoculating the microtiter trays with various dilutions of overnight cultures. Thus, following incubation, the biofilms that were started with a high number of bacteria were more mature than the biofilms that were started with a lower number of bacteria. Consequently, the various biofilm phases can be displayed in a plot that shows biofilm biomass as a function of the dilution of the inoculum (see Fig. S1 in the supplemental material). We grew the biofilms in microtiter trays for 8 h and then added either an arabinose inducer or water without the inducer as a control, and after 1 or 2 h of subsequent incubation, the amount of biofilm in the wells was quantified via crystal violet staining. In the absence of arabinose induction, the six P. aeruginosa strains initiated the intrinsic dispersal phase at different population sizes (Fig. 3 and 4), probably due to differences in the basal levels of c-di-GMP. However, the induction of either PA2133 or bifA resulted in biofilm dispersal (Fig. 3 and 4). The induction of PA2133 resulted in the efficient dispersal of both wild-type and wspF biofilms already after 1 h of induction (Fig. 3C and Fig. 4C), whereas the induction of bifA resulted in a substantial dispersal of the wild-type biofilms after 1 and 2 h (Fig. 3B and E) but only little dispersal of the wspF biofilms (Fig. 4B and E). The differences in the extents of biofilm dispersal are probably a consequence of differences in the efficiencies of the PA2133 and BifA PDEs. Importantly, however, the experiments clearly show that the induction of a native PDE leads to the dispersal of P. aeruginosa biofilms.
FIG 3.

Biofilms of P. aeruginosa (A and D), P. aeruginosa PBAD-bifA (B and E), and P. aeruginosa PBAD-PA2133 (C and F) were grown in microtiter tray wells for 8 h, after which an arabinose inducer (+) or a water control (−) was added, and incubation was continued for 1 h (A to C) or 2 h (D to F). The biofilms were initiated with various dilutions of the overnight cultures in order to establish biofilms with different degrees of maturity. The numbers at the x axis refer to the dilution of the inoculum. Biofilm biomass was determined by the use of a crystal violet staining assay. Average amounts of biofilm biomass from three replicate cultures are shown. The bars indicate standard deviations, and the asterisks indicate significant differences (*, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001).
FIG 4.

Biofilms of P. aeruginosa wspF (A and D), P. aeruginosa wspF PBAD-bifA (B and E), and P. aeruginosa wspF PBAD-PA2133 (C and F) were grown in microtiter tray wells for 8 h, after which an arabinose inducer (+) or a water control (−) was added, and incubation was continued for 1 h (A to C) or 2 h (D to F). The biofilms were initiated with various dilutions of the overnight cultures in order to establish biofilms with different degrees of maturity. The numbers at the x axis refer to the dilution of the inoculum. Biofilm biomass was determined by the use of a crystal violet staining assay. Average amounts of biofilm biomass from four replicate cultures are shown. The bars indicate standard deviations, and the asterisks indicate significant differences (*, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001).
Induction of PBAD-PA2133 or PBAD-bifA leads to dispersal of P. aeruginosa biofilms grown in flow cells.
We also employed a flow cell system to study the effect of the induction of native PDEs on P. aeruginosa biofilms. In order to image the biofilms by confocal laser scanning microscopy (CLSM), the strains were tagged with mini-Tn7-gfp. In the flow cell system, the biofilm formation defect of the P. aeruginosa PBAD-PA2133 strain was even more pronounced than that in the microtiter trays. Only a few cells were able to attach to the glass surface, and microcolonies were not formed upon subsequent incubation (Fig. S2). For the flow cell experiments, we therefore focused on the wspF strains. Initially, we quantified the amounts of biofilm formed by the P. aeruginosa wspF, P. aeruginosa wspF PBAD-PA2133, and P. aeruginosa wspF PBAD-bifA strains in the flow cells. We used automated CLSM to capture images every 60 min at three different locations in three developing biofilms for each of the three strains. The amount of biofilm biomass was subsequently quantified using the Measurement Pro feature of Imaris image analysis software. In accordance with the results of the microtiter tray biofilm assays, the P. aeruginosa wspF strain formed more biofilm than the P. aeruginosa wspF PBAD-bifA strain, whereas the P. aeruginosa wspF PBAD-PA2133 strain formed the smallest amount of biofilm (Fig. 5). The biofilm biomasses of all three strains seemed to reach a plateau after around 60 h (Fig. 5). In our PDE induction experiments, we added an arabinose inducer to the medium after 68 h of flow cell biofilm cultivation, and the amount of biomass was subsequently quantified every 14 min by the use of CLSM and image analysis. As shown in Fig. 6 and 7, the induction of PA2133 resulted in substantial biofilm dispersal, whereas the induction of BifA resulted in more modest biofilm dispersal. However, again, the results clearly show that the induction of a native PDE results in the dispersal of P. aeruginosa biofilms.
FIG 5.
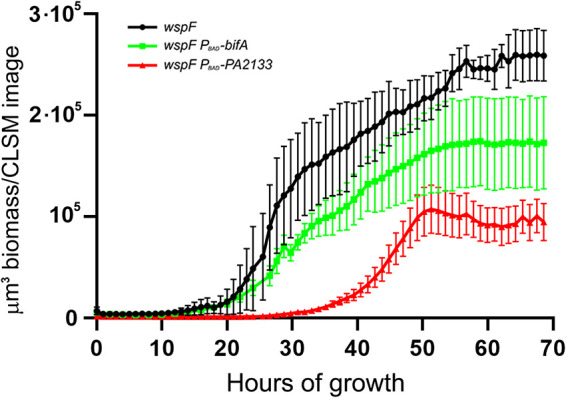
Biofilms of P. aeruginosa wspF, P. aeruginosa wspF PBAD-bifA, and P. aeruginosa wspF PBAD-PA2133 were grown in flow cells for 68 h. Automated CLSM was used to capture images every 60 min at three different locations in three biofilms of each of the three strains. The amount of biofilm biomass was subsequently quantified using the Measurement Pro feature of Imaris. The average biomass/CLSM image of the three P. aeruginosa wspF, P. aeruginosa wspF PBAD-bifA, and P. aeruginosa wspF PBAD-PA2133 biofilms as a function of time is shown. Bars indicate standard deviations. The final biomass (after 68 h of cultivation) was used to assess statistical significance. The biomass of the wspF biofilm differed significantly from that of the wspF PBAD-bifA biofilm (P = 0.048), the biomass of the wspF biofilm differed significantly from that of the wspF PBAD-PA2133 biofilm (P = 0.0001), and the biomass of the wspF PBAD-bifA biofilm differed significantly from that of the wspF PBAD-PA2133 biofilm (P = 0.040).
