

Abstract
Mutations in tubulins affect microtubule (MT) dynamics and functions during neuronal differentiation and their genetic interaction provides insights into the regulation of MT functions. We previously used Caenorhabditis elegans touch receptor neurons to analyze the cellular impact of tubulin mutations and reported the phenotypes of 67 tubulin missense mutations, categorized into three classes: loss-of-function (lf), antimorphic (anti), and neomorphic (neo) alleles. In this study, we isolated 54 additional tubulin alleles through suppressor screens in sensitized backgrounds that caused excessive neurite growth. These alleles included 32 missense mutations not analyzed before, bringing the total number of mutations in our collection to 99. Phenotypic characterization of these newly isolated mutations identified three new types of alleles: partial lf and weak neo alleles of mec-7/β-tubulin that had subtle effects and strong anti alleles of mec-12/α-tubulin. We also discovered complex genetic interactions among the tubulin mutations, including the suppression of neo mutations by intragenic lf and anti alleles, additive and synthetic effects between mec-7 neo alleles, and unexpected epistasis, in which weaker neo alleles masked the effects of stronger neo alleles in inducing ectopic neurite growth. We also observed balancing between neo and anti alleles, whose respective MT-hyperstablizing and -destabilizing effects neutralized each other.
INTRODUCTION
Understanding how the regulation of microtubules (MTs) affects neurite growth at the single cell level is important for deciphering the molecular mechanisms of neuronal morphogenesis. The α/β-tubulin heterodimer is the building block of MTs, and mutations in the tubulin genes have profound effects on MT dynamics and cellular functions (Basciano et al., 2015; Ti et al., 2016). Clinical studies in the past decades have identified over 100 tubulin missense mutations associated with a wide range of neurodevelopmental disorders, including microcephaly, lissencephaly, pachygyria, polymicrogyria, and other cortical malformations, as well as the agenesis or hypoplasia of the corpus callosum, internal capsule, commissural fibers, and corticospinal tracts (Tischfield et al., 2011). The diseases caused by mutations in tubulin genes are collectively termed “tubulinopathies” (Bahi-Buisson et al., 2014). Besides their severe clinical symptoms, the cellular impact of those tubulin missense mutations on individual neurons is not well characterized. Moreover, the genetic interaction among the tubulin mutations in the context of neuronal differentiation, especially in axonal growth, is not well studied.
We previously established a platform to study the impact of tubulin mutations on neuronal growth using the touch receptor neurons (TRNs) of the nematode Caenorhabditis elegans (Zheng et al., 2017). The TRNs have simple morphology and can be visualized by cell-specific expression of fluorescent proteins in live and free-moving animals. The two major TRN subtypes, ALM and PLM neurons, have well-defined cell shapes and neurite growth patterns (Figure 1, A and B). ALM neurons grow a single, anteriorly directed neurite, termed ALM-AN, and PLM neurons grow two neurites toward the anterior and the posterior, which are termed PLM-AN and PLM-PN, respectively. The simplicity of this neuronal structure offers the opportunity to quantitatively measure changes in neurite growth.
FIGURE 1:

Partial lf mutations in mec-7 suppressed ectopic neurite growth and led to the loss of the synaptic branch. (A) Schematic diagram of ALM and PLM morphologies in mec-7 and mec-12 lf, anti, and neo mutants, as well as the three newly identified types of alleles—partial lf and weak neo alleles of mec-7 and strong anti alleles of mec-12. (B–D) TRN morphology was visualized by TagRFP expressed from the TRN-specific mec-17 promoter. Compared with the WT animals, mec-7(u278neo) animals had a very long, ectopic ALM-PN (arrow), whose generation was suppressed by intragenic partial lf mutation R121K. Arrowhead indicates the absence of ALM-PN. (E, F) Length of ALM-PN and PLM-PN in young adults of various strains. “Col” means colchicine treatment of the WT animals. Double asterisks indicate statistically significant differences (p < 0.01) in a Dunnett’s test. (G) PLM-PN was significantly shortened in u278(C303Y neo) u1169(G146E lf) double mutants and mec-7(ok2152) deletion mutants but not in u278 u1156(R121K partial lf) and unk64(R121K) mutants. (H) Some unk64 mutants did not extend a synaptic branch from PLM-AN. (I) The percentage of PLM neurons that lost the synaptic branch in various mec-7 mutants. “Col” means colchicine treatment and “Tax” means taxol treatment. (J) The number of responses to five anterior gentle touches in young adults of indicated mec-7 mutants.
In our previous study, we analyzed the effects of 67 missense mutations in mec-12/α-tubulin and mec-7/β-tubulin, which are specifically expressed in the TRNs, on the neurite growth patterns of TRNs (Zheng et al., 2017). Based on the phenotypes, we categorized those mutant alleles into three classes: loss-of-function (lf), antimorph (anti), and neomorph (neo). The lf mutations rendered the tubulin inactive and caused mild neurite growth defects likely due to the compensation from other tubulin isotypes; antimorphic gain-of-function mutations acted as dominant-negatives to block MT polymerization and caused severe neurite growth defects; and neomorphic gain-of-function mutations resulted in increased MT stability and excessive neurite growth, in particular the production of an ectopic ALM posterior neurite, termed ALM-PN (Zheng et al., 2017). Structure–function analysis showed that lf mutations likely affected tubulin folding, anti mutations mostly affected residues in the GTP binding site and the intradimer and interdimer interfaces, and neo mutations, which were mapped to the exterior surface of the α/β-tubulin dimmer, were hypothesized to affect the interaction with MT-associated proteins (MAPs). Because of the hyperstable MTs and the generation of an ectopic ALM-PN in the neo strains, they provide a sensitized background that can be used to identify genes that contribute to MT stability and neurite growth, especially for genes whose mutations may not lead to significant changes in neuronal morphology in the otherwise wild-type (WT) background. Such suppressor screens may also identify mutations whose products interact genetically with the tubulin neomorphs.
mec-12 and mec-7 are not the only genes that can be mutated to cause excessive neurite growth of the TRNs. For example, lf mutations in a MT-depolymerizing kinesin-13 gene klp-7 resulted in the generation of an ectopic ALM-PN, presumably due to elevated MT stability (Zheng et al., 2017). Moreover, the loss of α-tubulin acetyltransferase gene mec-17 also induced the production of an ALM-PN, although this regulation of neurite growth was independent of the enzymatic activity of MEC-17 (Topalidou et al., 2012). Those mutants can also be used in suppressor screens to identify genes regulating MT dynamics and neurite growth.
In this study, we describe two forward genetic screens that identified suppressors of ectopic ALM-PN growth in mec-7(neo) and mec-17(lf) mutants, respectively. The two screens yielded 54 alleles in mec-7 and mec-12, of which 46 are missense alleles. These mutations included 32 new missense mutations. The addition of these new alleles brings the total number of tubulin missense mutations in our collection to 99. The phenotypic classification and structure–function analysis of these mutations provide a resource for understanding the functional impact of tubulin mutations on neuronal morphogenesis at the cellular level. The analysis of the newly isolated alleles defined three new types of tubulin alleles: partial lf alleles in mec-7 that specifically disrupted the growth of the PLM synaptic branch, weak mec-7(neo) alleles that caused the overextension of PLM-PN but did not induce the growth of an ectopic ALM-PN, and strong mec-12(anti) alleles that shortened all TRN neurites. Moreover, we found unexpected genetic interactions among the tubulin alleles, including the epistatic and synthetic interactions among the mec-7(neo) alleles, as well as a counterbalance between the effects of neo and anti mutations.
RESULTS
Genetic screens for suppressors of the ALM-PN phenotype yielded many mec-7 and mec-12 alleles
Among the tubulin neo mutations, mec-7(u278) produced the strongest phenotype with a very long ALM-PN that often extended to the tail region (Figure 1C). Leveraging this clear phenotype, we conducted a suppressor screen to search for mutations that can suppress the growth of the ectopic ALM-PN in mec-7(u278neo) mutants. We screened 53,600 haploid genomes and isolated 30 suppressors. Genetic mapping and whole-genome resequencing identified the phenotype-causing mutations in these suppressors, including 19 intragenic mutations in mec-7 (two nonsense alleles, one deletion allele, and 16 missense alleles), nine mutations in mec-12 (one deletion allele and 8 missense alleles), one mutation in mec-15, and one mutation in mbl-1 (Supplemental Table S1). mec-15 codes for an F-box domain and WD40 repeats-containing protein, whose role in regulating MT organization and neurite growth was previously reported (Zheng et al., 2020). mbl-1 codes for an RNA splicing regulator, whose function in the TRNs will be discussed elsewhere. In this study, we focus on the effects of mec-7 and mec-12 missense mutations on neuronal morphogenesis.
