

Abstract
Adsorption of biomolecules onto material surfaces involves a potentially complex mechanism where molecular species interact to varying degrees with a heterogeneous material surface. Surface adsorption studies by atomic force microscopy (AFM), Sum Frequency Generation (SFG) spectroscopy, and solid state NMR (ssNMR), detect the structures and interactions of biomolecular species that are bound to material surfaces and which, in the absence of a solid liquid interface, do not exchange rapidly between surface-bound forms and free molecular species in bulk solution. Solution NMR has the potential to complement these techniques by detecting and studying transiently bound biomolecules at the liquid-solid interface. Herein we show that dark-state exchange saturation transfer (DEST) NMR experiments on gel-stabilized TiO2 nanoparticle (NP) samples detect several forms of biomolecular adsorption onto titanium (IV) oxide surfaces. Specifically, we use the DEST approach to study the interaction of amino acids arginine (Arg), lysine (Lys), leucine (Leu), alanine (Ala), and aspartic acid (Asp) with TiO2 rutile nanoparticle surfaces. Whereas Leu, Ala, and Asp display only a single weakly interacting form in the presence of TiO2 nanoparticles , Arg and Lys displayed at least two distinct bound forms: a species that is surface bound and retains a degree of reorientational motion, and a second more tightly bound form characterized by broadened DEST profiles upon addition of TiO2 nanoparticles. Molecular Dynamics simulations indicate different surface bound states for both Lys and Arg depending on the degree of TiO2 surface hydroxylation, but only a single bound state for Asp regardless of the degree of surface hydroxylation, in agreement with results obtained from analysis of DEST profiles.
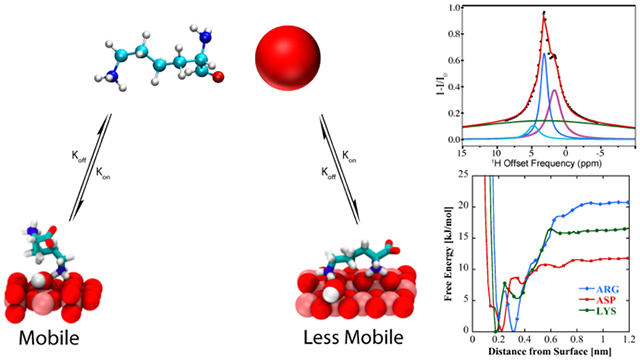
Graphical Abstract
INTRODUCTION
The interaction of biomolecules with titanium (IV) oxide (TiO2) nanoparticles (NP’s) is a topic relevant to a variety of fields including medical and dental implants, biosensors, chromatography, and catalysis.1-12 Fundamental to our understanding of how biomolecules interact with TiO2 NP’s is knowledge of the structures of these molecules at NP surfaces. However, detailed structural information of surface-adsorbed peptides is only now emerging,13-15 and the nature of protein-mineral surface interactions has yet to be clarified even for small monomeric amino acids and small mineral-binding peptides. Methods used to detect and characterize surface-bound molecular species include Atomic Force Microscopy (AFM),16-18 Sum Frequency Generation (SFG) Spectroscopy,19-20 Solid State NMR (ssNMR),21 to name a few. These methods detect the presence of partly or entirely immobilized biomolecular species at material surfaces where the amino acid side chains provide points of surface contact.
The mechanism of surface adsorption of biomolecules onto TiO2 NP’s is complex and may involve, prior to final attachment and immobilization on the surface, formation of biomolecular species that interact with and are only partly immobilized near the NP surface. In addition, the material surface may be heterogeneous, resulting in a variation in binding affinity over the surface and in multiple forms of bound species. Because they are the monomeric constituents of proteins, adsorption of amino acids onto metallic and oxide surfaces has been widely studied and recently reviewed.22 Although thermodynamic studies of lysine23,24 and histidine25 adsorption onto TiO2 NP’s fitted data using a simple Langmuir model, which assumes a single affinity constant and a single independently bound form, spectroscopic studies have identified more complex scenarios for adsorption of some amino acids on TiO2 NP’s. A IR spectroscopic study of the binding of glutamic acid and aspartic acid to TiO2 NP’s found that while at all pH’s aspartic acid binds in a single form, glutamic acid binds in at least two forms.26 A very recent thermodynamic study of the adsorption of amino acids onto TiO2 NP’s found that the BET model fits the binding of most amino acids to TiO2, with the binding being endothermic and thus entropy driven.27 In the same study computations showed surface interactions via hydrogen binding between the α-ammonium group and surface hydroxyl oxygens, while basic and acid amino acids can also interact with the surface via their side chains.
In view of the complex nature of amino acid and peptide adsorption onto TiO2 NP’s, it is useful to apply experimental techniques that can detect several types of adsorbed species, under the same sample conditions. Dark-state exchange saturation transfer (DEST) NMR experiments have provided thermodynamic and kinetic information on the binding of small proteins to aggregates and large molecular machines.28-30 DEST relies on slow exchange of nuclear spins between sites with very different values of the transverse relaxation rate R2, as would occur for example when a freely tumbling molecular species in solution with a small R2, adsorbs onto a TiO2 NP surface with the resulting immobilized species displaying a much larger R2. In addition to R2 values for free and bound molecular species, simulation of the DEST saturation profile yields further information, including the relative populations of the free and adsorbed species, and the kinetic constants that quantify the rate of exchange between free and adsorbed species. In an initial demonstration of this approach, Egner et al.31 applied 1H DEST and relaxation dispersion (RD) to the study of the adsorption of phenol and cholic acid onto cerium oxide NP’s. By direct solution of the Bloch-McConnell equations and subsequent simulation of the 1H DEST saturation profiles, Egner et al showed that while cholic acid adsorbs from bulk solution to a weakly-bound state that does not constitute a rigid adduct with the NP, phenol adsorption proceeds via an intermediate, weakly bound species to a state that is rigidly bound to the NP surface. Therefore, the Egner et al. study afforded not only populations of free and bound species, but also a kinetic mechanism for the adsorption process.