FIG 6.
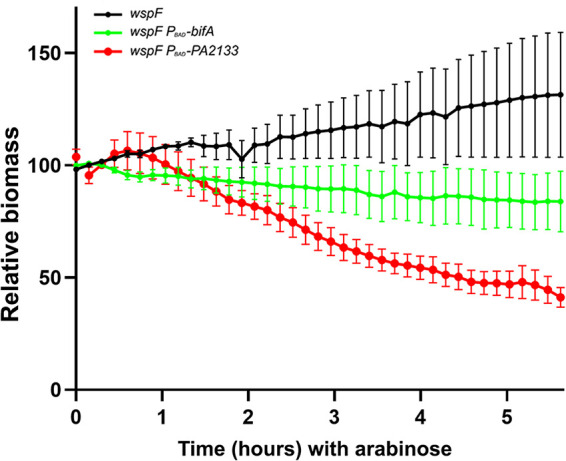
Biofilms of P. aeruginosa wspF, P. aeruginosa wspF PBAD-bifA, and P. aeruginosa wspF PBAD-PA2133 were grown in flow cells for 68 h, after which the medium was shifted to arabinose-containing medium. Subsequently, automated CLSM was used to capture images every 14 min at three different locations in three biofilms of each of the three strains. The amount of biofilm biomass/CLSM image was quantified using the Measurement Pro feature of Imaris. The relative biomasses of the three P. aeruginosa wspF, P. aeruginosa wspF PBAD-bifA, and P. aeruginosa wspF PBAD-PA2133 biofilms after the shift to arabinose-containing medium are shown. Bars indicate standard deviations. The biomass after 5.5 h of PDE induction was used to assess statistical significance. The biomass of the wspF biofilm differed significantly from that of the wspF PBAD-bifA biofilm (P = 0.033), the biomass of the wspF biofilm differed significantly from that of the wspF PBAD-PA2133 biofilm (P = 0.0001), and the biomass of the wspF PBAD-bifA biofilm differed significantly from that of the wspF PBAD-PA2133 biofilm (P = 0.046).
FIG 7.

Biofilms of P. aeruginosa wspF, P. aeruginosa wspF PBAD-bifA, and P. aeruginosa wspF PBAD-PA2133 were grown in flow cells for 68 h, after which the medium was shifted to arabinose-containing medium. Subsequently, automated CLSM was used to capture images every 14 min at three different locations in three biofilms of each of the three strains. Representative images of the biofilms immediately before the shift to arabinose-containing medium and 2.5 and 5.5 h after the shift to arabinose-containing medium are shown.
DISCUSSION
The results presented here are in contrast to the results of Chambers et al. (11), who did not see a dispersal of P. aeruginosa biofilms in response to the induction of the PDE PA2133 or DipA. However, Chambers et al. used expression of PBAD-PA2133 and PBAD-dipA from plasmids, whereas we used expression of PBAD-PA2133 and PBAD-bifA from chromosomal CTX insertions. Our findings indicate that leaky expression of PBAD-PA2133 from a chromosomal CTX insertion severely reduced the ability of the P. aeruginosa PAO1 wild type to form biofilm. The use of a plasmid-borne PBAD-PA2133 fusion with a high gene dosage should make the biofilm deficiency even stronger. Chambers et al. (11) grew biofilms with P. aeruginosa PAO1 containing plasmid-borne PBAD-PA2133 and PBAD-dipA fusions for 5 days under flowing conditions in a tube reactor and then added arabinose to induce the expression of the PDEs. There is a strong selection in flowthrough biofilm systems for mutants or plasmid-free cells with enhanced biofilm formation abilities and decreased biofilm dispersal abilities (20), which might have contributed to the contrasting results between our work and the work of Chambers et al. (11). However, we cannot exclude that the different experimental outcomes could be caused by differences in strains, media, and biofilm models.
Since most Gram-negative bacteria encode multiple DGCs and PDEs, it has been argued that output specificity may be governed by specific interactions between DGCs, PDEs, and effector proteins (12, 21, 22). While this may be true, our results indicate that the depletion of the global bacterial c-di-GMP content can overrule the requirement for specific protein-protein interactions and induce biofilm dispersal. Previously, we provided evidence that the induction of the E. coli PDE YhjH in P. aeruginosa resulted in biofilm dispersal (7). However, YhjH (also designated PdeH) has been shown to be a strongly expressed master PDE that eradicates the global effects of several DGCs by draining the global c-di-GMP pool (12). Therefore, it was a possibility that biofilm dispersal mediated by the induction of YhjH was a special case. However, in the present study, we provide evidence that the induction of the native PDEs BifA and PA2133 in P. aeruginosa leads to biofilm dispersal. This suggests that chemical compounds that have the ability to deplete the global c-di-GMP content in a bacterium may function as biofilm-dispersing drugs and that the activation of PDEs may be a particularly attractive drug target to achieve this.