Deletion of mec-17, which encodes an α-tubulin acetyltransferase (Akella et al., 2010), alters MT structure and induces the growth of an ectopic ALM-PN (Topalidou et al., 2012). Compared with mec-7(u278neo) animals, the ALM-PN in mec-17(lf) mutants was much shorter and only occurred in L4 and adult animals (Figure 1E and Supplemental Supplemental Figure S1A). We reason that the late onset of the ALM-PN phenotype in mec-17(lf) mutants may allow us to isolate suppressors different from the suppressors of mec-7(u278neo). In the mec-17(lf) suppressor screen, we isolated 16 mutations in mec-7 (two nonsense alleles, one splice variant, and 13 missense alleles), 10 mutations in mec-12 (one start-loss allele and nine missense alleles; Supplemental Table S2), and five nontubulin mutations that will be described elsewhere. Thus, from the two suppressor screens, we identified 29 mec-7 missense alleles and 17 mec-12 missense alleles in total. The identification of mec-7 and mec-12 mutations as suppressors of the ALM-PN phenotype suggested that the generation of ectopic neurites in TRNs may require the incorporation of the MEC-12/MEC-7 (α/β tubulin) heterodimers into the MTs.
An updated catalogue of tubulin missense mutations affecting neurite growth
We previously reported the effects of 48 mec-7 and 19 mec-12 missense mutations on the growth of TRN neurites and categorized them into three classes (lf, anti, and neo), each with distinct morphological phenotypes (Zheng et al., 2017). The screens described in this study added 20 new mec-7 and 12 new mec-12 missense mutations to this collection (Table 1). Since these mutations suppressed the growth of ectopic neurites, we expected them to generally reduce MT stability, although there were a few exceptions (see below).
TABLE 1:
The mec-7 and mec-12 mutations analyzed for the effects on TRN neurite growth.
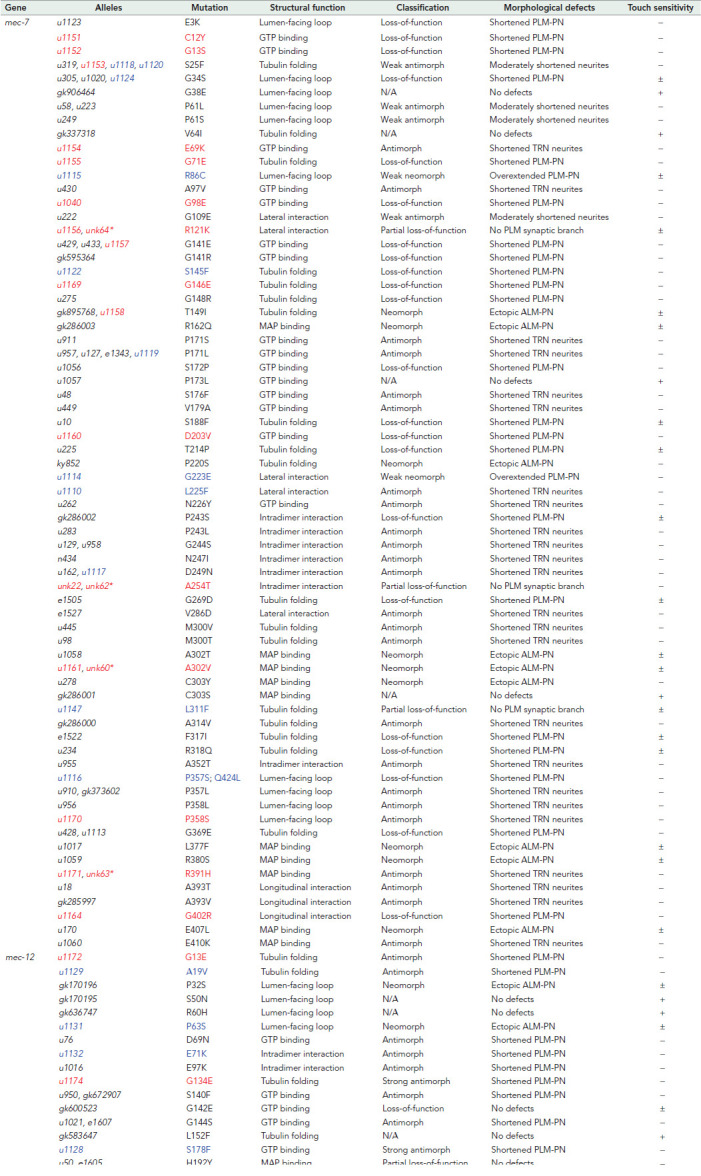 |
Alleles in black were previously analyzed (Zheng et al., 2017); alleles in red were isolated from the mec-7(u278 neo) suppressor screen; alleles in blue were isolated from the mec-17(-) suppressor screen. For touch sensitivity, (+) indicates the average response to five anterior stimuli is above 4; (±) indicates the average is between 4 and 1; (−) indicates the average is below 1. Asterisks indicate the alleles created through CRISPR/Cas9-mediated genome editing. Amino acid residues affected by the missense mutations were mapped to the B. taurus α/β-tubulin structures (Nogales et al., 1998).
To study the effect of each mutation on TRN morphogenesis, we removed mec-7(u278neo) from the mec-12 alleles obtained in the mec-7(neo) suppressor screen and removed mec-17(lf) mutations from the mec-7 and mec-12 alleles isolated in the mec-17(lf) suppressor screen through outcrossing. We then examined how the tubulin mutations affected TRN neurite growth in the otherwise WT background. For the intragenic mec-7 mutants isolated from the mec-7(neo) suppressor screen, it was technically impossible to remove the mec-7(u278neo) background through outcross, but we assumed the mec-7 suppressor alleles could mask the effects of mec-7(u278neo), allowing us to examine the single mutants’ phenotype. For example, G141E mutation was previously identified as a mec-7 lf allele (Table 1), and u278(C303Y) u1157(G141E) double mutants showed the same lf phenotype (shortening of PLM-PN) as the G141E single mutants. This strategy was effective in identifying the effects of most mec-7 intragenic suppressors except for four alleles, whose phenotype appeared to be ambiguous and did not fall into any phenotypical classification we previously established. To study the phenotypes of those four mec-7 missense mutations by themselves, we recreated them in the WT animals using CRISPR-mediated genome editing and examined their effects on TRN morphology (asterisks in Table 1).
A few mec-7 and mec-12 missense mutations were repeatedly isolated. For example, antimorphic S25F mutation in MEC-7 and G246E mutation in MEC-12 was isolated in previous screens using WT background and in both suppressor screens of this study. Overall, 11 missense mutations were recovered in more than one screen. Importantly, alleles that carry the same mutation but were independently isolated showed the same phenotype, suggesting that our system of phenotyping generated consistent results.
In summary, among the 20 newly found mec-7/β-tubulin missense alleles, 10 were lf alleles with clearly shortened PLM-PN and three were partial lf alleles that caused the loss of PLM synaptic branch but had normal length of PLM-PN (those partial lf alleles were not identified before and thus defined a new phenotypic category). Four were anti alleles that caused the shortening of all TRN neurites. The remaining three mutations were neo alleles that caused either the growth of ectopic ALM-PN or overextension of PLM-PN (two weak neo alleles had very low penetrance of ectopic ALM-PN phenotype and mostly caused the production of longer PLM-PNs, thus defining a new phenotypic category). The 12 newly isolated mec-12/α-tubulin missense alleles included 10 anti alleles that led to the shortening of PLM-PN, one neo allele that induced the growth of an ectopic ALM-PN, and one allele that appeared not to cause any phenotype by itself (Table 1). Moreover, lf alleles were mostly recessive, whereas anti and neo alleles were often dominant or semidominant.
Combining this study with our previous work (Zheng et al., 2017), we report the neuronal phenotype of a total of 99 tubulin missense mutations (68 β-tubulin and 31 α-tubulin mutations) represented by 127 alleles isolated or engineered independently (Table 1). Given the very high evolutionary conservation of tubulin amino acid sequences across species (identity > 95%), this large collection of tubulin mutants and their phenotypic characterization can serve as a resource for understanding the relationship of tubulin dimer structure with MT stability and neurite development in general.
