

The interplay of SMARCAD1-mediated active fork and BRCA1-mediated stalled fork stability pathways maintains genome integrity.
Abstract
The stalled fork protection pathway mediated by breast cancer 1/2 (BRCA1/2) proteins is critical for replication fork stability. However, it is unclear whether additional mechanisms are required to maintain replication fork stability. We describe a hitherto unknown mechanism, by which the SWI/SNF-related matrix-associated actin-dependent regulator of chromatin subfamily-A containing DEAD/H box-1 (SMARCAD1) stabilizes active replication forks, that is essential to maintaining resistance towards replication poisons. We find that SMARCAD1 prevents accumulation of 53BP1-associated nucleosomes to preclude toxic enrichment of 53BP1 at the forks. In the absence of SMARCAD1, 53BP1 mediates untimely dissociation of PCNA via the PCNA-unloader ATAD5, causing frequent fork stalling, inefficient fork restart, and accumulation of single-stranded DNA. Although loss of 53BP1 in SMARCAD1 mutants rescues these defects and restores genome stability, this rescued stabilization also requires BRCA1-mediated fork protection. Notably, fork protection-challenged BRCA1-deficient naïve- or chemoresistant tumors require SMARCAD1-mediated active fork stabilization to maintain unperturbed fork progression and cellular proliferation.
INTRODUCTION
Most breast cancer (BRCA)–mutated cancers acquire resistance toward chemotherapeutic agents such as cisplatin and poly(adenosine 5′-diphosphate–ribose) polymerase inhibitors (PARPi) (1). At present, besides the restoration of homologous recombination (HR), loss of poly(adenosine 5′-diphosphate–ribose) glycohydrolase or acquired protection of stalled replication forks provides a mechanism that can promote drug resistance in a BRCA-deficient genetic background (1–4). However, identification of additional mechanisms underlying resistance to chemotherapeutics can provide a real opportunity to improve therapies in BRCA-deficient cancer patients.
BRCA proteins play a genetically separable role at the site of double-stranded breaks (DSBs) where they mediate an error-free HR repair and at replication forks where they facilitate protection of reversed forks from extensive nuclease-mediated degradation, to maintain genome stability (2, 3, 5–7). Similarly, the factors of nonhomologous end joining (NHEJ), an error-prone pathway, along with their role in repair of DSBs have been shown to associate with stalled forks either for their protection or to promote their restart (2, 8, 9). However, the factors involved in limiting fork stalling and subsequent restarting of forks upon endogenous or exogenously induced replication stress are poorly understood.
Proliferating cell nuclear antigen (PCNA) is a DNA clamp that associates with the active replication forks and functions as a processivity factor for DNA polymerases to carry out the DNA synthesis process but dissociates from stalled forks via an active unloading mechanism (8, 10, 11). During replication, PCNA rings are repeatedly loaded and unloaded by the replicating clamp loader replication factor C (RFC) complex and an alternative PCNA ring opener, ATAD5 (ELG1 in yeast)–RFC-like complex (ATAD5-RLC). ATAD5-RLC unloads replication-coupled PCNA after ligation of Okazaki fragment and termination of DNA replication (12–14). Maintenance of the delicate balance of PCNA levels onto DNA is crucial because PCNA levels can influence chromatin integrity (15, 16) and persistent PCNA retention on DNA causes genome instability (17–19). However, mechanisms by which PCNA levels are regulated on replicating chromatin and the factors involved in this process still remain elusive.
Here, we uncover a novel function of human SMARCAD1 in regulating the fine control of PCNA levels at forks, which is required for the maintenance of replication stress tolerance and genome stability. SMARCAD1, a DEAD/H box helicase domain protein, belongs to a highly conserved adenosine 5′-triphosphate–dependent SWI/SNF (switch/sucrose non-fermentable) family of chromatin remodelers. Adenosine triphosphatase (ATPase) remodeling activity of SMARCAD1 is crucial for its function in HR repair and in maintenance of histone methyl marks for reestablishment of heterochromatin (20, 21).
In this study, we generated a separation-of-function mutant of human SMARCAD1, efficient in its HR function but defective in its interaction with the replication machinery. This strategy led to uncover a previously unrecognized role of SMARCAD1 in maintaining stability of active (unperturbed and restarted) replication forks, which is responsible for mediating resistance toward replication poisons. In the absence of SMARCAD1, replication fork progression requires BRCA1 to maintain the integrity of stalled forks to allow their restart. Furthermore, SMARCAD1 maintains replication fork stability and cellular viability in BRCA1-deficient naïve or chemoresistant mouse breast tumor organoids, highlighting its essential role in the survival of tumor cells. Our results suggest a conserved role of SMARCAD1 and BRCA1 proteins at replication forks, SMARCAD1 at active forks while BRCA1 at stalled forks, to safeguard replication fork integrity and ensure genome stability.
RESULTS
SMARCAD1 is preferentially enriched at unperturbed replication forks
Most factors associated with the active replisome are required to maintain the stability of the replication forks and could also be important for mediating efficient restart after stalling. To specifically identify novel factors involved in the stability of unperturbed forks, we performed isolation of proteins on nascent DNA (iPOND) coupled to stable isotope labeling with amino acids in cell culture (SILAC)–based quantitative mass spectrometry (8, 9). Mouse embryonic stem cells (mESCs) were used to compare the proteins present at unperturbed active replication forks versus hydroxyurea (HU)–induced stalled replication fork (fig. S1A). In total, 1443 common proteins were identified from two independent experiments (fig. S1, B and C). Consistent with previous reports, we observed a greater than twofold increase in replication stress response proteins, including RAD51 and BRCA1, at stalled forks (Fig. 1A) (8, 9). Levels of core components of the replicative helicase, such as mini-chromosome maintenance protein 6 (MCM6), remained largely unchanged during early replication stress (Fig. 1A). As shown previously (8), PCNA was enriched ~2-fold at the unperturbed forks when compared to the stalled forks, confirming that PCNA associates preferentially with active forks and showing proof of principle of this approach (Fig. 1A and fig. S1B). Among 66 proteins showing preferential enrichment at unperturbed replication forks (fig. S1C), we identified SMARCAD1, a conserved SWI/SNF chromatin remodeler (Fig. 1A and fig. S1B). KAP1/TRIM28 (KRAB-associated protein 1/tripartite motif-containing 28), a previously reported SMARCAD1 interacting partner, showed no preferential enrichment, a behavior that is similar to that of the MCM6 helicase, suggesting an additional and independent role of SMARCAD1 in replication fork dynamics (Fig. 1A) (8).
Fig. 1. SMARCAD1 associates with active replication forks via N-terminal PCNA-interacting domain.
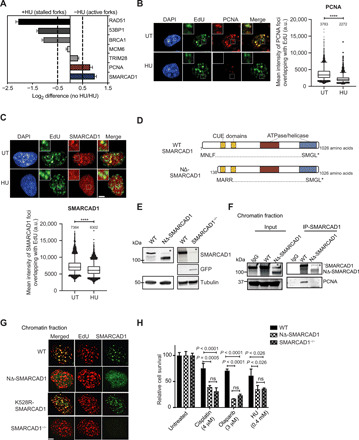
(A) Bar graph showing fold up-regulation of selected proteins under unperturbed (no HU)/HU-treated conditions based on SILAC H:L ratios. (B and C) Representative high-content microscopy images showing the colocalization of chromatin-bound (B, left) PCNA or (C, top) SMARCAD1 with 5-ethynyl-2’-deoxyuridine (EdU) under given conditions. Box plots showing the mean intensity of (B, right) PCNA or (C, bottom) SMARCAD1 foci overlapping with EdU are shown (note that for HU condition, EdU labeling was performed before HU treatment). Numbers above each scatterplot indicate the mean intensity. n > 3000 mid-late S phase cells; ****P ≤ 0.0001, unpaired t test. a.u., arbitrary units. Scale bar, 5 μm. (D) Schematic overview of protein domains in full-length and NΔ-SMARCAD1. Asterisk represents the stop codon. (E) Immunoblot showing SMARCAD1 levels under indicated conditions. Tubulin is the loading control. Asterisk represents a nonspecific band. (F) Chromatin immunoprecipitation (IP) of SMARCAD1 in wild-type (WT) and NΔ-SMARCAD1 cells, followed by immunoblot for SMARCAD1 and PCNA. Asterisk represents a nonspecific band. IgG, immunoglobulin G. (G) Representative images showing the expression of SMARCAD1 and EdU in WT, NΔ-SMARCAD1, K528R-SMARCAD1, and SMARCAD1−/− cells. Scale bar, 5 μm. (H) Quantification of colony survival assay (means + SD; n = 3) in WT, NΔ-SMARCAD1, and SMARCAD1−/− cells is shown for given conditions. HU was given for 48 hours. ns, nonsignificant; unpaired t test.
To confirm our iPOND-SILAC mass spectrometry data and to assess whether the preferential enrichment of SMARCAD1 and PCNA at unperturbed replication forks is conserved across species, we performed immunofluorescence assays to measure the localization of these proteins with respect to the sites of replication in MRC5 human fibroblast cells. Sites of active DNA replication were labeled with 5-ethynyl-2′-deoxyuridine (EdU), and localization of the chromatin-bound fraction of SMARCAD1, PCNA, and RAD51 within the sites of replication was measured in the presence or absence of HU using single cell–based, high-content microscopy. Consistent with the results of iPOND-SILAC mass spectrometry in mESCs, we observed that chromatin-bound SMARCAD1 and PCNA foci specifically colocalized with EdU. However, upon HU treatment, both these proteins showed a significant decrease in intensity at replication sites, suggesting that both SMARCAD1 and PCNA associate with unperturbed replication forks but dissociate from stalled forks (Fig. 1, B and C). As expected, RAD51 was found to be enriched significantly at replication sites upon HU treatment, suggesting a positive enrichment at stalled forks in contrast to PCNA and SMARCAD1 (fig. S1D) (8).
NΔ-SMARCAD1 lacks PCNA interaction and, thereby, association with replication forks
The N-terminal region of SMARCAD1 has been shown to be responsible for the PCNA-mediated localization of SMARCAD1 to replication forks (21, 22). To explore the role of this interaction at replication forks, we generated a SMARCAD1 mutant, using MRC5 cells, in which the canonical start site is disrupted and translation begins downstream at the next available start codon (Fig. 1D). Expression of this mutant gene results in a 137–amino acid N-terminally truncated product, designated as NΔ-SMARCAD1 that lacks the region responsible for its interaction with PCNA (22). The NΔ-SMARCAD1 protein is approximately 100 kDa in size (Fig. 1E) and retains the downstream CUE1, CUE2, ATPase, and helicase domains (fig. S1E), crucial for chromatin remodeling and DNA repair functions (21, 23), intact. For comparative analysis, we also generated a complete SMARCAD1 knockout (SMARCAD1−/−) by replacing the SMARCAD1 gene with an mClover [a green fluorescent protein (GFP) variant] reporter gene (Fig. 1E). Both quantitative reverse transcription polymerase chain reaction (qRT-PCR) assays of the SMARCAD1 coding region and RNA sequencing (RNA-seq)–based transcriptome analysis of cells containing the full length [wild-type (WT)] and those containing the truncated form (NΔ-SMARCAD1) confirmed that expression levels of the two SMARCAD1 alleles were nearly identical (fig. S1, E and F). As expected, cells containing the knockout, SMARCAD1−/−, showed a lack of transcripts specific to the coding region of the gene.
