

ABSTRACT
Lymph node (LN) is a key tissue in the pathophysiology of mature blood cancers, especially for chronic lymphocytic leukemia (CLL). Within the multiple de-regulated pathways affecting CLL homeostasis, the CC-chemokine receptor 7 (CCR7) grants homing of CLL cells into the LN where protective environments foster tumor progression. To cover the lack of specific therapies targeting the CCR7-dependence of CLL to enter into the LN, and aiming to displace the disease from LN, we generated CAP-100, an antibody that specifically binds to hCCR7 and neutralizes its ligand-binding site and signaling. In various in vitro and in vivo preclinical models CAP-100 strongly inhibited CCR7-induced migration, extravasation, homing, and survival in CLL samples. Moreover, it triggered potent tumor cell killing, mediated by host immune mechanisms, and was effective in xenograft models of high-risk disease. Additionally, CAP-100 showed a favorable toxicity profile on relevant hematopoietic subsets. Our results validated CAP-100 as a novel therapeutic tool to prevent the access of CLL cells, and other neoplasia with nodal-dependence, into the LN niches, thus hitting a central hub in the pathogenesis of cancer. The first-in-human clinical trial (NCT04704323), which will evaluate this novel therapeutic approach in CLL patients, is pending.
KEYWORDS: CCR7, CLL, antibody, CAP-100, immunotherapy, lymph node
Introduction
Lymph nodes (LN) function as a major immunological hub, essential for immune homeostasis,1 yet LNs are also the first site of metastasis for many cancers and a key tissue in the progression and treatment failure of chronic lymphocytic leukemia (CLL), the most common leukemia in western countries.2 A hallmark of this still incurable disease is the accumulation in peripheral blood (PB) of malignant cells in a cell cycle-arrested phase whereas cells proliferate in proliferation centers (PCs) mainly found in LN, and to a lesser extent in the spleen and bone marrow (BM).3,4 Therefore, the pathophysiology of CLL is entwined with the LN tumor microenvironment (TME) in which accessory cells or soluble factors promote disease progression and prevent spontaneous or drug-induced cell death.
In homeostasis and cancer, the chemokine receptor (CKR) CCR7, but not others, specifically drives cell homing into LN and other secondary lymphoid organs (SLO).5–7 This receptor orchestrates cell trafficking-positioning and activation upon binding the ligands CCL19 and CCL21, constitutively expressed by stroma cells in SLOs.8,9 In accordance with the LN dependence of CLL, CCR7 surface over-expression has been consistently reported in nearly all patients, regardless of the stage at diagnosis, adverse prognostic factors (e.g., del17p, IgVH mutational status) or previous treatments.10,11,12,13.14 CCR7 surface levels are higher than other targets such as CD2012 and, opposite to this lineage-specific antigen, CCR7 over-expression is critical for CLL cell trafficking, firm arrest, and extravasation through high endothelial venules (HEVs).10,11,12–17 CCR7 also guides CLL cells within the LN parenchyma toward niches where they find protective stimuli provided by accessory cells, including CCR7 ligands.7,13,18–20 Therefore, it is not surprising that increased CCR7 expression, ligand plasma levels, or high CCR7-induced migratory responses all strongly correlated with lymphadenopathy and aggressive CLL.10,11,13,21 This collection of evidence suggested that tools targeting CCR7 would be appealing to inhibit cell interaction with the LN microenvironment, thus hitting the “Achilles’ heel” of CLL. Similarly, driving leukemic cells out of LN to induce “death by neglect” is the mode of action (MOA) of ibrutinib, although complete response to this compound is only achieved in a small proportion of patients, and an increasing number do not respond or they relapse.2,22,23
CCR7 inhibition can be achieved by means of pharmacological inhibition with monoclonal antibodies (mAb)24 or small molecules.25 The limited specificity and affinity of small molecules, their inability to induce host anti-tumor responses, and their short serum half-life26 had earlier led us to hypothesize that a higher efficacy at reducing LN tumor burden would be achieved by a neutralizing anti-CCR7 mAb able to immobilize cancer cells and to elicit cell killing.12,24,27 Nonetheless, raising blocking antibodies against human CCR7 (hCCR7) is a challenging task because of its complex transmembrane structure; the necessity of targeting specific epitopes involved in ligand binding; and the high sequence homology between human and mouse CCR7 that impairs immunogenicity.26 Here, we used synthetic peptide mimics of an optimal CCR7 immunogen to generate CAP-100, a novel humanized IgG1 anti-hCCR7 blocking antibody, specifically aimed for cancer therapy, and evaluated its anti-tumor activity and elucidated its main MOA.
Results
Generation of CAP-100
A therapeutic anti-CCR7 antibody designed to displace tumor cells from LN must block the target/ligand interaction, through the antigen binding fragment (Fab). To secure CCR7-specific antagonist antibodies, we generated a library of linear and native epitope mimics (immunogens) containing the ligand binding site in the extracellular N-terminal domain of hCCR7 (amino acid sequence 36–52: NTTVDY{Z}TLFESLCSKKK; GenBank-EAW60669.1). Since this binding site includes a tyrosine residue in position 41 that can be found sulfated (sY = Z), a variant with this post-translational modification was synthesized for each epitope. Following mice immunization, antigens with Y41 produced non-blocking antibodies whereas sulfation of this residue yielded strong antagonists, especially with the linear antigen pEDEVTDDZIGDNTTVDZTLFESLCSKKDVRNK-NH2 (termed SYM-1899), which allowed the isolation of a series of high-affinity mAbs. Of these, the mouse 729 parental mAb was selected and its variable regions were engrafted into a hIgG1k backbone to generate humanized candidates, including CAP-100, which demonstrated the most potent binding to SYM-1899 by ELISA (EC50 of 1.78 ng/ml) (Figure 1(A)) and Biacore (KD ~0.8 nM). Epitope mapping of CAP-100 in the ligand binding site (36- NTTVDZTLFESLCSKKK-52) demonstrated the strongest binding to the N-terminal sequence 41-ZTLFESLCSKK-51 while the putative core epitope was determined to be 41-Z{Y}TLFE-45. Subsequent fine epitope mapping showed that substitutions in Z, leucine, phenylalanine or glutamic residues negatively impacted on their binding ability, suggesting critical roles for these residues, especially Z41 (Figure 1(B)).
Figure 1.
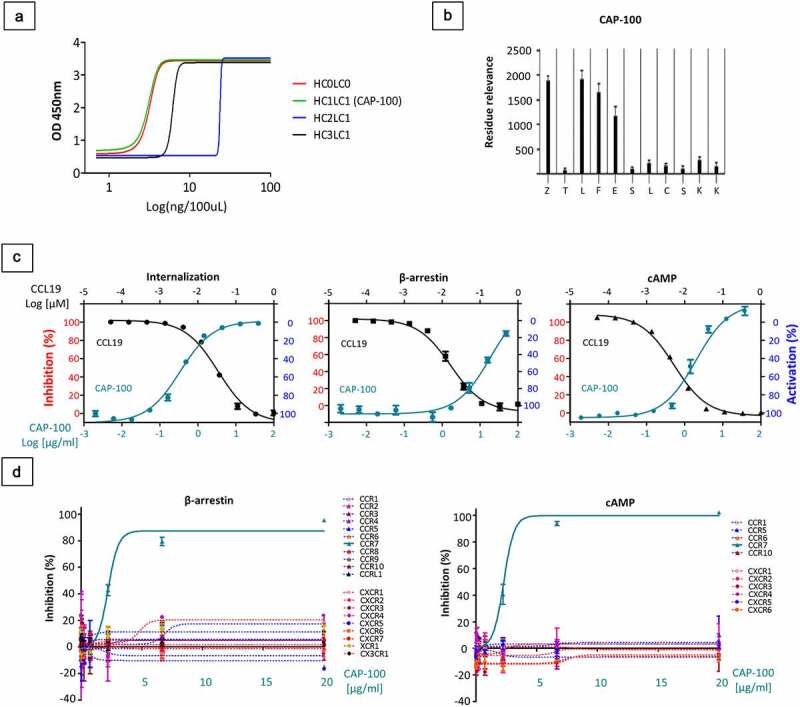
CAP-100 specifically blocks CCR7-pathways with high affinity and selectivity. A) Concentration dependent binding of humanized-729 candidates to 100 ng of CCR7 immunogen SYM1899, the sulfated peptide derived from the N-terminus of human CCR7. The graph shows the OD 450 nm measured by ELISA of 1:1 serial dilutions of antibody to a minimum concentration of 0.0078 μg/ml. B) Epitope mapping of CAP-100 mAb. Peptides were generated bearing single amino acid substitutions at each position of the wild-type sequence. Values of binding to each peptide were assayed by ELISA (residue relevance). The graph shows the OD-405 nm measurements. At the bottom of the plot the native sequence is indicated. Z, sulfated Tyr. C) CAP-100 inhibits CCL19-induced CCR7 internalization, β-arrestin recruitment, or Gi-mediated cAMP reduction. The percentage of inhibition was determined in CCR7+ CHO-K1 cells incubated with CAP-100 antibody and EC80 CCL19. The concentration of CAP-100 [μg/mL] and CCL19 [nM] are plotted on the x-axes versus inhibition (left y-axis) or activation (right y-axis). Data were normalized to the minimal and maximal response observed in the presence of vehicle and EC80 CCL19. The mean ± SD of triplicates is shown. D) CAP-100 mediates specific neutralization of CCR7. Selective inhibition by CAP-100 was evaluated in β-arrestin and cAMP assays in a panel of 20 chemokine receptors stably expressed by CHO-K1 cells. The following pairs of ligand/receptor were included: CCL3/CCR1; CCL27/CCR10; CCL2/CCR2; CCL13/CCR3; CCL22/CCR4; CCL3/CCR5; CCL20/CCR6; CCL1/CCR8; CCL25/CCR9; CCL19/CCR7; CCL19/CCRL1; fractalkine/CX3CR1; CXCL8/CXCR1; CXCL8/CXCL2; CXCL11/CXCR3; CXCL12/CXCR4; CXCL13/CXCR5; CXCL16/CXCR6; CXCL12/CXCR7; Lymphotactin/XCR1. The mean ± SD of triplicates is shown
CAP-100 neutralizes CCR7-triggered signaling
CCR7-downstream pathways start with β-arrestin recruitment to the receptor and G-protein activation, resulting in internalization and inhibition of cAMP production, respectively.9 To evaluate CAP-100 antagonism, we studied these processes, as representative readouts, in CCR7+ CHO-K1 cells stimulated with CCL19, the ligand triggering the most efficient receptor internalization.9 In all cases, CAP-100 dose-dependently inhibited ligand-induced activation, reaching maximal inhibitions close to 100 (Figure 1(C)). This antagonist was highly specific for CCR7 since CAP-100 did not interfere with β-arrestin activation or cAMP production in CHO-K1 cells transfected with empty vectors or a panel of 20 CKR (Figure 1(D)).
