

Graphical abstract

Keywords: ELF, LOL, NCI, SARS- CoV-2, Drug likeness
Abstract
In this work novel antiviral compound 4-(Dimethylamino) Pyridinium 3, 5-dichlorosalicylate was synthesized and characterized by UV–vis, FT-IR, FT-Raman, 1H NMR and 13C NMR spectra. Quantum chemical computations were carried out by Density functional theory methods at B3LYP level. Electronic stability of the compound arising from hyper conjugative interactions and charge delocalization is investigated using natural bond orbital analysis. Assignments of vibrational spectra have been carried out with the aid of Normal coordinate analysis following the SQMFF methodology. TD-DFT approach was applied to assign the electronic transition observed in UV visible spectrum measured experimentally. Frontier molecular orbital energy gap affirms the bioactivity of the molecule and NCI analysis gives information about inter and intra non covalent interactions. ESP recognises the nucleophilic and electrophilic regions of molecule and the chemical implication of molecule was explained using ELF, LOL. The reactive sites of the compound were studied from the Fukui function calculations and chemical descriptors define the reactivity of the molecule. Molecular docking done with SARS and MERS proteins endorses the bioactivity of molecule and drug likeness factors were calculated to comprehend the biological assets of DADS.
1. Introduction
Pyridine is pondered to be privileged ligands in medicinal chemistry since their derivatives or analogues have been publicised to exhibit a surfeit of biological deeds, including anti-cancer, antiviral, antibacterial, antifungal, antioxidant, antimicrobial, antidiabetic and anti-inflammatory properties [1]. Biological exploit of these molecules is mainly attributable to formation of hydrogen bond among the target and electrons of lone pair in a sp2 hybrid orbital of nitrogen atom [2]. Pyridine moieties are often used in drugs owing to their characteristics such as basicity, water solubility, stability, hydrogen bond-forming ability and their small molecular size. Replacement of Pyridine moieties by amines, amides, heterocyclic rings containing nitrogen atoms and benzene rings are important in drug discovery owing to their ability to act as bioisoters [3]. Dichlorosalicylic acid a colourless crystalline organic acid is used as a Keratolytic, Comedolytic and Bactereolytic agent in treatment of warts, Psoriasis, acne, ringworm and dandruff [4]. Salicylic acid derivatives are most widely used as an anti-inflammatory drug [5] and also found in plants with role such as plant growth, photosynthesis transpiration, ion uptake and transport [6].
A deep knowledge of intrinsic properties of titled compound is required to understand the reactivity involved in binding reactions. A thorough survey of the literature reveals that so far there is no detailed experimental and theoretical study on 4-Dimethylamino Pyridinium 3, 5- Dichlorosalicylate (DADS). Fascinating biological applications of pyridine derivatives inspires to do the Quantum chemical calculations in order to confirm its biological activities. Redistribution of electron density (ED) in various bonding, antibonding orbitals and E(2) energies are calculated by natural bond orbital (NBO) analysis. Electronic properties of DADS are scrutinised using frontier molecular orbital (FMO) energies and UV spectral analysis using Time dependent (TD) DFT (Density Functional Theory) method. DADS have been characterized by FT-IR, FT-Raman, NMR (Nuclear Magnetic Resonance) techniques to identify and study the activity of organic compound. Topology analyses were made on the basis of ELF (electron localization function) [7] and LOL (localized orbital locator) maps obtained from Multiwfn 3.4.1., a wave function analyser [8]. Fukui functions and chemical reactivity descriptors were calculated to study the reactivity of molecule and most reactive sites were also revealed. Drug likeness and molecular docking approach are enabled to check pharmaceutical potential and bioactivity of DADS.
2. Experimental details
2.1. Synthesis
Analytical grade, 4-(dimethylamino)pyridine (DA) and 3,5-dichlorosalicylic acid (DS) were taken in 1:1 stoichiometric proportions are dissolved homogeneously using methanol as a solvent and reaction mixture was thoroughly mixed together using mechanical stirrer up to 6 h at 30˚C. The synthetic scheme of DADS was depicted in Fig. 1 . The clear pale yellow coloured solution obtained was filtered to remove the impurities and was kept aside for about a week to get the crude product.
Fig. 1.

Synthetic scheme for DADS.
Slow evaporation- solution growth technique was used to grow single crystals. The pure raw material was dissolved in minimum quantity of methanol, stirred for a few hours, filtered through a quantitative filter paper (Whatman no.40) and filtrate was kept aside without any disturbance for the growth of crystals in a dust free environment at ambient temperature. Bright, transparent, pale yellow coloured DADS crystals were obtained after 10 days and the crystals were collected carefully from the mother liquor. The harvested crystals were recrystallized repeatedly to get superior quality crystals.
2.2. Characterization
Electronic absorption spectrum was measured in methanol using JASCO UV–Vis spectrophotometer in the range of 200–800 nm. In order to confirm the functional groups crystal was subjected to FT-IR spectral analysis by Perkin Elmer FT-IR 8000 spectrophotometer in the range of 4000–400 cm−1 using the KBr pellets method and FT-Raman spectral analysis by Bruker RFS 27: Standalone FT-Raman Spectrometer with Nd: YAG laser source at 1064 nm in the region 4000–50 cm−1. To confirm the molecular structure of the compound the 1H and 13C NMR spectra were recorded employing a Bruker AV III 400 MHz spectrometer in Deuterated Dimethyl Sulphoxide (DMSO) as solvent using TMS as an internal standard.
3. Computational details
Spectroscopic profiling of DADS was carried out with Becke3-Lee-Yang-Parr (B3LYP) level with 6–311 G** basis set [9] using Gaussian’09 program package [10]. NBO analysis has been performed on a molecule at the same level using second order Fock matrix and atomic natural charges is also been calculated by NBO method [11]. Normal coordinate analysis (NCA) had been executed using MOLVIB program version 7.0 written by Sundius in order to obtain thorough interpretation of the fundamental modes [12, 13]. In accordance to scaled quantum mechanical procedure (SQM) using selective scaling in the natural internal co-ordinate representation, scaling of the force field was performed to obtain a better concurrence between the theory and experiment [14]. NMR chemical shifts have been calculated with the gauge including atomic orbital (GIAO) approach and compared with experimental spectra to know about chemically significant area. Gauss view 5.0 software [15] is used to obtain highest occupied and lowest unoccupied molecular orbital maps (HOMO-LUMO), band energy gap and molecular electrostatic potential map (MEP) for identifying the potential region. Non Covalent Interaction (NCI) analysis, ELF, LOL, Fukui function were carried out by Multiwfn [8] which is a multifunctional wave function analysis program and all isosurface map were rendered by VMD program [16]. Thermodynamical properties for various temperatures are calculated using the perl script Thermo.pl [17]. Autodock suite 4.2.1 was used to find the minimum binding energy, Inhibition constant and various parameters of ligand–protein interactions [18], [19].
4. Results and discussion
4.1. Molecular geometry
The optimized geometric parameters computed using DFT method of DS, DA, DADS are portrayed in Table 1 & dihedral angles in Table S1 whereas atomic numbering scheme of DS, DA, DADS molecule obtained by DFT computation is exhibited in Fig. 2 . Self-consistent force field energies for DS, DA and DADS were obtained as 1415.4025, −382.3537, −1797.7807 eV respectively. BSSE and counterpoise corrected energy for DADS have been calculated as 0.003969 and −1797.7767 eV. Phenyl ring appears to be little distorted ascribed to substitution of chlorine atoms which slightly proliferates the bond angle C4-C5-C6 (121°) than hexagonal angle 120°. Bond angles C1-C2-O11 (123°) and C3-C2-O11 (118°) significantly deviate from the contemplated trigonal angle (120°) as an effect of attachment of electron donating hydroxyl group instead of hydrogen atoms.
Table 1.
Optimized bond length and bond angle of DS, DA and DADS.
| Bond Length |
Theoretical DADS (Å) |
Theoretical |
Bond Angle | Theoretical DADS (°) |
Theoretical |
||
|---|---|---|---|---|---|---|---|
| DS(Å) | DA(Å) | DS(°) | DA(°) | ||||
| C1-C2 | 1.402 | 1.4025 | – | C2-C1-C6 | 121.25 | 121.29 | – |
| C1-C6 | 1.388 | 1.388 | – | C2-C1--H7 | 118.84 | 118.71 | – |
| C1-H7 | 1.084 | 1.0831 | – | C6-C1-H7 | 119.91 | 119.98 | – |
| C2-C3 | 1.407 | 1.4072 | – | C1-C2-C3 | 117.65 | 117.57 | – |
| C2-O11 | 1.337 | 1.3361 | – | C1-C2-O11 | 123.55 | 123.56 | – |
| C3-C4 | 1.395 | 1.3947 | – | C3-C2-O11 | 118.80 | 118.86 | – |
| C3-Cl10 | 1.750 | 1.7506 | – | C2-C3-C4 | 121.74 | 121.77 | – |
| C4-C5 | 1.396 | 1.3956 | – | C2-C3-Cl10 | 118.42 | 118.42 | – |
| C4-C13 | 1.506 | 1.5068 | – | C4-C3-Cl10 | 119.84 | 119.79 | – |
| C5-C6 | 1.388 | 1.3881 | – | C3-C4-C5 | 118.82 | 118.80 | – |
| C5-C9 | 1.761 | 1.7609 | – | C5-C4-C13 | 120.80 | 120.8 | – |
| C6-H8 | 1.083 | 1.0822 | – | C4-C5-C6 | 120.64 | 120.65 | – |
| O11-H12 | 0.997 | 0.9996 | – | C4-C5-Cl9 | 120.19 | 120.14 | – |
| H16-N22 | 1.556 | 1.552 | – | C6-C5-Cl9 | 119.17 | 119.20 | |
| C13-O14 | 1.200 | 1.2001 | – | C1-C6-C5 | 119.90 | 119.89 | – |
| C13-O15 | 1.347 | 1.3475 | – | C1-C6-H8 | 120.33 | 120.37 | – |
| O15- H16 | 0.991 | – | – | C5-C6-H8 | 119.77 | 119.72 | – |
| C17-C18 | 1.382 | 1.3822 | C2-011-H12 | 112.32 | 112.07 | – | |
| C17-N22 | 1.341 | 1.3411 | O15-H16-N22 | 173.49 | – | ||
| C17-H23 | 1.086 | 1.0858 | C4-C13-O14 | 124.79 | 124.63 | – | |
| C18-C19 | 1.416 | 1.4156 | C4-C13-O15 | 111.38 | 111.44 | – | |
| C18-H24 | 1.081 | 1.0803 | O14-C13-O15 | 123.83 | 123.92 | – | |
| C19-C20 | 1.416 | 1.4156 | C13-O15-H16 | 107.08 | 106.57 | – | |
| C19-N27 | 1.366 | 1.3655 | C18-C17-N22 | 124.23 | 124.19 | ||
| C20-C21 | 1.382 | 1.382 | C18-C17-H23 | 119.80 | 119.77 | ||
| C20-H25 | 1.081 | 1.0803 | N22-C17-H23 | 115.97 | 116.03 | ||
| C21-N22 | 1.341 | 1.3413 | C17-C18-C19 | 119.63 | 119.60 | ||
| C21-H26 | 1.086 | 1.0858 | C17-C18-H24 | 118.81 | 118.90 | ||
| N27-H28 | 1.456 | 1.4569 | C19-C18-H24 | 121.56 | 121.48 | ||
| N27-C32 | 1.456 | 1.457 | C18-C19-C20 | 115.82 | 115.8 | ||
| C28-H29 | 1.096 | 1.0962 | C18-C19-N27 | 122.10 | 122.07 | ||
| C28-H30 | 1.088 | 1.0881 | C20-C19-N27 | 122.08 | 122.04 | ||
| C28-H31 | 1.095 | 1.0954 | C19-C20-C21 | 119.62 | 119.60 | ||
| C32-H33 | 1.095 | 1.0959 | C19-C20-H25 | 121.58 | 121.50 | ||
| C32-H34 | 1.088 | 1.088 | C21-C20-H25 | 118.80 | 118.89 | ||
| C32-H35 | 1.095 | 1.0956 | C21-C20-N22 | 124.25 | 124.20 | ||
| C20-C21-H26 | 119.87 | 119.82 | |||||
Fig. 2.

Optimized molecular structure of a) DS, b) DA and c) DADS.