MD simulations have also provided valuable insights into the binding mechanism of amino acids and their analogs on the surface of TiO2.32-36 Bowen et al. found that the adsorption of amino acids with polar sidechains on a negative rutile (110) surface is a function of both backbone and sidechain binding.32 Walsh and coworkers studied the binding of arginine, aspartate and lysine analogs on two variants of the rutile interface – negative and neutral; they report that the arginine analog adsorbed the strongest to both interfaces, followed by the lysine and aspartate analogs.35,36 Recently, Schelokov et al. described the adsorption of – amino acid on nanocrystalline anatase particles using QSPR and MD simulations, they find that the binding occurs primarily through the formation of two – three hydrogen bonds via sidechain or backbone groups that are charged.27
In this paper we apply 1H DEST techniques to the study of the adsorption of small biomolecules, i.e. amino acids, to TiO2 rutile nanocrystals. Application of DEST methods to studying the binding of biomolecules to mineral surfaces has the same requirements detailed in the study of Egner et al.31 Namely, the NMR-visible molecules (i.e. the free, unbound molecules) and the NMR-invisible molecules (i.e. the surface-bound molecules) have to remain homogeneously suspended in the NMR sample throughout the NMR measurement period. We followed the procedure described in Egner et al. and used 1 wt% agarose to prevent nanoparticle sedimentation. By introducing TiO2 nanocrystals into agarose gel suspensions, we investigated the binding of arginine (Arg), lysine (Lys), leucine (Leu), aspartate (Asp), and alanine (Ala) amino acid. We chose amino acids as model systems for initial study because they contain the same side chain functional groups used by peptides and proteins to adsorb onto TiO2 NP surfaces,16-18 yet their small size limits the number of surface-bound forms that may be present. Lorentzian deconvolution of 1H DEST saturation profiles indicate for Arg and Lys the existence of multiple forms of adsorbed molecules, distinguished by differing degrees of residual molecular motion. Simulation of the 1H DEST profiles by direct solution of the Bloch-McConnell equation provides quantitative information including relative populations of free and adsorbed species as well as kinetic constants that quantify rate of exchange between free and adsorbed species. Finally, molecular dynamics simulations also demonstrate the existence of multiple binding states of Arg and Lys on rutile surfaces, and investigate the role played by surface hydroxylation in mediating these interactions. Overall this paper demonstrates how the application DEST NMR experiments and MD calculations in a concerted fashion can elucidate both thermodynamic and kinetic aspects of the adsorption of biomolecules at liquid-solid interfaces.
EXPERIMENTAL
Materials.
Amino acids alanine, leucine, arginine, lysine, and aspartic acid were purchased from Sigma-Aldrich (St. Louis, MO) and used without purification. TiO2 (product number 637262; rutile titanium (IV) oxide nanopowder with a reported particle size <100 nm) nanoparticles were purchased from Sigma-Aldrich (St. Louis, MO) with a BET measured surface area of 28.6 m2/g. Agarose were purchased from Sigma Aldrich and used without further purification. Deuterated solvents were purchased from Sigma Aldrich and used without further purification.
Preparation of NMR samples.
10 mM Amino acids (Arg, Lys, Leu, Pro, Asp, and Ala) NMR samples were prepared in 20 mM phosphate buffer pH7 with 99.9% D2O.
Samples without TiO2 nanopowder in the presence of agarose gel were prepared by mixing 1% w/w agarose in 20 mM phosphate buffer (pH 7, 99.9% D2O). The mixture was placed in a boiling water bath for 5 min, then removed to a block heater to cool. When the temperature of equilibrated to between 50 and 60 °C, amino acid solution was added, resulting in a final concentration of 10 mM. The warm solution was transferred to an NMR tube and allowed to cool at room temperature.
Samples that contained TiO2 nanopowder in the presence of gel were prepared by mixing 1% w/w TiO2 nanopowder and agarose in 20 mM phosphate buffer (pH 7, 99.9% D2O). The mixture was vortex mixed and sonicated for 5 min, then placed in a boiling water bath for 5min. The sample was then removed to a block heater and its internal temperature allowed to equilibrate between 50-60 °C. The respective amino acid solution was added, resulting in a final concentration of 10mM. The warm solution was transferred to an NMR tube and allowed to cool to room temperature.
NMR spectroscopy.
The NMR measurements were performed at 25 °C on a Bruker Avance III 700 MHz NMR instrument equipped with a 5 mm Broadband Observe (BBO) probe. 1H-DEST experiments were measured at multiple saturation fields (50 Hz, 100 Hz, 150 Hz, 200 Hz, 250 Hz and 300 Hz) using a 1H DEST pulse scheme.31 1D 1H spectrums were recorded in steps of 0.25 or 2.5 ppm with the position of the 1H B1 field ranging from −100 ppm to +100 ppm, and an offset of −100 ppm was used for normalization. The saturation field was applied for 1 s, and a repetition delay of 3 s. NMR spectra were processed using Topspin 4.0.2 and Mnova NMR (http://mestrelab.com/software/mnova/nmr/). Spectra were analyzed using Bruker dynamics center (https://www.bruker.com/products/mr/nmr/nmr-software/software/dynamics-center/overview.html.) and Mnova NMR.
Analysis of data.
Data were processed with Peak Analyzer in OriginPro (OriginLab Corporation, Northampton, MA, USA). Z-spectra (I/I0) (or DEST profile) were normalized by the signal with RF irradiation at −100 ppm (I0). For the conventional fitting method, multi-pool Lorentzian fitting of the Z-spectra was applied to estimate the DEST effects from different pools.37-39 Briefly, the inverted Z-spectra (1-I/I0) were fitted as the sum of multiple Lorentzian functions with the following equation:
| (1) |
where ω is the frequency offset from the interest resonance, Ai, ωi, and σi are the amplitude, frequency offset and linewidth of the DEST peak for the ith proton pool respectively. In the DEST phantom, we employed a four-pool or five-pool Lorentzian model of magnetization transfer (MT), and Nuclear Overhauser enhancement (NOE) effects from bound water and/or nearby hydrogens in molecules.40-41
Global fitting with a homogenous form of McConnell equations using Matlab code DESTfit (https://spin.niddk.nih.gov/clore/Software/software.html),28 where a single spin in exchange between an observable free state with low R2 and a two types of bound with larger R2 values (A <==> the mixture of B and C).28 The cross-relaxation between two spins was incorporated in the McConnell model: the cross-relaxation rate σA between H1 (the observed signal) and H2 (coupled with H1 by cross relaxation) in the free amino acid is assumed to be −0.5 s−1, and the cross-relaxation rate σB between H1 and H2 of amino acid bound on particle is assumed to be −500 s−1.28
Molecular Dynamics Calculations
The effects of molecular (non-hydroxylated TiO2) and dissociated (hydroxylated TiO2) surface water were investigated for two variants of the rutile (110), using the force field developed by Předota et al.42 The surface dimensions are approximately 5.5 × 5.3 × 1.8 nm3, and both surfaces are negatively charged with a charge density of −0.103 C/m2, corresponding to a pH of 8. Three amino acids were chosen for this study – aspartic acid (Asp), lysine (Lys), and arginine (Arg); in the pH range of 7.5–8.0, the charges of the amino acid sidechains are −1, +1 and +1, respectively. To remain comparable with the 1H NMR experiment, amino acid termini have a deprotonated carboxylate group and a protonated amine group. The amino acids were modeled using the CHARMM36 forcefield.43 A water slab 8nm thick consisting of ~7500 molecules of SPC/E water was added above the surface. System equilibration was carried out using a Dona dio-Bussi-Parrinello44 and a Parrinello-Rahman45 barostat to maintain a temperature and pressure of 300K and 1 bar, respectively. After equilibrating the system at 1bar and 300K, well-tempered metadynamics46 was employed to calculate binding free energy profiles by biasing the vertical distance of the peptide from the surface (see Supporting Information for details). Simulations were performed using GROMACS 5.1.2,47 along with the PLUMED plugin48 for enhanced sampling.
