

Abstract
Objectives:
Obesity, a risk factor for pancreatic adenocarcinoma (PDAC), is often accompanied by a systemic increase in lipopolysaccharide (LPS; metabolic endotoxemia), which is thought to mediate obesity-associated inflammation. However, the direct effects of LPS on PDAC cells are poorly understood.
Methods:
The expression of toll like receptor-4 (TLR-4), the receptor for LPS, was confirmed in PDAC cell lines. AsPC-1 and PANC-1 cells were exposed to LPS and differential gene expression was determined by RNA-seq. Activation of the phosphoinositide 3 kinase (PI3K)/protein kinase B (Akt)/ mammalian target of rapamycin (mTOR) pathway by LPS in PDAC cells was assessed by Western blotting.
Results:
Expression of TLR-4 was confirmed in all PDAC cell lines. Exposure to LPS led to differential expression of 3083 genes (426 ≥5-fold) in AsPC-1 and 2584 (339 ≥5-fold) in PANC-1. A top canonical pathway affected by LPS in both cell lines was PI3K/Akt/mTOR. Western blotting confirmed activation of this pathway as measured by phosphorylation of the ribosomal protein S6 and Akt.
Conclusions:
Exposure of PDAC cells to LPS led to differential gene expression. A top canonical pathway was PI3K/Akt/mTOR, a known oncogenic driver. Our findings provided evidence that LPS can directly induce differential gene expression in PDAC cells.
Keywords: pancreatic cancer, obesity, lipopolysaccharide, RNA-seq
Introduction
Pancreatic cancer is a significant cause of cancer related mortality and is expected to become the second leading cause in the United States by 2030.1 Obesity is a known risk factor for PDAC.2 In the United States, approximately 40% of all adults are considered obese with rates increasing for both children and adults.3 The National Institutes of Health recently reported that 16.9% of PDAC cases in the United States are attributed to obesity, making it the number one modifiable risk factor for PDAC development, ahead of cigarette smoking.4 Not only has obesity been associated with PDAC development, but it has also been linked to the development of chemo-resistance.2 Additionally, those who are overweight or obese at the time of their diagnosis tend to have an overall worse prognosis.2
There are several mechanisms by which obesity can accelerate PDAC development, including chronic inflammation, hyperinsulinemia and changes in adipokine production amongst others.2 In addition, obesity is associated with intestinal dysbiosis and increased gut permeability,5 which lead to enhanced translocation of lipopolysaccharide (LPS), a component of the outer wall of intestinal gram-negative bacteria, across the intestinal barrier and an elevation of LPS in the blood (metabolic endotoxemia). Metabolic endotoxemia is thought to at least partially contribute to the insulin resistance and other metabolic disturbances seen in obesity.5
Lipopolysaccharide binds to TLR-4 on cell surfaces, leading to an inflammatory cascade.6 Mechanistically, the binding of LPS to TLR-4 in complex with the myeloid differentiation 2 protein is aided by the cluster of differentiation 14 glycoprotein,7 leading to receptor dimerization and recruitment of downstream molecules such as myeloid differentiation protein 88 and myeloid differentiation protein 88 adapter-like protein.8 This initiates an inflammatory cascade that includes factors such as the interleukin-1 receptor associated kinase, the tumor necrosis factor receptor-associated factor, transforming growth factor B- associated kinase 1, the c-Jun N-terminal kinase and I kappa B kinase, ultimately resulting in an inflammatory signature, which contributes to the development of metabolic diseases such as insulin resistance.8
Although the importance of LPS in metabolic diseases has been studied extensively, there has been very limited data on the direct effects of LPS on PDAC cells. Previous studies have shown that LPS can accelerate PDAC tumor progression and invasion,9–11 decrease tumor suppressor activity9 and increase migratory ability10 and tissue invasion through the Nuclear Factor kappa-light-chain-enhancer of activated B-cells (NF-kB) pathway.10,11 The aim of this present study was to investigate direct global effects of LPS on PDAC cells.
MATERIALS AND METHODS
Cell Lines and Reagents
The human pancreatic cancer cell lines PANC-1, AsPC-1, Capan-2, HPAF-II, BxPC-3, and the colorectal cancer cell line SW-480 were purchased from the American Type Culture Collection (Manassas, Va). Lipopolysaccharide, derived from Escherichia coli (E. Coli) 026:B6, was obtained from Thermo Fisher (West Hills, Calif).
Cell Culture
PANC-1 cells were cultured in Dulbecco’s Modified Eagle Media. AsPC-1, Capan-2, and BxPC-3 were cultured in Roswell Park Memorial Institute medium. HPAF-II was cultured in Eagle’s Minimum Essential Medium. All media were obtained from Gibco (Gaithersburg, Md). Each medium was supplemented with 10% FBS and 1% penicillin/streptomycin combination, both purchased from Thermo Fisher. Cells were grown in 10 cm culture dishes in an incubator maintained at 37°C with either 5% or 10% carbon dioxide.
