

Abstract
Background
Genetic systems have been developed for Chlamydia but the extremely low transformation frequency remains a significant bottleneck. Our goal is to develop a self-replicating transposon delivery vector for C. trachomatis which can be expanded prior to transposase induction.
Methods
We made E. coli/ C. trachomatis shuttle vectors bearing the Himar1 C9 transposase under control of the tet promoter and a novel rearrangement of the Himar1 transposon with the β-lactamase gene. Activity of the transposase was monitored by immunoblot and by DNA sequencing.
Results
We constructed pSW2-mCh-C9, a C. trachomatis plasmid designed to act as a self-replicating vector carrying both the Himar1 C9 transposase under tet promoter control and its transposon. However, we were unable to recover this plasmid in C. trachomatis following multiple attempts at transformation.
Therefore, we assembled two new deletion plasmids pSW2-mCh-C9-ΔTpon carrying only the Himar1 C9 transposase (under tet promoter control) and a sister vector (same sequence backbone) pSW2-mCh-C9-ΔTpase carrying its cognate transposon. We demonstrated that the biological components that make up both pSW2-mCh-C9-ΔTpon and pSW2-mCh-C9-ΔTpase are active in E. coli. Both these plasmids could be independently recovered in C. trachomatis.
We attempted to perform lateral gene transfer by transformation and mixed infection with C. trachomatis strains bearing pSW2-mCh-C9-ΔTpon and pSW2-RSGFP-Tpon (a green fluorescent version of pSW2-mCh-C9-ΔTpase). Despite success in achieving mixed infections, it was not possible to recover progeny bearing both versions of these plasmids.
Conclusions
We have designed a self-replicating plasmid vector pSW2-mCh-C9 for C. trachomatis carrying the Himar1 C9 transposase under tet promoter control. Whilst this can be transformed into E. coli it cannot be recovered in C. trachomatis. Based on selected deletions and phenotypic analyses we conclude that low level expression from the tet inducible promoter is responsible for premature transposition and hence plasmid loss early on in the transformation process.
Keywords: transposon, Chlamydia, transformation, Himar1
Introduction
Chlamydia trachomatis is a major human pathogen, causing trachoma, the most prevalent infectious blinding disease 1, and is also the main bacterial agent of sexually transmitted infections worldwide 2. C. trachomatis is an obligate intracellular pathogen with a unique developmental cycle which involves two distinct forms 3. The extracellular infectious form is known as an elementary body (EB). The EB attaches to a susceptible eukaryotic host cell and is then taken up within a membrane structure known as an inclusion 4. The early chlamydial inclusion evades phagolysosomal fusion as the EBs secrete and synthesise proteins that modify the inclusion membrane subverting the normal cellular vesicle pathway. Once an EB has been taken up within a nascent inclusion it will, over several hours (the process takes 8–12 hours depending on strain and host cell and culture conditions) differentiate into a reticulate body (RB). RBs are the non-infectious form of the microorganism and these then follow a normal bacterial growth curve dividing by binary fission for 8 to 10 generations 5. During the exponential growth phase of C. trachomatis L2 the RBs multiply at a rate of one bacterial division/chromosomal replication every 4 to 6 hours and during the process most asynchronously differentiate back to become EBs. Once the inclusion reaches a critical late phase of development, the host cell lyses. At this point of maturation the infected cell contains around 500 – 1000 EBs which are released to infect further cells and commence a new developmental cycle 5.
Progress in studying the molecular biology and genetics of Chlamydia has been severely hampered because it is not possible to unravel the stages of the intracellular developmental cycle from its close co-ordination with the host. There remains no routine cell-free culture system, nor systems to allow arrest/recommencement of the developmental cycle, although cell-free extracts have been described that support RB metabolism, but not replication 6. Thus the ability to analyse and purify components of chlamydial development lags far behind what is possible with the free-living bacteria that can be cultivated on defined media. Ten years ago a robust plasmid-based transformation system was developed in our laboratory by inducing bacterial competence with CaCl 2 and using the endogenous chlamydial plasmid as a part of an E. coli shuttle/cloning vector 7. Since then a great deal of progress has been made in studying chlamydial genetics 8. It is now possible to make targeted insertions and gene deletions in the chlamydial chromosome. However, a very signficant difficulty remains - the extremely low transformation frequency of Chlamydia. This makes chlamydial genetics both time consuming and technically demanding, especially so for mutagenesis, since overall gene essentiality is unknown and thus in the absence of any biological data target genes chosen for study could be impossible to ‘knock out’. This problem is further compounded as it is not possible to provide conditionally-controlled complementary copies of genes on a separate vector whilst making knock outs of ‘essential’ genes since there is only type one delivery vector (they are all based on the endogenous plasmid) and different variants of the same chlamydial plasmid are not tolerated in the same host, presumably due to incompatibility issues 7, 9.
The most efficient and straightforward way to construct knock-out mutants is by transposon mutagenesis 10. Transposon mutagenesis is a powerful technique that allows random insertion of DNA into the bacterial chromosome 11. Our ultimate aim is to construct a saturation gene knock out library which would be an enormously useful resource for the Chlamydia research community. This will enable the accurate identification of all genes essential to chlamydial growth/infectivity – namely, those genes absent from the library, as mutants with insertions in essential genes will not survive being passaged.
In other bacterial species library generation has been routinely achieved by combining purified transposase with a vector carrying the transposon and then transforming the target host with this mixture 12, 13. Transposition occurs, and the transposase would then be diluted away. Given the very low frequency of transformation this approach is not feasible in Chlamydia. As an alternative, transposon mutagenesis has recently been achieved albeit with a non replicating vector 14, 15. This was done by transforming with an E. coli based vector (incapable of replication in Chlamydia) carrying the Himar1 C9 transposase under control of a chlamydial promoter and a modified transposon bearing the RSGFP-cat fusion. Whilst transposon mutants were obtained, this was only possible at very low frequency and required multiple transformation experiments and painstaking selection of mutants. This approach is not suitable or efficient for generating mutant libraries at scale.
Our aim was to develop a self-replicating chlamydial vector carrying both the transposon and the transposase under tightly-regulated inducible control so the vector can be used to generate many different libraries under diverse conditons thereby allowing analysis of essential gene functions.