To test the interaction between PCNA and the NΔ-SMARCAD1 mutant, we generated a heterogeneously expressed GFP-tagged PCNA allele in both WT and NΔ-SMARCAD1 genetic backgrounds (fig. S1G). Cross-linked chromatin immunoprecipitation (IP) of GFP-tagged PCNA confirmed that although NΔ-SMARCAD1 associates with chromatin, it did not interact with GFP-PCNA, whereas the full-length WT SMARCAD1 protein retains this interaction (fig. S1H), as previously reported (21). Similarly, reverse chromatin IP of WT-SMARCAD1 and NΔ-SMARCAD1 protein confirmed the lack of interaction between PCNA and NΔ-SMARCAD1 protein (Fig. 1F). The interaction between SMARCAD1 and PCNA was also confirmed by immunoprecipitating PCNA under native conditions, again verifying the loss of interaction between NΔ-SMARCAD1 and PCNA (fig. S1I). To determine whether a SMARCAD1 interaction with PCNA is required for its association with replication sites, we performed an immunofluorescence analysis to measure the localization of SMARCAD1 mutants at sites of DNA replication marked with EdU. Our data show that chromatin-bound foci of full-length SMARCAD1 colocalized with EdU+ sites, as previously reported (Fig. 1G) (21). As expected, no specific SMARCAD1 signal could be seen in SMARCAD1−/− cells. Consistent with our IP data (Fig. 1F and fig. S1H), NΔ-SMARCAD1 showed nuclear localization but no colocalization with EdU signals (Fig. 1G), suggesting that NΔ-SMARCAD1 associates with chromatin but is not enriched at sites of replication.
Role of SMARCAD1 at the replication fork and not in HR mediates tolerance to replicative stress
Next, we sought to determine whether loss of SMARCAD1 association with replication forks affects cellular resistance to fork stalling agents such as HU, cisplatin, or the PARPi, olaparib. Both NΔ-SMARCAD1 and SMARCAD1−/− cells showed significant sensitivity to the replication poisons, suggesting that the presence of SMARCAD1 at replication forks is crucial for resistance to replication stress (Fig. 1H). To further explore the role of SMARCAD1 during DNA replication, we analyzed S phase progression by measuring EdU incorporation using high-content microscopy. We imaged >2000 cells and plotted for quantitative image-based cytometry analysis (QIBC) to obtain single cell–based cell cycle profile (24). Both NΔ-SMARCAD1 and SMARCAD1−/− cells displayed reduction in EdU intensities relative to WT cells, suggesting that loss of SMARCAD1 at forks causes DNA replication defects (fig. S1J).
Because the loss of SMARCAD1 causes defects in HR repair of DSBs due to inefficient DNA end resection (20, 23, 25), we next tested whether cells expressing NΔ-SMARCAD1 also exhibited defects in HR repair. We measured HR efficiency using a direct repeat–green fluorescent protein (DR-GFP) reporter assay (26). NΔ-SMARCAD1 cells had an HR efficiency similar to that of WT (Fig. 2A). However, HR efficiency was significantly reduced in both WT and NΔ-SMARCAD1 cells when SMARCAD1 was knocked down in these cells using small interfering RNA (siRNA), similar to that observed for BRCA1 knockdown (Fig. 2A). These data suggest that, although the complete loss of SMARCAD1 results in defective HR, expression of the truncated NΔ-SMARCAD1 retains HR proficiency. In addition, chromatin fractionation and observation of RAD51 focus formation by immunofluorescence using high-content microscopy both showed a remarkable increase in chromatin-bound RAD51 upon olaparib treatment in both WT and NΔ-SMARCAD1 but not in SMARCAD1-deficient cells (Fig. 2, B and C). These data further confirm that NΔ-SMARCAD1 cells are proficient in the loading of RAD51 in response to DNA damage unlike SMARCAD1−/−. Furthermore, NΔ-SMARCAD1 cells are also proficient in RAD51 foci formation similar to WT upon ionizing radiation (IR)–induced DSB formation (fig. S2A). Consistently, NΔ-SMARCAD1 does not show sensitivity to IR treatment in contrast to HR-defective BRCA1-depleted cells or NHEJ-defective p53-binding protein 1 (53BP1)–depleted cells (fig. S2, A and B). Surprisingly, however, both NΔ-SMARCAD1 and SMARCAD1 complete knockout showed similar sensitivity toward drugs, causing replication stress, olaparib, cisplatin, and HU (Figs. 1H and 2D and fig. S2C), arguing in favor of an uncoupling between HR repair function and resistance to replication stress in the NΔ-SMARCAD1 cells, corroborating it to be a separation-of-function mutant.
Fig. 2. SMARCAD1 provides resistance toward replication poisons, independent of its role in HR repair pathway.
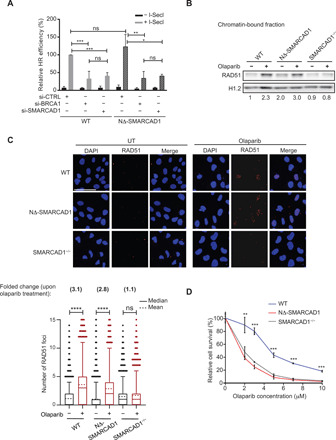
(A) Quantification of HR efficiency using DR-GFP reporter assay. DR-GFP reporter and pcBASceI constructs were cotransfected into WT and NΔ-SMARCAD1 MRC5 cells. Relative HR efficiency representing the percentage of GFP+ cells normalized to the transfection efficiency of the respective cell line is plotted. The means and SD are represented. n = 3; ***P ≤ 0.001, **P ≤ 0.01, and *P ≤ 0.05, unpaired t test. (B) Immunoblot showing the chromatin-bound fraction of RAD51 upon 7 μM olaparib treatment for 24 hours in WT, NΔ-SMARCAD1, and SMARCAD1−/− cells. H1.2 is used as a loading control. The numbers below the blots show the fold change of RAD51 after normalization with H1.2, as compared to WT untreated samples, for the given blot (n = 3). (C) Top: Representative high-content microscopy images depicting RAD51 foci formation upon 7 μM olaparib treatment for 24 hours in WT, NΔ-SMARCAD1, and SMARCAD1−/− cells. Scale bar, 50 μm. Bottom: Quantification of RAD51 foci upon 7 μM olaparib treatment for 24 hours using high-content microscopy. A total of 4700 cells were analyzed under each condition. ****P ≤ 0.001, one-way analysis of variance (ANOVA). Number above represents the fold change of RAD51 foci upon olaparib treatment compared to its own untreated samples. (D) Quantification of colony survival assay in WT, NΔ-SMARCAD1, and SMARCAD1−/− cells treated with different concentrations of olaparib. Error bars stand for ±SD (n = 3). ***P ≤ 0.001 and **P ≤ 0.01, unpaired t test.
We also performed transcriptome analysis to test whether the drug sensitivity observed in SMARCAD1 mutant cells could be a result of transcription deregulation of DNA damage response (DDR) genes in these cells, because transcription may be affected by its chromatin remodeling role. We observed a mild dysregulation in a subset of non-DDR genes (≥1.5-fold change in expression) in either NΔ-SMARCAD1 or SMARCAD1−/− cells, whereas almost no anomalous expression was observed in either mutant for a set of DDR genes (n = 179) (27), which included both HR and NHEJ DDR genes (fig. S2D). This suggests that the function of SMARCAD1 in promoting drug tolerance is unrelated to its role in heterochromatin maintenance or in transcriptional regulation. Furthermore, the efficient loading of RAD51 and the HR proficiency of cells expressing NΔ-SMARCAD1, in contrast to those lacking SMARCAD1, is most likely not due to a differential transcriptome or cell cycle profile but due to the presence of intact CUE and ATPase/helicase domains in NΔ-SMARCAD1 that are essential for its HR function (20, 25). The loss of PCNA interaction and association with the fork is the main cause for SMARCAD1-depleted cells to show sensitivity toward replication stress–inducing drugs.
SMARCAD1 facilitates normal replication fork progression and efficient restart upon replication stress
SMARCAD1 mutants displayed moderate but significant defects in progression through S phase (fig. S1J). To further monitor the dynamics of individual replication forks, we performed DNA fiber assay. We sequentially labeled WT and SMARCAD1 mutants (NΔ-SMARCAD1 and SMARCAD1−/−) cells with CldU (red) and IdU (green), followed by track length analysis. NΔ-SMARCAD1 cells exhibited a significant difference in the track lengths of both 5-chloro-2′-deoxyuridine (CldU) and 5-iodo-2′-deoxyuridine (IdU) in comparison to WT but similar to SMARCAD1−/− cells (Fig. 3A). To test the possibility that accumulation of DNA damage over time in the mutant cells was causing the replication fork defect observed, we also analyzed fork progression in cells in which SMARCAD1 was depleted transiently with siRNA. The transient knockdown of SMARCAD1 resulted in similar fork progression defects than the one observed in NΔ-SMARCAD1 and SMARCAD1−/− (Fig. 3A). This suggests that SMARCAD1 directly facilitates the progression of replication forks.
Fig. 3. SMARCAD1 is required for proper fork progression, fork restart, and genome stability.
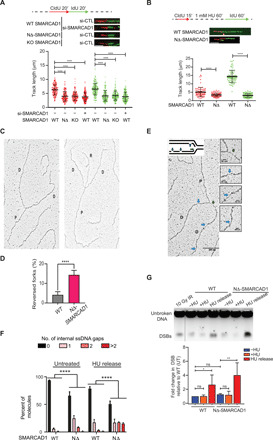
(A) Top: Schematic of replication fork progression assay with CldU and IdU labeling in WT, NΔ-SMARCAD1, and SMARCAD1−/− (KO) cells. Representative DNA fibers for each condition are shown below the schematic. Scale bar, 5 μm. Bottom: CldU (red) and IdU (green) track length (in micrometers) distribution for the indicated conditions. n = 3; ****P ≤ 0.0001, Kruskal-Wallis followed by Dunn’s multiple comparison test. (B) Top: Schematic of replication fork restart assay. Representative DNA fibers for each condition are shown below the schematic. Scale bar, 5 μm. Bottom: CldU (red) and IdU (green) track length (in micrometers) distribution for the indicated conditions. ****P ≤ 0.0001, unpaired t test. All DNA fiber experiments presented here were repeated three times with similar outcomes. (C) Representative image of a normal (left) and a reversed replication fork (right) observed by electron microscopy (EM). D, daughter strand; P, parental strand; R, reversed arm. (D) Bar chart representing the percentage of fork reversal in WT and NΔ-SMARCAD1 cells under untreated condition. n = 3; ****P ≤ 0.0001, unpaired t test. (E) Representative electron micrographs of single-stranded DNA (ssDNA) gaps. Green and blue arrows point toward ssDNA gaps at the fork and behind the fork, respectively. (F) Bar chart representing the distribution of ssDNA gaps behind the fork in WT and NΔ-SMARCAD1 under untreated condition and 1 hour after release from 1 mM HU treatment. Chi-square test of trends was performed to assess the significance of internal ssDNA gaps between WT and NΔ-SMARCAD1. n = 3; ****P < 0.0001. (G) Top: Pulsed-field gel electrophoresis (PFGE) analysis for DSBs shows WT and NΔ-SMARCAD1 cells with and without 4 mM HU treatment for 3 hours and upon 16 hours of release after the HU treatment. Bottom: Quantification from the three independent experiments showing DSB levels.
Because SMARCAD1 deficiency displayed significant replication defects during unperturbed replication (Fig. 3A and fig. S1J), we wondered whether SMARCAD1 also plays a role in the progression after fork stalling. To assess the overall rate of DNA synthesis upon replication stress, we treated cells with 1 mM HU for an hour. The replication rate after stress was measured by allowing the EdU incorporation for various time points after release from HU and EdU intensities that were measured in >3000 cells using high-content microscopy. Upon 30 min of release from HU, we observed a mild reduction in EdU incorporation in NΔ-SMARCAD1 cells. However, the reduction in EdU incorporation became more evident at later time points in NΔ-SMARCAD1 cells (fig. S2E). To further verify this, we performed a fork restart assay using DNA fiber analysis. Cells were labeled with CldU, followed by a mild dose of HU (1 mM) treatment for an hour to stall the forks and subsequently released into IdU. Consistently, we observed significant defects in CldU track lengths, representing an internal control for unperturbed forks (Fig. 3B) similar to those observed in the fork progression assay performed in Fig. 3A. However, analysis of IdU track lengths representing stressed forks revealed an even higher shortening of the track lengths in NΔ-SMARCAD1 cells, suggesting a more severe defect in the progression or restart of stalled forks (Fig. 3B). In addition, upon analysis of fork restart efficiency, we observed a significant difference between stalled and restarted forks in NΔ-SMARCAD1 cells (25% restarted) when compared to WT cells (60% restarted) after 15 min of release from HU stress, whereas this difference significantly reduced after 30 min of release from HU (86% WT and 74% NΔ-SMARCAD1) (fig. S2F, left), but the progression of restarted forks remained severely defective in NΔ-SMARCAD1 cells (fig. S2F, right). These data suggest that forks restart in absence of SMARCAD1 with moderate delay but further show severe defects in progression of stressed forks. Thus, SMARCAD1 mediates both the efficient restart and progression of replication forks, which also supports the finding that cells lacking SMARCAD1 are sensitive to replication stress–inducing agents.
