

In situ cancer vaccination by lipidoid nanoparticles enhances antigen cross-presentation and STING activation.
Abstract
In situ vaccination is a promising strategy for cancer immunotherapy owing to its convenience and the ability to induce numerous tumor antigens. However, the advancement of in situ vaccination techniques has been hindered by low cross-presentation of tumor antigens and the immunosuppressive tumor microenvironment. To balance the safety and efficacy of in situ vaccination, we designed a lipidoid nanoparticle (LNP) to achieve simultaneously enhancing cross-presentation and STING activation. From combinatorial library screening, we identified 93-O17S-F, which promotes both the cross-presentation of tumor antigens and the intracellular delivery of cGAMP (STING agonist). Intratumor injection of 93-O17S-F/cGAMP in combination with pretreatment with doxorubicin exhibited excellent antitumor efficacy, with 35% of mice exhibiting total recovery from a primary B16F10 tumor and 71% of mice with a complete recovery from a subsequent challenge, indicating the induction of an immune memory against the tumor. This study provides a promising strategy for in situ cancer vaccination.
INTRODUCTION
The development of cancer vaccines has been researched for several decades and is considered to be a powerful strategy for cancer treatment (1). Although this approach shows considerable promise for the eradication of cancer, only one cell-based vaccine has been approved by the U.S. Food and Drug Administration (FDA) to date, and the overall rate of clinical benefit is still low (2, 3). Two major obstacles have hindered the development of cancer vaccines: the high variability of tumor-associated antigens (TAAs) in different tumors and even in different patients with the same tumor and the immunosuppressive tumor microenvironments (4). Although the personalized vaccines based on neoantigens and immune checkpoint inhibitor have been developed to overcome these challenges, these procedures were complex, costly, and only effective in small populations (5, 6). In situ vaccination is considered a promising alternative cancer vaccination strategy, which does not require the identification and isolation of patient-specific TAAs (7). By local administration of therapeutic or immunomodulatory agents, in situ vaccination aims to activate the immunological response in the tumor microenvironment, reversing the immunosuppressive tendencies of the tumor, and to harness the abundant TAAs already found in the tumor. As the TAAs naturally present on the tumors are used directly, this approach completely avoids the challenges of identifying a patient’s unique individual TAAs and synthesizing a custom drug strategy, thus offering the possibility of much wider therapeutic application than has previously been possible with cancer vaccines.
Activation of the tumor immunological response via delivery of the oncolytic virus has been the most common method for in situ vaccination and has reached the stage of clinical trials for the treatment of metastatic melanoma (8, 9). However, the excessive systemic activation of the immune system by the oncolytic virus, which might lead to severe side effects such as the cytokine release syndrome (CRS), has become a major concern about the application of this strategy in humans (10). To date, some other in situ vaccination strategies, including photothermal therapy (PTT), radiation therapy (RT), agonist immunotherapy, and even in situ chemotherapy, were also shown to induce effective immune response (11–15). However, the PTT, RT, and chemotherapy strategies were only efficient in the production of tumor antigens, achieved via induction of immunogenetic cell death. The delivery and presentation of these released antigens are still limited by immune tolerance of the tumor microenvironments (16). The in situ application of immune agonists can boost the immune response but is deficient in the production of tumor antigens (17). An optimal therapeutic strategy would be able to combine these two effects. To generate an effective antitumor immune response, an in situ vaccine should ideally be able to induce immunogenic cancer cell death, facilitate the release of TAAs, enhance antigen presentation, and promote the activation of antitumor T cell responses.
Herein, we designed a lipidoid-based nanosystem that can enhance the effect of in situ vaccination by both promoting cross-presentation of TAAs and activating the so-called stimulator of interferon (IFN) genes (STING) pathway. As shown in Fig. 1, the primary tumors are injected with a small dose of doxorubicin (DOX), which induces tumor immunogenetic death and the release of TAAs (18). Then, lipid nanoparticles are formulated and loaded ex vivo with 2′5′-3′5′ cyclic guanosine monophosphate-adenosine monophosphate (cGAMP), an agonist of the STING pathway. The cGAMP-loaded lipidoid nanoparticles (LNPs/cGAMP) are injected to the apoptotic site of tumor. We hypothesize that in situ, the LNP/cGAMP could capture the released tumor antigens via electrostatic interaction, and subsequently, the tumor antigens and cGAMP-loaded LNPs are internalized by the antigen-presenting cells (APCs) through enhanced endocytosis, where the cargo TAAs and cGAMP are released into cytoplasm of the APCs via endo/lysosome escape effect of LNPs. The released tumor antigens are degraded by ubiquitin-proteasome system and presented by the major histocompatibility complex (MHC) class I to activate T cells (19), while the released cGAMP in the cytoplasm activates the STING pathway and induces the production of type I IFN and other proinflammatory cytokines, which also promotes activation of T cells (20). The integration of both enhanced antigen cross-presentation and STING activation can promote the in situ vaccination for tumor immunotherapy. Comparing with oncolytic virus–based cancer immunotherapy, the synthetic lipid nanoparticles have considerable advantages, including a better safety profile, and ease of production scale-up. Comparing with other non–viral-based in situ vaccination systems, the present system has the advantage of antigen capturing, delivery, and cross-presentation and thus provides better therapeutic effects.
Fig. 1. The scheme illustration of LNP system–mediated antigen capturing, cross-presentation, and STING activation.
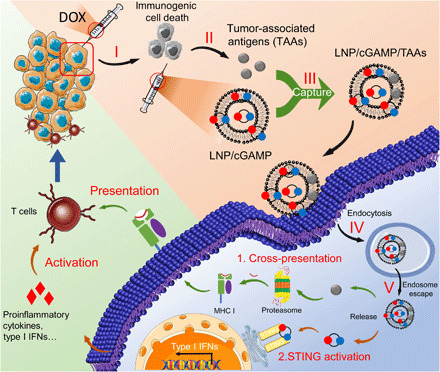
(I) Low dose of DOX-induced immunogenic cancer cell death. (II) TAAs were released after the administration of low dose of DOX. (III) The released TAAs were captured by lipidoid nanoparticle (LNP)/2′5′-3′5′ cyclic guanosine monophosphate-adenosine monophosphate (cGAMP). (IV) The TAAs and cGAMP encapsulated in LNPs were delivered into APCs via endocytosis. (V) The TAAs and cGAMP escaped from endo/lysosomes to cytoplasm for further cross-presentation and STING activation.
RESULTS
Combinatorial library screening of LNPs with adjuvant effect and enhanced cross-presentation
First, we selected a lipidoid to serve as the basis of the cancer vaccine. The composition and structure of lipidoids greatly influence the adjuvant effect of the LNPs, including the antigen delivery and immunostimulation properties of the nanoparticles. We have been using the combinatorial library strategy to develop synthetic lipidoids with various structures and properties for drug delivery (21–23). An ideal lipid nanoparticle for cancer immunotherapy should be able to (i) capture the released tumor antigen and deliver into APCs with enhanced cross-presentation and (ii) generate immunostimulatory effect. To identify the effective lipidoids, a rough screening of a selected library was carried out by evaluating the antibody response in C57BL/6 mice after immunization with the model antigen ovalbumin (OVA) formulated with different LNPs (Fig. 2A). On the basis of our previous experience with these lipidoids, we chose 18 lipidoids with varied heads and tails (fig. S2A) to formulate with OVA. Mice were immunized via a prime-boost immunization strategy with two injections at days 1 and 14, respectively (24). As shown in fig. S2B, lipopolysaccharide (LPS)/OVA was used as the positive control and showed extremely positive immunoglobulin G1 (IgG1) antibody response. However, OVA alone without LNP showed very low antibody response due to its low immunogenicity. Two LNPs from our library screening showed a particularly strong antibody response; both of these lipidoids compose the amine head 93 (fig. S2A), and we named the successful lipidoids as 93-O17S-F and 93-O17O-F. We further evaluated the detailed IgG1 and IgG2c antibody response of these lipidoid formulations. As shown in Fig. 2 (B and C), the total IgG and IgG1 response is consistent with the result from rough screening. The three leading LNPs showed much stronger antibody response than OVA alone, as well as the FDA-approved adjuvant alum (25). However, only the 93-O17S-F generated high IgG2c response, representing the activation of T helper 1 cells (TH1) (Fig. 2D). Moreover, the excipients in the LNP formulation also greatly affected the antibody production. Without the helper lipids including cholesterol and dioleoylphosphatidylethanolamine (DOPE), the 93-O17O showed much less antibody response compared with the formulated one (fig. S2B).
Fig. 2. The adjuvant effect and enhanced cross-presentation of LNP.
