

Abstract
Purpose of Review:
To summarize key advances in our understanding of the role of interleukin 17A (IL-17A) in the pathogenesis of hypertension and highlight important areas for future research and clinical translation.
Recent Findings:
While T helper 17 (Th17) cells are major producers of IL-17A, there are several additional innate and adaptive immune cell sources including gamma-delta T cells, innate lymphoid cells, and natural killer cells. IL-17A promotes an increase in blood pressure through multiple mechanisms including inhibiting endothelial nitric oxide production, increasing reactive oxygen species formation, promoting vascular fibrosis, and enhancing renal sodium retention and glomerular injury. IL-17A production from Th17 cells is increased by high salt conditions in vitro and in vivo. There is also emerging data linking salt, the gut microbiome, and intestinal T cell IL-17A production. Novel therapeutics targeting IL-17A signaling are approved for the treatment of autoimmune diseases and show promise in both animal models of hypertension and human studies.
Summary:
Hypertensive stimuli enhance IL-17A production. IL-17A is a key mediator of renal and vascular dysfunction in hypertensive mouse models and correlates with hypertension in humans. Large randomized clinical trials are needed to determine whether targeting IL-17A might be an effective adjunct treatment for hypertension and its associated end-organ dysfunction.
Keywords: interleukin 17A, hypertension, inflammation, cytokines, Th17, vascular disease
Introduction
Hypertension is the leading risk factor for worldwide morbidity and mortality and is a key contributor to cardiovascular disease [1]. In the United States alone, hypertension affects 46% of the adult population with prevalence rising dramatically with increasing age [2]. Approximately 50% of individuals with hypertension have uncontrolled blood pressures. While the proportion of individuals with controlled hypertension improved from 1999–2010, there has been no further improvements since 2010, highlighting the need for novel treatment strategies for this global silent killer [3, 4].
Hypertension is a complex medical condition characterized by persistently elevated blood pressure in the arteries that is associated with damage to the vasculature, kidneys, and brain, all of which are involved in blood pressure regulation. While immune cells have been observed in hypertensive organs for decades, it is only in recent years that we have obtained a more mechanistic understanding of how the innate and adaptive immune systems are activated and contribute both to the elevation of blood pressure and the end-organ damage that accompanies hypertension. In response to stimuli such as angiotensin II, salt, and microbial factors, there is mounting evidence that dendritic cells present neo-antigens to T cells that lead to T cell proliferation and cytokine secretion [5, 6]. T cells may also be activated directly in response to environmental stimuli and gut microbe metabolites. We and others have shown that interleukin 17A (IL-17A), a proinflammatory cytokine released by CD4+ T helper 17 (Th17) cells as well as other potential innate and adaptive immune cells, is an important mediator of hypertension in experimental animal models and in humans, mediating an increase in blood pressure through vascular dysfunction, renal damage, and disruption of sodium homeostasis (Figure 1) [7–9]. The purpose of this review is to summarize recent advances in our understanding of the role of IL-17A in the pathogenesis of hypertension and highlight important areas for further investigation. Research in this area has important clinical implications for future immune modulation as a potential adjunct treatment for hypertension.
Figure 1.
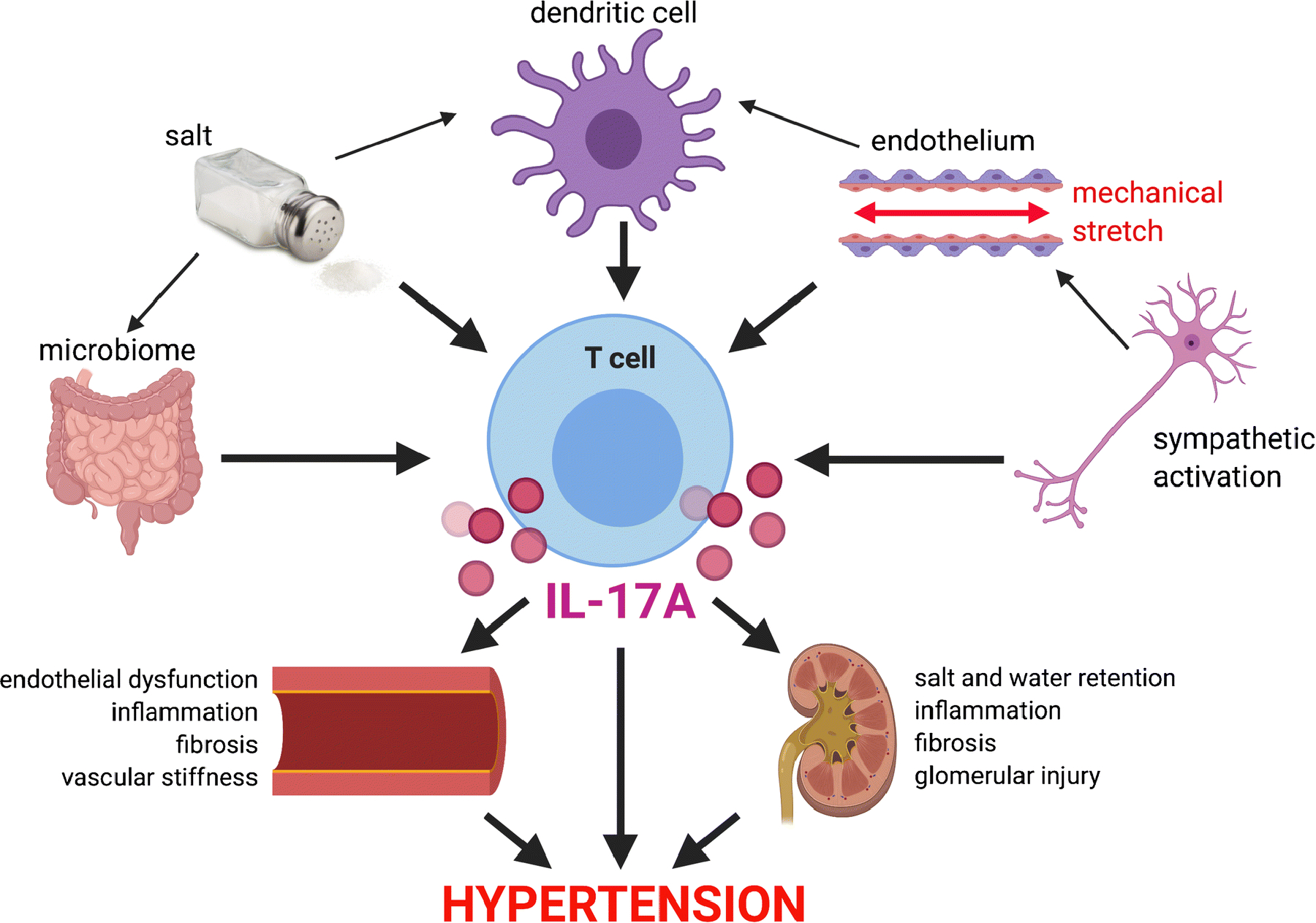
Schematic of how hypertensive stimuli increase T cell production of IL-17A which in turn promotes renal and vascular damage in hypertension.