Methods
Ethics statement
All genetic manipulations and containment work was approved under the UK Health and Safety Executive Genetically Modified Organisms (contained use) regulations, notification number GM57, 10.1 entitled ‘Genetic transformation of Chlamydiae’.
Study design and setting
Our study was designed to construct a self-replicating chlamydial vector carrying a transposon and transposase under tightly-regulated inducible control. This work was conducted from March 2017 to February 2020 at the Chlamydia Research Laboratory, Southampton General Hospital.
Cell culture and chlamydia infection
We selected the strain C. trachomatis L2P- 16 for these studies, which is a naturally occurring plasmid free strain that we have used as a plasmid recipient in multiple studies. McCoy cells (NCTC, Public Health England, UK) were grown in DMEM supplemented with 10% foetal calf serum. Cells were infected with EBs by centrifugation at 754xg (Beckman Coulter Allegra X-15R centrifuge) for 30 minutes at room temperature in T 25 tissue culture flasks or 96 well trays for infectivity assays. The infected cells were cultured with medium containing cycloheximide (1μg/ml) and gentamicin (20μg/ml) and incubated at 37°C in 5% CO 2.
Stocks of C. trachomatis L2P- were prepared as above and titres were determined as described below. C. trachomatis and McCoy cells were routinely tested for mycoplasma contamination by fluorescence microscopy and using the Lookout Mycoplasma PCR detection kit (Sigma, UK).
Infectivity assay using X-gal staining
The infectivity assay has been described in detail by Skilton et al. 17. A mouse monoclonal primary antibody for genus-specific LPS (Chlamydia Biobank Cat. No. #CT601 RRID: AB2721933) was diluted 1:1000 and incubated with methanol-fixed infected cells in 96 well trays for 16h at 4°C. These cells were washed with PBS and incubated with an anti-mouse antibody conjugated with β-galactosidase (Calbiochem) for 1 hour at 37°C. For staining, 100μl of a staining solution [5.0mM K 3Fe(CN) 6, 5.0mM K 4Fe(CN) 6·3H 2O, 2.0mM MgCl 2·6H 2O, 0.25M 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-Gal)] was added per well and incubated for 4 hours at 37°C. The chromogenic X-Gal substrate generated blue-stained C. trachomatis inclusions, which were then counted and titres were calculated.
Transformation of C. trachomatis
Transformation of C. trachomatis L2P- was performed using the calcium chloride based transformation protocol previously described 7 with the following minor modification - selection of transformants in T 25 flasks at passage 1. The experimental procedures were as follows: 3×10 7 IFU of L2P- EBs were pelleted and resuspended in 150μl CaCl 2 buffer (10 mM Tris pH 7.4 and 50 mM CaCl 2). 6µg plasmid DNA was diluted in 100μl CaCl 2 buffer and the two were mixed in a total volume of 250µl, then incubated for 30 mins at room temperature. This mixture was then split equally across 2×T 25 flasks with 1.5ml/flask of CaCl 2 and incubated at RT for 20 mins. 5mls/flask of DMEM +10% FCS (with cycloheximide and spectinomycin at 50µg ml -1or penicillin at 10 units ml -1) was then added and the flasks centrifuged at 754xg for 30 mins at RT. The flasks were returned to the incubator for 40 – 48hrs. Infected cells were scraped off the flasks and harvested (as described earlier) into 1ml 10% PBS plus 1ml 4SP for freezing at -80°C. This sample was called T 0. The selection of transformants was performed by infection of McCoy cells in 1×T 25 flasks using all of T 0 (passage 1) and selected with either spectinomycin at 50µg ml -1 or 10 units/ml of penicillin. Two days after infection, the sample was harvested from this flasks as ‘T 1’. All T 1 was used to infect McCoy cells in a T 25 flask with selection relevant to the transforming vector. Passaging was continued in T 25 flasks with selection until normal inclusions were recovered. The transformants were routinely recovered in passages 2 or 3.
Microscopy
Cells in culture and cells infected with C. trachomatis were routinely visualized by phase contrast microscopy using a Nikon eclipse TS100 inverted microscope with 10x, 20x and 40x objectives and fluorescence accessories. Images were captured using a Nikon DS-Fi1 camera head. Counting of inclusion forming units (IFU) to quantify chlamydial infectivity was performed at 20x magnification on serial dilutions of C. trachomatis in monolayers of McCoy cells grown in 96 well trays. For this assay inclusions were immunostained as described in the section “infectivity assay using X-gal staining”.
E.coli strains, plasmid purification and transformation
E. coli strain DH5α 18, 19 was used for construction of plasmid vectors using standard CaCl 2 treatment to render the cells competent 20. E. coli C2925 (NEB) is a Dam-/Dcm- strain, and was used to prepare unmethylated DNA for transformation of C. trachomatis. pRPF215 was purchased from Robert Fagan & Neil Fairweather (Addgene plasmid # 106377) 21. Plasmids for C. trachomatis transformation were purified from E. coli C2925 using the Invitrogen midi preparation kit as per the manufacturer’s instructions and assayed for DNA yield by Nanodrop. Plasmids were sequence-verified using the complete plasmid sequencing service using Next-Generation sequencing technology at Massachusetts General Hospital CCIB DNA Core, Cambridge MA, USA. The complete sequence of all the final plasmid constructs are available as FastA files 22.
Immunodetection of C9 transposase by Western blot
Proteins were separated by 12% SDS-PAGE and transferred to a polyvinylidene diflouride (PVDF) Immobilon membrane (EMD Millipore) in Pierce Fast Semi-Dry Buffer (ThermoFisher Scientific) using a Pierce Fast Semi-Dry Blotter. After blocking in 10% skimmed milk/PBS-T solution (Tesco, UK) for 1 hour at room temperature (RT), membranes were incubated with 1:5000 dilution of primary mouse polyclonal antisera to purified C9 transposase 23 in 1% skimmed milk/PBS-T for 1 hour at RT. Membranes were washed three times in PBS-T and incubated with secondary antibody (HRP-labelled goat anti-mouse IgG (Bio-Rad)) diluted 1:2000 in 1% skimmed milk/PBS-T for 1 hour at RT. Membranes were finally washed three times and visualised using Pierce enhanced chemiluminescence (ECL) system Western blotting substrate (ThermoFisher Scientific) and developed onto photographic film.
