

Summary
Glioblastoma stem cells (GSCs) resist current glioblastoma (GBM) therapies. GSCs rely highly on oxidative phosphorylation (OXPHOS), whose function requires mitochondrial translation. Here we explore the therapeutic potential of targeting mitochondrial translation and report the results of high-content screening with putative blockers of mitochondrial ribosomes. We identify the bacterial antibiotic quinupristin/dalfopristin (Q/D) as an effective suppressor of GSC growth. Q/D also decreases the clonogenicity of GSCs in vitro, consequently dysregulating the cell cycle and inducing apoptosis. Cryoelectron microscopy (cryo-EM) reveals that Q/D binds to the large mitoribosomal subunit, inhibiting mitochondrial protein synthesis and functionally dysregulating OXPHOS complexes. These data suggest that targeting mitochondrial translation could be explored to therapeutically suppress GSC growth in GBM and that Q/D could potentially be repurposed for cancer treatment.
Keywords: glioblastoma, glioblastoma stem cells, OXPHOS, mitoribosome, mitochondrial translation, high-content screening, cryo-EM, quinupristin, dalfopristin, drug repurposing
Graphical abstract
Highlights
-
•
Screen of putative mitoribosome inhibitors identifies Q/D as effective on GSCs
-
•
Q/D selectively inhibits growth of GSCs
-
•
Treatment with Q/D decreases clonogenicity, blocks cell cycle, and induces apoptosis
-
•
Q/D binds to mitoribosomes and inhibits mitochondrial translation and therefore OXPHOS
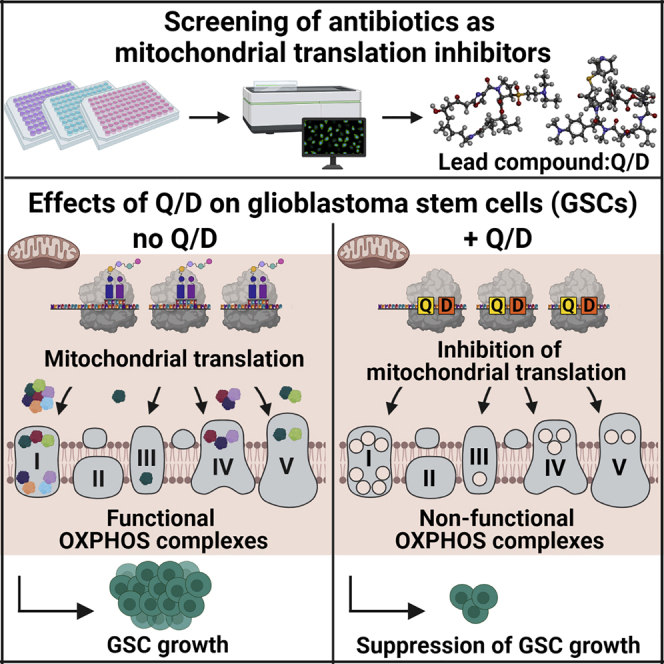
In a screen of antibiotics as putative mitochondrial translation inhibitors, Sighel et al. identify Q/D as a suppressor of glioblastoma stem cell growth. Q/D inhibits mitochondrial translation, leading to OXPHOS dysregulation and, consequently, decreasing clonogenicity, blocking the cell cycle, and inducing apoptosis.
Introduction
Glioblastoma (GBM) is the most common malignant primary brain tumor in adults. Despite extensive treatment involving surgery, radiotherapy, and chemotherapy, the survival of individuals with GBM remains extremely poor. The search for new effective therapies has proven challenging throughout the last decades, with temozolomide (TMZ), the only available drug for newly diagnosed GBM, increasing median survival from 12.1 to 14.6 months (Stupp et al., 2005). This is partially due to the presence of GBM stem cells (GSCs), which are responsible for resistance to standard treatments as well as disease recurrence (Auffinger et al., 2014; Chen et al., 2012). Therefore, further research is needed to identify novel effective therapeutic agents targeting GSCs.
For decades, tumor metabolism was believed to rely heavily on aerobic glycolysis, a phenomenon known as the Warburg effect (Warburg et al., 1927), thought to be caused by decreased or damaged oxidative phosphorylation (OXPHOS) (Warburg, 1956). However, over the past years, several studies have demonstrated that mitochondria and OXPHOS play an essential role in tumorigenesis and tumor progression (Porporato et al., 2018). Rho0 cells, which are devoid of mitochondrial DNA, undergo a dramatic reduction in tumorigenic potential and tend to acquire mitochondrial DNA from neighboring normal cells to rescue tumorigenesis (Cavalli et al., 1997; Tan et al., 2015). Furthermore, OXPHOS remains the major source of ATP in many cancer types, even in the presence of enhanced rates of glycolysis (Zu and Guppy, 2004). A growing body of literature reports that OXPHOS plays an important role in addressing the energy demands of cancer stem cells (CSCs) (Skrtić et al., 2011; Viale et al., 2014; Vlashi et al., 2014), including GSCs (Janiszewska et al., 2012; Vlashi et al., 2011). Moreover, chemotherapeutic genotoxic drugs induce a shift in cancer cell metabolism toward upregulated OXPHOS and mitochondrial biogenesis (Haq et al., 2013; Roesch et al., 2013).
Assembly of OXPHOS complexes in the inner mitochondrial membrane is largely orchestrated by mitoribosomes that synthesize 13 transmembrane proteins. The mitochondrial translation machinery is upregulated in a subset of human tumors (Kim et al., 2017; Sotgia et al., 2012), and breast CSCs show metabolic reliance on mitoribosome-synthesized proteins (Vlashi et al., 2014). Because human mitoribosomes, being descendants of bacterial ribosomes, differ from their cytosolic counterparts (Amunts et al., 2015; Petrov et al., 2019), they could, in principle, be targeted selectively to inhibit energy production. Indeed, mitoribosome sensitivity to antibiotics directed to bacterial ribosomes is a common side effect of these molecules (Zhang et al., 2005) and has been exploited preliminarily to target leukemia cells (Skrtić et al., 2011).
Given the emerging evidence of GSC addiction to OXPHOS, we systematically investigated whether inhibition of mitochondrial translation could be exploited to treat GBM by suppressing GSC growth. We performed high-content screening using a focused, custom-made library of compounds known to affect bacterial ribosome translation, hypothesizing that they could also act on mitoribosomes, given the structural similarity of the ribosomal core. This screening identified a US Food and Drug Administration (FDA)-approved combination of two molecules, quinupristin/dalfopristin (Q/D; 30:70 w/w), as the most effective compound on GSCs. We present mechanistic evidence that Q/D acts by inhibiting mitochondrial translation, leading to dysregulation of OXPHOS and suppression of GSC growth.
Results
Screening of bacterial ribosome targeting antibiotics identifies Q/D as the most effective compound on GSCs
To identify potent compounds effective on GSCs, we performed focused high-content screening using a custom-made library of potential mitochondrial translation inhibitors (Figure 1A). We mined the pharmacology of antibiotics targeting bacterial translation and compiled a custom-made library containing 71 compounds (Table S1). The majority of these compounds belong to eight molecular classes (tetracyclines, streptogramins, aminoglycosides, lincosamides, macrolides, amphenicols, oxazolidinones, and pleuromutilins), and most of them are FDA approved for treatment of bacterial infections in humans. We grew two GSC lines, COMI and VIPI, as adherent cultures on laminin using a method documented to preserve their stemness properties (Pollard et al., 2009). We then showed that both lines depend on OXPHOS by assessing their viability upon treatment with known inhibitors of the respiratory chain complexes or glycolysis (Figure S1A). The OXPHOS inhibitors rotenone, antimycin A, and oligomycin (which inhibit complex I, complex III, and complex V, respectively) significantly decreased the viability of both lines. In contrast, the glycolysis inhibitors sodium dichloroacetate (DCA) and 2-deoxy-D-glucose (2-DG) did not have a significant effect, in line with a previous report on GSCs (Janiszewska et al., 2012).
Figure 1.

Q/D is the most effective prospective mitochondrial translation inhibitor in GSCs
(A) An outline of the screening workflow. Each treatment was performed in a technical triplicate.
(B) Identification of hit classes based on class score calculation.
(C) Representative dose-response curves for all tested compounds belonging to the three hit classes validated in COMI and VIPI cells; n = 4 technical replicates, mean ± SD.
(D) The GI50 values of the tested compounds.
(E) The chemical structures of the lead compound Q/D (30:70 w/w) and virginiamycin M1, the product of dalfopristin hydrolysis.
See also Figures S1 and S2 and Tables S1–S3.
We treated COMI and VIPI cells with the aforementioned custom-made library of potential mitochondrial translation inhibitors for 24 h at 100 μM and stained them with two dyes: DRAQ5, a DNA dye that allowed us to quantify the number of cells displaying morphological features related to cytotoxicity, and MitoTracker Orange (MTO), which accumulates in healthy mitochondria, allowing observation of features related to mitotoxicity. Staining was followed by data acquisition using a high-content imaging system and data mining. As positive controls, we used three well-known OXPHOS disruptors at doses and times inducing clear phenotypes in GSCs. Long-term treatment (100 μM for 24 h) with rotenone, oligomycin A, or carbonyl cyanide p-(trifluoromethoxy) phenylhydrazone (FCCP) was a paradigm of late cellular effects of OXPHOS inhibition, whereas acute treatment with FCCP (100 μM for 30 min) accounted for immediate mitochondrial toxicity without effects on cell viability (Figure S1B). We phenotypically characterized the cells by extracting six non-redundant morphological features (Figure S1C; Table S2), which were informative of cellular or mitochondrial toxicity, as specified. Next, the informative content of each feature was assessed by testing whether the positive controls behaved as expected (Figure S2A). The late cellular effects were described by the cell number, nucleus area, and nucleus roundness, whereas mitochondrial toxicity was described by the area of disrupted mitochondria, number of MTO-positive spots detected in the cytoplasmic region of cells (mitochondrial spot number), and area covered by mitochondrial spots (mitochondrial spot area). We evaluated the effects of each library compound according to these six features (Figure S2B). A feature was defined as affected by a given compound when it deviated from the median value of all compounds by a distance larger than 1.5 times the median absolute deviation. To identify the most effective classes of drugs, we first calculated a compound score for each compound based on the number of features affected, and then the scores of all compounds in a given class were averaged to obtain the class score (Figure 1B; Table S3). On the basis of these results, we selected three classes with the highest class score for further evaluation: streptogramins, pleuromutilins, and tetracyclines.
Streptogramins are a class of antibiotics consisting of a mixture of two structurally different compounds: group A (also called M) streptogramins, which are polyunsaturated macrolactones, and group B (also called S) streptogramins, which are cyclic hexadepsipeptides (Mast and Wohlleben, 2014). Streptogramins A and B are known to act synergistically against bacteria (Di Giambattista et al., 1989) and are used in combination at a fixed 70:30 (w/w) ratio. We therefore assayed streptogramins as combinations of group A and B compounds, whereas tetracyclines and pleuromutilins were tested as single agents. We validated a number of compounds from the three hit classes by constructing dose-response curves and calculating the growth inhibition 50 (GI50) values. For this purpose, we treated COMI and VIPI cells with a range of drug concentrations for 48 h and assessed viability following staining with the Hoechst 33342 and propidium iodide (PI) DNA dyes (Figures 1C and 1D). This analysis pointed to streptogramins used as a combination of group A and B compounds as the most cytotoxic class for GSCs and in particular to the combination of quinupristin/virginiamycin M1 (Q/VM1) and Q/D as the most potent. Dalfopristin is a semisynthetic derivative of virginiamycin M1 (Figure 1E), and it rapidly hydrolyzes to virginiamycin M1 at pH 7.4 and 37°C (Noeske et al., 2014), which could account for the similar activity of Q/D and Q/VM1. Q/D (30:70 w/w) is an FDA-approved antibiotic for treatment of skin infections (Delgado et al., 2000) and is traded as Synercid. Given the cytotoxicity induced by Q/D and the potential for its repurposing for cancer treatment, we selected this molecule combination for further investigation.
Q/D selectively inhibits the growth of GSCs at clinically relevant concentrations
We assessed the activity of Q/D on a panel of GSCs composed of 21 lines derived from 18 individuals with variable clinical features (Table S4; cell line characterization in Marziali et al., 2016). The GI50 values spanned 2.5–32.5 μM after 48 h of treatment, narrowing to a range varying from 1.7–12.2 μM after 72 h of treatment (Figure 2A). These values are in the range of the maximal blood concentration values achievable in individuals treated with Q/D for bacterial infection, which varies between 14 and 32 μM (corresponding to 10.7 and 24.2 μg/mL) upon administration of 12.6 and 29.4 mg/kg, respectively (Bergeron and Montay, 1997). These results emphasize that the inhibitory effect of Q/D is time and dose dependent and is exerted at clinically relevant concentrations. We then attempted to correlate Q/D sensitivity with clinical parameters of the individuals from whom these GSC lines were derived and with GBM markers, such as EGFRvIII or PTEN status, but no statistically significant correlations emerged (Table S5). This negative result suggests that Q/D affects GSCs irrespective of the individual’s clinical and basic molecular profile, although, to unequivocally answer this question, a study of a larger panel of lines will have to be conducted.
Figure 2.

Q/D selectively inhibits growth of GSCs at clinically relevant concentrations, is effective under hypoxic conditions, and is more potent than TMZ
(A) GI50 values of a panel of 21 GSC lines derived from 18 tumor samples 48 and 72 h after Q/D treatment; n = 4 technical replicates.
(B) Q/D GI50 values for 14 GSC lines compared with Q/D GI50 values for astrocytes derived from human fetal neural stem cells (CB660, HNPC#13, and U3), human lung fibroblasts (MRC5), and human skin fibroblast (Hs68); n = 3 biological replicates, mean ± SD.
(C) Representative immunofluorescence images for SOX2, NESTIN, and GFAP staining of COMI, GB7, and VIPI cells grown under stemness (Stem) and differentiation (Diff) conditions. Scale bar, 100 μm.