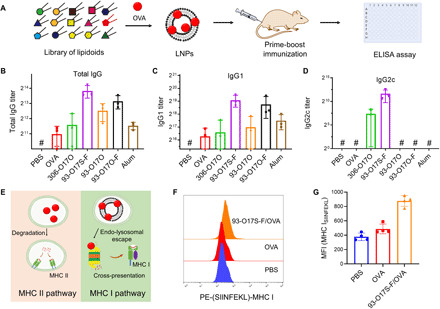
(A) The approach for the screening of LNP library by prime-boost route. (B to D) The OVA-specific immunoglobulin G (IgG) (B), IgG1 (C), and IgG2c (D) antibody titers after immunization with OVA-loaded LNPs. n = 3. #The titer was lower than the minimal dilution. (E) The enhanced cytoplasmic delivery of antigens by LNPs up-regulated the cross-presentation. (F) Typical flow cytometry data of the expression of SIINFEKL–MHC I complex on DC2.4 cells after incubation of different formulation of OVA. (G) The MFI of labeled SIINFEKL–MHC I complex calculated by flow cytometry. n = 4.
In addition to the successful activation of both CD4+ TH1 and TH2, a successful LNP cancer vaccine should also enhance activation of CD8+ T cell via cross-presentation. Briefly, as shown in Fig. 2E, it is understood that free antigens, such as OVA, may be internalized by APCs, degraded by enzymes in the lysosome and bound to the MHC class II complex, where it can be presented on the surface of the APC to stimulate CD4+ T cells and generate a primarily antibody-based immune response. By contrast, if the antigen can be delivered to the cytosol of the APCs, then it could instead be degraded by the proteasome and incorporated into the MHC class I molecules, where it is cross-presented to cytotoxic CD8+ T cells instead. The CD8+ T cell response is known to be critical to cancer immunotherapy. To assess the ability of the 93-O17S-F LNPs to stimulate CD8+ T cells, we used an antibody staining assay, with an antibody (H-2Kb bound to SIINFEKL) that specifically binds only to MHC class I molecules presenting a fragment of OVA (26). We delivered model OVA antigen to DC2.4 cells using 93-O17S-F in vitro. The DC2.4 cells treated with free OVA were used as control. Twenty-four hours after antigen delivery, cells were stained with fluorescent-tagged anti–H-2Kb-SIINFEKL antibodies, and the fluorescence intensity was measured via flow cytometry. A rightward shift of the fluorescence peak, indicating the increased fluorescence, was observed in the 93-O17S-F/OVA–treated cells compared with free OVA–treated cells (Fig. 2F). The mean fluorescence intensity (MFI) of cells was also calculated by flow cytometry, which confirmed the enhanced expression of SIINFEKL–MHC class I molecules in 93-O17S-F/OVA–treated cells (Fig. 2G), with 93-O17S-F/OVA–treated cells showing an MFI of about 1.8 times higher than the MFI of the free OVA group.
Cytoplasmic delivery of cGAMP by 93-O17S-F to promote STING activation
To further enhance the immune stimulation for cancer immunotherapy, we chose cGAMP, an agonist for the STING pathway, to be encapsulated in the 93-O17S-F for intracellular delivery. The recognition of cGAMP by STING has been demonstrated to result in the activation of APCs, the production of IFNs, and the priming of CD8+ T cells against tumor antigens, which was shown to be critically important in cancer immunotherapy (27, 28). However, cGAMP itself is not able to freely cross the cell membrane to reach the STING promoters on the endoplasmic reticulum. We hypothesize that ionized LNPs such as 93-O17S-F can serve as the carrier for cGAMP through electrostatic interaction and facilitate its intracellular delivery to activate the STING pathway (Fig. 3A). To test the hypothesis, we studied the intracellular distribution of cGAMP by confocal laser scanning microscopy using the fluoresceinyl-labeled cGAMP (cGAMPFluo). The cGAMPFluo was encapsulated into 93-O17S-F by simple mixing and then added into the medium of RA264.7 and DC2.4 cells in vitro at the dose equivalent to cGAMPFluo (200 ng/ml). Figure 3B showed the enhanced endocytosis and endo/lysosome escape of cGAMP delivered by 93-O17S-F. After 4 hours of incubation, cells treated with cGAMPFluo encapsulated in 93-O17S-F showed a strong green signal throughout the cytoplasm in both RAW264.7 and DC2.4 cells. However, there is almost no green signal of the cGAMPFluo in the cells treated with free cGAMPFluo owing to its low cell membrane permeability. Notably, cells were counterstained with LysoTracker, which visualizes the endo/lysosome. Figure 3B demonstrates that the green signal (indicating the subcellular distribution of cGAMPFluo) was not limited to the red-labeled endo/lysosome and instead was distributed throughout the cytosol. This demonstrates that the cGAMP could escape from the endo/lysosome into the cytoplasm of both RAW264.7 and DC2.4 cells.
Fig. 3. Enhanced STING activation by cytoplasmic delivery of cGAMP in vitro.
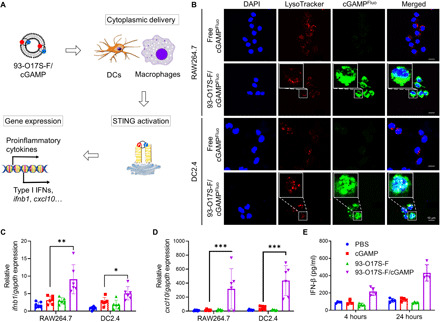
(A) The activation of STING pathway by cytoplasmic delivery of cGAMP using 93-O17S-F. (B) Subcellular distribution of cGAMPFluo and lysosome in RAW264.7 and DC2.4 cells after incubation of free cGAMPFluo or 93-O17S-F/cGAMPFluo for 4 hours. (C and D) Relative expressions of ifnb1 (C) and cxcl10 (D) genes in RAW264.7 and DC2.4 cells after incubation of 93-O17S-F/cGAMP for 4 hours. n = 6, *P ≤ 0.05, **P ≤ 0.01, and ***P ≤ 0.001. (E) The concentration of IFN-β in the medium of DC2.4 cells after incubation of 93-O17S/cGAMP for 4 and 24 hours. n = 4.
To evaluate the enhanced STING activation by the cytoplasmic delivery of cGAMP using the LNPs, we measured the expression of ifnb1 and cxcl10 genes by real-time polymerase chain reaction (RT-PCR) in RAW264.7 and DC2.4 cells. The ifnb1 and cxcl10 genes are two of the main genes related to the activation of STING, and their expression results in abundant secretion of type I IFNs and proinflammatory cytokines (29). As shown in Fig. 3C, the expressions of ifnb1 genes in both RAW264.7 and DC2.4 cells treated with 93-O17S-F/cGAMP were about 6.9- and 6.4-fold higher than those of the cells treated with phosphate-buffered saline (PBS). Free cGAMP only showed modest increase in the expression of ifnb1 due to its low penetration of cell membrane. The similar trend was also observed in the expression of cxcl10 gene in Fig. 3D. The 93-O17S-F/cGAMP generated markedly increased expression (more than 100-fold) of cxcl10 gene, further confirming the activation of STING pathway. Last, the secretion of IFN-β, a typical type I IFN, was measured after the treatment cGAMP to cells in Fig. 3E. Consistent with the gene expression data, the 93-O17S-F/cGAMP–treated cells induced higher concentration of secreted IFN-β, when compared with cells treated with PBS, free cGAMP, or empty 93-O17S-F LNPs. Furthermore, the secretion of IFN-β by 93-O17S-F/cGAMP–treated DC2.4 cells continued to increase over the course of 24 hours, while the concentration of IFN-β in other groups remained almost unchanged.
Improved humoral and cellular immune response by co-encapsulation of cGAMP into LNPs
We showed that lipidoid formulations enabled the intracellular delivery of both OVA protein (Fig. 2) and STING agonist cGAMP (Fig. 3). We speculate that these LNP formulations can facilitate the codelivery of cGAMP and OVA, which could further enhance the immune response against OVA. We chose 93-O17O-F and 93-O17S-F, produced the LNP/OVA formulation with or without cGAMP, and immunized the mice using the same prime and boost vaccination procedure shown in Fig. 2A. OVA prepared in the adjuvant alum, with and without cGAMP, were also used as controls. The serum antibody levels against OVA in immunized mice were evaluated using enzyme-linked immunosorbent assay (ELISA), as shown in Fig. 4A. Similar to the results shown in Fig. 2B, the mice immunized with LNP/OVA formulations showed higher antibody titers in IgG, IgG1, and IgG2c than with OVA with alum adjuvant. Incorporating cGAMP into the vaccine formulation generally increased the IgG, IgG1, and IgG2c antibody responses in all groups. Notably, however, the relative efficacies of each of the delivery formulations remained the same after the incorporation of cGAMP. For example, the LNP 93-O17S-F was the most effective formulation for eliciting an immune response in both the OVA single-delivery paradigm and in the OVA/cGAMP codelivery paradigm. Similarly, 93-O17O-F was the intermediate condition in both paradigms, and alum was the least effective condition in both paradigms. Both alum-formulated and 93-O17O-F–formulated OVA single deliveries failed to stimulate IgG2c production, yet successfully elicited IgG2c response when cGAMP was included in the vaccine formulation. The enhanced IgG2c response showed the inclusion of cGAMP in the vaccine formulation up-regulates CD4+ TH1 activation, which is important for the generation of memory T cells and the activation of macrophages (30).