PCR amplification for cloning experiments
DNA fragments for all cloning experiments were amplified by PCR from prepared plasmid DNA templates. PCR reactions were set up as follows using primer pairs from Table 1; 1x Phusion Flash MasterMix (ThermoScientific), 0.5μM each primer and 1ng plasmid DNA. PCR conditions were 10 seconds at 98°C, followed by 35 cycles of 2 seconds at 98°C, 5 seconds of required annealing temperature of primer pair (see Table 1), and 15 seconds/kb at 72°C, and then a final step of 1 minute at 72°C. PCR was performed using the Veriti 96 Well Thermal Cycler (Applied Biosystems). Products were purified using Promega Wizard SV Gel and PCR Clean-Up System, and concentration determined by NanoDrop 1000 spectrophotometer.
Table 1. Primers used in the cloning strategies of the E.coli/C. trachomatis shuttle plasmids described in Figure 1, Figure 4 and Figure 6.
| Primer
Number |
Primer Name | Sequence 5’-3’ | Restriction
Site |
Annealing
Temp |
Target
Plasmid |
Function |
|---|---|---|---|---|---|---|
|
1
2 |
Erm_del_F
Erm_del_R |
AAAAAA
GCGGCCGCCACACTCTTAAGTTTGCTTCTGTC
AAAAAA CTCGAGGGAGGAAATAATTCTATGAGTCGC |
NotI
XhoI |
52°C
|
pRPF215
|
To delete
erm gene
|
|
3
4 |
Bla_F
Bla_R2 |
AAAAAA
CTCGAGGAGTAAACTTGGTCTGACAGT
AAAAAA GCGGCCGCTGGTTTCTTAGACGTCAGGTGGCA |
XhoI
NotI |
52°C
|
pGFP::SW2
|
To amplify
bla gene
|
|
5
6 |
Divergent_F1
Divergent_R1 |
GGTGGT
GGCCGGCCAACCTCCTAGTATTATTGAGC
GGTGGT ACGCGTGGCCTCGGATCCTATAAG |
FseI
MluI |
58°C
|
pRPF215-Bla
|
To delete
Tpase(wt) gene
|
|
7
8 |
C9_F1
C9_R1 |
GGTGGT
GGCCGGCCATGGAAAAAAAGGAATTTCGTG
GGTGGT ACGCGTTTATTCAACATAGTTCCCTTCAAG |
FseI
MluI |
58°C
|
pMALC9
|
To amplify
Tpase(C9) gene
|
|
9
10 |
Transposon_SalI_F2
Transposon_SalI_R2 |
GGTGGT
GTCGACCACCTCCTTTTTGACTTTA
GGTGGT GTCGACGTCCCCATGCGCTCCATCAA |
SalI
SalI |
54°C
|
pRPF215-Bla-
C9Tpase |
To amplify tetR/transposon-C9
transposase unit |
|
11
12 |
Transposon_Deletion_F1
Transposon_Deletion_R1 |
ATATAT
CGCCGGCGGCATCTTTTTATTTAGGGATTTCTCAC
ATATAT CGCCGGCGGTTACCAGTGTGCTGGAATTC |
MreI
MreI |
56°C
|
pSW2-mCh-C9
|
To delete transposon unit |
|
13
14 |
BreakingC9_F1
BreakingC9_R1 |
ATATAT
CGCCGGCGGGTGTATTACGAGTTAACAGCTGC
ATATAT CGCCGGCGGTTCTCAGACCTCAAAAGGATGC |
MreI
MreI |
56°C | pSW2-mCh-C9 | To delete active site of
Tpase(C9) gene |
|
15
16 |
Short_Transposon_F
Transposon_R2 |
GGTGGT
TCCGGGAGGCTTCTCTCATGAGAAGTC
TTTTTT TCCGGGAGTCCCCATGCGCTCCATCAA |
PfoI
PfoI |
58°C
|
pRPF215-Bla
|
To amplify transposon unit |
Figure 1. Cloning strategy for the construction of the C. trachomatis/E. coli transposon-transposase shuttle vector pSW2-mCh-C9.

A: Replacement of the erythromycin resistance ( erm) gene within the transposon unit with the β-lactamase ( bla) gene. The starting point is plasmid pRFP215 21 which carries erm as a marker within the transposon. Since erythromycin is used to treat chlamydial infections use of this marker is not permissible under our genetic manipulations licence GM157. The erm gene was ‘excised’ by PCR using primers 1 and 2 ( Table 1). The β-lactamase ( bla) gene was amplified from our chlamydial shuttle vector pGFP::SW2 7 using primers 3 and 4 ( Table 1). These fragments were ligated to construct plasmid pRPF215-Bla. B: Replacement of the Clostridia codon-optimised transposase gene with the C9 transposase gene. The original transposase gene which had been codon-optimised for Clostridia located in pRPF215-Bla was removed by PCR with primers 5 and 6 ( Table 1). The C9 transposase gene was amplified by PCR with primers 7 and 8 ( Table 1) using plasmid pMALC9 as template. These PCR fragments were ligated to form plasmid pRPF215-Bla-C9Tpase. C: Construction of the C. trachomatis/E. coli shuttle vector carrying the C9 transposase-transposon. Primers 9 and 10 ( Table 1) were used to amplify the C9 transposase-transposon unit along with its regulatory control region ( TetR) from plasmid pRPF215-Bla-C9Tpase. These primers introduce a SalI restriction site at both ends of the PCR product. This was then cloned into p2TK2-SW2-mCh (kindly provided by Dr Isabelle Derre) via its unique SalI site to generate plasmid pSW2-mCh-C9.
Figure 4. Cloning strategy for the construction of the C. trachomatis/E. coli individual transposon and transposase shuttle vectors pSW2-mCh-C9-ΔTpase and pSW2-mCh-C9-ΔTpon.

A: Deleting the active site of the C9 transposase from pSW2-mCh-C9. The active site of the C9 transposase in pSW2-mCh-C9 was deleted by PCR using primers 13 and 14 ( Table 1), adding MreI sites to both ends. The resulting PCR product was re-ligated to form pSW2-mCh-C9-ΔTpase. This deletion (426 bp) is designed to inactivate the transposase generating a truncated protein of predicted molecular weight 17.4kDa ( Figure 5C). The scissor symbol indicates the location of the deletion. B: Deletion of the transposon unit from pSW2-mCh-C9. The transposon unit was deleted from pSW2-mCh-C9 by PCR using primers 11 and 12 ( Table 1), adding MreI sites to both ends. The resulting PCR product was re-ligated to form pSW2-mCh-C9-ΔTpon. The scissor symbol indicates the location of the deletion.