(D) Quantification of the fluorescence intensity of SOX2, NESTIN, and GFAP immunostaining (left) and Q/D GI50 values for COMI, GB7, and VIPI cells grown under Stem and Diff conditions (right). For immunostaining quantification, n = 6,000 objects for stem cells and n = 1,000 objects for differentiated cells; mean ± SEM. Values for differentiated cells were normalized to those of stem cells (dashed line). The GI50 values were calculated using 4–7 biological replicates; mean ± SD.
(E) Viability of COMI and VIPI cells grown under normal and hypoxic conditions with different doses of Q/D; n = 4 technical replicates, mean ± SD. Shown are representative results of 3 biological replicates.
(F) Representative dose-response curves to Q/D and temozolomide (TMZ) for COMI and VIPI cells; n = 3 biological replicates, mean ± SD.
To assess the selectivity of Q/D for GSCs, we evaluated its cytotoxicity in several normal diploid cells (astrocytes differentiated from three different human fetal neural stem cell lines [CB660, HNPC#13, and U3], a lung fibroblast cell line [MRC5], and a skin fibroblast cell line [Hs68]) and compared it with Q/D effects on a panel of 14 GSC lines. We treated the cells with a range of Q/D concentrations and assessed their viability (Figure 2B). The GI50 values for human fetal neural stem cell-derived astrocytes and fibroblast cells ranged from 68.3 ± 15.5 μM to 231.6 ± 30.7 μM, which are substantially higher than the GI50 of the other 14 GSC lines tested.
We then examined the effect of Q/D treatment on the differentiated GSC progeny. We exposed three GSC lines to a pro-differentiation environment by culturing them in medium deprived of growth factors and supplemented with 10% fetal bovine serum (FBS) for 14 days and then assessed their status by checking the expression of stemness (SOX2 and NESTIN) and differentiation (GFAP) markers by immunofluorescence (Figure 2C). COMI and GB7 cells differentiated to a greater extent than VIPI cells, as evidenced by a higher increase in expression of GFAP and decrease in SOX2 and NESTIN at the end of the treatment (Figure 2D). The GI50 values of the differentiated COMI and GB7 cells were higher than those of their stem counterparts, whereas the less differentiated VIPI cells showed a smaller increase in sensitivity to Q/D than their stem counterparts (Figure 2D). This suggests that well-differentiated GSCs are less sensitive to Q/D treatment.
These results indicate that Q/D selectively inhibits GSCs growth at clinically achievable concentrations.
Q/D is active under hypoxic conditions, and it is more effective than TMZ
GSCs are known to reside in dedicated tumor niches (i.e., anatomical regions defined by a unique microenvironment), which preserves them in low oxygen and represses their differentiation. Despite their enrichment in these hypoxic niches (Li et al., 2009), as little as 1% oxygen is sufficient for effective OXPHOS in GSCs (Janiszewska et al., 2012). In this context, we evaluated the activity of Q/D on GSCs grown under normoxia and hypoxia (21% and 1% oxygen, respectively) by assessing cell viability after Hoechst 33342 and PI staining (Figure 2E). At 1% O2, the viability of untreated cells was not affected, and the cells still responded to Q/D treatment in a dose-dependent manner, supporting Q/D efficacy on GSCs under hypoxic conditions.
The only chemotherapy drug used in the clinic to treat newly diagnosed GBM is TMZ, a pro-drug that is spontaneously converted to the alkylating agent 5-(3-methyltriazen-1-yl)imidazole-4-carboxamide at physiological pH (Friedman et al., 2000). The most widely studied mechanism of resistance to TMZ in GBM is expression of O6-methylguanine DNA-methyltransferase (MGMT), a DNA repair protein able to remove methyl adducts from O6-methylguanine and repair DNA damage. Because GSCs tend to be resistant to TMZ (Chen et al., 2012), we compared the cytotoxicity of Q/D with that of TMZ in COMI and VIPI cells (Figure 2F). In COMI cells, where the MGMT gene is silenced epigenetically by promoter methylation, the GI50 value for Q/D was 6.5 ± 1.1 μM, whereas that for TMZ was 96.5 ± 15.2 μM. In VIPI cells, where the MGMT promoter is unmethylated, the GI50 value for Q/D was 20.2 ± 1.4 μM, whereas that for TMZ was 337.7 ± 35.5 μM. These experiments demonstrate that Q/D is 15 times more effective than TMZ in terms of growth inhibition of GSCs, independent of MGMT promoter methylation status. Therefore, Q/D is equally able to affect GSCs in normoxia and hypoxia and is over an order of magnitude more potent than TMZ in vitro.
Q/D decreases clonogenicity, blocks cell cycle progression, and promotes apoptosis
We next investigated the extent of growth inhibition induced by Q/D in GSCs grown as gliomaspheres. Ten COMI cells were seeded per well in media with various Q/D concentrations, and the area of gliomaspheres was measured over the course of 9 days (Figures 3A and 3B). We observed that the 1 μM Q/D treatment only slightly inhibited gliomasphere formation but that the 5 and 10 μM treatments, concentrations achievable in the blood of affected individuals, nearly completely abolished gliomasphere formation.
Figure 3.

Q/D decreases clonogenicity, dysregulates the cell cycle, and promotes apoptosis
(A) Effects of Q/D treatment on COMI cells grown in suspension. Shown are example images from days 0, 4, and 9. Scale bar, 100 μm.
(B) Sphere area measured over the course of the 9-day experiment. The data were normalized to day 0. n = 30 technical replicates, mean ± SEM. One representative experiment is shown; n = 3 biological replicates.
(C) Representative images of the gliomasphere formation assay; scale bar, 1,000 μm.
(D) Quantification of the number of spheres larger than 100 μm; n = 20 technical replicates, mean ± SEM. ∗∗∗p < 0.001, unpaired two-tailed t test. One representative result is shown; n = 3 biological replicates.
(E) Representative fluorescence-activated cell sorting (FACS) analysis of the effects of Q/D on the cell cycle in COMI cells assayed using EdU incorporation and PI staining.
(F) Quantification of the percentage of cells in each cell cycle phase; n = 3 biological replicates, mean ± SD. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, unpaired two-tailed t test.
(G) Representative FACS analysis of apoptosis upon treatment with Q/D in COMI cells, as evaluated by Annexin V and PI staining.
(H) Quantification of the percentage of Annexin V-positive cells; n = 6 biological replicates, mean ± SD. ∗∗p < 0.01, unpaired two-tailed t test.
See also Figure S3.
Given the inhibitory growth effect of Q/D on GSCs, we wanted to find out whether this drug could affect GSC self-renewal and clonogenic maintenance. To assess the effects of Q/D on GSC clonogenic potential, we measured the gliomasphere formation ability of COMI cells after Q/D treatment. COMI cells were grown in suspension with several concentrations of Q/D for 72 h, and then the gliomaspheres were dissociated and seeded at a density of 10 or 100 cells per well in the absence of the drug. After 10 days, the clonogenic potential was evaluated by counting the number of gliomaspheres reformed (Figures 3C and 3D). Q/D decreased gliomasphere formation ability in a dose-dependent manner, confirming a substantial effect on GSC maintenance.
We then investigated the functional effects induced on the GSC cell cycle by performing EdU-PI staining upon treatment with Q/D and by measuring the percentage of cells in each phase (Figures 3E and 3F). Increasing concentrations of Q/D led to a marked dose-dependent decrease in the number of cells in S phase, indicating inhibition of proliferation. In addition, we observed a significant increase in the number of cells in G0/G1 phase at 5 μM Q/D, indicating an accumulation of cells in this phase, followed by an increase in the number of cells in G2/M phase starting at 5 μM and culminating at 10 μM Q/D (Figure 3F).
Because Q/D disrupts GSC maintenance and dysregulates the cell cycle, we next investigated whether Q/D induces apoptosis. We performed a flow cytometry analysis using Annexin V and PI staining and estimated the percentage of apoptotic cells by calculating the percentage of Annexin V-positive cells (Figures 3G and 3H). Treatment with 6.5 and 10 μM Q/D led to an increase in the percentage of apoptotic cells even though the effect was significant only at 10 μM.
Because autophagy can be induced upon drug treatment in GSCs (Angeletti et al., 2016), we investigated its induction upon Q/D treatment. We treated cells with Q/D alone or in combination with autophagy inhibitors to investigate autophagic flux and performed immunofluorescence and immunoblotting experiments using an anti-LC3 antibody, which suggested that Q/D causes an increase in autophagic flux (Figures S3A–S3D). We next investigated whether this increase could have a pro-death or pro-survival function by simultaneously treating GSCs with Q/D and the autophagy inhibitor chloroquine. A small but significant decrease in viability was observed upon combination treatment (Figure S3E), indicating that autophagy is a pro-survival mechanism. These data are in line with those reported for other molecules, such as the pan-histone deacetylase inhibitor givinostat (Angeletti et al., 2016).
This phenotypic analysis of GSCs treated with Q/D reveals loss of clonogenic potential because of cell cycle arrest accompanied by apoptosis.
Q/D inhibits mitochondrial translation and induces OXPHOS dysregulation
To investigate the molecular mechanism of action of Q/D, we determined the cryo-EM structure of the human mitoribosome from HEK293 cells treated with Q/D (Figure S4; Table S6). To ensure that the cryoelectron microscopy (cryo-EM) experiment reflects physiological conditions, Q/D was not added to any of the purification steps upon cell lysis (STAR Methods). The overall resolution of the reconstruction was 3.9 Å, and the local resolution of ∼3.4 Å allowed identification of both molecules in the large ribosomal subunit—dalfopristin in the cryo-EM density at the peptidyl-transferase center (PTC) and quinupristin at the entrance to the exit tunnel for nascent polypeptides (Figure 4A). The density for dalfopristin was of a quality comparable with the previously reported X-ray crystal structures of the bacterial counterparts (Harms et al., 2004; Noeske et al., 2014), which was sufficient to detect that the compound was hydrolyzed with loss of the diethylaminoethylsulfonyl group to form virginiamycin M1 (Figure 4B), an effect also reported in a pharmacology study of Q/D (Delgado et al., 2000). By comparing the Q/D-bound structure with the previously published unbound state of the human mitoribosome (Amunts et al., 2015), we identified no protein conformational changes and only a number of rRNA rearrangements induced by Q/D binding. The most prominent is the 90° shift of A2725 (A2062 in E. coli) (Figure 4C). Other residues affected are U2993 (U2506 in E. coli) and U3072 (U2585 in E. coli), but the latter was not well resolved in the unbound structure (EMD2876), probably because of inherent flexibility. Comparison with the E. coli and D. radiodurans ribosome structures with Q/D (Harms et al., 2004; Noeske et al., 2014) shows that the bacterial counterpart of A2725 (A2062 in E. coli) adopts a similar shift, whereas the equivalent of U3072 (U2585 in E. coli) is displaced by nearly 180 degrees, which was not observed in the mitoribosome because of the absence of the diethylaminoethylsulfonyl group of dalfopristin. In addition, we observed a shift in the mitoribosomal U2993 (U2506 in E. coli) that was not observed in the bacterial counterpart. Because Q/D was added directly to the cells, the data suggest that Q/D is able to pass the mitochondrial membranes and bind to the mitoribosome.
Figure 4.

Cryo-EM of the mitoribosome from Q/D-treated cells
(A) A model of the mitoribosomal large subunit with quinupristin and virginiamycin M1 (the hydrolysis product of dalfopristin). The two compounds are found in the entrance of the exit tunnel and the peptidyl transferase center (PTC).
(B) View of the cryo-EM density at an overall resolution of 3.9 Å around quinupristin (purple) and virginiamycin M1 (green). The two compounds interact with the surrounding rRNA.
(C) Comparison of the human mitoribosomal RNA when bound to quinupristin and virginiamycin M1 (black) and when unbound (PDB: 3J9M; light gray). Red arrows indicate rRNA movement to accommodate the molecules.
We next evaluated the effects induced by Q/D on mitochondrial translation and OXPHOS functionality. To determine whether Q/D specifically affects mitoribosomal function, we assayed for de-novo-synthesized proteins by mitochondrial or cytosolic ribosomes. We conducted metabolic labeling with 35S-methionine on COMI cells treated with Q/D for 24 h (Figure 5A). Q/D was very effective in inhibiting mitochondrial translation and nearly completely abolished it at 0.5 μM. Importantly, no effects on cytosolic translation were noted at this or even higher Q/D concentrations (up to 2.6 μM). We further confirmed the effects on proteins synthesized in the mitochondria and cytosol by performing immunoblotting and immunofluorescence analysis on COMI and VIPI cells treated with Q/D for 48 h (Figure 5B; Figures S5A and S5B). Cytochrome c oxidase subunit I (COX1) is synthesized in mitochondria, whereas cytochrome c oxidase subunit IV (COX4), succinate dehydrogenase complex flavoprotein subunit A (SDHA), and β-tubulin are translated in the cytosol. Expression of COX1 was decreased markedly upon treatment with Q/D, whereas expression of COX4, SDHA, and β-tubulin remained unchanged. The decrease in COX1 protein levels upon Q/D treatment was also confirmed at 1% O2 (Figure S5C), suggesting that, at 1% O2, Q/D behaves in the same manner as at 21%. We then assessed the effects of Q/D on mRNAs encoding COX1 and COX4, but we did not observe any significant changes (Figure 5C). Therefore, Q/D acts specifically on mitochondrial translation.
Figure 5.

Q/D inhibits mitochondrial translation and negatively affects OXPHOS functionality
(A) 35S metabolic labeling assay of mitochondrial (left) and cytosolic (right) translation on COMI cells after 24-h treatment with Q/D. One representative result is shown; n = 3 biological replicates.
(B) Effects of increasing concentrations of Q/D on COX1, COX4, SDHA, and β-tubulin proteins in COMI and VIPI cells after 48-h treatment, as assayed by immunoblotting. One representative result is shown; n = 2 biological replicates.
(C) Effects of Q/D on COX1 and COX4 mRNA levels on COMI and VIPI cells after 48-h treatment, assessed by qRT-PCR. Data are presented as fold change over control. n = 4–5 biological replicates, mean ± SD. Unpaired two-tailed t test.