Fig. 4. Enhanced humoral and cellular immune response by codelivery of cGAMP.
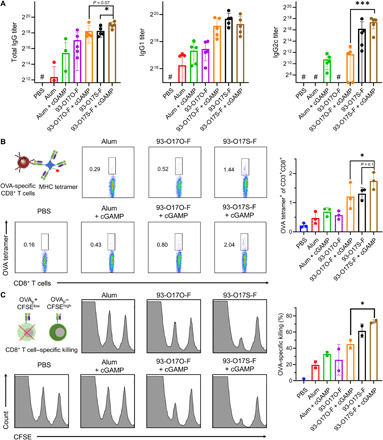
(A) The OVA-specific IgG, IgG1, and IgG2c antibody titers after immunization. #The titer was lower than the minimal dilution. n = 5, *P ≤ 0.05, ***P ≤ 0.001. (B) The representative flow images and the quantitated percentages of OVA-peptide (OVAp)–specific CD8+ T cells in spleen of the vaccinated mice. n = 3, *P ≤ 0.05. (C) The representative flow images and the quantitated percentages of OVA-specific killing by CD8+ T cells in spleen of the vaccinated mice. n = 2, *P ≤ 0.05.
To generate effective immunotherapy against cancer, it is crucial to induce cellular immune response mediated by CD8+ T cells after vaccination (31). SIINFEKL-specific H-2Kb tetramer can be used to label OVA-specific CD8+ T cells, which then can be counted using flow cytometer. Using this approach, we analyzed the percentage of OVA-specific CD8+ T cells in the spleen of the mice immunized with various formulations. As shown in Fig. 4B, mice immunized with OVA in alum, alum + cGAMP, and 93-O17O-F showed very low population (less than 0.6%) of OVA-specific CD8+ T cells in spleen. The inclusion of cGAMP in 93-O17O-F/OVA slightly increased the percentage (0.8%) of OVA-specific CD8+ T cells. However, the percentage of OVA-specific CD8+ T cells in the spleen of the mice immunized with OVA in 93-O17S-F or 93-O17S-F + cGAMP significantly increased and reached to about 1.4 and 1.8%, respectively. These results showed that 93-O17S-F enables the efficient CD8+ T cell activation, which can be further enhanced by codelivering cGAMP in the vaccine formulation.
We further evaluated the capability of the OVA-specific CD8+ T cells in lysing SIINFEKL peptide–loaded splenocytes in vivo, using an approach described in literature (32). The splenocytes harvested from naive mice were divided into two groups. One group was pulsed with SIINFEKL peptide and labeled with low concentration of carboxyfluorescein succinimidyl ester (CFSE). The other group was labeled with high concentration of CFSE only. We then injected both groups of splenocytes back to the mice immunized with OVA in different formulations. The OVA-specific CD8+ T cell could specifically kill the group of splenocytes pulsed with SIINFEKL peptide (lower fluorescence signal due to low concentration of CFSE) but not the unpulsed group of splenocytes (higher fluorescence signal due to high concentration of CFSE). Figure 4C showed the flow cytometer results of the fluorescent cell populations in the spleen of immunized mice. We found that the mice immunized with OVA formulated in alum, alum + cGAMP, and 93-O17O-F showed less than 30% efficiency in lysing SIINFEKL-pulsed splenocytes, although the addition of cGAMP in OVA/93-O17O-F did increase the cell lysing capability (still lower than 50%) in the immunized mice. However, the mice immunized with OVA in 93-O17S-F in single delivery or codelivered with cGAMP showed significant (60 and 70%, respectively) OVA-specific cell killing. These cell killing results are consistent with the tetramer labeling study (Fig. 4B), both showing that the 93-O17S-F enables the efficient CD8+ T cell activation, which can be further enhanced by addition of the cGAMP into the vaccine formulation.
In vivo antigen uptake and immune activation by 93-O17S-F/cGAMP
One of the distinguishable advantages of this design is that the LNP can also capture and deliver the tumor antigens released from the tumor cells after the treatment with small amount of chemotherapeutics (Fig. 5A). The ability of the LNPs to capture TAAs was initially investigated in vitro, by incubating the LNPs with tumor lysate and analyzing the subsequent changes in size and zeta potential of the LNP. LNPs were mixed with tumor lysate at a range of w/w ratios, from 10:0 (100% LNP, no lysate) to 10:10 (equal weights LNP and lysate). As shown in Fig. 5B, the size of the LNP tumor lysate complex increased markedly as the weight fraction of the tumor lysate was increased. The maximum particle size was observed at a weight ratio of 10:6; subsequent addition of tumor lysate was unable to further increase the particle size. This is likely due to achieving the maximum complexation capacity of the LNPs. Figure S5 shows the transmission electron microscopy images of blank 93-O17S-F alone or complexed with tumor lysate. The blank 93-O17S-F showed well-separated spherical morphology, while the nanoparticles formed clusters after mixing with tumor lysates. This demonstrates a direct physical interaction between the LNP and tumor lysates. Furthermore, the zeta potential of 93-O17S-F decreased from positive charge to negative charge with the increase in weight ratio of tumor lysate. Notably, the capture of the tumor lysate did not abolish the capability of the LNP for the delivery of cGAMP. As shown in fig. S6, the cellular uptake of cGAMPFluo by RAW264.7 or DC2.4 cells did not change significantly when the weight ratio of lysate to LNP was under 0.6. When the weight ratio increased to 0.6 or higher, the MFI of cGAMPFluo only decreased about 20 and 10% in RAW264.7 and DC2.4 cells, respectively, indicating that the capture of antigens did not significantly affect the LNP-mediated intracellular delivery of cGAMP. Moreover, compared with free cGAMPFluo, 93-O17S-F showed the enhanced delivery of cGAMPFluo to the draining lymph node (DLN) (fig. S6C).
Fig. 5. LNP enhances STING activation and shifts immunocellular composition of the tumor microenvironment in vivo.
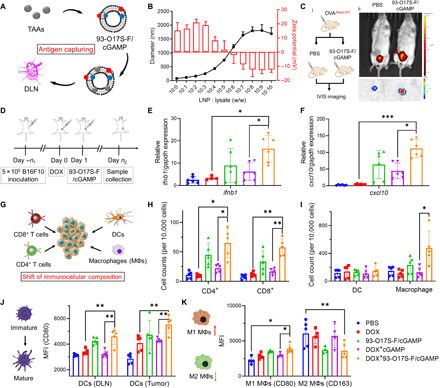
(A) Capture of the tumor antigens by 93-O17S-F. (B) The diameters and zeta potentials of 93-O17S-F and tumor lysate complex at different weight ratio. (C) Enhanced delivery of OVAAlexa647 to DLNs after being captured by 93-O17S-F in vivo. Photo credit: J.J. Chen, Tufts University. (D) The route of the in vivo STING activation experiments. (E and F) The relative expression of ifnb1 and cxcl10 genes in B16F10 tumors after the administration of 93-O17S-F/cGAMP for 6 hours. n = 6, *P ≤ 0.05 and ***P ≤ 0.001. (G) The activation of STING pathway recruited the immune cells to tumor sites. (H) The cell numbers of CD4+ and CD8+ T cells at tumor sites after the administration of 93-O17S-F/cGAMP for 48 hours. n = 5, *P ≤ 0.05 and **P ≤ 0.01. (I) The cell numbers of dendritic cells (DCs) and macrophages at tumor sites after the administration of 93-O17S-F/cGAMP for 48 hours. n = 5, *P ≤ 0.05. (J) The MFI of CD80 expressed on CD11c+MHC II+ DCs at DLNs and tumor sites. n = 5, **P ≤ 0.01. (K) The polarization of macrophages at tumor site determined by the MFI of CD80 and CD163 among CD11b+F4/80+ cells. n = 5, *P ≤ 0.05 and **P ≤ 0.01.