SMARCAD1 prevents accumulation of under-replicated regions and consequent genome instability
To investigate whether the delayed restart and poor fork progression upon release from HU stress results in increased single-stranded DNA (ssDNA) levels in the NΔ-SMARCAD1 cells, we analyzed RPA32, a surrogate for ssDNA, by chromatin fractionation. Upon HU treatment, the replication protein A, 32 kDa subunit (RPA32) signals were enhanced in WT cells (fig. S2G). Untreated NΔ-SMARCAD1 cells showed a marked increase in chromatin-associated RPA32 compared to untreated WT cells, suggesting that the accumulation of under-replicated regions in the genome could be due to defects in normal fork progression (Fig. 3A and fig. S2G). However, a significant increase in RPA32 levels could be seen upon HU treatment and upon release from HU-mediated block in NΔ-SMARCAD1 cells, suggesting that loss of SMARCAD1 at forks causes significant accumulation of under-replicated regions (fig. S2G). Furthermore, unperturbed NΔ-SMARCAD1 cells showed significant phosphorylation of checkpoint kinase 1 (CHK1) but not ataxia-telangiectasia-mutated (ATM) protein, suggesting that absence of SMARCAD1 at the unperturbed forks specifically leads to activation of ataxia telangiectasia and Rad3-related protein (ATR) – mediated checkpoint pathway, further corroborating replication stress in these cells (fig. S2H).
DNA replication stress, exogenous or endogenous, results in reversal of forks (28–31), and we hypothesized that slower fork progression and accumulation of RPA in NΔ-SMARCAD1 mutants under unperturbed conditions could be a result of frequent fork stalling that stabilizes into reversed forks. To test this hypothesis, we visualized replication intermediates formed in vivo using electron microscopy (EM) (9) in WT and NΔ-SMARCAD1 mutant cells. We observed a higher frequency of reversed forks in NΔ-SMARCAD1 than in WT cells, suggesting frequent stalling and remodeling of forks even under unperturbed conditions (Fig. 3, C and D). Moreover, we also observed an increase in the percentage of ssDNA gaps accumulated in daughter strands behind the fork of NΔ-SMARCAD1 cells relative to WT, which further enhanced markedly upon release from HU-mediated stress (Fig. 3, E and F). We also quantified the length of ssDNA at the fork that determines nascent strand processing activity at the fork, which showed no significant difference in NΔ-SMARCAD1 than compared to WT (fig. S2I). Together, these data further corroborate that the role of SMARCAD1 is critical in limiting fork stalling under unperturbed conditions and promoting efficient fork restart and fork progression globally upon replication stress.
We further investigated whether the increased accumulation of ssDNA upon replication stress leads to an increase in DSBs that would contribute to genome instability. To evaluate the accumulation of DNA damage, we performed pulsed-field gel electrophoresis (PFGE) to measure the physical presence of DSBs. There was no obvious increase in the level of DSBs upon the stalling of forks induced by HU treatment in either WT or NΔ-SMARCAD1 cells, suggesting that forks stalled for 3 hours with HU treatment do not immediately collapse and convert into DSBs. These data were further supported by the efficient loading of RAD51 observed at stalled forks induced upon HU treatment in NΔ-SMARCAD1 similar to WT (fig. S2J). However, after release from replication stress for 16 hours, a marked increase in the signal of broken DNA fragments can be observed in NΔ-SMARCAD1 cells in comparison to WT cells (Fig. 3G). Together, these data suggest a role of SMARCAD1 at replication forks that is crucial to maintain genome integrity upon replication stress.
SMARCAD1 maintains PCNA levels at replication forks, especially upon fork restart
Because NΔ-SMARCAD1 lacks interaction with PCNA (Fig. 1F and fig. S1H) and also display defects in fork progression (Fig. 3, A and B), we wondered whether the loss of SMARCAD1 at replication fork affects the PCNA clamp that acts as processivity factor for efficient DNA synthesis. We therefore measured the chromatin-bound PCNA levels in replicating cells labeled with EdU to observe the dynamics of PCNA localization during DNA synthesis. QIBC analysis showed significant reduction in chromatin-bound PCNA levels in replicating cells of NΔ-SMARCAD1 in comparison to WT (Fig. 4A), whereas the total levels of PCNA protein were not affected (Fig. 4B). These data suggest that absence of SMARCAD1 at forks affect PCNA levels at the forks. A similar reduction in PCNA levels at replication sites was observed in SMARCAD1−/− cells, suggesting that NΔ-SMARCAD1 behaves similar to the complete loss of SMARCAD1 protein and that NΔ-SMARCAD1 does not display a dominant negative phenotype (fig. S3A). We further monitored the impact of HU-mediated replication stress on PCNA recovery. Because PCNA dissociates from HU-mediated stalled forks (Fig. 1, A and B) (8), we hypothesized that aggravated defects in fork restart in NΔ-SMARCAD1 were due to poor recovery of PCNA at the forks upon release from HU. Using QIBC analysis, we simultaneously assessed the EdU incorporation and PCNA recovery upon HU stress using an average of 3000 cells per condition (Fig. 4C). WT replicating cells showed significantly reduced PCNA levels upon 1 mM HU treatment for an hour and had recovered to their untreated levels by 45 min of release from HU stress (Fig. 4C and fig. S3B). Consistently, we observed reduced PCNA levels and reduced EdU incorporation in NΔ-SMARCAD1 cells in comparison to WT cells under the untreated condition. NΔ-SMARCAD1 cells showed severe defects in recovery of PCNA levels and reduced EdU incorporation upon release from HU-mediated replicative stress (Fig. 4C and fig. S3, B and C). The significantly reduced EdU incorporation is consistent with the results of the DNA fiber assay of fork restart upon HU stress, which revealed severe defects in the progression of restarted forks in NΔ-SMARCAD1 cells (Fig. 3B). These data suggest that SMARCAD1 participates in the maintenance of PCNA levels at the unperturbed forks. Moreover, under stressed conditions, the absence of SMARCAD1 results in poor recovery of PCNA at restarting stalled forks, which subsequently causes inefficient fork restart and severe defects in fork progression upon replication stress.
Fig. 4. SMARCAD1 maintains PCNA level at replication forks.
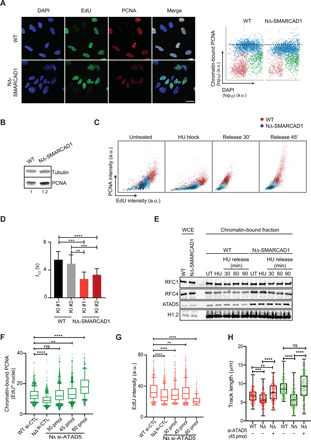
(A) Left: Representative confocal images showing chromatin-bound PCNA (red) in EdU+(green) WT and NΔ-SMARCAD1 MRC5 cells. Nucleus was stained with 4′,6-diamidino-2-phenylindole (DAPI) (blue). Scale bar, 20 μm. Right: QIBC analysis of the chromatin-bound PCNA in WT and NΔ-SMARCAD1 cells. G0–1, S, and G2-M phase cells are labeled in red, blue, and green, respectively. Dashed lines represent the mean chromatin-bound PCNA intensity of S phase cells in WT cells. (B) Immunoblot showing the total level of PCNA in WT and NΔ-SMARCAD1 cells. Tubulin is used as a loading control. Numbers below represent the quantification of PCNA level after normalized to the loading control. (C) QIBC analysis of PCNA versus EdU is shown in WT and NΔ-SMARCAD1 cells in untreated, 1 mM 1-hour HU block, and 45-min release after HU conditions (note that for the HU block condition, EdU labeling was performed before HU treatment). A total of >1800 S phase cells were plotted under each condition. The color gradient represents the density of the cells. (D) Quantification of half-life of the GFP-PCNA fluorescence decay in GFP-tagged PCNA knock-in (KI) WT and NΔ-SMARCAD1 clones, means ± S.D. ****P ≤ 0.0001, ***P ≤ 0.001, and **P ≤ 0.01, unpaired t test. (E) Immunoblot showing the whole-cell extract (WCE) and chromatin-bound fraction of RFC1, RFC4, and ATAD5 in WT and NΔ-SMARCAD1 cells. H1.2 is used as a loading control. (F and G) Foci analysis of (F) chromatin-bound PCNA intensity and (G) EdU intensity in si-control and various concentrations of si-ATAD5–treated WT and NΔ-SMARCAD1 cells. ****P ≤ 0.0001 and **P ≤ 0.01, unpaired t test. (H) CldU (red) and IdU (green) track length (in micrometers) distribution for replication fork restart assay. ****P ≤ 0.0001, ***P ≤ 0.001, and **P ≤ 0.01, Kruskal-Wallis followed by Dunn’s multiple comparison test.
We further determined the dynamics of PCNA in replicating WT and NΔ-SMARCAD1 cells using an inverse fluorescence recovery after photobleaching (iFRAP) live-cell imaging assay. iFRAP is an adapted FRAP approach optimized to analyze differences of dissociation rates (Koff) and involves continuous bleaching to quench the total nuclear fluorescence of a GFP-tagged protein with the exception of a small predefined area. Using this approach, we could determine the residence time of GFP-PCNA at the replication foci (unbleached area) as a direct readout of its turnover (fig. S3D). We performed iFRAP on GFP-tagged PCNA expressed from its endogenous allele in both WT and NΔ-SMARCAD1 cell types (fig. S1G). We observed nearly twofold shorter residence times for GFP-tagged PCNA foci in NΔ-SMARCAD1 cells compared to WT cells (Fig. 4D and fig. S3D). These data clearly suggest that the turnover of PCNA at replication forks is severely increased in the absence of SMARCAD1 at the forks, which may be caused by either a defect in the loading or unloading of PCNA in the absence of SMARCAD1 at the replication forks.
To further test this hypothesis, we performed chromatin fractionation to observe the chromatin-associated fraction of subunits of the PCNA loader, RFC (RFC1/RFC2-5), and of the unloader, RLC (ATAD5/RFC2-5) complex subunits (13, 32). We observed no obvious change in the level of RFC1, a major subunit of the RFC complex, in either cell type with or without HU treatment (Fig. 4E). The chromatin association of RFC4, a subunit shared between the RFC and RLC complexes, and that of ATAD5, a major subunit of the RLC complex, were found to be significantly enhanced in chromatin-bound fraction of NΔ-SMARCAD1 cells, while the total level of these proteins as shown in whole-cell extracts remains similar to WT, which is also supported by the transcriptome analysis of these proteins (Fig. 4E and fig. S3E). This finding suggests that the increased chromatin binding of the PCNA-unloader ATAD5-RLC causes the increased release of PCNA in the absence of SMARCAD1. Next, we tested whether depleting ATAD5 levels might restore normal PCNA chromatin association and reduce replication defects in NΔ-SMARCAD1 cells. Consistent with previous reports (33), we observed enhanced PCNA levels at replicating sites in WT cells and retention time of PCNA using iFRAP, upon strong ATAD5 knockdown (fig. S3, F to H). However, as previously reported (33), the strong reduction of ATAD5 significantly reduced the overall EdU incorporation even in WT cells, suggesting that the enhanced accumulation of PCNA at forks also affects overall DNA synthesis (fig. S3I). Therefore, we titrated the knockdown of ATAD5 in NΔ-SMARCAD1 cells to bring PCNA levels equivalent to WT, using lower concentrations of si-ATAD5 (Fig. 4F and fig. S3J). We observed that 30- and 45-pmol concentrations of si-ATAD5 resulted in PCNA and EdU levels in NΔ-SMARCAD1 similar to WT levels (Fig. 4, F and G). Further, using the controlled depletion of ATAD5 (45 pmol of si-ATAD5), we also observed the rescue in fork progression and fork restart efficiency (Fig. 4H). Similarly, the enhanced accumulation of PCNA and lower EdU incorporation with stronger depletion of ATAD5 could also be rescued by ectopically expressing Flag-tagged ATAD5 in WT and NΔ-SMARCAD1 cells (fig. S3, K and L) (33). Together, these data suggest that fine regulation of ATAD5 levels at replication forks is required to maintain fine-controlled PCNA levels that maintain efficient DNA synthesis in cells.