CAP-100 preferentially binds to CCR7 expressed on CLL
CLL cells from patients with adverse cytogenetic and/or refractory to standard-of-care (SOC) were used to test binding by flow cytometry to endogenously expressed CCR7 (Figure 2(A)). CAP-100 showed high levels of binding outweighing rituximab. Similar binding profiles were observed in other CCR7-expressing blood cancers, but not in conditions with lack or low target expression (Figure S1-A). We also tested binding to CCR7 expressed by healthy PB and BM hematopoietic cells, including B cells, T cells, natural killer (NK) cells, and dendritic cells (DCs).8,9 CAP-100 bound most mature circulating B cells, 65% of T cells, 40% of DCs, and 20% of NK cells, as well as 55% immature CD10+CD20− BM B cells (Figure 2(B)). These proportions likely included binding to CCR7-positive cells such as naïve T cells (TN), central memory T cells (TCM), regulatory CD16−CD56+ NK cells, and mature DCs. In these cell types, and contrary to tumor counterparts, binding was seen only at the highest concentrations (Table SM1). Finally, CAP-100 did not bind to monocytes, granulocytes, BM CD71+ erythroblasts, BM plasma cells (Pc), or BM CD34+ precursors.
Figure 2.
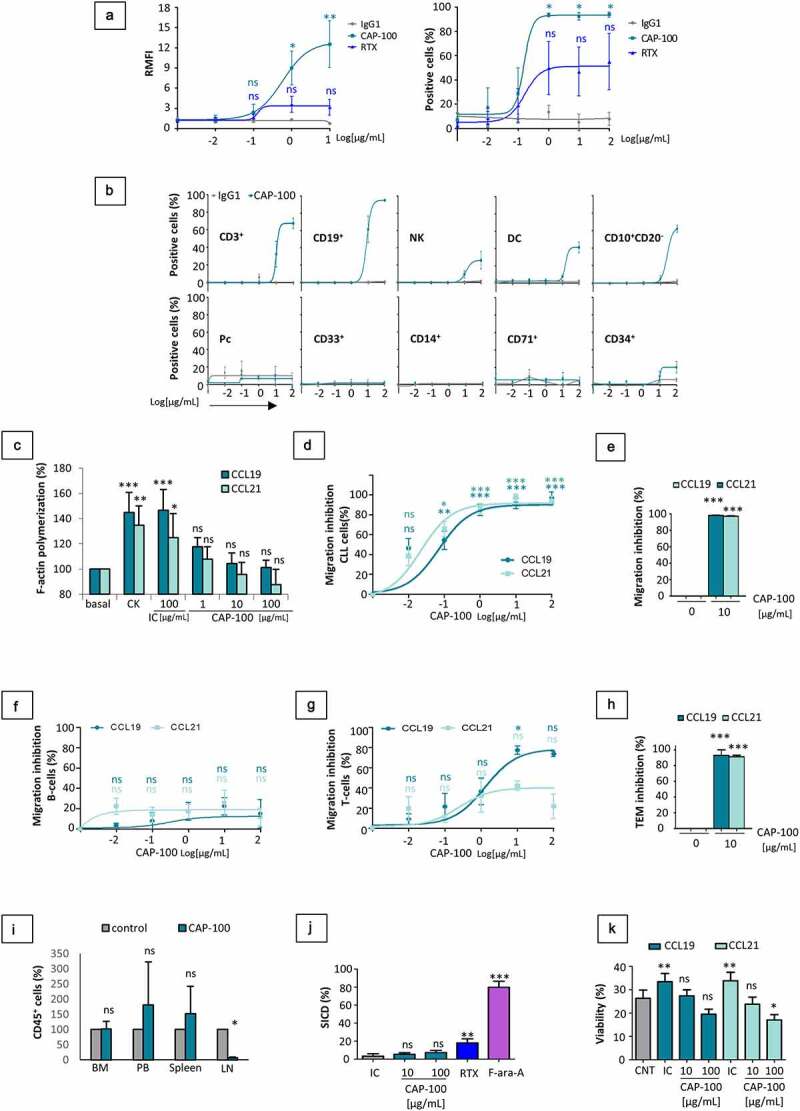
CAP-100 Fab region binds to CCR7 expressed in CLL cells and neutralizes target-induced signaling, chemotaxis, TEM, homing and survival. A) CAP-100 binds to CCR7 expressed in CLL cells from patients with high-risk samples (with del17p and/or refractory disease to anti-CD20 therapy or ibrutinib). Binding was tested by flow cytometry at the indicated concentration range. The therapeutic anti-CD20 rituximab (RTX) was used as reference antibody. Results are expressed as the relative median fluorescence intensity (RMFI, arbitrary units) normalized to the IgG1 isotype control (trastuzumab), or percentage of positive cells. Dots represent mean ± SEM (n = 4 patients). B) CAP-100 binds to CCR7+ non-tumor cells from healthy donors at high experimental concentrations. Cells from PB or BM were incubated with CAP-100 or trastuzumab (matched hIgG1) at different final concentrations and analyzed by flow cytometry. The graphs show analysis in PB (CD3+ T cells, CD19+ B cells, NK cells, DCs, CD14+ monocytes, CD33+ granulocytes), and in BM (immature CD19+CD10+CD20− B cells, CD19+ plasma cells (Pcs), CD71+ erythroblasts, and CD34+ precursors). Dots represent mean ± SEM (n = 3 donors). C) CAP-100 neutralizes CCR7-induced signaling on CLL cells. F-actin polymerization (% relative to baseline) was used as surrogate marker of CCR7-signaling and migration. CLL cells were pre-incubated with trastuzumab (IC, 100 µg/ml), CAP-100 (1, 10, and 100 µg/ml) or left untreated prior to stimulation with CCR7 ligands (1 µg/ml for 0.5 minutes). Polymerization was detected by flow cytometry. Bars represent the mean ± SD (n = 5 patients). D) CAP-100 neutralizes CCR7-mediated CLL cells migration in response to CCL19 or CCL21. Chemotaxis of CLL cells was assayed in uncoated transwell chambers for 4 h. CCL19 and CCL21 (1 µg/mL) were used as chemoattractant in the bottom chamber. Data were normalized to the maximal and minimal response observed in the presence of ligand and medium (basal/spontaneous migration), respectively. The graph shows the mean percentage of inhibition ± SEM (n = 16 patients). E) CAP-100 neutralizes CCR7-mediated T-cell prolymphocytic leukemia (T-PLL) cells migration toward CCR7 ligands. The specific blocking of CCR7-ligand interactions, expressed as the percentage of inhibition toward 1 µg/mL CCL19/21-induced migration is shown for T-PLL cells incubated with CAP-100 or a matched IC (10 µg/mL). Bars represent the mean ± SEM (n = 3 patients). F) CAP-100 minimally impairs CCR7-mediated migration in healthy B cells. The specific blocking of CCR7-ligand interactions, expressed as the percentage of inhibition of CCL19/21-induced migration is shown for B cells. Graph shows mean ± SEM (n = 3 HD). G) CAP-100 moderately inhibits migration in healthy T cells. The specific blocking of CCR7-ligand interactions, expressed as the percentage of inhibition of CCL19/21-induced migration is shown for T cells. Graph shows mean ± SEM (n = 3 HD). H) CAP-100 inhibits target-mediated CLL cells migration across endothelium. CLL cells migration was assayed in transwell filters coated with TNFα-activated HUVEC, in the presence or absence of CAP-100 (10 µg/mL) and CCR7 ligands (1 µg/mL). The percentage of inhibition (mean ± SEM) is shown (n = 5 patients). I) CAP-100 reduces in vivo homing to LN. In this model, CCR7-expressing lymphocytes were pre-incubated with CAP-100 or a matched IC (trastuzumab) at 10 µg/ml before tail vein injection into irradiated NSG mice (n = 5 per group). One hour later, mice were sacrificed and target cells found in LN, BM from femurs, spleen and PB enumerated by flow cytometry. The graph shows the mean proportion ± SD of hCD45+ lymphocytes in 106 cell suspensions from each tissue (normalized to control group). J) CAP-100 does not trigger specific-induced cell death (SICD) upon binding to CCR7 expressed on CLL cells. Leukemic cells were either treated with CAP-100 (10 or 100 µg/ml), an IgG1 control antibody (trastuzumab, 100 µg/ml), rituximab (RTX, 100 µg/ml), or with fludarabine (F-ara-A; 10 µmol/L) for 24 hours followed by flow cytometry analysis. The graph shows % SICD for each compound. Mean ± SD is shown (n = 13 patients). K) CAP-100 inhibits CCR7-induced survival in CLL. Cells were incubated with an isotype control (IC, 100 µg/ml) or with CAP-100 (10 µg/ml or 100 µg/ml) before long-term culture in 1% FBS medium alone or supplemented with CCR7 ligands (1 µg/mL). Cells incubated in medium alone, without chemokines or antibodies, were used as controls (CNT). Cell viability (%) was determined after 72 hours. Graph shows mean ± SEM (n = 8 HD). For all graphs: ns, not significant; * p < .05; ** p < .01; *** p < .001
CAP-100 neutralizes CCR7-mediated migration, extravasation and LN homing
To evaluate the potential of CAP-100 to prevent leukemic dissemination to CCL19/CCL21-producing locations, first we studied F-actin polymerization as a surrogate marker of cancer cell migration.28 In CLL cells, F-actin significantly increased upon CCL19/CCL21 stimulation whereas CAP-100 impaired this process (Figure 2(C)). Accordingly, in chemotaxis assays with CLL cells, CAP-100 demonstrated a strong dose-related inhibitory activity against CCR7-driven migration toward 1 µg/mL of ligands (Figure 2(D)), a concentration within the range estimated in T-zones of LN.29 CAP-100 (1–100 µg/ml) reached ~100% inhibition regardless of patient-to-patient variability and clinical features. Similar results were seen in other CCR7-expressing blood cancers (Figures 2(E) and S1-B), but not in healthy B cells or T cells (Figure 2(F,G)). Contrary to leukemic B cells, CAP-100 maximal inhibition in B cells reached values of 20% against both ligands, suggesting a preferential blocking activity on migration of CLL cells. This fact is likely explained by a different binding profile (Figure 2(B)) and indicated that CAP-100 Fab-mediated activities relied on a certain threshold of surface target density, as we reported with commercial antibodies.12,27 Indeed, quantification of CCR7 surface receptors showed CLL cells to display approximately 5 and 16 times more targets than normal T or B cells, respectively (Figure S1-C).