To evaluate whether such in antigen capture and delivery to DLN occurs in the complex in vivo environment, we developed an in vivo experiment for antigen capture and delivery using the Alexa Fluor 647–conjugated OVA (OVAAlexa-647) as a model antigen. Mice were injected subcutaneously with free OVAAlexa-647 in the right flank and immediately thereafter injected with either 93-O17S-F/cGAMP or PBS control at the same location [Fig. 5C (i)]. We hypothesize that the capture of free OVAAlexa-647 by our LNPs will serve to accurately model the ability of the LNPs to capture free TAAs after tumor cell death. Five hours after the second injection, the mice were imaged using the In Vivo Imaging System (IVIS; PerkinElmer). As shown in Fig. 5C (ii), when the second injection contained only PBS, the OVAAlexa-647 was mainly found in the bladder, suggesting the rapid clearance of the soluble protein through urine. By contrast, when the second injection contained 93-O17S-F/cGAMP, the fluorescence signal of OVAAlexa-647 was found in the DLNs. The DLNs of the two groups were harvested and imaged ex vivo using the IVIS [Fig. 5C (ii)]. The fluorescence intensity of the DLN from mouse treated with free OVAAlexa-647 followed by 93-O17S-F/cGAMP injection was much higher than that from mouse treated with free OVAAlexa-647 followed by PBS injection on the same spot. This result demonstrates that the 93-O17S-F can capture free proteins in the tissue and carry them to the DLN. We expect that these LNPs could similarly capture the tumor antigens from tumor lysate and present them to APCs residing in the DLN, which may enhance the antitumor effect and reduce the immune escape.
We further evaluated the in vivo STING activation using this LNP formulation in B16F10 subcutaneous tumor-bearing C57BL/6 mice, as illustrated in Fig. 5D. To generate the tumor model, 5 × 105 B16F10 cells were injected at the right flank of 4- to 6-week-old C57BL/6 mice. The tumors were allowed to grow up to 60 to 80 mm3 in volume, and then the mice were divided into five groups, each containing the same average tumor volume. At day 0, free DOX was injected directly into the tumors of mice from three groups to induce immunogenetic death and release large amount TAAs. PBS was injected into the other two groups. At day 1, DOX-treated tumor groups were injected with 93-O17S-F, free cGAMP, or 93-O17S-F/cGAMP formulation, injected into the same site where DOX was injected (these groups were termed as DOX, DOX + cGAMP, and DOX + 93-O17S-F/cGAMP, respectively). The other two groups were injected with PBS or 93-O17S-F/cGAMP (termed as PBS and 93-O17S-F/cGAMP, respectively). After 6 hours following the second injection, the tumors were collected and the expressions of ifnb1 and cxcl10 genes were analyzed by RT-PCR. As shown in Fig. 5 (E and F), comparing with tumor treated with DOX only, the expression of both ifnb1 and cxcl10 did not change significantly for tumor injected with PBS 1 day after the DOX injection. The tumor treated with free cGAMP 1 day after the DOX injection showed modest increase of these two genes, indicating that in situ administration of STING agonist only activates the STING pathway modestly. However, the 93-O17S-F/cGAMP–treated groups all showed increased expression of ifnb1 and cxcl10 genes compared with free cGAMP, regardless of whether DOX was administrated before the second injection or not. The administration of DOX also enhanced the activation of STING pathway by 93-O17S-F/cGAMP to some extent, which might be due to the released tumor antigens.
The successful activation of STING pathway by the LNP system could also change the immunocellular composition of the tumor microenvironment. As shown in Fig. 5G, it has been reported that the activation of STING pathway stimulated the secretion of proinflammatory factors, including multiple chemokines that could recruit various immune cells to tumor sites (33). To further evaluate the changes of the cellular composition after the treatment, we collected the tumors at 48 hours after the second injection. The tumors were dissociated to single cells, stained with antibodies, and analyzed by flow cytometry. The treatment with DOX alone did not make significant changes to the population of either CD4+ or CD8+ T cells (Fig. 5H). The free cGAMP–treated tumors exhibited a modest increase in both CD4+ and CD8+ T cells populations. However, after encapsulation into 93-O17S-F, significant increase in T cell populations was observed in 93-O17S-F/cGAMP–treated tumors, especially in the DOX-pretreated group. Moreover, the population of macrophages also showed similar trend with the population of T cells at tumor sites (Fig. 5I). However, the population of dendritic cells (DCs) showed no significant changes in any treated group.
The maturation of DCs (in both DLN and tumor) and polarization of macrophages (in tumor) were also evaluated (Fig. 5, J and K). The maturation of DCs in DLN and tumor was evaluated by the MFI of CD80 among the CD11c+MHC II+ cells (Fig. 5J). Comparing with the PBS-treated control group, all the treatment groups showed increased intensity of CD80 within the CD11c+MHC II+ cells inside the tumor. We found that only the group of mice treated with 93-O17S-F/cGAMP showed increased maturation of DCs in DLN, owing to the delivery of TAAs and cGAMP by LNPs in the DLN. The polarization of macrophages in tumor is important to cancer immunotherapy. We quantified the macrophage polarization inside tumor by labeling the macrophages with CD80 (proinflammatory M1-like macrophage) and CD163 (anti-inflammatory M2-like macrophage) among the CD11b+CD11c+ cells. The MFI was measured and shown in Fig. 5K. Among all treatment groups, 93-O17S-F/cGAMP in combination with the pretreatment with DOX led to the highest number of M1 macrophages and lowest number of M2 macrophages in tumor, which is consistent with the activation level of STING pathway. The enhanced maturation of DCs and the shifted M1-polarized macrophages both benefit the generation of strong adaptive immunity against tumor cells.
Therapeutic effect by in situ vaccination of 93-O17S-F/cGAMP
Last, the overall antitumor effect of the LNP system was evaluated in B16F10 allograft tumor model. As shown in Fig. 6A, B16F10 cells were injected subcutaneously into the right flank of the back of C57BL/6 mice. When the tumors reached 60 to 80 mm3 in volume, the mice were divided into five groups. Three groups were pretreated with DOX at day 0, and subsequently treated with either PBS, free cGAMP, or 93-O17S-F/cGAMP on days 1 and 5 (these groups were termed as DOX, DOX + cGAMP, and DOX +93-O17S-F/cGAMP, respectively). The other two groups did not receive a DOX pretreatment and only received PBS or 93-O17S-F/cGAMP on days 1 and 5 (termed as PBS and 93-O17S-F/cGAMP, respectively). On day 6, the mice were photographed to qualitatively assess the tumor progression (Fig. 6B). We observed small ulcerations on the tumors after treatment in every group, especially in the DOX + 93-O17S-F/cGAMP group. Among all experimental groups, the tumors in the DOX + 93-O17S-F/cGAMP group were the smallest, with some tumors reduced so small as to be undetectable by eye. The tumor volume was measured using calipers at 48-hour intervals from days 0 to 10 (Fig. 6C). The detailed tumor volumes of each mouse were shown in Fig. 6D. The tumor volumes of PBS-treated mice exceed 2000 mm3, the humane experimental end point, within 12 days. The free DOX only slightly postponed the rapid growth compared with that in the PBS group, but the tumor volumes still eventually reached 2000 mm3, and the tumor volume was not statistically reduced compared with the PBS group on day 10. The treatment with 93-O17S-F/cGAMP without pretreatment with DOX showed modest inhibition of tumor before day 8 but ultimately could not significantly reduce the tumor volume, and qualitatively, the tumors remained in an exponential growth paradigm. Mice pretreated with DOX that received cGAMP without the LNP carrier showed very similar results. By contrast, in the DOX + 93-O17S-F/cGAMP group, most of the tumor growth was significantly inhibited. At day 6, the tumor volume was significantly smaller than all other groups, and for two mice, the tumors were completely undetectable at the end of the experiment. The survival rate of mice during the treatment was shown in Fig. 6E. The mice treated with PBS all reached the humane end points within 14 days. The administration of free DOX, 93-O17S-F/cGAMP, or DOX + cGAMP only exhibited modest improvement of the survival rate, and all the mice reached the humane end points within 25 days (i.e., 0% survival rate). The mice treated with DOX + 93-O17S-F/cGAMP showed significantly extended survival rate and two of the seven mice showed eradication of tumors within 30 days (i.e., 28.6% survival rate). Together, these data confirm our hypothesis. Stimulation of the STING pathway via cGAMP delivery, either with or without pretreatment with DOX, provided modest but insufficient treatment for the tumor. Efficient antitumor activity was only achieved when DOX was pretreated to induce the release of TAAs, followed by delivery with our LNP to capture the TAAs for delivery to APCs, and codelivery with cGAMP to simultaneously activate the STING pathway.