Quantitative Reverse Transcription Polymerase Chain Reaction
Messenger ribonucleic acid (RNA) levels of TLR-4 were determined using quantitative reverse transcription polymerase chain reaction (qRT-PCR). Extraction of RNA was performed using a Total RNA Purification Plus Kit from Biomiga (San Diego, Calif). We performed reverse transcription using 0.75 μg total input RNA and heated sample mixtures to 25°C for 5 min, 46°C for 20 min, 95°C for 2 min, and subsequently held it at 4°C. All reagents were obtained from Bio-Rad (Hercules, Calif). The following primers were used: human TLR4 (forward: GGTCAGACGGTGATAGCGAG and reverse: TAGGAACCACCTCCACGCA) and as an internal control human 18S ribosomal RNA, both from Life Technologies (Carlsbad, Calif).
RNA Sequencing
Cells were seeded in a 50 mm cell culture plate in complete media. The following day, media was replaced with serum free media overnight. Cells were then exposed to 1 μg/mL of LPS, a concentration consistent with similar experiments in the literature,10 and treated with RNeasy Lysis Buffer from Qiagen (Germantown, Md) after 6 hours of incubation. The integrity of total RNA was examined by the Agilent 4200 TapeStation System (Agilent, Santa Clara, Calif). Libraries for RNA-Seq were constructed with KAPA stranded mRNA library construction kit (Roche, Indianapolis, Ind) to generate strand-specific RNA-seq libraries. The workflow consisted of poly(A) RNA selection, RNA fragmentation and double-stranded cDNA generation using a mixture of random and oligo(dT) priming, followed by end repair to generate blunt ends, adaptor ligation, strand selection and PCR amplification to produce the final libraries. Amplified libraries were quantified by Qubit dsDNA HS (High Sensitivity) Assay Kit (Thermo Fisher) and quality-checked by the Agilent 4200 TapeStation System. Different index adaptors were used for multiplexing samples in one sequencing lane. Sequencing was performed with Illumina HiSeq 3000 sequencer (Illumina, San Diego, Calif) to produce 50 base-pair single-end reads (1 × 50 bp). The STAR software (v2.7, Alexander Dobin) was used for alignment, the Partek Flow software (v10.0, Partek, St. Louis, Mo) for normalization and differential gene expression and the Ingenuity Pathway Analysis software (winter 2020 release, Qiagen) for pathway analysis.
Western Blot Analysis
PANC-1 cells were seeded in a 6-well plate and allowed to grow overnight in complete Dulbecco’s Modified Eagle Media. The following day, the medium was changed to serum free medium and incubated overnight. Cells were then again exposed to 1 μg/mL of LPS and protein was extracted at 15 min, 1 hour, and 2 hours after LPS treatment using an ice-cold lysis buffer mixed with a protease inhibitor cocktail, both from Thermo Fisher. After determining protein concentrations using the Bicinchoninic Acid kit from Thermo Fisher, Sodium Dodecyl Sulfate Polyacrylamide Gel Electrophoresis was performed. Proteins were then transferred to a nitrocellulose membrane and probed overnight at 4°C with primary antibodies against total S6, phospho-S6 (ser235/236), total Akt and phospho-Akt (ser473), all purchased from Cell Signaling Technologies (Danvers, Mass). The following day, an anti-rabbit secondary antibody from Cell Signaling Technologies was applied and the membrane was incubated at room temperature for one hour. Immunoreactivity was detected with a chemiluminescence kit from Thermo Fisher. Blots were quantified using the Image J software (v1.53g, National Institutes of Health, Bethesda, Md) with glyceraldehyde 3-phosphate dehydrogenase as a loading control.
RESULTS
First, we confirmed the expression of the TLR-4 receptor in several human PDAC cell lines. Compared to SW-480, a colonic adenocarcinoma cell line with documented response to LPS,12 TLR-4 mRNA expression levels were detected in all PDAC cell lines (Fig. 1). The highest mRNA expressions, as compared to SW-480, were seen in AsPC-1 (3.3-fold) and BxPC-3 (2.9-fold). PANC-1 (0.1-fold) and HPAF-II (0.04-fold) showed lower transcript levels compared to SW-480. To investigate global direct effects of LPS on PDAC cell lines, we performed RNA-seq analysis using AsPC-1 and PANC-1 as cell lines with high and low levels of TLR-4 mRNA expression, respectively.
FIGURE 1.
