

Abstract
Amyloid beta (Aβ) is a major component of amyloid plaques, which are a key pathological hallmark found in the brains of Alzheimer’s disease (AD) patients. We show that statins are effective at reducing Aβ in human neurons from nondemented control subjects, as well as subjects with familial AD and sporadic AD. Aβ is derived from amyloid precursor protein (APP) through sequential proteolytic cleavage by BACE1 and γ-secretase. While previous studies have shown that cholesterol metabolism regulates APP processing to Aβ, the mechanism is not well understood. We used iPSC-derived neurons and bimolecular fluorescence complementation assays in transfected cells to elucidate how altering cholesterol metabolism influences APP processing. Altering cholesterol metabolism using statins decreased the generation of sAPPβ and increased levels of full-length APP (flAPP), indicative of reduced processing of APP by BACE1. We further show that statins decrease flAPP interaction with BACE1 and enhance APP dimerization. Additionally, statin-induced changes in APP dimerization and APP-BACE1 are dependent on cholesterol binding to APP. Our data indicate that statins reduce Aβ production by decreasing BACE1 interaction with flAPP and suggest that this process may be regulated through competition between APP dimerization and APP cholesterol binding.
INTRODUCTION
The brains of patients with Alzheimer’s disease (AD) exhibit two key pathological hallmarks: accumulation of extracellular plaques primarily composed of amyloid beta peptide (Aβ) and accumulation of intracellular hyperphosphorylated Tau protein (pTau) (O’Brien and Wong, 2011).
Aβ is derived from sequential proteolytic cleavage of amyloid precursor protein (APP). APP can be cleaved in two major pathways (see Figure 8A later in this article). In the amyloidogenic pathway, APP is first cleaved by the β-secretase BACE1 to generate two protein fragments: soluble APPβ (sAPPβ) and a β-C-terminal fragment (βCTF). sAPPβ is released into the extracellular space. βCTF is cleaved by γ-secretase, which releases the APP intracellular domain (AICD) into the cytosol and generates Aβ peptides of various lengths. Aβ40, a 40-amino-acid peptide, is the major species generated. Aβ42, a slightly longer and more hydrophobic peptide, is produced in smaller quantities but is a major component found in amyloid plaques and is thought to be more prone to oligomerization into toxic Aβ species (O’Brien and Wong, 2011).
FIGURE 8:

Cholesterol/CE-lowering drugs inhibit APP processing by promoting flAPP dimerization. (A) Model of APP processing in untreated cells. (B) Diagram summarizing APP processing changes observed after statin treatment. flAPP dimerizes and accumulates while sAPPα and sAPPβ decrease. These changes are accompanied by reduced APP-BACE1 interaction, suggesting reduced BACE1 cleavage. Levels of α-CTF and β-CTF are not reduced after statin treatment despite the reduction in flAPP processing, indicating that statins also regulate CTF turnover through a secondary mechanism.
In the alternative, nonamyloidogenic pathway, APP is first cleaved by α-secretase to generate soluble APPα (sAPPα) and α-C-terminal fragment (αCTF). sAPPα is released into the extracellular space. αCTF is cleaved by γ-secretase, which releases the AICD into the cytosol and generates a small fragment called p3 (O’Brien and Wong, 2011).
A large body of evidence supports a link between cholesterol metabolism and AD. Statins inhibit cholesterol biosynthesis, and epidemiology studies have shown that statin users are at a decreased risk for AD (Jick et al., 2000; Wolozin et al., 2000; Haag et al., 2009; Li et al., 2010; Lin et al., 2015; Zissimopoulos et al., 2017). Statins alter APP processing and reduce Aβ levels (Simons et al., 1998; Kojro et al., 2001; Ostrowski et al., 2007), and the βCTF region of APP contains a transmembrane domain that can bind cholesterol (Barrett et al., 2012). We previously showed that neuronal lines carrying mutations that impair the APP cholesterol-binding domain are insensitive to statin-induced alterations in Aβ levels (van der Kant et al., 2019). We also showed that the effect of statins on Aβ production in these neurons is likely mediated by cholesterol esters (CE) or a localized pool of subcompartmentalized cholesterol, as statin treatment did not measurably alter free cholesterol levels but did cause a decrease in cellular CE levels (van der Kant et al., 2019). Cholesterol and CE have both previously been shown to influence APP processing (Bodovitz and Klein, 1996; Simons et al., 1998; Kojro et al., 2001; Puglielli et al., 2001; Wahrle et al., 2002; Ehehalt et al., 2003; Hutter-Paier et al., 2004; Huttunen et al., 2007, 2009, 2010; Xiong et al., 2008; Cossec et al., 2010; Marquer et al., 2011). CE has been reported to regulate Aβ production and accumulation in vivo (Hutter-Paier et al., 2004; Huttunen et al., 2010). Mechanistically, reduction of CE has been shown to delay maturation of APP, causing retention of APP in the endoplasmic reticulum (ER) and reduced processing of APP to Aβ (Huttunen et al., 2007, 2009).
APP processing is also dependent on APP dimerization (Rossjohn et al., 1999; Scheuermann et al., 2001; Munter et al., 2007; Eggert et al., 2009). The glycine-XXX-glycine (GXXXG) amino acid sequence is a common motif in transmembrane helices that can mediate helix–helix interactions and dimerization of proteins (Russ and Engelman, 2000; Kleiger et al., 2002). APP contains three of these GXXXG motifs in the juxtamembrane and transmembrane region, which are thought to mediate βCTF and flAPP dimerization (Munter et al., 2007; Kienlen-Campard et al., 2008; Khalifa et al., 2010; Decock et al., 2015, 2016; Higashide et al., 2017). In addition to these domains, the APP ectodomain can also contribute to dimerization of flAPP (Khalifa et al., 2010). Drug-induced dimerization of a flAPP-FKBP chimera causes an increase in flAPP and a decrease in sAPPβ (Eggert et al., 2009, 2018), suggesting that dimerization may inhibit processing of flAPP by BACE1. Similar to the effects of lowering CE, induced dimerization of flAPP causes its retention in the ER (Eggert et al., 2018). The relationship of cholesterol and CE to APP dimerization is not well understood. The three GXXXG motifs overlap with the transmembrane cholesterol-binding domain (Barrett et al., 2012), and in a purified reconstituted system, binding of cholesterol to APP prevents APP dimerization (Beel et al., 2010). Whether APP dimerization is also affected by cholesterol or CE under physiological conditions in neurons is not known.
Here we used human iPSC-derived neurons and transfected HEK cells to study the effects of statins on APP dimerization and processing. We used a bimolecular fluorescence complementation (BiFC) split Venus–based approach to show that statins increase flAPP dimerization in a manner that is dependent on the APP cholesterol-binding site. This is accompanied by a concurrent decrease in flAPP interaction with BACE1, which is also dependent on APP cholesterol binding. As a result of reduced processing by BACE1, statins increase flAPP protein levels and decrease sAPPβ and Aβ levels. Our data elucidate a pathway by which alterations in cholesterol metabolism can reduce APP processing to Aβ by enhancing APP dimerization and reducing APP interaction with BACE1. These changes appear to occur in a manner that is dependent on the APP cholesterol-binding site. Our data support the hypothesis that APP cholesterol binding and APP dimerization act in competition to modulate processing by BACE1.
RESULTS
Cholesterol-lowering drugs cause a reduction of secreted Aβ42 and Aβ40 in hiPSC-derived neurons
Previously, we showed that statin treatment reduces Aβ42 and Aβ40 secretion from neurons derived from one iPSC line from a nondemented control individual. Here we show that this effect also occurs in cell lines derived from a variety of individuals with unique genetic backgrounds. Simvastatin treatment significantly reduced both Aβ42 (Figure 1A and Supplemental Figure S1, A–I) and Aβ40 (Figure 1B and Supplemental Figure S2, A–I) levels in neurons derived from four nondemented control (NDC) and five sporadic Alzheimer’s disease (SAD) individuals. Simvastatin and atorvastatin also reduced Aβ42 (Figure 1C) and Aβ40 (Figure 1D) in hiPSC-derived neurons from an individual with familial AD caused by an APP duplication (APPdp). These results indicate that statin-induced reduction of Aβ secretion is conserved across neurons derived from familial Alzheimer’s disease (FAD), SAD, and NDC subjects.
FIGURE 1:

Cholesteryl ester–lowering drugs cause a reduction of secreted Aβ42 and Aβ40. (A, B) Effect of 5 d simvastatin (10 μM) treatment on (A) Aβ42 levels and (B) Aβ40 levels in iPSC-derived neurons from SAD (sporadic AD) and NDC (nondemented control) subjects. Each dot represents an average of n ≥ 3 measurements from one individual (NDC n = 4 individuals, SAD n = 5 individuals). (C, D) Mixed culture neurons derived from an individual with an AD causing APP duplication were treated with inhibitors of specific steps in the mevalonate pathway; atorvastatin (10 μM), simvastatin (10 μM), YM-53601 (10 μM) for 5 d. For the indicated conditions, mevalonate (MVA; 0.5 mM) was added to the media at t = 0. (C) Aβ42 levels and (D) Aβ40 levels were determined by Meso Scale Discovery electrochemiluminescence (MSD-ECL) (mean ± SEM, n ≥ 3). (E) Diagram of the mevalonate pathway and compounds used in this study.
