

Abstract
Inflammatory bowel disease (IBD) is a chronic inflammatory disease of the gut. Genetic association studies have identified the highly variable human leukocyte antigen (HLA) region as the strongest susceptibility locus for IBD and specifically DRB1*01:03 as a determining factor for ulcerative colitis (UC). However, for most of the association signal such as delineation could not be made because of tight structures of linkage disequilibrium within the HLA. The aim of this study was therefore to further characterize the HLA signal using a transethnic approach. We performed a comprehensive fine mapping of single HLA alleles in UC in a cohort of 9272 individuals with African American, East Asian, Puerto Rican, Indian and Iranian descent and 40 691 previously analyzed Caucasians, additionally analyzing whole HLA haplotypes. We computationally characterized the binding of associated HLA alleles to human self-peptides and analyzed the physicochemical properties of the HLA proteins and predicted self-peptidomes. Highlighting alleles of the HLA-DRB1*15 group and their correlated HLA-DQ-DR haplotypes, we not only identified consistent associations (regarding effects directions/magnitudes) across different ethnicities but also identified population-specific signals (regarding differences in allele frequencies). We observed that DRB1*01:03 is mostly present in individuals of Western European descent and hardly present in non-Caucasian individuals. We found peptides predicted to bind to risk HLA alleles to be rich in positively charged amino acids. We conclude that the HLA plays an important role for UC susceptibility across different ethnicities. This research further implicates specific features of peptides that are predicted to bind risk and protective HLA proteins.
Introduction
Ulcerative colitis (UC) is a chronic inflammatory disease of the gut. Like Crohn’s disease (CD), the other main subphenotype of inflammatory bowel disease (IBD), it is most likely caused by an abnormal reaction of the immune system to microbial stimuli with environmental factors also playing a role. Currently, >240 genetic susceptibility loci have been associated with IBD in Caucasians. The majority of these loci are shared between UC and CD (1–4). Strong genetic association signals with both diseases have been identified in the human leukocyte antigen (HLA) region. The HLA is mapped to the long arm of chromosome 6 between 29 and 34 Mb and moderates complex functions within the immune system. One of the major tasks of the HLA is the presentation of antigens to the host immune system. While HLA class I proteins usually present peptides derived from the cytosol (i.e. peptides derived from intracellularly replicating viruses), HLA class II proteins present peptides from extracellular pathogens that have entered the cell e.g. by phagocytosis. In Caucasian IBD a large percentage of the phenotypic variation is explained by variants within the HLA class II locus, with DRB1*01: 03 being the most significant risk allele for UC [P = 2.68 × 10−119, odds ratio (OR) = 3.59; 95% confidence interval (CI) = 3.22–4.00] (5), specifically by alleles of the HLA-DR and -DQ loci, although tight structures of linkage disequilibrium (LD) have hindered the assignment of the causal variants. Additionally, a systematic comparison across ethnicities for the HLA association in UC has not been performed, also because of the lack of HLA imputation panels that could accurately infer HLA alleles for transethnic genetic data sets (5–14). Recently, we created such a transethnic HLA imputation reference panel including dense single nucleotide polymorphism (SNP) fine mapping data typed on Illumina’s ImmunoChip, covering a large proportion of the HLA, within eight populations of different ethnicities (15). Here we report the first transethnic fine mapping study of the HLA in UC and some biological implications of the results.
Materials and Methods
Cohort description
A detailed description of the cohorts and recruitment sites can be found in the Supplementary Methods and Supplementary Material, Table S1. In brief, a total of 52 550 individuals (including 18 142 UC patients and 34 408 controls) were used in this study, of which 10 063 (3517 UC cases and 6546 controls) were of non-Caucasian origin. The Caucasian, Iranian, Indian and Asian data set (from which we extracted Japanese and Chinese individuals) are of part of the data freeze published in (2), whereas individuals of African American (16), Korean (17), Maltese and Puerto Rican descent were added. The recruitment of study subjects was approved by the ethics committees or institutional review boards of all individual participating centers or countries. Written informed consent was obtained from all study participants.
Genotyping and quality control
All individuals were typed on the Illumina HumanImmuno BeadChip v.1.0 or the Illumina Infimum ImmunoArray 24 v2.0 (Malta). Genotypes of the study subjects were quality controlled as described in the Supplementary Methods. A median of 8555 SNPs were extracted from the extended HLA region (chromosome 6, 25–34 Mb) and submitted to SNP Phasing and imputation and HLA allele imputation (Workflow in Supplementary Material, Fig. S1).
Phasing of single nucleotide variants
Using SHAPEIT2 (18) version r727, we phased quality-controlled genotype data on chromosome 6, 25–34 Mb of the respective cohorts using variants with a minor allele frequency (MAF) >1%. We excluded SNPs that did not match 1000 Genomes Phase III (19) (October 2014) alleles [published with the SNP imputation tool IMPUTE2 (20,21)] and ATCG variants that did not match the AFR, EUR, SAS, EAS or AMR populations (+ strand assumed for both). AFR (used for comparison with our African American samples), EUR (Caucasian, Iranian, Maltese), SAS (Indian), EAS (Chinese, Korean, Japanese) and AMR (Puerto Rican). Using default values of SHAPEIT2 (--input-thr 0.9, --missing-code 0, --states 100, --window 2, --burn 7, --prune 8, --main 20 and –effective-size 18 000), we first generated a haplotype graph and, as suggested by the authors of SHAPEIT2, calculated a value of phasing certainty on the basis of 100 haplotypes generated from the haplotype graph for each population separately. Then, we excluded SNPs with a median phasing certainty <0.8 within each population separately.
Imputation of single nucleotide variants
To increase the density of single nucleotide variants (SNVs, including variants with MAF <1%) within the HLA region, we used publicly available nucleotide sequences of HLA alleles and further imputed SNVs on the basis of the HLA alleles imputed for each individual using IMPUTE2 (22) with the 1000 Genomes Phase III (19) individuals as a reference (October 2014) using parameters: -Ne 20,000, -buffer 250, -burnin 10, -k 80, -iter 30, -k_hap 500, -outdp 3, -pgs_miss, -os 0 1 2 3, allowing additionally for the imputation of large regions (-allow_large_regions). Imputation of the Caucasian data set was performed in batches of 10 000 samples. Imputation quality control was performed post-imputation excluding variants with an IMPUTE2 info score < 0.8. For the Caucasian data set, we excluded variants with a median IMPUTE2 info score < 0.8 and a minimum IMPUTE2 info score < 0.3. Additionally, we imputed SNPs into the data set using imputed HLA allele information (i.e. translated imputed HLA information into real nucleotide information at each position of the allele) (Supplementary Material, Methods).
HLA imputation
QC-ed genotype data for each cohort were imputed using Beagle version 4.1 (22,23) on the basis of the corresponding genotype variants observed in the respective cohort to fill in missing genotypes with the study reference itself serving as a reference. We imputed HLA alleles at loci HLA-A, -C, -B, -DRB3, -DRB5, -DRB4, -DRB1, -DQA1, -DQB1, -DPA1 and -DPB1 at full context four-digit level using the IKMB reference published in (15) and the imputation tool HIBAG (24). Imputation of the Caucasian panel was additionally performed with the HLARES panel published with HIBAG (ImmunoChip-European_HLARES-HLA4-hg19.RData). Alleles were not excluded by setting a posterior probability threshold. However, we took the sensitivity and specificity measures we generated as previously reported (15) into consideration during interpretation.
Generation of HLA haplotypes
HLA haplotypes were generated by comparing SNP haplotypes generated by SHAPEIT2 (18) for each individual and SNP haplotypes stored for the alleles within the classifiers of the HLA reference model (15) for the alleles that were imputed for each individual at a given locus. For 10 random classifiers, we calculated the minimal distance between the SNP haplotypes stored for the allele of interest in the HLA reference model and the SNP haplotypes generated by SHAPEIT2. We assigned alleles to a parental haplotype on the basis of how often this allele had minimal difference to the haplotype. Phasing certainty was calculated as the percentage of times an allele was correctly assigned to the chosen parental haplotype. In cases no decision could be made or both alleles were assigned to the same haplotype, phasing certainty was set to 0. If an individual was homozygous at a locus, phasing certainty was set to 1. Only SNPs present in both the classifier and the SNP haplotypes generated by HIBAG were used after aligning the alleles to alleles stored in the HIBAG model.
HLA haplotype benchmark
We tested the generation of HLA haplotypes with the above method, using genotype information of trio samples [Utah Residents (CEPH) with Northern and Western European Ancestry (CEU) and Yoruba in Ibadan, Nigeria (YRI)] extracted from the Hapmap Phase 3 project and HLA allele information published for these individuals in the 1000 Genomes HLA diversity panel (25) (extracted from ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/technical/working/20140725_hla_genotypes/20140702_hla_diversity.txt) using the most common allele for ambiguous calls. The relationship between the individuals was extracted from the file relationships_w_pops_121708.txt published with the HapMap data. In total, 27 CEU samples and 24 YRI samples and their parents were analyzed. SNP genotype data were downloaded from the HapMap Phase 3 Server (version 2015-05) and positions present on the Illumina ImmunoChip were extracted. We applied the procedure described above for phasing of HLA alleles. The results are shown in Supplementary Material, Table S2. In brief, we phased the HLA alleles of an individual manually using the information on the parental HLA allele genotypes (g.mom, g.dad g.child) as shown in Supplementary Material, Table S2. We also phased the HLA alleles using the approach described above. The phasing certainty was calculated from assignments across 10 classifiers and the number of positions analyzed per classifier were noted.