Figure 6. Cloning strategy for the construction of the C. trachomatis/E. coli transposon shuttle vector pSW2-RSGFP-Tpon.

Primers 15 and 16 ( Table 1) were used to amplify the transposon unit carrying the β-lactamase gene (yellow) from plasmid pRPF215-Bla. These primers introduce a PfoI restriction site at both ends of the PCR product. This was then cloned into pRSGFP-5KO-ΔBla via its unique PfoI site to generate plasmid pSW2-RSGFP-Tpon.
Results and discussion
1. Design, construction and testing of a replication-proficient chlamydial transposon delivery vector
It has previously been demonstrated that a basic E. coli cloning plasmid (incapable of replication in Chlamydia) can be used to deliver an active transposase/transposon system to both C. trachomatis and C. muridarum 14, 15. These studies used the C9 Himar1 (horn fly) transposase 24 coupled with custom-built transposons designed to allow selection of mutants resistant to penicillin 14 or chloramphenicol and expressing the green fluorescent protein 15. However, due to the very low transformation frequency and the absence of an optimisation protocol for transposase induction, selection of the mutants was a time consuming process. Nevertheless, it provided proof-of-principle that the Himar1 system functioned in Chlamydia.
Here, our overall goal was to develop a self-replicating transposon delivery vector that can be transformed into C. trachomatis and then expanded prior to transposase induction at a controlled time point. This would circumvent the severe restriction imposed by low chlamydial transformation frequencies and ultimately make it possible, once we obtained a high titre of transformants, to generate a large pool of mutants.
Our experimental design was to build a C9 Himar1 transposase/transposon unit organised contiguously on the same plasmid in cis. In this construct we engineered the tet promoter to regulate transposase expression so that it could be induced at different times during chlamydial development. The tet promoter system has previously been shown to be active and its regulatory function operational in C. trachomatis, indeed this is the only ‘foreign’ inducible promoter system to date that has been identified as functional in C. trachomatis 25.
The tet promoter was included in the design to allow finely tuned transposase induction/expression using the soluble inducer anhydrotetracycline (ATc). The whole functional unit ( tet promoter/transposase/transposon) was constructed using existing biologically active vector systems derived from well-studied alternate systems and it was cloned into the chlamydial shuttle vector p2TK2-SW2-mCh 26 to give plasmid pSW2-mCh-C9. The cloning strategy and final vector maps are shown in Figure 1 and Figure 2.
Figure 2. Plasmid map of the C. trachomatis/E. coli transposon-transposase shuttle vector pSW2-mCh-C9 showing the essential features for plasmid selection and transposition.

The key coding sequences (CDS) and their direction of transcription are represented by the boxes and arrows in the outer circle. The spectinomycin resistance gene ( aadA) for selection of plasmid transformants is coloured purple. The β-lactamase gene ( bla) is yellow and flanked by the repeat sequences defining the limits of the transposon (pink). The C9 transposase gene (orange) is under inducible control of the tet promoter: the tetracycline repressor gene ( TetR) is coloured magenta. E. coli transformants bearing this plasmid fluoresce red under UV illumination due to the constitutive expression of mCherry (red). The inner circle is a scale bar with 1kb increments, chlamydial plasmid sequences are blue, vector sequences are black and the transposon unit is pink.
Plasmid pSW2-mCh-C9 was transformed into E. coli strain DH5α. The clones were stable and inducible expression of the C9 transposase was confirmed by Western blot ( Figure 3). This plasmid was then transformed into dam-/dcm- E. coli strain C2925 and plasmid DNA prepared for transformation of C. trachomatis L2P-. Despite nine separate transformation attempts, with standard chlamydial shuttle vector p2TK2-SW2-mCh as a control (which worked each time) it was not possible to recover transformants with the transposase/transposon bearing plasmid pSW2-mCh-C9 using spectinomycin selection.
Figure 3. Western blot showing the induction of the C9 transposase in E. coli.
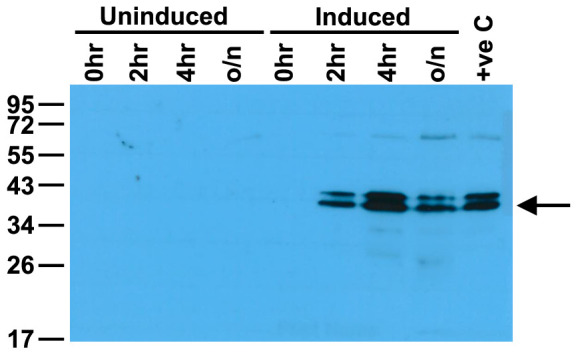
Equal amounts of protein were loaded in each gel lane. E. coli DH5α containing the pSW2-mCh-C9 shuttle vector were grown in liquid culture and induced with ATc and samples taken at the timepoints indicated. Samples were separated by 12% SDS-PAGE gel and the presence of the C9 transposase (arrowed at 40.7kDa) was detected by Western blot with polyclonal antisera to purified C9 transposase.
These results were confounding and unexpected since plasmid pSW2-mCh-C9 was stable in E. coli and the complete absence of any transformants in C. trachomatis strongly suggests the pSW2-mCh-C9 shuttle vector is inherently unstable (once it is transformed into C. trachomatis) .
The chlamydial transformation process requires initial addition of the transforming plasmid DNA to EBs in the presence of calcium chloride and then infection of host cells and the application of selection in subsequent rounds of infection. The routine success of transformation is confirmed by the detection of inclusions by fluorescence microscopy - with mCherry as a marker, transformed EBs will give rise to inclusions that fluoresce red. These are never seen in the first round (T 0) and thus it is not possible to give a transformation frequency to the process. Several passages are required to amplify a transformed chlamydium before inclusions reach a threshold where they are easily detected. The most likely explanation for the inability to select pSW2-mCh-C9 in C. trachomatis is that transposition is occurring from the vector, causing its loss early in the process.