We asked whether the effects of statins on Aβ secretion can be explained by their effects on cholesterol metabolism, or whether they can be attributed to off-target effects of statins on pathways that branch off from the cholesterol biosynthetic pathway. The mevalonate pathway is the primary pathway responsible for cholesterol biosynthesis. Statins inhibit 3-hydroxy-3-methylglutaryl-CoA reductase (HMGCR), which catalyzes conversion of HMG-CoA to mevalonate (MVA) (Figure 1E) (Buhaescu and Izzedine, 2007). Supplementation with MVA rescued the effect of simvastatin treatment on both Aβ42 (Figure 1C) and Aβ40 (Figure 1D) levels, indicating that the effects of statins on Aβ are due to inhibition of HMGCR. After synthesis of farnesyl-pyrophosphate (farnesyl-PP), the mevalonate pathway branches off into several pathways, leading to synthesis of various end products, one of which is cholesterol. Squalene synthase catalyzes the first step of the pathway that is committed exclusively to cholesterol biosynthesis (Figure 1E) (Buhaescu and Izzedine, 2007) and can be inhibited by the squalene synthase inhibitor YM-53601 in iPSC-derived neurons as characterized previously (van der Kant et al., 2019). YM-53601 treatment significantly reduced both Aβ42 (Figure 1C) and Aβ40 (Figure 1D) secretion. Consistent with findings in other model systems (Hutter-Paier et al., 2004; Cole et al., 2005; Huttunen et al., 2007, 2009, 2010), these data indicate that Aβ42 and Aβ40 generation and secretion in human neurons is regulated by the cholesterol-biosynthetic arm of the mevalonate pathway and that statins can be used as tool compounds to modify this pathway. Statins are widely used drugs with well characterized effects on cholesterol metabolism. Thus, we focus the rest of our investigation on the mechanistic effects of cholesterol metabolism on Aβ levels using statins as a tool compound.
Statin-mediated reduction in Aβ secretion is Tau-independent
Previously, we showed that statins also reduce pTau in iPSC-derived neurons. We previously showed that the effects of statins on pTau levels do not require Aβ and APP (van der Kant et al., 2019). We hypothesized that the reverse could be true, that statin-induced changes in pTau occur upstream of changes in APP and Aβ in a shared, cholesterol-dependent pathway. To test whether the effect of statins on Aβ is Tau-dependent, we used CRISPR/Cas9 to generate a MAPT KO iPSC line with an insertion-induced stop codon at the MAPT locus, which encodes the Tau protein (Figure 2A). Western blot was used to confirm a lack of Tau protein expression in the MAPT KO cell lines (Figure 2B). WT and MAPT KO iPSC lines can be differentiated to MAP2-positive neurons with neuronal morphology (Figure 2C)
FIGURE 2:

Statin-mediated reduction in Aβ secretion is Tau-independent. (A) We used CRISPR/Cas9 to create MAPT KO iPSC lines by creating an insertion-induced stop codon at the MAPT locus. (B) Western blot confirming knockout of Tau protein in the MAPT KO iPSC lines. (C) WT and MAPT KO iPSCs were differentiated to neurons and then fixed, permeabilized, stained for MAP2 (neuron cell body marker), and then imaged. WT and MAPT KO lines both differentiate to MAP2-positive neurons with similar neuronal morphologies. (D, E) WT or Tau KO neurons were treated with DMSO or 10 μM atorvastatin or simvastatin for 5 d. (D) Aβ42 levels and (E) Aβ40 levels were determined by MSD-ECL (mean ± SEM, n ≥ 3).
We treated both WT and MAPT KO iPSC derived neurons with dimethyl sulfoxide (DMSO), atorvastatin, or simvastatin and measured the effects on Aβ levels. Simvastatin and atorvastatin reduced Aβ42 (Figure 2D) and Aβ40 (Figure 2E) levels in both wild-type (WT) and MAPT KO neurons to similar extents. These results show that the effects of statins on APP processing to Aβ occur independently of their effects on Tau. Together with our previous findings (van der Kant et al., 2019), this shows that while cholesterol is an important regulator of both pTau and Aβ, cholesterol-dependent changes in pTau and Aβ occur independently of one another.
Statins reduce processing of flAPP to sAPPβ and sAPPα and processing of APP CTFs to Aβ
We hypothesized that statins reduce Aβ by interfering with APP processing. To test this, we characterized the effects of atorvastatin on APP fragment composition in WT hiPSC-derived neurons. We observed a decrease in both sAPPβ (Figure 3A) and sAPPα (Figure 3B) in conditioned media from WT neurons after atorvastatin treatment. We also observed an increase in flAPP after statin treatment (Figure 3, C and D). A similar cholesterol-dependent increase of flAPP has been reported previously in other model systems (Cole et al., 2005). This increase in flAPP was mostly driven by an increase in immature APP, consistent with ER retention of APP after statin treatment as previously suggested (Huttunen et al., 2009). The two WT lines we used had slightly different baseline expression levels of BACE1; however, BACE1 protein levels were not altered after statin treatment in either line (Figure 3, E and F), indicating that increased levels of flAPP and decreased Aβ production are not caused by a reduction in BACE1 levels. We also measured the effects of statins on APP CTF levels. We observed a strong reduction in βCTF/flAPP (Supplemental Figure S3A) and αCTF/flAPP (Supplemental Figure S3B) after statin treatment, which is in line with reduced flAPP processing. However, when normalized to actin, βCTF levels were unchanged while αCTF levels slightly increased (Supplemental Figure S3, D and E), indicating that statins stabilize CTF levels. This is consistent with a two-step regulation of APP processing by statins (Cole et al., 2005; Zhou et al., 2008): cholesterol-dependent regulation of flAPP processing and isoprenoid-dependent regulation of CTF processing to Aβ. Here, we focused our efforts on understanding the cholesterol-dependent effects of statins on flAPP processing.
FIGURE 3:
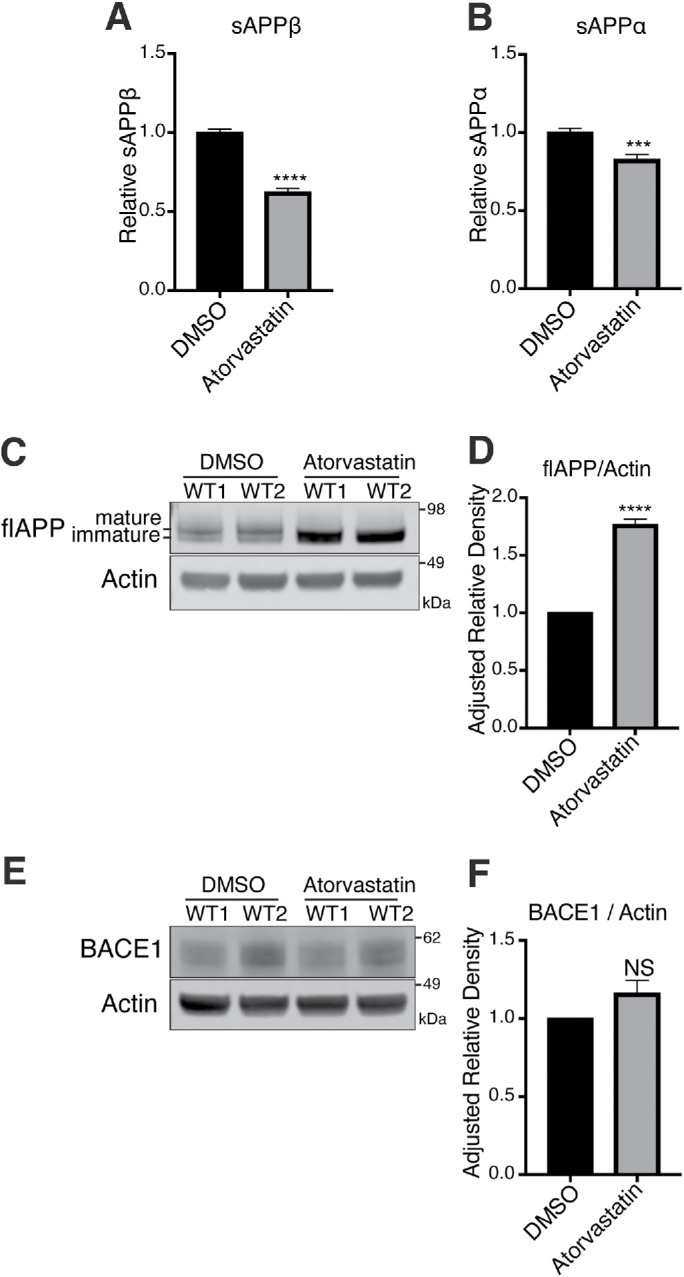
Statins reduce processing of flAPP to sAPPβ and sAPPα without altering BACE1 levels. (A–F) Neurons derived from two distinct WT iPSC lines were treated with DMSO or 10 μM atorvastatin for 5 d. Levels of (A) sAPPβ and (B) sAPPα were determined by MSD-ECL. (C) Levels of flAPP were determined by Western blot and quantified in D. (E) Levels of BACE1 were determined by Western blot and quantified in (F) (mean ± SEM, n ≥ 3).