Fig. 6. Antitumor therapeutic effect of 93-O17S-F/cGAMP.
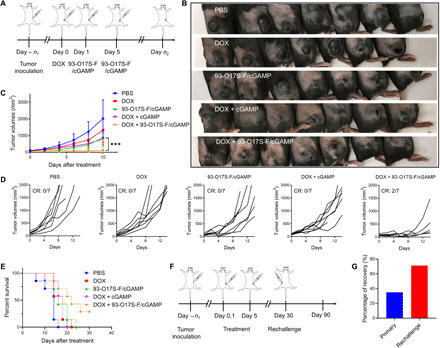
(A) The route of in situ vaccination by 93-O17S/cGAMP. (B) The photographs of B16F10 allograft model tumors at day 6. Photo credit: J.J. Chen, Tufts University. (C) The tumor volumes of B16F10 allograft model tumors after the treatment with different formulations. n = 7, ***P ≤ 0.001. (D) The individual tumor volumes after the treatment with different formulations. (E) The survival rates of mice bearing B16F10 allograft model tumors. (F) The route of tumor rechallenge assay. (G) The percentage of total recovery of primary and rechallenged tumor inoculated mice. For primary tumor, n = 20. For rechallenge, n = 7.
To investigate whether our LNP system could generate long-term immune memory, we conducted the rechallenge experiment shown in Fig. 6F. B16F10 tumor was established as previously described and allowed to grow up to 50 to 80 mm3. Then, the mice were pretreated with DOX at day 0 and treated with 93-O17S-F/cGAMP on days 1 and 5, as before. The tumors were monitored every 2 days and the mice that were demonstrated to be tumor free by day 30 were selected for the rechallenge experiment. As shown in Fig. 6G, the total recovery rate of the mice bearing primary tumors (that is, the percentage of mice that were demonstrated to be tumor free by day 30) was only about 35%, which is in line with the results from our initial antitumor experiment. We hypothesize that this low recovery rate is due to the well-known phenomenon of immune escape upon immunotherapy. However, among these mice who recovered from the initial tumor and were rechallenged with a new tumor, 71% of these exhibited total recovery 30 days after the rechallenge.
DISCUSSION
There are two major obstacles limiting the efficacy of the current in situ vaccination strategies, namely, low cross-presentation of tumor antigens and the immunosuppressive tumor microenvironment. Although the oncolytic virus is regarded as one of the most promising methods for in situ vaccination for cancer treatment, use of the oncolytic virus has been demonstrated to result in excessive activation of immune system and could result in severe side effects such as the CRS (34). Other in situ strategies, such as PTT, RT, agonist immunotherapy, and even in situ chemotherapy have limited effect on the cross-presentation of tumor antigens (35). Thus, developing a safe and effective in situ vaccination strategy integrated to both enhance the cross-presentation of antigens and activated tumor environment is highly desired for effective cancer immunotherapy.
In this work, we designed LNP-based in situ vaccination system with both enhanced antigen cross-presentation and STING activation. By screening a small library of cationic synthetic LNPs for delivering a model antigen OVA, we identified the 93-O17S-F, which showed excellent IgG1 and IgG2c antibody response. The IgG1 antibody response is mainly induced by CD4+ TH2, while IgG2c antibody is generated by CD4+ TH1 (36). TH1 activation is desired to activate macrophages and produce memory T cells, which is important for the killing of tumor cells, and thus IgG2c activity is an important marker for the development of a cancer vaccine. Moreover, 93-O17S-F LNP could also facilitate the cross-presentation of OVA on MHC class I molecules and subsequently activates cytotoxic CD8+ T cells. The CD8+ T cell response is critical for successful cancer immunotherapy. These results showed the excellent adjuvant effect and enhanced cross-presentation of antigen (OVA) delivered in 93-O17S-F.
STING pathway is shown to be critically important in cancer immunotherapy (37). Recently, many researchers focused on the delivery of cGAMP (STING agonist) to tumor site for the activation of tumor suppressive environment. However, the penetration of cGAMP to reach the STING promoters on the endoplasmic reticulum remains a challenge. Because of the cationic nature, the 93-O17S-F LNPs efficiently encapsulate cGAMP through electrostatic interaction and serve as a vehicle for its cytoplasmic delivery. In in vitro study using both RAW264.7 and DC2.4 cells, the cytoplasmic delivery of cGAMP using 93-O17S-F leads to the up-regulation of STING-activation related genes (ifnb1 and cxcl10) and proinflammatory factors (IFN-β). Further in vivo vaccination of 93-O17S-F/cGAMP also induces the increased expression of ifnb1 and cxcl10 genes, demonstrating that the 93-O17S-F could enhance the activation of STING pathway via cytoplasmic delivery of cGAMP. Furthermore, the encapsulation of cGAMP in LNPs enhances both the humoral and cellular immune response to the model antigen. 93-O17S-F/cGAMP showed the best antibody response, increased OVA-specific CD8+ T cell populations, and excellent OVA-specific T cell killing.
93-O17S-F is an ionizable LNP with positive charge, which could capture the tumor antigens released from the dead tumor cells via electrostatic interaction. To ensure a sufficient concentration of free tumor antigens at the site of the LNP injection, our strategy relies on pretreatment of the tumors with a small local injection of the chemotherapeutic DOX. The DOX treatment kills a small number of tumor cells directly at the site of injection, allowing the release of the tumor antigens for subsequent capture by the LNPs. We showed that 93-O17S-F could capture the tumor lysate and facilitate its delivery to the DLNs. Compared with other antigen-capturing systems, our LNP system showed both enhanced cross-presentation and STING activation (38–40). Thus, such in situ strategy would be more effective in inducing antitumor effect. We observed the significant increase in the number of various tumor infiltrating immune cells, including CD4+ T cells, CD8+ T cells, and macrophages, after sequential intratumoral injection of the DOX followed by 93-O17S-F/cGAMP. Moreover, up-regulated maturation of DCs at both tumor site and DLN was observed in the 93-O17S-F/cGAMP–treated group. The polarization of macrophages in tumor also shifted from M2- to M1-like subsets.
The therapeutic effect of the in situ vaccination strategy was evaluated in B16F10 melanoma tumor allograft model. Our data demonstrate that the low dose of DOX used was not alone sufficient to effectively treat the tumors here. Furthermore, without the DOX pretreatment to release tumor antigens in situ, simple LNP delivery of the STING agonist cGAMP was not sufficient to generate a strong antitumor immune environment. Only with the combination of pretreatment with DOX followed by 93-O17S-F/cGAMP LNP treatment did we observe total tumor reduction and mouse recovery, emphasizing that this LNP system does successfully attack the tumor on both the antigen presentation and the tumor microenvironment fronts. When using this sequential codelivery, the total recovery rate of the mice bearing primary tumors reaches to about 35% within 30 days. Notably, this is achieved without the coadministration of any additional drugs such as immune checkpoint inhibitors. After the rechallenge of the recovered mice with a new B16F10 tumor, 71% of these mice remained free of tumor for more than 60 days, confirming in situ vaccination by 93-O17S-F/cGAMP had generated strong immune memory. The excellent antitumor efficacy of our LNP system further confirmed the superiority of 93-O17S-F/cGAMP for in situ vaccination owing to the enhanced cross-presentation and STING activation.
In conclusion, as a proof of concept, we demonstrated an in situ vaccination by LNP system with enhanced cross-presentation and STING activation for anticancer immunotherapy. 93-O17S-F showed excellent adjuvant effect and enhanced cross-presentation of OVA antigen. Moreover, 93-O17S-F also exhibited enhanced STING activation of APCs owing to the increased cytoplasmic delivery of cGAMP both in vitro and in vivo. Last, our LNP system significantly inhibited the tumor growth of B16F10 allograft model tumor model and also showed excellent immune memory. Although interesting due to its simplicity with potential clinical translation, there are still a few aspects of our approach that can be improved in future to broaden its therapeutic applications. For example, in the current system, the DOX (for inducing the tumor antigen release) and LNP/cGAMP (for STING activation and tumor antigen capture and delivery) are administered sequentially. It will be desired to generate one pot nanoparticle formulation to simplify the treatment procedure. This in situ vaccination is currently administrated by intratumoral injection, which is not convenient to treat the hard-to-reach tumors. Further improvement in LNP formulation and delivery to the hard-to-reach tumor sites using systemic injection is desired. The possibility of inducing serious autoimmune diseases might be a major safety concern of such in situ vaccination approach for therapeutic cancer vaccines. However, there was no case report of the generation of autoimmune disease in human body by this strategy yet. The phase 1/2 clinical trial of intratumoral injection of SD-101, a TLR9 agonist, induced systemic responses in patients with indolent lymphoma without inducing significant autoimmune toxicities (41). Nonetheless, the in situ vaccination by 93-O17S-F showed great superiority to traditional in situ vaccination, providing a promising strategy for cancer immunotherapy.