Suppression of ectopic ALM-PN growth by tubulin lf and anti alleles
Among the 16 mec-7 intragenic missense mutations that suppressed the long ectopic ALM-PN caused by mec-7(u278) allele, 10 are lf alleles, four are antimorphs, and two are neomorphs. The eight mec-12 missense alleles that suppressed mec-7(u278) included four lf alleles and four anti alleles. Among the missense mutations that suppressed ALM-PN growth caused by the mec-17 deletion, there were six lf alleles, five anti alleles, and two neo alleles of mec-7; and one lf allele, six anti alleles, and one neo allele of mec-12 (Table 1). Thus, 89% of the suppressors are either tubulin lf or anti alleles, which down-regulate MT stability and limit neurite growth.
Interestingly, three mec-7 mutations, R121K, A254T, and L311F, defined a new type of lf alleles (R121K and A254T were recreated in the otherwise WT animals through CRISPR gene editing). The three mutations suppressed the ALM-PN production (Figure 1, D and E) but did not show a clear lf phenotype by themselves. For example, they did not cause the shortening of PLM-PN as seen in the mec-7 deletion alleles (Figure 1, F and G). They did, however, cause the loss of synaptic branch in PLM-AN and the reduction of touch sensitivity (Figure 1, H and J), which were also seen with the mec-7 deletion alleles. Because only part of the lf phenotypes was observed in these mutants, we classified them as partial lf alleles. The loss of the synaptic branch also led to disrupted transport of GFP-fused synaptic vesicle protein RAB-3. For PLM-ANs that had no clear branching point, we did not observe GFP::RAB-3 signal along the neurite; for PLM-ANs that had a very short branch, weak GFP::RAB-3 signal was observed at the branching site (Supplemental Figure S2, A and B).
We also found a new type of mec-12 anti alleles in this study. Previously, we classified mec-12 anti alleles as mutants that mostly shortened PLM-PN but not ALM-AN and PLM-AN and thought that mec-12 anti mutations had a weaker effect than mec-7 anti mutations, which caused the shortening of all TRN neurites (Figure 1A). Eight of the 10 new mec-12 anti alleles isolated in the suppressor screens indeed fit this phenotypic classification, but the other two alleles, u1174(G134E) and u1128(S178F), produced much stronger phenotypes, showing severe defects in the extension of ALM-AN, PLM-AN, and PLM-PN, similar to the phenotype of mec-7 anti alleles (Figure 2, A–C). Both mutations also caused excessive sprouting from PLM-AN, possibly the result of increased MT dynamics (Figure 2B). MT depolymerization induced a general down-regulation of protein expression in TRNs (Bounoutas et al., 2011). As a result, the level of TagRFP proteins (measured by fluorescent intensity) expressed from a TRN-specific promoter was down-regulated in tubulin mutants compared with the WT. We found that the TagRFP level was much lower in u1174 and u1128 mutants than in other mec-12 anti mutants, consistent with further reduced MT stability (Figure 2D). Since the phenotypes caused by these changes were more severe than the mec-12 anti alleles characterized before, we classified them as strong antimorphs (Table 1). These results suggest that, unlike previously thought, dominant-negative MEC-12 mutants can be as disruptive as the MEC-7 anti mutants in affecting TRN neurite growth.
FIGURE 2:

Strong anti alleles of mec-12 led to severe shortening of TRN neurites. (A) mec-12 anti mutations mostly shortened PLM-PN (arrowhead) but not PLM-AN. Arrow points to the terminal of PLM-AN and dashed line indicates the position of the vulva. (B) Strong anti mutations of mec-12 resulted in the shortening of not only PLN-AN (arrowed head) but also PLM-AN and ALM-AN (arrows). PLM-AN failed to reach the vulva (dashed line) and had excessive sprouting (asterisks). (C) Distance from the PLM-AN terminals to the vulva in various mec-12 mutants. Positive numbers mean that PLM-AN grew pass the vulva and negative numbers mean that PLM-AN did not reach the vulva. The “+ T” means taxol treatment of specific mutants. Double asterisks indicate a significant difference (p < 0.01) compared with the WT in a Dunnett’s test. (D) Fluorescent intensity of PLM neurons expressing the same mec-17p::TagRFP transgene measured under the same condition in various mutants. Intensity is in arbitrary units (A.U.).
Pharmacological studies supported that the TRN morphological defects in tubulin lf and anti mutants resulted from decreased MT stability. First, treatment with the MT-destabilizing agent colchicine in the WT animals shortened PLM-PN and caused the loss of PLM synaptic branch, similar to mec-7(lf) phenotypes (Figure 1, F and I). Second, MT-stabilizing drug paclitaxel (taxol) rescued the branching defects in mec-7 partial lf mutants (Figure 1I and Supplemental Figure S3). Third, taxol treatment also partially suppressed the severe neurite growth defects in mec-7 and mec-12 anti alleles (Figure 2C and Supplemental Figure S3).
Moreover, we also analyzed MT dynamics by tracking the MT-plus end binding protein EBP-2. MTs in WT animals are very stable and we could only observe a few EBP-2::GFP tracks in PLM neurites. However, mec-7(lf) mutants showed an increased number of EBP-2 tracks in PLM-AN and PLM-PN, and mec-7(anti) mutants had even more tracks (Figure 3, A and B). Furthermore, we found that MTs in PLM-AN maintained a largely uniform “plus-end-out” polarity and MTs in PLM-PN showed mixed polarity in WT animals, which is consistent with a previous observation (Hsu et al., 2014). Interestingly, MTs in both PLM-AN and PLM-PN had mixed polarity in mec-7 lf and anti mutants (Figure 3A and B). Increased MT dynamics and altered MT polarity in these mutants may have caused the defects in neurite extension.
FIGURE 3:

MT dynamics and polarity in mec-7 lf, anti, and neo mutants. (A) Representative kymographs of MT dynamics in the PLM-AN and PLM-PN of adult WT, mec-7(u1122lf; S145F), and mec-7(u1110anti; L225F) animals. (B) The total number of EBP-2::GFP comets in a 60-μm segment of PLM-AN and a 25-μm segment of PLM-PN from the cell body in WT, mec-7(u1122lf), and mec-7(u1110anti) animals. The fraction of MTs with plus-end-out (anterograde direction comets) and “minus-end-out” (retrograde direction comets) polarity in PLM-AN and PLM-PN. (C) Representative kymographs of MT dynamics in WT and mec-7(u278neo; C303Y) animals, which were treated with 0.125 mM colchicine for 8 h at L4 stage and then recovered on regular NGM plates for 1 h. (D) The number of EBP-2::GFP comets and the fraction of the comets in anterograde and retrograde directions in the treated WT and mec-7(u278neo) animals.
Structure–function relationships of tubulin mutations
Our previous studies revealed that the different types of tubulin missense mutations affected distinct parts of the molecules (Zheng et al., 2017). For example, the amino acids altered by lf mutations tend to be distributed throughout the molecules and often likely affected tubulin folding, whereas anti mutations altered residues involved in GTP/GDP binding and intradimer or interdimer interactions. This pattern is also supported by newly isolated mec-7 and mec-12 alleles. In total, 13 amino acids in the GDP binding pocket of MEC-7/β-tubulin (the E site) and five amino acids in the GTP binding pocket of MEC-12/α-tubulin (the N site) were affected by the lf or anti alleles (Figure 4, A and B; red for lf mutations and blue for anti mutations); C12Y, G13S, G98E, and D203V mutations in MEC-7 and S178F in MEC-12 were identified in this study.
FIGURE 4:

Positions of amino acid residues changed in mec-7 and mec-12 lf and anti mutants. The structure of the α/β-tubulin dimer (1jff.pdb) was visualized using ChimeraX with β-tubulin in cyan, α-tubulin in wheat, and GTP/GDP in green. (A, B) Amino acid substitutions around the GTP (green) binding pocket in mec-7 and mec-12 lf (blue) and anti (red) alleles. For the entire figure, substitutions without an asterisk were previously analyzed and substitutions with an asterisk are from this study. (C) C12Y mutation leads to clashes with V169 residue and G13S mutation leads to clashes with A9 and H137 residues. (D) G141E and G141R mutations lead to clashes with the GTP molecules. (E) mec-7 and mec-12 lf (blue) and anti (red) mutations mapped to the intradimer interface. (F) E71K substitution in MEC-12 leads to clashes with N247 on MEC-7. Two possible orientations of the substituting lysine are shown. In C, D, and F, gray sticks show the substituting residues and purple dashed lines indicate steric clashes. (G, H) mec-7 and mec-12 lf (blue) and anti (red) mutations mapped to interdimer interface. G223E substitution (yellow) was found in a weak neo allele of mec-7.