Statins decrease APP-BACE1 interaction
The observed reduction in APP processing and subsequent increase in flAPP in statin-treated neurons led us to hypothesize that statin treatment may alter APP-BACE1 interaction and APP dimerization. To understand the effect of statin treatment on APP processing and dimerization in more detail, we modified a bimolecular fluorescence complementation (BiFC) assay described in Das et al. (2016) to measure APP-BACE1 interactions (see Figure 5A later in this article) and APP-APP interactions (see Figure 6A later in this article).
FIGURE 5:
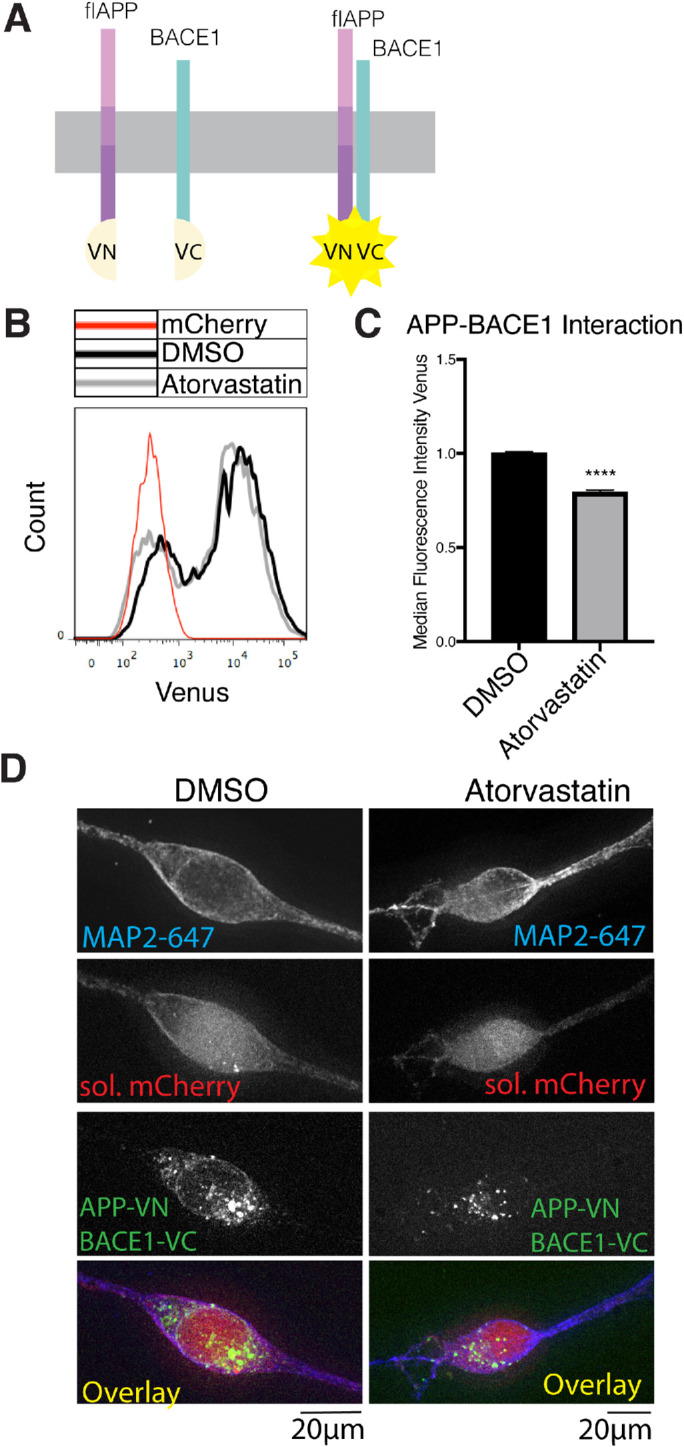
Statins decrease APP-BACE1. Interaction. (A) Principle of the APP-BACE1 interaction assay. The C-terminus of Venus fluorescent protein (vc) was tagged to the C-terminus of BACE1 to generate the BACE1vc plasmid. The N-terminal half of Venus (vn) was tagged to the C-terminus of flAPP to generate the APPvn plasmid. To test the interaction of APP and BACE1, both plasmids are cotransfected into the cells along with a soluble mCherry plasmid for cell selection. When APP and BACE1 interact, vn and vc fragments reconstitute into a functional Venus protein and emit fluorescence, which can be detected by microscopy or flow cytometry. (B, C) HEK cells were plated in DMSO or 10 μM atorvastatin and then transiently transfected 24 h later. Cells were transfected with wtAPPvn, wtAPPvc, and mCherry unless otherwise indicated. mCherry-positive cells were analyzed 16 h after transfection. (B) Histograms showing flow cytometry analysis of BACE1-flAPP interaction (Venus median fluorescence intensity). A negative control transfected with only mCherry is shown in red. (C) Quantification of flow cytometry analysis of BACE1-flAPP interaction (Venus median fluorescence intensity) (mean ± SEM, n ≥ 3). (D) iPSC-derived WT neurons were plated in DMSO or 10 μM atorvastatin. Four days later, neurons were transiently transfected with wtAPPvn, BACE1vc, and mCherry. Cells were fixed, permeabilized, and stained with MAP2 16 h post-transfection. Cells were selected for imaging based on positive MAP2 and mCherry signal and neuronal morphology. Imaging and image processing were performed blind.
FIGURE 6:

Statins increase flAPP dimerization. (A) Principle of the flAPP dimerization assay. Two flAPP constructs were tagged to complementary N-terminus (vn) or C-terminus (vc) fragments of Venus fluorescent protein (VFP). Dimerization of APP gives rise to Venus fluorescence, which can be detected by microscopy or flow cytometry. To test APP dimerization, both plasmids are cotransfected into the cells along with a soluble mCherry plasmid. Analysis of Venus median fluorescence intensity (APP dimerization) was performed only on transfected cells. (B, C) HEK cells were plated in DMSO or 10 μM atorvastatin and then transiently transfected with wtAPPvn, wtAPPvc, and mCherry 24 h later. Cells were analyzed 16 h after transfection. (B) Histograms showing flow cytometry analysis of dimerization (Venus median fluorescence intensity) in transfected cells. A negative control transfected with only mCherry is shown in red. (C) Quantification of flow cytometry analysis of APP dimerization (Venus median fluorescence intensity) (mean ± SEM, n ≥ 3). (D) iPSC-derived WT neurons were plated in DMSO or 10 μM atorvastatin. Four days later, neurons were transiently transfected with wtAPPvn, wtAPPvc, and mCherry. Cells were fixed, permeabilized, and stained with MAP2 16 h after transfection. Cells were selected for imaging based on positive MAP2 and mCherry signal and neuronal morphology. Imaging and image processing were performed blind.
We developed this system in HEK293 (HEK) cells and validated that APP processing in this system is regulated in a manner similar to that in iPSC-derived neurons. All APP fragments measured were much higher in wtAPPvc transfected cells compared with untransfected cells (Figure 4, A–E), indicating that most of the measured fragments are derived from processing of the transfected construct rather than from endogenous APP. Treatments with a γ-secretase inhibitor (compound E) and a BACE1 inhibitor (BIV) both reduced Aβ42 (Figure 4A) and Aβ40 (Figure 4B). Treatment with BIV reduced sAPPβ (Figure 4C) and did not significantly change sAPPα (Figure 4D). These data show that transfected APP can be processed by endogenous BACE1 and γ-secretase. Atorvastatin treatment reduced Aβ42 (Figure 4A), Aβ40 (Figure 4B), sAPPβ (Figure 4C), and sAPPα (Figure 4D) and increased levels of full-length wtAPPvc (visualized with an anti-GFP [green fluorescent protein] antibody) in the transfected HEK cells (Figure 4E). The changes in APP fragments that we observe in transfected HEK cells match the changes we see in hiPSC-derived neurons after atorvastatin treatment, indicating that the changes modeled in the split Venus transfection system are similar to those modeled in human neurons and can be used to gain mechanistic insights on this pathway.
FIGURE 4:

Statins reduce processing of transfected APP in HEK cells. To test whether transfected HEK cells can be used to model drug-induced changes in APP processing, we compared levels of APP proteolytic fragments in untransfected cells and transfected cells. We additionally tested whether statins and secretase inhibitors changed the composition of APP proteolytic fragments in transfected cells. (A–E) HEK cells were plated in DMSO, 10 μM atorvastatin, 4 μM BACE1 inhibitor (BIV), or 200 nM γ-secretase inhibitor (compound E) and then transiently transfected 24 h later. Cells and conditioned media were collected 16 h after transfection. (A–D) MSD-ECL based measurements of (A) Aβ42, (B) Aβ40, (C) sAPPβ, and (D) sAPPα from media of wtAPPvc transfected cells. (E) Western blot visualization of APPvc (visualized with an anti-GFP antibody that binds to the Venus c-terminus [vc] tag) and actin.
To robustly quantify Venus signal (APP-BACE1 interaction) in a high-throughput manner, we developed a flow cytometry assay that quantitatively measures thousands of cells (Supplemental Figures S4, A–E, and S5, A–D). We confirmed that the Venus signal observed in this assay is specific to the APP-BACE1 interaction rather than nonspecific fluorescence of the split Venus constructs or nonspecific reconstitution of the Venus signal (Supplemental Figure S7, A–D).