MATERIALS AND METHODS
General
Lipidoids were synthesized according to our previous reported work (21–23). Imject OVA, LPS, alum, and 96-well high-binding plates were purchased from Thermo Fisher Scientific (Pittsburgh, PA, USA). The STING agonist, 2′3′-cGAMP, was purchased from Sigma-Aldrich. The primers for RT-PCR were designed by quantitative PCR (qPCR) Assay Design software (Eurofins) and synthesized by Integrated DNA Technologies Inc. (Coralville, IA, USA). B16F10 murine melanoma cells were purchased from American Type Culture Collection (ATCC) (Manassas, VA, USA) and cultured in Dulbecco’s modified Eagle’s medium (DMEM; Sigma-Aldrich) with 10% fetal bovine serum (FBS; Sigma-Aldrich) and 1% penicillin-streptomycin (Gibco). RAW264.7 cells were a gift from D. L. Kaplan (Biomedical Engineering, Tufts University) and cultured in cultured in complete DMEM. DC2.4 cells were purchased from ATCC and cultured in RPMI 1640 medium with 10% FBS and 1% penicillin-streptomycin. H-2Kb (OVA257–264)–SIINFEKL-PE (phycoerythrin) tetramer was purchased from MBL International Corporation (Woburn, MA, USA). Fc receptor blocker was bought from Innovex Biosciences (Richmond, CA, USA). Alexa Fluor 647–conjugated OVA was purchased from Invitrogen. Cyclic (3′-O-(6-[fluoresceinyl]aminohexylcarbamoyl)guanosine-(2′-5′)-monophosphate-adenosine-(3′-5′)-monophosphate) (cGAMPFluo) was bought from BIOLOG Life Science Institute (Bremen, Germany). Mouse IFN-β DuoSet ELISA kit was purchased from R&D Systems (Minneapolis, MN, USA).
Prime-boost vaccination for screening of LNP
For library screening, the unformulated LNPs were prepared by dissolving the lipidoids into 25 mM sodium acetate solution (pH 5.2) (Gibco). The formulated LNPs were prepared by dropwise adding the ethanol solution containing the mixture of active lipidoid, cholesterol, and DOPE at the weight ratio of 16:4:1 to 25 mM sodium acetate solution. The mixed solution was then dialyzed using Thermo Fisher Scientific Slide-A-Lyzer MINI Dialysis Device (3.5K MWCO) to obtain the blank LNPs. The LNP/OVA was prepared by simply mixing of blank LNP and OVA at the weight ratio of 10:3 by in PBS solution. To prepare cGAMP- and OVA-loaded LNPs, 100 μg of blank LNPs were first mixed with 10 μg of cGAMP and then mixed with 30 μg of OVA in 100 μl of PBS.
All animal procedures were performed with ethical compliance and approval by the Institutional Animal Care and Use Committee at the Tufts University. Four- to 6-week-old female C57BL/6 mice (Charles River) received the prime vaccination by subcutaneous injection at tail base with 100 μl of LNP/OVA at the doses equivalent to 100 μg of LNP and 30 μg of OVA. The alum was mixed with the antigen solution in 50:50 μl ratio. LPS was used at a dose of 30 μg per mice. Two weeks later, the mice received the boost vaccination with the same dose of LNP/OVA. After 1 week of the second injection, 50 μl of blood was collected from the cheek, and the serum was isolated by centrifugation at 3000 rpm for 10 min at room temperature.
ELISA assays
The high-binding ELISA plates were covered with 50 μl of OVA at 20 μg ml−1 in sodium carbonate solution (pH 8.0) at 4°C overnight. The plates were then washed by PBST (PBS with 0.5% Tween 20) for three times and blocked by 5% bovine serum albumin solution (Sigma-Aldrich). The serum collected from immunized mice was diluted in triplicate from 1:100 and then added into the plates for 2 hours at room temperature. Then, the plates were washed three times and incubated with 100 μl of horseradish peroxidase–conjugated IgG, IgG1, and IgG2c antibodies (1:10,000 dilution) for another 2 hours. The plates were washed by PBST for three times and incubated with 100 μl of 3,3′,5,5′-tetramethylbenzidine substrate (Sigma-Aldrich). The reaction was stopped by 0.16 M sulfuric acid solution. The optical density (OD) values at 450 nm were read in a BioTex microplate reader. As shown in fig. S3, the end point titer is defined as the reciprocal of the highest dilution of a serum that gives a reading above the cutoff.
Detection of MHC class I complex bound to SIINFEKL
DC2.4 cells (5 × 104) were cultured in 24-well plates for 24 hours. Then, the cells were incubated with OVA, 93-O17S-F, or 93-O17S-F/OVA at doses equivalent to OVA (1 μg ml−1) for 24 hours. The cells were collected, washed with PBS, and stained with PE anti-mouse H-2Kb bound to SIINFEKL antibody in flow cytometry staining buffer (eBioscience) at 4°C for 2 hours. The stained cells were washed by PBS and then detected by an Attune NxT flow cytometer (Thermo Fisher Scientific). The data were analyzed by FlowJo-v10.
Cellular uptake of cGAMP
RAW264.7 and DC2.4 cells (5 × 104) were cultured in the chambered coverslip with four wells. cGAMPFluo-loaded LNP was prepared by simply mixing of blank LNP and cGAMPFluo in water. The encapsulation rate of cGAMPFluo by 93-O17S-F was measured to be 85% by fluorescence spectrum. The free cGAMPfluo or 93-O17S-F/cGAMPFluo was added to the medium at doses equivalent to cGAMP (200 ng/ml). After 4 hours of incubation, the medium was replaced and incubated by fresh medium containing LysoTracker Red DND-99 for another 1 hour. Then, the cells were washed by PBS and fixed in 4% paraformaldehyde solution for 10 min. Then, the slices were covered by Fluoroshield with 4′,6-diamidino-2-phenylindole (Sigma-Aldrich). The images were captured using Leica TCS SP8 microscopes.
In vitro evaluation of ifnb1 and cxcl10 gene expressions by RT-PCR
RAW264.7 and DC2.4 cells (5 × 105) were cultured in six-well plates for 24 hours. cGAMP-loaded 93-O17S-F was prepared by simply mixing the blank 93-O17S-F and cGAMP in water. Then, the cells were treated with PBS, free cGAMP, 93-O17S-F, or 93-O17S-F/cGAMP for 4 hours at doses equivalent to 150 nM cGAMP. The total mRNA was isolated by a TriPure reagent (Sigma-Aldrich) following the manufacturer’s manual. The complementary DNA (cDNA) was synthesized using a high-capacity cDNA reverse transcription kit (Applied Biosystems). The RT-PCR was carried out using Applied Biosystems PowerUp SYBR green master mix and detected in BioRad CFX96 touch RT-PCR detection system. The relative gene expressions were analyzed by CFX maestro software.
In vivo cytotoxic T cell assay
The in vivo cytotoxic T cell assay was carried out following the protocol with minor modification (42). Nine days after the boost injection, naive C57BL/6 mice were euthanized and splenocytes were collected. Half of the splenocytes were pulsed with OVA257–263 (SIINFEKL) peptides (0.2 μM) for 1 hour in complete RPMI 1640 medium at 37°C. The unpulsed and peptide-pulsed cells were labeled with 0.5 or 0.05 μM CFSE, respectively, in PBS for 15 min. Equal numbers (1 × 107) of CFSElow (OVA pulsed) and CFSEhigh (OVA unpulsed) cells were mixed and injected intravenously into the immunized mice. After 20 hours, spleen from treated mice was isolated, suspended into single cells by 70-μm cell strainer (Corning, NY, USA), and analyzed by flow cytometry (Attune NxT Flow Cytometer, Thermo Fisher Scientific). The ratios of CFSEhigh and CFSElow cells were determined and used to calculate the percentage of OVA peptide–pulsed target cell killing.
In vitro capture of tumor cell lysate
Tumor cell lysate was prepared via a freeze-thaw process. The cultured B16F10 cells were frozen quickly on dry ice for 5 min and immediately thawed at 37°C. The solution was then vortexed to mix well. This procedure was repeated twice. The final solution was centrifugated at 13,000 rpm for 10 min. The supernatant was collected, and the concentration of lysate was measured by bicinchoninic acid (BCA) assay method. The blank LNP was mixed with the cell lysate at different weight ratio. The size and zeta potential were characterized by Zeta-PALS zeta potential and particle size analyzer (Brookhaven Instruments).