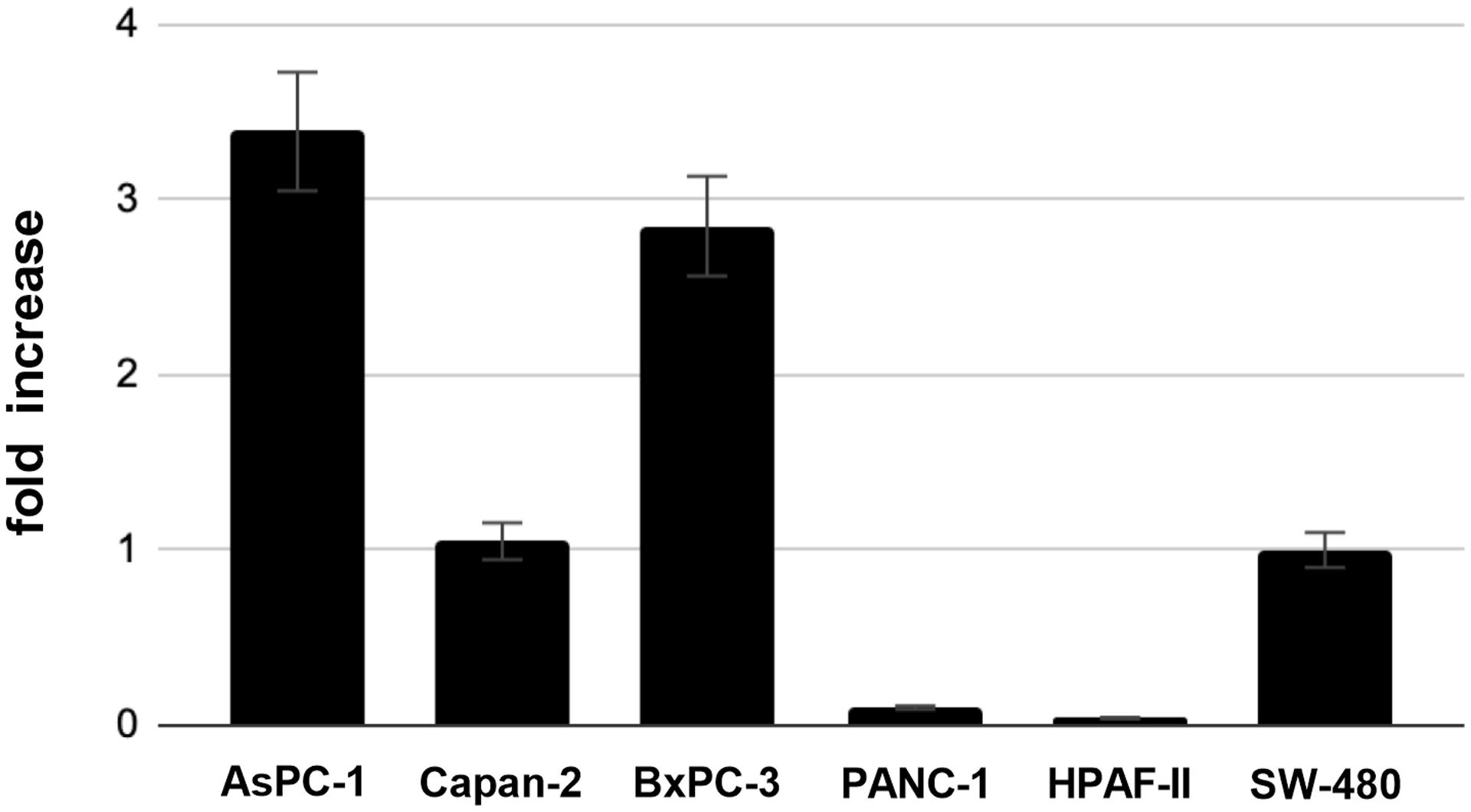
Messenger-RNA expression of TLR-4 in several PDAC cell lines as determined by RT-qPCR. SW-480 cells served as positive control.
Exposure of AsPC-1 to LPS for 6 hours resulted in 3083 differentially expressed genes, 341 of which had more than a 5-fold change in expression (225 with increased expression and 116 with decreased expression). The top five upregulated genes were 60S ribosomal protein L22 (RPL22, 14,210-fold), glutamine-fructose-6-phosphate transaminase 1 (GFPT1, 504-fold), Zinc Finger Protein 692 (ZNF692, 192-fold), Sorting Nexin 6 (SNX6, 167-fold), and Chromodomain-helicase-DNA-binding protein 3 (CHD3, 52-fold). The top five down regulated genes were: WW Domain Containing Adaptor With Coiled-Coil (WAC, 644-fold), Paxillin (PXN, 71-fold), Thyroid Adenoma Associated protein (THADA, 49-fold), Zinc Finger AN1-Type Containing 5 (ZFAND5, 35-fold) and Integrator Complex Subunit 6 (INTS6, 32-fold) (Table 1). Pathways analysis revealed that the top canonical pathways affected by LPS exposure in AsPC-1 cells were: protein ubiquitination, Huntington’s Disease signaling, regulation of eukaryotic initiation factor 4 and p70S6 kinase signaling, mTOR signaling and eukaryotic initiation factor 2 signaling (Table 2). PANC-1 cells exposed to LPS for 6 hours resulted in 2584 differentially expressed genes, 271 of which had a greater than 5-fold difference in expression (148 with increased expression and 123 with decreased expression). The top five upregulated genes were: Tumor Suppressor 2 (TUSC2, 653-fold), ADAM metallopeptidase With Thrombospondin Type I Motif 13 (ADAMTS13, 348-fold), Cell Division Cycle 25 Homolog C (CDC25C, 189-fold), Phospholipase C Delta 1 (PLCD1, 124-fold) and Prune Exopolyphosphatase 1 (PRUNE, 70-fold). The top five down regulated genes were: Baculoviral IAP Repeat Containing 2 (BIRC2, 124-fold), Small Nuclear Ribonucleoprotein Polypeptide A (SNRPA, 84-fold), Spermine Synthase (SMS, 78-fold), Gem Nuclear Organelle Associated Protein 4 (GEMIN4, 64-fold) and Eukaryotic Translation Initiation Factor 4E (EIF4E, 52-fold) (Table 1). Pathway analysis revealed that the top canonical pathways affected by LPS exposure in PANC-1 cells were: Phosphatase And