Using the structure editing function of ChimeraX, which is based on a smoothed backbone-dependent rotamer library (see Materials and Methods; Shapovalov and Dunbrack, 2011), we found that the amino acid substitutions in those missense alleles often caused steric clashes with the GDP molecule and/or other residues in the binding pocket. For instance, C12Y mutation led to direct clashes with the guanine on the GDP molecule and I16 and V169 surrounding the pocket; G13S led to clashes with H137 and A9 residues (Figure 4C). Similarly, previously identified lf mutations G141E and G141R led to severe clashes with the phosphate groups from the GDP molecules (Figure 4D). Thus, those amino acid substitutions likely rendered the MEC-7 mutants incapable of binding to GTP/GDP, thus creating a lf phenotype. The substitutions in the anti alleles (Figure 4) may cause MEC-7 to bind GTP/GDP in an abnormal way that blocks further polymerization, thus creating dominant-negative phenotypes.
Our study also identified seven more residues involved in tubulin intradimer or interdimer interactions that are altered in lf and anti alleles, bringing the total number of such residues to 20 (Figure 4, E–H). For example, at the intradimer interface, previous work found that P243L, G244S, N247I, and D249N mutations affecting the H7-to-H8 (the seventh helix to the eight helix) loop of MEC-7 all caused the anti phenotype with severely shortened TRN neurites (Zheng et al., 2017). In this study, we found that mutation of E71 in MEC-12, which makes direct contact with N247 in MEC-7, also creates anti phenotypes. The E71K substitution is predicted to cause clashes with N247 on β-tubulin (Figure 4F), which might lead to the formation of misshapen tubulin dimers and the termination of polymerization. We also found anti mutations affecting longitudinal interdimer interactions (T257K and G350E in MEC-12) and lateral interactions (L225F in MEC-7). These defects support the hypothesis that disrupting tubulin dimer stacking can generate dominant-negative effects. Two mec-7 partial lf alleles affected intradimer interaction (A254T) and lateral interdimer interaction (R121K), and one lf allele altered a residue involved in longitudinal interaction (G402R); these mutant dimers may be structurally compromised and might reduce MT stability when incorporated. The third mec-7 partial lf allele had L311F substitution, which was mapped to the interior of the tubulin structure and may affect tubulin folding.
Epistasis among the mec-7 neo mutations
The isolation of neo mutations from the mec-7(u278) suppressor screen looking for suppressors of ALM-PN was rather surprising. Two mec-7 intragenic mutations, T149I and A302V, were found to strongly suppress the growth of the long ALM-PN induced by mec-7(u278) allele (Figure 5, A and B). T149I substitution was previously characterized as a neo mutation because the gk895768 allele carrying the same mutation caused the growth of an ectopic ALM-PN; A302V was recreated in the WT background using CRISPR gene editing and was found to induce the production of an ALM-PN by itself. In both cases, the length of ALM-PN induced by T149I and A302V mutations alone was much shorter than the long ALM-PN induced by u278(C303Y) and similar to the mutants carrying double missense mutations (Figure 5, A and B). Thus, T149I and A302V mutations appeared to be intramolecularly epistatic to C303Y mutations in their effects on inducing excessive neurite growth.
FIGURE 5:

Epistatic and synthetic interaction between mec-7 neomorphs. (A) Short ALM-PNs in animals carrying T149I and A302V single mutations of mec-7 and the T149I C303Y and A302V C303Y double mutations. (B) Length of ALM-PNs in young adults of various mutants. Double asterisks indicate significant difference (p < 0.01). T149I and A302V single mutants were compared with WT. (C) Elongated PLM-PN in mec-7 weak neo alleles carrying G223E or R86C mutation. (D) Quantification of PLM-PN length in young adults of indicated mutants. Double asterisks indicate significant difference (p < 0.01) compared with WT. (E) Length of ALM-PNs in young adults of mec-17(-), mec-7(R86C), mec-7(G223E), and mec-12(P63S) single mutants and mec-17(-) double mutants with these mec-7 and mec-12 mutations. Double asterisks indicate significant difference (p < 0.01); mec-12(P63S) was compared with WT in B. (F) Anterior overextension of PLM-AN in mec-7(C303Y A302V) double mutants but not in mec-7(A302V) single mutants. (H) Distance from the PLM-AN terminals to the vulva in mec-7 neo single and double mutants. (G) Schematic diagram of ALM and PLM morphology in mec-7 neo single and double mutants. (I) The percentage of PLM-ANs that did not have synaptic branches in the indicated strains.
Pharmacological studies suggest that the neo mutations induce the growth of excessive ALM-PN likely by elevating MT stability. The MT-stabilizing agent taxol induced the growth of a short ALM-PN, and treatment with the MT-destabilizing drug colchicine eliminated the ALM-PN in mec-7(neo) mutants (Figure 5B). Moreover, mec-7(neo) mutants have reduced MT dynamics and higher resistance to colchicine compared with the WT animals. Using a mild colchicine treatment (0.125 mM for 8 h) followed by a 1-h recovery, we were able to significantly increase MT dynamics and observed more EBP-2::GFP tracks in the WT animals. mec-7(neo) mutants showed much fewer tracks than the WT animals under the same condition (Figure 3, C and D). Interestingly, mec-7(neo) mutants showed a more uniform (plus-end-out) MT polarity in PLM-PN compared with the mixed polarity in the WT animals. Reduced dynamics and uniform polarity may lead to the elongation of PLM-PN in mec-7(neo) animals. We were not able to observe consistent EBP-2 comets in ALM neurites in WT and mec-7(neo) mutants with or without the colchicine treatment.
With regard to structure–function relationships, neo mutations might increase MT stability by affecting the binding of MAPs. For example, neo mutations L377F, R380S, and E407L affected residues on helices H11 and H12, which are the major landing surface for MAPs (Nogales et al., 1998); R162Q was mapped to the H4-to-B5 loop, which is exposed to the exterior of MTs. As the strongest neo allele with the longest ALM-PN, u278(C303Y) altered C303 in the H9-to-B8 loop, which is adjacent to H11 and is also exposed on the exterior surface of MTs (Figure 6A). Structural modeling suggested that C303Y substitution would lead to steric clashes between the benzene ring of the tyrosine and E376 and L377 on the H11 with 60% probability or clashes with M267 on B7 (the seventh β-strand) with 18% probability, depending on the orientation of the phenyl group (Figure 6C). In contrast, C303S substitution did not generate any clashes in our analysis (Figure 6D), which is consistent with the findings that the gk286001(C303S) allele did not cause any TRN morphological defects. Thus, we hypothesize that the bulkiness of tyrosine might have pushed H11 out of its normal position, which altered the binding of certain MAPs and resulted in abnormally elevated MT stability.
FIGURE 6:

Structural modeling of the tubulin neo mutations. (A) Positions of the amino acids affected by mec-7 neo alleles. C303Y (yellow) is the strongest mec-7 neo alleles. Others (orange) are much weaker than C303Y mutation. (B) Positions of the amino acids affected by mec-12 neo alleles. In A and B, mutations with asterisks were isolated in this study and not analyzed before. (C) Modeling of the C303Y substitution with two possible orientations. Left panel shows a configuration with 60% possibility according to ChimeraX, which creates clashes (purple dashed lines) with L377 and E376 residues. Right panel shows another configuration with 18% possibility, leading to clashes with M267. (D) C303S substitution did not lead to any clashes with surrounding residues. (E) A302T and A302V substitutions led to clashes with D203 residue. C303 and A302 are labeled in yellow and orange, respectively.
The substitution of A302, which is next to C303 on the H9-to-B8 loop, with either threonine (T) or valine (V) produced the characteristic ectopic ALM-PN of the neo phenotype. Since A302 is exposed on the MT surface (Figure 6A), its alteration may affect the interaction with certain MAPs. On the other hand, both A302T and A302V mutations led to clashes with D203 on H6, which helps form the GTP binding pocket (Figure 6E); thus, these substitutions may also affect GTP binding or the structural rearrangement related to GTP hydrolysis. T149 is located on H4 in the interior of β-tubulin (Figure 6A), and the neomorphic T149I mutation likely altered tubulin folding or heterodimer stability. Thus, the finding that T149I and A302V mutations could mask the effects of C303Y in promoting the growth of long ALM-PN might be explained by structural changes that led to the reshaping of the MAP binding surface or the alteration of overall heterodimer stability.