Calculation of marginal probabilities for each allele
Since HIBAG stores a matrix of all posterior probability values of each allele combination per individual, we calculated the marginal sums of posterior probability for each allele per individual. The overall marginal probability of an allele was then calculated as the mean of the marginal sums of the posterior probability calculated for alleles predicted to carry this allele.
Association analysis
Subsequently, we performed a standard logistic regression association analysis on single alleles and SNVs. HLA alleles were coded as present (P) or absent (A) with genotype dosages (PP = 2, AP = 1 and AA = 0) by simply counting the number of times an allele occurred for a specific individual. SNPs imputed with IMPUTE2 were included as dosages. SNVs inferred from HLA alleles were coded as 0, 1, 2 on the minor allele. Additive association analyses for each marker were performed using.
log(oddsi) = β0 + β1xi + β2U1i + β3U2i + β4U3i + β5U4i + β6U5i +β (β7bi).
for individual i = 1,…,N, genotype dose or call (x) and eigenvectors (U1–U5). For the analysis of the Puerto Rican and Indian cohort, we additionally adjusted for batch (b) (Supplementary Methods; batches duringQC).
Meta-analysis
We performed a random effects (RE) meta-analysis of association statistics from the nine analyzed cohorts using the tools RE2 and RE2C (26,27). Classical fixed-effects (FE) meta-analyses are not optimal for the analysis across study estimates where underlying allele frequencies are different between cohorts or similar only for some of the analyzed cohorts (28) as in the case of transethnic analyses. Using RE2 (26) and REC2 (27), tools optimized for the analysis of heterogeneous effects, we combined the association statistics for all nine cohorts for SNPs and HLA alleles with MAF (SNPs) and allele frequency (AF) (HLA alleles) >1% in the respective cohorts to calculate a combined P-value. For the analysis with RE2C, we set the correlation between studies to uniform. We report both FE and RE measures in the Supplementary Tables.
Clustering according to preferential peptide binders
Using NetMHCIIpan-3.2 (29), we predicted binding affinities for five sets of the 200 000 unique random 15mer peptides (Supplementary Methods) for all alleles that were significant in the meta-analysis and had a frequency of >1% in at least one of the nine populations. We give the amino acid distribution of these sets in Supplementary Material, Table S3. We selected the top 2% (strong binders) preferential peptide binders as given by the NetMHCIIpan-3.2 software for each allele and calculated the pairwise Pearson correlation between alleles on the basis of the affinity values of the top 2% binders shared by the respective allele combinations (29) using R (version 3.3.1) creating a matrix of correlations. Clustering was performed on this matrix using hclust of the R package stats.
We additionally analyzed correlations between cluster dendrograms produced for each of the five sets of 200 000 unique random peptides using the R packages corrplot (version 0.84) and dendextend (version 1.12). Here, the correlation between cluster dendrograms (i.e. the concordance of the tree-structure) is calculated with a value of 0 signifying dissimilar tree-structures and 1 signifying highly similar tree-structures. Dendrograms were plotted using the ape (version 5.3) package, for HLA-DQ andDRB1.
Generation of combined peptide motifs
On the basis of the clusters generated above for the human peptides, we grouped the risk alleles and protective alleles into two clusters each (Supplementary Methods). For each of the five peptide sets, we concatenated the top 2% ranked binders (percentile rank of NetMHCIIpan-3.2) for alleles within each protective and risk group and excluded peptides that were among the 10% top-ranked binders (percentile rank of NetMHCIIpan-3.2) in two or more of the groups. On the basis of this, we generated peptide binding motifs using Seq2Logo (30) for each of the groups and also plotted the position-specific scoring matrix scores for chosen amino acids within a group.
Clustering according to physico-chemical properties
Clustering of HLA proteins was performed using five different numerical scores: the Atchley scores F1 and F3 (31), residue-volume (32) and self-defined parameters charge and hydrogen-acceptor capability (Supplementary Material, Table S4). The amino acid sequence of each respective allele was extracted at positions noted in Supplementary Material, Table S5 for Pockets 1, 4, 6, 7 and 9 from HLA allele protein sequences that were retrieved from the IMGT/HLA database (version3.37.0) (33) and aligned using MUSCLE (34). The alpha chain, of the HLA-DR locus is invariable and was not considered in the analysis of this locus. For the respective analysis, each amino acid was assigned its numerical score. Clustering was then performed on the scores using the hclust function of the R (version 3.3.1) package stats and Euclidian distances.
Results
Here we imputed HLA alleles for a total of nine cohorts (Supplementary Material, Fig. S2, Supplementary Material, Table S1) within three HLA class I (HLA-A, -C and -B) and eight class II loci (HLA-DRB3, -DRB5, -DRB4, -DRB1, -DQA1, -DQB1, -DPA1 and -DPB1) at full context four-digit level utilizing a median of 8 555 SNP genotypes (located within extended HLA between 25 and 34 Mb on chromosome 6p21) from Illumina’s ImmunoChip. After QC, a total of 17 276 UC cases and 32 975 controls remained. 13 927 cases and 26 764 controls were previously reported Caucasians (5) and 3251 cases and 6021 controls were non-Caucasian individuals (2). After SNP imputation and respective quality control a median of 88 087 SNVs with INFO score > 0.8 were additionally analyzed.
In line with our previous study in Caucasians (5), we observed strong, consistent association signals for SNPs and HLA alleles within the HLA class II region, featuring HLA-DRB1, HLA-DQA1 and HLA-DQB1, for all UC case-control panels except the small-sized Puerto Rican and Maltese cohorts (Fig. 1 and Supplementary Material, Fig. S3). The strongest association signal was seen for SNP rs28479879 (PVALUE_RE2 = 5.25 × 10−156, I2 = 79.56), located in the HLA-DR locus, including HLA-DRB1 and HLA-DRB3/4/5. In the Japanese and Korean panels, we further observed a ‘roof-top’-like association signal spanning the HLA class I and II loci (Fig. 1) that, as we subsequently demonstrated, was caused by strong LD between the most disease-associated class II alleles DRB1*15:02, DQA1*01:03 and DQB1*06:01 and the class I alleles B*52:01 and C*12:02. The ‘roof-top’-like signal disappeared when conditioning on class I and class II alleles separately (Supplementary Material, Fig. S4). Likely due to the lack of statistical power, e.g. for the Maltese data set, and/or diversity of the population, e.g. for the Puerto Ricans, association P-values for these populations did not achieve the genome-wide significance threshold (P < 5 × 10−8).
Figure 1.
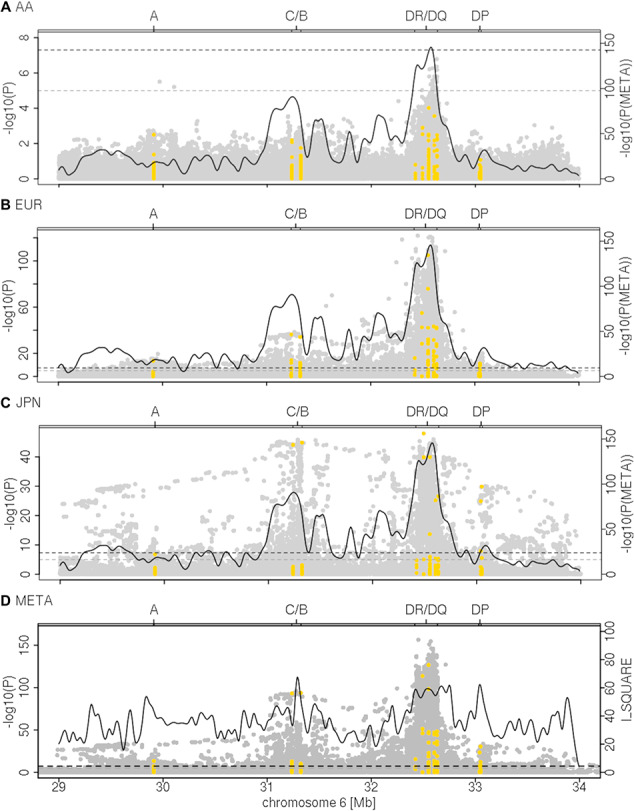
HLA regional association plots. Association analysis results for imputed and genotyped SNVs (gray) and four-digit HLA alleles (yellow) are shown for (A) 373 African American cases and 590 controls (AA), (B) 13 927 Caucasian cases and 26 764 controls (EUR) and (C) 709 Japanese cases 3169 and controls (JPN) as well as (D) the meta-analysis (META) results from the analysis with RE2 (26) at variants with a MAF > 1% in the respective cohorts (including 17 276 cases and 32 975 controls from nine different cohorts). The association plots for the remaining populations are provided in Supplementary Material, Fig. S3. The curves in (A–C) show the P-value of the meta-analysis (PVAULE_RE2). In (D), the overlying curve shows the I2 as a measure of heterogeneity in the meta-analysis indicating the heterogeneity of effects and allele frequencies in that region. Dashed lines indicate the thresholds of genome-wide (P = 5 × 10-8) and nominal significance (P = 10−5) The association analyses indicate HLA class II as the most associated susceptibility region across the different populations. In the Korean and the Japanese populations, a strong association signal is also seen for B*52:01 and C*12:02, both alleles being in strong LD with the HLA class II loci DRB1*15:02, DQA1*01:02 and DQB1*06:01, i.e. another population-specific haplotype association in these ethnicities exists.