2. Uncoupling of the transposon/transposase unit by functional deletion allows recovery of the individual transposon- and transposase-bearing plasmids in C. trachomatis
Chlamydial transformation frequencies are so low (possibly only 1–5 transformants per microgramme) therefore, at the very first stage in the process only a single chlamydium will be transformed by a single molecule of the shuttle vector. This transformed bacterium develops into an RB and under selection this will grow and form an inclusion. Several passages under selection are required to amplify that transformant and subsequent single inclusion to a point where it is visually detectable. If transposition occurs due to leaky low level induction of the transposase when there is just one plasmid molecule within an RB, this will lead to excision of the Himar1 transposon and loss of the delivery vector. This is because active transposase leaves a double stranded DNA break in the donor plasmid when the transposon is excised so the plasmid cannot replicate, causing its loss early in the process 27, 28. This will be especially so in the cis configuration with both transposase and transposon on the same delivery vector.
Whilst the tet promoter is highly repressed in E. coli, and currently is the best choice for finely controlling genes in Chlamydia, it seems that very low basal expression of the transposase may occur (in Chlamydia) even in the absence of induction (below the level of detection by Western blot) due to very slight leakiness of the tet promoter 25.
To test this notion we made ‘functional’ deletions in the pSW2-mCh-C9 plasmid.
2(a). A deletion mutant crippled for active transposase activity
If the expression of the C9 transposase in the presence of its target functional transposon is the cause of pSW2-mCh-C9 instability/loss in C. trachomatis then a simple inactivating mutation in the transposase should allow recovery of the deleted construct. Therefore, a deletion of 426 bp was made of the Himar1 C9 transposase gene of pSW2-mCh-C9 by PCR. This deletion removed the whole of the catalytic domain of the C9 transposase causing it to be prematurely truncated when expressed. The cloning strategy is shown in Figure 4A and the final vector map for pSW2-mCh-C9-ΔTpase is shown in Figure 5A. In stark contrast to pSW2-mCh-C9, the deleted transposase version was routinely and easily recovered by transformation of C. trachomatis L2P- and selection with spectinomycin ( Figure 5B). Western blots of transformed L2P- carrying pSW2-mCh-C9-ΔTpase induced with ATc showed induction of the truncated transposase ( Figure 5C). These data confirmed that our inability to recover pSW2-mCh-C9 in C. trachomatis is due to the presence of the active C9 transposase in that vector since all other parts of the plasmid pSW2-mCh-C9-ΔTpase remain identical to pSW2-mCh-C9.
Figure 5. Design and functional analysis of the individual transposon and transposase vectors.

A: Plasmid maps showing the individual C. trachomatis/E. coli shuttle vectors pSW2-mCh-C9-ΔTpase and pSW2-mCh-C9-ΔTpon carrying either the transposon or the C9 transposase respectively. The shared sequence coloured in the outer circle shows the key CDS for chlamydial plasmid replication and maintenance (blue), spectinomycin selection (purple) and the mCherry marker (red) for visualisation of inclusions. In pSW2-mCh-C9-ΔTpase the active site of the C9 transposase has been deleted, in pSW2-mCh-C9-ΔTpon the entire transposon unit has been deleted. The inner circle is a scale with 1kb increments, the transposon sequence in this circle is coloured pink. B: Red fluorescent inclusions in McCoy cells infected by C. trachomatis L2P- transformed with either pSW2-mCh-C9-ΔTpase or pSW2-mCh-C9-ΔTpon vectors and grown under spectinomycin selection. Phase contrast and UV light images are from the same field at 48h.p.i. Left-hand panels are from L2P- containing pSW2-mCh-C9-ΔTpase and right-hand panels are from L2P- containing pSW2-mCh-C9-ΔTpon. Scale bar represents 20μm. C: Western Blot showing the induction on the C9 transposase in C. trachomatis L2P- transformed with either pSW2-mCh-C9-ΔTpase or pSW2-mCh-C9-ΔTpon vectors. Equal amounts of protein loaded in each gel lane. McCoy cells were infected with C. trachomatis L2P- transformed with either the pSW2-mCh-C9-ΔTpase or pSW2-mCh-C9-ΔTpon vectors. They were induced with ATc at 24h.p.i. and harvested at 48h.p.i. Non-induced and un-transformed L2P- were used as controls. Samples were separated by 12% SDS-PAGE gel and the presence of the truncated C9 transposase (arrowed at 17.4kDa) or C9 transposase (arrowed at 40.7kDa) was detected by Western blot with polyclonal antisera to purified C9 transposase.
2(b). Inactivation of the transposon
If our hypothesis about the active C9 transposase in the context of the transposase/transposon combination on plasmid pSW2-mCh-C9 is correct then inactivation of the transposon will also allow recovery of stably transformed C. trachomatis able to carry (and express) the complete transposase. Deletion of the whole transposon unit (repeat sequences and bla gene) was performed by PCR (shown in Figure 4B) and the resulting plasmid, pSW2-mCh-C9-ΔTpon ( Figure 5A), was used to transform C. trachomatis L2P-. These transformants were stable and easily expandable ( Figure 5B). Following induction with ATc, Western blotting showed that the complete C9 transposase was both inducible and expressed well in C. trachomatis ( Figure 5C).
3. Lateral gene transfer of the transposon
We have demonstrated that it is possible to recover two independent stable transformants in Chlamydia, one able to carry the transposon and a second able to express the transposase under inducible control (apart from the specific deletions both these plasmids share identical backbones). These observations are consistent with our working hypothesis that it is not possible to recover the combined transposase/transposon plasmid because there is low level leakage/basal expression of the transposase in C. trachomatis from the tet promoter.
In bacterial systems with no, or extremely low-frequency genetic transformation systems, an alternate way of introducing transposon plasmids is by conjugation 29, 30. Unfortunately, there is no conjugative system for Chlamydia. However, it is possible to exchange DNA between Chlamydia either within the same species 31 and also between different species 32 by co-infection, a process known as lateral gene transfer. The mechanism(s) that allow Chlamydia to exchange DNA in the lateral gene transfer process have not been characterised. Co-infection of cells by two strains, each carrying a different chromosomally-located antibiotic-selectable marker allows recovery of hybrid progeny following dual detection; these ‘offspring’ have undergone chromosomal recombination and the progeny carry both selectable markers on the same chromosome. We sought to investigate whether we could introduce the individual transposase and transposon bearing plasmids into the same RBs by mixed infection and dual selection. As a first step, we needed to construct a second vector with different fluorescent markers and different selectable genes - these were needed to monitor for mixed infection and then to select for progeny carrying both plasmids.