(D) Effects of Q/D on the functionality of OXPHOS complexes, as assessed using BN-PAGE and an in-gel activity assay on COMI and VIPI cells after 48, 72, and 96 h of drug treatment. Coomassie staining served as the loading control. Shown are representative results of 2 biological replicates.
(E) Oxygen consumption of COMI and VIPI cells upon treatment with Q/D for 48 h, as measured using Oxygraph-2k. Cells were evaluated for routine (R), complex I (CI), complexes I and II (CI&II), uncoupled (ETS), and complex II (CII) respiration. Shown are representative results of 3 biological replicates.
(F) Quantification of the changes in mitochondrial membrane potential (MMP), as assessed by JC-1 staining in COMI cells. FCCP treatment was used as a positive control; n = 4 biological replicates, mean ± SD. ∗p < 0.05, unpaired two-tailed t test compared with the non-treated control.
(G) Effects of increasing concentrations of Q/D on L-lactate production in COMI and VIPI cells after 48 h of Q/D treatment. The L-lactate levels were normalized to the number of cells; n = 3 technical replicates, mean ± SD. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, unpaired two-tailed t test. Shown are representative results of 3 biological replicates.
See also Figure S5.
Because the 13 proteins synthesized by mitoribosomes are an essential part of the OXPHOS complexes, we tested the effects of Q/D on the functionality of these complexes by performing blue native polyacrylamide gel electrophoresis (BN-PAGE) followed by in-gel activity assays on COMI and VIPI cells. The activity of complexes I, IV, and V, which are composed of proteins mitochondrial and cytosolic in origin, was decreased upon Q/D treatment, whereas the activity of complex II, which is composed entirely of nuclearly encoded proteins, was unaffected (Figure 5D). In parallel, we assessed the amount of these complexes by BN-PAGE followed by immunoblotting and found that the levels of complexes I, IV, and V, but not those of complex II, were decreased (Figure S5D).
To verify whether the altered stoichiometry of the OXPHOS complexes led to decreased mitochondrial respiratory capacity upon treatment with Q/D, we tested mitochondrial respiration using high-resolution respirometry. For COMI and VIPI cells, the functionality of complex I and complex II was decreased upon 1 μM Q/D treatment, even when this did not have an apparent effect on basal respiration capacity (R). When we measured the maximal oxygen consumption (ETS) through injection of the FCCP uncoupler, we found that it was decreased, indicating substantial loss of reserve respiratory capacity (Figure 5E).
Because functional OXPHOS is necessary to maintain mitochondrial membrane potential (MMP), we investigated whether Q/D induced a consequent loss in MMP. We used the JC-1 dye to stain cells treated with Q/D at various concentrations and analyzed them by flow cytometry. In this experimental setting, mitochondrial depolarization is indicated by a shift from red to green fluorescence. We determined the percentage of cells with normal MMP (having high red fluorescence and low green fluorescence) as well as the percentage of cells with lower MMP (having low red fluorescence and high green fluorescence) and quantified these changes (Figure 5F). We observed few changes upon 2.5 μM Q/D treatment, whereas, from 5 μM onward, the percentage of cells with disrupted MMP increased in a dose-dependent manner, suggesting that indeed Q/D affects the MMP.
Another process by which cells can fulfill their ATP requirements is glycolysis, and switching to glycolysis is a potential mechanism of bioenergetic flexibility for OXPHOS targeting drugs in cancer cells. To verify whether GSCs switch to glycolysis upon treatment with Q/D, we measured the levels of L-lactate, a product of the glycolytic pathway. We observed a small but significant increase in L-lactate production starting from 1 μM Q/D treatment for COMI and VIPI cells (Figure 5G), indicating a tendency to switch to glycolysis.
Finally, we checked the correlation of the relative quantity of mitochondrial mass with the Q/D treatment response in GSC lines. We assessed eight GSC lines by treating them with different concentrations of Q/D for 48 h (Figure S5E). The data show that viability after Q/D treatment is correlated negatively with mitochondrial mass at 2.5 μM (r = −0.882, p < 0.001), 5 μM (r = −0.758, p < 0.05), and 10 μM treatment (r = −0.682, p = 0.06), indicating that GSCs with more mitochondria are more susceptible to Q/D-induced death.
These results clearly demonstrate that Q/D acts by selectively interfering with mitochondrial translation and, consequently, dysregulating OXPHOS in GSCs.
Genetic inhibition of mitochondrial translation suppresses GSC growth, recapitulating Q/D effects
To further assess whether mitochondrial translation is the target of Q/D activity on GSCs, we investigated whether genetic strategies would lead to a similar effect as Q/D treatment. To address this question, we used CRISPR/Cas9 to knock out genes involved in mitochondrial translation. We first generated Cas9-expressing COMI and VIPI cells and then transduced them with lentiviral vectors bearing sgRNAs specific for TUFM or MRPS18A and expressing GFP. A non-targeting sequence (sgNT) was used as a negative control. TUFM is a mitochondrial translation elongation factor whose shRNA-mediated knockdown has been shown to inhibit mitochondrial translation in acute myeloid leukemia cells (Skrtić et al., 2011). MRPS18A is a mitoribosome small subunit protein that has a zinc-binding function and has been shown to be upregulated in breast cancer (Sørensen et al., 2017).
We evaluated the effects of two sgRNAs targeting each gene on cell growth using a competition assay (Figure 6A). Both guides showed a similar inhibitory effect on cell growth, and we chose one guide for further validation (sgTUFM_1 and sgMRPS18A_1). We verified the efficiency of Cas9 cutting by sequencing the target site (Figure 6B) and performing immunoblotting (Figure 6C). To confirm that TUFM and MRPS18A knockout resulted in mitochondrial translation inhibition, we measured the expression of COX1 and COX4 proteins. Expression of COX1 was decreased markedly, whereas expression of COX4 and β-tubulin remained unchanged (Figure 6D).
Figure 6.

Genetic inhibition of mitochondrial translation suppresses GSC growth, recapitulating Q/D effects
(A) Competition assay in VIPI cells transduced with viruses expressing sgRNAs for target genes. Data were normalized to day 1. n = 4 biological replicates, mean ± SD.
(B) Percentage of insertions or deletions (indels) analyzed by decomposition (tracking of indels by decomposition [TIDE]) analysis in Cas9-expressing COMI and VIPI cells following lentiviral transduction of the sgRNAs selected in (A) (sgTUFM_1 and sgMRPS18A_1). n = 3–4 biological replicates, mean ± SD.
(C) Effects of TUFM and MRPS18A knockout on TUFM, MRPS18A, and β-tubulin proteins in COMI and VIPI cells, as assayed by immunoblotting. One representative result is shown; n = 2 biological replicates.
(D) Effects of TUFM and MRPS18A knockout on COX1, COX4, and β-tubulin proteins in COMI and VIPI cells, as assayed by immunoblotting. One representative result is shown; n = 2 biological replicates.
(E) Effects of TUFM and MRPS18A knockout on COMI and VIPI cells grown in suspension. Shown are example images from days 0, 4, 10, and 15; scale bar, 200 μm.
(F) Sphere area measured over the course of the 15-day experiment. The data were normalized to day 0. n = 15 technical replicates, mean ± SEM. One representative experiment is shown; n = 3 biological replicates.
(G) Effects of TUFM and MRPS18A knockout on COMI and VIPI proliferation when grown as adherent cultures. The data were normalized to day 0. n = 3 biological replicates; mean ± SD.
(H) Quantification of the number of spheres greater than 100 μm for COMI and VIPI cells; n = 20 technical replicates; mean ± SEM. ∗∗∗∗p < 0.0001, unpaired two-tailed t test. One representative result is shown; n = 3 biological replicates.
Next we evaluated the effects of TUFM and MRPS18A knockout on GSCs grown as gliomaspheres by measuring the area of the sphere formed over the course of 15 days. We observed that genetic inhibition of mitochondrial translation nearly completely abolished gliomasphere formation (Figures 6E and 6F). The same effect was also observed on COMI and VIPI cells grown as adherent cultures (Figure 6G). To assess the effect of genetic inhibition of mitochondrial translation on GSC self-renewal and clonogenic maintenance, we measured the gliomasphere formation ability of TUFM and MRPS18A knockout COMI and VIPI cells. COMI and VIPI cells were seeded at two different cell densities, and the clonogenic potential was evaluated by counting the number of gliomaspheres formed after 10–15 days (Figure 6H). TUFM and MRPS18A knockout substantially decreased the gliomasphere formation ability, confirming a strong effect on GSC maintenance.
These results show that genetic inhibition of mitochondrial translation almost completely suppresses GSC proliferation, strongly affecting GSC self-renewal and clonogenic potential maintenance, recapitulating the effects observed upon Q/D treatment.
Discussion
OXPHOS targeting leads to destabilization of energy homeostasis in CSCs, providing a possible target in oncology (Weinberg and Chandel, 2015). At least 14 inhibitors of one of the OXPHOS complexes have been evaluated in vivo or in the clinic, whereas at least six are in preclinical testing (Ashton et al., 2018). The anti-diabetic drug metformin has been shown to reversibly inhibit complex I (Wheaton et al., 2014), resulting in cytotoxic effects in different CSC types, including GSCs (Würth et al., 2013). Metformin is currently in more than 300 ongoing clinical trials in combination with standard treatments (Weinberg and Chandel, 2015). In GBM, high OXPHOS levels measured by complex IV activity are an independent negative prognostic factor (Griguer et al., 2013), suggesting that molecules able to affect OXPHOS activity might be an interesting new therapeutic strategy for this tumor type. Two different small molecules have been demonstrated to efficiently target complex IV function in GSCs in vitro and in vivo (Oliva et al., 2016, 2017). Recently, IACS-010759 and Gboxin, two inhibitors of complex I and complex V, respectively, have been shown to inhibit proliferation of GSCs in vitro and in vivo (Molina et al., 2018; Shi et al., 2019).
Here we adopted a strategy of OXPHOS targeting by selectively suppressing its assembly. We hypothesized that mitochondrial translation inhibition would hamper the formation and functionality of complexes I, III, IV, and V, leading to pronounced detrimental effects on cell viability. By performing high-content, imaging-driven screening on a library encompassing all known bacterial ribosome-targeting antibiotic classes, we identified the streptogramins Q/D as the most potent combination leading to significant growth inhibition of GSCs. We showed by cryo-EM that Q/D added to actively dividing cells binds to the mitoribosomal large subunit stalling polypeptide synthesis. This leads to inhibition of mitochondrial translation, which, in turn, dysregulates OXPHOS, leading to loss of clonogenic potential, cell cycle arrest, and death by apoptosis. Our data are consistent with the effect of other ribosome-targeting antibiotics, which have been shown to decrease tumor sphere formation in cell lines from eight different tumor types (Lamb et al., 2015). Among them, tigecycline, a tetracycline antibiotic, has been reported to be cytotoxic for acute myeloid leukemia stem cells (Kuntz et al., 2017; Ravà et al., 2018; Skrtić et al., 2011). Therefore, our results add to the increasing evidence showing that targeting OXPHOS, and in particular mitochondrial translation, could be clinically exploitable in cancer therapy.
Q/D is approved by the FDA for treatment of persistent bacterial infections and could be repurposed for other uses. The suitability of Q/D repurposing for GBM is supported by the fact that the range of GI50 values obtained in vitro matches blood concentration values achievable in individuals treated with Q/D for bacterial infections (Bergeron and Montay, 1997), but further in vivo studies will be necessary to determine whether such concentrations are sufficient to exert desired effects on tumors. We further explored the possibility of repurposing Q/D for GBM by demonstrating that Q/D preferentially targets GSCs rather than astrocytes or primary fibroblasts, suggesting a suitable therapeutic window. In vitro, GSCs are more sensitive to Q/D compared with their differentiated progeny, revealing preferential GSC targeting for Q/D, but further experiments will have to be conducted to understand the molecular basis of the preferential targeting of GSCs. In addition, a pre-clinical mouse model will have to be established to demonstrate Q/D effectiveness in vivo. GBM tumors are largely hypoxic, and GSCs have been identified in GBM perivascular areas and hypoxic regions (Calabrese et al., 2007; Li et al., 2009). Important for GBM therapy is that Q/D can exert its cytotoxic effects and inhibit mitochondrial translation under hypoxic conditions. Moreover, because drugs targeting the OXPHOS complexes alleviate or even eradicate tumor hypoxia (Ashton et al., 2018), Q/D could have an important, indirect antitumor effect in GBM by promoting oxygenation and downregulating neoangiogenesis. Further experiments are needed to investigate this possibility.
Similar to any newly proposed anticancer drug, a major issue for favoring transfer of Q/D to the clinical setting is the possibility to stratify affected individuals. We proved that Q/D decreased the cell viability of GSCs grown as adherent cultures and as gliomaspheres. We observed the effect on a large panel of GSCs and did not find any correlation between the sensitivity and the molecular features of the cells or the variable clinical features of the individuals from whom these lines were established. This suggests that Q/D could be used extensively on individuals with GBM, even though these data need to be verified on a larger panel of lines to account for tumor variability. Some CSCs, including GSCs (Vlashi et al., 2011), have a certain degree of metabolic flexibility after inhibition of glycolysis or OXPHOS, upregulating one of the two metabolic pathways to compensate for inhibition of the other. GSCs from individuals with GBM bearing a homozygous deletion of the key glycolytic enzyme enolase 1 (ENO1) (3.3% of individuals) have a reduced capacity for compensatory glycolysis and, thus, increased sensitivity to the complex I inhibitor IACS-010759 (Molina et al., 2018). Other molecular alterations can produce a similar effect; for example, the FGFR3-TACC3 gene fusion (3% of individuals with GBM) confers special sensitivity to OXPHOS inhibitors by activating mitochondrial metabolism (Frattini et al., 2018). Very recently, a mitochondrial GBM subtype was identified using a computational approach for unbiased characterization of core biological features from single cells and bulk tumor transcriptome analysis (Garofano et al., 2021). This GBM subtype was shown to rely exclusively on OXPHOS for energy production and to exhibit a marked vulnerability to OXPHOS inhibitors. Therefore, despite the fact that we did not observe a marked variability in the GI50 spectrum in GSCs from 18 different individuals, a search for people who are more responsive to Q/D based on genome features is feasible, exploiting single genotypes as ENO1 loss of function or the FGFR3-TACC3 translocation or more complex molecular signatures able to predict high reliance on OXPHOS or glycolysis impairment.