Loss of 53BP1 restores PCNA stability, fork restart, and drug tolerance in NΔ-SMARCAD1 cells
Having established the role of SMARCAD1 at the replication forks, we further investigated the mechanism of how SMARCAD1 promotes replication fork progression. Earlier studies have shown a role for SMARCAD1 in displacing 53BP1 from the site of DSBs to promote HR repair (20). Moreover, SMARCAD1 and 53BP1 show contrasting enrichments at unperturbed versus stalled replication forks, shown by iPOND-SILAC mass spectrometry (Fig. 1A and table S1) (8). We further validated the enrichments of 53BP1 at stalled forks versus restarted forks using fluorescence microscopy in WT cells (Fig. 5A). The data clearly showed 53BP1 colocalization with EdU mainly upon HU treatment, suggesting its enrichment at stalled forks in WT cells, whereas upon release from HU stress, the EdU-labeled sites representing restarted forks show clear displacement between 53BP1 and EdU foci (Fig. 5A). We hypothesized that, similar to DSBs (20), SMARCAD1 might prevent 53BP1 to accumulate at active or restarted replication forks by promoting its displacement from the stalled forks. To test this hypothesis, we measured the levels of 53BP1 protein in replicating cells (EdU+) of NΔ-SMARCAD1 compared to WT, in untreated and in cells released from HU stress. We observed a mild but significant increase in 53BP1 levels in replicating cells of NΔ-SMARCAD1, and notably, a significantly higher accumulation of 53BP1 levels could be seen in cells released from HU stress (fig. S4A). We further measured the localization of 53BP1 protein relative to EdU-marked replication sites in NΔ-SMARCAD1 compared to WT cells. Upon HU block, a significant percentage of replicating WT cells showed an overlap between EdU and 53BP1 foci, which significantly reduced upon release from HU stress (Fig. 5B). Significantly higher percentage of NΔ-SMARCAD1 cells showed colocalization of EdU and 53BP1 foci in HU block cells, which remained remarkably higher even upon release from HU stress (Fig. 5B). Supporting this observation, the Pearson’s overlap coefficient and Manders’ (M1/M2) overlap coefficients estimating the significance of overlap between EdU and 53BP1 foci were found to be significantly higher in NΔ-SMARCAD1 than in WT (fig. S4B). Together, these data suggest that SMARCAD1 is required to displace 53BP1 from stalled replication forks possibly to allow their restart.
Fig. 5. SMARCAD1 displaces 53BP1 from forks to maintain fine PCNA levels, fork progression, and genome stability.
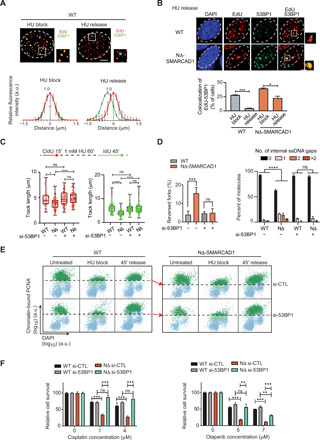
(A) Top: Representative image showing 53BP1 (green) and EdU (red) in WT cells treated with 4 mM HU for 3 hours (HU block) and 1-hour release after HU block (HU release). Bottom: The average distance between EdU and 53BP1 foci is measured. Error bars represent ±SD. (B) Top: Representative images showing 53BP1 foci in EdU-positive cells under indicated conditions. Bottom: Quantification of percentage of cells showing colocalization between EdU and 53BP1 is shown. ***P ≤ 0.001 and *P ≤ 0.05, unpaired t test. (C) Top: Schematic of fork restart assay. Bottom: CldU and IdU track length (in micrometers) distribution for the indicated conditions. n = 3; ****P ≤ 0.0001 and *P ≤ 0.05, Kruskal-Wallis with Dunn’s multiple comparison test. (D) Left: The frequency of reversed forks quantified using EM for indicated conditions. Right: Bar chart showing the distribution of ssDNA gaps behind the fork for the indicated conditions. n = 3; ***P ≤ 0.001, unpaired t test;****P ≤ 0.0001, chi-square test. (E) QIBC analysis of chromatin-bound PCNA and DAPI is shown under given conditions. Cells above dashed lines represent the S phase cells. The red arrows compare the PCNA level. (F) Quantification of colony survival assay under the indicated conditions. means + SD, n = 3; ***P ≤ 0.001 and **P ≤ 0.01, unpaired t test.
This observation led us to hypothesize that loss of 53BP1 may allow the normal progression of forks in NΔ-SMARCAD1 cells, which shows frequent fork stalling even under unperturbed conditions (Fig. 3C). We, therefore, first investigated the progression rate of unperturbed forks using si-53BP1 in NΔ-SMARCAD1 using a DNA fiber assay. Transient knockdown of 53BP1 completely rescued the fork progression defects of NΔ-SMARCAD1 cells (fig. S4, C and D). In addition, we also performed fork restart assay and found that both IdU track lengths and CldU track lengths, representing stressed (after HU treatment) and nonstressed forks (before HU treatment), respectively, showed complete restoration of fork progression rates in NΔ-SMARCAD1 (Fig. 5C). Consistently, we observed a rescue in accumulation of reversed forks and reduced accumulation of ssDNA gaps behind the fork in NΔ-SMARCAD1 cells upon 53BP1 knockdown condition (Fig. 5D). As the severe defects in restart of replication forks in NΔ-SMARCAD1 were correlated with the poor recovery of PCNA, we next sought to determine whether 53BP1 knockdown would also restore PCNA levels in NΔ-SMARCAD1 cells. Consistently, QIBC analysis showed that upon HU-mediated block, PCNA levels were significantly reduced in replicating cells even upon 53BP1 knockdown. However, PCNA showed a significant recovery in NΔ-SMARCAD1 similar to WT, when released from HU-mediated block (Fig. 5E and fig. S4E) under these conditions. Consistent with the restoration of PCNA levels, we also observed a marked reduction in chromatin-bound ATAD5 levels upon knockdown of 53BP1 in NΔ-SMARCAD1 (fig. S4F), suggesting that 53BP1 further promotes PCNA unloading in absence of SMARCAD1 at forks through ATAD5 activity. The potential protein-protein interaction between 53BP1 and ATAD5 was further confirmed by yeast two-hybrid assay (fig. S4G) and by chromatin IP of 53BP1 (fig. S4H), showing positive interaction in WT cells that further enhances under either HU-induced replication stress conditions in WT cells or under unperturbed conditions of NΔ-SMARCAD1 cells, both of which shows enhanced accumulation of stalled forks (Fig. 3C). We also noticed that the higher molecular weight band of ATAD5 was mainly immunoprecipitated with 53BP1 in chromatin IPs, which was further confirmed by notable reduction in signal of potentially phosphorylated ATAD5 band in cells targeted with si-ATAD5 (fig. S4H). The ATR-mediated phosphorylated form of ATAD5 has been reported to interact with RAD51 at stalled/regressed forks previously (34, 35). Together, these data suggest that 53BP1 interaction with ATAD5 regulates PCNA levels at stalled forks. Because loss of 53BP1 rescued genome instability, as monitored by the reduction of accumulated ssDNA gaps in NΔ-SMARCAD1 (Fig. 5D), we next determined whether 53BP1 knockdown rescues the sensitivity of NΔ-SMARCAD1 cells toward replication poisons. We observed a significant restoration of resistance toward cisplatin and olaparib treatment after the depletion of 53BP1 in NΔ-SMARCAD1 cells (Fig. 5F). Together, these data imply that SMARCAD1 maintains fine PCNA levels by suppressing unscheduled 53BP1 accumulation at the active replication forks and thereby maintains genome stability and replication stress tolerance in the cells.
From these data, we further hypothesized that chromatin remodeling activity of SMARCAD1 is possibly required to displace 53BP1-associated nucleosomes to suppress the untimely accumulation of 53BP1-ATAD5 complex at replication forks. To investigate this, we generated knock-ins of complementary DNA (cDNA)–SMARCAD1 that were either WT or contained an ATPase-disabling K528R mutation that can interact with replication forks but is defective in nucleosome remodeling activity, unlike NΔ-SMARCAD1 that does not associate with replication forks at all (Fig. 1G) (20). As expected, we observed a rescue in fork progression defects in NΔ-SMARCAD1 cells when corrected with fully functional SMARCAD1 but not with ATPase-dead K528R SMARCAD1 (fig. S4I). Moreover, K528R SMARCAD1 showed significant defects in fork progression and PCNA levels similar to NΔ-SMARCAD1 (fig. S4, I and J). We further confirmed that defects of 53BP1 displacement at restarted forks observed in NΔ-SMARCAD1 also existed in ATPase-dead SMARCAD1, detected by proximity ligation assay (PLA) approach between EdU and 53BP1 (fig. S4K). Furthermore, we also detected higher levels of 53BP1-associated ubiquitinated histone H2A lysine 15 (H2AK15Ub) nucleosomes at restarted forks in both ATPase-dead and NΔ-SMARCAD1 cells (fig. S4L). These data strongly suggest that the chromatin remodeling activity of SMARCAD1 is required to evict 53BP1-associated nucleosomes to displace 53BP1-ATAD5 complex, preventing PCNA recovery at restarted forks, causing replication fork restart and progression defects.
SMARCAD1-mediated active fork stability confers survival in BRCA1-mutated tumors, irrespective to their HR status
Our data imply that SMARCAD1-mediated replication fork stability contributes to genome stability in a manner independent of its role in HR repair of DSBs. Similarly, HR-independent roles in the protection of stalled forks during replication stress have been uncovered for BRCA1 and BRCA2 (2, 3, 5–7). To further test whether SMARCAD1 also protects stalled forks, similar to BRCA1, we investigated fork degradation using DNA fiber assay. Loss of BRCA1 resulted in stalled fork degradation upon 3 hours of exposure to 4 mM HU, while NΔ-SMARCAD1 showed no significant defects in fork protection similar to WT (Fig. 6A). Furthermore, as shown previously, longer exposure of cells to 4 mM HU (up to 8 hours) resulted in a moderate but significant processing of forks in WT cells (36), and we observed similar effects in NΔ-SMARCAD1, while loss of BRCA1 led to severe fork degradation (Fig. 6A). Further, these data also suggest that SMARCAD1 is not defective in the processing of stalled forks, as proposed for its fission yeast homolog (37). Thus, these data along with fork progression data (Fig. 3, A and B), taken together, suggest that replication defects observed in absence of SMARCAD1 are due to defective active replication fork stability and not due to defective stalled fork protection or fork processing activities. Furthermore, in the absence of SMARCAD1, unperturbed cells showed frequent stalling of replication forks without subsequent accumulation of DSBs (Fig. 3, C and G), which could possibly be due to BRCA-mediated fork protection in SMARCAD1 mutant cells. To test this hypothesis, we knocked down BRCA1 transiently from MRC5 WT, NΔ-SMARCAD1, and SMARCAD1−/− cells to analyze replication fork dynamics. As previously reported, si-BRCA1 in WT cells showed no significant defects in the progression rate of unperturbed forks (2). However, in NΔ-SMARCAD1 and SMARCAD1−/− cells, loss of BRCA1 resulted in significantly shorter track length (fig. S5A), which could not be rescued by loss of 53BP1 (fig. S5B). These data suggest that upon loss of SMARCAD1, BRCA1 is required to maintain progression of forks, possibly by protecting stalled forks from DNA nuclease–mediated degradation to allow their restart. To test whether indeed loss of BRCA1 in SMARCAD1 mutants leads to increased DNA damage, we performed QIBC analysis for the DNA-damage marker γH2AX and observed significantly enhanced accumulation of DNA damage upon BRCA1 knockdown in both NΔ-SMARCAD1 and SMARCAD1−/− mutants compared to single mutants or WT cells (Fig. 6B), suggesting that BRCA1 could be required to protect stalled forks from degradation to prevent DNA damage accumulation.