CAP-100 also impaired CCR7-induced migration of CLL cells in trans-endothelial migration (TEM) assays (Figure 2(H)). Since this approach emulated cell extravasation across HEVs toward the LN,10 next we aimed to corroborate CAP-100 activity against CCR7-driven homing to the LN in irradiated NSG mice, a model suited to study Fab-mediated inhibition without contribution of crystallizable fragment (Fc)-triggered depletion. However, we failed to demonstrate CLL cells homing to LN (data not shown). Therefore, we used hCD45+CCR7+ lymphocytes from a healthy donor (HD) instead. Cells were pre-incubated in vitro either with 10 µg/ml of CAP-100 or an isotype control (IC), and tail vein transferred into recipients (5 mice/group). After sacrifice, cells migrated to LN, BM, spleen, and PB were counted by flow cytometry. Compared to controls, CAP-100 significantly reduced the proportion of CCR7-expressing cells in LN (Figure 2(I)). Concomitantly, a non-significant increment of human cells was observed in PB and spleen of CAP-100-treated mice, suggesting that by preventing LN homing CAP-100 forces target cell accumulation in blood.
CAP-100 does not mediate direct specific-induced cell death but impairs CCR7-mediated survival
When leukemic cells were incubated with CAP-100, specific-induced cell death (SICD) did not differ from the negative control whereas cell death was detected upon treatment with positive controls fludarabine (F-ara-A) and rituximab (Figure 2(J)). Similarly, no SICD was seen after incubation with CCR7-expressing blood cancer cell lines (Figure S1-D).
In CLL, CCR7 supports cell survival.19,20,30 To study whether CAP-100 could impair this protective function in CLL, we analyzed cell survival in long-term suspension cultures of CLL cells exposed to CCL19 or CCL21, in the presence of either CAP-100 or an IC (Figure 2(K)). As expected, cell viability decreased over the days,31 but in cultures exposed to CCL19/CCL21 a moderate but statistically significant increase of viability was seen. This rescue from spontaneous apoptosis was abrogated by CAP-100.
CAP-100 triggers a potent antibody-dependent cell-mediated cytotoxicity
Besides strong Fab activities, another desirable feature of a therapeutic anti-CCR7 mAb is to elicit cell killing through Fc-mediated host effector mechanisms such as antibody-dependent cell-mediated cytotoxicity (ADCC) or complement-dependent cytotoxicity (CDC). Biacore studies on the binding of CAP-100 to the human Fc gamma receptors (hFcγR) family (Table 1) showed higher affinity for activator receptors (FcγRI, FcγRIIA, FcγRIIIA, FcγRIIIB) than for the inhibitory FcγRIIB, irrespective of the polymorphism tested. Additionally, binding to neonatal hFcRn indicated that CAP-100 plasma half-life should be similar to a canonical IgG1. Next, we performed ADCC assays with CLL samples. With human peripheral blood mononuclear cells (hPBMC) as effector cells, CAP-100 mediated a strong cell killing, irrespective of treatment status or high-risk features (Figure 3(A,B)). Indeed, lytic activity was confirmed against CLL samples harboring aberrant cytogenetic (e.g., del17p, del11q, tri12, unmutated IGVH) and alemtuzumab-refractory T-cell leukemia (Figure 3(C,D,E)). In these settings, CAP-100 outperformed rituximab and alemtuzumab regardless of the effector-to-target (E:T) ratios used. Similarly, with recombinant NK92-CD16+ cells as effectors, CAP-100 at 10 µg/ml was ~7 times more active against CLL cells than rituximab in the same concentration (Figure 3(F)). CDC is another main Fc-mediated mechanism to kill target cells. We confirmed here CAP-100 interactions with hC1q, the first component of the classical complement pathway (Figure 3(G)). Nonetheless, in two different in vitro systems CAP-100 did not show CDC against CLL or HuT-78 target cells (Figure 3(H,I)).
Table 1.
Binding of CAP-100 to FcR
| KD(M)/relative binding* | |||
|---|---|---|---|
| Fc Receptor | CAP-100 | Control IgG1 | Control IGg4 |
| CD64 | 1.38E-09 (++++) | 3.28E-09 (++++) | 9.16E-09 (++++) |
| CD16A (176Phe)/FcγRIIIA176Phe ** | 3.01E-07 (+++) | 4.68E-06 (++) | - |
| CD16A (176Val)/FcγRIIIA176Val | 1.46E-07 (+++) | 1.51E-06 (++) | - |
| CD16B/FcγRIIIB | 7.92E-07 (+++) | 6.17E.06 (++) | - |
| CD32A (167Arg)/FcγRIIA167Arg | 3.47E-06 (++) | 1.03E-05 (+) | - |
| CD32A (167His)/FcγRIIA167His | 5.54E-06 (++) | 1.03E-05 (+) | - |
| CD32B/FcγRIIB | 8.24E-06 (++) | 1.91E-05 (+) | 3.26E-05 (+) |
| FcRn (pH 6.0) | 1.27E-06 (++) | 1.34E-06 (++) | 3.38E-06 (++) |
| FcRn (pH 7.4) | - | - | - |
*The relative binding affinity scale is defined as follows: ++++ (10−8–10−9); +++ (10−7); ++ (10−6); + (10−5); ± (detectable binding); – (no detectable binding). As controls (both positive and negative, depending on the receptor and the assay) wild-type IgG1 and IgG4 were included. ** Position 176, or 167 including the signal peptide in the amino acid count. For FcγR, tests conducted at 5 point three-fold dilution of purified antibodies titrated 0.411 nM to 33.33 nM. For FcRn, at seven point two-fold dilutions from 4000 nM to 62.5 nM (4000 nM to 1000 nM for pH 6.0). For high affinity receptors (FcγRI), 1:1 kinetic analysis was used. For low affinity FcγRII and FcγRIII receptors, and for FcRn, steady state affinity analysis was used.
Figure 3.
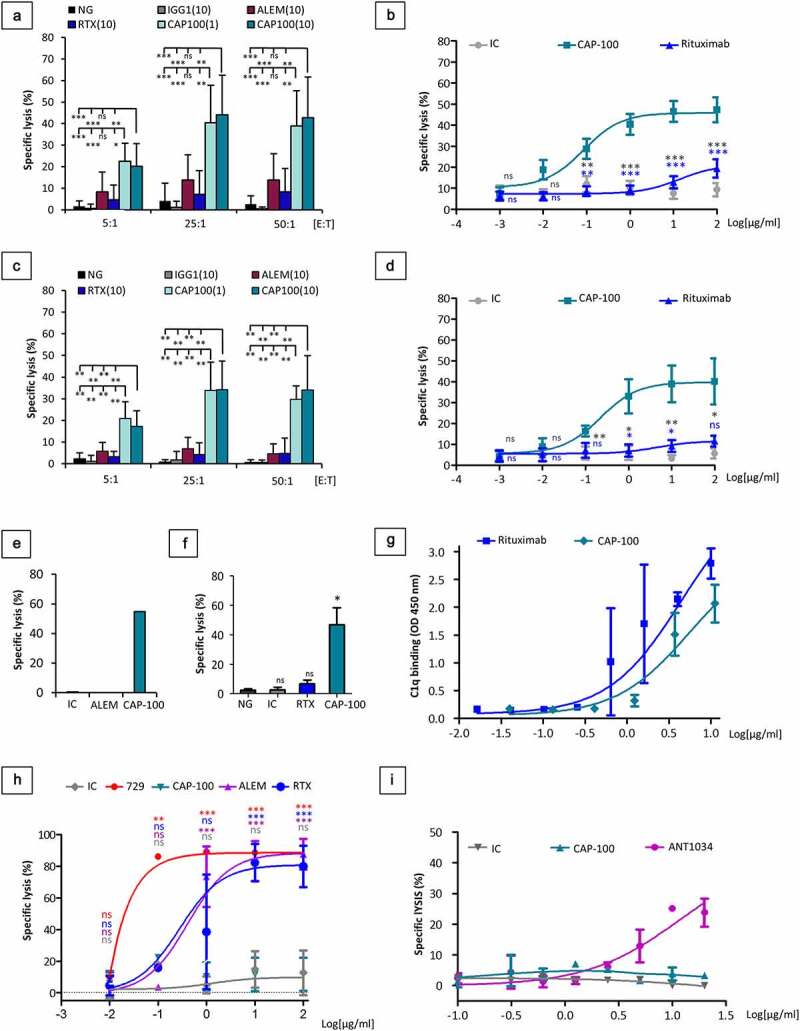
CAP-100 activates strong CLL cell killing through Fc-mediated host immune effector mechanisms. A) ADCC activity was assayed in CLL cells from patients with different known previous treatments and different cytogenetic. CLL cells were incubated in medium alone (NG) or with an IC (10 µg/ml, trastuzumab, IgG1), rituximab (10 µg/ml, RTX), alemtuzumab (10 µg/ml, ALEM) or CAP-100 (1 or 10 µg/ml). Isolated PBMCs were used as effector cells at different E:T ratios (5:1; 25:1; 50:1). The mean ± SD of the antibody-induced specific lysis (%) is shown (n = 13 patients). B) ADCC was assayed in CLL samples incubated with a concentration range of an IC (trastuzumab), rituximab (RTX), or CAP-100. PBMCs were used at 10:1 E:T ratio. The mean ± SEM of the antibody-induced specific lysis (%) is shown (n = 17 patients). C) The mean proportion of specific lysis induced by the antibodies ± SD (n = 6 patients) is shown for CLL with cytogenetic abnormalities incubated along with different E:T ratios. D) The mean proportion of specific lysis induced by the antibodies ± SEM ratios (n = 7 patients) is shown for CLL with cytogenetic abnormalities incubated along with concentration range of CAP-100 or control antibodies at a single E:T of 10:1. E) CAP-100-induced ADCC is effective against alemtuzumab refractory T-cell prolymphocytic leukemia (T-PLL). ADCC was assayed in cells from one patient with alemtuzumab-refractory T-cell prolymphocytic leukemia incubated with 10 µg/ml of IC (trastuzumab), alemtuzumab (ALEM) or CAP-100. PBMCs were used as effector cells at E:T ratio of 10:1. F) CAP-100 activates human NK-92-CD16+ cells to induce a strong ADCC on target CLL cells. Samples were incubated in medium alone (NG) or with µg/ml trastuzumab (IC), rituximab (RTX) or CAP-100. Effector cells were used at E:T ratio of 10:1. Columns represent the mean ± SD of 3 independent experiments. G) CAP-100 binds to C1q, the first component in the classical pathway of complement activation. Binding of antibodies (3-fold serial dilutions) to human C1q was measured by standard HRP/TMB-based ELISA. Rituximab (RTX) was used as positive control. The mean ± SD of duplicates is shown. H) CAP-100 does not induce CDC in CLL cells. Samples from CLL patients were incubated with different concentrations of CAP-100, alemtuzumab (ALEM), rituximab (RTX), mouse 729–2A (729) or an IC (trastuzumab, IgG1) and exposed to rabbit complement (30%). The mean percentage of specific lysis ± SD (n = 4 patients) is shown. I) CAP-100 does not induce CDC on HuT-78 CCR7-expressing cells. Assays were performed using normal human serum (25%) as source of complement. A matched IC and ANT1034, a human anti-CD52 (target expressed on HuT-78 cells), were used as negative and positive controls. Data points represent the mean specific lysis ± SEM of duplicates. ns, not significant; * p < .05; ** p < .01; *** p < .001
CAP-100 monotherapy extends survival in in vivo models of SOC refractory disease
Due to the lack of CAP-100 binding to mCCR7, to evaluate the utility of CAP-100 therapy in CLL we conducted in vivo proof-of-concepts in systemic xenogeneic models selected on a close resemblance to high-risk CLL. To evaluate the in vivo anti-tumor efficacy of CAP-100, we used as hosts the SCID strain, which preserves functional NK cells,32 thus providing a good background to test the proposed Fab and Fc MOA in one go. First, we used the aggressive Granta-519-luc+ model, insensitive to ibrutinib and venetoclax monotherapies33,34 and with defects in p53/17p and ataxia telangiectasia-mutated (ATM/11q), two frequent alterations in CLL and associated with poor prognosis and lymphadenopathy.35,36 CAP-100 was challenged in a post-implantation therapeutic set-up thus mirroring a clinical scenario. Treatment started at day +14 after tumor inoculation, once bioluminescence quantification confirmed engraftment (Figure S2-A). Group G1 received 200 µg/mouse (~10 mg/kg) control hIgG1k, whereas G2, G3, G4 and G5 received CAP-100 at 25, 50, 100, and 200 µg/mouse, respectively (Q4dx8). CAP-100 significantly prolonged median overall survival (mOS) regardless the dose (Figure 4(A)): G1 (mOS: 45 days; range: 44–49] vs G2 (80 days; 60–101; p < .0001), G3 (64 days; 57–101; p < .0001), G4 (67 days; 57–101; p < .0001), and G5 (86 days; 57–101; p < .0001). No statistical differences were found between CAP-100 groups and all mice in G2-G5 remained alive at the time when the last control was euthanized; 27% of these animals reached day +101, when the study was terminated.