We observed that statin treatment reduced the median fluorescence intensity of the Venus signal, indicating less flAPP-BACE1 interaction (Figure 5, B and C). Atorvastatin treatment did not significantly affect the transfection efficiency as measured by the percentage of transfected singlets (Supplemental Figure S6, A and B). These data show that statin treatment reduces APP-BACE1 interaction in transfected HEK cells. We confirmed these findings in iPSC-derived neurons transfected with the split Venus constructs using microscopy (Figure 5D). These results show that reduced sAPPβ production after statin treatment is indeed caused by a reduction of APP-BACE1 interaction, which might be expected when APP is retained in the ER.
Statins increase APP dimerization
Previous studies have suggested that full-length APP dimerization can alter APP processing by BACE1 (Kaden et al., 2008; Bhattacharyya et al., 2016). To determine whether statins affect APP dimerization, we modified the split Venus BiFC assay described above to measure APP dimerization (Figure 6A) (Chen et al., 2006; Ben Khalifa et al., 2011; So et al., 2013). Cells were gated and compensated in the same manner as described for the APP-BACE1 interaction assay (Supplemental Figures S4, A–E, and S5, A–D). We confirmed that the Venus signal observed in this assay is specific to APP dimerization rather than nonspecific fluorescence of the split Venus tags or nonspecific reconstitution of the Venus signal (Supplemental Figure S8, A–D).
Statin treatment increased Venus median fluorescence intensity (APP dimerization) as quantified by high-throughput flow cytometry analysis in HEK cells (Figure 6, B and C). These results were confirmed in transfected iPSC-derived neurons by microscopy (Figure 6D). Overall, these data indicate that statins, through a reduction of cholesterol biosynthesis, enhance APP dimerization and decrease APP-BACE1 interaction, explaining reduced Aβ production under these conditions.
The APP cholesterol-binding site mediates statin-induced changes in APP dimerization and APP-BACE1 interaction
One previous study showed that in a purified, reconstituted system, binding of cholesterol to APP acts in competition with APP dimerization (Song et al., 2013). Whether APP dimerization is affected by cholesterol binding in live cells is not known. To test this, we used site-directed mutagenesis to generate mutant APPvn and APPvc constructs with mutations in the cholesterol-binding site that are known to interfere with APP cholesterol binding (E693A and F691A) (Barrett et al., 2012; Nierzwicki and Czub, 2015). We will refer to these mutations collectively as Δcholesterol-APP mutants. We tested whether statin treatment causes alterations in dimerization in Δcholesterol-APP–transfected cells. We observed that compared with the strong increase in dimerization observed in the wtAPP condition, statin treatment has a blunted effect on APP dimerization in cells transfected with split Venus pairs containing the APP E693A mutation (eaAPPvn and eaAPPvc) and no effect on APP dimerization in cells transfected with split Venus pairs containing both the APP E693A and F691A mutations (eafaAPPvn and eafaAPPvc) (Figure 7A). This indicates that the influence of statins on APP dimerization is dependent on the APP cholesterol-binding site in cultured cells. To our knowledge, this is the first time this has been shown in live cells.
FIGURE 7:

The influence of statins on APP dimerization and APP-BACE1 interaction is dependent on the APP cholesterol-binding site. We made point mutations in the APP cholesterol-binding site in APPvn and APPvc plasmids. These Δcholesterol APP mutations were previously shown to obliterate APP cholesterol binding (eaAPPvn = APP E693A) (eafaAPPvn = APP E693A+F691A). HEK cells were plated in DMSO or 10 μM atorvastatin and then transiently transfected 24 h later. (A) Cells were transfected with WT or Δcholesterol mutant split Venus pairs and mCherry. mCherry-positive cells were analyzed by flow cytometry 16 h after transfection. Venus median fluorescence intensity was quantified as a measure of APP dimerization (mean ± SEM, n ≥ 3) (B) Cells were transfected with WT or Δcholesterol mutant APPvn, WT BACE1vc, and mCherry. mCherry-positive cells were analyzed by flow cytometry 16 h after transfection. Venus median fluorescence intensity was quantified as a measure of APP-BACE1 interaction (mean ± SEM, n ≥ 3).
We hypothesized that statin-induced changes in APP-BACE1 interaction were also dependent on APP binding to cholesterol. To test this, we asked whether APP-BACE1 interaction is affected by statin treatment in cells cotransfected with Δcholesterol-APP and BACE1 split Venus pairs. Compared with the strong decrease in APP-BACE1 interaction observed in the wtAPP condition, statin treatment has a blunted effect on APP-BACE1 interaction in cells transfected with eaAPPvn and BACE1vc and no effect on APP-BACE1 interaction in cells transfected with eafaAPPvn and BACE1vc. (Figure 7B). This indicates that the influence of statins on APP-BACE1 interaction is dependent on the APP cholesterol-binding site. Together, these data support the model that cholesterol binding and dimerization act in competition to regulate APP processing by BACE1 (Figure 8, A and B).
DISCUSSION
We probed the mechanisms of how statins regulate APP processing to Aβ. We observed that statins reduce Aβ secretion in neurons derived from NDC, SAD, and FAD individuals. Statins cause a reduction of APP interaction with BACE1 and drive the accumulation of flAPP in a process dependent on the APP cholesterol-binding domain and APP dimerization. These changes occur in a Tau-independent manner.
Statins reduce APP processing to Aβ by reducing APP interaction with β-secretase and altering APP dimerization
Together, the changes in APP proteolytic fragment composition along with the decreases in APP-secretase interaction that we observe after statin treatment suggests that statins reduce APP processing to Aβ42 and Aβ40 by increasing APP dimerization and reducing APP interaction with BACE1. These changes are tightly correlated with statin-induced increases in flAPP dimerization. We propose a model in which statin treatment inhibits processing by BACE1 by reducing APP cholesterol binding and inducing flAPP dimerization (Figure 8B). In a secondary process, statins also influence APP CTF turnover and processing (Supplemental Figure S3, A–E) in a process that is likely isoprenoid-dependent rather than cholesterol-dependent (Cole et al., 2005; Zhou et al., 2008). We focused on the cholesterol-dependent effect of statins on flAPP dimerization and showed that statin treatment induces flAPP dimerization and reduces APP processing by BACE1. Very few studies have looked at the effects of flAPP dimerization on APP processing by BACE1 (Kaden et al., 2008; Bhattacharyya et al., 2016). Inducing dimerization of flAPP using a chimeric APP-FKBP protein that can be induced to dimerize by addition of an FKBP-binding drug led to an increase in flAPP and a reduction in sAPPβ (Eggert et al., 2009). This suggests that flAPP dimerization may decrease flAPP processing by BACE1. Later studies from the same group showed that induced dimerization increased APP localization in the ER, possibly explaining dimerization-induced decreases in APP processing by BACE1 (Eggert et al., 2018). One previous in vitro study showed that binding of cholesterol to APP may prevent APP dimerization (Beel et al., 2010). Our data expand on these results by showing that APP dimerization is enhanced by inhibiting cholesterol metabolism under physiological conditions in live cells and neurons. We additionally show the importance of the APP cholesterol-binding site in mediating the effects of statins on APP dimerization, supporting the notion that dimerization and cholesterol binding are intimately connected. Statin-induced increases in dimerization were accompanied by decreases in APP-BACE1 interaction and an increase in flAPP that was primarily driven by increased immature APP. This was consistent with a previous study showing that statin treatment causes APP retention in the ER, resulting in reduced maturation of APP and thus limited availability of APP for proteolytic processing by the secretases (Huttunen et al., 2009). We propose that atorvastatin treatment induces dimerization and inhibits APP processing by BACE1, possibly through statin-induced alterations in ER localization and APP maturation. Another possibility is that the induction of flAPP dimerization that we observe after statin treatment makes APP impervious to BACE1 interaction. It is plausible that dimerization could obscure amino acids involved in APP-secretase interactions or obscure APP cleavage sites. More work is needed to determine whether statin-induced dimerization alters APP processing and APP-secretase interactions by altering APP trafficking or by obscuring amino acids that are essential to APP processing. BACE1 also forms dimers, trimers, and tetramers (Liebsch et al., 2017), and statins have also been shown to reduce BACE1 dimerization and BACE1-mediated APP processing (Parsons et al., 2006). Thus, statins may inhibit APP processing by BACE1 through multiple mechanisms. Developing pharmacological approaches to target APP dimerization or APP cholesterol binding may lead to novel therapeutic strategies for AD treatment.