We found that the induction of PA2133 resulted in the dispersal of biofilms formed by a P. aeruginosa wspF mutant strain. The P. aeruginosa wspF mutant overproduces Psl and Pel exopolysaccharides (15), and evidence has been provided that such mutants show enhanced persistence in the lungs of cystic fibrosis patients (16, 23, 24) and in chronic wounds (25). Therefore, in relation to the development of novel antibiofilm drugs, it is interesting that a decrease in the c-di-GMP level induces the dispersal of biofilms formed by a P. aeruginosa wspF mutant.
Microbial biofilms are the cause of problematic infections in various medical settings, and they are also causing severe problems in a variety of industrial plants. Therefore, any research avenue that might provide a solution to the biofilm problem should be explored. A decade of research has shown that c-di-GMP signaling is a central regulator of bacterial biofilm formation (5), and manipulation of the c-di-GMP level in bacteria has been suggested as a means of controlling biofilms (7, 8). Recently, however, evidence has been presented that the induction of native c-di-GMP phosphodiesterases does not lead to a dispersal of P. aeruginosa biofilms (11). The latter result may discourage attempts to use c-di-GMP signaling as a target for the development of antibiofilm drugs. However, our results suggest that the induction of the P. aeruginosa c-di-GMP phosphodiesterases PA2133 and BifA indeed results in the dispersal of P. aeruginosa biofilms in both a microtiter tray assay and a flow cell system. Thus, our study supports that manipulation of the c-di-GMP level in bacteria is a viable strategy for biofilm control.
MATERIALS AND METHODS
Bacterial strains, plasmids, primers, and growth media.
The P. aeruginosa and Escherichia coli strains used in this study are listed in Table 1. The growth medium employed for the propagation of the strains was either ABTrace medium (26) or lysogeny broth (LB). When necessary, growth media were supplemented with the following antibiotics: gentamicin at 10 μg/ml for E. coli and 60 μg/ml for P. aeruginosa, tetracycline at 10 μg/ml for E. coli and 60 μg/ml for P. aeruginosa, and carbenicillin at 200 μg/ml for P. aeruginosa. Plasmids and primers used in this study are listed in Tables 2 and 3, respectively.
TABLE 1.
Strains
| Strain | Description | Source or reference |
|---|---|---|
| E. coli | ||
| DH5α | F− endA1 glnV44 thi-1 recA1 relA1 gyrA96 deoR nupG purB20 ϕ80dlacZΔM15 Δ(lacZYA-argF)U169 hsdR17(rK− mK+) λ− | Laboratory collection |
| HB101 | recA thi pro leu hsdRM+ Smr | 36 |
| SM10-λpir | Kmr thi-1 thr leu tonA lacY supE recA::RP4-2-Tc::Mu pir | 37 |
| RK600 | HB101 carrying pRK600 | 36 |
| P. aeruginosa | ||
| PAO1 | P. aeruginosa wild type | 38 |
| wspF | wspF deletion mutant of PAO1 | 17 |
| wspF psl pel | wspF psl pel deletion mutant of PAO1 | 17 |
| JBAMG565 | P. aeruginosa araC-PBAD-bifA | This study |
| JBAMG621 | P. aeruginosa araC-PBAD-PA2133 | This study |
| JBAMG573 | P. aeruginosa wspF araC-PBAD-bifA | This study |
| JBAMG625 | P. aeruginosa wspF araC-PBAD-PA2133 | This study |
| MTR998 | P. aeruginosa psl pel wspF araC-PBAD-bifA | This study |
| MTR999 | P. aeruginosa psl pel wspF araC-PBAD-PA2133 | This study |
| JBAMG639 | P. aeruginosa Tn7-gfp | This study |
| JBAMG642 | P. aeruginosa araC-PBAD-bifA Tn7-gfp | This study |
| JBAMG654 | P. aeruginosa araC-PBAD-PA2133::Tn7-gfp | This study |
| JBAMG645 | P. aeruginosa wspF Tn7-gfp | This study |
| JBAMG648 | P. aeruginosa wspF araC-PBAD-bifA::Tn7-gfp | This study |
| JBAMG651 | P. aeruginosa wspF araC-PBAD-PA2133::Tn7-gfp | This study |
TABLE 2.
Plasmids
| Plasmid | Description | Reference or source |
|---|---|---|
| pJN105 | Arabinose-inducible expression vector | 14 |
| pCdrA-gfp | pUCP22Not-PcdrA-gfp(mut3)-T0-T1; Ampr Gmr | 17 |
| pJBAMG10 | bifA cloned into the EcoRI-XbaI sites of pJN105 | This study |
| pJN2133 | Gene PA2133 cloned into pJN105 | 15 |
| pDONRminiCTX2 | Gateway-compatible version of mini-CTX2 with a 2.9-kb attP-flanking fragment from Gateway donor vector pDONR221 (Invitrogen) inserted into the HindIII site of the multiple-cloning site; Tcr Cmr | H. Almblad and J. J. Harrison |
| pJBAMG13 | araC-PBAD-bifA fusion of pJBAMG10 inserted into the Gateway-compatible integration vector pDONRminiCTX2 | This study |
| pENTRYminiCTX::PBAD-PA2133 | araC-PBAD-PA2133 fusion of pJN2133 inserted into the Gateway-compatible integration vector pDONRminiCTX2 | H. Almblad and J. J. Harrison |
| pFlp2 | Source of Flp2 recombinase; Ampr | 39 |
| pRK600 | Mobilization plasmid; Cmr | 36 |
| pUX-BF13 | Tn7 transposase-encoding plasmid | 31 |
| pBK-miniTn7-gfp2 | Mini-Tn7-gfp2 delivery vector | 30 |
TABLE 3.