The most strongly and consistently associated class II risk alleles within the meta-analysis were alleles of the DRB1*15 group (PVALUE_RE2 = 1.10 × 10−116, I2 = 92.13) (Fig. 2, Supplementary Material, Table S6), observed to be located on the same haplotype as DQA1*01:02/03 and DQB1*06:01/02 (Fig. 3, Supplementary Material, Table S7). DRB1*15:02 was most frequent in the Asian populations (Japanese, Korean), whereas DRB1*15:03 was specific to the African American population and DRB1*15:01 had the stronger association and higher allele frequency in the Chinese and Caucasian population (Fig. 2, Supplementary Material, Table S6), which is consistent to data published in the HLA allele frequency database (35). Since effect sizes were heterogeneous across populations, we did not compute a combined score, but rather show the OR in Supplementary Material, Fig. S5. Other associated class II alleles included DQA1*03 alleles (PVALUE_RE2 = 3.51 × 10−81, I2 = 6.47) that were observed to be located on a haplotype with DRB1*04 (PVALUE_RE2* = 1.37 × 10−55, I2 = 0.00), DRB1*07:01 (PVALUE_RE2 = 3.66 × 10−35, I2 = 68.44) or DRB1*09:01 (PVALUE_RE2* = 1.65 × 10−12, I2 = 0.00). DRB1*04/07/09 alleles are all located on the same haplotype as HLA-DRB4 alleles (15), therefore absence of HLA-DRB4, hereafter named DRB4*00:00, was significantly associated with high risk (PVALUE_RE2 = 7.43 × 10−128, I2 = 27.74). Along the same line HLA-DRB5 is located on the same haplotype as DRB1*15. Its absence was therefore observed to be protective. We identified DRB1*10:01 as a novel association signal (PVALUE_RE2 = 6.41 × 10−7, I2 = 15.08). It was observed to be most frequent in the Iranian (3.2% controls and 1.6% cases) and Indian (6.7% controls and 3.3% cases) populations and rare in other populations (Supplementary Material, Table S6), which is most likely why it has not been described before. Among population-specific signals, we also observed significant association of UC with DRB1*14:04 (P = 0.004, OR = 1.64; 95%CI: 1.18–2.29) in the Indian population (Fig. 2). Overall, alleles of 11 of the 13 known HLA-DRB1 two-digit groups and all 5 known -DQB1 groups were associated with UC across the different cohorts (Fig. 2, Supplementary Material, Table S6, Supplementary Material, Fig. S6), with more HLA-DRB1 alleles conferring protection than risk. Effect sizes in the larger Caucasian and Japanese populations were observed to be moderate (0.5 < OR < 2.0 for alleles with AF > 1%, with the exception of DRB1*15:02 (OR = 2.87; 95%CI: 2.46–3.36 in the Japanese population). The comparison of beta estimates also showed that Japanese and Korean effects estimates were most similar (weighted correlation of 0.84, P = 1.3 × 10−30), whereas Iranian and Indian effects estimates correlated better with those of the Caucasian population (weighted correlation of 0.65, P = 1.0 × 10−16 and 0.69, P = 2.0 × 10−17, Supplementary Material, Fig. S7, Supplementary Methods). Notably, we identified DRB1*01:03, which was identified as the strongest association signal for IBD in our previous fine mapping analysis (5) to be population specific. It was not present in the Asian populations and was only observed with a frequency of <0.1% in the African American and Puerto Rican populations. Detailed analysis of the geographic distribution of the DRB1*01:03 allele showed that it seemingly occurs in Western Europe (Great Britain, Ireland, France, Spain) and former Western colonies with AF > 1%, whereas it seems to be infrequent in the Eastern parts of Europe. We therefore hypothesize that this allele is linked to the history of Western European countries (Fig. 6). Within this study, the frequency of DRB1*01:03 in the Caucasian population is likely underestimated and therefore not the top associated signal in the Caucasian analysis (i.e. DRB1*01:03 was imputed as DRB1*01:01 or DRB1*01:02 because of similarities in SNP haplotype between these alleles) because of applying a reference panel containing mostly non-Caucasian individuals and European individuals from Germany only. Indeed, using the European HLARES imputation panel, which contains a more diverse Caucasian population, we re-established the signal. The frequency of the remaining alleles imputed with our transethnic reference data set highly correlated with our original study in the Caucasian population (Supplementary Material, Fig. S8). Other DRB1*15, for instance DRB1*15:06, did not show association with UC. Interestingly, however, DRB1*15:06 has the same amino acid sequence as DRB1*15:01 in the peptide binding groove and may therefore biologically indeed play a role in IBD. With low overall global frequency of the DRB1*15:06 allele, it was not statistically associated with UC. It was most frequent in the Indian population (AF = 2.1%, OR = 1.27, 95%CI: 0.77–2.11). The theoretical power to detect an effect at the given sample size 1621, with OR 1.27 and AF 2.1% is estimated to be 0.50 for a significance level of 0.05. This is also true for other alleles listed in Supplementary Material, Table S8. The deviation from non-additivity of effects at the HLA locus observed in (5) could not be replicated in this study (data not shown).
Figure 2.
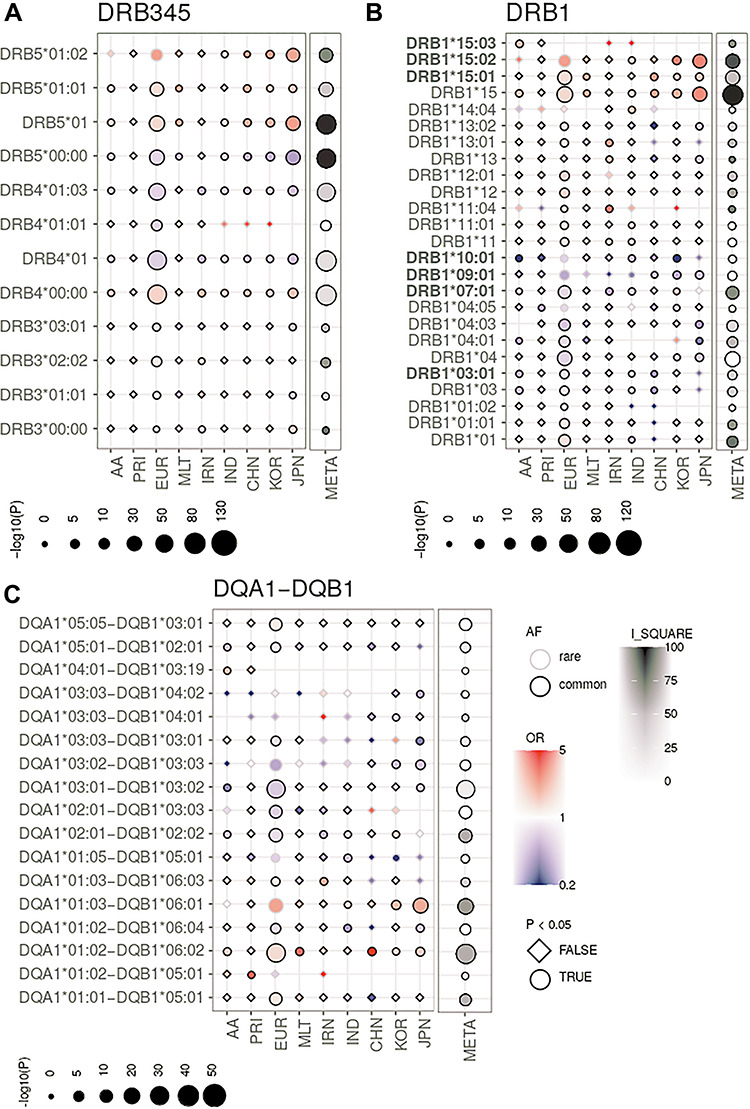
HLA single allele association analysis results at 2- and 4-digit resolution for MHC class II loci (A) HLA-DRB3/4/5, (B) HLA-DRB1 and (C) HLA-DQA1-DQB1. (AF; common defined as AF > 1%), OR, P-value (P) and whether an allele had a P-value < 0.05 (circle symbol) is shown for the respective population (e.g. circles with black boundary and red color represent an allele that is common and associated with risk). We depict association results of the analysis of the African American (AA), Puerto Rican (PRI), Caucasian (EUR), Maltese (MLT), Iranian (IRN), North Indian (IND), Chinese (CHN), Korean (KOR) and Japanese (JPN) cohorts and the meta-analysis (META) with I_SQUARE as an indicator of allelic heterogeneity and the P-value of association (PVALUE_RE2), combined here with single study P-values. Only HLA alleles, which are significant in the meta-analysis, have an AF > 1% in at least one population and have a marginal post-imputation probability > 0.6 are shown. The strongest association signals in the meta-analysis are for risk alleles of the DRB1*15 group, i.e. DRB1*15:01, DRB*15:02 and DRB1*15:03 and the alleles located on the same respective haplotype (Fig. 3). Alleles with OR > 5.0 or OR < 0.2 (rare and nonsignificant alleles may have larger/smaller OR) values were ‘ceiled’ at 5.0 and 0.2, respectively. The ‘consistent alleles’ that are highlighted in Figure 3 are highlighted in bold type on the left side. Null alleles at the HLA-DRB3/4/5 loci are described as DRB3*00:00, DRB4*00:00 and DRB5*00:00, respectively.