Cholesterol-dependent regulation of APP processing
In addition to statins, we also used a squalene synthase inhibitor to confirm that APP processing to Aβ is regulated by the cholesterol biosynthetic arm of the mevalonate pathway. We previously demonstrated that statin treatment of iPSC-derived neurons causes a reduction in CE but no measurable reduction in cholesterol levels (van der Kant et al., 2019). This supports the hypothesis that the effect of statins on flAPP and Aβ levels is mediated by changes in CE or by localized changes in cholesterol levels in subcellular organelles that are not detectable in whole cell lysates. Our observation that cholesterol homeostasis influences amyloidogenic APP processing to Aβ is in agreement with many previous studies (Bodovitz and Klein, 1996; Simons et al., 1998; Frears et al., 1999; Kojro et al., 2001; Ehehalt et al., 2003; Xiong et al., 2008; Cossec et al., 2010; Marquer et al., 2011). In addition, we observed a second step of regulation of CTF processing and turnover that likely depends on the isoprenoid pathway (Cole et al., 2005; Zhou et al., 2008). Understanding the regulation of APP processing by cholesterol is of high importance as cholesterol has been strongly implicated in AD pathogenesis. Several studies have shown that CE (the storage form of cholesterol) is enriched in AD patient brains and in transgenic AD mouse models (Chan et al., 2012; Tajima et al., 2013; Yang et al., 2014). Mutations that cause FAD have been associated with altered cholesterol homeostasis (Cho et al., 2019). And elevated LDL cholesterol has been associated with early-onset AD (Wingo et al., 2019). Changes in cholesterol homeostasis that are driven by FAD or SAD genetic risk factors could influence AD pathogenesis. For example, APOEe4 (the major genetic risk factor for SAD) is a protein involved in the transport of cholesterol. APOE is one of the major apolipoproteins in the CNS (Elliott et al., 2010) and shuttles lipids from glia to neurons and vice versa (Liu et al., 2017). The APOEe4 allele interferes with this lipid transport (Liu et al., 2017) and can cause cholesterol accumulation in different cell types (Lin et al., 2018). As we show that neuronal cholesterol and APP processing to Aβ in neurons are tightly coupled, it would be very interesting to investigate how neuronal cholesterol metabolism is subsequently regulated by glia and how this glia-dependent regulation of neuronal cholesterol might be impacted by APOE and/or other genetic risk factors for AD such as TREM2 and PLCγ2, which also have been shown to regulate brain lipid metabolism (Andreone et al., 2020; Nugent et al., 2020). Furthermore, new neuronal-cholesterol–lowering approaches, for example, based on activation of CYP46A1 and conversion of cholesterol to 24-hydroxycholesterol have been developed and are currently in clinical trials for AD (van der Kant et al., 2020). Overall, our work supports the relevance of targeting neuronal and brain cholesterol metabolism to treat AD and provide mechanistic insight into the influence of cholesterol on AD pathogenesis.
Statins reduce Aβ through a Tau-independent mechanism
Previously, we published that statins reduce pTau levels by a mechanism that is independent of APP and Aβ (van der Kant et al., 2019). We show here that statins also cause a reduction of Aβ levels that is independent of Tau by showing that simvastatin reduces Aβ in both WT and MAPT KO neurons. This suggests that in addition to previously defined pathways in which Aβ and Tau can work together in the same pathway, Aβ and pTau levels can both be closely coregulated by changes in cholesterol metabolism but through separate independent pathways. Finding drugs that target early pathogenic changes that occur upstream of Tau and AD pathology may be key to treating the disease.
General conclusions and future directions
Gaining an understanding of the upstream pathways that regulate Aβ may lead us to a better understanding of APP function and AD pathogenesis and ultimately to improved drug targets. Further investigation into targeting cholesterol metabolism, APP-cholesterol binding, and APP dimerization using pharmacological approaches may lead to novel therapeutic strategies to treat AD.
MATERIALS AND METHODS
Request a protocol through Bio-protocol.
Human iPSC-derived cell lines
The WT iPSC lines used in this study include the control lines WT1(B10) (RRID:CVCL_VR50) and WT2(B11) (RRID:CVCL_VR51), which are two unedited control lines derived from the CVB iPSC line (RRID:CVCL_1N86), which was generated and characterized in Gore et al. (2011). WT1(B10) and WT2(B11) were generated and characterized in van der Kant et al., (2019).
SAD and NDC iPSC lines used are SAD2 (RRID: CVCL_EJ93), SAD3 (RRID: CVCL_UB91), SAD4 (RRID: CVCL_UB92), SAD5 (RRID: CVCL_UB93), SAD6 (RRID: CVCL_UB94), NDC1 (RRID:CVCL_EJ84), NDC3 (RRID: CVCL_UB88), NDC4 (RRID: CVCL_UB89), and NDC5 (RRID: CVCL_UB90) described and characterized in Young et al. (2015).
The iPSC line from the AD patient with an APP duplication is APPdp1.1 (RRID:CVCL_EJ96) in Israel et al. (2012). See Israel et al. (2012) for characterization of this cell line and for patient details.
The generation of the MAPT KO iPSC line as well as its isogenic WT control is described below.
iPSC genome editing
iPSCs (iPSC CVI line) derived from fibroblasts obtained from a biopsy from J. Craig Venter (Gore et al., 2011) were pretreated with 10 μM Rock Inhibitor (RI; Y-27632 dihydrochloride; Abcam) before nucleofection. To obtain single cells, iPSCs were dissociated with Accutase (Innovative Cell Technologies) and filtered twice through 100 μM filters. Cells (8 × 105) were nucleofected using the Amaxa Human Stem Cell Nucleofector Kit I (Lonza) with 5 μg pSpCas9(BB)-2A-GFP (PX458) vector (Ran et al., 2013), containing guide RNA (gRNA) (5′-TCACGCTGGGACGTACGGGT-3′) targeting MAPT exon 2. After culturing the iPSCs in the presence of RI for 48 h, 1 × 104 GFP + IPSCs were FACS sorted (FACS Aria IIu; BD Biosciences) and plated on 10 cm MEF-feeder plates in the presence of RI. After 1 wk, single colonies were manually picked, cultured in 96-well plates, and expanded. DNA from single clones was harvested using QuickExtract DNA Extraction Solution (Epicentre), and the MAPT gene was PCR amplified using Phusion High-Fidelity DNA Polymerase (NEB) and primers (TAU5F; GGC CAA CTG TTA GAG AGG GT and TAU5R; TCT GGA TGC AAA CTG TTC CCG), purified using Exo-SAP-IT PCR Product Cleanup Reagent (ThermoFisher Scientific), and Sanger sequenced. Sequencing results were aligned against the MAPT wild-type sequence to determine the presence of disruption in the vicinity of the gRNA/Cas9 predicted cutting site. Possibly edited clones were expanded, resequenced, and digitally karyotyped by hybridization to the Infinium CoreExome-24 BeadChip (Illumina). One line was identified carrying multiple insertions in MAPT exon 2, predicted to generate a premature stop codon; this line was expanded and differentiated to neural progenitor cells (NPCs) and then differentiated to neurons. To test whether premature stop codon formation had induced nonsense-mediated decay of MAPT mRNA and thus loss of Tau protein, we examined Tau protein levels in this clone compared with a WT, an unedited subclone that underwent the genome editing process but was not modified. Anti–total Tau T6402 (1:1000; Sigma) failed to detect any Tau protein in the edited clone, suggesting that CRISPR/Cas9-induced insertions prevented translation of Tau protein generating a MAPT null line.
Generation of NPCs
NPCs were generated from iPSCs as described in Yuan et al. (2011). Briefly, 2 × 105 FACS-purified iPSC TRA1-81+ cells were seeded onto two 10 cm plates that were seeded the previous day with 5 × 105 PA6 cells and were cultured in PA6 differentiation media (450 ml Glasgow DMEM, 50 ml KO serum replacer, 5 ml sodium pyruvate, 5 ml nonessential amino acids) + 10 µm SB431542 + 0.5 µg/ml Noggin. On the 11th day, cells were dissociated with Accutase, and ∼5 × 105 CD184+CD24+CD44−CD271− NPCs were FACS-purified and plated onto poly-l-ornithine/laminin–coated plates and cultured with NPCbase + 20 ng/ml bFGF (Fibroblast Growth Factor basic protein) (Millipore). From here, cells were expanded and frozen down for use in future experiments.
Cell culture and generation of neurons
iPSCs were cultured on a MEF feeder layer in HUES medium (400 ml KO DMEM [Life Technologies 10829] + 50 ml Knockout serum [Thermo Fisher 10828028] + 50 ml plasmanate [Chapin] + 5 ml Pen/Strep [Life Technologies 15140-122] + 5 ml nonessential amino acids [Life Technologies 11140-050] + 5 ml glutamax [Life Technologies 35050-061] + 1 ml β-mercaptoethanol [Life Technologies 21985-023] + 20 ng/ml FGF [R&D Systems 233-FB-1]) as described in Israel et al. (2012). iPSCs were passaged with Accutase (Innovative Cell Technologies).
MEFS were plated in MEF media (450 ml DMEM high glucose [Life Technologies 11965] + 50 ml fetal bovine serum [Mediatech 35-011-CV] + 5 ml Pen/Strep [Life Technologies 15140-122] + 5 ml glutamine [Life Technologies 250303]) on plates coated with 0.1% gelatin.
NPCs were cultured on poly-l-ornithine (0.02 mg/ml) and laminin (5 µg/ml) (Sigma)-coated plates in NPC base media (DMEM:F12 + glutamax, 0.5× N2, 0.5× B27, Pen/Strep [all Life Technologies]) + 20 ng/ml FGF. Culture medium was changed three times weekly. NPCs were passaged with Accutase.
To differentiate NPCs to neurons, NPCs were grown to confluence, after which FGF was withdrawn from the NPC base culture media. Media was changed twice weekly for differentiating NPCs. After 3 wk of differentiation, neurons were replated in NPC base + BDNF/GDNF/cAMP and cultured for an additional 2 wk.
HEK cell culture
HEK293T cells were obtained from the American Type Culture Collection (CRL-3216). HEK cells were expanded on uncoated 10 cm plates in MEF media. HEK cells were passaged with trypsin. HEK cell transfections were performed with the Effectene Transfection Reagent kit using the manufacturer instructions (Qiagen 301425).