IL-17 Isoforms
IL-17A was discovered in 1993 by Rouvier et al. and initially named cytotoxic T lymphocyte-associated antigen 8 (CTLA-8) [10]. Since the discovery of IL-17A, five additional family members have been identified and designated IL-17B through IL-17F. IL-17A and IL-17F are the most closely related in terms of structure, function, and expression [11]. IL-17F shares the highest amino acid identity with IL-17A (50%) and is located in close proximity to IL-17A in the genome (chromosome 1 in mice and chromosome 6 in humans). IL-17A and IL-17F are produced by similar cell types and bind as homo- or heterodimers to the IL-17 receptor complex composed of IL-17RA and IL-17RC subunits [11]. IL-17A is the most widely studied isoform. In addition to its role in host defense against bacterial and fungal infections, IL-17A is a key cytokine in the pathogenesis of many autoimmune and inflammatory diseases such as rheumatoid arthritis, multiple sclerosis, inflammatory bowel disease, and periodontal disease [12, 11]. We discovered in 2010 that IL-17A also plays a critical role in hypertension and its associated complications [7].
Sources of IL-17A
CD4+ Th17 cells are a major source of IL-17A. Th17 differentiation is controlled by the master transcriptional regulator RORγt (RORC is the human ortholog of RORyt in mice) and STAT3. Th17 differentiation is induced by transforming growth factor beta (TGFβ) and IL-6 [13]. In addition, IL-21 is produced by Th17 cells and drives IL-17A production in a STAT3-dependent manner [14]. Recent data indicates that not all Th17 cells are pathogenic. Generation of pathogenic Th17 cells requires IL-23 stimulation in addition to TGFβ and IL-6. Pathogenic Th17 cells exhibit a distinct gene expression profile compared to non-pathogenic Th17 cells, suggesting the existence of functionally diverse Th17 subpopulations [13]. Analogous to CD4+ T helper cells, a subset of CD8+ cytotoxic T cells (Tc17 cells) produce IL-17A but generally at lower levels than CD4+ T cells [15, 16].
A subset of innate-like T cells called gamma-delta (γδ) T cells produce IL-17A and in some conditions are the predominant source of IL-17A production. These cells, which are rare in lymphoid tissues and blood of adult humans and rodents, are enriched in mucosal and epithelial tissues where they help eliminate invading pathogens and preserve barrier integrity [13, 17]. Like Th17 cells, IL-17A producing γδ T cells are characterized by expression of RORγt and are activated by IL-23 [18].
IL-17A, IL-21, and IL-23 are elevated in experimental animal models of hypertension and in hypertensive humans [19, 20]. We found that in hypertensive mouse kidneys and aortas, the major T cell sources of IL-17A are Th17 cells and γδ T cells with lesser contribution from other T cell subsets [16]. Of note, IL-17A can also be produced by innate immune cells including innate lymphoid cell 3 (ILC3) found at mucosal surfaces, natural killer cells, and macrophages [13, 21], but the relative contribution of these cells to IL-17A production in hypertension is not known.
Role of IL-17A in the Vasculature in Hypertension
IL-17A acts on both endothelial cells and smooth muscle cells in the vasculature to contribute to the hypertensive phenotype. We showed in 2010 that IL-17A−/− mice develop blunted hypertension in response to chronic angiotensin II (Ang II) infusion and are protected from Ang II-induced endothelial dysfunction, vascular superoxide production, and vascular inflammation [7]. Nguyen et al. demonstrated that IL-17A induces phosphorylation of the inhibitory site threonine 495 on endothelial nitric oxide synthase leading to decreased nitric oxide production through a Rho-kinase dependent pathway [22]. They also showed that in vivo infusion of IL-17A alone is sufficient to increase blood pressure. In vascular smooth muscle cells, IL-17A induces inflammatory cytokine and chemokine expression and promotes aortic stiffening via increased collagen synthesis [7, 23]. More recently, Orejudo et al investigated the effect of IL-17A on small resistance arteries and demonstrated that IL-17A infusion increases blood pressure and induces inward hypertrophic remodeling and stiffness of small mesenteric arteries. By treating with hydralazine and hydrochlorothiazide to prevent the blood pressure increase, these authors showed that the effects of IL-17A on remodeling and stiffness were still present, indicating that these effects are direct and independent of blood pressure elevation [24]. In addition, they showed that neutralizing antibodies to IL-17A reduced small mesenteric artery remodeling in Ang II-induced hypertension.
Using a unique mouse model of T cell specific IL-17A overexpression (CD4-IL-17Aind/+), Schuler et al. showed that mice with constitutive overexpression of IL-17A in T cells exhibit impaired endothelium-dependent relaxation of aortic rings and mildly increased aortic fibrosis, but no increase in blood pressure or infiltration of inflammatory cells into the aortic wall [25]. There are several possible explanations for this. First, the levels of IL-17A produced in this model may not be comparable to that produced by infusion of the cytokine via osmotic minipump and without a hypertensive stimulus, these cells may not infiltrate tissues to release IL-17A locally. Second, these cells may be more similar to the “non-pathogenic” Th17 cells that don’t express the full repertoire of factors expressed by pathogenic Th17 cells. Finally, these data suggest that we should also consider non-T cell sources of IL-17A which may be important in the full development of hypertension. Nevertheless, this study confirms that IL-17A production from T cells has direct effects on the vasculature independent of blood pressure.