Suppressors of mec-17(-) define a new type of mec-7 neomorphs
Among the mutants isolated in the mec-17(-) suppressor screen, two mec-7 alleles, u1115(R86C) and u1114(G223E), showed overextension of PLM-PN (a phenotype seen in mec-7 neo alleles) but not generation of an ectopic ALM-PN in single mutants (Figure 5, C–E). We classified this phenotype, which had not been seen before, as weak neo. R86 is located on the H2-to-B3 loop facing the lumen of MTs; G223 is located on H7 and is involved in lateral interaction. Neither of the two altered residues are mapped to the exterior surface of MTs and their substitutions did not have strong effects by themselves. Thus, the R86C and G223E mutations may increase MT stability slightly by affecting the tubulin folding or the lateral stacking of heterodimers. Their suppression of ALM-PN growth in mec-17(-) mutants was rather unexpected (Figure 5E).
In addition to being an α-tubulin acetyltransferase, MEC-17 has a genetically separable function in restricting neurite growth that is independent of its enzymatic activity (Topalidou et al., 2012). Because MEC-17 acetylates MEC-12 at K40 localized on the H1-to-B2 loop (Akella et al., 2010), one appealing hypothesis is that MEC-17 also plays a structural role by binding to the lumen-facing loops of MEC-12 to provide structural support for the MT organization and stability. In mec-17(-) mutants, the disorganization of the MTs may cause the growth of ectopic ALM-PN at the late L4 and early adult stages. The suppression of this excessive growth by the lf and anti alleles of mec-7 and mec-12 could be explained by their effects in inhibiting MT formation and neurite extension, whereas the suppression by the mec-7 neo alleles may be attributed to a moderate correction of the MT disorganization and instability. In fact, in addition to the ALM-PN phenotype, the swelling and bending of the TRN neurites in mec-17(-) mutants were also rescued by the R86C and G223E neo mutations in mec-7 (Supplemental Figure S1B).
The mec-17(-) suppressor screen also yielded a mec-12 neo mutation u1131(P63S), which caused the growth of an ectopic ALM-PN in the mec-17(+) animals (Figure 5E). Surprisingly, the mec-17(-) mec-12[u1131(P63S)] double mutants had essentially no or very short ALM-PN, whereas the two singe mutants both had substantial ALM-PN growth. P63 is located in the interior lumen-facing H1-to-B2 loop, where K40 is also located (Figure 6B). Among the previously identified mec-12 neo mutations, P32S is also mapped to this H1-to-B2 loop. A third neo mutation V323I is mapped to the B8-to-H10 loop facing the lumen.
We suspect that these mutations may compensate for the destabilization induced by the loss of MEC-17 in the lumen, thus suppressing ALM-PN growth in mec-17(-) mutants. Two other previously identified mec-12(neo) mutations E196K and V260I were mapped to the exterior surface of MTs.
Synthetic effects between mec-7 neo mutations
In addition to the suppression of mec-7(C303Y)-induced long ALM-PN by the intragenic A302V mutation, we surprisingly observed a remarkable overextension of PLM-AN in the animals carrying both A302V and C303Y mutation (Figure 5, F and G). This overextension was not observed in animals carrying either mutation alone. In fact, u278(C303Y) mutants had a slightly underextended PLM-AN; unk60(A302V) mutants, recreated in WT animals, had a PLM-AN with normal length (Figure 5H). To make sure this phenotype is not caused by a background mutation, we outcrossed the u278u1161(C303Y; C302V) mutants obtained from the screen five times and the overextended PLM-AN phenotype always segregated with the genotype of homozygous u278u1161. We also recreated C302V in the TU4879 [mec-7(u278); uIs115(mec-17p::TagRFP)] animals, the starter strain for the suppressor screen, through CRISPR gene editing, and obtained similar suppression of ALM-PN and overextension of PLM-AN. Thus, we conclude that the PLM-AN overextension is a synthetic phenotype of the A302V and C303Y mutations in mec-7.
We also observed an additive loss of PLM-AN synaptic branch. Around 15 and 40% of PLM-AN did not have a synaptic branch in mec-7 animals carrying the A302V and C303Y mutation, respectively, whereas 100% of the double mutants did not grow the branch (Figure 5I). Similarly, some A302V and C303Y single mutants showed the mislocalization of synaptic vesicle protein GFP::RAB-3 into the neurite endings of ALM-PN and PLM-PN, whereas almost 100% of the double mutants had the posterior mistargeting phenotype (Supplemental Figure S2, C and D). The coexistence of epistatic, synthetic, and additive effects between mec-7 neomorphs indicated that their genetic interaction is complex, which might result from the altered interaction with multiple MAPs in the double mutants.
Balancing and synthetic effects between mec-7 neo and anti mutations
Among the mec-7 intragenic suppressors, u278u1171(C303Y; R391H) mutants had a strong or complete suppression of ALM-PN outgrowth but did not show any shortening of PLM-PN (Figure 7A). Thus, we were not able to estimate the effects of R391H single mutation based on the phenotype of the double mutant. We then recreated the R391H mutation in the otherwise WT background and found that this substitution alone led to a strong anti phenotype with all TRN neurites severely shortened (Figure 7B). Many sprouts and protrusions emanated from the PLM cell body and its short axon in unk63(R391H) mutants, likely a result of highly dynamic MTs (Figure 7B). This finding was rather unexpected, because we assumed that dominant-negative effects of anti mutations would be epistatic to the effects of the neo mutation. For example, both u278u1154(C303Y; E69K) and u278u1170(C303Y; P358S) double missense mutants showed the shortening of all TRN neurites similar to the typical anti alleles (Supplemental Figure S4, A and B; in addition to the P358S mutation found in this study, P357L and P358L mutations we previously isolated also caused anti phenotypes). Similarly, the mec-7(u278); mec-12(anti) double mutants showed the same phenotype (shortening of PLM-PN or PLM-AN) as the mec-12(anti) alleles alone (Supplemental Figure S4, C and D). Thus, the unexpected lack of neurite-shortening phenotype in u278u1171(C303Y; R391H) animals suggested that the MT-destabilizing effect of R391H substitution may be balanced by the stabilizing effect of C303Y mutation (Figure 7C).
FIGURE 7:

Balancing and synthetic interactions between mec-7 neo and anti alleles. (A) TRN morphology of animals carrying C303Y R391H double mutations in mec-7. Arrowhead points to the absence of long ALM-PN. (B) R391H single mutation caused an anti phenotype with severely shortened TRN neurites. Arrows point to the premature termination of ALM-AN and PLM-AN. The lower panels show the excessive short sprouts growing out of PLM-AN or PLM cell body in mec-7(R391H) mutants. (C) Schematic diagram of ALM and PLM morphologies in mec-7 neo and anti single mutants, as well as their double mutants. (D) The positions of R391 (red) and C303 (yellow) mutated in the two alleles. (E) Overextension of PLM-PN and excessive neurite sprouting from PLM cell body in the mec-7(C303Y R391H) double mutants. The diagrams on the right illustrate the abnormal PLM morphology at the tail.
R391 is located at the end of H11 (the 11th helix) and is exposed on the exterior surface of MTs (Figure 7D), which is unusual because most anti alleles affected residues involved in GTP binding and intradimer or interdimer interactions. We suspect that R391H substitution may disrupt the binding with certain MT-stabilizing MAPs or strengthen the interaction with some MT-destabilizing MAPs, which resulted in a strong reduction of MT stability. This reduction may be counterbalanced by C303Y substitution, which could have pushed H11 and R391 out of its normal position, so that the R391H substitution no longer has a disruptive effect on MT stability. Nevertheless, the suppression of ALM-PN growth by R391H mutation in u278(C303Y) animals suggested that neither of the two mutations is completely epistatic to the other. Their effects appeared to cancel each other out and a balance on the regulation of MT stability may have been achieved in the double mutants.
Although ∼65% of the PLM neurons in the u278u1171 double mutants had normal morphology, 21% of them had overextended PLM-PN that grew to the tip of the tail, turned, and then extended anteriorly for a short length, and 14% of the PLMs had one or two ectopic neurites emanating from the cell body (Figure 7E). These phenotypes were not observed in either u278(C303Y) or unk63(R391H) single mutants and appeared to be synthetic, indicating that the double substitution may lead to altered MT stability or dysregulation in neurite guidance in PLM neurons. Moreover, the fact that excessive neurite growth is suppressed in ALM neurons but exacerbated in PLM neurons in the u278u1171 double mutants compared with u278 single mutants suggests that MT regulation might differ in the two TRN subtypes.