Primers
| Primer name | Nucleotide sequence | Reference |
|---|---|---|
| BifA_EcoRI_RBS_TTG_up | 5′-AAAAAAGAATTCACAAGGAAGGCCCCTTGAAACTG-3′ | This study |
| BifA_down_XbaI | 5′-TATATATCTAGACTGTTCAGCCGCCTGTTCTCA-3′ | This study |
| BBctxPBAD::attB1 | 5′-GGGGACAAGTTTGTACAAAAAAGCAGGCTTAACGCAATTAATGTGAGTT-3′ | 40 |
| BBctxPBAD::attB2 | 5′-GGGGACCACTTTGTACAAGAAAGCTGGGTAATTTCCATTCGCCATTCA-3′ | 40 |
| Pser-up | 5′-CGAGTGGTTTAAGGCAACGGTCTTGA-3′ | 27 |
| Pser-down | 5′-AGTTCGGCCTGGTGGAACAACTCG-3′ | 27 |
Cloning of bifA in pJN105.
The bifA gene (PA4367) of P. aeruginosa PAO1 was initially PCR amplified from chromosomal DNA as a 2.1-kb EcoRI-XbaI fragment using the primer pair BifA_EcoRI_RBS_up/BifA_down_XbaI and Phusion high-fidelity DNA polymerase (catalog number F-530L; Thermo Scientific) as described by the manufacturer. Subsequently, the 2.1-kb fragment was purified on PCR cleanup columns (Wizard SV gel and PCR cleanup system; Promega, USA), restricted with EcoRI and XbaI (Fermentas), and ligated into EcoRI/XbaI-restricted pJN105 (14) using T4 DNA ligase (Invitrogen) to create pJBAMG10. The resulting ligation mixture was electroporated into chemically competent cells of E. coli DH5α, and transformants were selected on LB plates supplemented with 10 μg/ml gentamicin. Next, plasmid DNA was extracted from selected transformants by employing a Qiagen spin prep kit (Qiagen GmbH, Germany), and the sequence of the constructed arabinose-inducible araC-PBAD-bifA expression cassette of pJBAMG10 was verified by sequencing.
Cloning of araC-PBAD-PDE expression cassettes in pDONRminiCTX2.
The araC-PBAD-bifA expression cassette of pJBAMG10 flanked by attB sites was PCR amplified with primer pair BBctxPBAD::attB1/BBctxPBAD::attB2 using Phusion high-fidelity DNA polymerase (catalog number F-530L; Thermo Scientific) as described by the manufacturer. Following purification on PCR cleanup columns (Wizard SV gel and PCR cleanup system; Promega, USA), the 3.9-kb PCR fragment was inserted into the Gateway-compatible integration vector pDONRminiCTX2 using Gateway BP Clonase (Invitrogen) as described by the manufacturer, to create pJBAMG13. The resulting BP Clonase reaction mixture was electroporated into chemically competent cells of E. coli DH5α, and transformants were selected on LB plates supplemented with 10 μg/ml tetracycline. Next, plasmid DNA was extracted from a few transformants by employing a Qiagen spin prep kit (Qiagen GmbH, Germany), and the arabinose-inducible araC-PBAD-bifA expression cassette in pJBAMG13 was verified by sequencing.
An integration vector carrying an araC-PBAD-PA2133 expression cassette was created using the same cloning strategy as the one outlined above. Initially, the araC-PBAD-PA2133 expression cassette of pJN2133 (15) was PCR amplified as a 3-kb fragment with the primer pair BBctxPBAD::attB1/BBctxPBAD::attB2, and subsequently, it was inserted into the integration vector pDONRminiCTX2, resulting in pENTR-araC-PBAD-PA2133.
Construction of strains with chromosomal insertions of araC-PBAD-PDE expression cassettes.
Strain PAO1 containing one chromosomal copy of the araC-PBAD-bifA expression cassette of pJBAMG13 was created in three steps. First, a transconjugant with plasmid pJBAMG13 inserted into the chromosomal ϕCTX attB site was constructed by three-parental mating using E. coli DH5α/pJBAMG13 as the donor, E. coli HB101/pRK600 as the helper, and P. aeruginosa PAO1 as the recipient. Second, the transconjugant was transformed with plasmid pFLP2 (encoding Flp recombinase [27]) to obtain transformants where the Flp recombination target (FRT)-flanked plasmid backbone of pJBAMG13 had been excised. Third, the resulting PAO1 strain containing one chromosomal copy of the araC-PBAD-bifA expression cassette was cured for plasmid pFLP2 using sucrose-based counterselection to create strain JBAMG565. Briefly, an 18-h-old culture of the recipient PAO1 strain (propagated in LB at 37°C) was diluted 2-fold into 42°C prewarmed LB medium and kept at 42°C for 4 h. Next, a mating solution, containing a 1:1:1 mixture of a heat-treated recipient PAO1 culture and late-exponential-phase cultures of the donor and helper strains, was prepared and subsequently spotted onto an LB plate. Following 20 h of mating at 30°C, the resulting mating spot was resuspended in 1 ml of 0.9% NaCl, and transconjugants were selected on ABTrace plates supplemented with 10 mM sodium citrate, 10 μM FeCl3, and 60 μg/ml tetracycline. Next, electrocompetent cells of the constructed transconjugant of PAO1 carrying plasmid pJBAMG13 in its chromosomal ϕCTX attB site were produced according to the protocol described previously by Choi et al. (28), and the bacteria were subsequently transformed with plasmid pFLP2 using an electroporator (E. coli Pulser; Bio-Rad, Denmark). Following 3 h of phenotypic expression in LB medium at 37°C, transformants were selected on LB plates supplemented with 200 μg/ml carbenicillin. Afterwards, transformants exhibiting resistance to carbenicillin but sensitivity to tetracycline were isolated. Next, the carbenicillin-resistant and tetracycline-sensitive transformants were cured for the plasmid by subjecting them to sucrose selection by means of repeated restreaking on both LB plates supplemented with 200 μg/ml carbenicillin and no-NaCl LB plates supplemented with 10% sucrose (29). Sucrose selection at 30°C was repeated until the emergence of sucrose-resistant and carbenicillin-sensitive transformants, to create strains cured for the pFLP2 plasmid. Finally, the chromosomal location of the araC-PBAD-bifA expression cassette of pJBAMG13 in strain PAO1 was verified by PCR using the primer pair Pser-up/Pser-down (27). Using the same protocol as the one outlined above, a PAO1 strain containing one chromosomal copy of the araC-PBAD-PA2133 expression cassette of pENTR-araC-PBAD-PA2133 was created and named JBAMG621.