Tensin Homolog signaling, PI3K/Akt signaling, hereditary breast cancer signaling, role of Breast Cancer Type 1 Susceptibility Protein in DNA damage response and molecular mechanisms of cancer (Table 2). It was noteworthy that both cell lines had the PI3K/Akt/mTOR signaling cascade as a top canonical pathway altered after LPS exposure. Additionally, this pathway is known to be a critical pro-oncogenic pathway in PDAC. As a result, we decided to confirm LPS induced activation of the PI3K/Akt/mTOR pathway using western blotting. Exposure of PANC-1 cells to LPS time-dependently increased phosphorylation of Akt and S6, indicating activation of this pathway in PANC-1 cells by LPS. Two hours after exposure to LPS, the levels of phospho-Akt (ser473) were increased by 6.4-fold, while the levels of phospho-S6 (ser235/236) were increased by 5.3-fold (Fig. 2).
TABLE 1.
Top Canonical Pathways With Differential Gene Expression on RNA Sequencing Analysis in AsPC-1 and PANC-1 Cell Lines Treated With LPS, Along With the Percent Overlap of Differentially Expressed Molecules in our Dataset With Molecules in Each Pathway
| Pathway | Percent Overlap, % |
|---|---|
| AsPC-1 | |
| Protein ubiquination | 19.4 |
| Huntington’s disease signaling | 20.6 |
| Regulation of eIF4 and p70S6K signaling | 25.9 |
| mTOR signaling | 27.0 |
| EIF2 signaling | 30.7 |
| PANC-1 | |
| PTEN signaling | 24.6 |
| PI3K/AKT signaling | 24.2 |
| Hereditary breast cancer signaling | 24.3 |
| Role of BRCA1 in DNA damage response | 30.0 |
| Molecular mechanisms of cancer | 18.1 |
TABLE 2.
Genes Exhibiting a Five-Fold or Greater Change in Expression on RNA Sequencing Analysis in AsPC-1 and PANC-1 Cell Lines Treated With LPS
| Gene | Fold Change |
|---|---|
| AsPC-1 | |
| Down-regulated | |
| WAC | −644.70 |
| PXN | −71.73 |
| THADA | −49.05 |
| ZFAND5 | −35.58 |
| INTS6 | −32.14 |
| Up-regulated | |
| RPL22 | 14,210.85 |
| GFPT1 | 504.30 |
| ZNF692 | 192.54 |
| SNX6 | 167.20 |
| HECTD1 | 101.92 |
| PANC-1 | |
| Down-regulated | |
| BIRC2 (ClAP1) | −134.7 |
| SNRPA | −84.32 |
| SMS | −78.55 |
| GEMIN4 | −64.53 |
| EIF4E | −52.79 |
| Up-regulated | |
| TUSC2 | 653.06 |
| ADAMTS13 | 348.87 |
| CDC25C | 189.14 |
| PLCD1 | 124.62 |
| PRUNE | 70.86 |
FIGURE 2.