DISCUSSION
A resource for phenotypic characterization of tubulin mutations
Understanding the cellular impact of tubulin mutations is of great interest not only to cell biologists but also to clinicians, given that over 100 tubulin missense mutations have been found to be associated with a range of cortical malformations, termed tubulinopathies (Romaniello et al., 2018). We previously proposed to use the C. elegans TRNs as a model to study the effects of tubulin mutation on MT stability and neuronal morphogenesis and reported the phenotype of 67 tubulin missense mutations on neurite growth (Zheng et al., 2017). This study, through suppressor screens, isolated and characterized 32 new mutations, thus bringing the total number of tubulin missense mutations in our collection to 99 (Table 1). To our knowledge, this is so far the largest collection of tubulin mutations with phenotypic characterization in neurons. These data may serve as a resource to help evaluate the effect of specific amino acid substitutions on MT functions during neuronal development.
Studies of the tubulin mutations also suggested that correct neurite growth pattern requires optimal MT stability. Reduction in MT stability (or increased dynamics) resulted in neurite growth defects in ALM and PLM neurons, and the level of the reduction correlated with the severity of the defects. For example, slight down-regulation of MT stability in mec-7 partial lf mutants caused the loss of the PLM synaptic branch first, further reduction in mec-7 lf mutants led to the shortening of PLM-PN, and strong destabilization of MTs in the anti mutants severally truncated all neurites, including ALM-AN, PLM-AN, and PLM-PN. The reason that PLM-PN is more sensitive to MT destabilization than ALM-AN and PLM-AN may be that PLM-PN has fewer MTs (∼17 MTs per cross section) than the other two neurites (∼30 MTs per cross section) (Chalfie and Thomson, 1979) and that PLM-PN has mixed MT polarity, whereas the other two neurites have uniform plus-end-out MT polarity (Figure 3). Furthermore, mixed MT polarity may be correlated with shorter neurites in the TRNs. mec-7 lf and anti mutations led to mixed polarity in PLM-AN and neurite growth defects.
On the other hand, elevated MT stability and reduced dynamics in mec-7 and mec-12 neo mutants led to the formation of the ectopic ALM-PN in ALM neurons. mec-7(neo) mutants also caused the elongation of PLM-PN, which was correlated with the conversion from mixed MT polarity to uniform plus-end-out polarity. This observation supports the connection between MT polarity and neurite extension. Nevertheless, PLM neurons did not seem to grow an extra neurite in the neo mutants. Normally ALM neurons are unipolar and PLM neurons are bipolar. Although ALM was able to extend a long neurite toward the posterior when MT became abnormally stable, the inability of PLM neurons to grow a third neurite suggested that neurite growth is under the control of various guidance cues (e.g., Wnt signals) and simple alteration of MT stability would not change the general neurite growth pattern along the longitudinal tracks of these neurons.
Connecting tubulin mutation phenotypes across species and to clinical symptoms
Recently, an online database called “The tubulin mutation database” was built to compile the phenotypes of missense tubulin mutations in different experimental systems, as well as clinical studies (Hussey and Fritz-Laylin, 2019; Pham and Morrissette, 2019). This database allowed us to compare phenotypes across species and to connect cellular defects to clinical symptoms. For example, among the newly identified mec-7/β-tubulin mutations, E3K and D203V caused the lf phenotype with a shortened PLM-PN and R121K led to a partial lf phenotype with the loss of the PLM synaptic branch. All three mutations likely decreased MT stability. Substitution of the same amino acids (E3A, D203A, and R121A) in yeast led to benomyl supersensitivity, which is indicative of less stable MTs (Reijo et al., 1994). Similarly, G146E mutation in mec-7 led to a lf phenotype in our studies and G146C mutation in mouse β1 tubulin rendered the protein incapable of forming heterodimers with α-tubulin (Zabala et al., 1996). Consistency across different organisms highlights the evolutionary conservation of tubulin functions and suggests that results from one organism can provide insight for studies in another.
Mutations newly identified in this study also have clinical relevance, since the same mutations or mutations affecting the same amino acids were also found in patients with neurological diseases. For example, the dominant, antimorphic R320C mutation in mec-12/α-tubulin was found among the human TUBA4A mutations associated with familial amyotrophic lateral sclerosis (Smith et al., 2014). Interestingly, when incorporated into the MTs, TUBA4A R320C mutants disrupted MT polymerization and stability through a dominant-negative mechanism in transfected COS7 cells (Smith et al., 2014). Thus, the effect of the human disease-associated mutation is highly consistent with its effect in C. elegans TRNs. Similarly, in human TUBB2B, G13A mutation was associated with cerebellar dysplasia (Oegema et al., 2015) and G98R was associated with cortical malformation, such as polymicrogyria and microlissencephaly (Cushion et al., 2013). These clinical symptoms are related to defects in neuronal differentiation and axonal growth, consistent with our findings that mutations affecting the same amino acids in mec-7 (G13S and G98E) led to lf phenotype. Moreover, R391H mutation in human TUBB4B caused Leber congenital amaurosis, a neurodegenerative disease of photoreceptor cells; this mutant TUBB4B can form α/β-heterodimers and incorporate into the MT lattice but would disrupt the MT dynamics and significantly dampen normal MT growth (Luscan et al., 2017). Very similarly, the same R391H mutation in mec-7 was antimorphic and severely impaired neurite growth.
The above examples support the utility of C. elegans TRNs as a testing platform to assess the cellular impact of disease-associated tubulin mutations and to connect the defects at the single cell level to clinical manifestation. Given the ease of gene editing in C. elegans, we envision that human tubulin mutations can be routinely installed in mec-7 and mec-12 and their effect on MT functions and neuronal growth can be quickly evaluated.
Genetic interactions among tubulin mutants
Ever since the pioneering work on the tubulins in Aspergillus nidulans (a filamentous fungus) by Ron Morris (Sheir-Neiss et al., 1978; Morris et al., 1979), molecular genetics approaches have been used to study the function of tubulins and MTs in various biological processes. For example, a missense mutation in the β-tubulin gene benA in A. nidulans called benA33 (Q134K) caused a temperature-sensitive hyperstabilization of kinetochore MTs, which inhibited the movement of chromosomes to the mitotic poles and blocked growth at 42°C (Oakley and Morris, 1981; Jung et al., 1998). This benA33 allele was used as a sensitized background to identify revertants that restored growth at the restrictive temperature, which led to the discovery of an α-tubulin gene tubA (all 14 extragenic suppressors were mapped to tubA; Morris et al., 1979). These results also suggested the interdependence of α- and β-tubulin genes. Similarly, our work focusing on neuronal MTs used a neo mutation of mec-7/β-tubulin that elevated MT stability in a revertant screen and identified both intragenic suppressors in mec-7 and extragenic suppressors in mec-12/α-tubulin. Our finding that lf and anti mutations in mec-7 and mec-12 are mostly epistatic to the neo mutation is consistent with earlier studies and support that the functions of α- and β-tubulin genes are dependent on each other.
In this study, we also observed additive or synergistic effects among the mec-7/β-tubulin neomorphs that presumably increased MT stability. The combination of A302V and C303Y mutations increased the penetrance for the loss of PLM synaptic branches and the mistargeting of synaptic vesicle proteins, compared with either single mutation. In a more extreme case, animals carrying the double mutations also showed a synthetic phenotype of PLM-AN overextension, which was not seen at all in the two single mutants. We suspect that the A302V; C303Y double mutations may affect the association of certain MAPs and render MTs resistant to MT-destabilizing signals at the growth cone during axonal termination, e.g., RPM-1 signaling (Borgen et al., 2017). Overall, our observations suggest that the independent effects of single tubulin mutations can be combined to create enhanced or sometimes synthetic effects.
Additive effects of tubulin mutations have also been seen in other organisms. For example, the combination of two pesticide-resistant α-tubulin mutations T239I and M268T found in the goosegrass Eleusine indica showed increased pesticide resistance compared with either mutation on its own (Anthony and Hussey, 1999). Similarly, compared with the single mutation E198A in β-tubulin, three double mutations E198A; M163I, E198A; F167Y, and E198A; F200S conferred significantly increased resistance to benzimidazoles in another filamentous fungi Corynespora cassiicola (Duan et al., 2019). In Drosophila, recessive lf mutations in the testis-specific β2-tubulin gene (B2t) failed to complement with the likely antimorphic V177M mutation in an α-tubulin gene for the male sterility phenotype (Hays et al., 1989), suggesting that additive effects also occur between mutations of α- and β-tubulin genes.