RESULTS AND DISCUSION
1H DEST NMR Studies of the interactions of amino acids Arg, Lys, Asp, Leu and Ala with TiO2 NPs in Agarose Gels.
The addition of TiO2 (1 wt%) to a 10 mM amino acid solution in 99.9% D2O resulted in extensive line broadening of the NMR resonances in all cases (Figure 1, and Figure S1), thus indicating that Arg, Lys, Asp, Leu and Ala all interact with TiO2 and exchange between a free and a bound state. A lesser degree of peak broadening in observed upon the addition of agarose gel to the sample, indicating that the amino acids are weakly interacting with the matrix, but retain the ability to diffuse and tumble freely.
Figure 1.

1H proton spectra of arginine neat (black), suspended in agarose gel (blue), and exposed to TiO2 nanoparticles in the presence of agarose gel (red). Addition of TiO2 to the sample creates distinct peak broadening not observed in the other two spectra, indicative of interactions with a slow tumbling object.
In DEST NMR experiments slow exchange and by inference contact with NP surfaces, is indicated by broadening of the DEST saturation profile upon addition of agarose gel and TiO2 NPs. Figure 2 shows the 1H DEST profiles of side chain protons for five monomeric amino acids (Arg, Lys, Leu, Asp, and Ala): 1) free in solution (10mM), 2) in the presence of agarose gel, and 3) in the presence of TiO2 NPs and agarose gel. These amino acids were chosen for their appearance in the hexamer peptide TBP-6, which has been shown to bind strongly to rutile TiO2.16 Mutation studies of the peptide have suggested that the three polar residues (Arg, Lys, and Asp) are involved in surface adhesion, while further studies have suggested that non-polar residues may also play a role in surface interactions.32-36 In the absence of TiO2 NP’s the DEST saturation profiles for Hε of Lys (Figure 2b) and Hδ of Arg (Figure 2a) are narrow and confined to <1 ppm region about the respective resonance frequencies. In both cases there is a slight broadening of the saturation profile upon addition of agarose gel (Figures 2a and 2b), indicating weak interactions of these basic amino acids with the gel. Upon addition of TiO2 NPs, the saturation profiles of both Arg and Lys become broadened and display broad “wings” indicative of slow exchange between the free state of the amino acid and a “dark”, i.e. surface-bound state, with a very large R2. The larger broadening of the Arg DEST profile upon TiO2 addition suggests that Arg has a greater affinity of binding to the nanoparticles than Lys.
Figure 2.

1H DEST profiles for Hδ of 10 mM Arg (a), Hε of 10 mM Lys (b), Hδ of 10 mM Leu (c), Hβ of Asp (d), and Hβ of 10 mM Ala (e) neat (black), with 1% w/w agarose gel (red), and with 1% w/w agarose gel and 1% w/w TiO2 nanoparticles (blue). Slight peak broadening of the DEST profile upon addition of agarose gel for Arg and Lys (b-c) indicates a weak interaction between the agarose gel and the amino acids, and broadening upon addition to TiO2 indicates an interaction with the oxide.
Figure 2c-e show analogous DEST saturation profiles for Hβ of Asp, Hδ of Leu, and Hβ of Ala respectively. In the three cases there is no broadening of the saturation profile upon addition of agarose gel (Figure 2c-d), indicating no interactions of these amino acids with the matrix. Interestingly, the Asp monomer as well as the nonpolar amino acids do not show broad “wings” in their DEST profiles upon addition of TiO2 NPs, indicating that these individual amino acid monomers retain considerable re-orientational degrees of freedom upon addition of TiO2 NPs and as a result have much shorter R2 values than is the case with Arg and Lys.
In the case of Arg and Lys the 1H DEST profiles in the presence of TiO2 NPs are asymmetric, indicating presence of spectral features in addition to the direct saturation lines. Information on the origins of the broad asymmetric DEST profiles for Arg (Figure 3) and Lys (Figure S2) may be obtained by a study of the DEST profiles as a function of 1H B1 field strength. Figure 3 shows 1H DEST profiles for the Hδ proton of Arg: (a) free in 10 mM solution; (b) 10 mM concentration in agarose gel; and (c) 10mM concentration in agarose gel and in the presence of TiO2 NPs, for B1 saturation fields ranging from 50-300 Hz. In all three figures a partial source of the asymmetry is traced in part to a line at a chemical shift of 1.68 ppm. In Figure 3a this is the small feature out of phase with the direct saturation line, while in Figure 3b and c the line at 1.68 ppm is in phase with the direct saturation line and is much more intense. This 1.68 ppm line is therefore an NOE to neighboring Hβ and Hγ protons. The fact that the NOE inverts from positive to negative upon addition of gel indicates a slowing of molecular reorientations, and the increase in the NOE intensity upon addition of TiO2 NPs indicates a further slowing of molecular reorientations due to strong interactions between the amino acid and the TiO2 NP surface. In fact, from the B1=50 Hz. data in Figure 3c at least three components to the 1H DEST saturation profile are discernable: 1) the direct Hδ saturation component at 3.16 ppm, 2) the aforementioned NOE to neighboring side chain protons at 1.68 ppm, and 3) the component at about 4.73 ppm. In addition to these three components, a fourth broad component is clearly observable in the 300Hz. profile. The component at 4.73 ppm is the NOE to the protons of surface-adsorbed water on TiO2 particles. In the DEST saturation profile of the Hδ proton of Arg, this broad component is centered at about 3.16 ppm and corresponds to a surface immobilized molecular species. This broad component and strong NOE peak is also observed in the 1H DEST profile for the Hδ proton of Arg (Figure 2a, and Figure 3) and the Hε proton of Lys (Figure 2b, and Figure S2), but not in the DEST profiles for the side chain protons in Leu, Asp, or Ala (see Figures 2c-e, Figures S3-S5). This component therefore is not associated with protons attached to the TiO2 surface but corresponds in the case of Arg to Hδ spins in Arg molecules that are strongly bound to and largely immobilized on the NP surface.