We describe identification and detailed characterization of Q/D action. Q/D selectively inhibits mitochondrial translation and, consequently, dysregulates OXPHOS. Q/D is an FDA-approved drug, and our study sets the stage for potentially repurposing Q/D for GBM treatment.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-OxPhos complex I antibody (39kDa subunit; clone 20C11), 1:1000 WB, Mouse | Thermo Fisher Scientific | Cat#A21344; RRID:AB_221505 |
| Anti-SDHA complex II antibody (2E3GC12FB2AE2), 1:5000 WB, Mouse | Abcam | Cat#ab14715; RRID:AB_301433 |
| Anti-OxPhos complex III antibody (core 1 subunit; clone 16D10), 1:5000 WB, Mouse | Thermo Fisher Scientific | Cat#A21362; RRID:AB_1501833 |
| Anti-OxPhos complex IV antibody (subunit 1; clone 1D6E1A8), 1:1000 WB, Mouse | Invitrogen | Cat#459600; RRID:AB_2532240 |
| Anti-ATP5A complex V antibody (15H4C4), 1:1000 WB, Mouse | Abcam | Cat#ab14748; RRID:AB_301447 |
| Anti-MTCO1 antibody (COX I), 1:1000 WB, 1:1000 IF, Mouse | Abcam | Cat#ab14705; RRID:AB_2084810 |
| Anti-COX IV (3E11) antibody, 1:1000 WB, Rabbit | Cell Signaling | Cat#4850; RRID:AB_2085424 |
| Anti-COX IV antibody (COX IV) in 1: 500 IF, Rabbit | Abcam | Cat#ab16056; RRID:AB_443304 |
| Anti-SDHA complex II antibody, 1:1000 WB, Rabbit | Cell Signaling | Cat#5839; RRID:AB_10707493 |
| Anti-TUFM antibody (CL2242), 1:1000 WB, Mouse | Thermo Fisher Scientific | Cat#MA5-31363 RRID:AB_2787000 |
| Anti-MRPS18A antibody, 1:500 WB, Rabbit | Thermo Fisher Scientific | Cat#PA5-57274 RRID:AB_2644184 |
| Anti-β-tubulin (3F3-G2) antibody, 1:10,000 WB, Mouse | Santa Cruz | Cat#sc-53140; RRID:AB_793543 |
| Anti-LC3 (D11) XP antibody, 1:1000 WB, 1:500 IF, Rabbit | Cell Signaling | Cat#3868S; RRID:AB_2137707 |
| Anti-GFAP antibody in 1:2000 IF, Rabbit | Sigma Aldrich | Cat#G3893; RRID:AB_477010 |
| Anti-nestin (10c2) antibody in 1:100 IF, Mouse | Santa Cruz Biotech. | Cat#sc-23927; RRID:AB_627994 |
| Anti-sox-2 (E-4) antibody in 1:100 IF, Mouse | Santa Cruz Biotech. | Cat#sc-365823; RRID:AB_10842165 |
| Anti-Rabbit IgG - Horseradish Peroxidase conjugated, 1:3000, Goat | Thermo Fisher Scientific | Cat#31460 |
| Anti-Mouse IgG - Horseradish Peroxidase conjugated, 1:3000, Goat | Santa Cruz Biotech. | Cat#sc-2005; RRID:AB_631736 |
| Anti-mouse IgG - Horseradish Peroxidase Linked, F(ab′)2 Fragment, 1:5000 | GE Healthcare | Cat#NA9310V |
| Anti-rabbit IgG, Horseradish Peroxidase Linked, F(ab′)2, 1:5000 | GE Healthcare | Cat#NA9340V |
| Anti-rabbit IgG (H+L) - Alexa Fluor 488, 1:250, F(ab’)2-Goat | Thermo Fisher Scientific | Cat#A11070; RRID:AB_2534114 |
| Anti-rabbit IgG (H+L) - Alexa Fluor 594, 1:250, Chicken | Thermo Fisher Scientific | Cat#A21442; RRID:AB_2535860 |
| Anti-mouse IgG (H+L) - Alexa Fluor 488, 1:250, Goat | Thermo Fisher Scientific | Cat#A11029; RRID:AB_2534088 |
| Anti-mouse IgG (H+L) - Alexa Fluor 488, 1:250, Rabbit | Thermo Fisher Scientific | Cat#A11059; RRID:AB_2534106 |
| Bacterial and virus strains | ||
| One Shot Stbl3 Chemically Competent E. coli | Thermo Fisher Scientific | Cat#C737303 |
| Chemicals, peptides, and recombinant proteins | ||
| Laminin | Thermo Fisher Scientific | Cat#23017015 |
| FCCP (carbonyl cyanide p-(trifluoromethoxy) phenylhydrazone) | Cayman Chemical | Cat#15218; CAS: 370-86-5 |
| MitoTracker Orange | Thermo Fisher Scientific | Cat#M7510 |
| Draq5 | Cell Signaling | Cat#4084 |
| Hoechst 33342 | Thermo Fisher Scientific | Cat#H1399 |
| Propidium Iodide (PI) | Sigma Aldrich | Cat#P4170 |
| Quinupristin-dalfopristin mesylate complex (Q/D) | Santa Cruz | Cat#sc-391726; CAS: 126602-89-9 |
| Calcein AM | Thermo Fisher Scientific | Cat#C3100MP |
| [35S]-methionine | Perkin Elmer | Cat#NEG709A005MC |
| HCS CellMask Deep Red Stain | Thermo fisher Scientific | Cat#H32721 |
| JC-1 | Abcam | Cat#ab113850 |
| Critical commercial assays | ||
| CellTiter-Glo® Cell Viability Assay | Promega | Cat#G7570 |
| Click-IT EdU Flow Cytometry Assay kit (Thermo Fisher Scientific) | Thermo Fisher Scientific | Cat#C10634 |
| Pierce™ BCA Protein Assay Kit | Thermo Fisher Scientific | Cat#23227 |
| Qubit Protein Assay Kit | Thermo Fisher Scientific | Cat#Q33212 |
| FITC Annexin V Apoptosis Detection Kit I (BD PharMingen) | BD Biosciences | Cat#556547 |
| Glycolysis Cell-Based Assay Kit | Cayman Chemical | Cat#600450 |
| QuickExtract DNA Extraction Solution | Lucigen | Cat#QE09050 |
| Deposited data | ||
| Cryo-EM map of human mitoribosome-D/Q | This paper | EMD-4434 |
| Coordinates of human mitoribosome-D/Q | This paper | 6I9R |
| Experimental models: cell lines | ||
| Human Glioblastoma stem cell COMI | Kindly provided by AntonioDaga’s lab | N/A |
| Human Glioblastoma stem cell VIPI | Kindly provided by AntonioDaga’s lab | N/A |
| Human Glioblastoma stem cell 030616 | Kindly provided by Rossella Galli’s lab | N/A |
| Human Glioblastoma stem cell GB6 | Kindly provided by Luciano Conti’s lab | N/A |
| Human Glioblastoma stem cell GB7 | Kindly provided by Luciano Conti’s lab | N/A |
| Human Glioblastoma stem cell GB8 | Kindly provided by Luciano Conti’s lab | N/A |
| Human Glioblastoma stem cell G144 | Kindly provided by Luciano Conti’s lab | N/A |
| Human Glioblastoma stem cell G166 | Kindly provided by Luciano Conti’s lab | N/A |
| Human Glioblastoma stem cell # 23p | Marziali et al., 2016 | N/A |
| Human Glioblastoma stem cell # 163 | Marziali et al., 2016 | N/A |
| Human Glioblastoma stem cell # 67 | Marziali et al., 2016 | N/A |
| Human Glioblastoma stem cell # 62 | Marziali et al., 2016 | N/A |
| Human Glioblastoma stem cell # 23C | Marziali et al., 2016 | N/A |
| Human Glioblastoma stem cell # 76 | Marziali et al., 2016 | N/A |
| Human Glioblastoma stem cell # 30PT | Marziali et al., 2016 | N/A |
| Human Glioblastoma stem cell # 28 | Marziali et al., 2016 | N/A |
| Human Glioblastoma stem cell # 148 | Marziali et al., 2016 | N/A |
| Human Glioblastoma stem cell # 83 | Marziali et al., 2016 | N/A |
| Human Glioblastoma stem cell # 1 | Marziali et al., 2016 | N/A |
| Human Glioblastoma stem cell # 30p | Marziali et al., 2016 | N/A |
| Human Glioblastoma stem cell # 74 | Marziali et al., 2016 | N/A |
| Human Glioblastoma stem cell # 120 | Marziali et al., 2016 | N/A |
| Human Glioblastoma stem cell # 61 | Marziali et al., 2016 | N/A |
| Human Glioblastoma stem cell # 70 | Marziali et al., 2016 | N/A |
| Human Glioblastoma stem cell # 83_2 | Marziali et al., 2016 | N/A |
| Human Glioblastoma stem cell # 112 | Marziali et al., 2016 | N/A |
| Human Glioblastoma stem cell # 151 | Marziali et al., 2016 | N/A |
| Human Glioblastoma stem cell # 147 | Marziali et al., 2016 | N/A |
| Human Glioblastoma stem cell # 68 | Marziali et al., 2016 | N/A |
| Human fetal neural stem cell CB660 | Kindly provided by Luciano Conti’s lab | N/A |
| Human fetal neural stem cell U3 | Kindly provided by Luciano Conti’s lab | N/A |
| Human fetal neural progenitor cell HNPC#13 | Ricci-Vitiani et al., 2004 | N/A |
| Human lung fibroblast cell line MRC5 | Kindly provided by HTS Facility, CIBIO | N/A |
| Human skin fibroblast cell line Hs68 | ATCC | ATCC® CRL-1635 |
| 293FT cell line | Thermo Fisher Scientific | Cat#R70007 |
| Oligonucleotides | ||
| COX1_F (qPCR primer) 5′-CTATACCTATTATTCGGCGCATGA-3′ |
Eurofins | N/A |
| COX1_R (qPCR primer) 5′-CAGCTCGGCTCGAATAAGGA −3′ |
Eurofins | N/A |
| COX4_F (qPCR primer) 5′-GCCATGTTCTTCATCGGTTTC-3 |
Eurofins | N/A |
| COX4_R (qPCR primer) 5′-GGCCGTACACATAGTGCTTCTG-3′ |
Eurofins | N/A |
| 18S _F (qPCR primer) 5′-GGACATCTAAGGGCATCACA-3′ |
Eurofins | N/A |
| 18S _R (qPCR primer) 5′-AGGAATTGACGGAAGGGCAC-3′ |
Eurofins | N/A |
| GAPDH _F (qPCR primer) 5′-CAACGAATTTGGCTACAGCA-3′ |
Sigma | N/A |
| GAPDH _R (qPCR primer) 5′-AGGGGTCTACATGGCAACTG-3′ |
Sigma | N/A |
| sgNT_F ACCGACGGAGGCTAAGCGTCGCAA |
IDT | N/A |
| sgNT_R AAACTTGCGACGCTTAGCCTCCGT |
IDT | N/A |
| sgTUFM_1_F ACCGACAGGCACTGCACCCCTCGA |
IDT | N/A |
| sgTUFM_1_R AAACTCGAGGGGTGCAGTGCCTGT |
IDT | N/A |
| sgTUFM_2_F ACCGTCGGGGTATCACCATCAATG |
IDT | N/A |
| sgTUFM_2_R AAACCATTGATGGTGATACCCCGA |
IDT | N/A |
| sgMRPS18A_1_F ACCGGGAAGAACACCGCAAGATCG |
IDT | N/A |
| sgMRPS18A_1_R AAACCGATCTTGCGGTGTTCTTCC |
IDT | N/A |
| sgMRPS18A_2_F ACCGCCGGGCCCGCTAGTAGCCCA |
IDT | N/A |
| sgMRPS18A_2_R AAACTGGGCTACTAGCGGGCCCGG |
IDT | N/A |
| TUFM_F (PCR primer) GTCTAAGCTCTGCCTCTAGC |
Metabion | N/A |
| TUFM_ (PCR primer) CATCTCAATGCCTAGGACGG |
Metabion | N/A |
| MRPS18A_F (PCR primer) GTGGAGGAAGTGATCTCAGC |
Metabion | N/A |
| MRPS18A_R (PCR primer) GGTAATAGACCTGAGTGGCG |
Metabion | N/A |
| Recombinant DNA | ||
| pLKO.1-puro U6 sgRNA BfuAI large stuffer plasmid | Phelan et al., 2018 | N/A |
| psPAX2 | Addgene | Cat#12260 RRID:Addgene_12260 |
| pMD2.G | Addgene | Cat#12259 RRID:Addgene_12259 |
| lenti-SpCas9 blast | Stringer et al., 2019 | Cat#104997 RRID:Addgene_104997 |
| Software and algorithms | ||
| Harmony Software 4.1 | Perkin Elmer | N/A |
| GraphPad Prism V.5 Software | GraphPad Prism | N/A |
| BD FacsDIVA V8.0.1 Software and V9.0 | BD Biosciences | N/A |
| FlowJo 10.7.1 | BD Biosciences | N/A |
| ImageQuant Software | Molecular Dynamics, GE Healthcare | N/A |
| DatLab Software | Oroboros Instruments | N/A |
| Image Lab 3.0 Software | Bio Rad | N/A |
| ImageJ Fiji Software | Softonic | N/A |
| CFX Manager | BioRad | N/A |
| Coot | MRC LMB | N/A |
| Phenix | N/A | N/A |
| MolProbity | N/A | N/A |
| UcsfDfCorr | UCSF | N/A |
| GCTF-0.5 | MRC LMB | N/A |
| RELION-2.1.b1 | MRC LMB/ Stockholm University | N/A |
| R | Free software environment for statistical computing and graphics | N/A |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Alessandro Quattrone (alessandro.quattrone@unitn.it).