High digression of bond length found in H16-N22 (1.53 Å (1.01 Å)) is due to intermolecular hydrogen bonding interaction between two moieties. Intermolecular hydrogen bonding interaction N22 - H16…O15 is corroborated by intermolecular hydrogen distance of N22….H16 (2.72 Å) which is shorter than van der Waals separation between N and H atom (3Å) [20]. Abatement in C17-N22-C21 angle (116.4°) is by dint of steric interaction which arises in virtue of intermolecular hydrogen bonding interactions. Bioactive molecules exert biological activity through hydrogen bonding interactions with their biological targets [21], [22]. N22 - H16 …O15 intermolecular hydrogen bonding is vital to get a closer perception into these interactions and design molecules with enhanced biological profile.
Bond length of C13 = O14 and C13-O15 found to be 1.200 Å and 1.3471 Å respectively is correlated with bond length for aromatic carboxylic group according to the international crystallography table. Computed C13 = O14 bond length 1.200 Å confirms the double bond characteristics of since it shows a deviation of about 0.03 Å when compared with literature value 1.23 Å [23]. Electron delocalization confirmed from the bond length of C19 = C20, increases from literature value 1.34 Å to 1.415 Å and C18-C19 decreases from literature value 1.415 Å to 1.54 Å due to the attachment of dimethyl amino group which donates electrons to bonds around 4th position of π deficient pyridine ring. Electron density of aromatic system is increased due to the hyper conjugation of dimethyl amino group with ring [24], [25].The heterocyclic ring appears to be crooked because of the dimethyl amino group substitution which is scrutinized from the bond angle of C20-C19-C18 (116°) smaller than typical hexagonal angle 120°. In pyridine ring, bond length of C atoms attached with amino group C19-C20, C19-C18 is found to be larger than the bond distances of C20-C21, C21-N22, N22-C17 and C17-C18 due to the effect of substitution of dimethyl amino group in the place of hydrogen.
C-N bond length calculated as 1.365 Å is shorter than the normal C-N bond length 1.480 Å and shortening of this C-N bond length reveal the effect of resonance in dimethyl amino group [26]. Internal C17-N22-C21 angle (116.4°) of protonated N atom is significantly less than other nearby two angles C20-C21-N22 (124.2°) and C18-C17-N22 (124.2°) within the ring in virtue of steric effect of the lone-pair electron predicted by valence-shell electron-pair repulsion theory, VSEPR [27].
The dimethyl amino group oriented in pyridine ring with antiperiplanar conformation as indicated by the torsional angle C18-C19-N27 -H31 (-179.3°). The carboxylic group of phenyl ring and dimethyl amino group of pyridine ring are found to be out of plane as indicated from the torsional angle C3-C3-C13-C14 (90.9°), C3-C4-C13-C15 (-89.36°), C5-C4-C13-C14 (−87.17°), C19-N27-C28-H29 (-61.403°), C19-N27-C28-H31 (60.08°), C19-N27-C32-H33(-60.41°)[28].
4.2. NBO analysis
NBO analysis is a cardinal tool which lucubrate the intra and intermolecular bonding interactions scrutinizes charge transfer or conjugative interactions in molecular systems. Second order Fock matrix was carried out at the B3LYP method to evaluate the donor- acceptor interactions in the NBO analysis is portrayed in Table 2a and composition of H-bonded NBOs in terms of natural atomic hybrids is portrayed in Table 2b .
Table 2a.
Second order perturbation theory analysis of Fock matrix in NBO basis.
| Donor | ED(i) (e) |
Acceptor | ED(j) (e) |
E(2) Kcalmol−1. |
E(j)-E(i) a. u |
F(i, j) a. u |
|---|---|---|---|---|---|---|
| σ(C1-C2) | 1.97229 | σ*(C1-C6) | 0.01647 | 3.10 | 1.28 | 0.056 |
| σ(C1-C2) | 1.97229 | σ*(C2-C3) | 0.04243 | 4.98 | 1.23 | 0.070 |
| π(C1-C2) | 1.61982 | π*(C3-C4) | 0.42943 | 18.29 | 0.28 | 0.064 |
| π(C1-C2) | 1.61982 | π*(C5-C6) | 0.41594 | 25.50 | 0.28 | 0.076 |
| σ(C1-C6) | 1.96801 | σ*(C1-C2) | 0.03047 | 2.82 | 1.25 | 0.053 |
| σ(C2-C3) | 1.97415 | σ*(C1-C2) | 0.03047 | 3.99 | 1.26 | 0.063 |
| σ(C3-C4) | 1.96327 | σ*(C4-C5) | 0.03853 | 6.41 | 1.28 | 0.081 |
| σ(C5-C6) | 1.97756 | σ*(C1-C6) | 0.01647 | 3.39 | 1.31 | 0.059 |
| σ(C17-C18) | 1.98072 | σ*(C18-C19) | 0.02437 | 3.39 | 1.25 | 0.056 |
| σ(C18-C19) | 1.97883 | σ*(C17-C18) | 0.02153 | 3.05 | 1.28 | 0.056 |
| σ(C19-C20) | 1.97016 | σ*(C20-C21) | 0.02154 | 3.05 | 1.28 | 0.058 |
| σ(C20-C21) | 1.98080 | σ*(C19-C20) | 0.02437 | 3.39 | 1.25 | 0.058 |
| σ(C21-N22) | 1.98688 | σ*(C17-N22) | 0.01851 | 1.32 | 1.36 | 0.038 |
| π(C3-C4) | 1.72240 | π*(C1-C2) | 0.39366 | 19.72 | 0.30 | 0.071 |
| π(C5-C6) | 1.69402 | π*(C3-C4) | 0.42943 | 23.47 | 0.29 | 0.076 |
| π(C17-C18) | 1.71705 | π*(C17-C18) | 0.29162 | 0.94 | 0.29 | 0.015 |
| π(C20-C21) | 1.71613 | π*(C20-C21) | 0.29055 | 0.93 | 0.29 | 0.015 |
| σ(C17- N22) | 1.98694 | σ*(C21-N22) | 0.01829 | 1.33 | 1.36 | 0.038 |
| LP(1)N22 | 1.86971 | π*(C17-C18) | 0.29162 | 60.91 | 0.22 | 0.116 |
| LP(1)Cl9 | 1.99202 | σ*(C4-C5) | 0.03853 | 1.75 | 1.47 | 0.046 |
| π*(C3-C4) | 0.42943 | σ*(C13-O14) | 0.02380 | 1.83 | 0.61 | 0.063 |
| π*(C3-C4) | 0.42943 | σ*(C13-O15) | 0.10455 | 3.26 | 0.34 | 0.058 |
| σ(O11-H12) | 1.98573 | σ*(C17-N22) | 0.01851 | 0.27 | 1.24 | 0.016 |
| σ(O11-H12) | 1.98573 | σ*(C21-N22) | 0.01829 | 0.20 | 1.24 | 0.007 |
| LP(1)N22 | 1.86971 | σ*(O15-H16) | 0.07224 | 108.99 | 0.79 | 0.137 |
E(2)means energy of hyper conjugative interactions.
E(j)-E(i) Energy difference between donor and acceptor i and j NBO orbitals.
F(i,j) is the Fock matrix element between i and j NBO orbitals.
Table 2b.
Composition of H-bonded NBO in terms of natural atomic hybrids.
| Bond (A-B) |
DS/DA | DADS | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| EDA % |
EDB % |
spn | A | B | EDA % |
EDB % |
spn | A | B | |||||
| s% | p% | s% | p% | s% | p% | s% | p% | |||||||
| O11-H12 | 73.7 | 26.2 | sp2.09 | 20.3 | 79.5 | 99.8 | 0.15 | 78.0 | 21.9 | sp1.96 | 26.1 | 73.8 | 99.8 | 0.22 |
| C17-N22 | 40.9 | 59.0 | sp1.96 | 32.3 | 67.5 | 35.3 | 64.6 | 40.0 | 59.9 | sp1.95 | 31.8 | 68.1 | 36.1 | 63.7 |
| C21-N22 | 40.9 | 59.0 | sp1.96 | 32.3 | 67.5 | 35.3 | 64.6 | 40.0 | 59.9 | sp1.95 | 31.7 | 68.1 | 35.9 | 63.9 |
Orbital overlap between LP (N22) → σ*(O15-H16) forms intermolecular hydrogen bonding which results in intermolecular charge transfer causing stabilization of H-bonded systems. Electron density of O15-H16 antibonding orbital upsurges due to this hydrogen bonding interactions and the stabilization energy concorded with this intermolecular interaction is found to be 108.99 Kcalmol−1. Intra-molecular interaction formed by orbital overlap between the bonding (C-C), (C-N), (C-H), (C-N) orbitals to antibonding orbitals is portrayed in Table 2a.
The σ and π electron bonds of (C1-C2), (C3-C4), (C5-C6) shows double bond characteristics whereas (C1-C6), (C2-C3) and (C4-C5) shows only single bond characteristics which was portrayed in Table 2a. Electron density of conjugated double as well as single bond of phenyl ring clearly demonstrates strong delocalization inside the molecule. Pyridine ring atoms have weaker stabilization energy than phenyl ring atoms which is evident from Table 2a, in virtue of negative inductive effect of nitrogen atom, electron density is not evenly distributed over the pyridine ring [29]. As per the resonance structure of pyridine, only C17-C18 and C20-C21 exhibit double band characteristics owing to the substitution of dimethyl amino group in the 4th position [30].
Occupancy of C-C bonds with σ and π (1.6 e) bonds are lesser than the C-C bonds with only σ (1.9 e) bonds since the electronegativity of sp2 hybridized carbon atom is greater than sp3 hybridized atom [31]. C-O bond of carboxyl group has lesser energy transfer to the antibonding C-O, since the oxygen atom of hydroxyl group has lost ‘full-control’ of this lone pair and the oxygen atom of the CO group has acquired a small share of this lone pair. The greater electron-attracting power of oxygen in a hydroxyl group over that of an oxygen atom alone is due to hydrogen atom which forms the bond, and thereby decreases the electron density on that oxygen atom [32]. In DADS a very resilient interaction has been observed between the lone electron pair LP (1) N22 → π*(O15-H16) with an energy of 108.99 Kcalmol−1 and interaction between π (C19-N27) → π*(C17-C18) with an energy of 89.47 Kcalmol−1 gives stronger stabilization to the system. The larger energy shows that the hyper conjugative interactions taking place between the electron donating groups to the acceptor group of the title molecules enhances the bioactivity.
NBO analysis of DADS in comparison with DS and DA clearly shows the evidences for formation of strong H-bonded interactions between oxygen lone electron pair and antibonding σ*(O11-H12) orbitals. The magnitude of charge transferred from lone pair (O14) → σ*(O11-H12) has been increased by 0.06507 e in DADS when compared with DA is ascribed to the weakening of bond strength and elongation of bond length. Hybrids related to central nitrogen N22 forming the NBOs of the protonated form have been nearly equal (sp1.96 and sp1.95) in DA and DADS.
The s-character of O11-H12 hybrid orbital which increases from sp1.96 to sp2.09 leads to the firming up of O11-H12 bond and its retrenchment evinces that there is negative impact in O11-H12 bond due to effect of rehybridization. The effect of rehybridization has been overcome by the hyperconjugative effect resulting in the retrenchment of O-H bond and a concomitant blue shift in O-H stretching frequency [33].
Acidity of the donor C-H group and its potential hydrogen bond donor strength increases in passing from sp3over sp2 to sp hybridized C atoms and is also increased by surrounding electron withdrawing atoms is confirmed in C17- H23 bond at C17 passing from sp2.48 to sp2.53 hybridized in DADS to DA respectively.
4.3. Vibrational spectral analysis
The aspiration of vibrational analysis is to find out vibrational modes associated with relevant and specific molecular structures of calculated compounds studied. DADS molecule has 35 atoms which undergo 99 normal modes of vibration and vibrational assignments of DADS were carried out with the aid of NCA. The theoretical assignments were initially made on the basis of unscaled frequency and then calibrated by the scaling factors so that the observed values are easily compared with the calculated value and the scaling factors are tabulated in Table S2 and S3. The assignments of DADS with potential energy distribution (PED) have been reported to know the maximum probability of vibrations in Table 3 and observed FT-IR and FT Raman spectrum along with the simulated spectra is shown in Fig. 3, Fig. 4 for visual comparison.
Table 3.