Hypertension is often associated with increased sympathetic outflow and release of catecholamines such as norepinephrine. In vitro studies by Xu et al. showed that exposure of dermal microvascular endothelial cells to norepinephrine followed by co-culture with antigen presenting cells (APCs), an antigen, and responsive T cells (expressing a transgenic receptor that recognizes the antigen) induces increased T helper cell production of IL-17A and upregulation of RORγT with downregulation of IFNγ. This effect did not require direct contact between endothelial cells and the immune cells and was prevented by siRNA inhibition of IL-6 in the endothelial cells [26]. Additional in vitro studies by Loperena et al. demonstrated that hypertensive mechanical stretch of endothelial cells increases endothelial IL-6 production and activates monocytes to produce Th17 polarizing cytokines [27]. Together these results demonstrate a crosstalk between the endothelium and immune cells whereby hypertensive stimuli lead to T cell IL-17A secretion which in turn promotes endothelial dysfunction (Figure).
Role of IL-17A in the Kidney in Hypertension
In contrast to other tissues in the body, the kidney has almost no regenerative capacity and no ability to generate new nephrons once they are lost. IL-17A plays a major role in hypertensive renal damage. We showed in an Ang II infusion (490 ng/kg/min) model that IL-17A deficient (but not IL-17F deficient) mice are protected from hypertension-induced albuminuria, which is indicative of damage to the glomerular filtration barrier [8]. To determine whether inhibition of IL-17A signaling can halt or reverse hypertension and end-organ damage in the setting of established hypertension, we infused Ang II for 4 weeks and administered neutralizing antibodies to IL-17A, IL-17F, or the IL-17RA receptor subunit during the final 2 weeks of Ang II-infusion. Interestingly, we found that neutralizing antibodies to IL-17A or IL-17RA, but not IL-17F, lowers blood pressure, reduces markers of inflammation and fibrosis, and halts the progression of albuminuria, suggesting isoform specific effects of IL-17 signaling and demonstrating the potential of using these inhibitors clinically for the treatment of established hypertension [16]. IL-17A also plays a critical role in models of calcineurin inhibitor-induced hypertension. Calcineurin inhibitors, including cyclosporine A and tacrolimus, are commonly used immunosuppressive medications after solid organ transplants. However, a major side effect of these drugs is renal toxicity and hypertension. If the immune system promotes hypertension and renal damage, it is apparently paradoxical that an immunosuppressant would cause hypertension. This apparent paradox can be explained by the observation that although calcineurin inhibitors reduce the total number of T cells, they increase IL-17A producing Th17 cells and reduce anti-inflammatory regulatory T cells. Chiasson et al. demonstrated that a neutralizing antibody to IL-17A prevented hypertension, endothelial dysfunction, and glomerular injury in several calcineurin inhibitor models [28]. Orejudo et al. subsequently showed that systemic infusion of IL-17A induces renal damage as evidenced by increased expression of neutrophil gelatinase-associated lipocalin (NGAL) and increased renal inflammation [29]. Furthermore, they demonstrated that IL-17A infusion upregulates the chemoattractant molecules Ccl-5/RANTES and Ccl-2/MCP1 in the kidney, both of which have been implicated in the pathogenesis of hypertension [30, 31]. Finally, consistent with our earlier findings [16], these authors showed that an anti-IL-17A neutralizing antibody attenuated renal damage, immune cell infiltration, and upregulation of inflammatory mediators in Ang II-infused mice [29].
In contrast, Marko et al. showed no effect of an IL-17A neutralizing antibody on blood pressure or albuminuria [32]. There are several potential explanations for this including differences in the dose of Ang II used (1000 ng/kg/min), the clone and frequency of antibody administration (only once per week in this study), and the duration of the study which was stopped at 14 days. We found that the major effect of IL-17A is during chronic (weeks 3–4) of Ang II infusion. There has been one study in which IL-17A deficiency actually worsened renal damage [33]. These authors used an extreme model of uninephrectomy, DOCA-pellet implantation, salt-feeding, and a high dose (1200 ng/kg/min) of Ang II. Under these conditions, there was no difference in blood pressure between wild type and IL-17A deficient mice, but the IL-17A deficient mice exhibited more albuminuria and glomerular injury that was associated with increased γδ T cell infiltration [33]. Thus, it is possible that compensatory changes in other immune cell populations resulted in worse renal injury in this setting. Taken together, the overwhelming majority of studies support a critical role for IL-17A in promoting renal injury and suggest that therapeutics targeting IL-17A may be beneficial in primary as well as calcineurin inhibitor-induced hypertension.
In addition to its role in promoting renal damage, we also showed that IL-17A regulates renal sodium excretion, leading to increased sodium and water retention and hypertension. During Ang II infusion, wild type mice lose their ability to appropriately excrete a saline load, but IL-17A deficient mice maintain their diuretic and natriuretic response to a saline challenge via modulation of renal sodium transporters [34]. Specifically, we noted that IL-17A deficiency results in a decrease in the proximal tubule sodium hydrogen exchanger 3 (NHE3) following 2 weeks of Ang II infusion and a blunting of distal transporter activation (sodium chloride co-transporter (NCC) and the epithelial sodium channel (ENaC)) following 4 weeks of Ang II infusion [34, 8]. Using cultured proximal and distal tubule cell lines, we showed that the effects of IL-17A on NHE3 expression and NCC activity, respectively, is dependent on serum and glucocorticoid regulated kinase 1 (SGK1). SGK1 inhibits Nedd4–2-mediated ubiquitination and degradation of NCC and ENaC in the distal convoluted tubule, thus enhancing expression of these transporters on the cell surface [35]. We showed that IL-17A induced phosphorylation of SGK1 at serine-78 and that an SGK1 inhibitor blocked the effects of IL-17A on NHE3 and NCC [8]. The ability to modulate sodium transporters is not unique to IL-17A. Additional cytokines have been shown to regulate sodium transport along various aspects of the nephron as reviewed in Norlander et al. [36]. Further work is needed to understand why and how inflammatory cytokines are able to fine tune sodium and water balance and how this process might become dysregulated in disease.