In a similar manner, one copy of either the araC-PBAD-bifA expression cassette of pJBAMG13 or the araC-PBAD-PA2133 expression cassette of pENTR-araC-PBAD-PA2133 was inserted into the chromosomal ϕCTX attB site of the P. aeruginosa wspF and P. aeruginosa wspF pel psl strains to create strains JBAMG573, JBAMG625, MTR998, and MTR999.
Construction of strains containing the cdrA-gfp fluorescent reporter.
To obtain strains capable of monitoring the intracellular c-di-GMP level, the reporter plasmid pCdrA-gfp (17) was electroporated into P. aeruginosa PAO1 and the derived P. aeruginosa araC-PBAD-bifA, P. aeruginosa araC-PBAD-PA2133, P. aeruginosa wspF pel psl, P. aeruginosa psl pel wspF araC-PBAD-bifA, and P. aeruginosa psl pel wspF araC-PBAD-PA2133 strains as outlined in the protocol reported previously by Choi et al. (28). Transformants of the respective strains were selected on LB plates supplemented with 60 μg/ml gentamicin, and one green fluorescent protein (GFP)-positive transformant of each strain was selected for c-di-GMP level assessments.
Assessment of c-di-GMP levels by use of the cdrA-gfp fluorescent reporter.
In 96-well microtiter plates, cultures of the pCdrA-gfp-containing strains grown overnight were diluted 200-fold in 100-μl ABTrace medium aliquots (26) supplemented with 0.2% glucose, 0.5% Casamino Acids, 60 μg/ml gentamicin, 1 μM FeCl3, and either no or 0.5% arabinose. To estimate the background fluorescence arising from the growth medium and the parent strain (autofluorescence), cultures of plasmid-free parent strains grown overnight were diluted 200-fold in 100-μl ABTrace medium aliquots (26) supplemented with 0.2% glucose, 0.5% Casamino Acids, 1 μM FeCl3, and either no or 0.5% arabinose. The microtiter plates were incubated at 37°C at 440 rpm in a Tecan reader (Infinite F200 Pro), and the corresponding values of cell density (optical density at 600 nm [OD600]) and GFP fluorescence (fluorescence units [FU]) were measured every 20 min for 24 h. All measured GFP fluorescence values were corrected for background fluorescence, and subsequently normalized by the corresponding cell density, to give relative c-di-GMP levels (FU/OD600). The fluorescence readouts presented in Fig. 2 display the relative c-di-GMP level (FU/OD600) of each of the tested strains following 10 h of cultivation (at the end of the exponential phase) in the absence or presence of 0.5% arabinose.
Construction of Tn7-gfp-tagged strains.
P. aeruginosa PAO1 strains carrying the mini-Tn7-gfp2 expression cassette of pBK-miniTn7-gfp2 (30) in their chromosomal Tn7 insertion site were constructed by four-parental mating between E. coli DH5α/pBK-miniTn7-gfp2 (donor), E. coli HB101/pRK600 (helper), SM10-λpir/pUX-BF13 (helper) (provider of the Tn7 transposase) (31), and P. aeruginosa PAO1 (recipient) as described previously by Koch and coworkers (30). Briefly, an 18-h-old culture of the recipient PAO1 strain (propagated in LB at 37°C) was diluted 2-fold into 42°C prewarmed LB medium and kept at 42°C for 4 h. Next, a mating solution containing a 1:1:1:1 mixture of the heat-treated recipient PAO1 culture and late-exponential-phase cultures of the donor and helper strains was prepared and subsequently spotted onto an LB plate. Following 20 h of mating at 30°C, the resulting mating spot was resuspended in 1 ml of 0.9% NaCl, and transconjugants were selected on ABTrace plates supplemented with 10 mM sodium citrate, 10 μM FeCl3, and 60 μg/ml gentamicin. Finally, gentamicin-resistant green fluorescent transconjugants were picked.
Microtiter plate biofilm experiments.
The impact of PDE expression on the biofilm formation of P. aeruginosa PAO1 strains in 96-well microtiter plates (Nunc) was assessed using a crystal violet assay as previously described (32, 33). Briefly, a 20-h-old culture of the respective PAO1 strain (cultivated at 37°C in ABTrace medium [26] supplemented with 0.2% glucose, 0.5% Casamino Acids, and 1 μM FeCl3) was diluted 100-fold into the same growth medium to create an inoculation culture. The inoculation culture was split in two, and arabinose (inducer of PDE expression) was added to one of the cultures to obtain a final concentration of 0.5% (wt/vol) arabinose. Next, three 100-μl aliquots of the inoculation cultures were transferred to the wells of microtiter plates. Subsequently, the microtiter plate was sealed with an air-permeable lid (sandwich cover CR1596; Enzyscreen, The Netherlands) and incubated on a rotary shaker (model HS501 digital; IKA, Germany) at 37°C at 160 rpm for the wild-type strains and 280 rpm for the wspF strains. Following 10 h of incubation, the culture supernatants were discarded, and the amount of biofilm present in the wells was quantified by crystal violet staining as described previously (33). To obtain series of wells containing biofilms of decreasing maturity, the inoculation step was modified so that 10-well dilution rows of 3-fold serial dilutions of the inoculation culture were employed.