Drug treatments
Compounds used are as follows: atorvastatin calcium salt (Sigma; PZ001), simvastatin (Sigma; S6196), mevalonolactone (mevalonic acid) (Sigma; M4667), YM-53601 (Cayman Chemicals; 18113), avasimibe (Sigma; PZ0190-5MG), compound E (Calbiochem CAS 209986-17-4), and BACE1 inhibitor (BIV) (Calbiochem CAS 797035-11-1).
Aβ, sAPPβ, and sAPPα MSD-ECL measurements
For Meso Scale Discovery electrochemiluminescence (MSD-ECL) measurements of conditioned media from iPSC-derived neurons, neurons were replated into 96 wells (2 × 105 living cells/well) after three wk of differentiation. Replated neurons were cultured for 2 wk in 200 µl NPC base + BDNF/GDNF/cAMP. After 2 wk, media was removed and fresh media (200 µl NPC base + BDNF/GDNF/cAMP) containing the tested compounds was added. The conditioned culture media was harvested from cells at the indicated time points.
For MSD-ECL measurements of conditioned media from HEK cells, HEK cells were plated on 12 well plates coated with poly-l-lysine (2 × 105 living cells/well) in the indicated drug treatment. On the second day, media was removed, the cells were washed once with phosphate-buffered saline (PBS), and media was replaced with fresh dilutions of the indicated treatment and cells were transfected. Media was harvested for analysis at the indicated time points (typically 16–24 h post-transfection).
For all Aβ measurements, 25 μl of the culture media was run on a V-PLEX Aβ Peptide Panel 1 (6E10) (K150SKE) kit. For all sAPP measurements, 25 µl of the culture media was run on a MULTI-SPOT sAPPα/sAPPβ plate (K15120E). All kits are from Meso Scale Discovery. Measurements were performed on the MSD imager MESO QuickPlex SQ 120.
Microscopy
Replated neurons derived from the CVB iPSC line (RRID:CVCL_1N86) on MatTek glass-bottom dishes were treated with 10 µM atorvastatin and DMSO for 5 d. Following treatment, cells were transfected with either APPvn/APPvc/sol. mCherry or APPvn/BACE1vc/sol. mCherry plasmids using Lipofectamine 2000 for 16–18 h. Cells were fixed in 4% PFA (paraformaldehyde) and permeabilized using Triton X-100. Immunostaining was performed using the MAP2 antibody.
Imaging and subsequent data analyses were done blinded. Images were acquired using an Olympus IX81 inverted epifluorescence microscope. Z-stack images were captured using a 100× objective (imaging parameters: 0.4 μm z-step, 400–800 ms exposure, and 1 × 1 binning). Neurons that were double-positive for MAP2 and soluble mCherry were selected for imaging. Captured images were deconvolved and subjected to a maximum-intensity projection using Metamorph Software (Molecular Devices).
Western blots
For Western blots of iPSC-derived neurons, neurons were replated after 3 wk of differentiation onto 24 well plates (2 × 106 live cells/well). Replated neurons were cultured for 2 wk in 200 µl NPC base + BDNF/GDNF/cAMP. After 2 wk, half of the media was removed and fresh media (500 µl NPC base + BDNF/GDNF/cAMP) containing the tested compounds was added. The cells were harvested for Western blot at the indicated time points.
For Western blots of HEK cell lysates, HEK cells were plated on 12 well plates coated with poly-l-lysine (2 × 105 living cells/well) in the indicated drug treatment. On the second day, media was removed, the cells were washed once with PBS, media was replaced with fresh dilutions of the indicated treatment, and cells were transfected. Cells were harvested for Western blot at the indicated time points (typically 16–24 h post-transfection).
Cells were lysed in RIPA lysis buffer (Millipore) with protease inhibitors (Halt) and phosphatase inhibitors (Halt 1861277). Protein concentrations were measured using a Pierce BCA Protein Assay Kit (Thermo Fisher), and samples were diluted to equal protein concentrations. LDS sample buffer (4×) (Invitrogen NP0007) + β-mercaptoethanol was added to the lysate to generate a final concentration of 1× sample buffer + 5% β-mercaptoethanol. Samples were boiled at 100°C for 5 min.
Samples were run on NuPAGE 4%–12% bis-Tris gels (Invitrogen). Gels were transferred to PVDF (polyvinylidene difluoride) membranes and then blocked for 1 h at room temperature in Odyssey Blocking Buffer (LI-COR). Blots were probed overnight at 4°C using the indicated primary antibody. The next day, blots were probed with IRDye secondary antibodies (LI-COR) at 1:5000. Images were acquired using a LICOR Odyssey imager. Bands were quantified using LICOR Image Studio Lite software.
Antibodies used were as follows: anti-actin (1:10,000; EMD Millipore), anti-BACE1 EPR3956 (1:1000; Abcam ab108394), anti-APP C-terminal 751-770 (1:500; Millipore 171610), and anti-GFP (1:1000; Abcam ab290).
Flow cytometry bimolecular fluorescence complementation assay
For flow cytometry analysis of transfected HEK cells, HEK cells were plated on 12 well plates coated with poly-l-lysine (2 × 105 living cells/well) in the indicated drug treatment. On the second day, media was removed, the cells were washed once with PBS, media was replaced with fresh dilutions of the indicated treatment, and cells were transfected. Cells were harvested for flow cytometry at the indicated time points (typically 16–24 h posttransfection).
To harvest cells for flow cytometry analysis, HEK cells were washed once in PBS and then incubated with trypsin to dissociate cells. Trypsinized cells were washed and filtered through 100 µm filters. Cells were centrifuged to pellet cells. Cell pellets were resuspended and filtered through 70 µm filters.
Cells were analyzed on a BD FACSAria II flow cytometer.
Plasmids
APPvn, BACE1vc, and APPStop40vn plasmids were reported in Das et al. (2016). Human WT APP695 isoform was PCR amplified and cloned at HindIII and SacII sites of pVC to generate the APPvc plasmid. Δcholesterol APP mutant plasmids were generated using the QuikChange XL Site-Directed Mutagenesis kit. eaAPPvn and eaAPPvc (APP E693A) were generated using the following mutagenic primers:
E693A forward: 5′-GGTGTTCTTTGCAGCCGATGTGGGTTCAAAC-3′
E693A reverse: 5′-GTTTGAACCCACATCGGCTGCAAAGAACACC-3′
eafaAPPvn and eafaAPPvc (APP E693A+F691A) were generated using the following mutagenic primers:
EAFA forward: 5′-GGTGTTCGCTGCAGCCGATGTGGGTTCAAAC-3′
EAFA reverse: 5′-GTTTGAACCCACATCGGCTGCAGCGAACACC-3′
All constructs were sequence verified.
Statistical analysis
Statistical analyses were performed using GraphPad Prism software. Statistical analyses comparing multiple groups were performed using a one-way analysis of variance with a Tukey’s multiple comparison test. Statistical analysis comparing two groups were calculated using two-tailed unpaired t tests. Data are depicted with bar graphs of the mean ± SEM of all values. Significance was defined as **** p < 0.0001, *** p < 0.001, ** p < 0.01, * p < 0.05. n indicates measurements from independent wells of cultured cells.
Supplementary Material
Acknowledgments
We thank Cody Fine and Jesus Olvera at the Sanford Consortium Stem Cell Core for helpful advice on flow cytometry data analysis. R.v.d.K. was supported by an Alzheimer Netherlands Fellowship (WE.15-2013-01) and an ERC Marie Curie International Outgoing fellowship (622444; APPtoTau). V.F.L. was supported by a National Institutes of Health T32 training grant (5T32AG000216-24). R.S.C. was supported by U.S. Department of Defense (DoD) Peer Reviewed Alzheimer’s Research Program (PRARP) grants (W81XWH-19-1-0315 and W81XWH-15-1-0561). U.D. and L.S.G. were supported by a National Institutes of Health grant (1R56AG054013). This work is supported by National Institute on Aging (1RF1AG048083-01) and California Institute for Regenerative Medicine (RB5-07011) grants to L.S.B.G.
Abbreviations used:
- Aβ
amyloid beta
- ACAT
Acyl-CoA cholesterol acyltransferase
- AD
Alzheimer’s disease
- AICD
APP intracellular domain
- APP
amyloid precursor protein
- α-CTF
α C-terminal fragment
- β-CTF
β C-terminal fragment
- BiFC
bimolecular fluorescence complementation
- CE
cholesterol esters
- FAD
familial Alzheimer’s disease
- farnesyl-PP
farnesyl-pyrophosphate
- flAPP
full-length APP
- geranyl-geranyl-PP
geranyl-geranyl-pyrophosphate
- GXXXG
glycine-XXX-glycine
- HMGCR
3-hydroxy-3-methylglutaryl-CoA reductase
- MSD-ECL
Meso Scale Discovery electrochemiluminescence
- MVA
mevalonate
- NDC
nondemented control
- NPC
neural progenitor cell
- PS
presenilin
- PS1
presenilin-1
- PS2
presenilin-2
- pTau
hyperphosphorylated Tau protein
- SAD
sporadic Alzheimer’s disease
- sAPPβ
soluble APPβ
- sAPPα
soluble APPα
- vn
Venus fluorophore N-terminus
- vc
Venus fluorophore C-terminus.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E20-05-0345) on December 9, 2020.