Salt modulates T cell IL-17A responses and hypertension
It has been known for decades that elevated dietary salt (NaCl) is linked to increased incidence of hypertension in humans, but the precise mechanism for this is still poorly understood [37]. In 2013, two groups demonstrated that increased NaCl concentration in vitro induces Th17 polarization and IL-17A production through a T cell SGK1 dependent pathway [38, 39]. Furthermore, they showed that a high salt diet worsens IL-17A mediated autoimmunity in mice. We subsequently showed that deletion of SGK1 in T cells attenuates Ang II and DOCA-salt induced hypertension and abrogates hypertension-induced renal and vascular inflammation and end-organ damage. T cell specific SGK1 deletion prevented the upregulation of IL-17A expressing T helper cells in the spleen in response to Ang II infusion [40]. It is also possible that salt indirectly activates T cells via its effects on antigen presenting cells. Van Beusecum et al. demonstrated that salt can activate dendritic cells through SGK1 and that SGK1 deletion in dendritic cells attenuates salt-sensitive hypertension [41]. Recently, salt has been shown to promote a Th17-like adaption in regulatory T cells [42]. Interestingly, the effects of salt on Th17 cells seems to be context dependent in that salt can promote either an anti-inflammatory or proinflammatory phenotype based on the cytokine milieu [43]. Major unresolved questions are whether immune cells in vivo actually encounter hypertonic NaCl concentrations and if not, exactly how does a high salt diet lead to immune activation. Titze and colleagues proposed that salt can be stored in the skin and skeletal muscle in the setting of hypertension and aging and that this hypertonic NaCl leads to immune activation [44, 45]. However, recent analyses call this concept into question and suggest that the apparent increase in tissue sodium seen by sodium MRI imaging may actually reflect isotonic edema [46]. Whether and how this isotonic edema influences immune cells is not known. Another way salt may activate IL-17A production is via modulation of the gut microbiome as discussed below.
Gut microbiome modulates IL-17A responses and hypertension
Gut microbiota play an important role in the normal function of the intestinal barrier, facilitating the production of essential nutrients, protection against pathogens, maintenance of the intestinal epithelium integrity, and modulation of immune responses [47]. Innate IL-17A producing cells serve as the first line of defense at barrier sites, and IL-17A is a key mediator of mucosal surveillance and barrier integrity via promoting the production of antimicrobial factors necessary to contain pathogens [48]. Accumulation of proinflammatory and anti-inflammatory immune cells is regulated by the various commensal microbiota populations within the gut. Resident gut microbiota such as segmented filamentous bacteria (SFB) induce Th17 polarization, while clostridium species and bacteroides fragilis seem to promote T regulatory cell polarization [49]. Alterations in the gut microbiota can lead to disruptions in immune cell activity contributing to gut dysbiosis. Gut dysbiosis is observed in autoimmune diseases such as rheumatoid arthritis and multiple sclerosis, both of which are associated with elevations in Th17 cells and IL-17A production [50–52].
There is an emerging role of the gut microbiome as a modulator of hypertension progression. While the precise mechanisms by which gut dysbiosis influence blood pressure are not fully understood, several studies found an altered gut microbiome composition and epithelial barrier dysfunction in subjects with essential hypertension compared to normotensive individuals [53–56].
Germ free mice have a blunted hypertensive response to Ang II that is associated with reduced RORγt expression in the aorta and reduced renal/vascular inflammation suggesting that gut microbiota facilitate Ang II-induced vascular dysfunction in part through IL-17A mediated immune cell infiltration [57]. A causal role for the microbiome in hypertension is supported by studies in experimental animal models of hypertension showing that germ free mice develop higher blood pressure after receiving fecal transplants from hypertensive compared to normotensive subjects [58]. Similarly, Toral et al. used fecal microbiota transplant between the spontaneously hypertensive rat (SHR) and Wistar Kyoto (WKY) controls. Transplant of SHR stool into WKY controls increased blood pressure and conversely, transplant of WKY stool into SHRs decreased blood pressure. SHR rats receiving WKY stool had a marked decrease in aortic expression of RORγt and IL-17A mRNA, while the converse was observed in WKY rats receiving SHR stool. Importantly, neutralizing IL-17A antibody administration reduced blood pressure and improved endothelial dysfunction in WKY rats that received SHR stool [59]. Wilck et al. demonstrated that mice fed a high-salt diet exhibit increased intestinal Th17 polarization mediated by decreased commensal Lactobacillus murinas and that supplementation of Lactobacillus murinas by gavage provided protection from high salt diet-induced blood pressure elevations and Th17 polarization [60].
IL-17A in human hypertension
Multiple studies by our group and others have demonstrated an association between plasma IL-17A levels or circulating T helper cell production of IL-17A and either blood pressure or hypertension diagnosis. We showed in 2010 that in a cohort of diabetic patients with or without hypertension, those with hypertension had significantly higher levels of plasma IL-17A [7]. Subsequently, Itani et al. showed that hypertensive humans have increased IL-17A producing T helper cells in blood [61]. Yao et al. demonstrated that circulating levels of IL-17A correlate with systolic blood pressure in people with pre-hypertension [62]. When we isolated peripheral blood T helper cells and analyzed cytokine production after 3 days in culture, we found that T cell production of IL-21 correlates with T cell production of IL-17A and systolic blood pressure at the time of blood draw [20]. Recently, Masenga et al. demonstrated that in people with HIV, those with hypertension have higher plasma levels of IL-17A and IL-6 compared to their normotensive counterparts [63]. Furthermore, Wang et al. investigated the relationship between IL-17A and blood pressure in patients with end-stage renal disease on hemodialysis and found that those with refractory hypertension had elevated circulating IL-17A compared to those with easy-to-control hypertension. Most notably, there was a linear relationship between circulating IL-17A and mean arterial pressure [64]. Ji et. al examined circulating IL-17A levels and frequency of Th17 cells in normotensive and hypertensive subjects and further subdivided hypertensive subjects into those with a nighttime drop of 10% or more in blood pressure values (“dippers”) and those with a nighttime drop of less than 10% (“nondippers”). A non-dipping blood pressure pattern is associated with increased cardiovascular risk. Th17 cell frequencies were significantly increased in hypertensives vs controls and in nondippers vs dippers. IL-17A levels were significantly increased in hypertensives vs controls and there was a trend for an increase in nondippers vs dippers [65]. Most studies in humans have been done primarily in blood due to ease of sample collection. However, Orejuda et al. demonstrated the presence of IL-17A producing T helper cells and γδ T cells in kidney biopsies of people with hypertensive nephropathy, consistent with our findings in the Ang II-hypertension mouse model [29, 16]. While most studies of IL-17A in human hypertension have been only correlative, some evidence of causality comes from a study by von Stebut et al. demonstrating in a randomized, double-blind, placebo-controlled trial that anti-IL-17A treatment with secukinumab improved endothelial function in patients with moderate-to-severe plaque psoriasis [66].