The impact of 1 h or 2 h of PDE expression on established P. aeruginosa PAO1 microtiter plate biofilms was assessed as follows. Initially, a 20-h-old culture of the respective strain (cultivated at 37°C in ABTrace medium [26] supplemented with 0.2% glucose, 0.5% Casamino Acids, and 1 μM FeCl3) was diluted 100-fold into the same growth medium to create an inoculation culture. To obtain series of wells containing biofilms of decreasing maturity, 10-well dilution rows of 3-fold serial dilutions of the inoculation culture were established in 100-μl ABTrace medium aliquots supplemented with 0.2% glucose, 0.5% Casamino Acids, and 1 μM FeCl3. Subsequently, the microtiter plate was sealed with an air-permeable lid (sandwich cover CR1596; Enzyscreen, The Netherlands) and incubated on a rotary shaker (model HS501 digital; IKA, Germany) at 37°C at 160 rpm for the wild-type strains and 280 rpm for the wspF strains. Following 8 h of biofilm formation, half of the 10-well dilution rows was supplemented with 5-μl aliquots of MilliQ water (control), while the remaining half of the 10-well dilution rows was supplemented with 5-μl aliquots of 10% arabinose (inducer of PDE expression). Next, the microtiter plates were sealed with an air-permeable lid, reinstalled on the rotary shaker, and further incubated for either 1 or 2 additional hours at 37°C with shaking. Finally, the culture supernatants were discarded, and the amount of biofilm present in the wells was quantified by crystal violet staining as described previously (33).
Flow cell biofilm experiments.
We used a continuous flow cell system described previously (34, 35) to assess the effect of PDE induction on P. aeruginosa PAO1 biofilms grown under hydrodynamic conditions. Cultures grown overnight were diluted to an OD450 of 0.01 in saline before inoculation into the flow channels. To ensure bacterial attachment, the flow cells were left for 2 h before the flow was initiated. The flow channels were irrigated at a rate of 3 ml h−1 with ABTrace minimal medium supplemented with 0.3 mM glucose. The biofilms were allowed to develop at 20°C for 68 h, during which three positions in each of the three flow cell channels were imaged every hour using a Zeiss LSM 880 confocal laser scanning microscope running ZEN 2.3. The biomass was CLSM imaged using a 488-nm laser for excitations and a 495- to 550-nm emission filter. Images were taken as 180-μm z-stacks with x-y dimensions of 135 by 135 μm using a 63×/1.4 oil objective.
After 68 h of cultivation, the flow was stopped, and the medium was changed to contain 0.2% (wt/vol) arabinose (Sigma-Aldrich, USA) for the induction of PDE gene expression. In addition, the 5 ml of medium contained in the bubble traps just upstream of the flow channels was also supplemented with arabinose to a final concentration of 0.2% (wt/vol). The flow was subsequently restarted, and the biofilms were irrigated with the arabinose-supplemented medium for 5.5 h. During the arabinose treatment, three positions in each of the three channels were imaged approximately every 14th minute. All images were obtained using constant laser power and exposure.
Images were processed in Imaris 9.5 (Bitplane; Oxford Instruments, United Kingdom) and were processed for qualitative presentation using the normal shading protocol for a shadow three-dimensional (3D) projection using constant parameters between images. The biomass was quantified by applying an isosurface over the biomass using constant parameters for the threshold. The volume of the biomass in each image at each time point was obtained using the Measurement Pro package for Imaris.
Statistical analysis.
Two-way analysis of variance (ANOVA) was used for assessment of the statistical significance of the quantified biofilm biomass in Fig. 1, 3, and 4 and for assessment of the statistical significance of c-di-GMP levels in Fig. 2. One-way ANOVA with Tukey’s multiple-comparison test was used for statistical analysis of the data shown in Fig. 5 and 6. P values of <0.05 were considered significant.
Supplementary Material
ACKNOWLEDGMENTS
We gratefully acknowledge Henrik Almblad and Joe J. Harrison (University of Calgary) for providing the pDONRminiCTX2 and pENTRYminiCTX::PBAD-PA2133 plasmids.
This work was supported by grants to M.G. and T.T.-N. from the Danish Council for Independent Research, the Danish Ministry of Higher Education and Science, the Lundbeck Foundation, and the Novo Nordisk Foundation.
Footnotes
Supplemental material is available online only.
REFERENCES
- 1.Hall CW, Mah TF. 2017. Molecular mechanisms of biofilm-based antibiotic resistance and tolerance in pathogenic bacteria. FEMS Microbiol Rev 41:276–301. doi: 10.1093/femsre/fux010. [DOI] [PubMed] [Google Scholar]
- 2.Ciofu O, Tolker-Nielsen T. 2019. Tolerance and resistance of Pseudomonas aeruginosa biofilms to antimicrobial agents—how P. aeruginosa can escape antibiotics. Front Microbiol 10:913. doi: 10.3389/fmicb.2019.00913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Costerton JW, Stewart PS, Greenberg EP. 1999. Bacterial biofilms: a common cause of persistent infections. Science 284:1318–1322. doi: 10.1126/science.284.5418.1318. [DOI] [PubMed] [Google Scholar]