Figure 3.
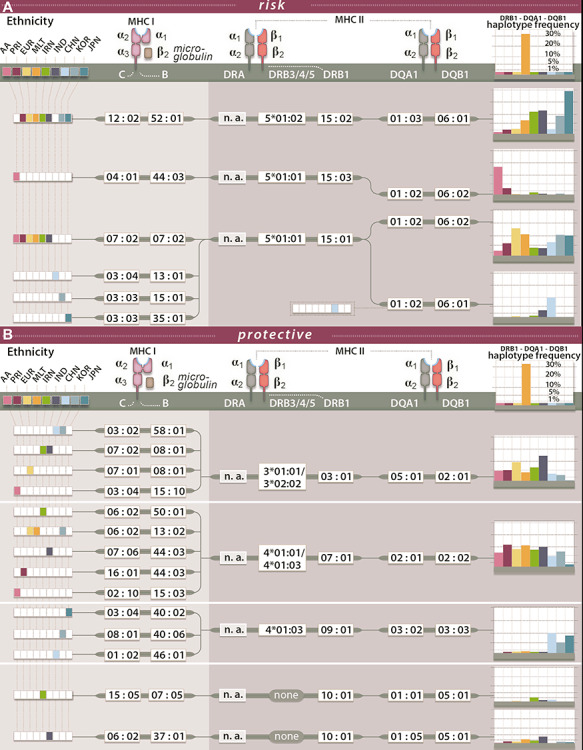
Haplotypes for associated HLA alleles. For a selection of associated HLA alleles, we show the most frequently observed risk (A) and protective (B) haplotypes in the respective populations. African American (AA), Puerto Rican (PRI), Caucasian (EUR), Maltese (MLT), Iranian (IRN), North Indian (IND), Chinese (CHN), Korean (KOR) and Japanese (JPN). Here we show only DRB1-DQA1-DQB1 haplotypes with a frequency > 1% in the case individuals in each respective population. The most frequently observed C-B alleles in each population were then added if the C-B-DRB1-DQA1-DQB1 haplotype occurred in more than or equal to five individuals. HLA-DRB3/4/5 alleles were taken from (15) and calculated on the basis of individuals hemizygous for HLA- DRB3/4/5 (i.e. carrying only one HLA-DRB1 observed with either HLA-DRB3, -DRB4 or -DRB5 and one DRB1*01, DRB1*08 or DRB1*10, which are not observed with any of the HLA- DRB3/4/5.)
Figure 6.
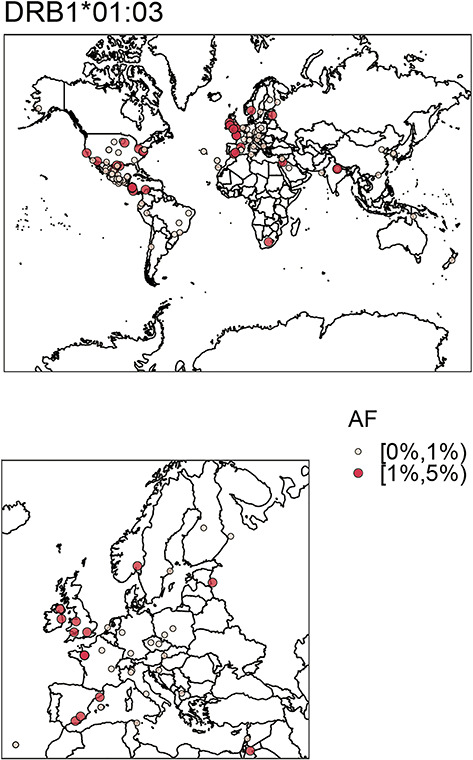
Frequency of DRB1*01:03 across populations available in the allele frequency net database. (A) ‘Worldmap’, (B) zoom into European continent. Frequencies are shown within different ranges noted by AF. Allele frequencies of DRB1*01:03 are lower across central Europe than in the UK, Spain, India, South Africa, USA and coastal regions of South America. Frequencies were binned according to allele frequency. The figures were created using the R-package rworldmap. Frequencies were extracted from the allele frequency network database (35) for populations >100 individuals. To plot the geographic locations, we converted assigned degree and minutes to decimal numbers. We deleted all non-Caucasian populations with USA coordinates prior to plotting.
To reduce the complexity of the HLA signal further and to identify the properties of potential culprit antigens leading to disease, we analyzed peptides preferentially bound by proteins, attributed risk and protection on the genetic level (Fig. 4). Additionally, we tried to identify shared physicochemical properties of these proteins. For this analysis, we only selected proteins for which the corresponding alleles had a significant P-value in the meta-analysis (PVALUE_RE2 < 0.05) and focused on the results of the DRB1 proteins (DQ shown in Supplementary Material, Fig. S9). First, we predicted the binding affinities for five sets of 200 000 random unique peptides sampled from the human proteome to the DRB1 proteins using NetMHCIIpan-3.2 (29) (Supplementary Material, Table S3, amino acid distribution). Next, we performed clustering analysis across all alleles using the top 2% ranked preferentially binding peptides. In brief, we correlated the affinity values of binders shared between combinations of HLA alleles and used these correlations for clustering. In general, we found DRB1-clustering (Fig. 4) to be more informative regarding separation of protective and risk alleles than DQ-clustering. Additionally, DRB1-clustering was more stable across the sets of random peptides (Supplementary Material, Fig. S9). Larger ‘risk clusters’ were identified for DRB1 including DRB1*15:01 and the newly identified DRB1*15:03. We defined two risk clusters including DRB1*11:01/04 and DRB1*13:01 (RISK 1) DRB1*12:01, DRB1*14:04 and DRB1*15:01/03 (RISK 2) and two protective clusters including DRB1*04:01/05, DRB1*07:01, DRB1*09:01 and DRB1*10:01 (PROT 1) and DRB1*04:03/06 (PROT 2). Within each cluster, we calculated a unique peptide binding motif by combining the top 2% of binders for each allele in the groups (Fig. 4). The peptide binding motifs of the two risk groups were enriched for basic amino acids (K and R) and depleted for acidic amino acids, whereas the peptide-binding motifs of the protective group were enriched for hydrophobic and polar amino acids. Interestingly, DRB1*01:03 clustered with protective alleles DRB1*04:01/05, DRB1*07:01, DRB1*09:01 and DRB1*10:01; however, a more detailed analysis of its physicochemical properties resulted in a predominant clustering with DRB1*15 (Fig. 5). Equally, DRB1*15:02 clustered with DRB1*13:02, whereas physicochemical properties resulted in a predominant clustering with the DRB1*15 group. In Supplementary Material, Figures 9–12, we show that this may be an artifact of NetMHCIIpan-3.2 caused by extrapolation of the DRB1*15:02 signal for unknown peptides from DRB1*13:02.
Figure 4.

Clustering of DRB1 proteins according to preferential peptide-binding and combined peptide-binding motifs. (MIDDLE CLUSTER): For five sets of 200 000 unique random human peptides the percentile rank scores of preferential peptide binding were calculated using NetMHCIIpan-3.2 (29) for all DRB1 proteins that were significant in the meta-analysis of genetic analysis of the HLA with and AF > 1% in at least one cohort. We additionally included DRB1*01:03. Within each set, the top 2% binders (according to NetMHCIIpan-3.2 threshold) were used to perform a clustering on the pairwise correlations between two alleles using complete observations only. We show clustering results for peptide set 2. Labels were colored according to risk (red) or protective (blue). (BINDING MOTIFS): Top 2% binders were combined for proteins (RISK 1) DRB1*11:01/04 and DRB1*13:01 DRB1*12:01, DRB1*14:04 and DRB1*15:01/03 (RISK 2), DRB1*04:01/05, DRB1*07:01, DRB1*09:01 and DRB1*10:01 (PROT 1) and DRB1*04:03/04/06 (PROT 2). For this analysis, shared peptides (10% top binders) between at least two of the groups were deleted from the set. Here we depict the results for human peptide set 2. Peptide motifs were plotted using Seq2Logo (30). The color scheme shows the chemistry of the amino acids. Red: positively charged amino acids, blue: negatively charged amino acids, green: polar amino acid, purple: neutral amino acid and black: hydrophobic amino acid.