Conclusions and Future Directions
In this review, we have outlined the importance of IL-17A in the development of hypertension and the associated end-organ damage in animal models as well as the accumulating evidence that IL-17A is important in human hypertension and vascular disease. T cell production of IL-17A can be stimulated directly or indirectly by gut microbial factors, salt, antigen presenting dendritic cells, mechanical stretch, and sympathetic activation (Figure). IL-17A in turn contributes to further increases in blood pressure and end-organ damage via effects on vascular and renal cells (Figure). The increase in Th17-like cells and IL-17A production observed in experimental animal models and humans with hypertension is similar to that seen in several autoimmune diseases such as multiple sclerosis, psoriasis, and systemic lupus erythematosus [67]. To date, several therapeutics have been developed to target IL-17A signaling in the setting of autoimmune diseases [68, 69], but to our knowledge, no long-term studies evaluating the effect of these drugs on hypertension or other cardiovascular endpoints has been conducted. Mehta and colleagues recently showed in a prospective observational study that biologic therapy including inhibitors of TNF-alpha, IL-23, and IL-17A is associated with favorable modulation of coronary plaque characteristics in one year, thus providing a rationale for larger, randomized controlled trials [70]. Future studies are needed to further delineate the precise cellular sources of IL-17A in hypertension, particularly the role of innate lymphoid cells in the gut, and how salt influences the activation and trafficking of IL-17A producing cells. Finally, large-scale clinical studies are needed to determine whether targeting IL-17A may provide an additional therapeutic option for the reduction of cardiovascular and renal complications in hypertensive individuals, supporting a role for immune modulation in hypertension.
Footnotes
Publisher's Disclaimer: This Author Accepted Manuscript is a PDF file of a an unedited peer-reviewed manuscript that has been accepted for publication but has not been copyedited or corrected. The official version of record that is published in the journal is kept up to date and so may therefore differ from this version.
Conflict of Interest
The authors declare no conflicts of interest relevant to this manuscript.
References
Papers of particular interest, published recently, have been highlighted as:
• Of importance
•• Of major importance
- 1.Collaborators GBDRF. Global, regional, and national comparative risk assessment of 79 behavioural, environmental and occupational, and metabolic risks or clusters of risks, 1990–2015: a systematic analysis for the Global Burden of Disease Study 2015. Lancet. 2016;388(10053):1659–724. doi: 10.1016/S0140-6736(16)31679-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Whelton PK, Carey RM, Aronow WS, Casey DE Jr., Collins KJ, Dennison Himmelfarb C et al. 2017 ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA Guideline for the Prevention, Detection, Evaluation, and Management of High Blood Pressure in Adults: Executive Summary: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Hypertension. 2018;71(6):1269–324. doi: 10.1161/HYP.0000000000000066. [DOI] [PubMed] [Google Scholar]
- •3.Foti K, Wang D, Appel LJ, Selvin E. Hypertension Awareness, Treatment, and Control in US Adults: Trends in the Hypertension Control Cascade by Population Subgroup (National Health and Nutrition Examination Survey, 1999–2016). Am J Epidemiol. 2019;188(12):2165–74. doi: 10.1093/aje/kwz177. [DOI] [PMC free article] [PubMed] [Google Scholar]; Comprehensive summary of trends in hypertension awareness, treatment, and control increased from 1999–2016, with few changes after 2010. However disparities in hypertension control are apparent between age, sex, race/ethnicity.
- 4.Fryar CD, Ostchega Y, Hales CM, Zhang G, Kruszon-Moran D. Hypertension Prevalence and Control Among Adults: United States, 2015–2016. NCHS Data Brief. 2017(289):1–8. [PubMed] [Google Scholar]
- 5.Elijovich F, Laffer CL, Sahinoz M, Pitzer A, Ferguson JF, Kirabo A. The Gut Microbiome, Inflammation, and Salt-Sensitive Hypertension. Curr Hypertens Rep. 2020;22(10):79. doi: 10.1007/s11906-020-01091-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Patrick DM, Van Beusecum JP, Kirabo A. The role of inflammation in hypertension: novel concepts. Curr Opin Physiol. 2021;19:92–8. doi: 10.1016/j.cophys.2020.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Madhur MS, Lob HE, McCann LA, Iwakura Y, Blinder Y, Guzik TJ et al. Interleukin 17 promotes angiotensin II-induced hypertension and vascular dysfunction. Hypertension. 2010;55(2):500–7. doi:HYPERTENSIONAHA.109.145094 [pii] 10.1161/HYPERTENSIONAHA.109.145094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Norlander AE, Saleh MA, Kamat NV, Ko B, Gnecco J, Zhu L et al. Interleukin-17A Regulates Renal Sodium Transporters and Renal Injury in Angiotensin II-Induced Hypertension. Hypertension. 2016;68(1):167–74. doi: 10.1161/HYPERTENSIONAHA.116.07493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Caillon A, Mian MOR, Fraulob-Aquino JC, Huo KG, Barhoumi T, Ouerd S et al. gammadelta T Cells Mediate Angiotensin II-Induced Hypertension and Vascular Injury. Circulation. 2017;135(22):2155–62. doi: 10.1161/CIRCULATIONAHA.116.027058. [DOI] [PubMed] [Google Scholar]
- 10.Rouvier E, Luciani MF, Mattei MG, Denizot F, Golstein P. CTLA-8, cloned from an activated T cell, bearing AU-rich messenger RNA instability sequences, and homologous to a herpesvirus saimiri gene. J Immunol. 1993;150(12):5445–56. [PubMed] [Google Scholar]
- 11.Iwakura Y, Ishigame H, Saijo S, Nakae S. Functional specialization of interleukin-17 family members. Immunity. 2011;34(2):149–62. doi: 10.1016/j.immuni.2011.02.012. [DOI] [PubMed] [Google Scholar]
- 12.Tesmer LA, Lundy SK, Sarkar S, Fox DA. Th17 cells in human disease. Immunol Rev. 2008;223:87–113. doi:IMR628 [pii] 10.1111/j.1600-065X.2008.00628.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jin W, Dong C. IL-17 cytokines in immunity and inflammation. Emerg Microbes Infect. 2013;2(9):e60. doi: 10.1038/emi.2013.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wei L, Laurence A, Elias KM, O’Shea JJ. IL-21 is produced by Th17 cells and drives IL-17 production in a STAT3-dependent manner. J Biol Chem. 2007;282(48):34605–10. doi: 10.1074/jbc.M705100200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huber M, Heink S, Pagenstecher A, Reinhard K, Ritter J, Visekruna A et al. IL-17A secretion by CD8+ T cells supports Th17-mediated autoimmune encephalomyelitis. J Clin Invest. 2013;123(1):247–60. doi: 10.1172/JCI63681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Saleh MA, Norlander AE, Madhur MS. Inhibition of Interleukin-17A, But Not Interleukin-17F, Signaling Lowers Blood Pressure, and Reduces End-Organ Inflammation in Angiotensin II–Induced Hypertension. JACC: Basic to Translational Science. 2016;1(7):606–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- •17.Khairallah C, Chu TH, Sheridan BS. Tissue Adaptations of Memory and Tissue-Resident Gamma Delta T Cells. Front Immunol. 2018;9:2636. doi: 10.3389/fimmu.2018.02636. [DOI] [PMC free article] [PubMed] [Google Scholar]; Review on gamma-delta T cells and their integral role in maintaining epithelial and mucosal barrier integrity, tissue homeostatis, and pathogen control. In certain disease states these cells can contribute to inflammation and disease progression.