- 4.Tolker-Nielsen T. 2015. Biofilm development. Microbiol Spectr 3:MB-0001-2014. doi: 10.1128/microbiolspec.MB-0001-2014. [DOI] [PubMed] [Google Scholar]
- 5.Jenal U, Reinders A, Lori C. 2017. Cyclic di-GMP: second messenger extraordinaire. Nat Rev Microbiol 15:271–284. doi: 10.1038/nrmicro.2016.190. [DOI] [PubMed] [Google Scholar]
- 6.Fazli M, Almblad H, Rybtke ML, Givskov M, Eberl L, Tolker-Nielsen T. 2014. Regulation of biofilm formation in Pseudomonas and Burkholderia species. Environ Microbiol 16:1961–1981. doi: 10.1111/1462-2920.12448. [DOI] [PubMed] [Google Scholar]
- 7.Christensen LD, van Gennip M, Rybtke MT, Wu H, Chiang WC, Alhede M, Hoiby N, Nielsen TE, Givskov M, Tolker-Nielsen T. 2013. Clearance of Pseudomonas aeruginosa foreign-body biofilm infections through reduction of the cyclic di-GMP level in the bacteria. Infect Immun 81:2705–2713. doi: 10.1128/IAI.00332-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tolker-Nielsen T. 2014. Pseudomonas aeruginosa biofilm infections: from molecular biofilm biology to new treatment possibilities. APMIS Suppl 122:1–51. doi: 10.1111/apm.12335. [DOI] [PubMed] [Google Scholar]
- 9.Gjermansen M, Ragas P, Tolker-Nielsen T. 2006. Proteins with GGDEF and EAL domains regulate Pseudomonas putida biofilm formation and dispersal. FEMS Microbiol Lett 265:215–224. doi: 10.1111/j.1574-6968.2006.00493.x. [DOI] [PubMed] [Google Scholar]
- 10.Gjermansen M, Nilsson M, Yang L, Tolker-Nielsen T. 2010. Characterization of starvation-induced dispersion in Pseudomonas putida biofilms: genetic elements and molecular mechanisms. Mol Microbiol 75:815–826. doi: 10.1111/j.1365-2958.2009.06793.x. [DOI] [PubMed] [Google Scholar]
- 11.Chambers JR, Cherny KE, Sauer K. 2017. Susceptibility of Pseudomonas aeruginosa dispersed cells to antimicrobial agents is dependent on the dispersion cue and class of the antimicrobial agent used. Antimicrob Agents Chemother 61:e00846-17. doi: 10.1128/AAC.00846-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sarenko O, Klauck G, Wilke FM, Pfiffer V, Richter AM, Herbst S, Kaever V, Hengge R. 2017. More than enzymes that make or break cyclic di-GMP—local signaling in the interactome of GGDEF/EAL domain proteins of Escherichia coli. mBio 8:e01639-17. doi: 10.1128/mBio.01639-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kuchma SL, Brothers KM, Merritt JH, Liberati NT, Ausubel FM, O’Toole GA. 2007. BifA, a cyclic-di-GMP phosphodiesterase, inversely regulates biofilm formation and swarming motility by Pseudomonas aeruginosa PA14. J Bacteriol 189:8165–8178. doi: 10.1128/JB.00586-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Newman JR, Fuqua C. 1999. Broad-host-range expression vectors that carry the L-arabinose-inducible Escherichia coli araBAD promoter and the araC regulator. Gene 227:197–203. doi: 10.1016/s0378-1119(98)00601-5. [DOI] [PubMed] [Google Scholar]
- 15.Hickman JW, Tifrea DF, Harwood CS. 2005. A chemosensory system that regulates biofilm formation through modulation of cyclic diguanylate levels. Proc Natl Acad Sci U S A 102:14422–14427. doi: 10.1073/pnas.0507170102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Starkey M, Hickman JH, Ma L, Zhang N, De Long S, Hinz A, Palacios S, Manoil C, Kirisits MJ, Starner TD, Wozniak DJ, Harwood CS, Parsek MR. 2009. Pseudomonas aeruginosa rugose small-colony variants have adaptations that likely promote persistence in the cystic fibrosis lung. J Bacteriol 191:3492–3503. doi: 10.1128/JB.00119-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rybtke MT, Borlee BR, Murakami K, Irie Y, Hentzer M, Nielsen TE, Givskov M, Parsek MR, Tolker-Nielsen T. 2012. Fluorescence-based reporter for gauging cyclic di-GMP levels in Pseudomonas aeruginosa. Appl Environ Microbiol 78:5060–5069. doi: 10.1128/AEM.00414-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rybtke M, Berthelsen J, Yang L, Hoiby N, Givskov M, Tolker-Nielsen T. 2015. The LapG protein plays a role in Pseudomonas aeruginosa biofilm formation by controlling the presence of the CdrA adhesin on the cell surface. Microbiologyopen 4:917–930. doi: 10.1002/mbo3.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lopez-Sanchez A, Jimenez-Fernandez A, Calero P, Gallego LD, Govantes F. 2013. New methods for the isolation and characterization of biofilm-persistent mutants in Pseudomonas putida. Environ Microbiol Rep 5:679–685. doi: 10.1111/1758-2229.12067. [DOI] [PubMed] [Google Scholar]
- 20.Gjermansen M, Ragas P, Sternberg C, Molin S, Tolker-Nielsen T. 2005. Characterization of starvation-induced dispersion in Pseudomonas putida biofilms. Environ Microbiol 7:894–906. doi: 10.1111/j.1462-2920.2005.00775.x. [DOI] [PubMed] [Google Scholar]
- 21.Chen G, Liang H. 2020. A novel c-di-GMP signal system regulates biofilm formation in Pseudomonas aeruginosa. Microb Cell 7:160–161. doi: 10.15698/mic2020.06.720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Massie JP, Reynolds EL, Koestler BJ, Cong JP, Agostoni M, Waters CM. 2012. Quantification of high-specificity cyclic diguanylate signaling. Proc Natl Acad Sci U S A 109:12746–12751. doi: 10.1073/pnas.1115663109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Evans TJ. 2015. Small colony variants of Pseudomonas aeruginosa in chronic bacterial infection of the lung in cystic fibrosis. Future Microbiol 10:231–239. doi: 10.2217/fmb.14.107. [DOI] [PubMed] [Google Scholar]