REFERENCES
- Andreone BJ, Przybyla L, Llapashtica C, Rana A, Davis SS, van Lengerich B, Lin K, Shi J, Mei Y, Astarita G, et al. (2020). Alzheimer’s-associated PLCγ2 is a signaling node required for both TREM2 function and the inflammatory response in human microglia. Nat Neurosci 23, 927–938. [DOI] [PubMed] [Google Scholar]
- Barrett PJ, Song Y, Van Horn WD, Hustedt EJ, Schafer JM, Hadziselimovic A, Beel AJ, Sanders CR (2012). The amyloid precursor protein has a flexible transmembrane domain and binds cholesterol. Science 336, 1168–1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beel AJ, Sakakura M, Barrett PJ, Sanders CR (2010). Direct binding of cholesterol to the amyloid precursor protein: an important interaction in lipid-Alzheimer’s disease relationships? Biochim Biophys Acta 1801, 975–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben Khalifa N, Tyteca D, Marinangeli C, Depuydt M, Collet J-F, Courtoy PJ, Renauld J-C, Constantinescu S, Octave J-N, Kienlen-Campard P (2011). Structural features of the KPI domain control APP dimerization, trafficking, and processing. FASEB J 26, 855–867. [DOI] [PubMed] [Google Scholar]
- Bhattacharyya R, Fenn RH, Barren C, Tanzi RE, Kovacs DM (2016). Palmitoylated APP forms dimers, cleaved by BACE1. PLoS One 11, e0166400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodovitz S, Klein WL (1996). Cholesterol modulates α -secretase cleavage of amyloid precursor protein. J Biol Chem 271, 4436–4440. [DOI] [PubMed] [Google Scholar]
- Buhaescu I, Izzedine H (2007). Mevalonate pathway: a review of clinical and therapeutical implications. Clin Biochem 40, 575–584. [DOI] [PubMed] [Google Scholar]
- Chan RB, Oliveira TG, Cortes EP, Honig LS, Duff KE, Small SA, Wenk MR, Shui G, Di Paolo G (2012). Comparative lipidomic analysis of mouse and human brain with Alzheimer disease. J Biol Chem 287, 2678–2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C-D, Oh S-Y, Hinman JD, Abraham CR (2006). Visualization of APP dimerization and APP-Notch2 heterodimerization in living cells using bimolecular fluorescence complementation. J Neurochem 97, 30–43. [DOI] [PubMed] [Google Scholar]
- Cho YY, Kwon O-H, Park MK, Kim T-W, Chung S (2019). Elevated cellular cholesterol in familial Alzheimer’s presenilin 1 mutation is associated with lipid raft localization of β-amyloid precursor protein. PLoS One 14, e0210535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole SL, Grudzien A, Manhart IO, Kelly BL, Oakley H, Vassar R (2005). Statins cause intracellular accumulation of amyloid precursor protein, β-secretase-cleaved fragments, and amyloid β-peptide via an isoprenoid-dependent mechanism. J Biol Chem 280, 18755–18770. [DOI] [PubMed] [Google Scholar]
- Cossec J-C, Simon A, Marquer C, Moldrich RX, Leterrier C, Rossier J, Duyckaerts C, Lenkei Z, Potier M-C (2010). Clathrin-dependent APP endocytosis and Abeta secretion are highly sensitive to the level of plasma membrane cholesterol. Biochim Biophys Acta 1801, 846–852. [DOI] [PubMed] [Google Scholar]
- Das U, Wang L, Ganguly A, Saikia JM, Wagner SL, Koo EH, Roy S (2016). Visualization of APP and BACE-1 approximation in neurons: new insights into the amyloidogenic pathway. Nat Neurosci 19, 55–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decock M, El Haylani L, Stanga S, Dewachter I, Octave J-N, Smith SO, Constantinescu SN, Kienlen-Campard P (2015). Analysis by a highly sensitive split luciferase assay of the regions involved in APP dimerization and its impact on processing. FEBS Open Bio 5, 763–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decock M, Stanga S, Octave J-N, Dewachter I, Smith SO, Constantinescu SN, Kienlen-Campard P (2016). Glycines from the APP GXXXG/GXXXA transmembrane motifs promote formation of pathogenic Aβ oligomers in Cells. Front Aging Neurosci 8, 107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggert S, Gonzalez AC, Thomas C, Schilling S, Schwarz SM, Tischer C, Adam V, Strecker P, Schmidt V, Willnow TE, et al. (2018). Dimerization leads to changes in APP (amyloid precursor protein) trafficking mediated by LRP1 and SorLA. Cell Mol Life Sci 75, 301–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggert S, Midthune B, Cottrell B, Koo EH (2009). Induced dimerization of the amyloid precursor protein leads to decreased amyloid-β protein production. J Biol Chem 284, 28943–28952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehehalt R, Keller P, Haass C, Thiele C, Simons K (2003). Amyloidogenic processing of the Alzheimer beta-amyloid precursor protein depends on lipid rafts. J Cell Biol 160, 113–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott DA, Weickert CS, Garner B (2010). Apolipoproteins in the brain: implications for neurological and psychiatric disorders. Clin Lipidol 51, 555–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frears ER, Stephens DJ, Walters CE, Davies H, Austen BM (1999). The role of cholesterol in the biosynthesis of β-amyloid. NeuroReport 10, 1699–1705. [DOI] [PubMed] [Google Scholar]
- Gore A, Li Z, Fung H-L, Young J, Agarwal S, Antosiewicz-Bourget J, Canto I, Giorgetti A, Israel M, Kiskinis E, et al. (2011). Somatic coding mutations in human induced pluripotent stem cells. Nature 471, 63–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haag MDM, Hofman A, Koudstaal PJ, Stricker BHC, Breteler MMB (2009). Statins are associated with a reduced risk of Alzheimer disease regardless of lipophilicity. The Rotterdam Study. J Neurol Neurosurg Psychiatry 80, 13–17. [DOI] [PubMed] [Google Scholar]
- Higashide H, Ishihara S, Nobuhara M, Ihara Y, Funamoto S (2017). Alanine substitutions in the GXXXG motif alter C99 cleavage by γ-secretase but not its dimerization. J Neurochem 140, 955–962. [DOI] [PubMed] [Google Scholar]
- Hutter-Paier B, Huttunen HJ, Puglielli L, Eckman CB, Kim DY, Hofmeister A, Moir RD, Domnitz SB, Frosch MP, Windisch M, et al. (2004). The ACAT inhibitor CP-113,818 markedly reduces amyloid pathology in a mouse model of Alzheimer’s disease. Neuron 44, 227–238. [DOI] [PubMed] [Google Scholar]
- Huttunen HJ, Greco C, Kovacs DM (2007). Knockdown of ACAT-1 reduces amyloidogenic processing of APP. FEBS Lett 581, 1688–1692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huttunen HJ, Havas D, Peach C, Barren C, Duller S, Xia W, Frosch MP, Hutter-Paier B, Windisch M, Kovacs DM (2010). The acyl-coenzyme A:cholesterol acyltransferase inhibitor CI-1011 reverses diffuse brain amyloid pathology in aged amyloid precursor protein transgenic mice. J Neuropathol Exp Neurol 69, 777–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huttunen HJ, Peach C, Bhattacharyya R, Barren C, Pettingell W, Hutter-Paier B, Windisch M, Berezovska O, Kovacs DM (2009). Inhibition of acyl-coenzyme A: cholesterol acyl transferase modulates amyloid precursor protein trafficking in the early secretory pathway. FASEB J 23, 3819–3828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Israel MA, Yuan SH, Bardy C, Reyna SM, Mu Y, Herrera C, Hefferan MP, Van Gorp S, Nazor KL, Boscolo FS, et al. (2012). Probing sporadic and familial Alzheimer’s disease using induced pluripotent stem cells. Nature 482, 216–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jick H, Zornberg GL, Jick SS, Seshadri S, Drachman DA (2000). Statins and the risk of dementia. Lancet 356, 1627–1631. [DOI] [PubMed] [Google Scholar]
- Kaden D, Munter L-M, Joshi M, Treiber C, Weise C, Bethge T, Voigt P, Schaefer M, Beyermann M, Reif B, et al. (2008). Homophilic interactions of the amyloid precursor protein (APP) ectodomain are regulated by the loop region and affect β-secretase cleavage of APP. J Biol Chem 283, 7271–7279. [DOI] [PubMed] [Google Scholar]
- Khalifa NB, Van Hees J, Tasiaux B, Huysseune S, Smith SO, Constantinescu SN, Octave J-N, Kienlen-Campard P (2010). What is the role of amyloid precursor protein dimerization? Cell Adh Migr 4, 268–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kienlen-Campard P, Tasiaux B, Hees JV, Li M, Huysseune S, Sato T, Fei JZ, Aimoto S, Courtoy PJ, Smith SO, et al. (2008). Amyloidogenic processing but not amyloid precursor protein (APP) intracellular C-terminal domain production requires a precisely oriented APP dimer assembled by transmembrane GXXXG motifs. J Biol Chem 283, 7733–7744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleiger G, Grothe R, Mallick P, Eisenberg D (2002). GXXXG and AXXXA: common α-helical interaction motifs in proteins, particularly in extremophiles. Biochemistry 41, 5990–5997. [DOI] [PubMed] [Google Scholar]