Vibrational assignments of DADS by NCA based on SQMFF calculations.
| Experimental wavenumber(cm−1) |
Calculated wavenumber (cm−1) |
Assignments (PED) |
|
|---|---|---|---|
| FT-IR | FT-Raman | ||
| 3450(s) | 3447(s) | 3447 | ʋ OH(1 0 0) |
| 3189 | ʋ asCH3(6 1), ʋ asCH3(3 3) | ||
| 3144 | δADOCH3(8 3), ʋ ssCH3 | ||
| 3115 | ʋasCH3 (6 0), ʋasCH3 (3 7) | ||
| 3091 | ʋ R1CH (9 9) | ||
| 3091 | ʋR1CH (9 9) | ||
| 3071(s) | 3065(s) | 3070 | ʋR1CH(9 9) |
| 3052(m) | 3052(m) | 3053 | ʋR1CH (9 9) |
| 3037(s) | 3036 | ʋR2CH(9 9) | |
| 3031(m) | 3033 | ʋR2CH(9 9) | |
| 3009 | ʋssCH3 (7 1), ʋasCH3(1 3), ʋssCH3(1 1) | ||
| 2975(s) | 2999 | ʋssCH3 (8 0) | |
| 2810(m) | 2972 | ʋasCH3 (9 0) | |
| 2794(s) | 2751(s) | 2748 | ʋ NH+ (6 6) |
| 1703(s) | 1708 | ʋssCO(39), ʋasCO(3 3), ʋR1CC (1 3) | |
| 1588(s) | 1588 | ʋR2CC (5 2), βR2CH (1 7), ʋR2CN(1 6) | |
| 1585(s) | 1580 | ʋR1CC (5 9) | |
| 1545(s) | 1550(s) | 1542 | ʋR1CC (6 6) |
| 1522 | ʋR2CC (3 6), ʋR2CN (3 2) | ||
| 1499 | ʋR2CN (3 6), βR2CH(1 4), δADCH3(1 2), ʋR2CC(12) | ||
| 1475 | δADCH3 (6 4) | ||
| 1472 | ʋasCH3 (8 9) | ||
| 1459 | ʋR1CC (3 5), βR1CH (1 7), βR1OH (1 1), ʋR1CO | ||
| 1451(m) | 1448(w) | 1451 | δADOCH3 (5 3), δADCH3 (1 9) |
| 1419 | δADCH3(6 7), δADOCH3(2 3) | ||
| 1417 | δSDCH3(3 9), βR2CH(2 2), δSDCH3(2 1) | ||
| 1411 | ʋR1CC(4 4), βR1CH(1 1), βR1OH(1 1) | ||
| 1401 | ʋR2CC(4 4), βR2CH(3 4) | ||
| 1385(m) | 1381(w) | 1385 | δSDCH3(4 9), δSDCH3(3 8) |
| 1336 | ʋR2CN (3 1), βR2CH (1 4), δSDCH3(1 1) | ||
| 1319 | ʋR1CO (2 2), ʋR1CC(2 0), βR1OH (1 6) | ||
| 1308 | ʋR1CC (3 8), βR1CH (1 5), βR1OH (1 0) | ||
| 1297 | βR2CH (7 5) | ||
| 1276(s) | 1274 | ʋR2CC (3 4), ʋR2CN (3 3), ʋas CN(1 1) | |
| 1273(s) | 1273 | βR1OH(4 9), ʋasCO(1 6) | |
| 1219(s) | 1211 | ʋR1CC (7 9) | |
| 1200 | ʋ as CN (3 2), ʋR2CC(2 6), βR2CH(1 5) | ||
| 1198 | βR2CH (6 8), ʋR2CN(1 4), | ||
| 1176(m) | 1179(m) | 1173 | ʋR1CC(3 8), βR1CH (3 2), βR1OH(1 3) |
| 1151 | ρ'CH3(2 2), ρ'CH3(1 9), ρCH3(12 | ||
| 1110(m) | 1110(m) | 1130 | βR1CH (6 0), ʋR1CC(2 8) |
| 1093 | δTDR1(2 6), ʋR1CCl(2 2), ʋR1CC(1 6), ʋasCO(11) | ||
| 1090 | ρCH3(4 2), ρCH3(2 4), ρ'CH3(1 3), | ||
| 1089 | βR2CH (4 3), ʋR2CC(1 8) | ||
| 1084 | ρ'CH3 (4 2), ρCH3(2 1), ρ'CH3(1 5), ρ'CH3(1 4) | ||
| 1058 | ʋR2CN(4 2), βR2CH(3 3), ʋR2CC(1 7) | ||
| 1042 | ʋssCO(2 5), ʋasCO(2 4), δ TD R(1 2), ʋR1CC(1 1), | ||
| 1022(m) | 1023 | ʋasNC(2 9), ρ'CH3(2 9), ρ'CH3(2 0) | |
| 994(s) | 999(s) | 993 | δTDR2 (6 4), ʋR2CC(1 5), ʋR2CN(1 3) |
| 960 | τ as R2 (4 3), RG2CH(3 1), τ'as R2(2 6) | ||
| 933 | R2P (4 5), τ'as R2(2 9), ωR2CH(2 5) | ||
| 939(w) | 939(w) | 931 | ʋR1CC(2 3), ʋssCO(2 0), ʋasCO(1 4), ʋR1CO(11) |
| 913 | ʋssNC(4 1), ʋR2CC(3 0) | ||
| 884 | τ HN(4 5), τ OH(4 4) | ||
| 869(s) | 871(s) | 871 | τ HN(4 6), τ OH(4 6) |
| 811 | ωR2CH (8 7) | ||
| 802(s) | 805(m) | 804 | τ' as R2 (6 1), R2P(2 2) |
| 788 | ʋR1CCl (3 9), δ AD R1(2 7), ωCO(1 1) | ||
| 751(w) | 765 | ωR1CH (7 5) | |
| 733(s) | 743 | ωCO (3 6), τOC(1 3), ʋR1CC(1 2), | |
| 733 | ʋssCN(3 9), δADOR2 (2 4), ʋR2CC(1 2), | ||
| 727(m) | 723 | R2P (5 5), τ'as R2(4 3) | |
| 701 | R1P (4 9), ωR1CCl (2 0), | ||
| 700(w) | 700(w) | 687 | R1P(2 0), τ OC(1 2), |
| 663 | δADR2 (4 7), δADOR2 (1 6), τ OH (1 4), τ HN(14) | ||
| 645 | COS (3 2), δ ADO R1(2 1) | ||
| 602 | τOC(4 1) | ||
| 595(m) | 594(m) | 596 | ωR1CO (3 2), τ' as R1(2 1), ωR1CCl (1 3) |
| 543 | R2P (6 0), τ as R2 (2 9) | ||
| 536 | NCS (3 1), δADOR2(2 4) | ||
| 525 | τ as R1(3 3), ωR1CCl (2 7), R1P(1 7) | ||
| 520 | βR1OC (2 3), τ OH(1 1) | ||
| 474(s) | 475(s) | 465 | ρ NC(4 7), βR2CN (2 7) |
| 436 | βR1ClC(2 3), ʋR1CC(2 1), ʋR1CCl(1 5) | ||
| 395(s) | 398 | τ asR2 (5 7), τ'as R2 (4 2) | |
| 390 | δ AD R1(3 0), ʋR1CCl(2 1), δ ADO R1(1 6) | ||
| 380 | NCS (3 9), τ as R2(1 6), τ'as R2(1 6) | ||
| 355(s) | 360(m) | 362 | ωR1CCl (2 8), τ'as R1(2 5), ρCO2 (2 0) |
| 330(s) | 332 | ωR1CCl (3 0), R1P(1 9), ωR1CO(1 8), ωR1CH(1 7) | |
| 316 | δADOR1 (2 3), ʋR1CCl (2 0), ʋR1C(1 6), | ||
| 280(w) | 280(w) | 285 | τ' as R2 (4 3), τ as R2 (1 2), βR1ClC(1 1) |
| 270 | τ' as R2 (6 8), τ as R2(21) | ||
| 203(s) | 240 | βR2CN (4 3), NCR(2 2), τ HN(1 3), τ OH(12) | |
| 182(m) | 184 | τ as R2(3 2), τ' as R2(3 0), βR1ClC(1 5) | |
| 184 | τ as R2(3 1), τ' as R2(2 6), βR1ClC(1 8), | ||
| 169(m) | 162 | ωR1CC(3 5), ρCO(1 6), ωR1CCl(1 4), τ as R (1 3) | |
| 144(s) | 147 | τ' as R2(6 9), τ as R2(1 8), PR2(1 1) | |
| 133 | βR1CC (6 0), ωCO(1 6) | ||
| 121 | τ' as R1(3 0), τ as R1(2 2), τ HN(1 7) | ||
| 80 | τ CN(4 1), τNC(1 5), τ NC(1 4), τ as R2 (1 2), τ' as R2 (1 1) | ||
| 74 | τ OC(1 5), τ' as R1 (1 4), ρCO (1 3), ωR1CC (1 2) | ||
| 64 | τ' as R2 (6 0), τ as R2 (1 4), R2P (1 2) | ||
| 58 | ʋ NH (6 7), ʋ OH (2 0) | ||
| 44 | τ HN (2 9), τ OH (2 6), τ' as R2 (1 1) | ||
| 15 | τ HN(5 7), τ' as R2 (1 2) | ||
w: weak, s: strong, m:medium, ν:stretching, νss: symmetric stretching, νas: asymmetric stretching, δ: deformations (SD: symmetric, AD: asymmetric, ADO : asymmetric out of plane RD: ring, TD: trigonal), τ: torsion, τ as : Asymmetric torsion, τ' as :Asymmetric torsion out of plane R1: Ring1, R2: Ring2, β: bending, ρ: rocking, ω:wagging, P: puckering, S: Scissoring.
Fig. 3.
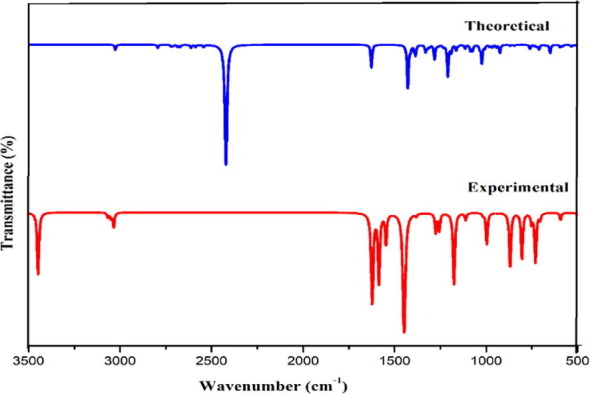
Experimental and simulated IR spectra.
Fig. 4.
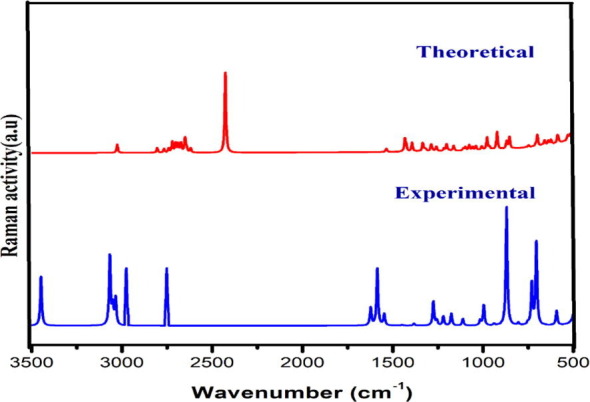
Experimental and simulated Raman spectra.
4.3.1. Phenyl ring vibrations
Aromatic structure shows the presence of C-H stretching vibrations in region 3100–3000 cm−1, the characteristic region for ready identification of C-H stretching vibrations [34], [35] are not affected appreciably by nature of substituents. The scaled wavenumber for C-H stretching fall in the range 3070 cm−1 and 3053 cm−1 with 99% PED is observed at 3071 cm−1, 3052 cm-1 in IR and at 3065 cm−1, 3052 cm−1 in Raman are assigned to C-H stretching vibrations.
The C-H in plane bending frequencies which is useful for the characterization purpose appears in the range 1000–1300 cm−1 is observed at 1110 cm−1,1176 cm−1 in FT-IR and at 1110 cm−1,1179 cm−1 in FT-Raman are assigned to bending vibration which coincides with the scaled values. The frequencies observed at 869 cm−1,751 cm−1 in FT- IR and 871 cm−1 and 733 cm−1 in FT-Raman spectra are assigned to out of plane bending which agrees with the scaled value by B3LYP method. Ring breathing modes due to stretching and contraction of C-C bonds in the ring normally occur in region 1620–1400 cm−1 [36] is observed as prominent peaks at 1585 cm−1,1545 cm-1 in FT-IR and 1550 cm−1 in FT-Raman agree with the scaled value.