3(a). Functional verification of engineered markers
Our experimental design was to use one of our existing chlamydial transformants as a donor for the mixed infection. Since both our constructs (pSW2-mCh-C9-ΔTpon and pSW2-mCh-C9-ΔTpase) were in the same plasmid backbone with the same selectable and fluorescent markers (both display red fluorescence from expression of the mCherry gene), one had to be redesigned. We chose the transformant bearing plasmid pSW2-mCh-C9-ΔTpon as parent 1 ( Figure 5A) since we have already demonstrated that the transposase is inducible, this strain is spectinomycin resistant and carries the mCherry marker. We designed plasmid pSW2-RSGFP-Tpon as parent 2. The cloning strategy is shown in Figure 6. Briefly, it is based on one of our existing cloning vector constructs for C. trachomatis which has ORF 5 deleted (a plasmid CDS not required for maintenance) to facilitate maximal insertion size, and also to enable differentiation of backbones in the event of recombination 33. The original shuttle plasmid carries the β-lactamase gene and separately a green fluorescence protein (RSGFP). RSGFP is contiguous with the chloramphenicol acetyl transferase gene and both expressed from a constitutive neisserial promoter. This allows selection of transformants by the presence of green inclusions that are resistant to chloramphenicol and/or penicillin. To avoid complications from gene duplication (the transposon carries an identical bla gene) the bla gene on the shuttle vector was deleted and plasmid pRSGFP-5KO-ΔBla was used as recipient of the bla gene within the transposon unit. Final vector map is shown in Figure 7A.
Figure 7. Design and functional analysis of the green version of the transposon shuttle vector.

A: Plasmid map of the C. trachomatis/E. coli transposon shuttle vector pSW2-RSGFP-Tpon showing the essential features for plasmid selection and transposition. The key CDS and their direction of transcription are represented by the boxes and arrows in the outer circle. The β-lactamase gene ( bla) is yellow and flanked by the repeat sequences defining the limits of the transposon (pink). E. coli transformants bearing this plasmid fluoresce green under UV illumination due to the constitutive expression of RSGFP (green). The inner circle is a scale bar with 1kb increments, chlamydial plasmid sequences are blue, vector sequences are black and the transposon unit is pink. B: Green fluorescent inclusions in McCoy cells infected by C. trachomatis L2P- transformed with pSW2-RSGFP-Tpon vector and grown under penicillin selection. The top panel shows McCoy cells infected at 48h.p.i. under phase contrast. The lower panel is of the same field under UV light illumination. The scale bar represents 20μm.
E. coli transformed by pSW2-RSGFP-Tpon was chloramphenicol and ampicillin resistant and fluoresced green under UV light compared with E. coli transformed with pSW2-mCh-C9-ΔTpon which was spectinomycin resistant only and fluoresced red.
A deficiency in the verification of the plasmid functions was that whilst we had shown the transposase was inducible (by Western blot with hyper immune sera), we had not shown that it was functional. Before we embarked on a complex series of genetic experiments in C. trachomatis it was necessary to demonstrate transposition in E. coli carrying both pSW2-mCh-C9-ΔTpon and pSW2-RSGFP-Tpon. We made competent cells of E. coli carrying pSW2-mCh-C9-ΔTpon - these cells were divided into two sets at exponential phase, one with ATc (induced) and one without (uninduced). Both sets were then transformed with pSW2-RSGFP-Tpon and tranformants selected on ampicillin plates. Transposon progeny (where the transposon has jumped from pSW2-RSGFP-Tpon to E. coli carrying pSW2-mCh-C9-ΔTpon) will be ampicillin resistant and fluoresce red. The parental pSW2-mCh-C9-ΔTpon strain will not grow under this selection and E. coli carrying pSW2-RSGFP-Tpon yields ‘green’ ampicillin resistant colonies.
Transformation of E. coli carrying pSW2-mCh-C9-ΔTpon with 10ng pSW2-RSGFP-Tpon gave approximately a thousand green colonies from both tranformations (with and without ATc induction) on ampicillin plates as expected, whereas red colonies (indicating transposition) were only recovered from the induced competent cells. A selection of these were used to make plasmid DNA which was re-transformed into E. coli to select for individual colonies that fluoresced red. Plasmid DNA from 10 individual colonies was purified and sequenced ( Figure 8). This showed transposition between pSW2-RSGFP-Tpon and pSW2-mCh-C9-ΔTpon in E. coli and confirmed the transposase was active.
Figure 8. Plasmid maps of the pSW2-mCh-C9-ΔTpon vectors with transposon insertions.

Plasmid maps showing the location of the integrated transposon units that were transposed from pSW2-RSGFP-Tpon into the pSW2-mCh-C9-ΔTpon plasmid giving rise to red, ampicillin resistance in E. coli colonies. Maps drawn from sequence data using SnapGene. The inserted ampicillin gene is coloured yellow and highlighted with an asterisk. Plasmid maps are orientated by the E. coli replication origin (dark blue). Plasmid sequences available as FastA files 22.
With the biological/ functional activities of all the vectors verified, our aim was to investigate whether we could introduce individual transposase and transposon plasmids into the same chlamydial host by lateral gene transfer, following co-infection. Plasmid pSW2-RSGFP-Tpon was transformed into C. trachomatis L2P- and green penicillin/chloramphenicol resistant inclusions were obtained, these were stable and grew well ( Figure 7B). Cells were then co-infected with C. trachomatis L2P- containing pSW2-mCh-C9-ΔTpon and pSW2-RSGFP-Tpon at MOI=1.0 (this was chosen to maximise chance of co-infection yet keeping below the threshold of over infectivity). We were consistently able to obtain mixed infections (as measured by dual fluorescing inclusions) ( Figure 9). Induction with ATc was performed at 24hrs post infection and cultures harvested for analysis at 48hrs.