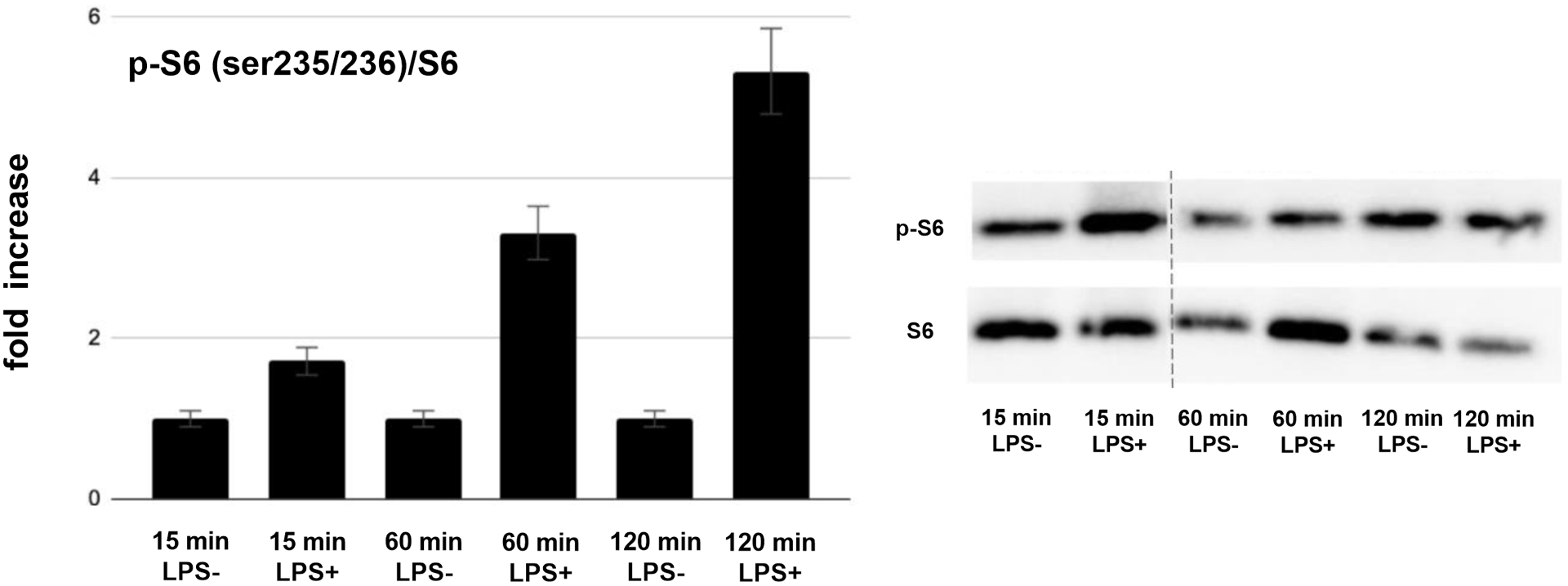
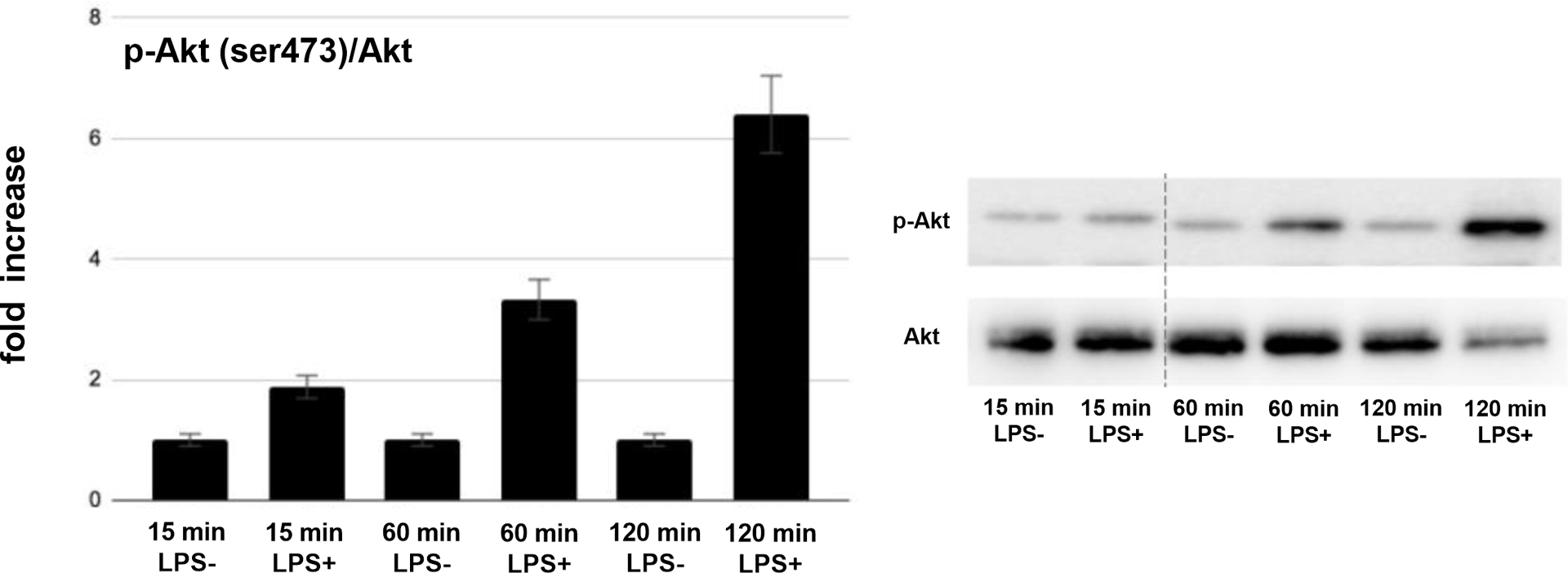
A, Quantification of phospho-S6 (p-S6) versus total S6 (S6) protein expression in PANC-1 cells treated with LPS for various time periods (fold increase over vehicle control; left panel) and representative western blot image (right panel). B, Quantification of phospho-Akt (p-Akt) versus total Akt (Akt) protein expression in PANC-1 cells treated with LPS for various time periods (fold increase over vehicle control; left panel) and representative western blot image (right panel).