Materials availability
Cell lines generated in this study can be requested from the lead contact.
Data and code availability
3D cryo-EM density maps and the coordinates of the atomic model have been deposited in the Worldwide Protein Data Bank under the accession numbers EMD-4434 and 6I9R, respectively.
Experimental model and subject details
Human glioblastoma stem cell lines COMI and VIPI were kind gifts from Antonio Daga (Azienda Ospedaliera Universitaria San Martino di Genova, Italy), and 030616 from Rossella Galli (H. S. Raffaele, Milan, Italy). COMI, VIPI and 030616 were cultured in DMEM/F-12 and Neurobasal media (Thermo Fisher Scientific, 1:1 ratio), supplemented with GlutaMAX (2 mM; Thermo Fisher Scientific), B27 supplement (1%; Thermo Fisher Scientific), Penicillin G (100 U/mL; Sigma Aldrich), recombinant human fibroblast growth factor-2 (bFGF) (10 ng/mL; R&D Systems), recombinant human epidermal growth factor (EGF) (20 ng/mL; R&D Systems) and heparin (2 μg/mL; Sigma Aldrich) at 37°C, 5% CO2. Cells were grown either as spheres in suspension or as adherent cultures on laminin-coated flasks where they maintain intact self-renewal capacity (Pollard et al., 2009).
Human glioblastoma stem cell lines GB6, GB7, GB8, G144, G166 and the human fetal neural stem cell lines CB660 and U3 were kind gifts from Luciano Conti (Department CIBIO, University of Trento, Italy) and were cultured as adherent cultures on laminin-coated flasks in Euromed-N media (Euroclone), supplemented with GlutaMax (2mM), B27 supplement (2%), N2 (1%; Thermo Fisher Scientific), Penicillin G (100 U/mL), recombinant human fibroblast growth factor-2 (bFGF) (20 ng/mL), and recombinant human epidermal growth factor (EGF) (20 ng/mL) at 37°C 5% CO2. Culture flasks were coated with laminin (10 μg/mL final, Thermo Fisher Scientific, cat. 23017015) and incubated for 3 h at 37°C or overnight at 4°C prior to use.
Human glioblastoma stem cell lines GSC23p, GSC163, GSC67, GSC62, GSC23C, GSC76, GSC30PT, GSC28, GSC148, GSC83, GSC1, GSC30p, GSC74, GSC120, GSC61, GSC70, GSC83_2, GSC112, GSC151, GSC147 and GSC68 were isolated from patients’ surgical samples at the Institute of Neurosurgery, Catholic University of Rome as previously described (Marziali et al., 2016). Human fetal neural stem cell line HNPC#13 was obtained from the brain of a 12-week-old embryo legally aborted, according to the ethical guidelines of the European Network for Transplantation (NECTAR), as previously described (Ricci-Vitiani et al., 2004). These lines were grown in suspension in DMEMF12 serum-free medium (Thermo Fisher Scientific) containing 2 mM L-glutamine, 0.6% glucose, 9.6 mg/mL putrescine, 6.3 ng/mL progesterone, 5.2 ng/mL sodium selenite, 0.025 mg/mL insulin, 0.1 mg/mL transferrin sodium salt (Sigma, St Louis, MO), EGF (20 ng/mL), bFGF (10 ng/mL) and heparin (2 μg/mL; Sigma Aldrich) at 37°C, 5% CO2.
Human lung fibroblasts, MRC5, were cultured in EMEM media (Thermo Fisher Scientific), supplemented with 10% FBS, GlutaMAX (2 mM; Thermo Fisher Scientific) and Penicillin G (100 U/mL; Sigma Aldrich) at 37°C, 5% CO2.
Human skin fibroblast, Hs68, were cultured in DMEM media (Thermo Fisher Scientific), supplemented with 10% FBS, GlutaMAX (2 mM; Thermo Fisher Scientific) and Penicillin G (100 U/mL; Sigma Aldrich) at 37°C, 5% CO2.
For GSCs differentiation, cells were grown on laminin-coated plates in the above media without growth factors and with the addition of 10% FBS (Thermo Fisher Scientific) for 14 days. For astrocyte differentiation, CB660, U3 and HNPC#13 cells were grown on laminin-coated plates in the above media without growth factors and with the addition of 5% FBS for 3 weeks.
For hypoxia and anoxia experiments, cells after treatment were grown in a hypoxic chamber (Invivo2 200, Baker Ruskinn, Hypoxic, 1% O2).
Method details
Cas9 cell line generation
sgRNA cloning
pLKO.1-puro U6 sgRNA BfuAI large stuffer plasmid where the puromycin gene was removed and replaced with a puro-GFP fusion protein as previously described by Phelan et al., 2018 was used (gift from Dr. Louis Staudt, National Cancer Institute, USA). The resulting plasmid was digested with BfuAI (NEB cat. R0701S). Complementary sgRNA sequences flanked by ACCG on the 5′ end, and CTTT on the 3′ of the reverse strand were annealed, diluted and ligated into the cut vector with T4 ligase (NEB cat. M0202M) according to the manufacturer’s instructions. All transformations were performed in Stbl3 bacteria (Thermo Fisher Scientific cat. C737303) and sequencing was performed to verify the correctness of the inserts. The sgRNA sequences are listed in the Key resources table.
Lentiviral vectors production
Lentiviral particles were produced in 293FT cells (Thermo Fisher Scientific cat. R70007) by transfecting Cas9 vector or sgRNA vectors with packaging vectors psPAX2 (Addgene cat. 12260) and pMD2.G (Addgene cat. 12259) in a 4:3:1 ratio in serum-free Opti-MEM. Trans-IT 293T (Mirus cat. MIR2705) was added and incubated for 15 min before adding the mixture dropwise to cells. Supernatants were harvested 48 and 72 h later, filtered through a 45μm filter and then incubated with Lenti-X concentrator (Takarabio cat. 631232). Virus was concentrated according to the manufacturer’s instructions, aliquoted and stored frozen at −80°C.
Cas9 cell line generation
COMI-Cas9 and VIPI-Cas9 cells were generated by transducing COMI and VIPI cells twice with lenti-SpCas9 blast (Addgene cat. 104997) and selected with 100 μg/ml blasticidin. The generated cell lines were tested for functional Cas9 cutting after transduction with sgRNA targeting the essential ribosomal subunit RPL6 and expressing a GFP reporter. The loss of GFP was assessed by FACS for 15-17 days. Since the efficiency of COMI-Cas9 cell pool cutting was not sufficient, this line was dilution cloned. Single-cell clones were picked and tested for functional Cas9 cutting and the best cutting clone was selected for further experiments.
Generation of knockout cell lines
COMI and VIPI Cas9-expressing cells were transduced with sgRNA lentiviral vectors targeting sgNT (negative control), sgTUFM and sgMRPS18A and expressing a GFP reporter and selected with 1 μg/ml puromycin for COMI and 1.25 μg/ml puromycin for VIPI for 72h. The transduction and selection were performed in biological triplicates. These cell lines were then used to perform the 3D viability, proliferation and gliomasphere formation assays.
Screening and analysis
A custom-made library was created using compounds available from the Selleck Translation Inhibitors and MS Discovery libraries, and additional compounds acquired from other companies (Table S1). All compounds were solubilized in dimethyl sulfoxide (DMSO, Sigma Aldrich) and stored frozen prior to use. COMI and VIPI cells were plated in 96-well microtiter plates in 150 μL of media at plating densities of 6,000 and 4,500 cells/well, respectively, and incubated for 24 h prior to drug treatment. The drugs were added to a final concentration of 100 μM and the plates were incubated for 24 h at 37°C. Each treatment was performed in a technical triplicate. The positive control wells were treated with FCCP, rotenone and oligomycin A at 100 μM for 24 h or with FCCP (Cayman Chemical, cat.15218) at 100 μM for 30 min at 37°C. Next, cells were stained with 200 nM MitoTracker Orange (Thermo Fisher Scientific, cat. M7510) and incubated for 30 min at 37°C. Subsequently, cells were fixed by direct addition of paraformaldehyde solution to a final concentration of 4% v/v and incubation for 15 min at room temperature, followed by a wash with 200 μL of PBS. The cells were then stained with 5 μM DRAQ5 (Cell Signaling, cat. 4084) for 20 min at room temperature protected from light. Drug dilution and addition were performed using Tecan Evo 200 liquid handler, whereas fixing, PBS washing, and dyes dispensation were performed using BioTek EL406 washer dispenser. The plates were read using the Operetta-High Content Imaging System (Perkin Elmer). Compound precipitation was visually assessed, and interfering compounds were excluded from the analysis.
In order to perform phenotypic profiling of the compound-induced toxicity effects, the image data were visualized and analyzed using the Harmony Software. The data were plate-wise normalized by computing the compound percentage of the vehicle control treatment (DMSO). Six informative features describing cellular compactness and intracellular fluorescence distribution were extracted to describe cellular or mitochondrial toxicity phenotypes (Table S2). The informative content of a feature was assessed by testing whether the feature values for the positive controls were significantly different from the vehicle treatment (t test, FDR < 0.05). For each library compound, we assessed the size of the effect induced on each feature. A feature for a given compound was defined as a hit when the feature deviated from the median value of all compounds by a distance larger than 1.5 times the median absolute deviation. Individual compounds were scored by the number of affected features. When a feature was affected by a compound, it accounted for one over the number of informative features N (in this case ⅙, since N = 6). Therefore, the maximal attainable score when all the features are affected for a given compound is one (or 100%). We next assigned a percentage score to each class of compounds by calculating the average of the scores attained by all the compounds included in each class.
Viability assays
For the evaluation of the effect of OXPHOS and glycolysis inhibitors on GSCs viability, cells were plated in a 96 laminin-coated well plate (2,500 cells/well of COMI and VIPI) and treated the following day with rotenone (1 μM, Sigma Aldrich, cat. R8875), Oligomycin (1 μM, Sigma Aldrich, cat. O4876), Antimycin A (1 μM, Sigma Aldrich, cat. A8674), 2-deoxyglucose (2-DG, 250 μM, Sigma Aldrich, D8375) and dichloroacetic acid (DCA, 5 mM, Sigma Aldrich, cat. 347795) for 72 h. Each treatment was performed in technical triplicate.
For the evaluation of the effect of streptogramins, tetracyclines and pleuromutilins on GSCs viability, cells were seeded into 96-well laminin-coated microtiter plates in 150 μL of media at plating densities ranging from 3,500-6,000 cells/well. The plates were incubated for 24 h prior to drug treatment. Serial drug dilutions were prepared in PBS to provide a total of seven drug concentrations plus control. 10 μL of these dilutions were added to each well, and the plates were incubated for additional 48 h. Each treatment was performed in technical quadruplicate. The following compounds were tested: Dalfopristin (Santa Cruz, cat. sc-362728), Quinupristin (Bioaustralis, cat. BIA-Q1354), Quinupristin-dalfopristin mesylate complex (Santa Cruz, cat. sc-391726), Virginiamycin M1 (Sigma Aldrich, cat. V2753), Virginiamycin B (Santa Cruz, cat. sc-362038), Virginiamycin S1 (Apollo Scientific, cat. BIV1004), Griseoviridin (Apollo Scientific, cat. BIBR1137), Viridogrisein (Apollo Scientific, cat. BIBR1132), Tetracycline (Sigma Aldrich, cat. 87128-25G), Methacycline HCl (Sigma Aldrich, cat.37906-100MG-R), Oxytetracycline (Sigma Aldrich, cat PHR1537-1G), Doxycycline HCl (Sigma Aldrich, cat. D3447-500MG), Demeclocycline HCl hydrate (Sigma Aldrich, cat. 46161-100MG), Meclocycline Sulfosalicylate (Sigma Aldrich, cat. M1388-500 MG), Minocycline HCl (Santa Cruz, cat. sc-203339), Pipacycline (Santa Cruz, cat. sc-391690), Tigecycline (Santa Cruz, cat. sc-394197), Valnemulin (MicroSource Discovery library) and Tiamulin (Sigma Aldrich, cat., 34044).
After the drug treatments, the cells were stained with Hoechst 33342 (1 μg/mL; Thermo Fisher Scientific, cat. H1399) and Propidium Iodide (PI, 1 μg/mL; Sigma Aldrich, cat. P4170) and incubated for 20 min shaking in the dark. The plates were then read using Operetta-High Content Imaging System (Perkin Elmer) and analyzed using the Harmony Software. The number of viable cells was calculated by subtracting PI positive cells from the total number of cells estimated by Hoechst 33342 staining and normalized to the non-treated control.
For cytotoxicity analysis on GSCs grown as gliomaspheres, the cells were mechanically dissociated and plated at a density of 20,000 cells/mL in a 96-well plate, in triplicate. Quinupristin/dalfopristin (Q/D) was added 24 h after cell plating. ATP levels were measured using the CellTiter-Glo® Luminescent Cell Viability Assay (Promega, cat. G7570) as per the manufacturer’s instructions after 48 h and 72 h of treatment. Percentage viability was calculated upon normalization on the non-treated control.
Dose-response curves were plotted, and growth inhibition 50 (GI50) values calculated using the GraphPad Prism software.
3D Viability assay
Ten COMI cells/well were plated in Ultra-Low attachment round bottom 96 well plates (Costar) and treated with desired Q/D concentrations. The cells were centrifuged at 300 g for 30 s, followed by the first acquisition using Operetta-High Content Imaging System (Perkin Elmer). The images were subsequently acquired over the course of 9-10 days. The area of the spheres formed was assessed using the Harmony Software.
Ten COMI-Cas9 cells/well and twenty-five VIPI-Cas9 cells/well transduced with the respective sgRNA lentiviral vectors were plated in Ultra-Low attachment round bottom 96 well plates (Costar). The experiment was then performed as described above. The images were acquired over the course of 15 days. The area of the spheres formed was assessed using the Harmony Software.