- Kojro E, Gimpl G, Lammich S, Marz W, Fahrenholz F (2001). Low cholesterol stimulates the nonamyloidogenic pathway by its effect on the alpha-secretase ADAM 10. Proc Natl Acad Sci USA 98, 5815–5820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G, Shofer JB, Rhew IC, Kukull WA, Peskind ER, McCormick W, Bowen JD, Schellenberg GD, Crane PK, Breitner JCS, et al. (2010). Age-varying association between statin use and incident Alzheimer’s disease. J Am Geriatr Soc 58, 1311–1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liebsch F, Aurousseau MRP, Bethge T, McGuire H, Scolari S, Herrmann A, Blunck R, Bowie D, Multhaup G (2017). Full-length cellular β-secretase has a trimeric subunit stoichiometry, and its sulfur-rich transmembrane interaction site modulates cytosolic copper compartmentalization. J Biol Chem 292, 13258–13270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin F-C, Chuang Y-S, Hsieh H-M, Lee T-C, Chiu K-F, Liu C-K, Wu M-T (2015). Early statin use and the progression of Alzheimer disease. Medicine (Baltimore) 94, e2143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y-T, Seo J, Gao F, Feldman HM, Wen H-L, Penney J, Cam HP, Gjoneska E, Raja WK, Cheng J, Rueda R, et al. (2018). APOE4 causes widespread molecular and cellular alterations associated with Alzheimer’s disease phenotypes in human iPSC-derived brain cell types. Neuron 98, 1141–1154.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, MacKenzie KR, Putluri N, Maletic‘-Savatic‘ M, Bellen HJ (2017). The glia-neuron lactate shuttle and elevated ROS promote lipid synthesis in neurons and lipid droplet accumulation in glia via APOE/D. Cell Metab 26, 719–737.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marquer C, Devauges V, Cossec J-C, Liot G, Lécart S, Saudou F, Duyckaerts C, Lévêque-Fort S, Potier M-C (2011). Local cholesterol increase triggers amyloid precursor protein-Bace1 clustering in lipid rafts and rapid endocytosis. FASEB J 25, 1295–1305. [DOI] [PubMed] [Google Scholar]
- Munter L-M, Voigt P, Harmeier A, Kaden D, Gottschalk KE, Weise C, Pipkorn R, Schaefer M, Langosch D, Multhaup G (2007). GxxxG motifs within the amyloid precursor protein transmembrane sequence are critical for the etiology of Aβ42. EMBO J 26, 1702–1712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nierzwicki Ł, Czub J (2015). Specific binding of cholesterol to the amyloid precursor protein: structure of the complex and driving forces characterized in molecular detail. J Phys Chem Lett 6, 784–790. [DOI] [PubMed] [Google Scholar]
- Nugent AA, Lin K, van Lengerich B, Lianoglou S, Przybyla L, Davis SS, Llapashtica C, Wang J, Kim DJ, Xia D, et al. (2020). TREM2 regulates microglial cholesterol metabolism upon chronic phagocytic challenge. Neuron 105, 837–854.e9. [DOI] [PubMed] [Google Scholar]
- O’Brien RJ, Wong PC (2011). Amyloid precursor protein processing and Alzheimer’s disease. Annu Rev Neurosci 34, 185–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostrowski SM, Wilkinson BL, Golde TE, Landreth G (2007). Statins reduce amyloid-β production through inhibition of protein isoprenylation. J Biol Chem 282, 26832–26844. [DOI] [PubMed] [Google Scholar]
- Parsons RB, Price GC, Farrant JK, Subramaniam D, Adeagbo-Sheikh J, Austen BM (2006). Statins inhibit the dimerization of β-secretase via both isoprenoid- and cholesterol-mediated mechanisms. Biochem J 399, 205–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puglielli L, Konopka G, Pack-Chung E, Ingano LAM, Berezovska O, Hyman BT, Chang TY, Tanzi RE, Kovacs DM (2001). Acyl-coenzyme A: cholesterol acyltransferase modulates the generation of the amyloid β-peptide. Nat Cell Biol 3, 905–912. [DOI] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F (2013). Genome engineering using the CRISPR-Cas9 system. Nat Protoc 8, 2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossjohn J, Cappai R, Feil SC, Henry A, McKinstry WJ, Galatis D, Hesse L, Multhaup G, Beyreuther K, Masters CL, et al. (1999). Crystal structure of the N-terminal, growth factor-like domain of Alzheimer amyloid precursor protein. Nat Struct Mol Biol 6, 327–331. [DOI] [PubMed] [Google Scholar]
- Russ WP, Engelman DM (2000). The GxxxG motif: a framework for transmembrane helix-helix association. J Mol Biol 296, 911–919. [DOI] [PubMed] [Google Scholar]
- Scheuermann S, Hambsch B, Hesse L, Stumm J, Schmidt C, Beher D, Bayer TA, Beyreuther K, Multhaup G (2001). Homodimerization of amyloid precursor protein and its implication in the amyloidogenic pathway of Alzheimer’s disease. J Biol Chem 276, 33923–33929. [DOI] [PubMed] [Google Scholar]
- Simons M, Keller P, De Strooper B, Beyreuther K, Dotti CG, Simons K (1998). Cholesterol depletion inhibits the generation of beta-amyloid in hippocampal neurons. Proc Natl Acad Sci USA 95, 6460–6464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- So PP, Khodr CE, Chen C-D, Abraham CR (2013). Comparable dimerization found in wildtype and familial Alzheimer’s disease amyloid precursor protein mutants. Am J Neurodegener Dis 2, 15–28. [PMC free article] [PubMed] [Google Scholar]
- Song Y, Hustedt EJ, Brandon S, Sanders CR (2013). Competition between homodimerization and cholesterol binding to the C99 domain of the amyloid precursor protein. Biochemistry 52, 5051–5064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajima Y, Ishikawa M, Maekawa K, Murayama M, Senoo Y, Nishimaki-Mogami T, Nakanishi H, Ikeda K, Arita M, Taguchi R, et al. (2013). Lipidomic analysis of brain tissues and plasma in a mouse model expressing mutated human amyloid precursor protein/tau for Alzheimer’s disease. Lipids Health Dis 12, 68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Kant R, Langness VF, Herrera CM, Williams DA, Fong LK, Leestemaker Y, Steenvoorden E, Rynearson KD, Brouwers JF, Helms JB, et al. (2019). Cholesterol metabolism is a druggable axis that independently regulates Tau and amyloid-β in iPSC-derived Alzheimer’s disease neurons. Cell Stem Cell 24, 363–375.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Kant R, Goldstein LSB, Ossenkoppele R (2020). Amyloid-β-independent regulators of tau pathology in Alzheimer disease. Nat Rev Neurosci 21, 21–35. [DOI] [PubMed] [Google Scholar]
- Wahrle S, Das P, Nyborg AC, McLendon C, Shoji M, Kawarabayashi T, Younkin LH, Younkin SG, Golde TE (2002). Cholesterol-dependent γ-secretase activity in buoyant cholesterol-rich membrane microdomains. Neurobiol Dis 9, 11–23. [DOI] [PubMed] [Google Scholar]
- Wingo TS, Cutler DJ, Wingo AP, Le N-A, Rabinovici GD, Miller BL, Lah JJ, Levey AI (2019). Association of early-onset Alzheimer disease with elevated low-density lipoprotein cholesterol levels and rare genetic coding variants of APOB. J Am Med Assoc Neurol 76, 809–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolozin B, Kellman W, Ruosseau P, Celesia GG, Siegel G (2000). Decreased prevalence of Alzheimer disease associated with 3-hydroxy-3-methyglutaryl coenzyme A reductase inhibitors. Arch Neurol 57, 1439–1443. [DOI] [PubMed] [Google Scholar]
- Xiong H, Callaghan D, Jones A, Walker DG, Lue L-F, Beach TG, Sue LI, Woulfe J, Xu H, Stanimirovic DB, et al. (2008). Cholesterol retention in Alzheimer’s brain is responsible for high beta- and gamma-secretase activities and Abeta production. Neurobiol Dis 29, 422–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang D-S, Stavrides P, Saito M, Kumar A, Rodriguez-Navarro JA, Pawlik M, Huo C, Walkley SU, Saito M, Cuervo AM, et al. (2014). Defective macroautophagic turnover of brain lipids in the TgCRND8 Alzheimer mouse model: prevention by correcting lysosomal proteolytic deficits. Brain 137, 3300–3318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young JE, Boulanger-Weill J, Williams DA, Woodruff G, Buen F, Revilla AC, Herrera C, Israel MA, Yuan SH, Edland SD, et al. (2015). Elucidating molecular phenotypes caused by the SORL1 Alzheimer’s disease genetic risk factor using human induced pluripotent stem cells. Cell Stem Cell 16, 373–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan SH, Martin J, Elia J, Flippin J, Paramban RI, Hefferan MP, Vidal JG, Mu Y, Killian RL, Israel MA, et al. (2011). Cell-surface marker signatures for the isolation of neural stem cells, glia and neurons derived from human pluripotent stemcells. PLOS One 6, e17540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Suram A, Venugopal C, Prakasam A, Lin S, Su Y, Li B, Paul SM, Sambamurti K (2008). Geranylgeranyl pyrophosphate stimulates γ-secretase to increase the generation of Aβ and APP-CTFγ. FASEB J 22, 47–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zissimopoulos JM, Barthold D, Brinton RD, Joyce G (2017). Sex and race differences in the association between statin use and the incidence of Alzheimer disease. J Am Med Assoc Neurol 74, 225–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.