Fig. 6. Smarcad1 is essential for fork progression and proliferation of BRCA1-deficient mouse tumor cells.
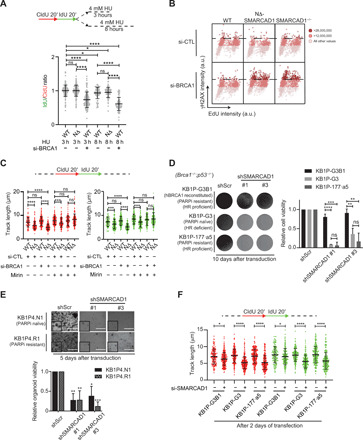
(A) Top: Schematic of replication fork degradation assay with CldU and IdU labeling. Bottom: Ratio of IdU to CldU tract length was plotted for the indicated conditions. ****P ≤ 0.0001 and *P ≤ 0.05, Kruskal-Wallis with Dunn’s multiple comparison test. (B) QIBC analysis of γH2AX versus EdU is shown in WT, NΔ-SMARCAD1, and SMARCAD1−/−cells under si-control and si-BRCA1 conditions. A total of >1000 cells were plotted under each condition. The color gradient represents the γH2AX levels in each cell. (C) Top: Schematic of replication fork progression assay with CldU and IdU labeling. Bottom: CldU (red) and IdU (green) track length (in micrometers) distribution for the indicated conditions. n = 3; ****P ≤ 0.0001 and ***P ≤ 0.001, Kruskal-Wallis followed by Dunn’s multiple comparison test. (D) Left: Representative images of KB1P (Brca1−/−;p53−/−) mouse tumor cells imaged at day 10, after transduction of scramble control shRNA and shSMARCAD1 #1 and #3. Right: Quantification of cell viability using crystal violet staining assay. Error bars stand for +SD. n = 3; ****P ≤ 0.0001, ***P ≤ 0.001, and **P ≤ 0.01, unpaired t test. (E) Top: Representative images of KB1P mouse tumor organoid. Image was taken 5 days after the transduction of scramble control shRNA and shSMARCAD1 #1 and #3. Scale bars, 1000 μm. Bottom: Quantification of cell viability using cell titer blue assay. Error bars stand for +SD. n = 3; ***P ≤ 0.001, **P ≤ 0.01, and *P ≤ 0.05, unpaired t test. (F) Top: Schematic of replication fork progression assay. Bottom: CldU (red) and IdU (green) track length (in micrometers) distribution in KB1P mouse tumor cells treated with si-control or si-SMARCAD1. n = 3; ****P ≤ 0.0001 and *P ≤ 0.05, Kruskal-Wallis followed by Dunn’s multiple comparison test.
As previously reported, BRCA1 protects stalled forks from degradation mediated by DNA nuclease Mre11 (7). Therefore, to test this hypothesis, we treated cells with Mirin, an inhibitor of DNA nuclease Mre11, and monitored fork progression using a DNA fiber assay. Notably, Mirin treatment completely rescues the severe fork progression defects observed upon loss of BRCA1 in the SMARCAD1 mutant (Fig. 6C). These data suggest that, in the absence of SMARCAD1 stalled forks indeed require BRCA1 protection to allow fork progression and maintain genome integrity.
Previously, SMARCAD1 was reported to play a critical role in the metastasis of triple-negative breast cancer (38, 39). To test whether differential levels of SMARCAD1 expression could be an indicator of patient responses to replication stress–inducing platinum chemotherapy, we analyzed patients with high-grade serous ovarian cancer (HGSOC) for their correlation between BRCA1 and SMARCAD1 expression levels to their response to chemotherapy. Survival analysis demonstrated that platinum-treated BRCA1-low patients, but not BRCA1-high patients, with low SMARCAD1 expression were correlated with a longer progression-free survival (PFS), while higher expression of SMARCAD1 correlated was with poor response to chemotherapy (fig. S5C). These data suggest that SMARCAD1 levels could be a biomarker for acquired resistance to platinum-based chemotherapy in BRCA1-low/deficient ovarian cancers.
To further verify this experimentally, we queried whether SMARCAD1 is required for fork progression in BRCA1-deficient tumor cells and whether its loss could hypersensitize HR-deficient BRCA1−/− mouse breast tumor cells generated using K14Cre;Brca1F/F;p53F/F (KB1P) mouse mammary tumor models (40). We generated short hairpin RNA (shRNA)–mediated knockdowns of SMARCAD1 in Brca1−/−;p53−/− defective mouse breast tumor–derived cell lines (fig. S5D). Unexpectedly, the loss of SMARCAD1 resulted in a significant reduction in colony formation in HR-defective BRCA1−/− (KB1P-G3; PARPi naïve) (41) tumor cells but not in KB1P-G3 tumor cells that were reconstituted with human BRCA1 (KB1P-G3B1) and proficient in HR (42), suggesting that loss of SMARCAD1 causes synthetic lethality in BRCA1-deficient tumor cells (Fig. 6D). These data indicate a potential role of SMARCAD1 in maintaining active fork stability, which may be the reason for the survival of BRCA1-deficient HR-defective tumor cells. Furthermore, we also tested whether BRCA1 and 53BP1 double-knockout tumor cells, which are proficient for HR and resistant to PARPi treatments (KB1P-177.a5; PARPi resistant) (41), require SMARCAD1 for proliferation. A SMARCAD1 knockdown, again, resulted in lethality in these cells, suggesting that SMARCAD1’s role is essential for the proliferation of BRCA-defective tumor cells, irrespective of their HR status (Fig. 6D). Furthermore, 53BP1 deficiency in BRCA1-defective genetic background could not rescue defects of SMARCAD1 knockdown, which suggests that fork protection mediated by BRCA1 becomes critical for cellular survival in the absence of SMARCAD1, similar to what we observed in human fibroblast cells (fig. S5, A and B). In addition, we tested the effect of SMARCAD1 knockdown on KB1P-derived, PARPi-naïve (KB1P4.N), and PARPi-resistant (KB1P4.R) tumor organoids grown in ex vivo cultures (43). Consistent with our results in KB1P tumor cell lines, we observed a synthetic lethality in the three-dimensional (3D) tumor organoids, suggesting that SMARCAD1 is essential for the survival of BRCA1-mutated tumors (Fig. 6E). These data strongly suggest a conserved and nonepistatic role of SMARCAD1 and BRCA1 at replication forks.
Because BRCA1-deficient cells show reduced fork protection and high levels of endogenous stress (7, 44), we hypothesized that the loss of SMARCAD1 further enhances replication stress due to the defective progression of forks, causing proliferation defects. To test this hypothesis, we used siRNA to transiently deplete SMARCAD1 protein (45) in KB1P 2D tumor-derived cell lines (fig. S5E) to monitor individual fork progression using DNA fiber assay. We sequentially labeled human BRCA1-reconstituted, KB1P-G3B1 cells as control, KB1P-G3 (HR deficient), and KB1P-177.a5 (chemoresistant; HR proficient) with CldU (red) and IdU (green), followed by track length analysis. In support to the survival assays, although sublethal SMARCAD1 knockdown affects only mildly the cell cycle of all three cell lines (fig. S5F), it led to a significantly shorter track lengths of both CldU and IdU in both KB1P-G3 and KB1P-177 cells in comparison to BRCA1-reconstituted KB1P-G3B1 cells, suggesting an essential role of SMARCAD1 in mediating fork progression in the absence of BRCA1 (Fig. 6F). Consistently, we also observed the reduction in PCNA levels and enhanced 53BP1 enrichments at the fork, using the PLA approach with EdU, upon loss of SMARCAD1 in BRCA1−/− mouse tumor cells, similar to human cells (fig. S5, G and H). Together, these results strongly suggest that the SMARCAD1-mediated stability of active replication forks is a physiologically important process for cellular proliferation of BRCA1-deficient tumors, irrespective of their HR status (fig. S6).
DISCUSSION
Our study has revealed a novel mechanism of active fork stability that has important implications in the survival of tumor cells.
A genetically distinct role of SMARCAD1 at active replication forks, from HR
As opposed to the commonly attributed role of DNA repair factors in replication fork protection (6, 7, 9, 46), here, we identify a new function of SMARCAD1 in maintaining the stability of active (unperturbed and restarted) replication forks, while its absence does not disturb stalled fork protection and fork processing activities (Figs. 3, A and B, and 6A and fig. S2I). Using a separation-of-function SMARCAD1 mutant (NΔ-SMARCAD1), we show that SMARCAD1’s role in stabilization of active replication forks is genetically separable from its role in HR repair and is critical in maintaining genome stability especially upon replication stress. The physical interaction between SMARCAD1 and PCNA, established using in vitro and in vivo assays (21), was suggested to be responsible for SMARCAD1’s association with replication machinery (21, 22). Our biochemical and immunofluorescence assays further confirm that the NΔ-SMARCAD1 protein, lacking initial 137 amino acids, can bind to chromatin but lacks the ability to interact with PCNA. This finding is consistent with the lack of association between NΔ-SMARCAD1 and replication forks, as previously suggested (22). However, other components may also be involved in promoting SMARCAD1’s association with replication machinery, such as phosphorylation of SMARCAD1 by cyclin-dependent kinase (CDK). A CDK phosphorylation site at the N terminus of SMARCAD1 is among the 137 amino acids that are missing in the NΔ-SMARCAD1 protein (47). Nonetheless, the CUE-dependent protein-protein interactions and ATPase-dependent chromatin remodeling activity, in the context of HR repair and nuclear association, seem to remain functional in the NΔ-SMARCAD1 protein. Notably, cells with a transient depletion of SMARCAD1, SMARCAD1-null (SMARCAD1−/−) genotype, and those expressing the NΔ-SMARCAD1 allele show similar defects in fork progression, suggesting that it is the direct effect of loss of protein at the replication forks and not the secondary effects of mutants accumulating damages that result in slower fork progression. Furthermore, the similar sensitivity toward replication poisons of HR-proficient NΔ-SMARCAD1 and HR-deficient SMARCAD1−/− cells argues that the role of SMARCAD1 at replication forks is, in fact, crucial in mediating resistance to replication stress–inducing drugs rather than its role in HR.
Furthermore, our data showed evidence of frequent accumulation of stalled forks and ssDNA gaps behind the replication forks in NΔ-SMARCAD1 cells. The accumulation of ssDNA and stalled forks could be indicative of a hindered replication fork progression through certain difficult-to-replicate regions, such as highly transcribing regions or repetitive regions of the genome (48). Alternatively, ssDNA accumulation could also be a resultant of the repriming events by PRIMPOL at stalled forks that in the process of reinitiating, the DNA synthesis leads to the accumulation of ssDNA gaps (49, 50). However, in BRCA1-challenged cells, PRIMPOL activity was shown to be responsible for DNA synthesis upon replication stress condition. Here, our study shows a unique pathway of active fork stabilization mediated by SMARCAD1, which is critical for fork progression in BRCA1-deficient cells even under unperturbed conditions. This implies that SMARCAD1-mediated active replication fork stability is a central and a separate pathway for stabilization of replication forks than from recently described PRIMPOL-mediated fork repriming or well-established BRCA1-mediated fork protection pathway (51).