Figure 3.

1H DEST profiles for Hδ of 10 mM Arg (a) in 10mM solution; (b) in 10mM solution + agarose gel; (c) in 10mM +agarose gel+ TiO2 NPs. The strong enhancement of NOE peak upon addition of TiO2 indicates a strong interaction between Arg and TiO2.
Inverted 1H DEST saturation profiles of Arg (i.e. 1-I/I0) were fitted to Equation 1 for B1=50-300Hz. as described in the data analysis section (Figure 4a-c and Table S1-S3). It is shown that NOE peaks and broad “wing” components enhance with increasing B1 saturation power; NOE peak intensity show relative less sensitive to B1 saturation power although broad “wing” components are much more sensitive to B1 saturation power (Table S1-S3). It was assumed that the chemical shifts ωi were constant for all B1 values while Ai and σi were varied. Several conclusions can be drawn from inspection of the fitted data. First, the goodness of the fits to the data validate the four-component assumption. Second, the direct saturation line is in phase with the NOEs, i.e. the NOEs are negative. This indicates that Arg is tumbling slowly due to interactions with the NP surface. Third, the negative NOE to water is likely not due to magnetization transfer to bulk water but rather involves water that is bound to the TiO2 NP surface. Finally, these simulations indicate presence of two bound forms of Arg: a weakly bound form which undergoes slow re-orientational motions and is closely associated with surface water molecules, and an immobilized form. A similar analysis has also been performed on the 1H DEST profile for the Hε proton of Lys with similar results (Figure S6).
Figure 4.

Fitted 1H DEST profiles for Hδ of 10 mM Arg at 50 Hz (a), 150 Hz (b), and 300 Hz (c). Z-spectrums were fitted as the sum of multiple Lorentzian functions (see text).
In contrast to Lys and Arg, the DEST profiles for the side chain protons of Asp, Leu and Ala lack the broad component feature (Figures 2c-2d) and NOE intensities to proximal protons are not observed or are much weaker than is the case for protons in Arg and Lys (Figures S3-S5). These data indicate that Asp, Leu, and Ala do not have multiple forms of surface attached species, these amino acids do not display a strongly surface-attached, immobilized form, and reorientational motions are much faster than is the case for Lys and Arg, indicating much weaker interactions with the TiO2 NP surface.
To obtain surface adsorption/desorption rates, transverse relaxation R2 rates and the populations of the bulk solution versus adsorbed species, DEST saturation profiles were simulated by numerical solution of the Bloch-McConnell equations.49 The multi-Lorentzian fittings to the inverted DEST saturation profiles for 10mM Arg in the presence of TiO2 NP’s indicate presence of at least two adsorbed forms of Arg. The simplest kinetic scheme consistent with the simulated profiles in Figure 4 is shown in Figure 5. Figure 5 is based on a three site exchange model where monomeric Arg in bulk solution (A) is in exchange with two physically distinct adsorbed forms: a partially mobile bound form B and an immobilized bound form C. Adsorption of the amino acid in bulk solution A to these bound forms B and C is characterized by the kinetic constants kAB and kAC, respectively. Desorption from B and C forms to A is characterized by kBA and kCA, respectively. Exchange between the surface-bound forms B and C is characterized by the kinetic constants kBC and kCB.
Figure 5.

Kinetic Scheme for adsorption of Arg onto TiO2 NP’s. A weakly adsorbed form which retains some freedom or motion is designated B and a strongly bound essentially immobilized form is designated C. Adsorption from the bulk to weakly bound and strongly bound form is designated by kinetic constants kAB and kAC, respectively. Desorption from the weakly bound and strongly bound forms is designated by kinetic constants kBA and kCA, respectively. Interconversion between bound forms B and C is quantified by kBC and kCB.
1H DEST profiles of 10mM Arg in 1 wt % TiO2 and 1 wt % agarose at a proton Larmor frequency of 700 MHz were simulated using the program DESTfit, according to protocols described in detail in reference 28 and 50. As expected from the multi- Lorentzian fits in Figure 4, the DEST profile for 10mM Arg is fitted best by a model involving exchange between the amino acid in bulk solution A and two adsorbed forms B and C. The best fit model is shown in Figure 6 and Figure S7, and corresponds to the pseudo-two site exchange described in reference 28 and 50, where A exchanges with a single kinetic off rate koff, between a mixture of B and C forms. Notably a two-site exchange model where A exchanges with a single bound form did not fit the data well. This case is included as Supplementary Information (Figure S8). A similar analysis of the 1H DEST profile for Lys adsorbed onto TiO2 NPs is also included in Supplementary Information (Figure S9).
Figure 6.

1 H DEST profiles for Hδ of 10 mM Arg in presence of 1 wt % TiO2 and 1 wt % agarose on a 700 MHz spectrometer with different B1 saturation fields 50 Hz, 100 Hz, 150 Hz, 200 Hz, 250 Hz, and 300 Hz, and global fitting with a homogenous form of McConnell equations using Matlab code DESTfit,28,50 where a single spin in exchange between an observable free state A with low R2 and two bound states (B and C) with larger R2 values (A <==> the mixture of B and C). The cross-relaxation rate σA between Hδ (the observed signal) and Hγ (coupled with Hδ by cross relaxation) in free Arginine is assumed to be −0.5 s−1, and cross-relaxation rate σB between Hδ and Hγ of Arginine bound on particle is assumed to be −500 s−1, the output of global fitting: R2 (strong binding) = 38785±119 s−1, R2 (weak binding) = 784±2s−1 with population weights of 0.296 and 0.704, respectively, koff = 36.9±0.1 s−1, kon = 2.9±0.0 s−1, total population of binding state = 0.073, population of free state=0.927, population of strong binding state=0.022, population of weak binding state=0.051.