- 18.Akitsu A, Iwakura Y. Interleukin-17-producing gammadelta T (gammadelta17) cells in inflammatory diseases. Immunology. 2018;155(4):418–26. doi: 10.1111/imm.12993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.••.Higaki A, Mahmoud AUM, Paradis P, Schiffrin EL. Role of Interleukin-23/Interleukin-17 Axis in T-Cell Mediated Actions in Hypertension. Cardiovasc Res. 2020. doi: 10.1093/cvr/cvaa257. [DOI] [PubMed] [Google Scholar]; A thorough review on the IL-23/IL-17 axis in hypertension. The IL-23/IL-17 axis may offer promising therepeutics in the treatment of hypertension.
- 20.Dale BL, Pandey AK, Chen Y, Smart CD, Laroumanie F, Ao M et al. Critical role of Interleukin 21 and T follicular helper cells in hypertension and vascular dysfunction. JCI insight. 2019;5:e129278. doi: 10.1172/jci.insight.129278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gladiator A, LeibundGut-Landmann S. Innate lymphoid cells: new players in IL-17-mediated antifungal immunity. PLoS Pathog. 2013;9(12):e1003763. doi: 10.1371/journal.ppat.1003763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nguyen H, Chiasson VL, Chatterjee P, Kopriva SE, Young KJ, Mitchell BM. Interleukin-17 causes Rho-kinase-mediated endothelial dysfunction and hypertension. Cardiovasc Res. 2013;97(4):696–704. doi: 10.1093/cvr/cvs422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wu J, Thabet SR, Kirabo A, Trott DW, Saleh MA, Xiao L et al. Inflammation and mechanical stretch promote aortic stiffening in hypertension through activation of p38 mitogen-activated protein kinase. Circ Res. 2014;114(4):616–25. doi: 10.1161/CIRCRESAHA.114.302157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.•.Orejudo M, Garcia-Redondo AB, Rodrigues-Diez RR, Rodrigues-Diez R, Santos-Sanchez L, Tejera-Munoz A et al. Interleukin-17A induces vascular remodeling of small arteries and blood pressure elevation. Clin Sci (Lond). 2020;134(5):513–27. doi: 10.1042/CS20190682. [DOI] [PubMed] [Google Scholar]; These studies demonstrate that IL-17A contributes to increased blood pressure through the remodeling of small mesenteric arteries and increased arterial stiffness.
- 25.Schuler R, Efentakis P, Wild J, Lagrange J, Garlapati V, Molitor M et al. T Cell-Derived IL-17A Induces Vascular Dysfunction via Perivascular Fibrosis Formation and Dysregulation of (.)NO/cGMP Signaling. Oxid Med Cell Longev. 2019;2019:6721531. doi: 10.1155/2019/6721531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xu L, Ding W, Stohl LL, Zhou XK, Azizi S, Chuang E et al. Regulation of T helper cell responses during antigen presentation by norepinephrine-exposed endothelial cells. Immunology. 2018;154(1):104–21. doi: 10.1111/imm.12871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Loperena R, Van Beusecum JP, Itani HA, Engel N, Laroumanie F, Xiao L et al. Hypertension and increased endothelial mechanical stretch promote monocyte differentiation and activation: Roles of STAT3, interleukin 6 and hydrogen peroxide. Cardiovasc Res. 2018. doi: 10.1093/cvr/cvy112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chiasson VL, Pakanati AR, Hernandez M, Young KJ, Bounds KR, Mitchell BM. Regulatory T-Cell Augmentation or Interleukin-17 Inhibition Prevents Calcineurin Inhibitor-Induced Hypertension in Mice. Hypertension. 2017;70(1):183–91. doi: 10.1161/HYPERTENSIONAHA.117.09374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Orejudo M, Rodrigues-Diez RR, Rodrigues-Diez R, Garcia-Redondo A, Santos-Sanchez L, Randez-Garbayo J et al. Interleukin 17A Participates in Renal Inflammation Associated to Experimental and Human Hypertension. Front Pharmacol. 2019;10:1015. doi: 10.3389/fphar.2019.01015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rudemiller NP, Patel MB, Zhang JD, Jeffs AD, Karlovich NS, Griffiths R et al. C-C Motif Chemokine 5 Attenuates Angiotensin II-Dependent Kidney Injury by Limiting Renal Macrophage Infiltration. Am J Pathol. 2016;186(11):2846–56. doi: 10.1016/j.ajpath.2016.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Alsheikh AJ, Dasinger JH, Abais-Battad JM, Fehrenbach DJ, Yang C, Cowley AW Jr. et al. CCL2 mediates early renal leukocyte infiltration during salt-sensitive hypertension. Am J Physiol Renal Physiol. 2020;318(4):F982–F93. doi: 10.1152/ajprenal.00521.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Marko L, Kvakan H, Park JK, Qadri F, Spallek B, Binger KJ et al. Interferon-gamma signaling inhibition ameliorates angiotensin II-induced cardiac damage. Hypertension. 2012;60(6):1430–6. doi: 10.1161/HYPERTENSIONAHA.112.199265. [DOI] [PubMed] [Google Scholar]
- 33.Krebs CF, Lange S, Niemann G, Rosendahl A, Lehners A, Meyer-Schwesinger C et al. Deficiency of the interleukin 17/23 axis accelerates renal injury in mice with deoxycorticosterone acetate+angiotensin ii-induced hypertension. Hypertension. 2014;63(3):565–71. doi: 10.1161/HYPERTENSIONAHA.113.02620. [DOI] [PubMed] [Google Scholar]
- 34.Kamat NV, Thabet SR, Xiao L, Saleh MA, Kirabo A, Madhur MS et al. Renal transporter activation during angiotensin-II hypertension is blunted in interferon-gamma−/− and interleukin-17A−/− mice. Hypertension. 2015;65(3):569–76. doi: 10.1161/HYPERTENSIONAHA.114.04975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ellison DH. Ubiquitylation and the pathogenesis of hypertension. J Clin Invest. 2013;123(2):546–8. doi: 10.1172/JCI66882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.••.Norlander AE, Madhur MS. Inflammatory Cytokines Regulate Renal Sodium Transporters: How, Where, and Why? Am J Physiol Renal Physiol. 2017:ajprenal 00465 2016. doi: 10.1152/ajprenal.00465.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]; This review summarizes how inflammatory cytokines regulate sodium transporters along the nephron.