SMARCAD1 regulates PCNA levels at active replication forks
Our findings suggest a hitherto unrecognized role for SMARCAD1 in maintaining the fine control of PCNA levels at the forks. In this study, along with previously published study (21, 22), we have strong evidence of a positive interaction between SMARCAD1 and PCNA, which is also responsible for SMARCAD1 association with replication machinery. A global reduction in chromatin-bound PCNA levels at the fork and a faster dissociation rate of PCNA foci in NΔ-SMARCAD1 cells further suggest a mutualistic interaction between SMARCAD1 and PCNA at the replication forks (Fig. 4, C and D). Consistently, an increase in PCNA unloading by the ATAD5-RLC complex was observed in NΔ-SMARCAD1 cells. A recent report demonstrated a critical role of ATAD5 in the removal of PCNA from stalled forks to promote the recruitment of fork protection factors (34). Consistent with this report, we observed reduced PCNA levels at replication forks, accompanied by an increased accumulation of ATAD5-RLC complex and increased frequency of reversed forks (protected stalled forks) in unperturbed NΔ-SMARCAD1 cells. Furthermore, a significant number of peptides arising from RFC2-5 protein subunits that are shared between PCNA loading (RFC) and unloading (ATAD5-RLC) complexes were obtained from SMARCAD1 coimmunopurification (21). These data may indicate the direct involvement of SMARCAD1 in regulating loading/unloading activity of PCNA at replication forks. However, an interesting finding from our study is that loss of 53BP1 results in a significant restoration of PCNA levels in NΔ-SMARCAD1 cells accompanied with a significant reduction in ATAD5 levels at replication forks. Furthermore, the direct interaction observed between 53BP1 and ATAD5 in WT cells is enhanced in NΔ-SMARCAD1 cells or HU-treated WT cells possibly due to ATR-mediated posttranslationally modified ATAD5. Whether the posttranslational modification of ATAD5 is solely ATR mediated or additional mechanisms play a role in its regulation (34) could distinguish between the physiological roles of ATAD5 in regulating PCNA dynamics that involves continuous loading/unloading events during normal fork progression versus the persistent unloading of PCNA from stalled forks.
SMARCAD1 prevents 53BP1 accumulation to mediate tolerance to replication stress
Our study shows an unforeseen role of SMARCAD1 in preventing 53BP1 accumulation at active restarted replication forks. Previously, 53BP1 has been shown to bind to H2AK15Ub nucleosomes at DSBs (52), while SMARCAD1 was proposed to displace 53BP1-associated nucleosomes at DSBs to promote HR repair (20). This observation is consistent with the finding that SMARCAD1 and its homologs in yeast can slide, evict, and exchange H2A-H2B dimer, also regulating histone turnover in replicating cells of fission yeast cells (48, 53–55). Consistent with these observations, it has been shown that the loss of SMARCAD1 results in a prolonged enrichment of 53BP1 at DSBs (20, 25). SMARCAD1 and 53BP1 also show contrasting enrichments at stalled versus unperturbed forks, suggesting that their coexistence is possibly also prohibited by SMARCAD1 at replication forks in a manner similar to that of their interaction at DSBs (Figs. 1, A and C, and 5A) (8). Notably, we found increased 53BP1 and the histone epigentic mark that it associates with, at restarted forks in NΔ-SMARCAD1 and ATPase-dead SMARCAD1. These data imply that both the ability of SMARCAD1 to localize to forks and its chromatin remodeling activity are required to evict the 53BP1-associated nucleosomes to prevent untimely 53BP1-ATAD5 accumulation on active forks. As shown previously, the ATR-mediated phosphorylation of ATAD5, upon HU-induced stalled fork accumulation, interacts with proteins at reversed forks proteins (34). We suggest that in the absence of SMARCAD1, enhanced ATAD5-RLC levels causing PCNA dissociation from forks lead to frequent fork stalling and, consequently, accumulation of reversed forks resulting in activation of ATR checkpoint. The chromatin remodeling activity of SMARCAD1 is required to evict 53BP1-bound H2AK15Ub nucleosomes at reversed arm of stalled forks for their restart. However, in the absence of SMARCAD1, enhanced accumulation of 53BP1 possibly further stabilizes ATR-mediated phosphorylated ATAD5 at the reversed forks, which leads to continuous PCNA unloading, causing severe defects in restart/progression of forks. In addition to loss of 53BP1, the controlled depletion of ATAD5 could also restore normal PCNA levels at the fork that rescued the overall DNA synthesis and replication fork restart efficiency (Fig. 4, F to H). This suggests that active replication fork stability is indeed regulated by maintaining fine-controlled PCNA levels at the forks.
Furthermore, it was previously suggested that the loss of 53BP1 restores HR in SMARCAD1-depleted cells, which is responsible for developing resistance to replication stress-inducing drugs (20). However, this study using a separation-of-function SMARCAD1 mutant, which is HR proficient but defective for fork stability, shows that the extent of damage generated upon replication stress is rather responsible for the cellular sensitivity and not unrepaired DSBs due to lack of HR. This further suggests that the role of SMARCAD1 at forks is crucial for tolerance to replication stress–inducing agents. We have, therefore, revealed a moonlighting function of SMARCAD1 at the replication forks in displacing 53BP1 to maintain replication fork progression and genome stability. Other NHEJ factors such as mammalian Rap1-interacting factor 1 (RIF1), Pax transactivation domain-interacting protein (PTIP), and others have also been found in association with replication forks. Therefore, it would be interesting to investigate whether 53BP1 works in complex with NHEJ machinery or have a separate role in association with ATAD5-RLC complex to regulate PCNA homeostasis and thereby fork dynamics.
An essential role of SMARCAD1 in the viability of BRCA1-defective tumors
BRCA1/2 factors, independent of their role in HR, protect replication forks and prevent their collapse into genome-destabilizing DSBs (6, 7). Although SMARCAD1and BRCA1 have been shown to act epistatically during HR repair (20, 25), here, we show contrasting differences in role of SMARCAD1 and BRCA1 at replication forks that can be observed by (i) differential enrichment of SMARCAD1 and BRCA1 at the replication forks, where SMARCAD1 preferentially associates with active forks, while BRCA1 associates with stalled forks (Fig. 1A) (8); (ii) stalled forks in the absence of SMARCAD1 remain protected and do not degrade unlike in absence of BRCA1; (iii) loss of SMARCAD1 but not BRCA1 causes defects in unperturbed replication fork progression (Fig. 3A and fig. S5A) (2); and last, (iv) loss of 53BP1 in BRCA1-deficient cells that restores HR repair capacity does not rescue sensitivity of BRCA1 mutants to cisplatin treatment (Fig. 5F) (56). However, loss of 53BP1 in SMARCAD1 mutant rescues cisplatin sensitivity, suggesting that replication stress sensitivity is uncoupled from HR repair and that SMARCAD1’s role at active replication forks is distinct from that of BRCA1’s role at stalled replication forks to maintain tolerance toward replication stress–inducing agents. Thus, loss of SMARCAD1 results in enhanced accumulation of replication fork–associated DNA damage and, ultimately, synthetic lethality in mouse BRCA1-defective tumors irrespective of their HR status. Loss of 53BP1 could not rescue severe replication fork progression defects observed under SMARCAD1 and BRCA1 double-mutant condition. This suggests that frequently accumulated stalled forks in the absence of SMARCAD1 essentially require BRCA1-mediated fork protection, which could only be rescued by Mre11 inhibition. Together, these data suggest a distinct role of SMARCAD1 and BRCA1 at replication forks, acting in two independent pathways, where SMARCAD1 mediates active fork stability, while BRCA1 mediates stalled fork protection. However, both the pathways are interdependent for maintaining replication fork integrity, which is also conserved across species, from mouse to human.
In summary, we have found a distinct pathway of active fork stabilization mediated by SMARCAD1 and have shown a conserved interplay between SMARCAD1 and BRCA1 in stabilization of replication forks to maintain genome integrity (fig. S6). Notably, SMARCAD1-mediated stabilization of unperturbed forks promotes cellular proliferation in BRCA1-deficient mouse breast tumor, cells, and organoids, independently of their HR and PARPi resistance status. Similarly, the correlation of reduced chances of survival after chemotherapy in cancer patients with enhanced expression of SMARCAD1 along with reduced expression of BRCA1 suggests that stabilization of active forks promotes tolerance toward chemotherapy in BRCA1-defective tumors. Last, the observation that SMARCAD1 becomes essential for genome stability and cellular survival in the absence of BRCA1 suggests that targeting the stability of active replication forks has the potential to be a clinically effective remedy for BRCA-deficient tumors, naïve or chemoresistant. It also suggests that SMARCAD1 could be a strong candidate for development of novel therapeutic treatment for BRCA1-deficient cancer patients.
MATERIALS AND METHODS
Cell line generation
Plasmid transfections were performed using X-tremeGENE 9 DNA transfection agent (Roche) according to the manufacturer’s protocol. To generate MRC5 NΔ-SMARCAD1 cells, MRC5 WT cells were transfected with pLentiCRISPR-V2 plasmid (Addgene #52961) containing a guide RNA (gRNA) sequence targeting exon 2 of SMARCAD1, followed by puromycin selection (1 μg/ml). To generate MRC5 SMARCAD1−/−, two gRNA sequences targeting exon 2 and exon 24 of SMARCAD1 were selected and cotransfected with a homolog repair template containing an mClover gene.
To express mClover-SMARCAD1 full-length/SMARCAD1 K528R–mutant cDNA, gRNAs targeting SMARCAD1 exon 2 and exon 24 were cotransfected with mClover-SMARCAD1 full-length/ SMARCAD1 K528R–mutant cDNA, respectively, in MRC5 WT and NΔ-SMARCAD1 cells. The K528R mutant was generated by site-directed mutagenesis of full-length SMARCAD1 cDNA.
To generate GFP-tagged PCNA knock-in MRC5 cells, a gRNA sequence targeting exon 2 of PCNA was selected and inserted into lentiCRISPR V2 (Addgene #52961). MRC5 WT and NΔ-SMARCAD1 cells were transfected with the gRNA and the FLAG-GFP-PCNA repair template and sorted by fluorescence-activated cell sorting (FACS). Sequences of gRNAs and mutagenesis primers are listed in table S3.
Cell culture
All MRC5 human fibroblasts were cultured in a 1:1 ratio of Dulbecco’s modified Eagle’s medium (DMEM) and Ham’s F10 (Invitrogen) supplemented with 10% fetal calf serum (FCS; Biowest) and 1% penicillin-streptomycin (PS; Sigma-Aldrich) at 37°C and 5% CO2 in a humidified incubator. KB1P-G3, KB1P-177.a5 (41, 42), and KB1P-G3B1 (42) have been described previously. All KB1P mouse tumor cell lines were cultured in DMEM/F12 and GlutaMAX (Gibco) containing insulin (5 μg/ml; Sigma-Aldrich), cholera toxin (5 ng/ml; Sigma-Aldrich), murine epidermal growth factor (EGF; 5 ng/ml; Sigma-Aldrich), 10% FCS, and 1% PS under low oxygen conditions (3% O2 and 5% CO2 at 37°C).
All tumor-derived organoid lines have been described before (43). KB1P4.N1 and KB1P4.R1 tumor organoids were derived from a mammary KB1P PARPi-naïve and PARPi-resistant tumor, respectively (female donor). Cultures were embedded in Cultrex Reduced Growth Factor Basement Membrane Extract Type 2 (BME; Trevigen; 40 ml of BME:growth medium 1:1 drop in a single well of 24-well plate) and grown in Advanced DMEM/F12 (Gibco) supplemented with 1 M Hepes (Gibco), GlutaMAX (Gibco), PS (50 U/ml), B27 (Gibco), 125 mM N-acetyl-l-cysteine (Sigma-Aldrich), and EGF (50 ng/ml). Organoids were cultured under standard conditions (37°C and 5% CO2).
mESCs were maintained in 2i medium deficient in lysine, arginine, and l-glutamine (PAA) at 37°C and 5% CO2 in a humidified incubator. For SILAC labeling, cells were grown in medium containing light [12C6]-lysine (73 μg/ml) and [12C6, 14N4]-arginine (42 μg/ml) (Sigma-Aldrich) or similar concentrations of heavy [13C6]-lysine and [13C6, 15N4]-arginine (Cambridge Isotope Laboratories).
Method details
siRNA transfection, shRNA transduction, and cell titer assay
siRNA transfection was performed with Lipofectamine RNAiMAX (Thermo Fisher Scientific) according to the manufacturer’s protocol. Details of siRNA oligomers and shRNAs used in this study are given in table S3.