Figure 9. Mixed infection of McCoy cells infected with C. trachomatis L2P- transformed with pSW2-RSGFP-Tpon vector and C. trachomatis L2P- transformed with pSW2-mCh-C9-ΔTpon vector, grown with no selection.

This figure shows an example of the same microscopic field observed under phase contrast (Panel A), UV light (green filter) (Panel B) and UV light (red filter) (Panel C). Panel D is a merged image of the phase, green (L2P- transformed with pSW2-RSGFP-Tpon) and red (L2P- transformed with pSW2-mCh-C9-ΔTpon). White arrows highlight inclusions showing both green and red fluorescence. Scale bar represents 20μm.
Despite repeated success in achieving mixed inclusions at optimal frequency, we were not able to select any Chlamydia that fluoresced red and were penicillin resistant. Since all the biological components of the vectors were functional, the inability to obtain transposition mutants strongly suggests that stable plasmid transfer/co-existence of pSW2-mCh-C9-ΔTpon and pSW2-RSGFP-Tpon in C. trachomatis is not possible. However, it has been possible to force chlamydial plasmid recombination by antibiotic selection following transformation 9. Therefore, in a final series of experiments we attempted to mimic the experiments performed with E. coli ( section 3 (b)) and tried to transform C. trachomatis L2P- carrying the pSW2-mCh-C9-ΔTpon plasmid with pSW2-RSGFP-Tpon but were unable to select any red penicillin resistant transformants. These observations support our hypothesis that it is not possible for the C9 transposase or transposon to exist on a naturally occurring recombinant plasmid in this expression system.
Conclusions
We have designed and assembled an ‘ideal’ chlamydial vector ‘pSW2-mCh-C9’ for transposition that is stable in E. coli but it has not proven possible to recover this vector in C. trachomatis. By selective deletion of pSW2-mCh-C9 we have demonstrated that the transposase and transposon are functional and are individually stable in C. trachomatis. C. trachomatis has only one type of plasmid and so for efficient delivery of transposons the transposase and transposon have to be on the same vector. However, our data suggests that leaky expression of the transposase is responsible for premature loss of the transposon plasmid, thereby preventing establishment of stable clones in C. trachomatis carrying this engineered vector configuration (pSW2-mCh-C9). Our previous studies have shown that when two ‘competing’ chlamydial plasmids are present in the same host one is either eliminated or recombination occurs. Therefore, we also attempted both co-infection and transformation to bring the two individually stable plasmids (pSW2-mCh-C9-ΔTpon and pSW2-RSGFP-Tpon) together in the same bacterium in the hope both would co-exist under dual selection and allow transposition. Whilst this approach worked in E. coli (by using direct transformation) the process was not a success in C. trachomatis.
The options for overcoming low-level basal expression of the transposase are limited. Guaranteeing binary complete on/off control of promoters remains a biotechnological challenge. Our understanding of gene regulation in Chlamydia lags far behind other bacterial systems and there are no bespoke chlamydial expression systems with high level regulatory features. We have used the highly regulated tet promoter system which is currently the best option for Chlamydia. It may be possible to tighten regulatory controls by increasing repressor concentration or operator binding affinity for the repressor protein. Therefore, the solution for repressing transposase expression lies in adding additional control sequences or trying other highly repressed bacterial expression systems that might work in Chlamydia. We hope that our results here are informative and will guide the field in finding a way to achieve saturation mutagenesis in Chlamydia.
Data availability
Underlying data
Open Science Framework: Progress towards an inducible, replication-proficient transposon delivery vector for Chlamydia trachomatis. https://doi.org/10.17605/OSF.IO/5F2PE 22.
This project contains the following underlying data:
-
-
The complete sequence of all the final plasmid constructs (FastA files)
-
-
Underlying images for Figures 3, 5, 7, 9
Data are available under the terms of the Creative Commons Zero "No rights reserved" data waiver (CC0 1.0 Public domain dedication).
Acknowledgements
We are indebted to Dr. Isabelle Derre (University of Virginia, USA) for the provision of plasmid p2TK2-SW2-mCH ahead of publication.
Funding Statement
This work was funded by Wellcome Trust grant number 202755/Z/16/Z entitled ‘Saturation transposon mutagenesis of Chlamydia trachomatis’.
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
[version 1; peer review: 2 approved]
References
- 1. Hu VH, Harding-Esch EM, Burton MJ, et al. : Epidemiology and control of trachoma: systematic review. Trop Med Int Health. 2010;15(6):673–91. 10.1111/j.1365-3156.2010.02521.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Newman L, Rowley J, Hoorn SV, et al. : Global Estimates of the Prevalence and Incidence of Four Curable Sexually Transmitted Infections in 2012 Based on Systematic Review and Global Reporting. PLoS One. 2015;10(12):e0143304. 10.1371/journal.pone.0143304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ward ME: Chlamydial classification, development and structure. Br Med Bull. 1983;39(2):109–115. 10.1093/oxfordjournals.bmb.a071800 [DOI] [PubMed] [Google Scholar]
- 4. Hybiske KAH: The Chlamydial Inclusion. In Chlamydia Biology: from Genome to Disease. M. Tan Hegemann, J.H. and Sutterlin, C Editor. Caister Academic Press: Norfolk, UK.2020;85–110. 10.21775/9781912530281 [DOI] [Google Scholar]
- 5. Lambden PR, Pickett MA, Clarke IN: The effect of penicillin on Chlamydia trachomatis DNA replication. Microbiology (Reading). 2006;152(Pt 9):2573–2578. 10.1099/mic.0.29032-0 [DOI] [PubMed] [Google Scholar]