In addition to the above genetic interactions, our work also identified some unexpected interactions among the tubulin mutations. One such interaction is the epistasis among the mec-7 neo mutations. We found that weaker neo changes, such as T149I and A302V mutations, could suppress the stronger neo mutation u278(C303Y). This result indicates that tubulin mutations producing similar phenotypes can not only be additive with each other but also be epistatic to each other. The molecular basis for this epistasis may result from the structural change caused by one mutation becoming functionally ineffective in the presence of another mutation, perhaps by the subsequent change of tubulin structures or interactions with MAPs. This intramolecular epistasis may fit into the category of “conformational epistasis” defined by Ben Lehner (2011).
Another unexpected interaction is the balancing effect between the mec-7 neo change C303Y and the anti change R391H. In most cases, we expected and observed anti mutations being epistatic to neo mutations, since anti mutations often cause structural alterations that render the tubulin protein toxic and block MT polymerization. R391H mutation may be an exception because R391 is exposed on the surface of MTs and its substitution might affect the interaction with certain MAPs. If the C303Y(neo) and R391H(anti) mutations impact the association of MAPs with opposite functions (e.g., MT-stabilizing vs. -destabilizing), one may expect their effects to neutralize each other.
The above examples reveal a previously underappreciated complexity in the genetic interactions among tubulin mutations. Since each mutation may alter tubulin structure or function in unique ways, structural modeling and functional characterization are needed to determine the combinatorial effects of different tubulin mutations.
MATERIALS AND METHODS
Request a protocol through Bio-protocol.
Strains and transgenes
C. elegans WT (N2) and mutant strains were maintained at 20°C as previously described (Brenner, 1974). Alleles used in this study are described in the main text. Most strains were provided by the Caenorhabditis Genetics Center, which is funded by the National Institutes of Health (NIH) Office of Research Infrastructure Programs (P40 OD010440). Transgenes uIs115[mec-17p::TagRFP] IV, uIs134[mec-17p::TagRFP] V, uIs31[mec-17p::GFP] III, and uIs110[mec-17p::TagRFP; sto-5p::GFP] were used to visualize TRNs. Transgene jsIs821[mec-7p::GFP::rab-3] X was used to visualize the transport of synaptic proteins.
Suppressor screens
For the mec-7(neo) suppressor screen, we used TU4879 [mec-7(u278) X; uIs115(mec-17p::TagRFP) IV] as the starter strain, which showed the growth of a long ALM-PN, and ethyl methanesulfonate (EMS) as the mutagen (Brenner, 1974). After screening through 53,600 haploid genomes, we isolated 30 mutants that showed very short or no ALM-PN and outcrossed the mutants against TU4879 and identified the phenotype-causing mutations through a combination of whole-genome resequencing, candidate gene sequencing, and complementation tests (Supplemental Table S1).
For mec-17(-) suppressor screen, we used TU6216 [mec-17(ok2109) IV; uIs110(mec-17p::TagRFP; sto-5p::GFP) II] as the starter strain, which grew a ALM-PN with moderate length in L4 animals and young adults. By screening through 15,000 haploid genomes, we isolated in total 31 mutants that had very short or no ALM-PN in day 1 adults and identified 16 mec-7 and 10 mec-12 mutations by targeted sequencing (Supplemental Table S2).
CRISPR/Cas9-mediated gene editing
To create mec-7 alleles, unk64(R121K), unk62(A254T), unk60(A302V), and unk63(R391H) in the WT background, we performed CRISPR/Cas9-mediated gene editing in TU4065 [uIs115(Pmec-17::RFP) IV] animals using a previously reported method (Dokshin et al., 2018). Briefly, sgRNAs targeting specific sequences in mec-7 were synthesized using the EnGen sgRNA Synthesis Kit (E3322V) from New England Biolabs (NEB; Ipswich, MA) and purified using NEB Monarch RNA Cleanup Kit (T2030L); 1 μg of the sgRNA was injected into C. elegans together with 20 pmol recombinant Cas9 (EnGen S. pyogenes Cas9 NLS from NEB, M0646T) and 1μg ssDNA repair donor for homologous recombination. Synonymous mutations were included in the repair template to avoid the digestion by Cas9. pCFJ104 (myo-3p::mCherry) was used as a co-injection marker and the transformants with red muscles were genotyped for successful incorporation of the repair template. CRISPR target sequences and repair donor sequences used to generate the above alleles are shown in Supplemental Table S3.
Structural modeling
Missense mutations in mec-7 and mec-12 were mapped to the structure of Bos taurus α/β tubulin (1JFF in PDB). Molecular graphics and analyses were performed with University of California, San Francisco (UCSF) ChimeraX (macOS 10.13; https://www.rbvi.ucsf.edu/chimerax/, developed by the Resource for Biocomputing, Visualization, and Informatics at UCSF with support from NIH R01-GM129325 and the Office of Cyber Infrastructure and Computational Biology, National Institute of Allergy and Infectious Diseases; Goddard et al., 2018). The effects of amino acid substitution were modeled using the “Structure Editing/Rotamer” function of ChimeraX and the Dunbrack rotamer library (Shapovalov and Dunbrack, 2011). Only the positions with high probability of substituted amino acids were shown. To model the clashes, pairs of atoms with VDW overlap ≥0.6 Å were identified; 0.4 Å were subtracted from the overlap for potentially H-bonding pairs. The numbering of α-helices (H1 to H12) and β-strands (B1 to B10) was based on the α/β tubulin dimer structure (Nogales et al., 1998).
Pharmacological treatment and EBP-2 tracking
To treat various animals with MT-destabilizing and -stabilizing agents, colchicine and paclitaxel (taxol) were added to the NGM plates for a final concentration of 1 and 1 μM, respectively. L1 animals were placed onto the plates and grew to day 1 adults to be analyzed.
To study MT dynamics, we crossed juIs338[mec-4p::ebp-2::GFP + ttx-3p::RFP], which expressed EBP-2::GFP in TRNs specifically, with various tubulin mutants. We then mounted day 1 adults on agarose pad and immobilized the animals with 100-nm polystyrene beads and imaged the TRNs for 1 min. The number of EBP-2::GFP tracks within 60 μm from the cell body were counted for PLM-AN and within 25 μm for PLM-PN. About 10 animals were imagined for each strain. For the colchicine treatment, L4 animals were placed onto a plate with 0.125 mM colchicine for 8 h and then recovered on a regular NGM plate for 1 h before imaging.
Phenotype scoring and statistical analysis
Fluorescent imaging was done on a Leica DMi8 inverted microscope equipped with a Leica DFC7000 GT monochrome camera. Measurements were made using the Leica Application Suite X (3.7.0.20979) software.
We measured the length of ALM-PN and PLM-PN in at least 20 day 1 adults grown at 20°C. Defects in PLM-AN growth were assessed by measuring the distance from the PLM-AN terminals to the center of the vulva. Positive numbers mean PLM-AN terminals are more anterior than the vulva; negative numbers mean PLM-AN did not reach the vulva; at least 20 cells were examined for each strain. All bar graphs show mean ± SD.
Fluorescence intensity of the TagRFP expressed from the uIs115[mec-17p::TagRFP] transgene in the PLM cell body was used to measure TRN protein levels; intensity was measured by first acquiring images using the identical exposure time and laser power and then manually subtracting the background from the fluorescent signal using Leica Application Suite X (3.7.0.20979). At least 20 cells were imaged and analyzed for each strain.
For statistical analysis, analysis of variance and the post hoc Dunnett’s test (comparison of each of many treatments with a single control) were performed using the GraphPad Prism (version 8.4.0 for Mac, GraphPad Software, La Jolla, CA; www.graphpad.com) to identify significant difference among samples. Single and double asterisks indicate p < 0.05 and p < 0.01, respectively. All measurements were repeated three times on different days and similar results were obtained; data from one representative trial were shown.
Supplementary Material
Acknowledgments
We thank Alice Niu and Mona Liu at Columbia University for helping with the mec-17(-) suppressor screen. We acknowledge support from the Research Grant Council of Hong Kong (ECS 27104219 to C.Z.), the Food and Health Bureau of Hong Kong (HMRF 07183186 to C.Z.), seed funds from the University of Hong Kong (201812159005 and 201910159087 to C.Z.), and the NIH (GM30997 and GM122522 to M.C.).
Abbreviations used:
- anti
antimorph
- lf
loss-of-function
- MAP
MT-associated protein
- MT
microtubule
- neo
neomorph
- PN
posterior neurite
- TRN
touch receptor neuron
- WT
wild type.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E20-07-0492) on December 30, 2020.