Molecular Dynamics Simulations
To investigate the nature and origins of structural diversity of adsorbed amino acids at liquid-TiO2 NP interfaces, and the role played by surface-adsorbed water and surface hydroxyl groups in molecular adsorption, metadynamics was used to compute the binding free energy as a function of amino acid center of mass distance from the non-hydroxylated (Figure 7a) and hydroxylated (Figure 8a) surfaces for Arg and Lys, both of which show broadened DEST profiles in the presence of TiO2 NP’s , as well as Asp, which does not show a broadened DEST profile in the presence of TiO2 NP’s. Calculation of binding free energy is described in the Supporting Information. On both surfaces, we see that Arg is the strongest binder, followed by Lys, and finally Asp. On the non-hydroxylated surface (Figure 7a), there are two free energy minima for Lys and Arg, whereas Asp only shows a single minimum. Interestingly, on the hydroxylated surfaces, although the binding free energies for Arg and Lys are comparable to the non-hydroxylated surface, there is only a single minimum in the binding free energy profile. Asp binds with a lower free energy on the hydroxylated surface compared to the non-hydroxylated surface. The binding free energies for Arg, Lys, and Asp on the non-hydroxylated and hydroxylated surfaces are given in Table 1.
Figure 7:

(a) Free energy profiles as a function of amino acid distance from the low-hydroxylated surface of TiO2. Dominant binding conformations represented by these curves are shown for arginine (b and c), lysine (d and e), and aspartic acid (f) on the low-hydroxylated surface.
Figure 8:

(a) Free energy as a function of amino acid distance from the hydroxylated surface of TiO2. Dominant binding conformations are shown for arginine (b), lysine (c), and aspartate (d) on the hydroxylated surface.
Table 1:
Binding free energies for the three amino acids on the two surfaces, in kJ/mol. The binding modes indicate binding poses (flat vs. extended) on the surface.
| Binding Free Energy (kJ/mol) Non-Hydroxylated Surface |
Binding Free Energy (kJ/mol) Hydroxylated Surface |
||
|---|---|---|---|
| Mode 1 (flat) | Mode 2 (extended) | ||
| Arginine | −17 | −20 | −17 |
| Lysine | −15 | −10 | −15 |
| Aspartate | −10 | – | −6 |
To understand the structural diversity of the amino acids on the surface, bound structures corresponding to the minima in the free energies for Arg (Figures 7 b-c, Figure 8b), Lys (Figures 7 d-e, Figure 8c), and Asp (Figure 7f, Figure 8d) are shown. On the non-hydroxylated surface, Arg and Lys adopt distinct flat (Figures 7b and 7d, respectively) and extended (Figures 7c and 7e, respectively) conformations. The flat conformation for both (Figures 7b and 7d) is mediated by the binding of the N-terminus, whereas the extended conformation occurs through the sidechain binding. On the hydroxylated surface, both amino acids adopt a flat conformation, mediated by the sidechain and C-terminus. This is in overall agreement with the features of the free energy curve in Figure 8. In contrast, Asp adopts a single conformation on both hydroxylated and non-hydroxylated surfaces. There is also good agreement with prior simulation studies, which report that binding occurs through backbone and side chain associations,54 as well as the fact that arginine binding is the strongest.32,33
The binding of amino acids with TiO2
Study of the binding of amino acids to inorganic oxide NP surfaces, and TiO2 NP surfaces in particular, is motivated by the fact that amino acids serve as models for the binding of more complicated polypeptides which are believed to interact with TiO2 surface hydroxyl groups via amino acid side chains. Numerous experimental studies, cited above, have characterized the binding affinity of amino acids to TiO2 NP surfaces. The present DEST NMR study, accompanied by a theoretical analysis of binding structures, not only characterizes the binding affinity of five amino acids (Arg, Lys, Leu, Asp, Ala) to TiO2 NP surfaces, but also the kinetics of binding. This is accomplished by simulation of the 1H DEST saturation profiles through solution of the Bloch-McConnell equations, which yields R2 values and populations as well as the kinetic constants that quantify exchange between the various free and bound species.
Kinetic information afforded by DEST simulations are useful both for interpreting the relationship between the free and multiple bound states as well as the degree to which interactions with the surface immobilizes the various bound molecular forms, where the transverse relaxation rate increases with the degree of immobilization of the molecule on the surface. It is interesting to compare the results of the DEST study to earlier studies of amino acids bound to TiO2 surfaces, and how kinetic information enhances our view of the binding mechanism. The 1H DEST NMR studies indicate that the amino acids Leu, Ala, and Asp have only a single partly mobile bound form in exchange with the freely tumbling amino acid in bulk solution. The Asp result is in accord with an earlier Reflectance IR study which detected at least two bound forms of glutamic acid (Glu) on TiO2 surfaces, but a single bound form for Asp.26 The fact that Asp is weakly adsorbed onto the TiO2 NP surface is indicated by its narrow DEST profile which indicates the occurrence of reorientational motion even in the bound form. This weak binding is also in accord with the fact that at pH 7 the net charge on Asp is negative and TiO2 has a negative surface charge.
From 1H DEST profiles of side chain protons however, both Arg and Lys have at least two bound forms on TiO2 NP surfaces, which display in both cases varying degrees of re-orientational motion as indicated by very different R2 values. An interesting conclusion of the DEST study of Arg involves the relationship between these weakly and strongly bound forms. In the DEST study of cholic acid and phenol to cerium oxide particles by Egner et al.31 analysis of DEST saturation profiles for cholic acid indicated only a single weakly bound form. However similar DEST studies of phenol indicated two bound forms: a weakly bound form displaying residual re-orientational motion, and a strongly bound form with a larger R2 relaxation rate indicating a greater degree of surface immobilization, a similar conclusion to that drawn by our DEST studies of Lys and Arg on TiO2 NP surfaces. However, the kinetic information derived from the Egner et al study affords a different view of the binding mechanism than is given by the present study of amino acid binding to TiO2 NP’s. With reference to the kinetic scheme in Figure 5, Egner et al. found a best fit to their DEST profile assuming kAC=kCA=0, which indicates that phenol does not directly attach from the bulk solution, where it freely reorients, to the surface, where it is in an immobilized state. Egner et al. also found a finite rate of exchange between the weakly and strongly bound forms, indicating that phenol binds strongly to cerium oxide NP’s via a weakly bound intermediate, which retains some reorientational degrees of freedom.