Figure 5.
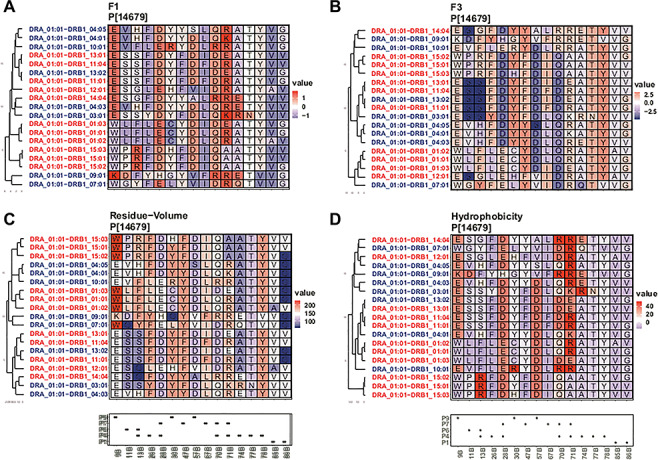
Clustering according to chosen physicochemical properties of amino acids within the peptide binding pockets. We only show sites with variable information in pockets (P) 1, 4, 6, 7 and 9 and only proteins for which the genetic analysis was significant (meta-analysis PVALUE_RE2 < 0.05) and for which at least one cohort had AF > 1%. We additionally show DRB1*01:03. Clustering was performed using the hclust function of the R package stats. The box below the cluster plot shows positions of P1, 4, 6, 7 and 9 of the beta (B) chain of the molecules (as defined in Supplementary Material, Table S5). Here we show combined scores F1 (A) and F3 (B) derived from a factor analysis of 54 unique amino acid properties (31). F1 captures polarity and hydrophobicity of the amino acid, whereas factor F3 captures amino acid size and bulkiness. For F1, high values indicate larger hydrophobicity, polarity and hydrogen donor abilities, whereas low values indicate nonpolar amino acids. For F3, high values indicate larger and bulkier amino acids, whereas low values indicate smaller, more flexible amino acids. We additionally show the residue-volume (C) as a measure of pocket size and defined a score ‘hydrogen acceptor’ (HB-acceptor) (D), which defines the ability of an amino acid to participate in hydrogen bonds and corresponds to the number of atoms within the sidechain that can accept a hydrogen. Additional information for the ‘charge’ parameter and the analysis for DQA1-DQB1 can be found in Supplementary Material, Fig. S9 and S10.
Discussion
Several conclusions can be drawn from this transethnic HLA fine mapping study in UC: HLA allele associations and their effect directions are broadly consistent across the different populations analyzed in this study, and signals previously observed in a Caucasian-only approach can be replicated in this context (5). Magnitudes of effects vary and are more similar across populations with regard to shared ancestry (e.g. effect magnitudes are more similar within distinct populations of Asian and European populations, respectively). Although not in every case the same HLA allele is implicated across the different populations, alleles of the same HLA allele group are associated with UC, as is the case for the HLA allele group DRB1*15, of which DRB1*15:01, DRB1*15:02 and DRB1*15:03 are all associated with the disease dependent on the HLA allele frequencies in each respective population. The frequencies for these alleles computed in this study were consistent to the frequencies stored in the allele frequency net database (35). Population-specific association signals largely correlate with the frequencies of these alleles in the respective cohorts (i.e. being frequent in this population, DRB*15:03 is associated with UC in the African American population, whereas DRB1*15:01, more frequent in the Caucasian populations, is associated here. Likewise, DRB1*09:01 is associated with UC in the Korean and Japanese population and newly identified DRB1*10:01 is very infrequent in the Caucasian population and thus previously not associated with UC in this population). Heterogeneity of effect sizes was observed; however, the accuracy of estimation of the effect sizes would increase with larger per-population sample sizes. As observed also in the Caucasian-only approach, HLA associations are correlated across different HLA genomic loci, especially for HLA-DRB1, -DRB3/4/5 and -DQ alleles, such that neither locus can be ruled out as disease relevant. Overall a high conservation of HLA-DQ-DR haplotypes was observed across different ethnicities. In the Japanese and Korean population and entire haplotype spanning class I and class II was observed for C*12:01-B*52:02-DRB5*01:02-DRB1*15:02-DQA1*01:03-DQB1*06:01. For South Western Asian (Iranian, Indian) individuals, other HLA associations were observed to be more dominant (i.e. HLA-DRB1*11 and HLA-DRB1*14). Overall, the HLA association was dependent on the frequency of the allele and the size of the study cohort (i.e. alleles with a sufficiently high frequency at the DRB1 locus were usually also associated with the disease, except for alleles of the HLA-DRB1*08 and HLA-DRB1*16 groups, which had frequency of 2.9% and 1.7%, respectively, in the Caucasian population). Associations at DPA1-DPB1 can most likely be ruled out, and associations may merely result from correlation with HLA-DQ-DRB1. In the analysis of peptide-binding preferences for HLA-DRB1 alleles, we observed clustering according to the effect’s direction in the genetic analysis, i.e. protective or risk, which may point more to HLA-DRB1 playing a role. However, an important limitation for the analogous DQ analysis is the limited availability of data present in models for HLA–peptide-binding prediction. The highly similar binding pockets of HLA-DRB1*13:01 and HLA-DRB1*13:02 suggest HLA-DQ alleles to mediate disease risk. DRB1*13:01, which was estimated to confer risk, is correlated with DQA1*01:03-DQB1*06:03, whereas DRB1*13:02, which was estimated to be protective, is correlated with DQA1*01:02-DQB1*06:04.
Alleles of the DRB1*15 group also play a role as risk factors in other immune-related diseases including multiple sclerosis (36–39) (a chronic inflammatory neurological disorder), systemic lupus erythematosus (40) and Dupuytrien’s disease (41,42) (both are disorders of the connective tissue). They have also been reported to be associated with adult-onset Still’s disease (43) (a systemic inflammatory disease), Graves’ disease (an autoimmune disease that affects the thyroid), pulmonary tuberculosis and leprosy (44,45) (a disease caused by Mycobacterium leprae that affects the skin). For multiple sclerosis, DRB1*15:03, like in our study, was observed to be specific for African American populations (36). DRB1*15 alleles have been reported to be strongly protective in type 1 diabetes (in which the autoimmune system attacks insulin-producing beta cells of the pancreas) and pemphigus vulgaris (a skin blistering disease). However, the functional consequences of HLA-DRB1*15 association with these diseases have not been addressed and for most of them the potential disease-driving antigens are not known. Exceptions are leprosy, in which M. leprae causes the disease and pemphigus vulgaris, in which the skin protein desmoglein is targeted. Krause-Kyora et al. (44) found that DRB1*15:01, among 18 contemporary DRB1 proteins, was predicted to ‘bind the second smallest number of potential M. leprae antigens’ and further hypothesized that limited presentation of the M. leprae antigens may impair the immune response against this pathogen. Here an important note should be, that DRB1*15:01 is on average also the most frequent HLA-DRB1 allele in the most analyzed British/Central American European populations as such has a higher statistical power to be detected in an association analysis.
Analysis of peptide-binding motifs showed that protective and risk alleles cluster stably and that risk and protective groups have peptide binding motifs that are distinguishable by their physicochemical properties. In this study, we opted for analyzing properties of the binding pockets, known to be important for peptide binding, which we believe is most accommodating to the biology of HLA–peptide binding, rather than analyzing single amino acid positions. The arginine (R) and lysine (K) content was observed to be increased in peptides bound by HLA–proteins that were assigned to confer risk on a genetic level. This was more prominent for risk cluster 1 than risk cluster 2. Interestingly, Dhanda et al., who compared 1,032 known T-cell epitopes from 14 different sources (including Mycobacterium tuberculosis, dengue fever, virus, zika virus, house mite and other allergens) and known nonepitopes from the same data set, showed that T-cell epitope amino acid motifs also are enriched in lysine and arginine content. The established motif is especially similar to the binding motif of risk cluster 1. Arginine is also found at an increased level in antimicrobial peptides (46–48). Antimicrobial peptides are made of cationic residues and are part of the innate immunity. They target the cell wall of bacteria or structures in the cytosol of bacteria (49). If and how this plays a role in the etiology of UC is however only to be speculated about.
One important limitation of the analysis of preferential HLA–peptide binding is the amount of data that is used to train machine learning algorithms, which was especially limited for the HLA-DQ proteins. In the future, larger data sets from peptidomics experiment will likely increase the accuracy of these predictions and increase confidence in the risk and protective motifs that may be indicative of culprit antigens in UC because of distinct features. Larger per-population patient collections will be needed in future studies to confirm our results and to obtain even more precise effect estimates of associated HLA alleles. In addition, we hope that IBD patient panels from other ethnicities will become available for genetic fine mapping studies. With typing of HLA alleles now being possible using next-generation sequencing methods, real typing rather than imputation analyzes should become standard, thereby avoiding possible imputation artifacts. The construction of haplotype maps will then likely be even more accurate.