Gliomasphere formation assay
COMI cells grown in suspension were plated at a density of 50,000 cells/mL and treated with Q/D for 72 h. The spheres were then dissociated, cells counted and plated at a density of 10 or 100 cells/well in a 96 well plate without the drug. After 10 days, the spheres formed were stained with 1 μM Calcein AM (Thermo Fisher Scientific, cat. C3100MP), incubated for 20 min at 37°C, after which the spheres were imaged using Operetta-High Content Imaging System (Perkin Elmer) and analyzed using the Harmony Software. Only spheres greater than 100 μm were quantified. The experiment was performed in a biological triplicate, with 20 technical replicates each.
COMI-Cas9 cells and VIPI-Cas9 cells transduced with the respective sgRNA lentiviral vectors were plated at two different cell densities (10 and 100 cells/well for COMI, and 100 and 250 cells/well for VIPI). After 10 days, the spheres formed were stained and analyzed as described above.
Cell cycle assay
Cells were plated 24 h prior to treatment (120,000 cells/well of COMI cells in a 6 well plate) and incubated with Q/D for additional 48h. Cell cycle analysis was conducted using FACS (BD FACSCanto II) after staining with the Click-IT EdU Flow Cytometry Assay kit (Thermo Fisher Scientific, cat.C10634) according to the manufacturer’s instructions. The experiment was performed in a biological triplicate. Data were processed by the BD FacsDIVA V8.0.1 software.
Apoptosis assays
Apoptosis was assessed using FITC Annexin V Apoptosis Detection Kit I (BD PharMingen, BD Biosciences, cat. 556547). Cells were plated 24 h prior to treatment (270,000 cells/6cm2 plate of COMI cells) and incubated with Q/D for further 48h. 200,000 cells were stained according to the manufacturer’s instructions and analyzed using FACS (BD FACSCanto II). Data were processed by BD FacsDIVA V8.0.1 software.
Autophagy assays
To assess the autophagic flux, cells were plated 24 h prior to treatment and incubated with 6.5 μM Q/D for further 48 h. In addition, for immunoblotting the cells were treated with 60 μM chloroquine for 24 h (Sigma Aldrich, cat. C6628), 6.5 nM bafilomycin for 3 h (Sigma Aldrich, cat. B1793) or 5 mM NH4Cl for 3 h (Sigma Aldrich, cat. A9434). Immunoblotting was performed as described in the Immunoblotting section. LC3B-II was quantified by densitometric analysis (Image Lab 2.0.1 software, Biorad) and normalized to β-tubulin as a loading control. For immunofluorescence analysis, the cells were treated with 60 μM chloroquine, 6.5 nM bafilomycin or 10 mM NH4Cl for 24 h. Immunofluorescence for LC3 staining was carried out according to the procedure described in the Immunofluorescence section. Cell morphology was determined by staining with CellMask Deep Red Stain (Thermo Fisher Scientific, cat. H32721).
In order to evaluate the role of autophagy in Q/D cytotoxic activity, COMI cells were seeded into 96-well microtiter plates in 150 μL of media at plating densities of 4,000 cells/well. The plates were incubated for 24 h prior to drug treatment. Cells were pretreated with chloroquine (5, 10 and 20 μM) for 3 h and then treated with 6.5 μM of Q/D for further 48 h. The cells were stained with Hoechst 33342 (1 μg/mL; Thermo Fisher Scientific, cat. H1399) and Propidium Iodide (PI, 1 μg/mL; Sigma Aldrich, cat. P4170) and incubated for 20 min shaking in the dark. The plates were then read using Operetta-High Content Imaging System (Perkin Elmer) and analyzed using the Harmony Software. The number of viable cells was calculated by subtracting PI positive cells from the total number of cells estimated by Hoechst 33342 staining and normalized to the control. Each treatment was performed in technical quadruplicate and in biological triplicate.
Cryo-EM data collection
Human mitoribosomes and grids were prepared as described previously (Brown et al., 2014) with modifications involving scaling down the prep in order to minimize the time from the antibiotic treatment to cryo-EM (Aibara et al., 2018). Briefly, 2 l HEK293S TetR GnTi- cells lacking N-acetyl-glucosaminyltransferase I were grown to a final concentration of 4.2 × 106 cell/mL. Prior to cell collection and mitoribosome isolation, the cells were treated with 5 μM Q/D for 24 h. Three μL of purified mitochondrial ribosomes at a concentration of 3.6 Abs at 280 nm were incubated for 30 s on glow-discharged holey carbon grids (Cu Quantifoil R2/2), onto which a home-made continuous carbon film had previously been deposited, prior to vitrification in a Vitrobot MKIV (3 s blot, 4°C, 100% humidity). Images were collected on a 300 kV FEI Titan Krios electron microscope, a slit width of 20 eV on a GIF-Quantum energy filter. A Gatan K2-Summit detector was used in counting mode at a magnification of 130,000 (yielding a pixel size of 1.05 Å), and a dose rate of ∼5.2 electrons per pixel per second. Exposures of 8 s (yielding a total dose of 38 e-Å-2) were dose-fractionated into 20 movie frames that were stacked into a single MRC stack using newstack. 4225 images were collected using EPU automatic data collection with defocus values ranging from 0.4 to 4.5 μm.
Cryo-EM image processing, model building and refinement
Collected micrographs were corrected for local-frame movement as well as dose filtered using UcsfDfCorr. The contrast transfer function parameters were estimated using GCTF-0.5, for all other image processing steps RELION-2.1.b1. 578,573 particles were picked using reference based picking, where bad particles were removed by particle sorting (546,701 particles retained) and subjected to reference-free 2D class averaging, resulting in 387,428 particles for 3D classification. 4 classes containing 138,030 particles were pooled and subjected to 3D auto-refinement, yielding a map of the human mitochondrial ribosome. However, since there was some heterogeneity visible in the reconstruction, the particles were subjected to further fine angular classification and 100,000 particles were selected for another 3D auto-refinement step. Since Q/D is known to interact with the bacterial large subunit and to improve the quality of the map, masked focused-refinement was used to focus on the mitoribosomal large subunit. The resolution of the final reconstruction was 3.9 Å.
Reported resolutions are based on gold-standard refinement applying the 0.143-criterion on the FSC between reconstructed half-maps. The FSC was corrected for the effects of a soft mask on the using high-resolution noise substitution. All 3D refinements used a 70 Å low-pass filtered initial model, the first of which was a previous low resolution reconstruction of the human mitochondrial ribosome (EMDB-2876; Amunts et al., 2015). Before visualization, all density maps were corrected for the modulation transfer function of the detector, and then sharpened by applying a negative B-factor that was estimated using automated procedures.
The quality of the map allowed unambiguous model building with Coot. Stereochemical refinement was performed using phenix.real_space_refine as implemented in the PHENIX suite. The final model was validated using MolProbity. Refinement statistics are given in Table S6.
Protein synthesis assay
Cells were plated 24 h prior to treatment (180,000 cells/well of COMI cells in a 6 well plate) and incubated with Q/D for further 24 h prior to 35S labeling. To assay for mitochondrial protein synthesis, growth medium was removed and cells were washed twice with methionine/cysteine-free DMEM medium, followed by an incubation in methionine/cysteine-free DMEM medium containing 96 μg/mL Cysteine, 1% B27 supplement, 1% GlutaMax; 1% Sodium Pyruvate, 10 ng/mL bFGF, 20 ng/mL EGF, 2 μg/mL heparin and 80 μg/mL emetine (Sigma Aldrich, cat. E2375) for 15 min at 37°C. Subsequently, [35S]-methionine (Perkin Elmer, cat. NEG709A005MC) was added to a final concentration of 166.6 μCi/mL and the labeling was performed for 20 min at 37°C. The cells were then detached and pelleted at 4,000 rpm for 5 min. The pellet was washed three times with 1 mL of PBS. Cell pellets were resuspended in protein lysis buffer containing protease inhibitors and 1.25 U/μL benzonase. Protein concentrations were measured with Pierce™ BCA Protein Assay Kit (Thermo Fisher Scientific, cat. 23227) and equal amount of protein samples were separated on SDS-PAGE gels (NuPAGE™ 12% Bis-Tris Protein Gels, Thermo Fisher Scientific, cat. NP0343BOX). The labeled proteins were visualized and quantified using a PhosphorImager system and ImageQuant software (Molecular Dynamics, GE Healthcare). To assay for cytosolic translation, the above procedure was used without the addition of emetine.
Immunoblotting
Total cell lysates were prepared from cells. Briefly, cells were washed with PBS and resuspended in lysis buffer (50 mM Tris-HCl pH 7.4, 150 mM NaCl, 1 mM EDTA, 0.25% NP-40, 0.1% Triton X-100, 0.1% SDS and supplemented with protease inhibitors). Protein concentrations were measured with Pierce™ BCA Protein Assay Kit (Thermo Fisher Scientific, cat. 23227). Equal amounts of protein were separated on SDS-PAGE and transferred to nitrocellulose or PVDF (for anti-LC3 antibody only) membrane. Membranes were probed with anti-MTCO1 (COX1, Abcam, cat. ab14705), anti-COX4 (Cell Signaling, cat. 4850), anti-SDHA (Cell Signaling, cat. 5839), anti-β-tubulin (Santa Cruz, cat. sc-53140), anti-TUFM (Thermo Fisher Scientific, cat. MA5-31363), anti-MRPS18A (Thermo Fisher Scientific, cat. PA5-57274), anti-LC3 (Cell Signaling, cat. 3868S), and secondary HRP-conjugated antibodies (Santa Cruz Biotechnology). Detection was performed using Amersham ECL Prime or Select Western Blotting Detection Reagent (GE Healthcare Life Sciences) and ChemiDoc Imaging System (Bio-Rad). Data were analyzed using ImageLab software.
Immunofluorescence
The cells were fixed either with paraformaldehyde solution (4% v/v final, 15 min incubation at room temperature) or with 100% ice-cold methanol (5 min incubation at room temperature, only for LC3B IF), followed by two washes with PBS. The cells were then permeabilized with 0.3% Triton X-100 - 3% BSA in PBS for 45 min at room temperature. The primary and secondary antibodies were diluted in 3% BSA-PBS solution. The incubation with the primary antibody was carried out at 4°C overnight, followed by a wash with 3% BSA-PBS solution and incubation with the secondary antibody for 1 h at room temperature. Cell morphology was determined by staining with HCS CellMask Deep Red Stain (Thermo Fisher Scientific, cat. H32721, 1:2000, 20 min, room temperature). The plates were then read either using Operetta-High Content Imaging System (Perkin Elmer) and analyzed by the Harmony Software or using the Leica TCS SP5 confocal microscope and processed by imaging softwares ImageJ (version v1.51w) and Photoshop. For the latter, z stack images were acquired and LC3 puncta quantification was performed on image stacks of region of interests containing single cells, using the “3D Maxima finder” plugin of ImageJ. Both size and intensity threshold constraints were applied to the quantification.
Confocal imaging
Images were acquired on a Leica TCS SP5 confocal microscope with a 63x oil immersion objective, 2x zoom, 1024x1024 resolution, 200Hz speed, lasers Argon 488 nm and Diode laser 633 nm, step 0.89 μm. Images were further analyzed and processed using imaging softwares ImageJ (Fiji) and Photoshop.
RNA extraction, reverse transcription and qPCR
Total RNA was extracted using QIAzol reagent (QIAGEN) according to the manufacturer’s instructions. Reverse transcription was performed using iScript Reverse Transcription Supermix (BioRad, cat. 170-8891) on C1000 Thermal Cycler (BioRad) and quantitative real-time PCR was performed using 2x qPCR SyGreen Mix Separate ROX (PCR Biosystems, cat. PB20.14-05) following the manufacturer’s instructions on CFX384 Real-Time System (BioRad). All assays were performed in triplicate in 4-5 independent experiments. Data was analyzed using the CFX Manager software (BioRad). Relative expression values of each target gene were normalized to GAPDH and 18S RNA level. The following primers were used at 500 nM concentration:
COX1: 5′- CTATACCTATTATTCGGCGCATGA-3′ and 5′-CAGCTCGGCTCGAATAAGGA −3′,
COX4: 5′-GCCATGTTCTTCATCGGTTTC-3′ and 5′-GGCCGTACACATAGTGCTTCTG-3′,
18S: 5′-GGACATCTAAGGGCATCACA-3′ and 5′-AGGAATTGACGGAAGGGCAC-3′,
GAPDH: 5′-CAACGAATTTGGCTACAGCA-3′ and 5′-AGGGGTCTACATGGCAACTG-3′.
BN-PAGE and in gel complex activity assay
Cells were plated in two T75 flasks (700,000 cells/flask) and treated with Q/D after 24 h for additional 48 h, 72 h and 96 h. Mitochondria were isolated in the following manner: cells were detached, pelleted and resuspended in 750 μL of MIB+BSA buffer (0.32 M sucrose, 1 mM EGTA pH 8, 20 mM Tris-HCl, pH 7.2, 0.1% fatty acid-free BSA). The cells were homogenized using Potter-Elvehjem homogenizer and centrifuged at 1,000 g for 5 min at 4°C. The supernatant was collected and the pellet resuspended in MIB+BSA, rehomogenized and centrifuged again. The supernatant was collected and pooled with the first one, then centrifuged at 12,000 g for 10 min at 4°C to pellet the mitochondria. The pellet of mitochondria was subsequently washed once with MIB+BSA, once with MIB, resuspended in 100 μL of ACNA buffer (1.5 M aminocaproic acid (Sigma Aldrich, cat. A2504), 50 mM BisTris, pH 7.00) and quantified using Qubit Protein Assay Kit (Thermo Fisher Scientific, cat. Q33212). Digitonin (Sigma Aldrich, cat. D5628-1G) was added to a final concentration of 1% w/v, the samples were vortexed and incubated on ice for 20 min, followed by a centrifugation at 14,000 g for 30 min at 4°C. The supernatant was mixed with the loading buffer, and 50 μg of protein was separated on Blue Native PAGE gels (NativePAGE™ 3%–12% BisTris Protein Gels, Thermo Fisher Scientific, cat. BN1001BOX). The gels were incubated overnight at room temperature with the respective complex substrates: Complex I: 2 mM Tris HCl, pH 7.4; 0,1 mg/mL NADH (Sigma Aldrich, cat. N8129-50MG); 2.5 mg/mL iodonitrotetrazolium chloride (Sigma Aldrich, cat. I8377-1G); Complex II: 4.5 mM EDTA, 0.2 mM phenazine methosulfate (Sigma Aldrich, cat. P9625-500MG), 84 mM succinic acid (Sigma Aldrich, cat. S2378-100G) and 0.5 mg/mL iodonitrotetrazolium chloride (Sigma Aldrich, cat. I8377-1G); Complex IV: 0.5 mg/mL 3.3′-diamidobenzidine tetrahydrochloride (DAB) (Sigma Aldrich, cat. D5637-1G), 50 mM phosphate buffer pH 7.4; 1 mg/mL cytochrome c (Sigma Aldrich, cat. C2506-1G), 0.2 M sucrose, 20 μg/mL (1 nM) catalase (Sigma Aldrich, cat. C9322-1G), Complex V: 3.76 mg/mL glycine, 5 mM MgCl2, Triton X-100, 0.5 mg/mL lead nitrate (Sigma Aldrich, cat. 203580), ATP (Sigma Aldrich, cat. A7699-5G), pH 8.4.