DISCUSSION
There are very few studies on direct effects of LPS on PDAC cells.9–11 Treatment with LPS has been shown to increase cell migration, decrease the expression of the tumor suppressors phosphatase and tensin homolog and Mitogen-Activated Protein Kinase Kinase 4,9 and increase the invasive ability of PDAC via a NF-kB related pathway.10 One study determined that LPS treatment can lead to increased expression of vascular endothelial growth factor, a marker of angiogenesis, and similar to our findings, phosphorylation of Akt; both effects which were abolished after TLR-4 silencing.13 A related study treated PDAC cells with palmitic acid, a component of saturated animal fat that can also act via the TLR-4 receptor.11 They found that the downstream effects of TLR-4 stimulation can lead to the generation of reactive oxidative species, activation of NF-kB, and stimulation of matrix metalloproteinase 9, a marker of increased cell invasion and migration.11 Lipopolysaccharide has also been shown to contribute to resistance to drugs, such as gemcitabine.14
Our study provides a first global transcriptome profile of PDAC cells treated with LPS, in order to help tailor and focus further studies. Our results indicate that the exposure of PDAC cell lines AsPC-1 and PANC-1 to LPS results in differential gene expression. Several of the differentially expressed genes have had a documented association with PDAC and/or other gastrointestinal cancers. In AsPC-1, these genes included RPL22, GFPT1(GFAT1), ZNF692, and SNX6 amongst those that were up-regulated and WAC, PXN, ZFAND5, and INTS6 amongst those that were down-regulated. In PANC-1, these genes included TUSC2, ADAMTS13, PLCD1, and PRUNE amongst those that were up-regulated and Birc-2, SNRPA, SMS, GEMIN4, and eIF4E amongst those that were down-regulated.
The gene that had the greatest increased differential expression in AsPC-1 was RPL22 (+14,210 fold change), which acts as a tumor suppressor interacting with the p53 pathway. It has been shown to be highly mutated in human cancers, such as colorectal cancer, becoming a driver of cell proliferation and growth.15 GFPT1 (GFAT1, +504 fold change) is a rate limiting enzyme in the glucose metabolism hexosamine biosynthesis pathway and its high expression is positively correlated with lymph node metastasis, tumor stage and a shorter overall survival in PDAC.16 ZNF692 (+192 fold change) an enzyme also involved in glucose metabolism, has been shown to be upregulated in colon adenocarcinoma and also contributes to metastasis and the development of a higher tumor stage of several other cancers.17 Also contributing to increased PDAC metastatic potential is SNX6 (+167 fold change), which is thought to disrupt the epithelial-mesenchymal transition.18 Amongst those that were down-regulated in AsPC-1, WAC (-644 fold change), PXN (-71 fold change) and INTS6 (-32 fold change) all act as tumor suppressors whose inactivation have been linked to PDAC and several other cancers.19–21 The zinc finger ZFAND5 (-35 fold change) has been previously associated with increased pathogenesis of hepatocellular cancer.22
Amongst the highest upregulated differentially expressed genes in PANC-1 that have been described in gastrointestinal oncology literature was ADAMTS13 (+348 fold change), a von Willebrand Factor cleavage molecule whose expression may be useful as a biomarker for early detection of hepatocellular carcinoma.23 The phospholipase PLCD1 (+124 fold change) has previously been found to be significantly overexpressed in PDAC tissues, leading to questions about its possible use as a prognostic marker.24 The PRUNE gene (+70 fold change) has been described as a promoter of invasion in many cancer types, including PDAC, colorectal, breast and anaplastic thyroid.25 Amongst genes down-regulated in PANC-1, Birc-2 (CAP1, −134 fold change) has been identified as a possible anti-apoptotic oncogene in PDAC.26 SNRPA (-84 fold change) has been linked to progression and poor prognosis of gastric cancer.27 SMS (-78 fold change) has been proposed as a possible hormonal therapy for PDAC, breast cancer and colon adenocarcinoma.28 GEMIN4 (-64 fold change), a regulator in the miRNA biogenesis process, has also been linked with an increased risk of cancer development.29 Finally, eIF4E (-52 fold change) is thought to interact with other signaling molecules, PHGDH included, and promote PDAC development.30
Interestingly, pathway analysis showed that one of the top canonical pathways altered upon exposure to LPS in both PDAC cell lines was the PI3K/Akt/mTOR signaling cascade. The PI3K/AKT/mTOR pathway is well-described in the literature and is thought to be a central regulator of cell metabolism, growth, proliferation and survival whose deregulation has been linked to PDAC tumor formation.31,32 Components of this pathway are activated by Kras, whose constitutively active mutation is present in almost all cases of PDAC.33 WAC and GFPT1, two of the genes whose expression was most upregulated are also thought to be upstream regulators of mTOR.34,35 WAC facilitates the joining of upstream complexes that promote mTOR activation and GFPT1 is a fructose and metabolism modulator enzyme, also upstream from mTOR.34,35 Activation of the PI3K/Akt/mTOR pathway by LPS, as suggested by the pathway analysis, was subsequently confirmed by western blot analysis of signaling molecules downstream in this pathway. Both phospho-S6 (ser235/236) and phosho-Akt (ser473) exhibited increased expression at 15 minutes and 1 and 2 hours, indicating an activation of this pathway after LPS treatment.