Figure 4.
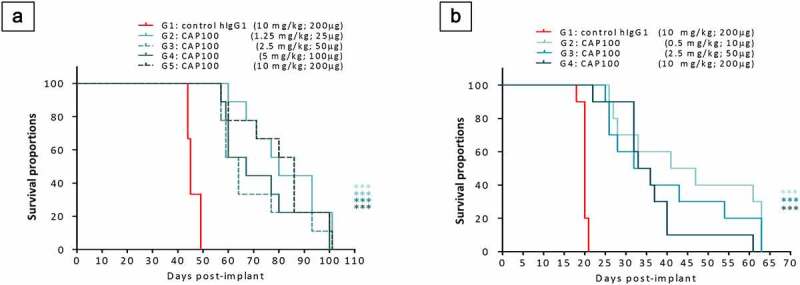
CAP-100 monotherapy is effective in systemic models of high-risk disease. A) Survival curve for Granta-519-luc+-bearing mice. In this model, irradiated recipients (n = 45 animals) were inoculated via tail vein with 2 × 106 cells. At day +14, the animals were allocated in 5 experimental groups (n = 9). Positive engraftment was monitored via bioluminescence detection. Treatment with CAP-100 or the isotype control was administrated IP, Q4x8 (first dose at day +14). B) Survival curve for JVM-3-bearing SCID mice. In the model, irradiated recipients (n = 40 animals) were inoculated via tail vein with 20 × 106 cells. At day +3, the animals were allocated in 4 experimental groups (n = 10). Treatment with CAP-100 or the isotype control was administrated IP, Q3x6 (first dose at day +3). In A and B, survival was estimated and compared by Kaplan-Meier survival curves and Log-Rank (Mantel-Cox) tests, respectively. ***, p < .001
In a second in vivo study we used a model with JVM-3 cells, also insensitive to ibrutinib37,38 and with tri12, which associates with lymphadenopathy and poor prognosis in CLL.16,17 Again, CAP-100 (G2: 10, G3: 50, and G4: 200 µg/mouse, Q3dx6) significantly extended lifespan when compared to IC (200 µg/mouse, G1) and irrespective of the dose (Figure 4(B)): G1 (mOS: 20 days; range: 18–21 days) vs G2 (44 days; 26–63; p < .001), G3 (34 days; 25–63; p < .001), and G4 (34.5 days; 22–61; p < .001). Once more, all mice in the CAP-100 groups were alive when the last control died, no statistical differences were found between CAP-100 groups, and 26.6% of these animals survived until the study conclusion (day +63).
CAP-100 reduces tumor burden and affects the TME
CCR7 is critical in enabling leukemic cells accessing and establishing TME.7 To evaluate whether CAP-100 could affect these processes, we traced tumor burden in the Granta-519-luc+ model by means of weekly whole-body bioluminescence quantifications. This approach showed delayed tumor onset and reduced tumor growth with CAP-100 therapy (Figures 5(A,B,C) and S2-B). At day +14, all animals had similar tumor burden while in weeks 3–7 significant differences were observed between control and CAP-100 groups. Quantification in CAP-100 groups remained low until the conclusion of the study, suggesting that CAP-100 impaired CCR7-mediated tumor dissemination and growth. Accordingly, postmortem analysis of selected tissues consistently showed a significant reduction of infiltrating tumor cells with CAP-100 therapy (Figure 6(A)). In addition, detailed analysis of tumor cells found in LN, BM, or spleen, all of them niches for CCR7-mediated survival/proliferation in leukemia/lymphoma,7 showed a strong CAP-100-mediated reduction in malignant cell viability and proliferation (Figure 6(B,C)). Finally, the impairment in LN infiltration, survival and proliferation translated into markedly smaller LN size than the control animals (Figure 6(D)).
Figure 5.
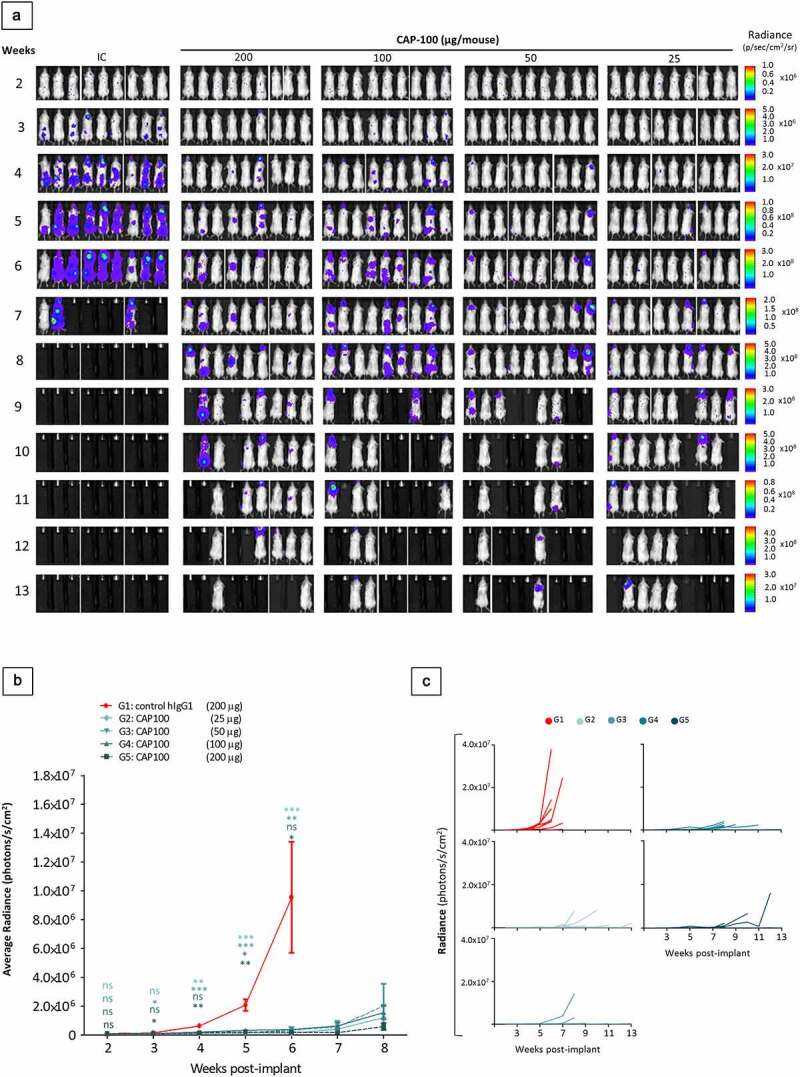
CAP-100 reduces tumor burden in the Granta-519-luc+ model. A) Tumor burden follow-up was determined by whole body (back and front) bioluminescence analyses on each mouse on weeks 2 to 13. Back images of each mouse are shown. B) Quantification of luciferase activity detected on the whole body of the animals (back + front). The average radiance is shown as mean ± SEM for total animals alive in each time and until the first removal of a mouse from a group (week 6 for IC and week 8 for CAP-100 treatment groups). C) Radiance on the whole body of the animals (back + front) is shown as dots and connecting lines for each individual mouse in each treatment group. ns, not significant; *, p < .05; **, p < .001; ***, p < .0001
Figure 6.