- 37.Elliott P, Dyer A, Stamler R. The INTERSALT study: results for 24 hour sodium and potassium, by age and sex. INTERSALT Co-operative Research Group. J Hum Hypertens. 1989;3(5):323–30. [PubMed] [Google Scholar]
- 38.Kleinewietfeld M, Manzel A, Titze J, Kvakan H, Yosef N, Linker RA et al. Sodium chloride drives autoimmune disease by the induction of pathogenic TH17 cells. Nature. 2013;496(7446):518–22. doi: 10.1038/nature11868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wu C, Yosef N, Thalhamer T, Zhu C, Xiao S, Kishi Y et al. Induction of pathogenic TH17 cells by inducible salt-sensing kinase SGK1. Nature. 2013;496(7446):513–7. doi: 10.1038/nature11984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.••.Norlander AE, Saleh MA, Pandey AK, Itani HA, Wu J, Xiao L et al. A salt-sensing kinase in T lymphocytes, SGK1, drives hypertension and hypertensive end-organ damage. JCI Insight. 2017;2(13). doi: 10.1172/jci.insight.92801. [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper demonstrates a mechanism by which T cells sense salt and describes how deletion of the salt-sensing kinase SGK1 in T cells provides significant protection from hypertenive end-organ damage.
- 41.•.Van Beusecum JP, Barbaro NR, McDowell Z, Aden LA, Xiao L, Pandey AK et al. High Salt Activates CD11c(+) Antigen-Presenting Cells via SGK (Serum Glucocorticoid Kinase) 1 to Promote Renal Inflammation and Salt-Sensitive Hypertension. Hypertension. 2019;74(3):555–63. doi: 10.1161/HYPERTENSIONAHA.119.12761. [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper describes a salt sensing mechanism in dendritic cells and how dendritic cells contribute to the development of hypertension.
- 42.••.Yang YH, Istomine R, Alvarez F, Al-Aubodah TA, Shi XQ, Takano T et al. Salt Sensing by Serum/Glucocorticoid-Regulated Kinase 1 Promotes Th17-like Inflammatory Adaptation of Foxp3(+) Regulatory T Cells. Cell Rep. 2020;30(5):1515–29 e4. doi: 10.1016/j.celrep.2020.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]; In an SGK1 dependent manner, high salt drives Treg cells to adapt a Th17-like cell phenotype. This paper highlights the plasticity of T cells and the role of salt in inflammation.
- 43.Matthias J, Heink S, Picard F, Zeitrag J, Kolz A, Chao YY et al. Salt generates antiinflammatory Th17 cells but amplifies pathogenicity in proinflammatory cytokine microenvironments. J Clin Invest. 2020;130(9):4587–600. doi: 10.1172/JCI137786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kopp C, Linz P, Dahlmann A, Hammon M, Jantsch J, Muller DN et al. 23Na magnetic resonance imaging-determined tissue sodium in healthy subjects and hypertensive patients. Hypertension. 2013;61(3):635–40. doi: 10.1161/HYPERTENSIONAHA.111.00566. [DOI] [PubMed] [Google Scholar]
- 45.Machnik A, Neuhofer W, Jantsch J, Dahlmann A, Tammela T, Machura K et al. Macrophages regulate salt-dependent volume and blood pressure by a vascular endothelial growth factor-C-dependent buffering mechanism. Nat Med. 2009;15(5):545–52. doi: 10.1038/nm.1960. [DOI] [PubMed] [Google Scholar]
- 46.Rossitto G, Mary S, Chen JY, Boder P, Chew KS, Neves KB et al. Tissue sodium excess is not hypertonic and reflects extracellular volume expansion. Nature communications. 2020;11(1):4222. doi: 10.1038/s41467-020-17820-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Douzandeh-Mobarrez B, Kariminik A. Gut Microbiota and IL-17A: Physiological and Pathological Responses. Probiotics Antimicrob Proteins. 2019;11(1):1–10. doi: 10.1007/s12602-017-9329-z. [DOI] [PubMed] [Google Scholar]
- 48.Abusleme L, Moutsopoulos NM. IL-17: overview and role in oral immunity and microbiome. Oral Dis. 2017;23(7):854–65. doi: 10.1111/odi.12598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hooper LV, Littman DR, Macpherson AJ. Interactions between the microbiota and the immune system. Science. 2012;336(6086):1268–73. doi: 10.1126/science.1223490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chu F, Shi M, Lang Y, Shen D, Jin T, Zhu J et al. Gut Microbiota in Multiple Sclerosis and Experimental Autoimmune Encephalomyelitis: Current Applications and Future Perspectives. Mediators Inflamm. 2018;2018:8168717. doi: 10.1155/2018/8168717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang X, Zhang D, Jia H, Feng Q, Wang D, Liang D et al. The oral and gut microbiomes are perturbed in rheumatoid arthritis and partly normalized after treatment. Nat Med. 2015;21(8):895–905. doi: 10.1038/nm.3914. [DOI] [PubMed] [Google Scholar]
- 52.Sun Y, Chen Q, Lin P, Xu R, He D, Ji W et al. Characteristics of Gut Microbiota in Patients With Rheumatoid Arthritis in Shanghai, China. Front Cell Infect Microbiol. 2019;9:369. doi: 10.3389/fcimb.2019.00369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.•.Kim S, Goel R, Kumar A, Qi Y, Lobaton G, Hosaka K et al. Imbalance of gut microbiome and intestinal epithelial barrier dysfunction in patients with high blood pressure. Clin Sci (Lond). 2018;132(6):701–18. doi: 10.1042/CS20180087. [DOI] [PMC free article] [PubMed] [Google Scholar]; This manuscript highlights the association of intestinal barrier and microbiome dysfunction in human hypertension. Targeting the gut microbiome may serve as a potential therapeutic avenue to treat hypertension.