- 24.Malone JG, Jaeger T, Spangler C, Ritz D, Spang A, Arrieumerlou C, Kaever V, Landmann R, Jenal U. 2010. YfiBNR mediates cyclic di-GMP dependent small colony variant formation and persistence in Pseudomonas aeruginosa. PLoS Pathog 6:e1000804. doi: 10.1371/journal.ppat.1000804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gloag ES, Marshall CW, Snyder D, Lewin GR, Harris JS, Santos-Lopez A, Chaney SB, Whiteley M, Cooper VS, Wozniak DJ. 2019. Pseudomonas aeruginosa interstrain dynamics and selection of hyperbiofilm mutants during a chronic infection. mBio 10:e01698-19. doi: 10.1128/mBio.01698-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pamp SJ, Tolker-Nielsen T. 2007. Multiple roles of biosurfactants in structural biofilm development by Pseudomonas aeruginosa. J Bacteriol 189:2531–2539. doi: 10.1128/JB.01515-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hoang TT, Kutchma AJ, Becher A, Schweizer HP. 2000. Integration-proficient plasmids for Pseudomonas aeruginosa: site-specific integration and use for engineering of reporter and expression strains. Plasmid 43:59–72. doi: 10.1006/plas.1999.1441. [DOI] [PubMed] [Google Scholar]
- 28.Choi KH, Kumar A, Schweizer HP. 2006. A 10-min method for preparation of highly electrocompetent Pseudomonas aeruginosa cells: application for DNA fragment transfer between chromosomes and plasmid transformation. J Microbiol Methods 64:391–397. doi: 10.1016/j.mimet.2005.06.001. [DOI] [PubMed] [Google Scholar]
- 29.Hmelo LR, Borlee BR, Almblad H, Love ME, Randall TE, Tseng BS, Lin C, Irie Y, Storek KM, Yang JJ, Siehnel RJ, Howell PL, Singh PK, Tolker-Nielsen T, Parsek MR, Schweizer HP, Harrison JJ. 2015. Precision-engineering the Pseudomonas aeruginosa genome with two-step allelic exchange. Nat Protoc 10:1820–1841. doi: 10.1038/nprot.2015.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Koch B, Jensen LE, Nybroe O. 2001. A panel of Tn7-based vectors for insertion of the gfp marker gene or for delivery of cloned DNA into Gram-negative bacteria at a neutral chromosomal site. J Microbiol Methods 45:187–195. doi: 10.1016/s0167-7012(01)00246-9. [DOI] [PubMed] [Google Scholar]
- 31.Bao Y, Lies DP, Fu H, Roberts GP. 1991. An improved Tn7-based system for the single-copy insertion of cloned genes into chromosomes of gram-negative bacteria. Gene 109:167–168. doi: 10.1016/0378-1119(91)90604-a. [DOI] [PubMed] [Google Scholar]
- 32.O’Toole GA, Kolter R. 1998. Flagellar and twitching motility are necessary for Pseudomonas aeruginosa biofilm development. Mol Microbiol 30:295–304. doi: 10.1046/j.1365-2958.1998.01062.x. [DOI] [PubMed] [Google Scholar]
- 33.Groizeleau J, Rybtke M, Andersen JB, Berthelsen J, Liu Y, Yang L, Nielsen TE, Kaever V, Givskov M, Tolker-Nielsen T. 2016. The anti-cancerous drug doxorubicin decreases the c-di-GMP content in Pseudomonas aeruginosa but promotes biofilm formation. Microbiology (Reading) 162:1797–1807. doi: 10.1099/mic.0.000354. [DOI] [PubMed] [Google Scholar]
- 34.Kragh KN, Hutchison JB, Melaugh G, Rodesney C, Roberts AE, Irie Y, Jensen PO, Diggle SP, Allen RJ, Gordon V, Bjarnsholt T. 2016. Role of multicellular aggregates in biofilm formation. mBio 7:e00237-16. doi: 10.1128/mBio.00237-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sternberg C, Tolker-Nielsen T. 2006. Growing and analyzing biofilms in flow cells. Curr Protoc Microbiol Chapter 1:Unit 1B.2. doi: 10.1002/9780471729259.mc01b02s00. [DOI] [PubMed] [Google Scholar]
- 36.Kessler B, de Lorenzo V, Timmis KN. 1992. A general system to integrate lacZ fusions into the chromosomes of gram-negative eubacteria: regulation of the Pm promoter of the TOL plasmid studied with all controlling elements in monocopy. Mol Gen Genet 233:293–301. doi: 10.1007/BF00587591. [DOI] [PubMed] [Google Scholar]
- 37.Simon R, Priefer UB, Puhler A. 1983. A broad host range mobilization system for in vivo genetic engineering: transposon mutagenesis in gram-negative bacteria. Nat Biotechnol 1:784–791. doi: 10.1038/nbt1183-784. [DOI] [Google Scholar]
- 38.Stover CK, Pham XQ, Erwin AL, Mizoguchi SD, Warrener P, Hickey MJ, Brinkman FS, Hufnagle WO, Kowalik DJ, Lagrou M, Garber RL, Goltry L, Tolentino E, Westbrock-Wadman S, Yuan Y, Brody LL, Coulter SN, Folger KR, Kas A, Larbig K, Lim R, Smith K, Spencer D, Wong GK, Wu Z, Paulsen IT, Reizer J, Saier MH, Hancock RE, Lory S, Olson MV. 2000. Complete genome sequence of Pseudomonas aeruginosa PAO1, an opportunistic pathogen. Nature 406:959–964. doi: 10.1038/35023079. [DOI] [PubMed] [Google Scholar]
- 39.Hoang TT, Karkhoff-Schweizer RR, Kutchma AJ, Schweizer HP. 1998. A broad-host-range Flp-FRT recombination system for site-specific excision of chromosomally-located DNA sequences: application for isolation of unmarked Pseudomonas aeruginosa mutants. Gene 212:77–86. doi: 10.1016/s0378-1119(98)00130-9. [DOI] [PubMed] [Google Scholar]
- 40.Almblad H, Harrison JJ, Rybtke M, Groizeleau J, Givskov M, Parsek MR, Tolker-Nielsen T. 2015. The cyclic AMP-Vfr signaling pathway in Pseudomonas aeruginosa is inhibited by cyclic di-GMP. J Bacteriol 197:2190–2200. doi: 10.1128/JB.00193-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.