Transductions were performed in duplicate in KB1P mouse tumor cells. After 3 days of selection, KB1P mouse tumor cells were expanded to 10-cm dishes. Five days after passage, samples were fixed with 4% formaldehyde and stained with 0.1% crystal violet, and quantification was carried out by determining the absorbance at 590 nm after extraction with 10% acetic acid.
3D tumor-derived organoids were transduced according to a previously established protocol (43). Puromycin selection (3 μg/ml) was carried out for three consecutive days after transduction. Pictures were taken at day 5. For quantification, cells were incubated with CellTiter-Blue (Promega) reagent at day 5.
Chromatin fractionation
Cells were lysed in lysis buffer [30 mM Hepes (pH 7.6), 1 mM MgCl2, 130 mM NaCl, 0.5% Triton X-100, 0.5 mM dithiothreitol, and EDTA-free protease inhibitor], at 4°C for 30 min. Chromatin-containing pellet was spinned down by centrifugation at 16,000g for 10 min and resuspended in lysis buffer supplemented with Benzonase (250 U/μl; Merck Millipore) and incubated for 15 min at 4°C.
Live-cell confocal imaging
Live-cell confocal laser scanning microscopy was carried out as described before (57), with minor adjustments. All live-cell imaging experiments were performed using a Leica TCS SP5 microscope equipped with HCX PL APO CS 63× oil immersion objective, at 37°C and 5% CO2. For iFRAP, GFP-PCNA–expressing WT and NΔ-SMARCAD1 MRC5 cells were continuously bleached at high 488-nm laser outside the selected GFP-PCNA foci, and the fluorescence decrease in the selected foci was determined over time. The resulting dissociation curves were background corrected and normalized to prebleach values, set at 1.
DR-GFP reporter assay
The procedure for DR-GFP reporter was described previously (26) and applied with minor alterations. After being seeded in a six-well plate overnight, cells were cotransfected with DR-GFP reporter plasmid (Addgene #26475) and I-Scel expression vector (Addgene #26477) or empty vector using X-tremeGENE 9 DNA transfection agent (Roche) according to the manufacturer’s protocol for two consecutive days. p-MAX-GFP plasmid (Addgene #16007) was transfected in parallel to assess transfection efficiency. On day 3, GFP expression was analyzed by flow cytometer.
DNA fiber analysis
Cells were sequentially pulse labeled with 30 μM CldU (MP Biomedicals) and 250 μM IdU (Sigma-Aldrich) according to the schematic in each figure. For Mirin treatment, 100 μM Mirin was added to the medium for 2 hours before the experiment. DNA fiber analysis was carried out according to the standard protocol as mentioned previously (30). Fibers were visualized and imaged by Axio Imager D2 microscope (Carl Zeiss). ImageJ software was used for the quantification. The Kruskal-Wallis test followed by Dunn’s multiple comparison test was applied for statistical analysis using the GraphPad Prism software. The combined summary of DNA fiber spread data analysis is given in table S2.
Immunoblot and antibodies
After lysis with radioimmunoprecipitation assay (RIPA) buffer (whole-cell lysate) or resuspended in chromatin fractionation lysis buffer (chromatin-bound proteins), samples were mixed with 2× Laemmli sample buffer, boiled for 5 min, loaded on Bis-Tris Gel, and transferred to a polyvinylidene difluoride membrane. Membranes were blocked with 5% nonfat milk in tris-buffered saline (TBS) for 1 hour and incubated with primary antibody diluted in 5% bovine serum albumin (BSA) in TBS overnight at 4°C. Membranes were then washed in 0.1% Tween 20 in TBS, incubated with a secondary antibody coupled to near-IR dyes CF 680/770, and visualized using Odyssey CLx infrared scanner (LI-COR). ImageJ software was used for quantification. Primary and corresponding secondary antibodies are listed in table S4.
Immunofluorescence staining
Cells were labeled with EdU (10 μM) for 30 min, unless otherwise mention. For HU-treated samples, EdU was labeled before the treatment. In analysis of chromatin-bound protein, cells were first preextracted with 0.1% Triton X-100 in cytoskeletal (CSK) buffer on ice and then fixed in 4% formaldehyde in phosphate-buffered saline (PBS) for 15 min at room temperature for SMARCAD1, 53BP1, RAD51, and γH2AX or 100% −20°C methanol for 10 min for PCNA. Subsequently, samples were permeabilized in 0.1% Triton X-100 in PBS for 10 min, blocked with 5% BSA in PBS, and stained with a primary antibody diluted in blocking buffer, followed by incubation in fluorescence-conjugated secondary antibody. EdU was visualized with a Click-IT reaction using Alexa Fluor 488 azide or Alexa Fluor 594 azide (Invitrogen) according to the manufacturer’s protocol. Samples were stained with 4′,6-diamidino-2-phenylindole (DAPI) and mounted on slides using ProLong Gold (Invitrogen).
Proximity ligation assay
Cells were washed once with 1× PBS, treated with 0.1% Triton X -100 in CSK buffer on ice, and fixed with ice-cold methanol for 10 min (PCNA) or with 4% formaldehyde (FA) in PBS for 15 min (53BP1 and H2AK15ub). Subsequently, cells were permeabilized with 0.1% Triton X-100 in PBS for 10 min and blocked with 5% BSA in PBS at room temperature for 1 hour. Afterward, cells were treated with Click-iT reaction according to the manufacturer’s protocol for 1 hour and were incubated with PCNA (PC10), 53BP1, H2AK15ub, and biotin at 4°C overnight in a humid chamber. After washes with PBS with 0.1%Tween-20 (PBST), cells were incubated with anti-mouse minus and anti-rabbit plus PLA probes (Sigma-Aldrich) at 37°C for 1 hour. Following the manufacturer’s instructions, the PLA reaction was performed with the Duolink In Situ Detection Reagents. Cells were stained with DAPI and mounted on slides using ProLong Gold. Images were captured using Metafer5 and quantified using MetaSystem.
Image acquisition and image analysis
Coverslip images were obtained using a LSM700 microscope equipped with a Plan-Apochromat 63×/1.4 oil objective (Carl Zeiss), MetaSystems5 equipped with an EC Plan-Neofluar 40×/0.75 objective (Carl Zeiss), or SP5 microscope equipped with HCX PL APO CS 63× oil objective (Leica). Detection of EdU-positive cells was performed in combination with the DAPI channel applying a cross entropy–based thresholding and binary watershed segmentation. The brightness and contrast adjustment was applied differently because of differential backgrounds in the indicated cell lines of Fig. 1G for the qualitative representation. To compute the Pearson and Manders’ overlap coefficients in fig. S4B, the 53BP1 foci in 488- and 568-nm channels for EdU-positive cells were segmented using an à trous wavelet transform with three scales, and the wavelet coefficients were thresholded at the level of 3-sigma (58). To measure the distance between 53BP1 and EdU foci in Fig. 5A, a line of 3 μm was drawn across the proximal foci, and the intensity of the two channels were measured using Multi Plot in ImageJ. Further analysis was performed using Microsoft Excel. For high-content imaging given in Figs. 1 (B and C), 2C, 4 (A and C), and 5E and figs. S1D and S2J, all the data were obtained using the Opera Phenix High-Content Screening System (PerkinElmer) with a 40× water objective (numerical aperture, 1.1) and analyzed with the Harmony v4.9 high-content imaging and analysis software (PerkinElmer). At least 75 fields were imaged as a Z-stack of eight planes (step size, 1 μm). In the maximum projection, nuclei were detected using DAPI. Selection of S phase cells was based on EdU signal under untreated (UT) and HU block condition. Under HU release conditions, S phase cells were determined by intensity of PCNA median. The pixel intensities (sum) were determined in DAPI, 488- and 568-nm channel for each nucleus. PCNA sum normalized to DAPI sum was shown in the bar chart. For quantification of EdU-positive foci in Fig. 1 (B and C) and fig. S1D, an additional mask was generated on the basis of the detection of local intensity maxima (region to spot intensity) in the EdU channel and was used for quantification of spot intensities together with spot contrast in the 488- and 568-nm channels. For quantification of foci in Fig. 2C and figs. S2A, S4 (K and L), and S5 (G and H), a mask was generated using the detection of spot contrast and intensity, with threshold for spot radius. The quantified values for each foci/cell were exported to the TIBCO Spotfire software.
RNA extraction, reverse transcription, real-time qPCR, and RNA-seq
Total RNA was extracted using the ReliaPrep RNA Miniprep Systems (Promega). One thousand nanograms of total RNA was used to synthetize cDNA using Moloney Murine Leukemia Virus Reverse Transcriptase, Ribonuclease H Minus, Point Mutant (Promega). qPCR was performed using the GoTaq qPCR Master Mix (Promega), and β-actin was used for normalization. Primers used for qPCR are listed in table S3.
Next-generation sequencing short reads were trimmed using fastp and processed using Kalliso, an RNA-seq quantification program that uses a pseudo-alignment method of assigning reads to genomic locations in lieu of a more costly traditional alignment (59). The human transcriptome, version GRCh38.p12, was indexed, the paired, trimmed reads were assigned to transcripts, and read counts were converted to transcripts per million (TPMs) by Kallisto. TPMs from transcripts originating from the same gene were aggregated, and relative expression levels were computed as the log2 fold change relative to the matched WT using an in-house script (available as a separate file in the Supplementary Materials). RPKM (reads per million kilobases) values were computed from TPMs using the median transcript length per gene.
Pseudo-alignments, output by Kallisto in a standard BAM format, were used to assess transcript structure such as the assignment of the transcription start for NΔ-SMARCAD1. Box plots and bar plots were produced using ggpubr and ggplot2, respectively, in the R program (the R Foundation).
iPOND-SILAC mass spectrometry
Light lysine and arginine labeled mESCs were incubated with 10 μM EdU for 10 min and treated with 4 mM HU for 3 hours. Heavy lysine and arginine labeled mESCs were incubated with 10 μM EdU for 10 min. iPOND mass spectrometry was performed essentially as described. At least two peptides were required for protein identification. Quantitation is reported as the log2 of the normalized heavy/light ratios. SILAC data were analyzed using MaxQuant. The resulting output tables of two independent experiments were merged and used as the input for calculating the average fold change to identify significantly up-regulated proteins in unperturbed forks and stalled forks based on the ratio of heavy and light peptides (H/L ratio) in the SILAC experiment in MaxQuant software (9).
Cross-linked IP
Cells were cross-linked in 1% formaldehyde in serum-free medium for 10 min at room temperature. Cross-linking reaction was quenched with 0.125 M glycine, and cells were washed with PBS. Cross-linked cells were scrapped, and chromatin was purified as described (57). Chromatin was sheared using a Bioruptor sonicator (Diagenode) using cycles of 20-s on, 60-s off during 15 min, after which samples were centrifuged. The supernatant containing cross-linked chromatin was used for IP. For native IP, cells were collected by trypsinization and lysed with lysis buffer [1 mM MgCl2, 150 mM NaCl, 1 mM EDTA, 0.5% NP-40, and 30 mM Hepes buffer (pH 7.6)] for 20 min at 4°C. Chromatin fraction was collected by spinning. DNA was fragmented by passing the lysed suspension 10 times through a needle attached to a 1-ml syringe, after which samples were centrifuged. The supernatant containing the chromatin fraction was used for IP.
For IP, extracts were incubated with GFP-Trap beads (ChromoTek), 53BP1 (1.8 μg), PCNA (1.8 μg), or SMARCAD1 (1.8 μg) antibody overnight at 4°C. For IP with PCNA, 53BP1, and SMARCAD1 antibodies, protein A agarose/salmon sperm DNA slurry (Millipore) was added for 4 hours at 4°C. Subsequently, beads were washed five times in RIPA buffer, and elution of the precipitated proteins was performed by extended boiling in 2× Laemmli sample buffer for immunoblotting analysis.