International IBD Genetics Consortium
A full list of members and affiliations appears in the Supplementary Material, Note.
MAAIS Recruitment Center
A full list of members and affiliations appears in the Supplementary Material, Note.
Data Availability
The ImmunoChip data used in this study are proprietary to the IIBGDC genetics consortium and may be requested from the consortium. Any data produced within this study, may be requested from the corresponding authors upon reasonable request including association statistics of imputed and genotypedSNVs.
Code Availability
Code used for analysis of data within this study is available from f.degenhardt@ikmb.uni-kiel.de upon reasonable request.
Funding
This project received infrastructure support from the DFG Excellence Cluster No. 306 ‘Inflammation at Interfaces’. M.W. and H.E. are supported by the German Research Foundation (DFG) through the Research Training Group 1743, ‘Genes, Environment and Inflammation’. E.E. received funding from the European Union Seventh Framework Program (FP7-PEOPLE-2013-COFUND; grant agreement No. 609020; Scientia Fellows). University Medical Center Groningen, Groningen, The Netherlands; Institute for Digestive System Disease, Tehran University of Medical Sciences, Tehran, Iran to S.A. Funding for the Multicenter African American IBD Study (MAAIS) samples, for the GENESIS samples and for the African Americans recruited by Cedars Sinai was provided by the U.S.A. National Institutes of Health (NIH) (DK062431 to S.R.B., DK 087694 to S.K. and DK062413 to D.P.B.M), respectively; BioBank Japan Project; Grant-in-Aid for Scientific Research (B) (26293180) funded by the Ministry of Education, Culture, Sports, Science, and Technology, Japan; Mid-career Researcher Program grant through the National Research Foundation of Korea (2017R1A2A1A05001119 to K.S.) funded by the Ministry of Science, Information & Communication Technology and Future Planning; Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI) (HI18C0094) funded by the Ministry of Health & Welfare, Republic of Korea. Funding for the Indian samples was provided by the Centre of Excellence in Genome Sciences and Predictive Medicine, Department of Biotechnology, Government of India (BT/01/COE/07/UDSC/2008). This work was supported by the EU's Horizon 2020 SYSCID program under the grant agreement No 733100.
Authors Contributions
S.R.B., T.H.K., J.D.R. and A.F. jointly supervised this work. F.D. performed statistical and computational analysis, G.B. contributed to statistical analysis. M.W. and H.E. performed computational analysis with contributions from D.E., M.Hü, S.L., A.M., T.L. and S.R. G.M. performed protein structure analysis and analysis of physicochemical properties with contributions from F.D. F.D. and M.W. set up the HLA imputation pipeline. S.J. performed HLA typing in contribution to the HLA reference panel. F.D., G.M., E.E., E.R. wrote or revised this manuscript. S.A., B.A., T.B.K., S-K.Y., B.D.Y., J.H.C., L.W.D., N.E.D., P.E., M.E., Y.F., D.P.B.M., T.H., M.Ho., G.J., E.S.J., M.K., S.K., R.M., V.M., S.C.N., D.T.O, J.S., S.S., K.S., A.S., A.T., E.A.T, J.U., H.V., R.K:W.,S.H:W., K.Y. were involved in study subject recruitment, contributed genotype data and or/phenotype data. F.D., T.H.K., J.D.R., S.R.B. and A.F. conceived, designed and managed the study. All authors reviewed, edited and approved the final manuscript.
Supplementary Material
Contributor Information
Frauke Degenhardt, Institute of Clinical Molecular Biology, Christian-Albrechts-University, 24105 Kiel, Germany.
Gabriele Mayr, Institute of Clinical Molecular Biology, Christian-Albrechts-University, 24105 Kiel, Germany.
Mareike Wendorff, Institute of Clinical Molecular Biology, Christian-Albrechts-University, 24105 Kiel, Germany.
Gabrielle Boucher, Research Center, Montréal Heart Institute, Université de Montréal and the Montréal Heart Institute, Montréal, Québec H1T 1C8, Canada.
Eva Ellinghaus, K.G. Jebsen Inflammation Research Centre, Institute of Clinical Medicine, University of Oslo, 0372 Oslo, Norway.
David Ellinghaus, Institute of Clinical Molecular Biology, Christian-Albrechts-University, 24105 Kiel, Germany; Norwegian PSC Research Center, Department of Transplantation Medicine, Division of Surgery, Inflammatory Diseases and Transplantation, Oslo University Hospital Rikshospitalet, 0372 Oslo, Norway.
Hesham ElAbd, Institute of Clinical Molecular Biology, Christian-Albrechts-University, 24105 Kiel, Germany.
Elisa Rosati, Institute of Clinical Molecular Biology, Christian-Albrechts-University, 24105 Kiel, Germany.
Matthias Hübenthal, Institute of Clinical Molecular Biology, Christian-Albrechts-University, 24105 Kiel, Germany; Department of Dermatology, Venerology and Allergy, University Hospital Schleswig-Holstein, 24105 Kiel, Germany.
Simonas Juzenas, Institute of Clinical Molecular Biology, Christian-Albrechts-University, 24105 Kiel, Germany.
Shifteh Abedian, Department of Epidemiology, University Medical Center Groningen, 9713 Groningen, The Netherlands; Digestive Disease Research Center, Digestive Disease Research Institute, Tehran University of Medical Sciences, Tehran 1411713135, Iran.
Homayon Vahedi, Digestive Disease Research Center, Digestive Disease Research Institute, Tehran University of Medical Sciences, Tehran 1411713135, Iran.
B K Thelma, Department of Genetics, University of Delhi South Campus, New Delhi, Delhi 110021, India.
Suk-Kyun Yang, Department of Gastroenterology, Asan Medical Center, University of Ulsan College of Medicine, Seoul 05505, Republic of Korea.
Byong Duk Ye, Department of Gastroenterology, Asan Medical Center, University of Ulsan College of Medicine, Seoul 05505, Republic of Korea.
Jae Hee Cheon, Department of Internal Medicine and Institute of Gastroenterology, Yonsei University College of Medicine, Seoul 03722, Republic of Korea.
Lisa Wu Datta, Harvey M. and Lyn P. Meyerhoff Inflammatory Bowel Disease Center, Department of Medicine, John Hopkins University School of Medicine, Baltimore, MD 21205, USA.
Naser Ebrahim Daryani, Department of Gastroenterology, Emam Hospital, Tehran University of Medical Sciences, Tehran 1419733141, Iran.
Pierre Ellul, Department of Gastroenterology, Mater Dei Hospital, Msida, Malta.
Motohiro Esaki, Department of Medicine and Clinical Science, Graduate School of Medical Sciences, Kyushu University, Fukuoka 812-8582, Japan.
Yuta Fuyuno, Department of Medicine and Clinical Science, Graduate School of Medical Sciences, Kyushu University, Fukuoka 812-8582, Japan; Laboratory for Genotyping Development, Center for Integrative Medical Sciences, Riken, Yokohama 351-0198, Japan.
Dermot P B McGovern, F. Widjaja Foundation Inflammatory Bowel and Immunobiology Research Institute, Cedars-Sinai Medical Center, Los Angeles, CA 90048, USA.
Talin Haritunians, F. Widjaja Foundation Inflammatory Bowel and Immunobiology Research Institute, Cedars-Sinai Medical Center, Los Angeles, CA 90048, USA.
Myhunghee Hong, Department of Biochemistry and Molecular Biology, University of Ulsan College of Medicine, Seoul, 136-701 Korea.
Garima Juyal, School of Biotechnology, Jawaharlal Nehru University, New Delhi, Delhi 110067, India.
Eun Suk Jung, Institute of Clinical Molecular Biology, Christian-Albrechts-University, 24105 Kiel, Germany; Department of Internal Medicine and Institute of Gastroenterology, Yonsei University College of Medicine, Seoul 03722, Republic of Korea.
Michiaki Kubo, RIKEN Center for Integrative Medical Sciences, Yokohama, 351-0198, Japan.
Subra Kugathasan, Department of Pediatrics, Emory University School of Medicine, Atlanta, GA 30322, USA; Pediatric Institute, Children's Healthcare of Atlanta, Atlanta, GA, USA.
Tobias L Lenz, Research Group for Evolutionary Immunogenomics, Max Planck Institute for Evolutionary Biology, 24306 Plön, Germany.
Stephen Leslie, Schools of Mathematics and Statistics and BioSciences and Melbourne Integrative Genomics, University of Melbourne, Victoria 3010, Australia.
Reza Malekzadeh, Digestive Disease Research Center, Digestive Disease Research Institute, Tehran University of Medical Sciences, Tehran 1411713135, Iran.
Vandana Midha, Dayanand Medical College and Hospital, Ludhiana, Punjab 141001, India.
Allan Motyer, Schools of Mathematics and Statistics and BioSciences and Melbourne Integrative Genomics, University of Melbourne, Victoria 3010, Australia.
Siew C Ng, Department of Medicine and Therapeutics, Institute of Digestive Disease, Chinese University of Hong Kong, Hong Kong.