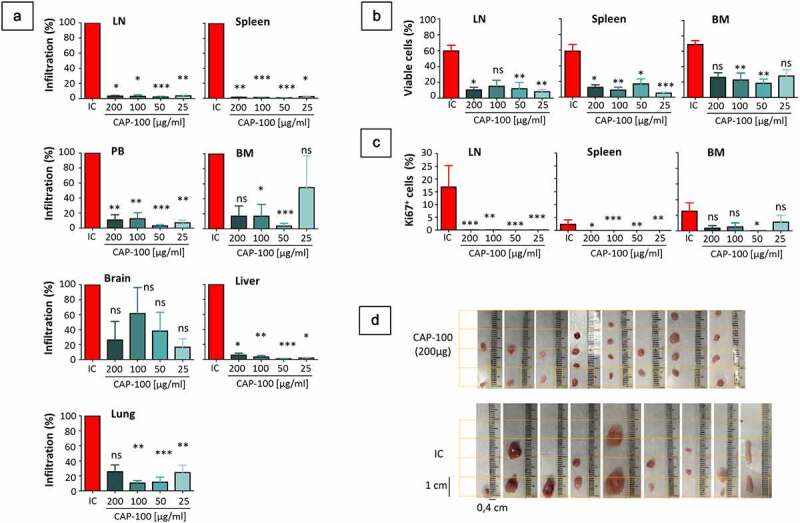
CAP-100 reduces LN infiltration, tissue survival and proliferation in the Granta-519-luc+ model. A) CAP-100 impairs tissue infiltration. Results are expressed as the proportion of human CD20+ tumor cells found in CAP-100 groups normalize to proportions found in the control group. The mean ± SEM is shown for a total of 45 brains, BM, spleens, livers and lungs, 44 PB, and 39 LN. B) CAP-100 reduces survival in protective niches. The proportion of viable cells (within the proportion of infiltrating CD20+ tumor cells found in LN, spleen, BM) was determined by flow cytometry by staining with annexin-V-FITC/7-AAD. The mean and SEM are shown. C) CAP-100 reduces proliferation in protective niches. The proportion of Ki67+ cells was determined within the proportion of viable CD20+ tumor cells in each tissue. The mean and SEM are shown. D) CAP-100 treatment shrinkages LN size. Representative images of LNs obtained in the necropsy of control or CAP-100 groups (200 μg/ml). *, p < .05; **, p < .001; ***, p < .0001; ns, not significant
CAP-100 pharmacodynamics on non-tumor immune cells
Our data on CAP-100 showed restricted binding to normal immune cells and a limited effect on healthy B-cell migration, thus suggesting a mild impact of CAP-100 on immune subsets. To address this question, we performed ex vivo depletion assays in whole PB from HD. In these tests, CAP-100 moderately reduced proportions of B cells, TN, TCM, and the regulatory CD56+brightCD16−dim NK cells, while it spared DC, monocytes, NK T cells, effector T cells (TEM, TEMRA), the cytotoxic CD56−dimCD16+bright NK cells, neutrophils, and Pc (Figure 7(A,B) and data not shown). Accordingly, in regulatory safety studies with non-human primates (NHP), repeated administrations of CAP-100 (Q4dx8) at dose levels of 10, 35, and 100 mg/kg only reduced CD4+ T cells, but not cytotoxic T cells or other immune cells (Figure 7(C)). In an additional non-clinical study, in which three healthy NHP were infused with a single bolus of CAP-100 (10 mg/kg), we confirmed a specific reduction of CCR7-expressing TN and TCM, whereas no changes were detected in the other T cell subsets or immune cells (Figure S3). Finally, in these studies no overt signs of toxicity or marked clinical/laboratory changes were observed after CAP-100 administration, even at high dose levels such as 35 or 100 mg/kg.
Figure 7.
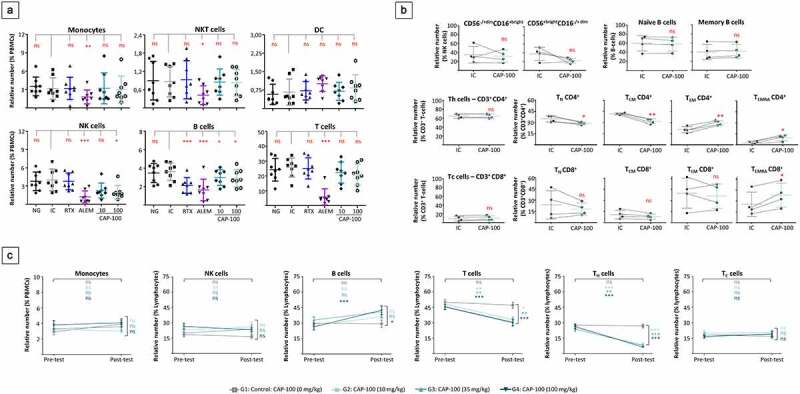
CAP-100 spares most non-tumor immune cells. A-B) Autologous ex vivo depletion assays. In A, heparinized whole blood samples from healthy human volunteers were incubated without antibody (NG), with CAP-100 (10 and 100 µg/ml), or 100 µg/ml of control antibodies trastuzumab (IC), alemtuzumab (ALEM) or rituximab (RTX). After 24 hours, relative numbers were analyzed by flow cytometry. The mean ± SD is shown (n = 8 donors). In B, heparinized whole blood samples from healthy human volunteers were incubated for 24 hours with CAP-100 or trastuzumab (IC), both at 100 µg/ml, and analyzed by flow cytometry. The mean ± SD is shown (n = 4 donors). C) Impact of repeated administrations of CAP-100, at different dose levels, in immune cells from NHP. Thirty-two animals (males and females in a ratio 1:1) were allocated in four groups: G1 (0 mg/kg; vehicle; n = 10), G2 (10 mg/kg; n = 6), G3 (35 mg/kg; n = 6); G4 (100 mg/kg; n = 10). CAP-100 was intravenously infused twice a week for 4 consecutive weeks. PB samples were collected in K2EDTA tubes and analyzed in two occasions: pre-dose and at day +29 (post-dose). The effect on relative numbers is given as mean ± SEM. The following abbreviations are used: Th, CD4+ helper T cells; Tc, CD8+ cytotoxic T cells; TN, naïve T cells; TCM, central memory T cells; TEM, effector and effector memory T cells; TEMRA, terminally differentiated effector memory T cells; ns, not significant; *, p < .05; **, p < .001; ***, p < .0001
Discussion
A hallmark of CLL is the strong dependence on LN environments that promote disease progression and relapses after therapy. In this work, we evaluated a novel strategy to prevent CLL cells homing into the LN and subsequent tissue interactions. Since these processes are tightly controlled by CCR7 and its ligands, which are, respectively, consistently highly expressed on CLL cells12 and over-produced within LN in patients,10,16 we developed CAP-100, an antagonist mAb targeting the N-terminal ligand binding site of CCR7 with high specificity and affinity. This novel antibody for CLL therapy is differentiated by its innovative, differentiated MOA targeting the Achilles` heel of CLL, the LN dependence (Figure 8).
Figure 8.
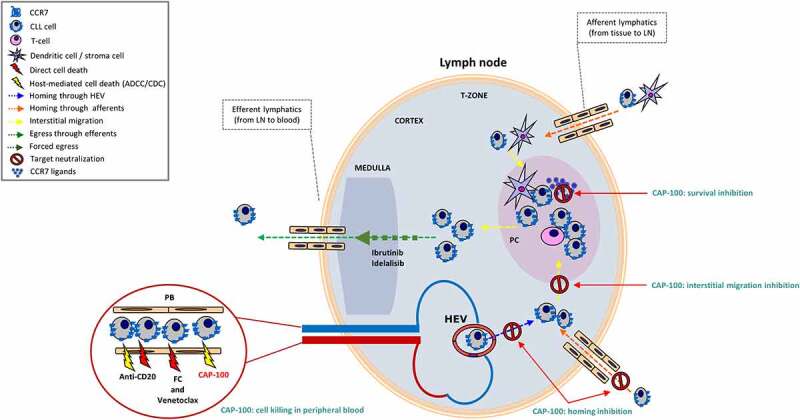
CAP-100 is a single molecule affecting both homed and circulating CLL cells. In CLL, CCR7 mediates migration and invasion to LN and other lymphoid organs. Here CCR7 also mediates positioning of tumor cells within protective niches where tumor cells find trophic factors, including CCL19 and CCL21. CAP-100 is presented as a novel and potential tool in the treatment of CLL due to its double and effective mechanism of action. CAP-100 blocks CLL cells in the bloodstream, preventing homing of CLL through HEV and afferents into lymphoid tissues where it also prevents tissue interactions with accessory cells or CCR7 cognate ligands. This way, CAP-100 forces accumulation of CLL cells in bloodstream where tumor cells are depleted by CAP-100 Fc-mediated immune effector mechanisms, and potentially by other drugs since CAP-100 activities suggest complementarity with other SOC in CLL. ADCC, antibody-dependent cell-mediated cytotoxicity; CDC, complement-dependent cytotoxicity; FC, fludarabine + cyclophosphamide; HEV, high endothelial venules; LN, lymph node; PC, proliferation center
Due to its Fab region, CAP-100 inhibits dissemination to LN and lymphadenopathy. We show here that Fab-mediated neutralization of CCR7 impaired ligand-driven cytoskeleton re-organization, chemotaxis, and extravasation of CLL cells, as well as in vivo homing to LN of CCR7+ lymphocytes. Ideally, LN homing should has been performed with CLL cells, but our pilot studies showed poor capacity of CLL to reach LN, in line with previous works of Hartmann et al.39 Even though, we can assume similar benefits in patients substantiated by broad evidence describing key roles of CCR7 in integrin-mediated arrest to HEVs and trans-endothelial extravasation of CLL cells.16,17,40,41 Additionally, in mouse models, CAP-100 reduced tumor cells infiltration in LN and nodal size, although in these models we cannot exclude a contribution from Fc-mediated depletion. In line with our results, LN of CCR7-deficient mice are devoid of lymphocytes, whereas these cells are markedly expanded in PB, spleen, and BM.5 CAP-100 reproduced this phenotype, thus confirming its ability to accumulate target cells in bloodstream that become more accessible to Fc-mediated depletion, other drugs, or even fostering resting CLL cells apoptosis by neglect since CAP-100, like commercial anti-CCR7 clones, did not induce direct target cell death.12,27
CAP-100 can also impair positioning of tumor cells in protective niches in the LN. CAP-100 significantly delayed disease onset and reduced tumor burden reproducing results by Rehm et al. who proposed the exclusion of CCR7-deficient lymphoma cells from tumor niches as a mechanism to impair protective environment formation in LN and spleen.7 Accordingly, CCR7 levels remain high on CLL cells following their SLOs homing and it is known to guide CLL cells toward niches where accessory cells provide reciprocal crosstalk and trophic factors that can promote tumor progression.7,18,42–44 In these niches, soluble CCL19/CCL21 are also available and activate CCR7-induced survival,19,20,30 a process inhibited by CAP-100 in vitro and in vivo, especially in the LN and spleen. Moreover, CAP-100 demonstrated activity reducing the proportion of Ki67+ Granta-519 cells in LN, spleen and BM, therefore anti-proliferative activities in patients may be expected.
Interestingly, sustained CCR7 over-expression disabled the S1P1-mediated egress from LN to PB45 and contributed to prolong CLL cells residency in protective niches.13 Therefore, it is plausible CAP-100 may induce egress. Although we did not study every option, it is known the lack of CCR7 signaling endowed lymphocytes with a higher propensity to leave lymphoid tissues.5,45 Similarly, CAP-100 may interfere with CCR7-dependent functions of some accessory cells that hold malignant cells within the LN or promote tumor tolerance and progression.31,46 Future preclinical and clinical work in these fields is assumed.