The DEST analysis of amino acid attachment to TiO2 NPs presented in this paper indicates a different mechanism of binding between basic amino acids Lys and Arg and rutile TiO2 NPs than occurs between phenol and cerium oxide NPs. Again with reference to Figure 5, a model that best fits DEST profiles assumes for both Lys and Arg the absence of exchange between the weakly and strongly bound species, i.e. kBC=kCB=0, and that Lys and Arg in solution interact directly with NP surfaces to form populations of weakly and strongly bound forms.
Because exchange between the two bound forms of Lys/Arg is absent in the best fit model to the DEST data, we cannot propose that the partially mobile forms of Lys or Arg are binding intermediates. So we turn to the surface chemistry of TiO2 as a possible source of variation in binding. It has long been known that dissociative adsorption of water onto rutile surfaces results in the formation of surface hydroxyl groups. 51,52 Dissociative adsorption of water and subsequent formation of surface hydroxyl groups is known to be face sensitive.53 The surface chemistry observed for TiO2 NPs is also known to vary with crystal face and surface area.54 Therefore, the occurrence of bound Lys and Arg amino acids with different R2 values may arise when amino acids attach to NP faces with different surface hydroxyl densities, resulting in different degrees of immobilization.
Molecular dynamics simulations were used to assess the degree to which the variation of surface water and surface hydroxyl groups on the TiO2 NP surface might account for these multiple bound forms of Lys and Arg. MD simulations in this paper treated two extreme cases: complete absence and presence of surface hydroxyl groups. Under these conditions good qualitative agreement was achieved with 1H DEST data. For example, on non-hydroxylated surfaces, Arg was found to occur in two forms distinguished by two modes of surface attachment: 1) attachment via the guanidinium group and 2) attachment via the amino group. In each case the opposite end of the amino acid was free to undergo restricted reorientational motions. We propose that these two forms would contribute to the so-called partly mobile bound form observed as a relatively narrow component of the DEST profile of the Hδ Arg proton. However, on hydroxylated TiO2 surfaces, Arg is attached via both its side chain and its amino group, essentially immobilizing the molecule or restricting its motions to a much greater degree than is observed on non-hydroxylated surfaces. This would account for the broad components observed in the 1H DEST profiles of Arg and Lys.
CONCLUSIONS
This paper describes an application of 1H DEST NMR techniques and Molecular Dynamics (MD) simulations to the study of the kinetics and thermodynamics of biomolecular adsorption onto rutile TiO2 surfaces. In this work basic amino acids are observed by 1H DEST experiments to adsorb onto rutile TiO2 surfaces in multiple forms distinguished by varying degrees of mobility on the surface. Molecular Dynamics simulations indicate that variation in surface hydroxyl group group density may be partly responsible for these observations.
At low saturation power, we can clearly observe the strong NOE peak of Arg or Lys, however the NOE peaks merge with the broad "wing" (the broad DEST profile) at high saturation power in presence of TiO2, the NOE peak indicates the strong binding of Arg or Lys with TiO2 particles; the study of DEST profile of substrates at low power could be a complementary to STD NMR where the target particle should be saturated and the interest hydrogens of target particle should not overlap with the hydrogens of substrates, however such limits can be ovoid in the study of substrate-target interaction with DEST NMR.
Although this study focused on the adsorption of monomeric amino acids onto TiO2 NP surfaces, 1H DEST will be useful in general for studying the effect of surface heterogeneity in the binding of larger peptides and proteins to inorganic oxide surfaces. For example, a widely studied surface-binding peptide is the 12 amino acid peptide aptamer i.e. TBP-1 (RKLPDAPGMHTW) developed by Sano and coworkers used peptide phage display methodology which electrostatically interacts with the oxidized surface of Ti16-18. The N-terminal heaxapeptide RKLPDA is sufficient for TiO2 . Even in the case of this relatively small peptide, diverse views have been reported for its interactions with TiO2 surfaces. Sano and coworkers proposed that the positively charged side chain of R1 binds to acidic (−O− ) hydroxyl sites while the negatively charged side chain of D5 binds to basic (−OH2+ ) hydroxyl sites.16 This binding model was supported by a subsequent adhesion force analysis using AFM.18 In contrast an NMR study by Suzuki et.al. of TBP-6 bound to TiO2 NP’s concluded that the peptide interacts with the NP surface via the side chains of R1 and K2, while the C-terminal amino acids do not display interactions with the surface.55
Based on the results of this study, it is possible that the results of both of these studies are valid. The structure observed by Suzuki et al. may a partly mobile form with a small R2, while the fully immobilized or dark state form reported by Sano and coworkers possesses a much larger R2. This situation, which may result from heterogeneity of the NP surface chemistry, could be identified by a 1H DEST study. We will report such a study in the near future.
Supplementary Material
Acknowledgements.
GPD acknowledges National Institutes of Health Grant R21 A126113, NASA grant NNX17AK86G (Exobiology), and National Science Foundation Grant MCB-1715123. GPD also acknowledges support from collaboration with the University of Oslo through the Research Council of Norway grant 231530.