David T Okou, Division of Gastroenterology, Hepatology and Nutrition, Department of Pediatrics, Emory University School of Medicine, Atlanta, GA 30322, USA.
Soumya Raychaudhuri, Center for Data Sciences, Brigham and Women's Hospital, Harvard Medical School, Boston, MA 02114, USA; Department of Medicine, Brigham and Women's Hospital, Harvard Medical School, Boston, MA 02114, USA; Department of Biomedical Informatics, Harvard Medical School, Boston, MA 02115, USA; Program in Medical and Population Genetics, Broad Institute, Cambridge, MA 02142, USA; Centre for Genetics and Genomics Versus Arthritis, Division of Musculosceletal and Dermatological Sciences, School of Biological Sciences, University of Manchester, Manchester, UK.
John Schembri, Department of Gastroenterology, Mater Dei Hospital, Msida, Malta.
Stefan Schreiber, Institute of Clinical Molecular Biology, Christian-Albrechts-University, 24105 Kiel, Germany; Department of Medicine, Christian-Albrechts-University, 24105 Kiel, Germany.
Kyuyoung Song, Department of Biochemistry and Molecular Biology, University of Ulsan College of Medicine, Seoul, 136-701 Korea.
Ajit Sood, Dayanand Medical College and Hospital, Ludhiana, Punjab 141001, India.
Atsushi Takahashi, Laboratory for Statistical and Translational Genetics, Center for Integrative Medical Sciences, Riken, Yokohama, 230-0045, Japan.
Esther A Torres, Department of Medicine, University of Puerto Rico Center for IBD, University of Puerto Rico School of Medicine, Rio Piedras, San Juan, PR 00936-5067, USA.
Junji Umeno, Department of Medicine and Clinical Science, Graduate School of Medical Sciences, Kyushu University, Fukuoka 812-8582, Japan.
Behrooz Z Alizadeh, Department of Epidemiology, University Medical Center Groningen, 9713 Groningen, The Netherlands.
Rinse K Weersma, Department of Gastroenterology and Hepatology, University of Groningen and University Medical Center Groningen, 9700 AB Groningen, The Netherlands.
Sunny H Wong, Department of Medicine and Therapeutics, Institute of Digestive Disease, Chinese University of Hong Kong, Hong Kong.
Keiko Yamazaki, Laboratory for Genotyping Development, Center for Integrative Medical Sciences, Riken, Yokohama 351-0198, Japan.
Tom H Karlsen, Norwegian PSC Research Center, Department of Transplantation Medicine, Division of Surgery, Inflammatory Diseases and Transplantation, Oslo University Hospital Rikshospitalet, 0372 Oslo, Norway; Research Institute for Internal Medicine, Division of Surgery, Inflammatory Diseases and Transplantation, Oslo University Hospital Rikshospitalet and University of Oslo, 0372 Oslo, Norway.
John D Rioux, Research Center, Montréal Heart Institute, Université de Montréal and the Montréal Heart Institute, Montréal, Québec H1T 1C8, Canada.
Steven R Brant, Harvey M. and Lyn P. Meyerhoff Inflammatory Bowel Disease Center, Department of Medicine, John Hopkins University School of Medicine, Baltimore, MD 21205, USA; Department of Medicine, Rutgers Robert Wood Johnson School of Medicine and Department of Genetics, Rutgers University Brunswick and Piscataway, NJ 08903-0019, USA.
Andre Franke, Institute of Clinical Molecular Biology, Christian-Albrechts-University, 24105 Kiel, Germany.
References
- 1. Jostins, L., Ripke, S., Weersma, R.K., Duerr, R.H., McGovern, D.P., Hui, K.Y., Lee, J.C., Schumm, L.P., Sharma, Y., Anderson, C.A.et al. (2012) Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature, 491, 119–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Liu, J.Z., vanSommeren, S., Huang, H., Ng, S.C., Alberts, R., Takahashi, A., Ripke, S., Lee, J.C., Jostins, L., Shah, T.et al. (2015) Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat. Genet., 47, 979–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ellinghaus, D., Jostins, L., Spain, S.L., Cortes, A., Bethune, J., Han, B., Park, Y.R., Raychaudhuri, S., Pouget, J.G., Hubenthal, M.et al. (2016) Analysis of five chronic inflammatory diseases identifies 27 new associations and highlights disease-specific patterns at shared loci. Nat. Genet., 48, 510–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. de Lange, K.M., Moutsianas, L., Lee, J.C., Lamb, C.A., Luo, Y., Kennedy, N.A., Jostins, L., Rice, D.L., Gutierrez-Achury, J., Ji, S.G.et al. (2017) Genome-wide association study implicates immune activation of multiple integrin genes in inflammatory bowel disease. Nat. Genet., 49, 256–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Goyette, P., Boucher, G., Mallon, D., Ellinghaus, E., Jostins, L., Huang, H., Ripke, S., Gusareva, E.S., Annese, V., Hauser, S.L.et al. (2015) High-density mapping of the MHC identifies a shared role for HLA-DRB1*01:03 in inflammatory bowel diseases and heterozygous advantage in ulcerative colitis. Nat. Genet., 47, 172–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Stokkers, P.C., Reitsma, P.H., Tytgat, G.N. and vanDeventer, S.J. (1999) HLA-DR and -DQ phenotypes in inflammatory bowel disease: a meta-analysis. Gut, 45, 395–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lappalainen, M., Halme, L., Turunen, U., Saavalainen, P., Einarsdottir, E., Farkkila, M., Kontula, K. and Paavola-Sakki, P. (2008) Association of IL23R, TNFRSF1A, and HLA-DRB1*0103 allele variants with inflammatory bowel disease phenotypes in the Finnish population. Inflamm. Bowel Dis., 14, 1118–1124. [DOI] [PubMed] [Google Scholar]
- 8. Lu, M. and Xia, B. (2006) Polymorphism of HLA-DRB1 gene shows no strong association with ulcerative colitis in Chinese patients. Int. J. Immunogenet., 33, 37–40. [DOI] [PubMed] [Google Scholar]
- 9. Okada, Y., Yamazaki, K., Umeno, J., Takahashi, A., Kumasaka, N., Ashikawa, K., Aoi, T., Takazoe, M., Matsui, T., Hirano, A.et al. (2011) HLA-Cw*1202-B*5201-DRB1*1502 haplotype increases risk for ulcerative colitis but reduces risk for Crohn’s disease. Gastroenterology, 141, 864–865. [DOI] [PubMed] [Google Scholar]
- 10. Myung, S.J., Yang, S.K., Jung, H.Y., Chang, H.S., Park, B., Hong, W.S., Kim, J.H. and Min, I. (2002) HLA-DRB1*1502 confers susceptibility to ulcerative colitis, but is negatively associated with its intractability: a Korean study. Int. J. Color. Dis., 17, 233–237. [DOI] [PubMed] [Google Scholar]
- 11. Mohammadi, M., Rastin, M., Rafatpanah, H., Abdoli Sereshki, H., Zahedi, M.J., Nikpoor, A.R., Baneshi, M.R. and Hayatbakhsh, M.M. (2015) Association of HLA-DRB1 alleles with ulcerative colitis in the City of Kerman, South eastern Iran. Iran J. Allergy Asthma Immunol., 14, 306–312. [PubMed] [Google Scholar]
- 12. Gao, F., Aheman, A., Lu, J.J., Abuduhadeer, M., Li, Y.X. and Kuerbanjiang, A. (2014) Association of HLA-DRB1 alleles and anti-neutrophil cytoplasmic antibodies in Han and Uyghur patients with ulcerative colitis in China. J. Dig. Dis., 15, 299–305. [DOI] [PubMed] [Google Scholar]
- 13. Uyar, F.A., Imeryuz, N., Saruhan-Direskeneli, G., Ceken, H., Ozdogan, O., Sahin, S. and Tozun, N. (1998) The distribution of HLA-DRB alleles in ulcerative colitis patients in Turkey. Eur. J. Immunogenet., 25, 293–296. [DOI] [PubMed] [Google Scholar]
- 14. Han, B., Akiyama, M., Kim, K.-K., Oh, H., Choi, H., Lee, C.H., Jung, S., Lee, H.-S., Kim, E.E., Cook, S.et al. (2018) Amino acid position 37 of HLA-DRβ1 affects susceptibility to Crohn’s disease in Asians. Hum. Mol. Genet, 27, 3901–3910. [DOI] [PubMed] [Google Scholar]
- 15. Degenhardt, F., Wendorff, M., Wittig, M., Ellinghaus, E., Datta, L.W., Schembri, J., Ng, S.C., Rosati, E., Hübenthal, M., Ellinghaus, D.et al. (2019) Construction and benchmarking of a multi-ethnic reference panel for the imputation of HLA class I and II alleles. Hum. Mol. Genet., 28, 2078–2092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Huang, C., Haritunians, T., Okou, D.T., Cutler, D.J., Zwick, M.E., Taylor, K.D., Datta, L.W., Maranville, J.C., Liu, Z., Ellis, S.et al. (2015) Characterization of genetic loci that affect susceptibility to inflammatory bowel diseases in African Americans. Gastroenterology, 149, 1575–1586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ye, B.D., Choi, H., Hong, M., Yun, W.J., Low, H.Q., Haritunians, T., Kim, K.J., Park, S.H., Lee, I., Bang, S.Y.et al. (2016) Identification of ten additional susceptibility loci for ulcerative colitis through immunochip analysis in Koreans. Inflamm. Bowel Dis., 22, 13–19. [DOI] [PubMed] [Google Scholar]
- 18. Delaneau, O., Marchini, J. and Zagury, J.F. (2011) A linear complexity phasing method for thousands of genomes. Nat. Methods, 9, 179–181. [DOI] [PubMed] [Google Scholar]