Related to the Fc region, CAP-100 effectively triggered leukemic cells killing through host effector mechanisms, especially ADCC (though C1q binding assays confirmed that CAP-100 is not discounted for CDC in vivo). CAP-100 outperformed rituximab and alemtuzumab, a highly active mAb against del17p CLL47 that is no longer used as a therapy for this disease, thus indicating that ADCC is a relevant MOA for CAP-100. Although we did not evaluate the specific contribution of Fab- and Fc-mediated MOA in the in vivo efficacy of CAP-100 in this study, in the context of the data presented in SCID mice both kind of mechanisms seems equally important and necessary to achieve the maximum therapeutic benefit. Future work is needed to uncover whether CAP-100 has prevailing, additive, or synergic MOA.
Interestingly, CAP-100-mediated activities were selective of tumor cells while sparing healthy counterparts. This effect is likely due to a lower apparent affinity of CAP-100 for CCR7 expressed in non-tumor cells and a lower target density. In this respect, we and others reported a strong, preferential chemotaxis of malignant CLL and mantle cell lymphoma (MCL) cells toward CCR7 ligands, and opposite to normal B cells that strongly migrated toward CXCR4 and CXCR5 ligands.11,48 Similarly, normal counterparts are less dependent on CCR7 than CLL cells for arrest on HEVs and homing.45,49,50 Related to depletion of non-tumor cells, CAP-100 had an impact on TN and TCM T cells, an effect previously reported with commercial mouse antibodies51,52 and in line with the well-documented restricted expression of CCR7 to certain immune cells.8,9 Together, these results lead us to expect a low-to-mild associated immunosuppression in patients. Although CAP-100 could impair new immunization processes dependent on TN cells, it should not affect memory effector responses, B-cell lymphopoiesis nor immunoglobulin secretory function.5,53 Indeed, anti-CCR7 therapy in preclinical syngeneic mouse models of cancer, autoimmunity, or inflammation did not uncover unwanted treatment-associated side effects,51,54,55 and CAP-100 toxicology studies in NHP did not reveal overt toxicities or autoimmune disease, indicating tolerability of this novel therapy.
In a clinical scenario, CAP-100 might provide opportunities to all CLL patients, particularly in the context of tackling bulky lymphadenopathy, relapsed/refractory disease, or minimal residual disease in SLOs.2,22,23 Indeed, CAP-100 was highly effective in relevant xenograft models of ibrutinib and venetoclax refractory disease. Strikingly, in these models CAP-100 showed no statistical differences between the distinct doses. We believe that factors related to CAP-100 pharmacodynamics or pharmacodynamics underlie this outcome. As a support, it has been very recently reported that intraperitoneal injection of 25 μg anti-CCR7 mAbs to recipient immunodeficient mice resulted in peak plasma antibody concentrations of ~ 30 μg/ml (200 nM),54 far higher than CAP-100 maximal in vitro efficacy, consistently observed within the range [0.1–10 µg/ml]. Therefore, it is plausible that CAP-100 plasma concentration for all tested doses (including the lowest of 10 and 25 µg/mouse) was sufficient to effectively act against target cells. Despite these results, and other limitations associated with this kind of model (e.g., lack of immunosuppressive side-effects), systemic xenogeneic models are predictive of clinical results in blood cancers.33 We can, therefore, envision CAP-100 as a promising monotherapy or combined with existing CLL drugs to supplement their activity or to reduce resistances.
Finally, CAP-100 may fulfill not only the need for novel rationally-based therapies in CLL, but also in MCL, diffuse large B-cell lymphoma, T-cell lymphoma/leukemia.24,56–58 Similarly, CAP-100 could be beneficial in certain solid tumors with tropism to LN,59 graft-versus host disease or autoimmune diseases.51,52,54 In the coming months, CAP-100 will be evaluated in a Phase 1 clinical trial in CLL (NCT04704323). The first data about tolerability and anti-tumor activity of this novel therapy will be available soon.
Materials/subjects and methods
Generation of CAP-100
A library of linear and native peptide-epitope mimics, corresponding to the N-terminal ligand binding site sequence of hCCR7, was designed by Pepscan (Lelystad, The Netherlands) using a proprietary software. Peptides were synthesized using standard Fmoc-based solid-phase synthesis and involved Chemically-Linked Peptides on Scaffolds (CLIPS) technology (Pepscan). Candidates were selected on the basis of apparent affinity studies and epitope mapping (as described below) with CCR7 ligands. Mouse anti-hCCR7-specific mAbs were obtained via hybridoma technology after immunization of Balb-c mice. Every mouse was immunized with a single candidate peptide. The V region sequences of selected mouse antibody 729 were complementary-determining region engrafted onto hIgG1κ backbones. Binding kinetics and affinity were assayed on a BiacoreTM T200 surface plasmon resonance biosensor (GE Life Sciences, Eindhoven, The Netherlands). Apparent affinity was measured by a horseradish peroxidase (HRP)-based ELISA using tetra-methyl-benzidine (TMB) as substrate (450 nm). For linear epitope mapping, the binding to synthesized peptides (wild-type sequence or variants containing single amino acid mutations) was tested using an HRP-ELISA with azino-bis-ethylbenzothiazoline-6-sulfonic-acid (ABTS) substrate (405 nm) (Pepscan).
Samples, cell lines and reagents
Patients included in this study were diagnosed for CLL according to World Health Organization consensus criteria.2 PBMC from patients or HD were isolated from fresh samples. Human umbilical vein endothelial cells (HUVECs) were stripped from freshly donated cords.13 Informed consent was obtained in accordance with the Declaration of Helsinki. Procedures were approved by the Institutional Board of the Hospital de La Princesa (HULP). Cell lines were purchased from commercial sources (Table SM2). The following therapeutics were provided by HULP Pharmacy Department: trastuzumab, used as irrelevant hIgG1 (IC) in Her-2-negative leukocytes (Roche-Genentech, CA); rituximab (Roche-Genentech); alemtuzumab (Genzyme, Cambridge, MA), and F-ara-A (Genzyme). CCL19 and CCL21 were purchased from PeproTech (Rocky-Hill, NJ). A detailed list of reagents is given in Table SM3.
In vitro approaches
Flow cytometry, migration, TEM, F-actin polymerization, SICD, cell survival, binding to Fc receptors and C1q, CDC, ADCC, or autologous ex vivo depletion were conducted as reported.11,12,15,20,27–28,60 Detailed procedures are shown in the Supplementary data and Tables SM4, SM5, SM6, and SM7.
Mouse in vivo studies
Procedures were carried out at Crown Bioscience (Jiangsu, China) following an approved IACUC protocol, and at CBM-SO (Madrid, Spain) in compliance with the guidelines approved by the Spanish National Research Council.
To study LN homing of CCR7+CD45+ PB lymphocytes, 107 cells were pre-incubated with antibodies (10 µg/ml) and injected into the tail vein of 8-weeks old pre-irradiated (2.5 Gy) NSG mice (NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ; Charles River, Barcelona, Spain). After one hour, animals were euthanized. Inguinal and mesenteric LNs, spleen, BM and PB were extracted, processed, and cell suspensions incubated with anti-hCD45-PerCP antibody (BD Biosciences) for flow cytometry analysis.
The Granta-519-luc+ MCL model was developed through tail vein implantation of 2 × 106 cells/animal into sub-lethally irradiated (2 Gγ), 6–8-weeks-old C.B-17 SCID mice (CB17/Icr-Prkdcscid/IcrIcoCrl, Charles River). The bioluminescent cell line was generated as reported.61 Randomization into study groups was based on both bioluminescence analysis and body weight. Antibodies were dosed intraperitoneally (IP). Further details on bioluminescence imaging tissue analysis can be found in the Supplementary data. For the JVM-3 CLL model, 2 × 107 cells were tail vein injected into sub-lethally irradiated 6–8 weeks old C.B17-SCID mice (Vital River Laboratories, Beijing, China). Randomization was based on body weight and antibodies dosed IP.
NHP studies
Cynomolgus monkeys were used to establish a former pharmacodynamics and toxicological profile at different dose levels. In two separate studies we evaluated by flow cytometry the effect CAP-100 on immune cells. Blood samples were extracted in K2EDTA tubes before and after CAP-100 dosing (Tables SM8 and SM9). The studies were conducted at CiToxLAB laboratories (France) in compliance with animal health regulations (Directive 2010/63/EU and French decret 2013–118). The Citoxlab Ethics Committee reviewed the study plans.
Statistics
Quantitative variables are expressed as measures of central tendency (mean) and dispersion (SD, SEM). Parametric variables were analyzed using t-test or ANOVA. For heteroscedasticity, un-paired samples were tested with Mann-Whitney-U or Kruskal-Wallis and paired samples with Wilcoxon or Friedman tests. Survival curves and mOS were analyzed by the Kaplan-Meier method and log-rank tests. The data were fitted with a non-linear regression. Significance was set at values of <0.05(*), <0.01(**) or <0.001(***).
Supplementary Material
Acknowledgments
The authors would like to thank Dr. Maria Luisa Toribio, Dr. Patricia Fuentes, Dr. Juan Ruiz, Dr. Ton Adang, Dr. Wiebe Olive, and Marijke Driessen for advice and technical assistance. The authors are also grateful to Dr. Francisco Sánchez-Madrid, Dr. Arantzazu Alfranca, and Dr. Maria Luisa Toribio for reagents and advice, and Lawrence Baron for proofreading and editing of the manuscript.
Disclosure statement
At the time this work was performed or written CCM was an employee of Catapult Therapeutics and of Immunological and Medical Products (IMMED S.L.), and is a shareholder in the same companies. JL has received honoraria from Abbvie, Janssen, and Astra-Zeneca. FT and WM were CEO of Catapult Therapeutics, and FT was CEO of IMMED.S.L. and is a shareholder in the same company and Catapult Therapeutics. CMC was a consultant for IMMED S.L., held a patent for the use of therapeutic antibodies targeting CCR7 in cancer and has received research funds from IMMED.S.L. and Catapult Therapeutics. She also holds shares in IMMED S.L. RJS was an employee of IMMED S.L. The other authors declare that they have no competing interests. None of the authors received grants for this work.
Competing interests:
At the time this work was performed or written CCM was an employee of Catapult Therapeutics and of Immunological and Medical Products (IMMED S.L.), and is a shareholder in the same companies. JL has received honoraria from Abbvie, Janssen, and Astra-Zeneca. FT and WM were CEO of Catapult Therapeutics, and FT was CEO of IMMED.S.L. and is a shareholder in the same company and Catapult Therapeutics. CMC was a consultant for IMMED S.L., held a patent for the use of therapeutic antibodies targeting CCR7 in cancer and has received research funds from IMMED.S.L. and Catapult Therapeutics. She also holds shares in IMMED S.L. RJS was an employee of IMMED S.L. The other authors declare that they have no competing interests.