What makes our study novel is that, to our knowledge, no other study has performed RNA sequencing analysis on LPS treated PDAC cells. Our study thus provides a unique global transcriptome profile of differentially expressed genes and pathways, as well as the further investigation of LPS effects on the mTOR/PI3K/Akt pathway. Many of the genes with differential expression as a result of LPS, such as WAC, PXN, INTS6, GFPT1, ZNF692, SNX6, PLCD1, and PRUNE exhibited a change consistent with increased oncogenicity. Other genes, such as ZFAND5, RPL22, Birc-2, SNRPA, GEMIN4, EIF4E, TUSC2, and ADAMTS13, exhibited a differential expression consistent with decreased oncogenicity. These findings provide further evidence of direct transcriptional effects of LPS on PDAC cells and provide a global transcriptome profile of genes that should be further explored in the context of obesity-related tissue inflammation and PDAC.
Financial Support and Conflicts of Interest:
National Institutes of Health T32 Training Grant (T32DK007180 to R.L.M.), National Cancer Institute (P01CA236585 to G.E.), H&H Lee foundation (to R.L.M.), Hirshberg Foundation for Pancreatic Cancer Research.
Footnotes
Disclosure: The authors declare no conflict of interest.
REFERENCES
- 1.Rahib L, Smith BD, Aizenberg R, et al. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014;74:2913–2921. [DOI] [PubMed] [Google Scholar]
- 2.Chang HH, Eibl G. Obesity-induced adipose tissue inflammation as a strong promotional factor for pancreatic ductal adenocarcinoma. Cells. 2019;8:673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hales CM, Carroll MD, Fryar CD, et al. Prevalence of obesity among adults and youth: United States, 2015–2016. NCHS Data Brief. 2017;288:1–8. [PubMed] [Google Scholar]
- 4.Islami F, Goding Sauer A, Miller KD, et al. Proportion and number of cancer cases and deaths attributable to potentially modifiable risk factors in the United States. CA Cancer J Clin. 2018;68:31–54. [DOI] [PubMed] [Google Scholar]
- 5.Boutagy NE, McMillan RP, Frisard MI, et al. Metabolic endotoxemia with obesity: Is it real and is it relevant? Biochimie. 2016;124:11–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Plociennikowska A, Hromada-Judycka A, Borzecka K, et al. Co-operation of TLR4 and raft proteins in lps-induced pro-inflammatory signaling. Cell Mol Life Sci. 2015;72:557–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kim SJ, Kim HM. Dynamic lipopolysaccharide transfer cascade to TLR4/MD2 complex via LBP and CD14. BMB Rep. 2017;50:55–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Saad MJA, Santos A, Prada PO. Linking gut microbiota and inflammation to obesity and insulin resistance. Physiology (Bethesda). 2016;31:283–293. [DOI] [PubMed] [Google Scholar]
- 9.Liu J, Xu D, Wang Q, et al. LPS induced miR-181a promotes pancreatic cancer cell migration via targeting PTEN and MAP2K4. Dig Dis Sci. 2014;59:1452–1460. [DOI] [PubMed] [Google Scholar]
- 10.Ikebe M, Kitaura Y, Nakamura M, et al. Lipopolysaccharide (LPS) increases the invasive ability of pancreatic cancer cells through the TLR4/MyD88 signaling pathway. J Surg Oncol. 2009;100:725–731. [DOI] [PubMed] [Google Scholar]
- 11.Binker-Cosen MJ, Richards D, Oliver B, et al. Palmitic acid increases invasiveness of pancreatic cancer cells AsPC-1 through TLR4/ROS/NF-kB/MMP-9 signaling pathway. Biochem Biophys Res Commun. 2017;484:152–158. [DOI] [PubMed] [Google Scholar]
- 12.Lee I, Bae E, Hyun Y, et al. Dextran sulfate sodium and 2,4,6-trinitrobenzene sulfonic acid induce lipid peroxidation by the proliferation of intestinal gram-negative bacteria in mice. J Inflamm (London). 2010;7:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sun Y, Wu C, Ma J, et al. Toll-like receptor 4 promotes angiogenesis in pancreatic cancer via PI3K/AKT signaling. Exp Cell Res. 2016;347:274–282. [DOI] [PubMed] [Google Scholar]
- 14.Ma JX, Sun YL, Yu Y, et al. Triptolide enhances the sensitivity of pancreatic cancer Panc-1 cells to gemcitabine by inhibiting TLR4/NF-kB signaling. Am J Transl Res. 2019;11:3750–3760. [PMC free article] [PubMed] [Google Scholar]