REFERENCES
- Akella JS, Wloga D, Kim J, Starostina NG, Lyons-Abbott S, Morrissette NS, Dougan ST, Kipreos ET, Gaertig J (2010). MEC-17 is an alpha-tubulin acetyltransferase. Nature 467, 218–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anthony RG, Hussey PJ (1999). Double mutation in eleusine indica alpha-tubulin increases the resistance of transgenic maize calli to dinitroaniline and phosphorothioamidate herbicides. Plant J 18, 669–674. [DOI] [PubMed] [Google Scholar]
- Bahi-Buisson N, Poirier K, Fourniol F, Saillour Y, Valence S, Lebrun N, Hully M, Bianco CF, Boddaert N, Elie C, et al. (2014). The wide spectrum of tubulinopathies: what are the key features for the diagnosis? Brain 137, 1676–1700. [DOI] [PubMed] [Google Scholar]
- Basciano PA, Matakas J, Pecci A, Civaschi E, Cagioni C, Bompiani N, Burger P, Christos P, Snyder JP, Bussel J, et al. (2015). beta-1 tubulin R307H SNP alters microtubule dynamics and affects severity of a hereditary thrombocytopenia. J Thromb Haemost 13, 651–659. [DOI] [PubMed] [Google Scholar]
- Borgen MA, Wang D, Grill B (2017). RPM-1 regulates axon termination by affecting growth cone collapse and microtubule stability. Development 144, 4658–4672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bounoutas A, Kratz J, Emtage L, Ma C, Nguyen KC, Chalfie M (2011). Microtubule depolymerization in Caenorhabditis elegans touch receptor neurons reduces gene expression through a p38 MAPK pathway. Proc Natl Acad Sci USA 108, 3982–3987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner S (1974). The genetics of Caenorhabditis elegans. Genetics 77, 71–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalfie M, Thomson JN (1979). Organization of neuronal microtubules in the nematode Caenorhabditis elegans. J Cell Biol 82, 278–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cushion TD, Dobyns WB, Mullins JG, Stoodley N, Chung SK, Fry AE, Hehr U, Gunny R, Aylsworth AS, Prabhakar P, et al. (2013). Overlapping cortical malformations and mutations in TUBB2B and TUBA1A. Brain 136, 536–548. [DOI] [PubMed] [Google Scholar]
- Dokshin GA, Ghanta KS, Piscopo KM, Mello CC (2018). Robust genome editing with short single-stranded and long, partially single-stranded DNA donors in Caenorhabditis elegans. Genetics 210, 781–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan Y, Xin W, Lu F, Li T, Li M, Wu J, Wang J, Zhou M (2019). Benzimidazole- and QoI-resistance in Corynespora cassiicola populations from greenhouse-cultivated cucumber: An emerging problem in China. Pestic Biochem Physiol 153, 95–105. [DOI] [PubMed] [Google Scholar]
- Goddard TD, Huang CC, Meng EC, Pettersen EF, Couch GS, Morris JH, Ferrin TE (2018). UCSF ChimeraX: Meeting modern challenges in visualization and analysis. Protein Sci 27, 14–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hays TS, Deuring R, Robertson B, Prout M, Fuller MT (1989). Interacting proteins identified by genetic interactions: a missense mutation in alpha-tubulin fails to complement alleles of the testis-specific beta-tubulin gene of Drosophila melanogaster. Mol Cell Biol 9, 875–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu JM, Chen CH, Chen YC, McDonald KL, Gurling M, Lee A, Garriga G, Pan CL (2014). Genetic analysis of a novel tubulin mutation that redirects synaptic vesicle targeting and causes neurite degeneration in C. elegans. PLoS Genet 10, e1004715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussey SP, Fritz-Laylin LK (2019). “ The missing link”: the tubulin mutation database connects over 1500 missense mutations with phenotypes across eukaryotes. Cytoskeleton (Hoboken) 76, 175–176. [DOI] [PubMed] [Google Scholar]
- Jung MK, May GS, Oakley BR (1998). Mitosis in wild-type and beta-tubulin mutant strains of Aspergillus nidulans. Fungal Genet Biol 24, 146–160. [DOI] [PubMed] [Google Scholar]
- Lehner B (2011). Molecular mechanisms of epistasis within and between genes. Trends Genet 27, 323–331. [DOI] [PubMed] [Google Scholar]
- Luscan R, Mechaussier S, Paul A, Tian G, Gerard X, Defoort-Dellhemmes S, Loundon N, Audo I, Bonnin S, LeGargasson JF, et al. (2017). Mutations in TUBB4B cause a distinctive sensorineural disease. Am J Hum Genet 101, 1006–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris NR, Lai MH, Oakley CE (1979). Identification of a gene for alpha-tubulin in Aspergillus nidulans. Cell 16, 437–442. [DOI] [PubMed] [Google Scholar]
- Nogales E, Wolf SG, Downing KH (1998). Structure of the alpha beta tubulin dimer by electron crystallography. Nature 391, 199–203. [DOI] [PubMed] [Google Scholar]
- Oakley BR, Morris NR (1981). A beta-tubulin mutation in Aspergillus nidulans that blocks microtubule function without blocking assembly. Cell 24, 837–845. [DOI] [PubMed] [Google Scholar]
- Oegema R, Cushion TD, Phelps IG, Chung SK, Dempsey JC, Collins S, Mullins JG, Dudding T, Gill H, Green AJ, et al. (2015). Recognizable cerebellar dysplasia associated with mutations in multiple tubulin genes. Hum Mol Genet 24, 5313–5325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pham CL, Morrissette NS (2019). The tubulin mutation database: A resource for the cytoskeleton community. Cytoskeleton (Hoboken) 76, 186–191. [DOI] [PubMed] [Google Scholar]
- Reijo RA, Cooper EM, Beagle GJ, Huffaker TC (1994). Systematic mutational analysis of the yeast beta-tubulin gene. Mol Biol Cell 5, 29–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romaniello R, Arrigoni F, Fry AE, Bassi MT, Rees MI, Borgatti R, Pilz DT, Cushion TD (2018). Tubulin genes and malformations of cortical development. Eur J Med Genet 61, 744–754. [DOI] [PubMed] [Google Scholar]
- Shapovalov MV, Dunbrack RL Jr (2011). A smoothed backbone-dependent rotamer library for proteins derived from adaptive kernel density estimates and regressions. Structure 19, 844–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheir-Neiss G, Lai MH, Morris NR (1978). Identification of a gene for beta-tubulin in Aspergillus nidulans. Cell 15, 639–647. [DOI] [PubMed] [Google Scholar]
- Smith BN, Ticozzi N, Fallini C, Gkazi AS, Topp S, Kenna KP, Scotter EL, Kost J, Keagle P, Miller JW, et al. (2014). Exome-wide rare variant analysis identifies TUBA4A mutations associated with familial ALS. Neuron 84, 324–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ti SC, Pamula MC, Howes SC, Duellberg C, Cade NI, Kleiner RE, Forth S, Surrey T, Nogales E, Kapoor TM (2016). Mutations in human tubulin proximal to the kinesin-binding site alter dynamic instability at microtubule plus- and minus-ends. Dev Cell 37, 72–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tischfield MA, Cederquist GY, Gupta ML Jr, Engle EC. (2011). Phenotypic spectrum of the tubulin-related disorders and functional implications of disease-causing mutations. Curr Opin Genet Dev 21, 286–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topalidou I, Keller C, Kalebic N, Nguyen KC, Somhegyi H, Politi KA, Heppenstall P, Hall DH, Chalfie M (2012). Genetically separable functions of the MEC-17 tubulin acetyltransferase affect microtubule organization. Curr Biol 22, 1057–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zabala JC, Fontalba A, Avila J (1996). Tubulin folding is altered by mutations in a putative GTP binding motif. J Cell Sci 109 (Pt 6), 1471–1478. [DOI] [PubMed] [Google Scholar]
- Zheng C, Atlas E, Lee HMT, Jao SLJ, Nguyen KCQ, Hall DH, Chalfie M. (2020). Opposing effects of an F-box protein and the HSP90 chaperone network on microtubule stability and neurite growth in Caenorhabditis elegans. Development 147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng C, Diaz-Cuadros M, Nguyen KCQ, Hall DH, Chalfie M (2017). Distinct effects of tubulin isotype mutations on neurite growth in Caenorhabditis elegans. Mol Biol Cell 28, 2786–2801. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.