- 54.Li J, Zhao F, Wang Y, Chen J, Tao J, Tian G et al. Gut microbiota dysbiosis contributes to the development of hypertension. Microbiome. 2017;5(1):14. doi: 10.1186/s40168-016-0222-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yan Q, Gu Y, Li X, Yang W, Jia L, Chen C et al. Alterations of the Gut Microbiome in Hypertension. Front Cell Infect Microbiol. 2017;7:381. doi: 10.3389/fcimb.2017.00381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dan X, Mushi Z, Baili W, Han L, Enqi W, Huanhu Z et al. Differential Analysis of Hypertension-Associated Intestinal Microbiota. Int J Med Sci. 2019;16(6):872–81. doi: 10.7150/ijms.29322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Karbach SH, Schonfelder T, Brandao I, Wilms E, Hormann N, Jackel S et al. Gut Microbiota Promote Angiotensin II-Induced Arterial Hypertension and Vascular Dysfunction. Journal of the American Heart Association. 2016;5(9). doi: 10.1161/JAHA.116.003698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jama HA, Kaye DM, Marques FZ. The gut microbiota and blood pressure in experimental models. Curr Opin Nephrol Hypertens. 2019;28(2):97–104. doi: 10.1097/MNH.0000000000000476. [DOI] [PubMed] [Google Scholar]
- 59.Toral M, Robles-Vera I, de la Visitacion N, Romero M, Sanchez M, Gomez-Guzman M et al. Role of the immune system in vascular function and blood pressure control induced by faecal microbiota transplantation in rats. Acta Physiol (Oxf). 2019;227(1):e13285. doi: 10.1111/apha.13285. [DOI] [PubMed] [Google Scholar]
- 60.Wilck N, Matus MG, Kearney SM, Olesen SW, Forslund K, Bartolomaeus H et al. Salt-responsive gut commensal modulates TH17 axis and disease. Nature. 2017;551(7682):585–9. doi: 10.1038/nature24628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Itani HA, McMaster WG Jr., Saleh MA, Nazarewicz RR, Mikolajczyk TP, Kaszuba AM et al. Activation of Human T Cells in Hypertension: Studies of Humanized Mice and Hypertensive Humans. Hypertension. 2016;68(1):123–32. doi: 10.1161/HYPERTENSIONAHA.116.07237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yao W, Sun Y, Wang X, Niu K. Elevated Serum Level of Interleukin 17 in a Population With Prehypertension. J Clin Hypertens (Greenwich). 2015;17(10):770–4. doi: 10.1111/jch.12612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Masenga SK, Elijovich F, Hamooya BM, Nzala S, Kwenda G, Heimburger DC et al. Elevated Eosinophils as a Feature of Inflammation Associated With Hypertension in Virally Suppressed People Living With HIV. J Am Heart Assoc. 2020;9(4):e011450. doi: 10.1161/JAHA.118.011450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wang Z, Shi W, Liang X, Wang W, Liang J. Association of interleukin 17 / angiotensin II with refractory hypertension risk in hemodialysis patients. Afr Health Sci. 2016;16(3):766–71. doi: 10.4314/ahs.v16i3.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ji Q, Cheng G, Ma N, Huang Y, Lin Y, Zhou Q et al. Circulating Th1, Th2, and Th17 Levels in Hypertensive Patients. Dis Markers. 2017;2017:7146290. doi: 10.1155/2017/7146290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.••.von Stebut E, Reich K, Thaci D, Koenig W, Pinter A, Korber A et al. Impact of Secukinumab on Endothelial Dysfunction and Other Cardiovascular Disease Parameters in Psoriasis Patients over 52 Weeks. J Invest Dermatol. 2019;139(5):1054–62. doi: 10.1016/j.jid.2018.10.042. [DOI] [PubMed] [Google Scholar]; Secukinumab, a monoclonal antibody against IL-17A, has shown high efficacy in treatment against psoriasis. This randomized, double-blinded, placebo-controlled trial indicates secukinumab may also have beneficial effects on cardiovascular risk through its effect on flow mediated dilation.
- 67.Han L, Yang J, Wang X, Li D, Lv L, Li B. Th17 cells in autoimmune diseases. Front Med. 2015;9(1):10–9. doi: 10.1007/s11684-015-0388-9. [DOI] [PubMed] [Google Scholar]
- 68.Hueber W, Patel DD, Dryja T, Wright AM, Koroleva I, Bruin G et al. Effects of AIN457, a fully human antibody to interleukin-17A, on psoriasis, rheumatoid arthritis, and uveitis. Sci Transl Med. 2010;2(52):52ra72. doi: 10.1126/scitranslmed.3001107. [DOI] [PubMed] [Google Scholar]
- 69.Papp KA, Langley RG, Sigurgeirsson B, Abe M, Baker DR, Konno P et al. Efficacy and safety of secukinumab in the treatment of moderate-to-severe plaque psoriasis: a randomized, double-blind, placebo-controlled phase II dose-ranging study. Br J Dermatol. 2013;168(2):412–21. doi: 10.1111/bjd.12110. [DOI] [PubMed] [Google Scholar]
- 70.Elnabawi YA, Dey AK, Goyal A, Groenendyk JW, Chung JH, Belur AD et al. Coronary artery plaque characteristics and treatment with biologic therapy in severe psoriasis: results from a prospective observational study. Cardiovasc Res. 2019;115(4):721–8. doi: 10.1093/cvr/cvz009. [DOI] [PMC free article] [PubMed] [Google Scholar]