- 15.Rao S, Peri S, Hoffmann J, et al. RPL22L1 induction in colorectal cancer is associated with poor prognosis and 5-FU resistance. PLoS One. 2019;14:e0222392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang C, Peng P, Li L, et al. High expression of GFAT1 predicts poor prognosis in patients with pancreatic cancer. Sci Rep. 2016;6:39044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xing Y, Ren S, Ai L, et al. ZNF692 promotes colon adenocarcinoma cell growth and metastasis by activating the PI3K/AKT pathway. Int J Oncol. 2019;54:1691–1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hu P, Liang Y, Hu Q, et al. SNX6 predicts poor prognosis and contributes to the metastasis of pancreatic cancer cells via activating epithelial-mesenchymal transition. Acta Biochim Biophys Sin (Shanghai). 2018;50:1075–1084. [DOI] [PubMed] [Google Scholar]
- 19.De la Rosa J, Weber J, Friedrich MJ, et al. A single-copy Sleeping Beauty transposon mutagenesis screen identifies new PTEN-cooperating tumor suppressor genes. Nat Genet. 2017;49:730–741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yan J, Jia Y, Chen H, et al. Long non-coding RNA PXN-AS1 suppresses pancreatic cancer progression by acting as a competing endogenous RNA of miR-3064 to upregulate PIP4K2B expression. J Exp Clin Cancer Res. 2019;38:390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lu KY, Zhao H, Qiu C, et al. Integrator complex subunit 6 (INTS6) inhibits hepatocellular carcinoma growth by Wnt pathway and serve as a prognostic marker. BMC Cancer. 2017;17:644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zheng Y, Yu K, Huang C, et al. Integrated bioinformatics analysis reveals role of the LINC01093/miR-96–5p/ZFAND5/NF-kB signaling axis in hepatocellular carcinoma. Exp Ther Med. 2019;18:3853–3860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Takaya H, Namisaki T, Kitade M, et al. VWF/ADAMTS13 ratio as a potential biomarker for early detection of hepatocellular carcinoma. BMC Gastroenterol. 2019;19:167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhou X, Liao X, Wang X, et al. Noteworthy prognostic value of phospholipase C delta genes in early stage pancreatic ductal adenocarcinoma patients after pancreaticoduodenectomy and potential molecular mechanisms. Cancer Med. 2020;9:859–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nambu J, Kobayashi T, Hashiomoto M, et al. h-prune affects anaplastic thyroid cancer invasion and metastasis. Oncol Rep. 2016;35:3445–3452. [DOI] [PubMed] [Google Scholar]
- 26.Hoskins JW, Jia J, Flandez M, et al. Transcriptome analysis of pancreatic cancer reveals a tumor suppressor function for HNF1a. Carcinogenesis. 2014;35:2670–2678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dou N, Yang D, Yu S, et al. SNRPA enhances tumour cell growth in gastric cancer through modulating NGF expression. Cell Prolif. 2018;51:e12484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Upp JR jr, Olson D, Poston GJ, et al. Inhibition of growth of two human pancreatic adenocarcinomas in vivo by somatostatin analog SMS 201–995. Am J Surg. 1988;155:29–35. [DOI] [PubMed] [Google Scholar]
- 29.Zhu W, Zhao J, He J, et al. Genetic variants in the microRNA biosynthetic pathway gemin3 and gemin4 are associated with a risk of cancer: A meta-analysis. Peer J. 2016;4:e1724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ma X, Li B, Liu J, et al. Phosphoglycerate dehydrogenase promotes pancreatic cancer development by interacting with eIF4A1 and eIF4E. J Exp Clin Cancer Res. 2019;38:66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hassan Z, Schneeweis C, Wirth M, et al. MTOR inhibitor-based combination therapies for pancreatic cancer. Br J Cancer. 2018;118:366–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.LaPlante M, Sabatini DM. mTOR signaling at a glance. J Cell Sci. 2009;122:3589–3594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mann KM, Ying H, Juan J, et al. KRAS-related proteins in pancreatic cancer. Pharmacol Ther. 2016;168:29–42. [DOI] [PubMed] [Google Scholar]
- 34.David-Morrison G, Xu Z, Rui YN, et al. WAC regulates mTOR activity by acting as an adaptor for the TTT and pontin/reptin complexes. Dev Cell. 2016;36:139–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu B, Huang ZB, Chen X, et al. Mammalian target of rapamycin 2 (MTOR2) and C-MYC modulate glucosamine-6-phosphate synthesis in glioblastoma (GBM) cells through glutamine: fructose-6-phosphate aminotransferase 1 (GFAT1). Cell Mol Neurobiol. 2019;39:415–434. [DOI] [PMC free article] [PubMed] [Google Scholar]