- 6. Omsland A, Sager J, Nair V, et al. : Developmental stage-specific metabolic and transcriptional activity of Chlamydia trachomatis in an axenic medium. Proc Natl Acad Sci U S A. 2012;109(48):19781–5. 10.1073/pnas.1212831109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wang Y, Kahane S, Cutcliffe LT, et al. : Development of a transformation system for Chlamydia trachomatis: restoration of glycogen biosynthesis by acquisition of a plasmid shuttle vector. PLoS Pathog. 2011;7(9):e1002258. 10.1371/journal.ppat.1002258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. O'Neill CE, Clarke IN, Fisher DJ: Chlamydia Genetics. In Chlamydia Biology: from Genome to Disease. M. Tan Hegemann, J.H. and Sutterlin, C, Editor. Caister Academic Press: Irvine, California.2020;241–262. 10.21775/9781912530281 [DOI] [Google Scholar]
- 9. Wang Y, Cutcliffe LT, Skilton RJ, et al. : The genetic basis of plasmid tropism between Chlamydia trachomatis and Chlamydia muridarum. Pathog Dis. 2014;72(1):19–23. 10.1111/2049-632X.12175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hayes F: Transposon-based strategies for microbial functional genomics and proteomics. Annu Rev Genet. 2003;37:3–29. 10.1146/annurev.genet.37.110801.142807 [DOI] [PubMed] [Google Scholar]
- 11. Polard P, Chandler M: Bacterial transposases and retroviral integrases. Mol Microbiol. 1995;15(1):13–23. 10.1111/j.1365-2958.1995.tb02217.x [DOI] [PubMed] [Google Scholar]
- 12. Kirby JR: In vivo mutagenesis using EZ-Tn5. Methods Enzymol. 2007;421:17–21. 10.1016/S0076-6879(06)21003-6 [DOI] [PubMed] [Google Scholar]
- 13. Riess T, Anderson B, Fackelmayer A, et al. : Rapid and efficient transposon mutagenesis of Bartonella henselae by transposome technology. Gene. 2003;313:103–109. 10.1016/s0378-1119(03)00636-x [DOI] [PubMed] [Google Scholar]
- 14. LaBrie SD, Dimond ZE, Harrison KS, et al. : Transposon Mutagenesis in Chlamydia trachomatis Identifies CT339 as a ComEC Homolog Important for DNA Uptake and Lateral Gene Transfer. mBio. 2019;10(4):e01343–19. 10.1128/mBio.01343-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wang Y, LaBrie SD, Carrell SJ, et al. : Development of Transposon Mutagenesis for Chlamydia muridarum. J Bacteriol. 2019;201(23):e00366–19. 10.1128/JB.00366-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Peterson EM, Markoff BA, Schachter J, et al. : The 7.5-kb plasmid present in Chlamydia trachomatis is not essential for the growth of this microorganism. Plasmid. 1990;23(2):144–148. 10.1016/0147-619x(90)90033-9 [DOI] [PubMed] [Google Scholar]
- 17. Skilton RJ, Cutcliffe LT, Pickett MA, et al. : Intracellular parasitism of chlamydiae: specific infectivity of chlamydiaphage Chp2 in Chlamydophila abortus. J Bacteriol. 2007;189(13):4957–4959. 10.1128/JB.00235-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hanahan D: Techniques for Transformation of E.coli. In DNA cloning. D.M. Glover, Editor. IRL Press: Oxford.1985;109–135. [Google Scholar]
- 19. Grant SG, Jessee J, Bloom FR, et al. : Differential plasmid rescue from transgenic mouse DNAs into Escherichia coli methylation-restriction mutants. Proc Natl Acad Sci USA. 1990;87(12):4645–4649. 10.1073/pnas.87.12.4645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Brown TA: Essential Molecular Biology: A Practical Approach. The Practical Approach Series. ed. T.A. Brown. IRL Press.1995;1. [Google Scholar]
- 21. Dembek M, Barquist L, Boinett CJ, et al. : High-throughput analysis of gene essentiality and sporulation in Clostridium difficile. mBio. 2015;6(2):e02383. 10.1128/mBio.02383-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Skilton R, O’Neill C, Thomson NR, et al. : Progress towards an inducible, replication-proficient transposon delivery vector for Chlamydia trachomatis.2021. 10.17605/OSF.IO/QSBYA [DOI] [PMC free article] [PubMed]
- 23. Akerley BJ, Lampe DJ: Analysis of gene function in bacterial pathogens by GAMBIT. Methods Enzymol. 2002;358:100–8. 10.1016/s0076-6879(02)58082-4 [DOI] [PubMed] [Google Scholar]
- 24. Lampe DJ, Akerley BJ, Rubin EJ, et al. : Hyperactive transposase mutants of the Himar1 mariner transposon. Proc Natl Acad Sci U S A. 1999;96(20):11428–11433. 10.1073/pnas.96.20.11428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wickstrum J, Sammons LR, Restivo KN, et al. : Conditional gene expression in Chlamydia trachomatis using the tet system. PLoS One. 2013;8(10):e76743. 10.1371/journal.pone.0076743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cortina ME, Ende RJ, Bishop RC, et al. : Chlamydia trachomatis and Chlamydia muridarum spectinomycin resistant vectors and a transcriptional fluorescent reporter to monitor conversion from replicative to infectious bacteria. PLoS One. 2019;14(6):e0217753. 10.1371/journal.pone.0217753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hagemann AT, Craig NL: Tn7 transposition creates a hotspot for homologous recombination at the transposon donor site. Genetics. 1993;133(1):9–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bender J, Kuo J, Kleckner N: Genetic evidence against intramolecular rejoining of the donor DNA molecule following IS10 transposition. Genetics. 1991;128(4):687–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Minton N, Carter G, Herbert M, et al. : The development of Clostridium difficile genetic systems. Anaerobe. 2004;10(2):75–84. 10.1016/j.anaerobe.2003.11.003 [DOI] [PubMed] [Google Scholar]
- 30. Smith CJ, Markowitz SM, Macrina FL: Transferable tetracycline resistance in Clostridium difficile. Antimicrob Agents Chemother. 1981;19(6):997–1003. 10.1128/aac.19.6.997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Demars R, Weinfurter J, Guex E, et al. : Lateral gene transfer in vitro in the intracellular pathogen Chlamydia trachomatis. J Bacteriol. 2007;189(3):991–1003. 10.1128/JB.00845-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Suchland RJ, Sandoz KM, Jeffrey BM, et al. : Horizontal transfer of tetracycline resistance among Chlamydia spp. in vitro. Antimicrob Agents Chemother. 2009;53:4604–4611. 10.1128/AAC.00477-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wang Y, Kahane S, Cutcliffe LT, et al. : Genetic transformation of a clinical (genital tract), plasmid-free isolate of Chlamydia trachomatis: engineering the plasmid as a cloning vector. PLoS One. 2013;8(3):e59195. 10.1371/journal.pone.0059195 [DOI] [PMC free article] [PubMed] [Google Scholar]