4.3.2. Pyridine ring vibration
Portrayal of C –C stretching pyridine ring and C –N stretching bands of aromatic ring typically observed in the same frequency range 1600–1450 cm−1 and 1450–950 cm−1 [37]. In present study C-N stretching mode scaled at 993 cm−1 and 1274 cm−1 coincides with the experimental wavenumber 999 cm−1 observed in FT-Raman and 994 cm−1 observed in FT-IR spectra. Scaled pyridine ring breathing vibrational modes at 1588 cm−1, 1522 cm−1, 1499 cm−1 concur with peak at 1588 cm−1 recorded in FT-Raman spectra. Aromatic C-H stretching vibrations of nitrogen heterocyclic aromatic compounds give rise to a band at 3100–3010 cm−1 [35] is perceived in FT-IR at 3071 cm−1 and in FT-Raman at 3052 cm−1 are in good concurrence with scaled value in the region 3091 cm-1and 3053 cm−1. Bands of variable intensity are observed in the regions 1300–1180 cm−1 and 1100–1000 cm−1 due to in- plane deformation vibrations and in DADS in plane deformation vibrations are obtained at 1297 cm−1, 1200 cm−1,1198 cm−1 by B3LYP method. C-H out of plane deformation vibrations which are the characteristics of the position of substitution are observed as strong bands in the region 850–690 cm−1. The ring out of plane deformation of DADS obtained theoretically at 811 cm−1 and 804 cm−1 coincides with the experimental wavenumber 802 cm−1 in FT-IR and 805 cm−1 at FT- Raman spectrum. NH + stretching vibrations involve in hydrogen bonding interaction provide clear evidence for charge transfer interaction. Bibi et al [40] reported NH + stretching vibration for Pyridinium around 2780 cm−1 as a strong band is observed in DADS at 2794 cm−1 in FT-IR and 2751 cm−1 in FT- Raman spectrum agrees with scaled value.
4.3.3. C-Cl vibrations
The vibrations related to the bond between the benzene ring and halogen atoms (Cl) are important here at where mixing of vibrations happens based on reduction in molecular symmetry and also based on the existence of heavy atoms on the periphery of molecule [38], [39]. According to literature C-Cl stretching vibrations give strong bands in region 580–730 cm−1 agrees with calculated value where strong bands are found at 645 cm−1 and 685 cm−1 FT-IR and FT-Raman band at 280 cm−1 assigned to in plane C-Cl bending concord well with theoretical value. The out plane bending vibrations observed at 355 cm−1 in FT-IR and at 360 cm−1 in FT-Raman agrees with the scaled value.
4.3.4. COO– vibrations
Vibrational analysis of carboxylic acid was carried out giving preference to carbonyl group which are significant for the antiviral activity of the compound. C = O stretch bands appear in region 1710–1680 cm−1 depends on the physical state, electronic, mass effect of neighbouring substituents, conjugation, intra-molecular and intermolecular hydrogen bonding [42] is observed at 1703 cm−1 in FT-IR agrees with the scaled value at 1708 cm1. Carboxylic acid also contains a C-O band and the C-O stretch of carboxylic acid appears between 1320 cm−1 and 1210 cm−1 and for DADS the C-O stretching vibrations is observed at 1273 cm−1 in FT-IR agrees with the scaled value at 1273 cm−1.
4.3.5. Hydroxyl vibrations
O-H bond is highly polar because of large electronegativity difference between oxygen and hydrogen. The spectral bandwidth of hydroxyl group is greater than other functional groups due to the chemical environment caused by hydrogen bonding [41]. The most diagnostics of these band, O-H stretching vibration is typically found in 335050 cm−1. O-H stretching vibrations observed as strong bands in FT-IR spectrum at 3450 cm-1with the counterpart in FT-Raman spectrum at 3447 cm−1 as strong band agrees well with the scaled value at 3447 cm−1 with 100% PED contributions. O-H group can bend in and out of the plane defined by the acid moiety and in general in plane O-H bending found in the range 1440 to 1395 cm−1 are usually broader than C-H bending bands making them easy to distinguish. The peaks obtained at 1385 cm−1 in FT IR spectra is assigned to the O-H bending vibrations which coincides with scaled value obtained at 1459 cm−1 and 1411 cm−1. The blue shift in wavenumber shows the spectral evidence for the formation of O…H-N hydrogen bonding interactions which endorses the bioactivity of compound. This has been validated from results of optimized geometry where there is an increase in bond length of O11-H12 (0.99 Å) than the normal O-H bond length (0.96 Å). Also, the up shift in stretching frequency has been substantiated by the second order perturbation energy that takes place between n1(O14) → σ* (O11–H12) with the stabilization energy of 29.02 kcal/mol and the occupancy of the interacting NBOs.
4.3.6. CH3 vibrations
DADS molecule possesses a CH3 group in 2 positions and nine fundamentals can be associated to each CH3 group for the assignments of CH3 group frequencies. Based on the orientation of DADS methyl groups are supposed to vibrate more elastically than pyridine ring. Normally CH3 asymmetric and symmetric stretching vibrations are found at 2962 cm−1 and 2872 cm−1 [43]. Strong absorptions in FT-IR at 2810 cm−1 is assigned to the asymmetric CH3 stretching modes and the symmetric stretching is identified in FT-IR at 2975 cm−1.
The changes in CH3 stretching mode is due to the influence of electronic effects resulting from hyper conjugation of methyl group with nitrogen atom and the aromatic ring system which points to change in polarizability and dipole moment due to electron delocalization [41], [23]. Thus the hyperconjugation of methyl group, causing changes in IR, clearly evinces that methyl hydrogen is directly involved in the donation of electronic charge.
Asymmetric and symmetric methyl deformation modes normally appear around 1460 cm−1 and 1375 cm−1 is observed in FT-IR at 1451 cm-1corresponds to the asymmetric deformation. Umbrella modes normally show up at 1375 ± 10 cm-1confirms the presence of methyl group of DADS. FT-IR band at 1385 cm−1 and FT-Raman band at 1381 cm−1 corresponds to the umbrella mode agree with the scaled value. The position of umbrella mode is less sensitive to changes in molecular structure than the C-H stretching. CH3 rocking mode vibration is expected to occur in region 1070–900 cm−1 [44], [45], [46] is observed in FT-Raman spectra 1023 cm−1.
4.4. Aromaticity
Aromaticity is an axiomatic concept in organic chemistry enacts an important role in ordaining the structure, stability and reactivity of molecules [47], [48]. The geometrical aromaticity of homocyclic and heterocyclic compounds can be evaluated by harmonic oscillator model of aromaticity (HOMA) index value.
HOMA index value for phenyl ring found to be 0.984 is slightly lower than the ideal value of aromaticity due to replacement of hydrogen atoms by chlorine and hydroxyl group in benzene ring. For the pyridine ring HOMA index value found to be 0.9350 and this slight accrual in aromaticity is due to the substitution of dimethyl amino group. HOMA index of pyridine is lower than phenyl ring ascribed to H-bonding interactions and the protonation-induced delocalization. From the HOMA index value of both the phenyl and pyridine ring it is concluded that both these rings are aromatic in nature.
4.5. Frontier orbital analysis
In order to evaluate the energetic behaviour of DADS the frontier orbital calculations were topped off by means of B3LYP method. The FMO's were delineated in Fig. 5 where positive phase is exposed in green and negative is depicted in red which embodies the charge transfer within the molecule.
Fig. 5.

HOMO-LUMO Plot of DADS.
Atom occupied by more densities of HOMO should have stronger ability to detach electron whereas the atom with occupation of LUMO should have the dexterity to gain electron [49]. It is grasped from the HOMO plot that charge density is localized over the Phenyl ring, Chlorine atom, hydroxyl group whereas LUMO is spread over the carbon atoms pyridine and phenyl ring.
HOMO and LUMO energy values reckoned to be −5.8771 eV and −0.9442 eV concludes that homo value is high in virtue of substitution of electron withdrawing chlorine group in phenyl ring and low LUMO value owing to the presence of electron donating dimethyl amino group. Frontier orbital energy gap is a critical parameter in determining molecular electrical transport properties, rigidness or suppleness of molecule. Molecule with large frontier orbital gap is less polarizable with high stability and a molecule with small frontier orbital gap is more polarizable with high chemical reactivity [50]. The frontier orbital gap of DADS calculated as 4.9329 eV confirms the bioactivity of DADS molecule as a result of intermolecular charge transfer. This small energy separation between donor and acceptor reflects high chemical reactivity, low molecular stability that demonstrates the more charges are excited easily from the ground state to excited state.
4.6. UV analysis
TD-DFT calculations on electronic absorption spectra are enacted to comprehend the nature of electronic transition within the DADS molecule. λmax value obtained from UV–visible spectra, calculated UV absorption maxima λmax, theoretical excitation energies, oscillator strengths are tabulated in Table 4 and UV spectra is depicted in Fig. 6 .
Table 4.
UV–vis excitation energy and oscillator strength.
| Experimental |
Energy (cm−1) |
Theoretical |
Oscillator Strength f |
Symmetry | Assignments |
||
|---|---|---|---|---|---|---|---|
| λmax (nm) |
Band Gap (eV) |
λmax (nm) |
Band Gap (eV) |
||||
| 35,401 | 282 | 4.397 | 0.0 | Singlet - A | HOMO → L + 1 (99%) | ||
| 35,524 | 281 | 4.412 | 0.0651 | Singlet -A | H-2 → L + 3 (11%), HOMO → L + 2 (88%) | ||
| 270 | 4.59 | 37,343 | 268 | 4.626 | 0.0204 | Singlet -A | H-1 → LUMO (91%) |
| 38,787 | 257 | 4.806 | 0.0016 | Singlet -A | HOMO → L + 4 (55%), HOMO → L + 5 (24%), HOMO → L + 6 (17%) |
||
| 41,822 | 239 | 5.188 | 0.0015 | Singlet -A | HOMO → L + 4 (39%), HOMO → L + 5 (30%), HOMO → L + 6 (28%) |
||
| 42,033 | 238 | 5.210 | 0.005 | Singlet -A | HOMO → LUMO (99%) | ||
Fig. 6.
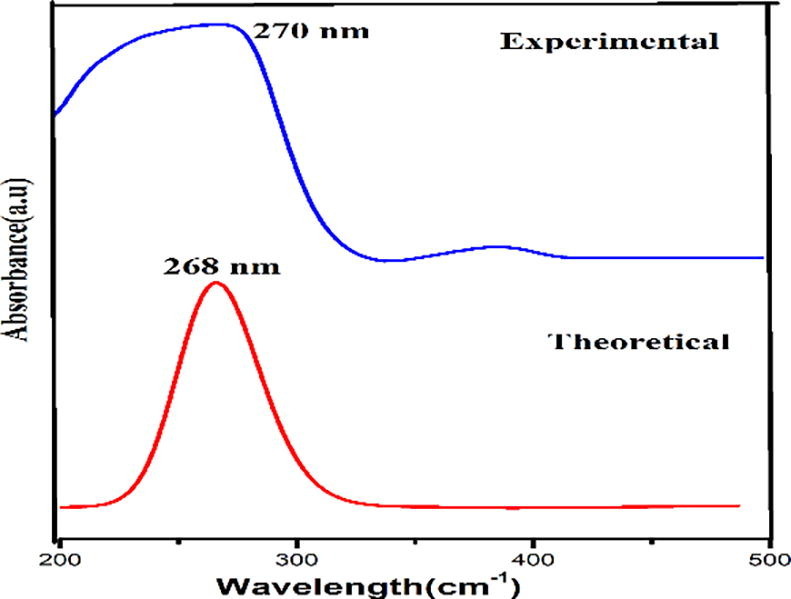
Experimental and theoretical UV spectra.
Oscillator strength f is a dimensionless quantity that describes the strength of an electronic transition [51] and the oscillator strength for the transition at λmax 268 nm is higher in magnitude than the other transitions which agrees with the experimental absorption value 270 nm corresponds to π -π * transition. The band energy gap for the observed λmax found to be 4.626 eV agrees with the frontier orbital band gap is found to be 4.9329 eV and the band energy gap of DADS molecule confirms its biological activity by intermolecular charge transfer [52].
Calculations of molecular orbital geometry show that the visible absorption maxima of this molecule correspond to the electron transition between frontier orbitals such as translation from HOMO to LUMO. From the calculated absorption spectra the maximum absorption wavelength corresponds to electronic transition from HOMO −2 to LUMO + 3 with 11% contribution and from HOMO to LUMO + 2 with 88% contribution. HOMO to LUMO + 2 has more contribution to excited state than HOMO −2 to LUMO + 3 due to the transition coefficients
4.7. Global reactivity descriptors
Electronegativity(χ), hardness (η), softness (S) are commonly used as global reactivity parameters within DFT. Energy of frontier molecular orbitals HOMO and LUMO, ΔE and global reactivity parameters for compound DADS calculated at B3LYP level are listed in Table 5 . Hardness of a molecule is related to gap between frontier molecular orbitals and global softness is inverse of global hardness [53]. Negative value of chemical potential evinces that compound is stable and does not decompose spontaneously into its elements. Stability of compound is endorsed by chemical hardness which measures 2.4664 eV and competency to attract the shared electrons which describes the electronegativity of compound is found to be 3.4106 eV. The scope of compound in medical and industrial field can be ascertained using the chemical softness value estimated to be 0.4054 eV indicates the non toxic nature of the molecule. Electrophilicity index italicizes the biological activity, confirms the pathway for molecular docking approach with different protein targets [54].