REFERENCES
- (1).Verket A; Tiainen H; Haugen HJ; Lyngstadaas SP; Nilsen O; Reseland JE Enhanced osteoblast differentiation on scaffolds coated with TiO2 compared to SiO2 and CaP coatings. Biointerphases 2012, 7, 36. [DOI] [PubMed] [Google Scholar]
- (2).Chen QZ; Thompson ID; Boccaccini AR 45S5 Bioglass®-derived glass–ceramic scaffolds for bone tissue engineering. Biomaterials 2006, 27, 2414–2425. [DOI] [PubMed] [Google Scholar]
- (3).Yuan H; de Bruijn JD; Zhang X; van Blitterswijk CA; de. Groot K Bone induction by porous glass ceramic made from bioglass® (45S5). J. Biomed. Mater. Res 2001, 58, 270–276. [DOI] [PubMed] [Google Scholar]
- (4).Forsgren J; Svahn F; Jarmar T; Engqvist H Formation and adhesion of biomimetic hydroxyapatite deposited on titanium substrates. Acta Biomater. 2007, 3, 980–984. [DOI] [PubMed] [Google Scholar]
- (5).Uchida M; Kim H; Kokubo T; Fujibayashi S; Nakamura T Structural dependence of apatite formation on titania gels in a simulated body fluid. J. Biomed. Mater. Res. Part A 2003, 64, 164–170. [DOI] [PubMed] [Google Scholar]
- (6).Tiainen H; Wohlfahrt JC; Verket A; Lyngstadaas SP; Haugen HJ Bone formation in TiO2 bone scaffolds in extraction sockets of minipigs. Acta Biomater. 2012, 8, 2384–2391. [DOI] [PubMed] [Google Scholar]
- (7).Haugen H; Will J; Köhler A; Hopfner U; Aigner J; Wintermantel E Ceramic TiO2-foams: Characterisation of a potential scaffold. J. Eur. Ceram. Soc 2004, 24, 661–668. [Google Scholar]
- (8).Tiainen H; Lyngstadaas SP; Ellingsen JE; Haugen HJ Ultra-porous titanium oxide scaffold with high compressive strength. J. Mater. Sci. Mater. Med 2010, 21, 2783–2792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Jones FH Teeth and bones: Applications of surface science to dental materials and related biomaterials. Surf. Sci. Rep 2001, 42, 75–205. [Google Scholar]
- (10).Linsebigler AL; Lu G; Yates JT Photocatalysis on TiO2 surfaces: Principles, mechanisms, and selected results. Chem. Rev 1995, 95, 735–758. [Google Scholar]
- (11).Varghese OK; Grimes CA Metal oxide nanoarchitectures for environmental sensing. J. Nanosci. Nanotechnol 2003, 3, 277–293. [DOI] [PubMed] [Google Scholar]
- (12).Wintermantel E; Eckert K-L; Huang N-P; Textor M; Brunette DM Titanium ceramics for cell-carriers and for medical applications; Springer Berlin Heidelberg, 2001; pp 649–671. [Google Scholar]
- (13).Vallee A; Humblot V; Pradier C Peptide interactions with metal and oxide surfaces. Acc. Chem. Res 2010, 43, 1297–1306. [DOI] [PubMed] [Google Scholar]
- (14).Buckle EL; Lum JS; Roehrich AM; Stote RE; Vandermoon B; Dracinsky M; Filocamo SF; Drobny GP Serine–lysine peptides as mediators for the production of titanium dioxide: investigating the effects of primary and secondary structures using solid-state NMR spectroscopy and DFT calculations. J. Phys. Chem. B 2018, 122, 4708–4718. [DOI] [PubMed] [Google Scholar]
- (15).Buckle EL; Prakash A; Bonomi M; Sampath J; Pfaendtner J; Drobny GP Solid-state NMR and MD study of the structure of the statherin mutant SNa15 on mineral surfaces. J. Am. Chem. Soc 2019, 141, 1998–2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Sano K; Shiba K A hexapeptide motif that electrostatically binds to the surface of titanium. J. Am. Chem. Soc 2003, 125, 14234–14235. [DOI] [PubMed] [Google Scholar]
- (17).Sano K; Sasaki H; Shiba K Specificity and biomineralization activities of Ti-Binding Peptide-1 (TBP-1). Langmuir 2005, 21, 3090–3095. [DOI] [PubMed] [Google Scholar]
- (18).Hayashi T; Sano K-I; Shiba K; Kumashiro Y; Iwahori K; Yamashita I; Hara M Mechanism underlying specificity of proteins targeting inorganic materials. Nano Lett. 2006, 6, 515–519. [DOI] [PubMed] [Google Scholar]
- (19).Chen Z; Shen YR; Somorjai GA Studies of polymer surfaces by sum frequency generation vibrational spectroscopy. Annu. Rev. Phys. Chem 2002, 53, 437–465. [DOI] [PubMed] [Google Scholar]
- (20).Weidner T; Castner DG SFG analysis of surface bound proteins: A route towards structure determination. Phys. Chem. Chem. Phys 2013, 15, 12516–12524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Shaw WJ Solid-state NMR studies of proteins immobilized on inorganic surfaces. Solid State Nucl. Magn. Reson 2015, 70, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Costa D; Savio L; Pradier C-M Adsorption of amino acids and peptides on metal and oxide surfaces in water environment: A synthetic and prospective review. J. Phys. Chem. B 2016, 120, 7039–7052. [DOI] [PubMed] [Google Scholar]
- (23).Okazaki S; Aoki T; Koichi T The adsorption of basic α-amino acids in an aqueous solution by titanium (IV) oxide. Bull. Chem. Soc. Jpn 1981, 54, 1595–1599. [Google Scholar]
- (24).Roddick-Lanzilotta AD; Connor PA; McQuillan AJ An in situ infrared spectroscopic study of the adsorption of lysine to TiO2 from an aqueous solution. Langmuir 1998, 14, 6479–6484. [Google Scholar]
- (25).Mudunkotuwa IA; Grassian VH Histidine adsorption on tio2 nanoparticles: An integrated spectroscopic, thermodynamic, and molecular-based approach toward understanding nano–bio interactions. Langmuir 2014, 30, 8751–8760. [DOI] [PubMed] [Google Scholar]
- (26).Roddick-Lanzilotta AD; McQuillan AJ An in situ infrared spectroscopic study of glutamic acid and of aspartic acid adsorbed on TiO2: Implications for the biocompatibility of titanium. J. Colloid Interf. Sci 2000, 227, 48–54. [DOI] [PubMed] [Google Scholar]
- (27).Shchelokov A, Palko N, Potemkin V, Grishina M, Morozov R, Korina E, Uchaev D, Krivtsov I, Bol’shakov O, Adsorption of native amino acids on nanocrystalline TiO2: Physical chemistry, QSPR, and theoretical modeling. Langmuir 2019, 35, 538–550. [DOI] [PubMed] [Google Scholar]