Funds
None of the authors received grants for this work.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website
List of abbreviations
- ADCC
antibody-dependent cell-mediated cytotoxicity
- ANOVA
analysis of variance
- ATM
ataxia telangiectasia-mutated
- BM
bone marrow
- CCL19/21
C-C chemokine ligand 19/21
- CCR7
C-C chemokine receptor 7
- CD
cluster of differentiation
- CDC
complement-dependent cytotoxicity
- CKR
chemokine receptor
- CLIPS
Chemically-Linked Peptides on Scaffolds
- CLL
chronic lymphocytic leukemia
- DCs
dendritic cells
- del
deletion
- E:T
effector-to-target
- Fab
fragment antigen-binding
- F-ara-A
fludarabine
- Fc
Fragment crystallizable
- FcγR
Fc gamma receptor
- HD
healthy donor
- HEV
high endothelial veins
- hPBMC
human peripheral blood mononuclear cells
- HRP
horseradish peroxidase
- HUVEC
human umbilical vein endothelial cells
- IC
isotype control
- IgVH
immunoglobulin variable heavy chain gene
- IP
intraperitoneally
- LN
lymph node
- luc
luciferase
- mAb
monoclonal antibody
- MCL
mantle cell lymphoma
- MOA
mode/mechanism of action
- mOS
median overall survival
- NHP
non-human primate
- NK
natural killer
- PB
peripheral blood
- Pc
plasma cells
- PC
proliferation centers
- SCID
severe combined immunodeficiency
- SD
standard deviation
- SEM
standard error of the mean
- SICD
specific-induced cell death
- SLO
secondary lymphoid organs
- SOC
standard-of-care
- TCM
central memory T cells
- TEM
trans-endothelial migration
- TEM
effector memory T cells
- TEMRA
terminally differentiated effector memory T cells
- TMB
tetramethylbenzidine
- TME
tumor microenvironment
- TN
naïve T cells
- TNF
tumor necrosis factor
- tri
trisomy
- Z
sulfated Tyr (sY)
References
- 1.Riedel A, Shorthouse D, Haas L, Hall BA, Shields J.. Tumor-induced stromal reprogramming drives lymph node transformation. Nat Immunol. 2016;17(9):1118–15. doi: 10.1038/ni.3492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hallek M, Cheson BD, Catovsky D, et al. iwCLL guidelines for diagnosis, indications for treatment, response assessment, and supportive management of CLL. Blood. 2018;131(25):2745–60. doi: 10.1182/blood-2017-09-806398. [DOI] [PubMed] [Google Scholar]
- 3.Burger JA. Nurture versus nature: the microenvironment in chronic lymphocytic leukemia. Hematology Am Soc Hematol Educ Program. 2011;2011(1):96–103. doi: 10.1182/asheducation-2011.1.96. [DOI] [PubMed] [Google Scholar]
- 4.Caligaris-Cappio F, Bertilaccio MT, Scielzo C. How the microenvironment wires the natural history of chronic lymphocytic leukemia. Semin Cancer Biol. 2014;24:43–48. doi: 10.1016/j.semcancer.2013.06.010. [DOI] [PubMed] [Google Scholar]
- 5.Forster R, Schubel A, Breitfeld D, et al. CCR7 coordinates the primary immune response by establishing functional microenvironments in secondary lymphoid organs. Cell. 1999;99(1):23–33. doi: 10.1016/S0092-8674(00)80059-8. [DOI] [PubMed] [Google Scholar]
- 6.HE Wiley, EB Gonzalez, Maki W, MT Wu, ST Hwang. Expression of CC chemokine receptor-7 and regional lymph node metastasis of B16 murine melanoma. J Natl Cancer Inst. 2001;93(21):1638–43. doi: 10.1093/jnci/93.21.1638. [DOI] [PubMed] [Google Scholar]
- 7.Rehm A, Mensen A, Schradi K, et al. Cooperative function of CCR7 and lymphotoxin in the formation of a lymphoma-permissive niche within murine secondary lymphoid organs. Blood. 2011;118(4):1020–33. doi: 10.1182/blood-2010-11-321265. [DOI] [PubMed] [Google Scholar]
- 8.Forster R, Davalos-Misslitz AC, Rot A. CCR7 and its ligands: balancing immunity and tolerance. Nat Rev Immunol. 2008;8(5):362–71. doi: 10.1038/nri2297. [DOI] [PubMed] [Google Scholar]
- 9.Hauser MA, Legler DF. Common and biased signaling pathways of the chemokine receptor CCR7 elicited by its ligands CCL19 and CCL21 in leukocytes. J Leukoc Biol. 2016;99(6):869–82. doi: 10.1189/jlb.2MR0815-380R. [DOI] [PubMed] [Google Scholar]
- 10.Till KJ, Lin K, Zuzel M, Cawley JC. The chemokine receptor CCR7 and alpha4 integrin are important for migration of chronic lymphocytic leukemia cells into lymph nodes. Blood. 2002;99:2977–84. [DOI] [PubMed] [Google Scholar]
- 11.Lopez-Giral S, Quintana NE, Cabrerizo M, et al. Chemokine receptors that mediate B cell homing to secondary lymphoid tissues are highly expressed in B cell chronic lymphocytic leukemia and non-Hodgkin lymphomas with widespread nodular dissemination. J Leukoc Biol. 2004;76(2):462–71. doi: 10.1189/jlb.1203652. [DOI] [PubMed] [Google Scholar]
- 12.Cuesta-Mateos C, Loscertales J, Kreutzman A, et al. Preclinical activity of anti-CCR7 immunotherapy in patients with high-risk chronic lymphocytic leukemia. Cancer Immunol Immunother. 2015;64(6):665–76. doi: 10.1007/s00262-015-1670-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Patrussi L, Capitani N, Martini V, et al. Enhanced chemokine receptor recycling and impaired S1P1 expression promote leukemic cell infiltration of lymph nodes in chronic lymphocytic leukemia. Cancer Res. 2015;75(19):4153–63. doi: 10.1158/0008-5472.CAN-15-0986. [DOI] [PubMed] [Google Scholar]
- 14.Richardson SJ, Matthews C, Catherwood MA, et al. ZAP-70 expression is associated with enhanced ability to respond to migratory and survival signals in B-cell chronic lymphocytic leukemia (B-CLL). Blood. 2006;107(9):3584–92. doi: 10.1182/blood-2005-04-1718. [DOI] [PubMed] [Google Scholar]
- 15.Redondo-Munoz J, Jose Terol M, Garcia-Marco JA, Garcia-Pardo A. Matrix metalloproteinase-9 is up-regulated by CCL21/CCR7 interaction via extracellular signal-regulated kinase-1/2 signaling and is involved in CCL21-driven B-cell chronic lymphocytic leukemia cell invasion and migration. Blood. 2008;111(1):383–86. doi: 10.1182/blood-2007-08-107300. [DOI] [PubMed] [Google Scholar]
- 16.Laufer JM, Lyck R, Legler DF. ZAP70 expression enhances chemokine-driven chronic lymphocytic leukemia cell migration and arrest by valency regulation of integrins. Faseb J. 2018;32(9):4824–35. doi: 10.1096/fj.201701452RR. [DOI] [PubMed] [Google Scholar]
- 17.Ganghammer S, Hutterer E, Hinterseer E, et al. CXCL12-induced VLA-4 activation is impaired in trisomy 12 chronic lymphocytic leukemia cells: a role for CCL21. Oncotarget. 2015;6(14):12048–60. doi: 10.18632/oncotarget.3660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Girbl T, Hinterseer E, Grossinger EM, et al. CD40-mediated activation of chronic lymphocytic leukemia cells promotes their CD44-dependent adhesion to hyaluronan and restricts CCL21-induced motility. Cancer Res. 2013;73(2):561–70. doi: 10.1158/0008-5472.CAN-12-2749. [DOI] [PubMed] [Google Scholar]
- 19.Ticchioni M, Essafi M, Jeandel PY, et al. Homeostatic chemokines increase survival of B-chronic lymphocytic leukemia cells through inactivation of transcription factor FOXO3a. Oncogene. 2007;26(50):7081–91. doi: 10.1038/sj.onc.1210519. [DOI] [PubMed] [Google Scholar]
- 20.Cuesta-Mateos C, Lopez-Giral S, Alfonso-Perez M, et al. Analysis of migratory and prosurvival pathways induced by the homeostatic chemokines CCL19 and CCL21 in B-cell chronic lymphocytic leukemia. Exp Hematol. 2010;38(9):756–64. 764 e751–754. doi: 10.1016/j.exphem.2010.05.003. [DOI] [PubMed] [Google Scholar]
- 21.Yan XJ, Dozmorov I, Li W, et al. Identification of outcome-correlated cytokine clusters in chronic lymphocytic leukemia. Blood. 2011;118(19):5201–10. doi: 10.1182/blood-2011-03-342436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brown JR, Relapsed CLL. sequencing, combinations, and novel agents. Hematology Am Soc Hematol Educ Program. 2018;2018(1):248–55. doi: 10.1182/asheducation-2018.1.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jain N. Selecting Frontline Therapy for CLL in 2018. Hematology Am Soc Hematol Educ Program. 2018;2018(1):242–47. doi: 10.1182/asheducation-2018.1.242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Somovilla-Crespo B, Alfonso-Perez M, Cuesta-Mateos C, et al. Anti-CCR7 therapy exerts a potent anti-tumor activity in a xenograft model of human mantle cell lymphoma. J Hematol Oncol. 2013;6(1):89. doi: 10.1186/1756-8722-6-89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jaeger K, Bruenle S, Weinert T, et al. Structural basis for allosteric ligand recognition in the human CC chemokine receptor 7. Cell. 2019;178(5):1222–30. e1210. doi: 10.1016/j.cell.2019.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hutchings CJ, Koglin M, Olson WC, Marshall FH. Opportunities for therapeutic antibodies directed at G-protein-coupled receptors. Nat Rev Drug Discov. 2017;16:1–24. [DOI] [PubMed] [Google Scholar]
- 27.Alfonso-Perez M, Lopez-Giral S, Quintana NE, Loscertales J, Martin-Jimenez P. Anti-CCR7 MC. Monoclonal antibodies as a novel tool for the treatment of chronic lymphocyte leukemia. J Leukoc Biol. 2006;79(6):1157–65. doi: 10.1189/jlb.1105623. [DOI] [PubMed] [Google Scholar]