Table 5.
Molecular orbital energies and global reactivity parameters of DADS.
| Global Reactivity Parameters | Calculated values(eV) |
|---|---|
| EHOMO | −5.8771 |
| ELUMO | −0.9442 |
| EHOMO-1 | −6.5612 |
| ELUMO+1 | −0.9387 |
| EHOMO-LUMO | 4.9329 |
| Ionization potential | 5.8771 |
| Electron affinity | 0.9442 |
| Electronegativity | 3.4106 |
| Global hardness | 2.4664 |
| Global softness | 0.4054 |
| Chemical potential | −3.4106 |
| Electrophilicity index | 2.358 |
4.8. Atomic natural charges
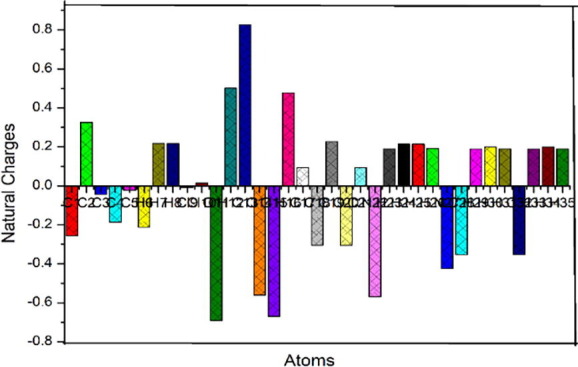
Atomic natural charges calculated by NBO method are portrayed in Table S4 and atomic natural charges are depicted in Fig. 7 . All carbon atoms extant in phenyl ring are positive except C2 atom, which is negative due to attachment of the hydroxyl group. Hydrogen atoms in the phenyl ring are found to be equally positive due to sharing of electrons with nearest carbon atoms. Electron withdrawing groups increase positive charge on the hydrogen atom of the interacting C-H bond, which increases attractive electrostatic interaction with phenyl ring. Charge of hydrogen H12 in O-H group is massive than other hydrogen atoms due to the hydrogen bonding interaction and the oxygen atom which take part in this interaction has more negative charge. C2 atom is negative due to the attachment of hydroxyl group in the place of hydrogen atom. Cl9 atom is found to have high negative charge and massively small positive charge is found in Cl10 atom since it is switched in the place of hydrogen atoms. The chlorine atoms are most preferable site for electrophilic attack and this is also confirmed from the Fukui analysis.
Fig. 7.
Atomic natural charges.
In the pyridine ring carbon atoms C17, C19 and C21 attached with the nitrogen atoms N22 and N27 are positive while other carbon atoms are negative. Influence of methyl group electronic effect resulting from hyper conjugation interaction of methyl group of the dimethyl amino pyridine moiety causes negative charge as noticed in C28 and C32 atoms.
4.9. MEP analysis
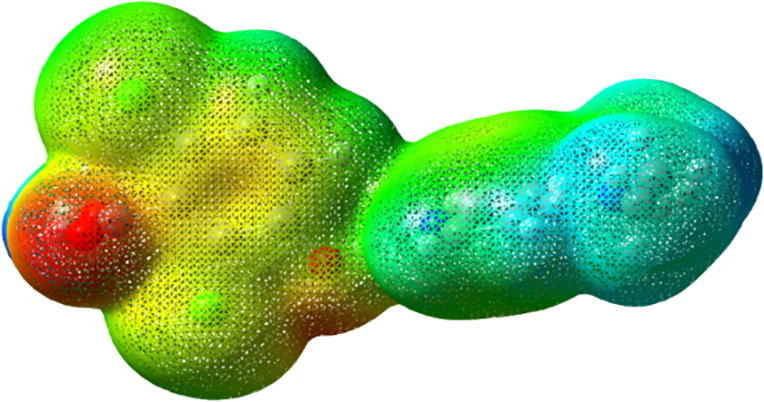
MEP is very important tool to investigate and correlate between the molecular structure and the physiochemical property relationship of molecules including biomolecules and drugs. Knowledge of charge distribution can be used to arbitrate how molecules interact with one another and also used to identify sites for intra and intermolecular interactions [55].
MEP plot for DADS calculated by B3LYP/6-311G** method depicted in Fig. 8 and colour range for the potential surface varies from −0.105 e to 0.105 e. Blue colour in MEP connotes the truancy of electrons in which electrostatic potential is high, red colour manifest the existence of electrons where electrostatic potential is low and green colour bespeak intermediate potential energy [56], [57].
Fig. 8.
Molecular Electrostatic potential plot of DADS.
MEP of DADS exhibit that positive potential is spread over the hydrogen atoms of dimethyl amino pyridine and negative potential is spread over carboxylic and hydroxyl group. Blue region is the locale for nucleophilic attack where strong repulsion takes place and electrophilic attack takes place around the oxygen atoms. From MEP of DADS it is concluded that electronegativity difference is small for DADS molecule since the molecule has many charges spread around the intermediate potential region. Reactive areas of the compound identified based on electron and proton interactions confirm the biological activity of DADS.
4.10. Fukui function
Electron density-based local reactivity descriptors such as Fukui functions were proposed to provide information about chemical selectivity or reactivity at a particular site of a chemical system [58]. Condensed Fukui functions are investigated to indicate the dexterity of each atom in a molecule to serve as a reactive site. Morrel et al have proposed a new dual descriptor Δf(r) for the identification of molecular site prone to electrophilic and nucleophilic attack [59].
The isosurface plot for f-, f+, f0 and dual descriptor are shown in Fig. 9 and Fukui functions are portrayed in Table S5. From the isosurface plot blue colour bespeak negative region prone to electrophilic attack and nucleophilic attack is represented by the positive green region. Phenyl ring is prone to electrophilic attack owing to the delocalized pi electrons which behaves as source of electrons and the chlorine atoms (Cl9 & Cl10) attached to the phenyl ring are the most favourable site for electrophilic attack. Due to the electronegative nitrogen atom pyridine ring is electron deficient and it is more prone to nucleophilic attack. MEP map also confirms that pyridine ring is prone to nucleophilic attack and nucleophilic attack takes around the oxygen atoms. The electronegativity of the carbon, oxygen and nitrogen atoms are greater than the hydrogen atoms, making the hydrogen atoms electron deficient and prone to nucleophilic attack [60]. But in DADS H7, H8, H12, H29, H33 & H35 attached to the carbon atoms of phenyl ring and amino group act as a nucleophile due to the electron density around the carbon atoms. The reactivity descriptors of DADS act as a forerunner to uphold the biological activity of the compound through molecular docking [54].
Fig. 9.

Isosurface plot for the f-,f+,f0 and dual descriptor.
4.11. ELF and LOL analysis
ELF and LOL are studies which predominantly manifest the surface of topological analysis. ELF and LOL reveal high probability of finding an electron pair on molecular surface are carried out by Multiwfn software package. The colour shaded map and relief map of ELF and LOL are portrayed in Fig. 10 where the relief map with large or contracted peak area delineates the electron environment around each atom.
Fig. 10.

Colour filled and Relief map for ELF and LOL.
ELF and LOL have homogenous chemical content because they hinge on kinetic energy density. ELF explains the electron pair density whereas LOL explains maximum localized orbitals overlapping due to gradient of orbitals [7]. ELF map is designed in the range 0.0 to 1.0; however region below 0.5 shows delocalized electronic region [60] and the LOL attains large values > 0.5 in regions where electron density is dominated by electron localization [61].
Colour shades of ELF and LOL maps confirm the existence of bonding and nonbonding electrons, where the red colour around hydrogen atoms with maximum value bespeak the presence of bonding and nonbonding electrons. High ELF or LOL values indicated by red colour around hydrogen atoms shows high localization of electrons due to the ubiety of covalent bond, a lone pair of electrons or a nuclear shell in that region. Blue colour around carbon atoms in phenyl and pyridine rings evince the delocalized electron cloud around it whereas blue circle around nitrogen and chlorine atoms bespeak the region of electronic depletion between the inner layer and valence layer. The central region of hydrogen atom in the LOL is white as the electron density exceeds the upper limit of colour scale.
4.12. NMR analysis
NMR technique elucidate about the unknown structural, chemical and magnetic properties of organic molecules [62]. The combined use of NMR and computer simulation methods offers a powerful way to interpret and predict the structure of large biomolecules [63]. Experimental 13C and 1H NMR spectra were recorded in DMSO solvent and calculated in the same solvent using GIAO method. The experimental 13C and 1H NMR spectra are depicted in Fig. 11 and chemical shift values are portrayed in Table 6 . Chemical shifts were recorded in DMSO using TMS as internal reference and isotropic shielding values of TMS are 31.882 ppm in 1H NMR and 180.466 ppm in 13C NMR.
Fig. 11.
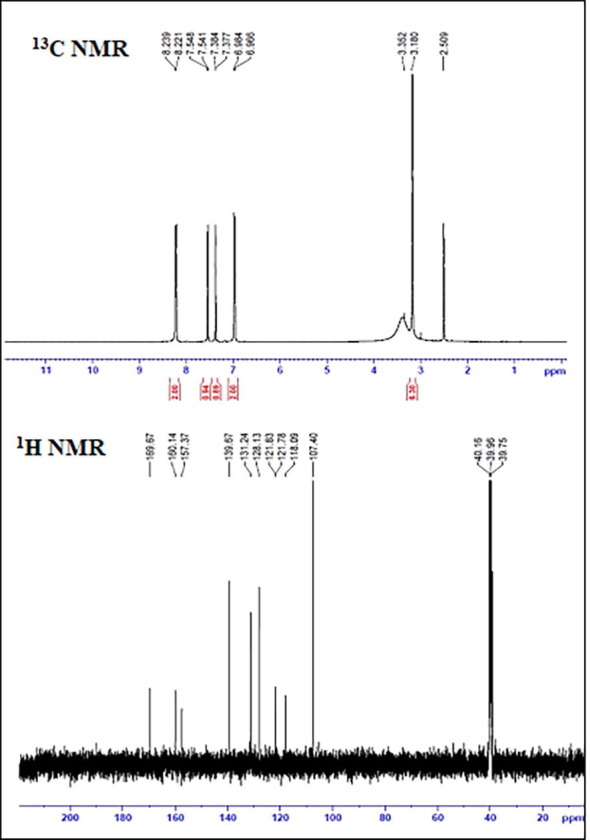
Experimental 13C NMR & 1H NMR isotropic chemical shift of DADS.
Table 6.
Experimental and calculated 1H and 13C NMR isotropic chemical shifts (ppm) of DADS.
| Atoms | Experimental(ppm) | Calculated(ppm) | Atoms | Experimental(ppm) | Calculated(ppm) |
|---|---|---|---|---|---|
| C13 | 160.07 | 169.67 | H12 | 13.23 | 8.23 |
| C2 | 160.75 | 160.14 | H23 | 8.27 | 8.22 |
| C19 | 157.88 | 157.37 | H26 | 8.27 | 7.54 |
| C21 | 150.68 | 154.38 | H7 | 7.07 | 7.54 |
| C17 | 150.62 | 144.78 | H8 | 7.07 | 7.38 |
| C4 | 138.02 | 139.67 | H24 | 6.56 | 7.37 |
| C6 | 133.08 | 131.24 | H16 | 6.56 | 6.98 |
| C5 | 128.97 | 128.13 | H25 | 6.56 | 6.96 |
| C3 | 127.20 | 121.83 | H35 | 3.07 | 3.35 |
| C1 | 119.51 | 121.78 | H31 | 3.07 | 3.35 |
| C20 | 109.79 | 118.09 | H29 | 3.07 | 3.18 |
| C18 | 109.69 | 107.40 | H33 | 3.07 | 3.18 |
| C28 | 40.45 | 40.16 | H30 | 2.85 | 2.50 |
| C32 | 40.24 | 39.96 | H34 | 2.85 | 2.50 |
NMR signals for same nuclei normally appear at different ppm values as each nucleus in the molecule is chemically different from other due to the surrounding electrons and the nearby atoms [64]. Literature reported that chemical shift value for aromatic carbon should be>100 ppm [65], [66] and in DADS there are 11 aromatic carbons present, six of which is present in benzene ring and remaining in pyridine ring lie within the range of 100–160 ppm. The carbon atoms attached to the carboxylic and hydroxyl group is found to have upfield chemical shift of 160.07(169.67) ppm and 160.75(160.14) ppm owing to removal of electron density. The chemical shift value of methylene carbon C28 and C32 found to be 40.45 (40.16) ppm and 40.24(39.96) ppm is highly shielded owing to the ubiety of electronegative atoms.