In vivo antigen capture and delivery
Fifty microliters of OVAAlexa-647 at the concentration of 1 μg/μl was injected subcutaneously into the right flank of BALB/c mice. Five minutes after the first injection, 50 μl of PBS or 93-O17S-F/cGAMP containing 200 μg of LNP was injected subcutaneously at the location of first injection. The distribution of OVAAlexa-647 and the isolated DLNs was monitored by IVIS.
In vivo evaluation of ifnb1 and cxcl10 gene expressions by RT-PCR
B16F10 cells (5 × 105) were inoculated in the right flank of 4- to 6-week-old C57BL/6 mice. The tumors were allowed to grow up to 60 to 80 mm3. Mice were divided into five groups including PBS, free DOX (noted as DOX), 93-O17S-F/cGAMP, free cGAMP after the administration of DOX (noted as DOX + cGAMP), and 93-O17S-F/cGAMP after the administration of DOX (noted as DOX + 93-O17S-F/cGAMP). At day 0, DOX was intratumorally injected into three groups of the tumors directly at doses equivalent to 0.1 mg/kg body weight [(kg BW)−1]. At day 1, the second injection including PBS, 93-O17S-F, free cGAMP, and formulated 93-O17S-F/cGAMP was intratumorally injected into the same site where DOX was injected at doses equivalent to 20 μg of cGAMP encapsulated 400 μg of 93-O17S-F per mouse. After 6 hours after the second injection, the tumors were collected, and the total mRNA was isolated using a TriPure reagent. The detailed RT-PCR was carried out in the same route as before.
General multicolor flow cytometry staining method
At predetermined time, the tumors or spleens were collected and suspended to single-cell suspensions by 70-μm strainer. The cells were washed with flow cytometry staining buffer (eBioscience) and incubated with Fc receptor blocker (Innovex Biosciences, CA, USA) for 10 min at room temperature. The cells were then incubated at 4°C with fluorescent antibodies chosen from table S1 for 30 min in recommended dilution provided by the manufacturers. After washing by staining buffer twice, the cells were kept at 4°C for the analysis on the same day. Fluorescent data were collected using LSR-II flow cytometer (BD Biosciences) or Attune NxT Flow Cytometer (Thermo Fisher Scientific). The data were analyzed by FlowJo-v10.
Treatment of B16F10 tumor by in situ vaccination
The tumor model was built as same as described above. Mice were also divided into five groups including PBS, DOX, 93-O17S-F/cGAMP, DOX + cGAMP, and DOX + 93-O17S-F/cGAMP. At day 0, DOX was intratumorally injected into three groups of the tumors directly at doses equivalent to 0.1 mg/kg body weight [(kg BW)−1]. At days 1 and 5, the second and third injections including PBS, 93-O17S-F, free cGAMP, and formulated 93-O17S-F/cGAMP were intratumorally injected into the same site where DOX was injected at doses equivalent to 20 μg of cGAMP encapsulated in 400 μg of 93-O17S-F per mouse. The length (L) and width (W) of the tumors were measure every other day, and the tumor volumes (V) were calculated by the equation: V = L × W2/2.
The experiment of tumor rechallenge was carried out as following. Twenty mice were treated as same as described before. The mice free of tumor for 30 days were reinoculated with 2 × 105 B16F10 cells and monitored up to 90 days.
Acknowledgments
Funding: We acknowledge the financial support by the NIH grants R01 EB027170-01. Author contributions: Q.X. generated the original idea about in situ vaccination by LNP, acquired the funding, supervised the project, and revised the manuscript. J.J.C. completed the final idea, designed and conducted the experiments, analyzed the data, and wrote the draft manuscript. M.Q. helped conduct the animal experiments and revised the manuscript. Z.Y. helped design the PCR primer and assisted the qPCR experiment. T.N. helped with the vaccination and the ELISA assay for lipid library screening. Y.L. helped the synthesis of lipidoids. Z.G. and J.Z.C. helped revise the manuscript. X.Z. and L.Y. gave valuable suggestions about the experiment. J.Z.C. gave valuable suggestion in the design of the experiments. Competing interests: Q.X. and J.J.C. are inventors on a pending patent related to this work filed by the Tufts University (63/122,229; filed 7 December 2020). The other authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/19/eabf1244/DC1
REFERENCES AND NOTES
- 1.Currie G. A., Eighty years of immunotherapy: A review of immunological methods used for the treatment of human cancer. Br. J. Cancer 26, 141–153 (1972). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kantoff P. W., Higano C. S., Shore N. D., Berger E. R., Small E. J., Penson D. F., Redfern C. H., Ferrari A. C., Dreicer R., Sims R. B., Xu Y., Frohlich M. W., Schellhammer P. F.; IMPACT Study Investigators , Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N. Engl. J. Med. 363, 411–422 (2010). [DOI] [PubMed] [Google Scholar]
- 3.Melero I., Gaudernack G., Gerritsen W., Huber C., Parmiani G., Scholl S., Thatcher N., Wagstaff J., Zielinski C., Faulkner I., Mellstedt H., Therapeutic vaccines for cancer: An overview of clinical trials. Nat. Rev. Clin. Oncol. 11, 509–524 (2014). [DOI] [PubMed] [Google Scholar]
- 4.Melief C. J. M., van Hall T., Arens R., Ossendorp F., van der Burg S. H., Therapeutic cancer vaccines. J. Clin. Invest. 125, 3401–3412 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Martins F., Sofiya L., Sykiotis G. P., Lamine F., Maillard M., Fraga M., Shabafrouz K., Ribi C., Cairoli A., Guex-Crosier Y., Kuntzer T., Michielin O., Peters S., Coukos G., Spertini F., Thompson J. A., Obeid M., Adverse effects of immune-checkpoint inhibitors: Epidemiology, management and surveillance. Nat. Rev. Clin. Oncol. 16, 563–580 (2019). [DOI] [PubMed] [Google Scholar]
- 6.Sahin U., Türeci Ö., Personalized vaccines for cancer immunotherapy. Science 359, 1355–1360 (2018). [DOI] [PubMed] [Google Scholar]
- 7.Hammerich L., Binder A., Brody J. D., In situ vaccination: Cancer immunotherapy both personalized and off-the-shelf. Mol. Oncol. 9, 1966–1981 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Monge B. M. C., Xie C., Steinberg S. M., Fioraventi S., Walker M., Mabry-Hrones D., Wood B. J., Kleiner D. E., Greten T. F., A phase I/II study of Pexa-Vec oncolytic virus in combination with immune checkpoint inhibition in refractory colorectal cancer. J. Clin. Oncol. 38, 117 (2020). [Google Scholar]
- 9.Fong Y., Expediting viral therapies for cancers to the clinic. Mol. Ther. 24, 1161–1162 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pelin A., Foloppe J., Petryk J., Singaravelu R., Hussein M., Gossart F., Jennings V. A., Stubbert L. J., Foster M., Storbeck C., Postigo A., Scut E., Laight B., Way M., Erbs P., Le Boeuf F., Bell J. C., Deletion of apoptosis inhibitor F1L in vaccinia virus increases safety and oncolysis for cancer therapy. Mol. Ther. Oncol. 14, 246–252 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen Q., Xu L., Liang C., Wang C., Peng R., Liu Z., Photothermal therapy with immune-adjuvant nanoparticles together with checkpoint blockade for effective cancer immunotherapy. Nat. Commun. 7, 13193 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shin H., Na K., In situ vaccination with biocompatibility controllable immuno-sensitizer inducing antitumor immunity. Biomaterials 197, 32–40 (2019). [DOI] [PubMed] [Google Scholar]