- 28.Kashyap MK, Amaya-Chanaga CI, Kumar D, et al. Targeting the CXCR4 pathway using a novel anti-CXCR4 IgG1 antibody (PF-06747143) in chronic lymphocytic leukemia. J Hematol Oncol. 2017;10(1):112. doi: 10.1186/s13045-017-0435-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Luther SA, Bidgol A, Hargreaves DC, et al. Differing activities of homeostatic chemokines CCL19, CCL21, and CXCL12 in lymphocyte and dendritic cell recruitment and lymphoid neogenesis. J Immunol. 2002;169(1):424–33. doi: 10.4049/jimmunol.169.1.424. [DOI] [PubMed] [Google Scholar]
- 30.Chunsong H, Yuling H, Li W, et al. CXC chemokine ligand 13 and CC chemokine ligand 19 cooperatively render resistance to apoptosis in B cell lineage acute and chronic lymphocytic leukemia CD23+CD5+ B cells. J Immunol. 2006;177(10):6713–22. doi: 10.4049/jimmunol.177.10.6713. [DOI] [PubMed] [Google Scholar]
- 31.Hopken UE, Rehm A. Homeostatic chemokines guide lymphoma cells to tumor growth-promoting niches within secondary lymphoid organs. J Mol Med (Berl). 2012;90(11):1237–45. doi: 10.1007/s00109-012-0906-z. [DOI] [PubMed] [Google Scholar]
- 32.Bertilaccio MT, Scielzo C, Simonetti G, et al. Xenograft models of chronic lymphocytic leukemia: problems, pitfalls and future directions. Leukemia. 2013;27(3):534–40. doi: 10.1038/leu.2012.268. [DOI] [PubMed] [Google Scholar]
- 33.Ackler S, Oleksijew A, Chen J, et al. Clearance of systemic hematologic tumors by venetoclax (Abt-199) and navitoclax. Pharmacol Res Perspect. 2015;3(5):e00178. doi: 10.1002/prp2.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ma J, Lu P, Guo A, et al. Characterization of ibrutinib-sensitive and -resistant mantle lymphoma cells. Br J Haematol. 2014;166(6):849–61. doi: 10.1111/bjh.12974. [DOI] [PubMed] [Google Scholar]
- 35.Verner J, Trbusek M, Chovancova J, et al. NOD/SCID IL2Rgamma-null mouse xenograft model of human p53-mutated chronic lymphocytic leukemia and ATM-mutated mantle cell lymphoma using permanent cell lines. Leuk Lymphoma. 2015;56(11):3198–206. doi: 10.3109/10428194.2015.1034701. [DOI] [PubMed] [Google Scholar]
- 36.Klanova M, Soukup T, Jaksa R, et al. Mouse models of mantle cell lymphoma, complex changes in gene expression and phenotype of engrafted MCL cells: implications for preclinical research. Lab Invest. 2014;94(7):806–17. doi: 10.1038/labinvest.2014.61. [DOI] [PubMed] [Google Scholar]
- 37.Loisel S, Ster KL, Quintin-Roue I, et al. Establishment of a novel human B-CLL-like xenograft model in nude mouse. Leuk Res. 2005;29(11):1347–52. doi: 10.1016/j.leukres.2005.04.017. [DOI] [PubMed] [Google Scholar]
- 38.Voltan R, Rimondi E, Melloni E, et al. Ibrutinib synergizes with MDM-2 inhibitors in promoting cytotoxicity in B chronic lymphocytic leukemia. Oncotarget. 2016;7(43):70623–38. doi: 10.18632/oncotarget.12139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hartmann TN, Grabovsky V, Wang W, et al. Circulating B-cell chronic lymphocytic leukemia cells display impaired migration to lymph nodes and bone marrow. Cancer Res. 2009;69(7):3121–30. doi: 10.1158/0008-5472.CAN-08-4136. [DOI] [PubMed] [Google Scholar]
- 40.Montresor A, Bolomini-Vittori M, Simon SI, Rigo A, Vinante F, Laudanna C. Comparative analysis of normal versus CLL B-lymphocytes reveals patient-specific variability in signaling mechanisms controlling LFA-1 activation by chemokines. Cancer Res. 2009;69(24):9281–90. doi: 10.1158/0008-5472.CAN-09-2009. [DOI] [PubMed] [Google Scholar]
- 41.De Rooij MF, Kuil A, Geest CR, et al. The clinically active BTK inhibitor PCI-32765 targets B-cell receptor- and chemokine-controlled adhesion and migration in chronic lymphocytic leukemia. Blood. 2012;119(11):2590–94. doi: 10.1182/blood-2011-11-390989. [DOI] [PubMed] [Google Scholar]
- 42.Haerzschel A, Catusse J, Hutterer E, et al. BCR and chemokine responses upon anti-IgM and anti-IgD stimulation in chronic lymphocytic leukaemia. Ann Hematol. 2016;95(12):1979–88. doi: 10.1007/s00277-016-2788-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Burger JA, Burger M, Kipps TJ. Chronic lymphocytic leukemia B cells express functional CXCR4 chemokine receptors that mediate spontaneous migration beneath bone marrow stromal cells. Blood. 1999;94(11):3658–67. doi: 10.1182/blood.V94.11.3658. [DOI] [PubMed] [Google Scholar]
- 44.Burger JA, Quiroga MP, Hartmann E, et al. High-level expression of the T-cell chemokines CCL3 and CCL4 by chronic lymphocytic leukemia B cells in nurselike cell cocultures and after BCR stimulation. Blood. 2009;113(13):3050–58. doi: 10.1182/blood-2008-07-170415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pham TH, Okada T, Matloubian M, Lo CG, Cyster JG. S1P1 receptor signaling overrides retention mediated by G alpha i-coupled receptors to promote T cell egress. Immunity. 2008;28(1):122–33. doi: 10.1016/j.immuni.2007.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Beyer M, Kochanek M, Darabi K, et al. Reduced frequencies and suppressive function of CD4+CD25hi regulatory T cells in patients with chronic lymphocytic leukemia after therapy with fludarabine. Blood. 2005;106(6):2018–25. doi: 10.1182/blood-2005-02-0642. [DOI] [PubMed] [Google Scholar]
- 47.Stilgenbauer S, Dohner H. Campath-1H-induced complete remission of chronic lymphocytic leukemia despite p53 gene mutation and resistance to chemotherapy. N Engl J Med. 2002;347(6):452–53. doi: 10.1056/NEJM200208083470619. [DOI] [PubMed] [Google Scholar]
- 48.Corcione A, Arduino N, Ferretti E, et al. CCL19 and CXCL12 trigger in vitro chemotaxis of human mantle cell lymphoma B cells. Clin Cancer Res. 2004;10(3):964–71. doi: 10.1158/1078-0432.CCR-1182-3. [DOI] [PubMed] [Google Scholar]
- 49.Okada S, Ngo VN, Ekland EH, et al. Chemokine requirements for B cell entry to lymph nodes and Peyer’s patches. J Exp Med. 2002;196(1):65–75. doi: 10.1084/jem.20020201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Forster R, Mattis AE, Kremmer E, Wolf E, Brem G, Lipp M. A putative chemokine receptor, BLR1, directs B cell migration to defined lymphoid organs and specific anatomic compartments of the spleen. Cell. 1996;87(6):1037–47. doi: 10.1016/S0092-8674(00)81798-5. [DOI] [PubMed] [Google Scholar]
- 51.Moschovakis GL, Bubke A, Friedrichsen M, et al. The chemokine receptor CCR7 is a promising target for rheumatoid arthritis therapy. Cell Mol Immunol. 2019;16(10):791–99. doi: 10.1038/s41423-018-0056-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cuesta-Mateos C, Portero-Sainz I, García-Peydró M, et al. Evaluation of therapeutic targeting of CCR7 in acute graft-versus-host disease. Bone Marrow Transplant. 2020;55(10):1935–45. doi: 10.1038/s41409-020-0830-8. [DOI] [PubMed] [Google Scholar]
- 53.Hopken UE, Achtman AH, Kruger K, Lipp M. Distinct and overlapping roles of CXCR5 and CCR7 in B-1 cell homing and early immunity against bacterial pathogens. J Leukoc Biol. 2004;76(3):709–18. doi: 10.1189/jlb.1203643. [DOI] [PubMed] [Google Scholar]
- 54.Fowler KA, Vasilieva V, Ivanova E, et al. R707, a fully human antibody directed against CC-chemokine receptor 7, attenuates xenogeneic acute graft-versus-host disease. Am J Transplant. 2019. July;19(7):1941–54. doi: 10.1111/ajt.15298. [DOI] [PubMed] [Google Scholar]
- 55.Shields JD, Kourtis IC, Tomei AA, Roberts JM, Swartz MA. Induction of lymphoidlike stroma and immune escape by tumors that express the chemokine CCL21. Science. 2010;328(5979):749–52. doi: 10.1126/science.1185837. [DOI] [PubMed] [Google Scholar]
- 56.Du H, Zhang L, Li G, et al. CXCR4 and CCR7 expression in primary nodal diffuse large B-cell lymphoma-A clinical and immunohistochemical study. Am J Med Sci. 2019;357(4):302–10. doi: 10.1016/j.amjms.2019.01.008. [DOI] [PubMed] [Google Scholar]
- 57.Sokolowska-Wojdylo M, Wenzel J, Gaffal E, et al. Circulating clonal CLA(+) and CD4(+) T cells in Sezary syndrome express the skin-homing chemokine receptors CCR4 and CCR10 as well as the lymph node-homing chemokine receptor CCR7. Br J Dermatol. 2005;152(2):258–64. doi: 10.1111/j.1365-2133.2004.06325.x. [DOI] [PubMed] [Google Scholar]
- 58.Hasegawa H, Nomura T, Kohno M, et al. Increased chemokine receptor CCR7/EBI1 expression enhances the infiltration of lymphoid organs by adult T-cell leukemia cells. Blood. 2000;95(1):30–38. doi: 10.1182/blood.V95.1.30.001k09_30_38. [DOI] [PubMed] [Google Scholar]
- 59.Legler DF, Uetz-von Allmen E, Hauser MA. CCR7: roles in cancer cell dissemination, migration and metastasis formation. Int J Biochem Cell Biol. 2014;54:78–82. doi: 10.1016/j.biocel.2014.07.002. [DOI] [PubMed] [Google Scholar]
- 60.Mossner E, Brunker P, Moser S, et al. Increasing the efficacy of CD20 antibody therapy through the engineering of a new type II anti-CD20 antibody with enhanced direct and immune effector cell-mediated B-cell cytotoxicity. Blood. 2010;115(22):4393–402. doi: 10.1182/blood-2009-06-225979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Garaulet G, Alfranca A, Torrente M, et al. IL10 released by a new inflammation-regulated lentiviral system efficiently attenuates zymosan-induced arthritis. Mol Ther. 2013;21(1):119–30. doi: 10.1038/mt.2012.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.