The proton NMR signal of organic compounds typically ranges 0–12 ppm [65] and for DADS the aromatic protons H8 and H7 found to have a ppm scale of 7.07 which agrees with the literature value. From the Table 6 it is found that some proton signals have same chemical shift because of same surrounding environment. The proton of hydroxyl group H12 have high ppm scale of 13.23(8.23) due to removal of electron density since these atoms are attached with high electronegative oxygen atom and the deviation in chemical shift is due to the hydrogen bonding interaction. H atom is smallest of all atoms and mostly localized on the periphery of molecule, therefore chemical shifts would be more susceptible to intermolecular interactions in the aqueous solution as compared to that of heavier atom. The methyl group attached to the ring have electron donating resonance effect and shows a substantial lowering in chemical shift [67].
4.13. NCI analysis
NCI analysis is implemented to explore the intra and inter non-covalent interaction in a molecular system. The region of these interactions and their graphical visualization is provided using the Reduced density gradient (RDG) analysis based on the electron density and its derivatives.
Graphical representation of ρ(r) versus sign (λ2) ρ, where sign (λ2) ρ is the second eigen value of electron density, provides convenient information regarding strength and nature of interactions. The repulsive, attractive and van der Waals (VDW) interactions correspond to sign(λ2) ρ > 0, sign(λ2) ρ < 0 and sign(λ2) ρ≈0, respectively [68], [69]. 2D scatter plot and the 3D RDG isosurface densities of DADS are depicted in Fig. 12 .
Fig. 12.
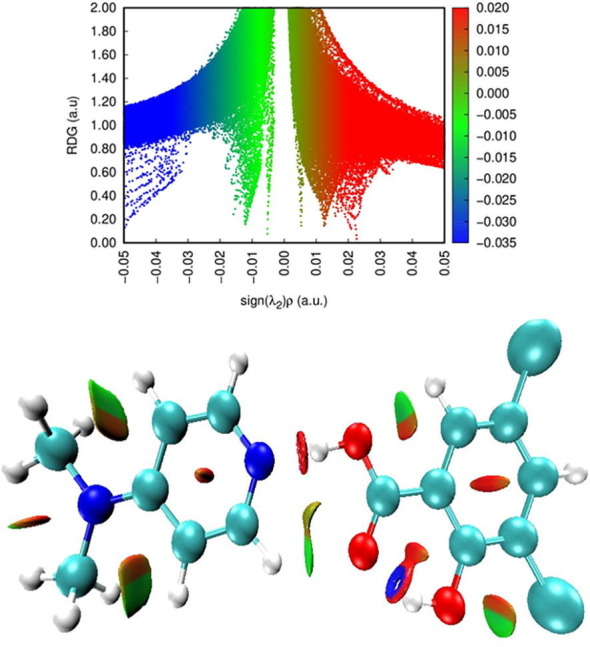
2D scatter and Isosurface density plots illustrating the non-bonded interactions of DADS.
From the RDG isosurface red flaky patches in the centre of phenyl ring and pyridine ring implies strong steric effect and the red spikes between 0.010 and 0.020 a. u in the scatter plot confirms this steric effect. Blue colour patches indicates stronger attractive interaction corresponding to strong hydrogen bonds (N-H…O and O-H…O) and this strong interaction is also seen from the RDG plot between the negative region −0.020 to −0.035 a.u. This hydrogen bond interaction is also evident from the bond length of N22-H16 …O15 and NBO analysis which transfers charge from n1 (N22) → σ*(O15–H16) confirms the biological activity of DADS molecule. Green flaky patches bespeak the presence of weak non-covalent H…H interaction which is also seen from the RDG graph where more green spikes appear between 0.005 to – 0.015 a. u.
4.14. Thermal analysis (TG-DTA)
Thermal characteristics of DADS crystal was studied by using TG-DTA under nitrogen atmosphere at a heating rate of 10 °C/min from 30 to 500 °C and TG-DTA thermogram is depicted in Fig. 13 . DADS were stable up to 180 °C and single stage decomposition observed from 200 to 275 °C. There is gradual weight loss taking place after 200 °C due to liberation of volatile substances such as CO, CO2, H2O, and CH4. Endothermic dip at 198 °C reveals the melting point of crystal which is equal to the melting point determined from conventional method. Broad endothermic dip at 275 °C divulges complete decomposition of compound. Thermal analysis validates the applicability of DADS for any biological applications.
Fig. 13.
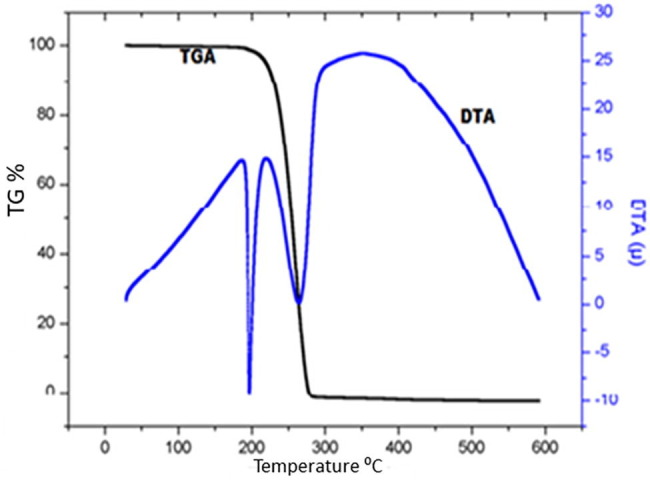
TG-DTA thermogram.
4.15. Thermodynamic properties
It is important to have knowledge on the temperature dependence of titled compound since the reactivity of compound may vary with temperature [70]. Standard statistical thermodynamic functions such as heat capacity, entropy, enthalpy changes were computed. Thermodynamic properties for various temperature from 100 K to 1000 K is tabulated in Table S6 and the correlation graph between these thermodynamic properties and temperature T are shown in Fig. 14 . It is observed from the table that gradual increase in thermodynamical parameters with respect to temperature is due to equipartition energy. The gradual increase in heat capacity shows that system attains more kinetic energy to improve the strength of the vibrations and enthalpy change produce the chemical reaction in DADS [71].
Fig. 14.
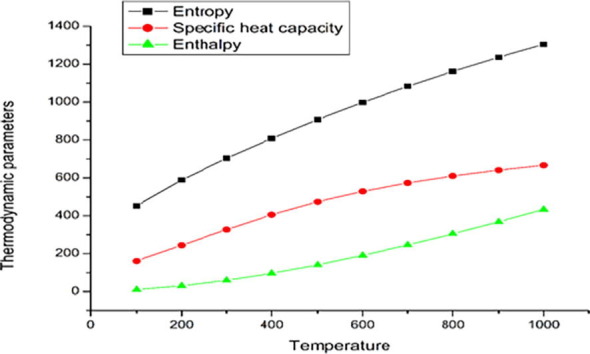
Correlation graph between thermodynamic properties and temperature.
The linear fitting equations with fitting factors (R2) are deduced for the three Thermodynamical parameters. The fitting equations are
Entropy y = 329.21 + 1.3408 T – 3.6921 10-4 T2 [R2 = 0.99974]
Heat capacity y = 57.6411 + 1.0423 T – 4.35275 10-4 T2 [R2 = 0.99969]
Enthalpy y = -12.6266 + 0.16756 T + 2.8213 10-4 T2 [R2 = 0.99961]
The thermodynamic data supply useful information for further study of DADS and systematic literature reports reveal that the entropy (S), which focuses on in a biomedical system, drives the direction of all chemical reactions towards disorder. The inferences obtained from above thermodynamic parameters (S, Cp, H) may help to obtain other relation of thermodynamic energies [72] respectively. Together, thermodynamic studies will progress fundamental understanding of molecular interactions and will enable new and improved strategies for successful drug discovery.
4.16. Molecular docking
Molecular docking is an alluring scaffold to know about the drug bio molecular interactions for the rational drug design and discovery. It predicts the binding mode and type of interaction between active site proteins and ligand along with their distance and vicinity of functional groups involved [73].
In order to study the antiviral activity of the DADS docking was done into the active sites of the target proteins namely SARS- CoV-2 proteins (6 M03, 6M2N, 7CHF), MERS virus protein (6VGY) and the docking parameters such as binding energy, inhibition constant are listed in Table 7 . PDB structures of the target protein are downloaded from the RCSB protein data bank (https://www.rcsb.org/pdb/home.do). The autodock tools graphical user interface was used to prepare the protein by removing water, adding polar hydrogens, required charges and the preparation of ligand with minimum energy for docking was done. Molecular docking of ligand DADS with the SARS-CoV-2 and MERS virus proteins are depicted in Fig. 15 .
Table 7.
Molecular Docking constraints of DADS with target proteins.
| Protein | Bonded residue | Bonded distance Å |
Binding energy kcal/mol |
Inhibition constant(Ki) milli Molar(mM) |
Reference RMSD Å |
|---|---|---|---|---|---|
| 6M2N | TRP’207 | 2.0 | −2.66 | 11.14 | 64.55 |
| 7CHF | TYR’198 | 2.1 | −2.4 | 17.42 | 34.91 |
| 6VGY | VAL’205 | 2.4 | −2.77 | 9.29 | 24.74 |
| 6 M03 | TRP’207 | 1.7 | −2.54 | 13.83 | 6.19 |
Fig. 15.

Molecular docking of DADS with SARS- CoV-2 and MERS proteins.
Docked conformations corresponding to the lowest binding energy was chosen to study the mode of binding. The binding energy of the DSDA molecule bounded with 6M2N, 7CHF, 6VGY and 6 M03 were found to be −2.66 kcal/mol, −2.4 kcal/mol, −2.77 kcal/mol and −2.54 kcal/mol respectively. It is evident from Fig. 13 that the hydrogen bonds are formed between the hydrogen and nitrogen atom of the ligand and proteins. The docking conformation of ligand with protein reveals that the electronegative nitrogen atom is the only site undergoing hydrogen bond formation. This hydrogen bond interaction which confirms the bioactivity of the compound is also evident from the deviation in bond length of N22 - H16….…O15, charge transfer from n1(N22) → σ* (O15–H16) in NBO analysis and also from NCI analysis. Charge transfer in frontier orbital molecules is also evident for the intermolecular interaction which confirms the bioactivity.
Ramachandran plot for four docked SARS-CoV-2 and MERS virus protein portrayed in Fig. 16 evinces 90% of the amino acids lie in allowed region represented by red colour and only very few lie in disallowed region which indicates the stability of the protein chosen for the binding interaction [74].
Fig. 16.

Ramachandran plot of SARS- CoV-2 proteins.
The antiviral activity of DADS is confirmed in virtue of its good binding affinity with SARS-CoV-2 and MERS virus proteins and so it is suggested that DADS can be a good candidate for the treatment of certain viral disease including Corona diseases.
4.17. Drug likeness
The feedback on drug likeness constraints directs the non-toxic and organically dynamic nature of the titled compound. Assessment of drug likeness is influential to find out the compounds which are not ample to be a dynamic drug. There are few important parameters used in identifying the drug likeness property of the compound such as hydrogen bond acceptors (HBA) number, hydrogen bond donors (HBD) number, rotatable bonds, logP, and molar refractivity [75]. All of these parameters constitute the drug likeness test and the values corresponding to the antiviral drug DADS is tabulated in Table 8 .
Table 8.
Drug likeness parameters of DADS.
| Descriptors | Calculated | Expected |
|---|---|---|
| Molecular mass(Dalton) | 329 | <500 |
| Hydrogen bond donor | 2 | <5 |
| Hydrogen bond acceptor | 5 | <10 |
| Log P | 2.9 | <5 |
| Molar refractivity | 81.05 | 40–130 |
The constraints of drug likeness were predictable to yield standards within the superior range conferring to Lipinski’s rule of five. The number of hydrogen bond donors and hydrogen bond acceptors for DADS found to be 2 and 5 respectively satisfies the Lipinski’s rule of five. The values of log P was recognised to be 2.9, which is a pointer of hydrophobic/lipophilic nature of compound and Molar refractivity found to be 81.05 is perceived to be within the appropriate range [76], [77].
Since DADS obeys the Lipinski’s rule of five it is confirmed that it is an active potential drug and has drug properties. So DADS is suggested to be used as an antiviral drug to treat certain viral diseases. Molecular docking studies also confirm that DADS is preferable to be used as an antiviral drug ascribed to its good binding affinity towards SARS- CoV-2 and MERS virus proteins.