- 19. Auton, A., Brooks, L.D., Durbin, R.M., Garrison, E.P., Kang, H.M., Korbel, J.O., Marchini, J.L., McCarthy, S., McVean, G.A. and Abecasis, G.R. (2015) A global reference for human genetic variation. Nature, 526, 68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Howie, B., Marchini, J. and Stephens, M. (2011) Genotype imputation with thousands of genomes. G3(1), 457–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Howie, B.N., Donnelly, P. and Marchini, J. (2009) A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet., 5, e1000529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Browning, B.L. and Browning, S.R. (2016) Genotype imputation with millions of reference Samples. Am. J. Hum. Genet., 98, 116–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Browning, S.R. and Browning, B.L. (2007) Rapid and accurate haplotype phasing and missing-data inference for whole-genome association studies by use of localized haplotype clustering. Am. J. Hum. Genet., 81, 1084–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zheng, X., Shen, J., Cox, C., Wakefield, J.C., Ehm, M.G., Nelson, M.R. and Weir, B.S. (2014) HIBAG-HLA genotype imputation with attribute bagging. Pharmacogenomics J., 14, 192–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gourraud, P.A., Khankhanian, P., Cereb, N., Yang, S.Y., Feolo, M., Maiers, M., Rioux, J.D., Hauser, S. and Oksenberg, J. (2014) HLA diversity in the 1000 genomes dataset. PLoS One, 9, e97282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Han, B. and Eskin, E. (2011) Random-effects model aimed at discovering associations in meta-analysis of genome-wide association studies. Am. J. Hum. Genet, 88, 586–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lee, C.H., Eskin, E. and Han, B. (2017) Increasing the power of meta-analysis of genome-wide association studies to detect heterogeneous effects. Bioinformatics, 33, i379–i388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Morris, A.P. (2011) Transethnic meta-analysis of genomewide association studies. Genet. Epidemiol., 35, 809–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Jensen, K.K., Andreatta, M., Marcatili, P., Buus, S., Greenbaum, J.A., Yan, Z., Sette, A., Peters, B. and Nielsen, M. (2018) Improved methods for predicting peptide binding affinity to MHC class II molecules. Immunology, 154, 394–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Thomsen, M.C.F. and Nielsen, M. (2012) Seq2Logo: A method for construction and visualization of amino acid binding motifs and sequence profiles including sequence weighting, pseudo counts and two-sided representation of amino acid enrichment and depletion. Nucleic Acids Res, 40, W281–W287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Atchley, W.R., Zhao, J., Fernandes, A.D. and Drüke, T. (2005) Solving the protein sequence metric problem. Proc. Natl. Acad. Sci. USA, 102, 6395–6400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Goldsack, D.E. and Chalifoux, R.C. (1973) Contribution of the free energy of mixing of hydrophobic side chains to the stability of the tertiary structure of proteins. J. Theor. Biol., 39, 645–651. [DOI] [PubMed] [Google Scholar]
- 33. Shah, T.S., Liu, J.Z., Floyd, J.A., Morris, J.A., Wirth, N., Barrett, J.C. and Anderson, C.A. (2012) optiCall: a robust genotype-calling algorithm for rare, low-frequency and common variants. Bioinformatics, 28, 1598–1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Edgar, R.C. (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res., 32, 1792–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gonzalez-Galarza, F.F., Takeshita, L.Y., Santos, E.J., Kempson, F., Maia, M.H., daSilva, A.L., Teles e Silva, A.L., Ghattaoraya, G.S., Alfirevic, A., Jones, A.R.et al. (2015) Allele frequency net 2015 update: new features for HLA epitopes, KIR and disease and HLA adverse drug reaction associations. Nucleic Acids Res., 43, D784–D788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hollenbach, J.A. and Oksenberg, J.R. (2015) The immunogenetics of multiple sclerosis: A comprehensive review. J. Autoimmun., 64, 13–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Alcina, A., Abad-Grau Mdel, M., Fedetz, M., Izquierdo, G., Lucas, M., Fernandez, O., Ndagire, D., Catala-Rabasa, A., Ruiz, A., Gayan, J.et al. (2012) Multiple sclerosis risk variant HLA-DRB1*1501 associates with high expression of DRB1 gene in different human populations. PLoS One, 7, e29819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Prat, E., Tomaru, U., Sabater, L., Park, D.M., Granger, R., Kruse, N., Ohayon, J.M., Bettinotti, M.P. and Martin, R. (2005) HLA-DRB5*0101 and -DRB1*1501 expression in the multiple sclerosis-associated HLA-DR15 haplotype. J. Neuroimmunol., 167, 108–119. [DOI] [PubMed] [Google Scholar]
- 39. Patsopoulos, N.A., Barcellos, L.F., Hintzen, R.Q., Schaefer, C., vanDuijn, C.M., Noble, J.A., Raj, T., IMSGC, ANZgene, Gourraud, P.A.et al. (2013) Fine-mapping the genetic association of the major histocompatibility complex in multiple sclerosis: HLA and non-HLA effects. PLoS Genet., 9, e1003926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hanscombe, K.B., Morris, D.L., Noble, J.A., Dilthey, A.T., Tombleson, P., Kaufman, K.M., Comeau, M., Langefeld, C.D., Alarcon-Riquelme, M.E., Gaffney, P.M.et al. (2018) Genetic fine mapping of systemic lupus erythematosus MHC associations in Europeans and African Americans. Hum. Mol. Genet., 27, 3813–3824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Brown, J.J., Ollier, W., Thomson, W. and Bayat, A. (2008) Positive association of HLA-DRB1*15 with Dupuytren’s disease in Caucasians. Tissue Antigens. [DOI] [PubMed] [Google Scholar]
- 42. DIAbetes Genetics Replication And Meta-analysis (DIAGRAM) Consortium, Asian Genetic Epidemiology Network Type 2 Diabetes (AGEN-T2D) Consortium, South Asian Type 2 Diabetes (SAT2D) Consortium, Mexican American Type 2 Diabetes (MAT2D) Consortium, Type 2 Diabetes Genetic Exploration by Next-generation sequencing in muylti-Ethnic Samples (T2D-GENES) Consortium, Mahajan, A., Go, M.J., Zhang, W., Below, J.E.et al. (2014) Genome-wide trans-ancestry meta-analysis provides insight into the genetic architecture of type 2 diabetes susceptibility. Nat. Genet., 46, 234–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Yap, L.M., Ahmad, T. and Jewell, D.P. (2004) The contribution of HLA genes to IBD susceptibility and phenotype. Best Pract. Res. Clin. Gastroenterol., 18, 577–596. [DOI] [PubMed] [Google Scholar]
- 44. Krause-Kyora, B., Nutsua, M., Boehme, L., Pierini, F., Pedersen, D.D., Kornell, S.C., Drichel, D., Bonazzi, M., Möbus, L., Tarp, P.et al. (2018) Ancient DNA study reveals HLA susceptibility locus for leprosy in medieval Europeans, Nat. Commun., 9, 1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zhang, F., Liu, H., Chen, S., Wang, C., Zhu, C., Zhang, L., Chu, T., Liu, D., Yan, X. and Liu, J. (2009) Evidence for an association of HLA-DRB115 and DRB109 with leprosy and the impact of DRB109 on disease onset in a Chinese Han population. BMC Med. Genet., 10, 133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Nguyen, L.T., Chau, J.K., Perry, N.A., deBoer, L., Zaat, S.A.J. and Vogel, H.J. (2010) Serum stabilities of short tryptophan- and arginine-rich antimicrobial peptide analogs. PLoS One, 5, e12684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Chan, D.I., Prenner, E.J. and Vogel, H.J. (2006) Tryptophan- and arginine-rich antimicrobial peptides: structures and mechanisms of action. Biochim. Biophys. Acta., 1758, 1184–1202. [DOI] [PubMed] [Google Scholar]
- 48. Cutrona, K.J., Kaufman, B.A., Figueroa, D.M. and Elmore, D.E. (2015) Role of arginine and lysine in the antimicrobial mechanism of histone-derived antimicrobial peptides. FEBS Lett., 589, 3915–3920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Brogden, K.A. (2005) Antimicrobial peptides: pore formers or metabolic inhibitors in bacteria? Nat. Rev. Microbiol., 3, 238–250. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The ImmunoChip data used in this study are proprietary to the IIBGDC genetics consortium and may be requested from the consortium. Any data produced within this study, may be requested from the corresponding authors upon reasonable request including association statistics of imputed and genotypedSNVs.