Immunoblotting after BN-PAGE
Proteins separated on Blue Native PAGE gels (NativePAGE™ 3%–12% BisTris Protein Gels, Thermo Fisher Scientific, cat. BN1001BOX) were transferred to PVDF membrane (25V, 4°C, overnight, wet-transfer, transfer buffer with 20% MeOH and 2 mL of 10% SDS in 1L). Membranes were stripped to remove the blue-staining (Restore PLUS Western Blot Stripping Buffer, Thermo Fisher Scientific, cat. 46430, 3 min) and probed with anti-OxPhos complex I (39kDa subunit; clone 20C11, Thermo Fisher Scientific, cat. A21344), anti-SDHA complex II (2E3GC12FB2AE2, Abcam, cat. ab14715), anti-OxPhos complex III (core 1 subunit; clone 16D10, Thermo Fisher Scientific, cat. A21362), anti-OxPhos complex IV (subunit 1; clone 1D6E1A8, Invitrogen cat. 459600), anti-ATP5A complex V (15H4C4, Abcam, cat. ab14748) and Amersham ECL secondary HRP-conjugated antibodies (GE Healthcare). Proteins detection was performed using Clarity Western ECL Substrate Detection Reagent (Biorad, cat. 1705061) and the film exposure time was adjusted according to signal intensity.
Respiration assay
High-resolution respirometry was performed using a 2 mL chamber OROBOROS Oxygraph-2k (Oroboros Instruments) at 37°C. Respiration rates were calculated as the negative time derivative of oxygen concentration measured in the closed respirometer and expressed per number of viable cells and corrected by residual oxygen consumption (ROX, after antimycin A addition). The amplified signal was recorded on a computer with online display of the calibrated oxygen concentration and oxygen flux (DatLab software for data acquisition and analysis; Oroboros Instruments). Cells were plated 24 h prior to treatment and incubated with Q/D for further 48 h. The cells were then detached and 1,000,000 cells were injected into each chamber. Oxygen consumption was evaluated for cellular ROUTINE respiration, and then cells were permeabilized with digitonin 4.1 uM (Fluka, cat. 37008) in MiR05 medium (10 mM KH2PO4, 60 mM lactobionic acid (Sigma Aldrich, cat. 153516), 20 mM HEPES, 3 mM MgCl2, 0.5 mM EGTA, 20 mM taurine (Sigma Aldrich, cat. T0625), 110 mM D-sucrose, and 1 mg/mL BSA fatty acid free). Complex I activity was measured after malate (2 mM, Sigma Aldrich, cat. M1000), glutamate (10 mM, Sigma Aldrich, cat. G1626) and ADP (5 mM, Calbiochem, cat. 117105) injection, and complex I&II activity after additional succinate (10 mM, Sigma Aldrich, cat. S2378) injection. The ETS capacity (maximum uncoupled respiration) was induced by stepwise titration of carbonyl cyanide p-(trifluoromethoxy) phenylhydrazone (FCCP, Sigma Aldrich, cat. C2920) (typically 3–4 steps, 1 ul each of 1 mM FCCP). Complex II activity was measured after the addition of rotenone (0.5 uM, Sigma Aldrich, cat. R8875). Residual respiration (ROX) was measured after inhibition with antimycin A (2.5 μM, Sigma Aldrich, cat. A8674).
Mitochondrial membrane potential assessment
Cells were plated 24 h prior to treatment (120,000 cells/well of COMI cells in a 6 well plate) and incubated with Q/D for further 48h. Positive control was treated with 100 uM FCCP for 10 min. Cells were detached and 500,000 cells were resuspended in fresh media containing 5 μg/mL JC-1 (Abcam, cat. ab113850), and incubated at 37°C for 30 min. Cells were then centrifuged at 400 g for 5 min and resuspended in 0.5 mL PBS and analyzed using FACS (BD FACSCanto II). Data were processed by BD FacsDIVA V8.0.1 software.
Lactate Assay
Cells were plated in a 96 well plate (4,000 cells/well of COMI and 3,500 cells/well of VIPI) and treated the following day with Q/D for 48h. Media was collected and the lactate production was measured using Glycolysis Cell-Based Assay Kit (Cayman Chemical, cat. 600450) according to the manufacturer’s instructions. The cells were then fixed with 4% v/v paraformaldehyde for 15 min at room temperature and stained with Hoechst 33342 (1 μg/mL). The nuclei were quantified using Operetta-High Content Imaging System (Perkin Elmer) and analyzed by the Harmony Software. The lactate production was normalized to the number of cells.
Mitochondrial mass
Mitochondrial mass was assayed by staining the mitochondria with MitoTracker Orange (Thermo Fisher Scientific) or anti-COX4 antibody (Abcam) as described in the immunofluorescence section. The number of mitochondria was estimated by counting the number of MitoTracker Orange or COX4 positive spots per area of cytoplasm using Operetta-High Content Imaging System (Perkin Elmer) and analyzed by the Harmony Software.
Competition assay
VIPI-Cas9 cells were transduced with sgRNA lentiviral vectors targeting TUFM and MRPS18A and expressing a GFP reporter. sgNT was used as a negative control. Three days after transduction, GFP levels were measured by flow cytometry on a BD LSR II using Diva version 9.0 and analyzed with FlowJo version 10.7.1. GFP levels were followed every 3-4 days for 21 days and normalized to the day 1 measurement.
Analysis of Cas9-induced indels
Genomic DNA was extracted using QuickExtract DNA Extraction Solution and the target site was PCR-amplified with Taq DNA polymerase (Meridian Bioscience).
The following primers were used at a concentration of 400 nM:
TUFM for: GTCTAAGCTCTGCCTCTAGC
TUFM rev: CATCTCAATGCCTAGGACGG
MRPS18A for: GTGGAGGAAGTGATCTCAGC
MRPS18A rev: GGTAATAGACCTGAGTGGCG
PCR products were purified and sequenced. To assess the Cas9-induced indels with each sgRNA, tracking of indels by decomposition (TIDE) analysis was performed as reported in Brinkman et al. (2014).
2D viability assay
COMI-Cas9 and VIPI-Cas9 cells transduced with the respective sgRNA lentiviral vectors were plated in 96 well plates at a cell density of 1500 cells/well. Every 24 h, five wells per condition were stained with Hoechst 33342 (1 μg/mL; Thermo Fisher Scientific, cat. H1399) and Propidium Iodide (PI, 1 μg/mL; Sigma Aldrich, cat. P4170) and incubated for 20 min shaking in the dark. The plates were then read using Operetta-High Content Imaging System (Perkin Elmer) and analyzed using the Harmony Software. The number of viable cells was calculated by subtracting PI positive cells from the total number of cells estimated by Hoechst 33342 staining and normalized to day 0.
Quantification and statistical analysis
Results were reported as mean ± SD (standard deviation) or mean ± SEM (standard error of the mean), as indicated in the figure legend. Details of each analysis are in figure legends. Statistical significance is determined by unpaired two-tailed t test (∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001; ns p ≥ 0.05). n represents number of biological or technical replicates, as indicated in figure legends. All the experiments with representative images (including immunoblotting and immunofluorescence) were repeated at least twice and representative images are shown. The association of GI50 values with continuous clinical features was computed by Pearson’s correlation coefficient and with categorical values by ANOVA test.
Acknowledgments
This work was supported by Fondazione Giovanni Celeghin Onlus and by a donation from Ivana and Enrico Zobele (to A.Q.). This work was also supported by the Swedish Foundation for Strategic Research (FFL15:0325), the Ragnar Söderberg Foundation (M44/16), the Swedish Research Council (NT_2015-04107), Cancerfonden (CAN 2017/1041), the European Research Council (ERC-2018-StG-805230), the Knut and Alice Wallenberg Foundation (2018.0080) (to A.A.), the FEBS Long-Term Fellowship Program (to S.A.), an EMBO short-term fellowship (to A.M.), a postdoctoral fellowship (CRISPR Screen Action) from the Canceropôle Provence-Alpes-Côte d’Azur, the French National Cancer Institute (INCa), the Provence-Alpes-Côte d’Azur Region (to J.R.), and the AIRC Foundation (IG 2019 Id.23154 to R.P.). The graphical abstract was created with BioRender. COMI and VIPI cells were a kind gift from Antonio Daga (Azienda Ospedaliera Universitaria San Martino di Genova, Italy) and 030616 from Rossella Galli (H.S. Raffaele, Milan, Italy). The human GSC lines GB6, GB7, GB8, G144, and G166 and the human fetal neural stem cell lines CB660 and U3 were kind gifts from Luciano Conti (Department CIBIO, University of Trento). We thank the HTS, CASF, CT, and the Imaging Core Facilities of CIBIO for assistance with experimentation. We thank Camilla Maffezzini at the Karolinska Institute for help with BN-PAGE and in-gel activity assay. The cryo-EM data were collected at the Swedish National Facility funded by the Knut and Alice Wallenberg Foundation, Family Erling Persson Foundation, and Kempe Foundation. We thank M. Carroni, J.M. de la Rosa Trevin, and S. Fleischmann for smooth data collection and processing.
Author contributions
Conceptualization, A.M. and A.Q.; methodology, D.S. and A.M.; formal analysis, A.R.; investigation, D.S., M.N., S.A., G.R., V.A., M.G., A.S., C.A., F.B., E.F.R., M.B., Q.G.D., S.G., S.P., R.P., A.A., and A.M.; writing – original draft, A.M.; writing – review & editing, A.M., D.S., and A.Q.; funding acquisition, A.M., A.Q., and A.A.; resources, A.Q., L.R.-V., R.P., J.R., A.A., S.R., and I.M.; supervision, A.M. and A.Q.
Declaration of interests
D.S., I.M., A.M., and A.Q. have applied for protection of intellectual property related to the results in the manuscript and to Q/D analogs in cancer therapy (Italian patent number 102020000012601).
Published: April 27, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.109024.
Contributor Information
Denise Sighel, Email: denise.sighel@unitn.it.
Angelika Modelska, Email: a.modelska@gmail.com.
Alessandro Quattrone, Email: alessandro.quattrone@unitn.it.