5. Conclusion
In the present work a complete study on the structural, vibrational, electronic, topology and thermodynamic properties of DADS was carried out with the aid of quantum chemical calculations. The structural optimization of DADS was conceded out by B3LYP method and geometrical parameters are calculated. Intermolecular interaction which confirms biological activity of compound is found from the deviation in bond length of N22-H16…O15. Presence of intermolecular hydrogen bonding which confirms the nature and properties of biomolecules is also studied by the NBO analysis. Detailed interpretation of vibrational assignments were done with the aid of NCA and the FT-IR, Raman spectra of DADS were recorded and analysed. The frontier orbital gap calculated as 4.9329 eV indicates that DADS is more polarizable and is generally associated with a high chemical reactivity. The charge transfer in molecular orbitals is responsible for the bioactive property for the biomedical compound. The electronic properties were studied theoretically using TD-DFT method and experimentally by observing UV spectrum band where the energy gap calculated from the UV spectrum agrees with the HOMO-LUMO energy gap. Electron distribution and reactive sites on the surface were analysed using ESP, ELF and LOL. HOMA index value confirms the aromatic character of the compound and the difference in HOMA index value in the phenyl ring and pyridine ring is due the substitutions in the place of hydrogen. From the correlation between the Thermodynamical parameters and temperature it is seen that thermodynamic parameters increase with increasing temperature since the molecular vibrational intensities increases with temperature. NCI approach analyses the weak attractive interactions, strong attraction, and steric repulsion existed in the title compound. Molecular docking of DADS is carried out with SARS-CoV-2 and MERS virus proteins affirms that DADS act as a good antiviral agent and further drug likeness studies also supports that DADS can be used as an active drug to treat certain viral diseases including Covid-19 virus disease.
CRediT authorship contribution statement
J.D. Deephlin Tarika: Conceptualization, Data curation, Writing - Original draft. X.D. Divya Dexlin: Data curation. S. Madhankumar: Provision of samples. D. Deva Jayanthi: Conceptualization. T. Joselin Beaula: Supervision, Writing - review & editing.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgement
The authors express their gratitude to Dr. I. Hubert Joe, Associate Professor, Department of Physics, University of Kerala for providing us Gaussian software for Computational works.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.saa.2021.119907.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- 1.A. N. Hussein, O. F. Abdul Rasheed, M.F. Mahdi, A.M.R. Raauf, Molecular Docking Studies for Design, Synthesis and Characterization of New Imatinib Analogues, Pharm. Anal. Acta. 10(4) (2019) 616,doi: 10.35248/2153-2435.19.10.616.
- 2.Hamada Y. InTech; 2018. Pyridine. 10.5772/intechopen.74719. [Google Scholar]
- 3.Altaf Ataf Ali, et al. A Review on the Medicinal Importance of Pyridine Derivatives. J. Drug Desi. Medi. Chemistry. 2015;1(1):11. doi: 10.11648/j.jddmc.20150101.11. [DOI] [Google Scholar]
- 4.Madan R.K., Levitt J. A review of toxicity from topical salicylic acid preparations. J. Am. Acad. Dermatol. 2014;70(4):788–792. doi: 10.1016/j.jaad.2013.12.005. [DOI] [PubMed] [Google Scholar]
- 5.Bandurska H. SALICYLIC ACID. Springer; Dordrecht: 2013. Salicylic acid: An update on Biosynthesis and Action in plant response to water deficit and performance under drought; pp. 1–14. [Google Scholar]
- 6.A.C. Vlot,A. Dempsey, D.F. Klessig, Salicylic Acid, a MultifacetedHormone to Combat Disease Annual review of phytopathology (2009) 177-206, https://doi.org/10.1146/annurev.phyto.050908.135202 [DOI] [PubMed]
- 7.Silvi B., Savin A. Classification of chemical bonds based on topological analysis of electron localization functions. Nature. 1994;371(6499):683–686. doi: 10.1038/371683a0. [DOI] [Google Scholar]
- 8.Lu T., Chen F. Multiwfn: a multifunctional wavefunction analyzer. J. Comput. Chem. 2012;33(5):580–592. doi: 10.1002/jcc.v33.510.1002/jcc.22885. [DOI] [PubMed] [Google Scholar]
- 9.Becke A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993;98(7):5648–5652. [Google Scholar]
- 10.M.J. Frisch, G.W. Trucks, H.B. Schlegel, G.E. Scuseria, M.A. Robb,J.R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G.A. Petersson,H. Nakatsuji, M. Caricato, X. Li, H.P. Hratchian, A.F. Izmaylov, J. Bloino,G. Zheng, J.L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda,J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven,J.A. Montgomery Jr., J.E. Peralta, F. Ogliaro, M. Bearpark, J.J. Heyd, E. Brothers,K.N. Kudin, V.N. Staroverov, T. Keith, R. Kobayashi, J. Normand,K. Raghavachari, A. Rendell, J.C. Burant, S.S. Iyengar, J. Tomasi, M. Cossi,N. Rega, J.M. Millam, M. Klene, J.E. Knox, J.B. Cross, V. Bakken, C. Adamo,J. Jaramillo, R. Gomperts, R.E. Stratmann, O. Yazyev, A.J. Austin, R. Cammi,C. Pomelli, J.W. Ochterski, R.L. Martin, K. Morokuma, V.G. Zakrzewski,G.A. Voth, P. Salvador, J.J. Dannenberg, S. Dapprich, A.D. Daniels, O. Farkas,J.B. Foresman, J.V. Ortiz, J. Cioslowski, D.J. Fox, Gaussian 09, Revision C.02,Gaussian Inc., Wallingford CT, 2010.
- 11.E.D. Glendening, A.E. Reed, J.E. Carpenter, F.Weinhold, NBO Version 3.1, TCI, Universityof Wisconsin, Madison, 1998.
- 12.Sundius T. Scaling of ab initio force fields by MOLVIB. Vib. Spectrosc. 2002;29(1-2):89–95. doi: 10.1016/S0924-2031(01)00189-8. [DOI] [Google Scholar]
- 13.Sundius T. Molvib - a flexible program for force field calculations. J. Mol. Struct. 1990;218:321–326. doi: 10.1016/0022-2860(90)80287-T. [DOI] [Google Scholar]
- 14.P. Pulay, G. Fogarasi, G. Pongor, J.E. Boggs, A. Vargha, Combination of theoretical ab initio and experimental information to obtain reliable harmonic force constants, J.Am. Chem. Soc. 105 (1983) 7037, https://doi.org/10.1021/ja00362a005.
- 15.GaussView, Version 6.1, Roy Dennington, Todd A. Keith, and John M. Millam, Semichem Inc., Shawnee Mission, KS, 2016.
- 16.Humphrey W., Dalke A., Schulten K. VMD: visual molecular dynamics. J. Mol. Graph. 1996;14(1):33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 17.K. K. Irikura, Thermo.pl. National Institute of Standards and Technology (2002).
- 18.Morris G.M., Huey R., Lindstrom W., Sanner M.F., Belew R.K., Goodsell D.S., Olson A.J. Autodock4 and AutoDockTools4: automated docking with selec- tive receptor flexiblity. J. Comput. Chem. 2009;16(2009):2785–2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.G.M. Morris, D.S. Goodsell, R.S. Halliday, R. Huey, W.E. Hart, R.K. Belew, A.J. Olson,Automated docking using a lamarckian genetic algorithm and empirical bindingfree energy function, J. Comput. Chem. 19 (1998) 1639–1662, https://doi.org/10. 1002/(SICI)1096-987X(19981115)19:14%3C1639.
- 20.Stato H., Dybal J., Murakami R., Noda I., Ozaki Y. Infrared and Raman spectroscopy and quantum chemistry calculation studies of C-H⋯O hydrogen bondings and thermal behavior of biodegradable polyhydroxyalkanoate. J. Mol.Struct. 2005;35:744–747. doi: 10.1016/j.molstruc.2004.10.069. [DOI] [Google Scholar]
- 21.P. Novak, T. Jednacak, in: Z. Mandic (Ed.), Physico Chemical Methods in DrugDiscovery and Development, IAPC Publishing, Zagreb 85(4) (2012) https://doi.org/10.5562/cca2123.
- 22.Abeydeera N. Dinuka, Chow Christine S. Synthesis and characterization ofmodified nucleotides in the 970 hairpin loop of Escherichia coli 16S ribosomalRNA. Bioorg. Med. Chem. 2009;17(16):5887–5893. doi: 10.1016/j.bmc.2009.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Beaula T. Joselin, Muthuraja P., Dhandapani M., Jothy V. Bena. Effect of charge transfer with spectral analysis on the antibacterial compound 4-(Dimethyl amino) pyridine: 3,5-Dinitrobenzoic acid: Experimental and theoretical perspective. J. Mol. Struct. 2018;1171:511–526. doi: 10.1016/j.molstruc.2018.06.026. [DOI] [Google Scholar]
- 24.J. Huheey,1972, Inorganic chemistry: Principles of Structure and Reactivity pps. A-21 to A-34
- 25.Cottrell T.L. second ed. Butterworths; London: 1958. The Strengths of Chemical Bonds. [Google Scholar]
- 26.Arslan Hakan, Flörke Ulrich, Külcü Nevzat, Binzet Gün. The molecular structure and vibrationalspectra of 2- Chloro-N-(diethylcarbamothioyl)benzamide by Hartree-Fock and density functional methods. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2007;68(5):1347–1355. doi: 10.1016/j.saa.2007.02.015. [DOI] [PubMed] [Google Scholar]
- 27.Gillespie R.J. The VSEPR model revisited. Chem. Soc. Rev. 1992;59:59–69. doi: 10.1039/CS9922100059. [DOI] [Google Scholar]
- 28.Velraj G., Soundharam S., Sridevi C. Investigation of structure, vibrational, electronic, NBO and NMR analyses of 2-chloro-4-nitropyridine (CNP), 2-chloro-4-methyl-5-nitropyridine (CMNP) and 3-amino-2-chloro-4-methylpyridine (ACMP) by experimental and theoretical approach’. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2015;137:790–803. doi: 10.1016/j.saa.2014.08.075. [DOI] [PubMed] [Google Scholar]
- 29.J. A Joule, K. Mills (2010). Heterocyclic Chemistry, 5th ed Chichester 2010.
- 30.https://en.wikipedia.org/wiki/Pyridine.
- 31.Balachandran V., Rajeswari S., Lalitha S. DFT computations, vibrational spectra, monomer, dimer, NBO and NMR analyses of antifungal agent: 3,5-Dibromosalicylic acidJ. Mol. Struct. 2012;1007:63–73. doi: 10.1016/j.molstruc.2011.10.014. [DOI] [Google Scholar]
- 32.Arivazhagan M., Kavitha R. FT-IR, FT-Raman and theoretical NBO, NLO and MESP analysis of 4-bromoveratrole by ab-initio HF and DFT methods. Ind. J. Pure Appl. Phys. 2012;50:709–718. http://hdl.handle.net/123456789/14778 [Google Scholar]
- 33.Jenepha Mary S.J., Siddique Mohd Usman Mohd, Pradhan Sayantan, Jayaprakash Venkatesan, James C. Quantum chemical insight into molecular structure, NBO analysis of the hydrogen-bonded interactions, spectroscopic (FT–IR, FT–Raman), drug likeness and molecular docking of the novel anti COVID-19 molecule 2-[(4,6-diaminopyrimidin-2-yl)sulfanyl]-N-(4-fluorophenyl)acetamide dimer. Spectrochim. Acta A. 2021;244 doi: 10.1016/j.saa.2020.118825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zacharias Adway Ouseph, Varghese Anitha, Akshaya K.B., Savitha M.S., George Louis. Louis George, DFT, spectroscopic studies, NBO, NLO and Fukui functional analysis of 1-(1-(2,4-difluorophenyl)-2-(1H–1,2,4-triazol-1-yl)ethylidene) thiosemicarbazide. J. Mol. Struct. 2018;1158:1–13. doi: 10.1016/j.molstruc.2018.01.002. [DOI] [Google Scholar]
- 35.Socretes G. third ed. Wiley; Chichester: 2001. Infrared and Raman characteristic Group Frequencies: Tables and Charts. [Google Scholar]
- 36.Chandra S., Saleem H., Sundaraganesan N., Sebastian S. The spectroscopic FT-IR gas phase, FT- IR, FT- Raman, polarizabilities analysis of Naphthoic acid by DFT methods. Spectrochim. Acta A. 2009;74(2009):704–713. doi: 10.1016/j.saa.2009.07.025. [DOI] [PubMed] [Google Scholar]
- 37.K.R. Santhy , M. Daniel Sweetlin, S. Muthu d, Christina Susan Abraham, M. Raja, Molecular structure, spectroscopic (FT-IR, FT-Raman) studies, Homo–Lumo and Fukui function calculations of 2-Acetyl amino-5-bromo- 4 methyl pyridine by density functional theory, Chem. Data Collec. 24 (2019) 100291, https://doi.org/10.1016/j.cdc.2019.100291.