- (28).Fawzi NL; Ying J; Torchia DA; Clore GM Kinetics of amyloid β monomer-to-oligomer exchange by NMR relaxation. J. Am. Chem. Soc 2010, 132, 9948–9951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Fawzi NL; Ying J; Ghirlando R; Torchia DA; Clore GM Atomic-resolution dynamics on the surface of amyloid-β protofibrils probed by solution NMR. Nature 2011, 480, 268–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Fawzi NL; Libich DS; Ying J; Tugarinov V; Clore GM Characterizing methyl-bearing side chain contacts and dynamics mediating amyloid β protofibril interactions using 13Cmethyl-DEST and lifetime line broadening. Angew. Chem. Int. Ed 2014, 53, 10345–10349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Egner TK; Naik P; Nelson NC; Slowing II; Venditti V Mechanistic insight into nanoparticle surface adsorption by solution NMR spectroscopy in an aqueous gel. Angew. Chem. Int. Ed 2017, 129, 9934–9938. [DOI] [PubMed] [Google Scholar]
- (32).Yazdanyar A; Aschauer U; Bowen P Adsorption free energy of single amino acids at the rutile (110)/water interface studied by well-tempered metadynamics. J. Phys. Chem. C 2018, 122, 11355–11363. [Google Scholar]
- (33).Kang Y; Li X; Tu Y; Wang Q; Ågren H On the mechanism of protein adsorption onto hydroxylated and nonhydroxylated TiO2 surfaces. J. Phys. Chem. C 2010, 114, 14496–14502. [Google Scholar]
- (34).Brandt EG; Lyubartsev AP Molecular dynamics simulations of adsorption of amino acid side chain analogues and a titanium binding peptide on the TiO2 (100) surface. J. Phys. Chem. C 2015, 119, 18126–18139. [Google Scholar]
- (35).Skelton AA; Liang T; Walsh TR Interplay of sequence, conformation, and binding at the peptide–titania interface as mediated by water. ACS Appl. Mater. Interfaces 2009, 1, 1482–1491. [DOI] [PubMed] [Google Scholar]
- (36).Sultan AM; Hughes ZE; Walsh TR Binding affinities of amino acid analogues at the charged aqueous titania interface: implications for titania-binding peptides. Langmuir 2014, 30, 13321–13329. [DOI] [PubMed] [Google Scholar]
- (37).Zhou IY; Wang E; Cheung JS; Zhang X; Fulci G; Sun PZ Quantitative chemical exchange saturation transfer (CEST) MRI of glioma using Image Downsampling Expedited Adaptive Least-squares (IDEAL) fitting. Sci. Rep 2017, 7, 84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Cai K; Singh A; Poptani H; Li W; Yang S; Lu Y; Hariharan H; Zhou XJ; Reddy R CEST signal at 2 ppm (CEST@2ppm) from Z-spectral fitting correlates with creatine distribution in brain tumor. NMR Biomed. 2015, 28, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Zaiss M; Windschuh J; Paech D; Meissner J-E; Burth S; Schmitt B; Kickingereder P; Wiestler B; Wick W; Bendszus M; Schlemmer H-P; Ladd ME; Bachert P; Radbruch A Relaxation-compensated CEST-MRI of the human brain at 7 T: Unbiased insight into NOE and amide signal changes in human glioblastoma. NeuroImage 2015, 112, 180–188. [DOI] [PubMed] [Google Scholar]
- (40).Zhang X-Y; Wang F; Afzal A; Xu J; Gore JC; Gochberg DF; Zu Z A new NOE-mediated MT signal at around −1.6 ppm for detecting ischemic stroke in rat brain. Magn. Reson. Imaging 2016, 34, 1100–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Zhang X; Wang F; Jin T; Xu J; Xie J; Gochberg DF; Gore JC; Zu Z MR imaging of a novel NOE-mediated magnetization transfer with water in rat brain at 9.4 T. Magn. Reson. Med 2017, 78, 588–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Předota M; Bandura AV; Cummings PT; Kubicki JD; Wesolowski DJ; Chialvo AA; Machesky ML Electric double layer at the rutile (110) surface. 1. Structure of surfaces and interfacial water from molecular dynamics by use of ab initio potentials. J. Phys. Chem. B 2004, 108, 12049–12060. [Google Scholar]
- (43).Huang J; MacKerell AD Jr CHARMM36 all-atom additive protein force field : Validation based on comparison to NMR data. J. Comput. Chem 2013, 34, 2135–2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Bussi G; Donadio D; Parrinello M Canonical sampling through velocity rescaling. J. Chem. Phys 2007, 126, 014101. [DOI] [PubMed] [Google Scholar]
- (45).Parrinello M; Rahman A Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys 1981, 52, 7182. [Google Scholar]
- (46).Barducci A; Bussi G; Parrinello M Well-tempered metadynamics: A smoothly converging and tunable free-energy method. Phys. Rev. Lett 2008, 100, 020603. [DOI] [PubMed] [Google Scholar]
- (47).Abraham MJ; Murtola T; Schulz R; Páll S; Smith JC; Hess B; Lindahl E GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX, 2015, 1–2, 19–25. [Google Scholar]
- (48).Tribello GA; Bonomi M; Branduardi D; Camilloni C; Bussi G PLUMED 2: New feathers for an old bird. Comput. Phys. Commun 2014, 185, 604–613. [Google Scholar]
- (49).Helgstrand M; Härd T; Allard P Simulations of NMR pulse sequences during equilibrium and non-equilibrium chemical exchange. J. Biomol. NMR 2000, 18, 49–63. [DOI] [PubMed] [Google Scholar]
- (50).Fawzi NL; Ying J; Torchia DA; Clore GM Probing exchange kinetics and atomic resolution dynamics in high-molecular-weight complexes using dark-state exchange saturation transfer NMR spectroscopy. Nat. Protoc 2012, 7, 1523–1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Connor PA; Dobson KD; McQuillan AJ Infrared spectroscopy of the TiO2/aqueous solution interface. Langmuir 1999, 15, 2402–2408. [Google Scholar]
- (52).Henderson MA The interaction of water with solid surfaces: fundamental aspects revisited. Surf. Sci. Rep 2002, 46, 1–308. [Google Scholar]
- (53).Henderson MA Structural sensitivity in the dissociation of water on TiO2 single-crystal surfaces. Langmuir 1996, 12, 5093–5098. [Google Scholar]
- (54).Bolis V; Busco C; Ciarletta M; Distasi C; Erriquez J; Fenoglio I; Livraghi S; Morel S Hydrophilic/hydrophobic features of TiO2 nanoparticles as a function of crystal phase, surface area and coating, in relation to their potential toxicity in peripheral nervous system. J. Colloid Interface Sci 2012, 369, 28–39. [DOI] [PubMed] [Google Scholar]
- (55).Suzuki Y; Shindo H; Asakura T Structure and dynamic properties of a Ti-binding peptide bound to TiO2 nanoparticles as accessed by 1H NMR spectroscopy. J. Phys. Chem. B 2016, 120, 4600–4607. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.