- 13.Nam J., Son S., Ochyl L. J., Kuai R., Schwendeman A., Moon J. J., Chemo-photothermal therapy combination elicits anti-tumor immunity against advanced metastatic cancer. Nat. Commun. 9, 1074 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Morris Z. S., Guy E. I., Francis D. M., Gressett M. M., Werner L. R., Carmichael L. L., Yang R. K., Armstrong E. A., Huang S., Navid F., Gillies S. D., Korman A., Hank J. A., Rakhmilevich A. L., Harari P. M., Sondel P. M., In situ tumor vaccination by combining local radiation and tumor-specific antibody or immunocytokine treatments. Cancer Res. 76, 3929–3941 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brody J. D., Ai W. Z., Czerwinski D. K., Torchia J. A., Levy M., Advani R. H., Kim Y. H., Hoppe R. T., Knox S. J., Shin L. K., Wapnir I., Tibshirani R. J., Levy R., In situ vaccination with a TLR9 agonist induces systemic lymphoma regression: A phase I/II study. J. Clin. Oncol. 28, 4324–4332 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huang L., Li Y., Du Y., Zhang Y., Wang X., Ding Y., Yang X., Meng F., Tu J., Luo L., Sun C., Mild photothermal therapy potentiates anti-PD-L1 treatment for immunologically cold tumors via an all-in-one and all-in-control strategy. Nat. Commun. 10, 4871 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dovedi S. J., Melis M. H. M., Wilkinson R. W., Adlard A. L., Stratford I. J., Honeychurch J., Illidge T. M., Systemic delivery of a TLR7 agonist in combination with radiation primes durable antitumor immune responses in mouse models of lymphoma. Blood 121, 251–259 (2013). [DOI] [PubMed] [Google Scholar]
- 18.Obeid M., Tesniere A., Ghiringhelli F., Fimia G. M., Apetoh L., Perfettini J.-L., Castedo M., Mignot G., Panaretakis T., Casares N., Métivier D., Larochette N., van Endert P., Ciccosanti F., Piacentini M., Zitvogel L., Kroemer G., Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat. Med. 13, 54–61 (2007). [DOI] [PubMed] [Google Scholar]
- 19.Oberli M. A., Reichmuth A. M., Dorkin J. R., Mitchell M. J., Fenton O. S., Jaklenec A., Anderson D. G., Langer R., Blankschtein D., Lipid nanoparticle assisted mRNA delivery for potent cancer immunotherapy. Nano Lett. 17, 1326–1335 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Su T., Zhang Y., Valerie K., Wang X.-Y., Lin S., Zhu G., STING activation in cancer immunotherapy. Theranostics 9, 7759–7771 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang M., Zuris J. A., Meng F., Rees H., Sun S., Deng P., Han Y., Gao X., Pouli D., Wu Q., Georgakoudi I., Liu D. R., Xu Q., Efficient delivery of genome-editing proteins using bioreducible lipid nanoparticles. Proc. Natl. Acad. Sci. U.S.A. 113, 2868–2873 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li Y., Yang T., Yu Y., Shi N., Yang L., Glass Z., Bolinger J., Finkel I. J., Li W., Xu Q., Combinatorial library of chalcogen-containing lipidoids for intracellular delivery of genome-editing proteins. Biomaterials 178, 652–662 (2018). [DOI] [PubMed] [Google Scholar]
- 23.Wang M., Sun S., Neufeld C. I., Perez-Ramirez B., Xu Q., Reactive oxygen species-responsive protein modification and its intracellular delivery for targeted cancer therapy. Angew. Chem. Int. Ed. 53, 13444–13448 (2014). [DOI] [PubMed] [Google Scholar]
- 24.Buchan S., Grønevik E., Mathiesen I., King C. A., Stevenson F. K., Rice J., Electroporation as a “prime/boost” strategy for naked DNA vaccination against a tumor antigen. J. Immunol. 174, 6292–6298 (2005). [DOI] [PubMed] [Google Scholar]
- 25.De Gregorio E., Tritto E., Rappuoli R., Alum adjuvanticity: Unraveling a century old mystery. Eur. J. Immunol. 38, 2068–2071 (2008). [DOI] [PubMed] [Google Scholar]
- 26.Jiang Y., Krishnan N., Zhou J., Chekuri S., Wei X., Kroll A. V., Yu C. L., Duan Y., Gao W., Fang R. H., Zhang L., Engineered cell-membrane-coated nanoparticles directly present tumor antigens to promote anticancer immunity. Adv. Mater. 32, 2001808 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Galluzzi L., Vanpouille-Box C., Bakhoum S. F., Demaria S., SnapShot: cGAS-STING signaling. Cell 173, 276–276.e1 (2018). [DOI] [PubMed] [Google Scholar]
- 28.Holm C. K., Jensen S. B., Jakobsen M. R., Cheshenko N., Horan K. A., Moeller H. B., Gonzalez-Dosal R., Rasmussen S. B., Christensen M. H., Yarovinsky T. O., Rixon F. J., Herold B. C., Fitzgerald K. A., Paludan S. R., Virus-cell fusion as a trigger of innate immunity dependent on the adaptor STING. Nat. Immunol. 13, 737–743 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Park G., Kim S. Y., Song Y.-J., Ester alkaloids from Cephalotaxus interfere with the 2′3′-cGAMP-induced type I interferon pathway in vitro. PLOS ONE 12, e0182701 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ruterbusch M., Pruner K. B., Shehata L., Pepper M., In vivo CD4+ T cell differentiation and function: Revisiting the Th1/Th2 paradigm. Annu. Rev. Immunol. 38, 705–725 (2020). [DOI] [PubMed] [Google Scholar]
- 31.van Montfoort N., Borst L., Korrer M. J., Sluijter M., Marijt K. A., Santegoets S. J., van Ham V. J., Ehsan I., Charoentong P., André P., Wagtmann N., Welters M. J. P., Kim Y. J., Piersma S. J., van der Burg S. H., van Hall T., NKG2A blockade potentiates CD8 T cell immunity induced by cancer vaccines. Cell 175, 1744–1755.e15 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Luo M., Wang H., Wang Z., Cai H., Lu Z., Li Y., Du M., Huang G., Wang C., Chen X., Porembka M. R., Lea J., Frankel A. E., Fu Y.-X., Chen Z. J., Gao J., A STING-activating nanovaccine for cancer immunotherapy. Nat. Nanotechnol. 12, 648–654 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gajewski T. F., Schreiber H., Fu Y.-X., Innate and adaptive immune cells in the tumor microenvironment. Nat. Immunol. 14, 1014–1022 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shen Y., Nemunaitis J., Fighting cancer with vaccinia virus: Teaching new tricks to an old dog. Mol. Ther. 11, 180–195 (2005). [DOI] [PubMed] [Google Scholar]
- 35.Hammerich L., Marron T. U., Upadhyay R., Svensson-Arvelund J., Dhainaut M., Hussein S., Zhan Y., Ostrowski D., Yellin M., Marsh H., Salazar A. M., Rahman A. H., Brown B. D., Merad M., Brody J. D., Systemic clinical tumor regressions and potentiation of PD1 blockade with in situ vaccination. Nat. Med. 25, 814–824 (2019). [DOI] [PubMed] [Google Scholar]
- 36.Cookenham T., Lanzer K. G., Gage E., Lorenzo E. C., Carter D., Coler R. N., Baldwin S. L., Haynes L., Reiley W. W., Blackman M. A., Vaccination of aged mice with adjuvanted recombinant influenza nucleoprotein enhances protective immunity. Vaccine 38, 5256–5267 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Corrales L., McWhirter S. M., Dubensky T. W. Jr., Gajewski T. F., The host STING pathway at the interface of cancer and immunity. J. Clin. Invest. 126, 2404–2411 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Min Y., Roche K. C., Tian S., Eblan M. J., McKinnon K. P., Caster J. M., Chai S., Herring L. E., Zhang L., Zhang T., De Simone J. M., Tepper J. E., Vincent B. G., Serody J. S., Wang A. Z., Antigen-capturing nanoparticles improve the abscopal effect and cancer immunotherapy. Nat. Nanotechnol. 12, 877–882 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang M., Song J., Zhou F., Hoover A. R., Murray C., Zhou B., Wang L., Qu J., Chen W. R., NIR-triggered phototherapy and immunotherapy via an antigen-capturing nanoplatform for metastatic cancer treatment. Adv. Sci. 6, 1802157 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang R., He Z., Cai P., Zhao Y., Gao L., Yang W., Zhao Y., Gao X., Gao F., Surface-functionalized modified copper sulfide nanoparticles enhance checkpoint blockade tumor immunotherapy by photothermal therapy and antigen capturing. ACS Appl. Mater. Interfaces 11, 13964–13972 (2019). [DOI] [PubMed] [Google Scholar]
- 41.Frank M. J., Reagan P. M., Bartlett N. L., Gordon L. I., Friedberg J. W., Czerwinski D. K., Long S. R., Hoppe R. T., Janssen R., Candia A. F., Coffman R. L., Levy R., In situ vaccination with a TLR9 agonist and local low-dose radiation induces systemic responses in untreated indolent lymphoma. Cancer Discov. 8, 1258–1269 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kim M. V., Ouyang W., Liao W., Zhang M. Q., Li M. O., Murine in vivo CD8+ T cell killing assay. Bio. Protoc. 4, e1172 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/19/eabf1244/DC1