Supplemental information
References
- Aibara S., Andréll J., Singh V., Amunts A. Rapid Isolation of the Mitoribosome from HEK Cells. J. Vis. Exp. 2018;140:57877. doi: 10.3791/57877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amunts A., Brown A., Toots J., Scheres S.H.W., Ramakrishnan V. Ribosome. The structure of the human mitochondrial ribosome. Science. 2015;348:95–98. doi: 10.1126/science.aaa1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angeletti F., Fossati G., Pattarozzi A., Würth R., Solari A., Daga A., Masiello I., Barbieri F., Florio T., Comincini S. Inhibition of the Autophagy Pathway Synergistically Potentiates the Cytotoxic Activity of Givinostat (ITF2357) on Human Glioblastoma Cancer Stem Cells. Front. Mol. Neurosci. 2016;9:107. doi: 10.3389/fnmol.2016.00107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashton T.M., McKenna W.G., Kunz-Schughart L.A., Higgins G.S. Oxidative Phosphorylation as an Emerging Target in Cancer Therapy. Clin. Cancer Res. 2018;24:2482–2490. doi: 10.1158/1078-0432.CCR-17-3070. [DOI] [PubMed] [Google Scholar]
- Auffinger B., Tobias A.L., Han Y., Lee G., Guo D., Dey M., Lesniak M.S., Ahmed A.U. Conversion of differentiated cancer cells into cancer stem-like cells in a glioblastoma model after primary chemotherapy. Cell Death Differ. 2014;21:1119–1131. doi: 10.1038/cdd.2014.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergeron M., Montay G. The pharmacokinetics of quinupristin/dalfopristin in laboratory animals and in humans. J. Antimicrob. Chemother. 1997;39(Suppl A):129–138. doi: 10.1093/jac/39.suppl_1.129. [DOI] [PubMed] [Google Scholar]
- Brinkman E.K., Chen T., Amendola M., van Steensel B. Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res. 2014;42:e168. doi: 10.1093/nar/gku936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown A., Amunts A., Bai X.C., Sugimoto Y., Edwards P.C., Murshudov G., Scheres S.H.W., Ramakrishnan V. Structure of the large ribosomal subunit from human mitochondria. Science. 2014;346:718–722. doi: 10.1126/science.1258026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabrese C., Poppleton H., Kocak M., Hogg T.L., Fuller C., Hamner B., Oh E.Y., Gaber M.W., Finklestein D., Allen M. A perivascular niche for brain tumor stem cells. Cancer Cell. 2007;11:69–82. doi: 10.1016/j.ccr.2006.11.020. [DOI] [PubMed] [Google Scholar]
- Cavalli L.R., Varella-Garcia M., Liang B.C. Diminished tumorigenic phenotype after depletion of mitochondrial DNA. Cell Growth Differ. 1997;8:1189–1198. [PubMed] [Google Scholar]
- Chen J., Li Y., Yu T.-S., McKay R.M., Burns D.K., Kernie S.G., Parada L.F. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature. 2012;488:522–526. doi: 10.1038/nature11287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delgado G., Jr., Neuhauser M.M., Bearden D.T., Danziger L.H. Quinupristin-dalfopristin: an overview. Pharmacotherapy. 2000;20:1469–1485. doi: 10.1592/phco.20.19.1469.34858. [DOI] [PubMed] [Google Scholar]
- Di Giambattista M., Chinali G., Cocito C. The molecular basis of the inhibitory activities of type A and type B synergimycins and related antibiotics on ribosomes. J. Antimicrob. Chemother. 1989;24:485–507. doi: 10.1093/jac/24.4.485. [DOI] [PubMed] [Google Scholar]
- Frattini V., Pagnotta S.M., Tala, Fan J.J., Russo M.V., Lee S.B., Garofano L., Zhang J., Shi P., Lewis G. A metabolic function of FGFR3-TACC3 gene fusions in cancer. Nature. 2018;553:222–227. doi: 10.1038/nature25171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman H.S., Kerby T., Calvert H. Temozolomide and treatment of malignant glioma. Clin. Cancer Res. 2000;6:2585–2597. [PubMed] [Google Scholar]
- Garofano L., Migliozzi S., Oh Y.T., D’Angelo F., Najac R.D., Ko A., Frangaj B., Caruso F.P., Yu K., Yuan J. Pathway-based classification of glioblastoma uncovers a mitochondrial subtype with therapeutic vulnerabilities. Nat. Can. 2021;2:141–156. doi: 10.1038/s43018-020-00159-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griguer C.E., Cantor A.B., Fathallah-Shaykh H.M., Gillespie G.Y., Gordon A.S., Markert J.M., Radovanovic I., Clement-Schatlo V., Shannon C.N., Oliva C.R. Prognostic relevance of cytochrome C oxidase in primary glioblastoma multiforme. PLoS ONE. 2013;8:e61035. doi: 10.1371/journal.pone.0061035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haq R., Shoag J., Andreu-Perez P., Yokoyama S., Edelman H., Rowe G.C., Frederick D.T., Hurley A.D., Nellore A., Kung A.L. Oncogenic BRAF regulates oxidative metabolism via PGC1α and MITF. Cancer Cell. 2013;23:302–315. doi: 10.1016/j.ccr.2013.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harms J.M., Schlünzen F., Fucini P., Bartels H., Yonath A. Alterations at the peptidyl transferase centre of the ribosome induced by the synergistic action of the streptogramins dalfopristin and quinupristin. BMC Biol. 2004;2:4. doi: 10.1186/1741-7007-2-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janiszewska M., Suvà M.L., Riggi N., Houtkooper R.H., Auwerx J., Clément-Schatlo V., Radovanovic I., Rheinbay E., Provero P., Stamenkovic I. Imp2 controls oxidative phosphorylation and is crucial for preserving glioblastoma cancer stem cells. Genes Dev. 2012;26:1926–1944. doi: 10.1101/gad.188292.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H.-J., Maiti P., Barrientos A. Mitochondrial ribosomes in cancer. Semin. Cancer Biol. 2017;47:67–81. doi: 10.1016/j.semcancer.2017.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuntz E.M., Baquero P., Michie A.M., Dunn K., Tardito S., Holyoake T.L., Helgason G.V., Gottlieb E. Targeting mitochondrial oxidative phosphorylation eradicates therapy-resistant chronic myeloid leukemia stem cells. Nat. Med. 2017;23:1234–1240. doi: 10.1038/nm.4399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamb R., Ozsvari B., Lisanti C.L., Tanowitz H.B., Howell A., Martinez-Outschoorn U.E., Sotgia F., Lisanti M.P. Antibiotics that target mitochondria effectively eradicate cancer stem cells, across multiple tumor types: treating cancer like an infectious disease. Oncotarget. 2015;6:4569–4584. doi: 10.18632/oncotarget.3174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z., Bao S., Wu Q., Wang H., Eyler C., Sathornsumetee S., Shi Q., Cao Y., Lathia J., McLendon R.E. Hypoxia-inducible factors regulate tumorigenic capacity of glioma stem cells. Cancer Cell. 2009;15:501–513. doi: 10.1016/j.ccr.2009.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marziali G., Signore M., Buccarelli M., Grande S., Palma A., Biffoni M., Rosi A., D’Alessandris Q.G., Martini M., Larocca L.M. Metabolic/Proteomic Signature Defines Two Glioblastoma Subtypes With Different Clinical Outcome. Sci. Rep. 2016;6:21557. doi: 10.1038/srep21557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mast Y., Wohlleben W. Streptogramins - two are better than one! Int. J. Med. Microbiol. 2014;304:44–50. doi: 10.1016/j.ijmm.2013.08.008. [DOI] [PubMed] [Google Scholar]
- Molina J.R., Sun Y., Protopopova M., Gera S., Bandi M., Bristow C., McAfoos T., Morlacchi P., Ackroyd J., Agip A.A. An inhibitor of oxidative phosphorylation exploits cancer vulnerability. Nat. Med. 2018;24:1036–1046. doi: 10.1038/s41591-018-0052-4. [DOI] [PubMed] [Google Scholar]
- Noeske J., Huang J., Olivier N.B., Giacobbe R.A., Zambrowski M., Cate J.H.D. Synergy of streptogramin antibiotics occurs independently of their effects on translation. Antimicrob. Agents Chemother. 2014;58:5269–5279. doi: 10.1128/AAC.03389-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliva C.R., Markert T., Ross L.J., White E.L., Rasmussen L., Zhang W., Everts M., Moellering D.R., Bailey S.M., Suto M.J., Griguer C.E. Identification of Small Molecule Inhibitors of Human Cytochrome c Oxidase That Target Chemoresistant Glioma Cells. J. Biol. Chem. 2016;291:24188–24199. doi: 10.1074/jbc.M116.749978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliva C.R., Zhang W., Langford C., Suto M.J., Griguer C.E. Repositioning chlorpromazine for treating chemoresistant glioma through the inhibition of cytochrome c oxidase bearing the COX4-1 regulatory subunit. Oncotarget. 2017;8:37568–37583. doi: 10.18632/oncotarget.17247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrov A.S., Wood E.C., Bernier C.R., Norris A.M., Brown A., Amunts A. Structural Patching Fosters Divergence of Mitochondrial Ribosomes. Mol. Biol. Evol. 2019;36:207–219. doi: 10.1093/molbev/msy221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phelan J.D., Young R.M., Webster D.E., Roulland S., Wright G.W., Kasbekar M., Shaffer A.L., 3rd, Ceribelli M., Wang J.Q., Schmitz R. A multiprotein supercomplex controlling oncogenic signalling in lymphoma. Nature. 2018;560:387–391. doi: 10.1038/s41586-018-0290-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard S.M., Yoshikawa K., Clarke I.D., Danovi D., Stricker S., Russell R., Bayani J., Head R., Lee M., Bernstein M. Glioma stem cell lines expanded in adherent culture have tumor-specific phenotypes and are suitable for chemical and genetic screens. Cell Stem Cell. 2009;4:568–580. doi: 10.1016/j.stem.2009.03.014. [DOI] [PubMed] [Google Scholar]
- Porporato P.E., Filigheddu N., Pedro J.M.B., Kroemer G., Galluzzi L. Mitochondrial metabolism and cancer. Cell Res. 2018;28:265–280. doi: 10.1038/cr.2017.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravà M., D’Andrea A., Nicoli P., Gritti I., Donati G., Doni M., Giorgio M., Olivero D., Amati B. Therapeutic synergy between tigecycline and venetoclax in a preclinical model of MYC/BCL2 double-hit B cell lymphoma. Sci. Transl. Med. 2018;10:eaan8723. doi: 10.1126/scitranslmed.aan8723. [DOI] [PubMed] [Google Scholar]
- Ricci-Vitiani L., Pedini F., Mollinari C., Condorelli G., Bonci D., Bez A., Colombo A., Parati E., Peschle C., De Maria R. Absence of caspase 8 and high expression of PED protect primitive neural cells from cell death. J. Exp. Med. 2004;200:1257–1266. doi: 10.1084/jem.20040921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roesch A., Vultur A., Bogeski I., Wang H., Zimmermann K.M., Speicher D., Körbel C., Laschke M.W., Gimotty P.A., Philipp S.E. Overcoming intrinsic multidrug resistance in melanoma by blocking the mitochondrial respiratory chain of slow-cycling JARID1B(high) cells. Cancer Cell. 2013;23:811–825. doi: 10.1016/j.ccr.2013.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y., Lim S.K., Liang Q., Iyer S.V., Wang H.Y., Wang Z., Xie X., Sun D., Chen Y.J., Tabar V. Gboxin is an oxidative phosphorylation inhibitor that targets glioblastoma. Nature. 2019;567:341–346. doi: 10.1038/s41586-019-0993-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skrtić M., Sriskanthadevan S., Jhas B., Gebbia M., Wang X., Wang Z., Hurren R., Jitkova Y., Gronda M., Maclean N. Inhibition of mitochondrial translation as a therapeutic strategy for human acute myeloid leukemia. Cancer Cell. 2011;20:674–688. doi: 10.1016/j.ccr.2011.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sørensen K.M.J., Meldgaard T., Melchjorsen C.J., Fridriksdottir A.J., Pedersen H., Petersen O.W., Kristensen P. Upregulation of Mrps18a in breast cancer identified by selecting phage antibody libraries on breast tissue sections. BMC Cancer. 2017;17:19. doi: 10.1186/s12885-016-2987-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotgia F., Whitaker-Menezes D., Martinez-Outschoorn U.E., Salem A.F., Tsirigos A., Lamb R., Sneddon S., Hulit J., Howell A., Lisanti M.P. Mitochondria “fuel” breast cancer metabolism: fifteen markers of mitochondrial biogenesis label epithelial cancer cells, but are excluded from adjacent stromal cells. Cell Cycle. 2012;11:4390–4401. doi: 10.4161/cc.22777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stringer B.W., Day B.W., D’Souza R.C.J., Jamieson P.R., Ensbey K.S., Bruce Z.C., Lim Y.C., Goasdoué K., Offenhäuser C., Akgül S. A reference collection of patient-derived cell line and xenograft models of proneural, classical and mesenchymal glioblastoma. Sci. Rep. 2019;9:4902. doi: 10.1038/s41598-019-41277-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stupp R., Mason W.P., van den Bent M.J., Weller M., Fisher B., Taphoorn M.J.B., Belanger K., Brandes A.A., Marosi C., Bogdahn U., European Organisation for Research and Treatment of Cancer Brain Tumor and Radiotherapy Groups. National Cancer Institute of Canada Clinical Trials Group Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005;352:987–996. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- Tan A.S., Baty J.W., Dong L.-F., Bezawork-Geleta A., Endaya B., Goodwin J., Bajzikova M., Kovarova J., Peterka M., Yan B. Mitochondrial genome acquisition restores respiratory function and tumorigenic potential of cancer cells without mitochondrial DNA. Cell Metab. 2015;21:81–94. doi: 10.1016/j.cmet.2014.12.003. [DOI] [PubMed] [Google Scholar]
- Viale A., Pettazzoni P., Lyssiotis C.A., Ying H., Sánchez N., Marchesini M., Carugo A., Green T., Seth S., Giuliani V. Oncogene ablation-resistant pancreatic cancer cells depend on mitochondrial function. Nature. 2014;514:628–632. doi: 10.1038/nature13611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlashi E., Lagadec C., Vergnes L., Matsutani T., Masui K., Poulou M., Popescu R., Della Donna L., Evers P., Dekmezian C. Metabolic state of glioma stem cells and nontumorigenic cells. Proc. Natl. Acad. Sci. USA. 2011;108:16062–16067. doi: 10.1073/pnas.1106704108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlashi E., Lagadec C., Vergnes L., Reue K., Frohnen P., Chan M., Alhiyari Y., Dratver M.B., Pajonk F. Metabolic differences in breast cancer stem cells and differentiated progeny. Breast Cancer Res. Treat. 2014;146:525–534. doi: 10.1007/s10549-014-3051-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warburg O. On the origin of cancer cells. Science. 1956;123:309–314. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- Warburg O., Wind F., Negelein E. The metabolism of tumors in the body. J. Gen. Physiol. 1927;8:519–530. doi: 10.1085/jgp.8.6.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberg S.E., Chandel N.S. Targeting mitochondria metabolism for cancer therapy. Nat. Chem. Biol. 2015;11:9–15. doi: 10.1038/nchembio.1712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheaton W.W., Weinberg S.E., Hamanaka R.B., Soberanes S., Sullivan L.B., Anso E., Glasauer A., Dufour E., Mutlu G.M., Budigner G.S., Chandel N.S. Metformin inhibits mitochondrial complex I of cancer cells to reduce tumorigenesis. eLife. 2014;3:e02242. doi: 10.7554/eLife.02242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Würth R., Pattarozzi A., Gatti M., Bajetto A., Corsaro A., Parodi A., Sirito R., Massollo M., Marini C., Zona G. Metformin selectively affects human glioblastoma tumor-initiating cell viability: A role for metformin-induced inhibition of Akt. Cell Cycle. 2013;12:145–156. doi: 10.4161/cc.23050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L., Ging N.C., Komoda T., Hanada T., Suzuki T., Watanabe K. Antibiotic susceptibility of mammalian mitochondrial translation. FEBS Lett. 2005;579:6423–6427. doi: 10.1016/j.febslet.2005.09.103. [DOI] [PubMed] [Google Scholar]
- Zu X.L., Guppy M. Cancer metabolism: facts, fantasy, and fiction. Biochem. Biophys. Res. Commun. 2004;313:459–465. doi: 10.1016/j.bbrc.2003.11.136. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
3D cryo-EM density maps and the coordinates of the atomic model have been deposited in the Worldwide Protein Data Bank under the accession numbers EMD-4434 and 6I9R, respectively.