- 38.Madhavan V.S., Varghese H.T., Mathew S., Vinsova J., Panicker C.Y. ‘FT IR, FT Raman and DFT calculation of 4 chloro 2-(3, 4- dichlorophenylcarbamoyl) phenyl acetate. Spectrochim. Acta A. 2009;72:547–553. doi: 10.1016/j.saa.2008.10.061. [DOI] [PubMed] [Google Scholar]
- 39.Hiremath C.S., Yenagi J., Tonannavar J. FT-Raman and infrared spectra and vibrational assignments for 3- chloro 4- methoxybenzaldehyde, as supported by ab initio, hybrid DFT and normal coordinate calculations. Spectrochim. Acta A. 2007;68:710–717. doi: 10.1016/j.saa.2006.12.050. [DOI] [PubMed] [Google Scholar]
- 40.Omidvar Bibi Amineh, Tayyari Sayyed Faramarz, Vakili Mohammad, Nekoei Abdo-Reza. Sayyed Faramarz Tayyari, Mohammad Vakili, Abdo-Reza Nekoei, ‘Vibrational spectra, normal coordinate analysis, and hydrogen bond investigation of pyridinium perchlorate’. Spectrochim. Acta A. 2018;191:558–565. doi: 10.1016/j.saa.2017.10.067. [DOI] [PubMed] [Google Scholar]
- 41.Brian C. A Systematic Approach, CRC Press; Infrared Spectral Interpretation: 1998. Smith. [Google Scholar]
- 42.Vein D.L., Colthup N.B., Fatley W.G., Grasselli J.G. Academic press; New York: 1991. The Handbook of Infrared and Raman Characteristic Frequencies of Organic Molecules. [Google Scholar]
- 43.Colthup N.B., Daly L.H., Wiberly S.E. Academic press; Newyork: 1990. Introduction to Infrared and RamanSpectroscopy. [Google Scholar]
- 44.Varsanyi G. Adam Hilger; London: 1974. Assignments for Vibrational Spectra of Seven Hundred Benzene Derivaties. [Google Scholar]
- 45.Stuart B.H. John Wiley & Sons; England: 2004. Infrared Spectroscopy: Fundamentals and Applications. [Google Scholar]
- 46.Hernandez V., Castiglioni C., Zerbi G. Zerbi, Hyperconjugation from infrared intensities:the case of methyl acetate and of its selectively deuterated derivatives. G. J. Mol. Struct. 1994;324(1-2):189–198. doi: 10.1016/0022-2860(94)08239-1. [DOI] [Google Scholar]
- 47.De Proft F., Geerlings P. Conceptual and computational DFT on the study of aromaticity’. Chem Rev. 2001;101:1451–1464. doi: 10.1021/cr9903205. [DOI] [PubMed] [Google Scholar]
- 48.Beaula T. Joselin, Muthuraja P., Sethuram M., Dhandapani M., Rastogi V.K., Jothy V. Bena. Biological and spectral studies of O-Tolyl Biguanide: Experimental andtheoretical approach. J. Mol. Struct. 2017;1128:290–299. doi: 10.1016/j.molstruc.2016.08.060. [DOI] [Google Scholar]
- 49.Pearson R.G. Absolute electronegativity and hardness correlated with molecular orbital theory. Proc. Natl. Acad. Sci. 1986;83(22):8440–8441. doi: 10.1073/pnas.83.22.8440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Barim E., Akman F. Synthesis, characterization and spectroscopic investigation of N-(2-acetylbenzofuran-3-yl)acrylamide monomer: Molecular structure, HOMO- LUMO study, TD-DFT and MEP analysis. J. Mol. Struct. 2019;1195:506–513. doi: 10.1016/j.molstruc.2019.06.015. [DOI] [Google Scholar]
- 51.Mariana Rocha, Alejandro Di Santo, Juan Marcelo Arias, Diego M. Gil , Aída Ben Altabe, Ab-initio and DFT calculations on molecular structure, NBO,HOMO–LUMO study and a new vibrational analysis of 4-(Dimethylamino) BenzaldehydeSpectrochim. Acta A,73 136 (2015) 635–643, https://doi.org/10.1016/j.saa.2014.09.077. [DOI] [PubMed]
- 52.Ravikumar C., Hubert Joe I., Jayakumar V.S. Charge transfer interactions and nonlinear optical properties of push pull chromophore benzaldehyde phenylhydrazone: a vibrational approach’. Chem. Phys. Lett. 2008;460(4–6):552–558. doi: 10.1016/j.cplett.2008.06.047. [DOI] [Google Scholar]
- 53.Yang Weitao., Mortier Wilfried J. The use of global and local molecular parameters forthe analysis of the gas-phase basicity of amines. J. Am. Chem. Soc. 1986;108(19):5708–5711. doi: 10.1021/ja00279a008. [DOI] [PubMed] [Google Scholar]
- 54.P. Manjushaa, Johanan Christian Prasana, S. Muthu, B. FathimaRizwana, Spectroscopic elucidation (FT-IR, FT-Raman and UV-visible) with NBO,NLO, ELF, LOL, drug likeness and molecular docking analysis on 1-(2-ethylsulfonylethyl)-2-methyl-5-nitro-imidazole: An antiprotozoal agent, Comput. Bio.Chemistry, 88 (2020) 107330, https://doi.org/10.1016/j.compbiolchem.2020.107330. [DOI] [PubMed]
- 55.P.Politzer, M.C.Concha,J.S.Murray,DFT study of dimers of dimethylnitramine, Int.J.quant.chem,80(2000)184-192,https://doi.org/10.1002/1097-461X(2000)80:2<184::AID-QUA12>3.0.CO;2-O.
- 56.Hussein W., Walker C.G., PeraltaInga Z., Murray J.S. Compound electrostatic potentials and average local ionization energies on the molecular surfaces of some teracyclines. Int. J. Quantchem. 2001;82:160–167. doi: 10.1002/qua.1031. [DOI] [Google Scholar]
- 57.Mansour A.M. Molecular structure and spectroscopic properties of novelmanganese (II) complex with sulfamethazine drug. J. Mol. Struct. 2013;1035:114–123. doi: 10.1016/j.molstruc.2012.09.048. [DOI] [Google Scholar]
- 58.Yang W., Parr R.G. Hardness, softness, and the fukui function in the electronic theory of metals and catalysis Proc. Natl. Acad. Sci. U. S. A. 1985;82(20):6723–6726. doi: 10.1073/pnas.82.20.6723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Morell Christophe, Grand André, Toro-Labbé Alejandro. New dual descriptor forchemical reactivity. J. Phys. Chem. A. 2005;109(1):205–212. doi: 10.1021/jp046577a. [DOI] [PubMed] [Google Scholar]
- 60.Fathima Rizwana B., Prasana J.C., Muthu S., Abraham C.S. Molecular docking studies, charge transfer excitation and wave function analyses (ESP, ELF, LOL) on valacyclovir: A potential antiviral drug Comput. Biol. Chem. 2019;78:9–17. doi: 10.1016/j.compbiolchem.2018.11.014. [DOI] [PubMed] [Google Scholar]
- 61.Jacobsen Heiko. Localized-orbital locator (LOL) profiles of chemical bonding. Can. J. Chem. 2008;86(7):695–702. doi: 10.1139/v08-052. [DOI] [Google Scholar]
- 62.Shoba D., Periandi S., Boomadevi S., Ramalingam S., Fereyduni E. FT-IR, FT-Raman, UV, NMR spectra, molecular structure, ESP, NBO and HOMO–LUMO investigation of 2-methylpyridine 1-oxide: A combined experimental and DFT study. Spectrochim. Acta A. 2014;118:438–447. doi: 10.1016/j.saa.2013.09.023. [DOI] [PubMed] [Google Scholar]
- 63.Cavalli A., Salvatella X., Dobson C.M., Vendruscolo M. Vendruscolo, Protein structure determination from NMR chemical shifts Proc. Natl. Acad. Sci. USA. 2007;104(23):9615–9620. doi: 10.1073/pnas.0610313104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Alam M., Lee D.U. Synthesis, spectroscopic and computational studies of 2- (thiophen-2-yl)-2, 3-dihydro-1H-perimidine: an enzymes inhibition study. Comput. Biol. Chem. 2016;64(2016):185–201. doi: 10.1016/j.compbiolchem.2016.06.006. [DOI] [PubMed] [Google Scholar]
- 65.Pu Yunqiao, Chen Fang, Ziebell Angela, Davison Brian H., Ragauskas Arthur J. NMR characterization of C3H and HCT down-regulated alfalfa lignin. Bio energ. Res. 2009;2(4):198–208. doi: 10.1007/s12155-009-9056-8. [DOI] [Google Scholar]
- 66.H.O.Kalinowski, S. Berger, S. Braun, Carbon 13 NMR Spectroscopy, John Wiley and sons, Chichester,1988.
- 67.Manju Pandey, S.Muthu,N.M. Nanje Gowda, Quantum mechanical and spectroscopic (FT-IR, FT-Raman,1H,13C NMR, UV-Vis) studies, NBO, NLO, HOMO, LUMO and Fukui function analysis of 5-Methoxy-1H-benzo[d]imidazole-2(3H)-thione by DFT studies, J.Mol.Struct, 1130(2017) 511-521, https://doi.org/10.1016/j.molstruc.2016.10.064.
- 68.Saleh G., Gatti C., Lo Presti L. Non-covalent interaction via the reduced density gradient: independent atom model vs experimental multipolar electron densities. Comput. Theor. Chem. 2012;998:148–163. doi: 10.1016/j.comptc.2012.07.014. [DOI] [Google Scholar]
- 69.Johnson Erin R., Keinan Shahar, Mori-Sánchez Paula, Contreras-García Julia, Cohen Aron J., Yang Weitao. Revealing noncovalent interactions. J. Am. Chem. Soc. 2010;132(18):6498–6506. doi: 10.1021/ja100936w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bopp F., Meixner J., Kestin J. Academic Press Inc.; (London) Ltd, New York: 1967. Thermodynamics and Statistical Mechanics, fifthed. [Google Scholar]
- 71.S. Aayisha, T.S.Renugadevi,S.Janani, S. Muthu, M.Raja, R. Hemamalini, Structural (PES), AIM, spectroscopic profiling (FT-IR, FT-Raman, NMR and UV), HOMO-LUMO and docking studies of 2,2-dimethyl-N-(2-pyridinyl)propanamide – a DFT approach, chemi. data collec. 24(2019) 100287, https://doi.org/10.1016/j.cdc.2019.100287.
- 72.Zamani M., Shafiee M., Keshavarz M.H. IR, 1H NMR and DFT studies of novel bis-Betti base derivatives of 2,6-dihydroxynaphthalene: thermodynamic control of diastereoselectivity and configurational preference. J. Mol. Struct. 2014;1075:139–146. doi: 10.1016/j.molstruc.2014.06.072. [DOI] [Google Scholar]
- 73.Arulaabaranam K., Mani G., Muthu S. Computational assessment on wave function (ELF, LOL) analysis, molecular confirmation and molecular docking explores on 2-(5-Amino-2- Methylanilino)-4-(3-pyridyl) pyrimidine Chem. Data Collec. 2020;29:100525. doi: 10.1016/j.cdc.2020.100525. [DOI] [Google Scholar]
- 74.Laskowski, Roman Aleksander, M. W. Macarthur, D. S. Moss,Thornton, Janet, PROCHECK: a program to check the stereochemical quality of protein structures. J.Appl. Crystallogr. 26 (2), 283–291.1993.https://doi.org/10.1107/S0021889892009944
- 75.Lipinski C.A. Lead-and drug-like compounds: the rule-of-five revolution. DrugDiscov. Technol. 2004;1(4):337–341. doi: 10.1016/j.ddtec.2004.11.007. [DOI] [PubMed] [Google Scholar]
- 76.Walters W.P., Murcko M.A. Prediction of drug-likeness. Adv. Drug Deliv. Rev. 2002;54(3):255–271. doi: 10.1016/s0169-409x(02)00003-0. [DOI] [PubMed] [Google Scholar]
- 77.Clark D.E., Pickett S.D. Computational methods for the prediction of drug-likeness. Drug Discov. Today. 2000;5(2):49–58. doi: 10.1016/s1359-6446(99)01451-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.