Clonogenic survival assay
Cells were seeded in triplicate in 10-cm culturing dish and treated with olaparib (Selleckchem), cisplatin (Sigma-Aldrich), or HU (Sigma-Aldrich) 1 day after seeding. HU was given at the indicated concentration for 24 or 48 hours, as indicated in the figure legend. Olaparib treatment was given throughout the whole experimental process. Different concentrations of cisplatin were given for 4 hours before being replaced with new medium, except the 1 μM cisplatin group in Fig. 5F, which were given throughout the whole experimental process.
After 1 week, colonies were fixed and stained in a mixture of 43% water, 50% methanol, 7% acetic acid, and 0.1% Brilliant Blue R (Sigma-Aldrich) and subsequently counted with GelCount (Oxford Optronix). The survival was plotted as the mean percentage of colonies detected following the treatment normalized to the mean number of colonies from the untreated samples.
Cell cycle analysis
Cells were grown to 70 to 80% confluency, labeled with EdU for 30 min, and fixed for 10 min in 4% formaldehyde in PBS at room temperature. Cells were then washed with 1% BSA/PBS and permeabilized in 0.5% saponin buffer in 1% BSA/PBS. EdU was labeled with the Click-iT reaction using Alexa Fluor 594 azide according to the manufacturer’s protocol (Invitrogen). DAPI was used to stain the DNA.
Electron microscope analysis
EM analysis was performed according to the standard protocol (9). For DNA extraction, cells were lysed in lysis buffer and digested at 50°C in the presence of proteinase K for 2 hours. The DNA was purified using chloroform/isoamyl alcohol, precipitated in isopropanol, given 70% ethanol wash, and resuspended in elution buffer. Isolated genomic DNA was digested with Pvu II high-fidelity restriction enzyme for 4 to 5 hours. After digestion, the DNA solution was transferred to a Microcon DNA fast flow centrifugal filter. The filter was washed with tris-EDTA (TE) buffer after spinning for 7 min. The benzyldimethylalkylammonium chloride method was used to spread the DNA on the water surface and then loaded on carbon-coated nickel grids, and last, DNA was coated with platinum using high-vacuum evaporator MED 010 (Bal-Tec). Microscopy was performed with a transmission electron microscope FEI Talos, with 4K by 4K complementary metal-oxide semiconductor camera. For each experimental condition, at least 200 replication fork intermediates were analyzed from three independent experiments, and MAPS software (Thermo Fisher Scientific) was used to analyze the images.
Pulsed-field gel electrophoresis
For HU-treated samples, cells were treated with 4 mM HU for 3 hours, following or not with a 16-hour release, before harvesting for PFGE assay. DSB detection by PFGE was performed as reported previously (9). The gel was stained with ethidium bromide and imaged on a Uvidoc-HD2 imager. ImageJ software was used for the quantification of broken DNA normalized to unbroken DNA for each lane.
Purification of SMARCAD1 and mass spectrometry
NΔ-SMARCAD1 protein was purified from whole-cell lysate using MRC5 NΔ-SMARCAD1 cell line. Cells were resuspended in the IP buffer, sheared 10 times as 15-s on and then 45-s off at mode high using a Bioruptor sonicator (Diagenode) at 4°C, and incubated with 500 U of Benzonase (Merck Millipore) for 60 min, after which samples were centrifuged. The supernatant was used for IP. For IP, extracts were incubated with SMARCAD1 (1.8 μg) antibody overnight at 4°C. Protein A agarose/salmon sperm DNA slurry (Millipore) was added for 2 hours at 4°C. Subsequently, beads were washed five times in IP buffer, and elution of the protein was performed by extensive boiling in 2× Laemmli sample buffer. Eluted protein was run on bis-tris gel, gel slices were trypsinized, and peptides were analyzed by mass spectrometry to determine the protein sequence as described previously (57).
Bioinformatic analysis on The Cancer Genome Atlas datasets
Disease-free survival curves of The Cancer Genome Atlas (TCGA) patients with HGSOC were generated by the Kaplan-Meier method, and differences between survival curves were assessed for statistical significance with the log-rank test. We divided the TCGA patients with ovarian carcinoma expressing replication stress markers (CCNE1 overexpression, CDKN2A-low expression, and/or RB1 deletion) into cohorts according to their BRCA1 mRNA expression levels: BRCA1 low (below median) and BRCA1 high (above median) (60). In each of these cohorts, we analyzed the correlation between SMARCAD1 expression and outcome. Normalization of expression values was performed using z score transformation, such that SMARCAD1-low expression with z score < 0.75 and SMARCAD1-high expression with z score > 0.75 (fig. S5C). Cohort with BRCA1-high and SMARCAD1-low expression, n = 66; BRCA1-low and SMARCAD1-high expression, n = 10. Cohort with BRCA1-low and SMARCAD1-low expression, n = 87; BRCA1-low and SMARCAD1-high expression, n = 10.
Yeast two-hybrid analysis
Human 53BP1 full length was fused to the LexA protein in pBTM116 and was coexpressed with human ATAD5 full length fused to the GAL4 activation domain in pGAD-HA in the yeast strain L40. Interactions were assayed using the (LexAop)4-HIS3 reporter system.
Quantification and statistical analysis
For all data, the means, SD, and SEM were calculated using either Microsoft Excel or GraphPad Prism 8.
Acknowledgments
We thank R. Kanaar, W. Vermeulen, and C. Wyman for stimulating discussions and sharing important reagents used in the manuscript; K. Myung and K. Lee for ATAD5 antibody and sharing technical information; D. Chowdhury for advice on PFS analysis; P. Zegerman for Y2H reagents; and E. Goggola for help with the initial phase of mouse tumor cells culture. We acknowledge infrastructural support from the Josephine Nefkens Precision Cancer Treatment Program. Funding: This work was supported by grant NWO-Vidi (193.131) and the Dutch Cancer Society funded grant (11008/2017-1) to A.R.C., the Oncode Institute partly financed by the Dutch Cancer Society funded grant (KWF grant 10506) to J.A.M. and J.J., the European Union’s Horizon 2020 research and innovation program under the Marie Sklodowska-Curie grant (agreement no. 722729) to J.J., Daniel den Hoed Stitching Young Investigator Award grant (DDHS no. 10834) to N.T., and start-up funds from the Erasmus MC to N.T. Author contributions: C.S.Y.L. conducted all the QIBC, FACS, and PFGE experiments. M.v.T. performed iFRAP and chromatin fractionation experiments and, with help from Y.Z., performed chromatin IP experiments. V.G. performed all the DNAfiber and immunofluorescence experiments related to ATAD5. M.P.D. performed all the cloning experiments and clonogenic assays using mouse tumor cells/organoids under the supervision of J.J. Y.Z. with the help of M.v.d.D. performed cloning experiments of cDNA-SMARCAD1. E.M.M. with help from C.S.Y.L. performed clonogenic assays with MRC5 cells and chromatin fractionations for RAD51. H.L. helped C.S.Y.L. and M.v.d.D. in cloning experiments in MRC5 cells. M.E.v.R., W.Z., and I.S. analyzed fluorescence microscopy data. The assistance to use high-content imaging microscope facility was provided by M.E.v.R and P.J.F. J.D. analyzed mass spectrometry data. J.G.S.C.S.G. analyzed TCGA ovarian BRCA data. D.W. analyzed RNA-seq data. J.A.M. supervised the iFRAP and chromatin fractionation experiments. A.R.C. supervised the iPOND experiments performed by C.M. and EM experiments performed by E.M.M. N.T. conceptualized the project, supervised it, and wrote the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors. NCBI BioProject accession number is as follows: PRJNA609878.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/19/eabe7804/DC1
REFERENCES AND NOTES
- 1.Lord C. J., Ashworth A., Mechanisms of resistance to therapies targeting BRCA-mutant cancers. Nat. Med. 19, 1381–1388 (2013). [DOI] [PubMed] [Google Scholar]
- 2.Chaudhuri A. R., Callen E., Ding X., Gogola E., Duarte A. A., Lee J. E., Wong N., Lafarga V., Calvo J. A., Panzarino N. J., John S., Day A., Crespo A. V., Shen B., Starnes L. M., de Ruiter J. R., Daniel J. A., Konstantinopoulos P. A., Cortez D., Cantor S. B., Fernandez-Capetillo O., Ge K., Jonkers J., Rottenberg S., Sharan S. K., Nussenzweig A., Erratum: Replication fork stability confers chemoresistance in BRCA-deficient cells. Nature 539, 456 (2016). [DOI] [PubMed] [Google Scholar]
- 3.Ding X., Chaudhuri A. R., Callen E., Pang Y., Biswas K., Klarmann K. D., Martin B. K., Burkett S., Cleveland L., Stauffer S., Sullivan T., Dewan A., Marks H., Tubbs A. T., Wong N., Buehler E., Akagi K., Martin S. E., Keller J. R., Nussenzweig A., Sharan S. K., Synthetic viability by BRCA2 and PARP1/ARTD1 deficiencies. Nat. Commun. 7, 12425 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gogola E., Duarte A. A., de Ruiter J. R., Wiegant W. W., Schmid J. A., de Bruijn R., James D. I., Llobet S. G., Vis D. J., Annunziato S., van den Broek B., Barazas M., Kersbergen A., van de Ven M., Tarsounas M., Ogilvie D. J., van Vugt M., Wessels L. F. A., Bartkova J., Gromova I., Andújar-Sánchez M., Bartek J., Lopes M., van Attikum H., Borst P., Jonkers J., Rottenberg S., Selective loss of PARG restores PARylation and counteracts PARP inhibitor-mediated synthetic lethality. Cancer Cell 33, 1078–1093.e12 (2018). [DOI] [PubMed] [Google Scholar]
- 5.Mijic S., Zellweger R., Chappidi N., Berti M., Jacobs K., Mutreja K., Ursich S., Ray Chaudhuri A., Nussenzweig A., Janscak P., Lopes M., Replication fork reversal triggers fork degradation in BRCA2-defective cells. Nat. Commun. 8, 859 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schlacher K., Christ N., Siaud N., Egashira A., Wu H., Jasin M., Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell 145, 529–542 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schlacher K., Wu H., Jasin M., A distinct replication fork protection pathway connects Fanconi anemia tumor suppressors to RAD51-BRCA1/2. Cancer Cell 22, 106–116 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dungrawala H., Rose K. L., Bhat K. P., Mohni K. N., Glick G. G., Couch F. B., Cortez D., The replication checkpoint prevents two types of fork collapse without regulating replisome stability. Mol. Cell 59, 998–1010 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mukherjee C., Tripathi V., Manolika E. M., Heijink A. M., Ricci G., Merzouk S., de Boer H. R., Demmers J., van Vugt M. A. T. M., Ray Chaudhuri A., RIF1 promotes replication fork protection and efficient restart to maintain genome stability. Nat. Commun. 10, 3287 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Moldovan G. L., Pfander B., Jentsch S., PCNA, the maestro of the replication fork. Cell 129, 665–679 (2007). [DOI] [PubMed] [Google Scholar]
- 11.Yu C., Gan H., Han J., Zhou Z. X., Jia S., Chabes A., Farrugia G., Ordog T., Zhang Z., Strand-specific analysis shows protein binding at replication forks and PCNA unloading from lagging strands when forks stall. Mol. Cell 56, 551–563 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kanellis P., Agyei R., Durocher D., Elg1 forms an alternative PCNA-interacting RFC complex required to maintain genome stability. Curr. Biol. 13, 1583–1595 (2003). [DOI] [PubMed] [Google Scholar]
- 13.Kang M. S., Ryu E., Lee S. W., Park J., Ha N. Y., Ra J. S., Kim Y. J., Kim J., Abdel-Rahman M., Park S. H., Lee K. Y., Kim H., Kang S., Myung K., Regulation of PCNA cycling on replicating DNA by RFC and RFC-like complexes. Nat. Commun. 10, 2420 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kubota T., Katou Y., Nakato R., Shirahige K., Donaldson A. D., Replication-coupled PCNA unloading by the Elg1 complex occurs genome-wide and requires Okazaki fragment ligation. Cell Rep. 12, 774–787 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mejlvang J., Feng Y., Alabert C., Neelsen K. J., Jasencakova Z., Zhao X., Lees M., Sandelin A., Pasero P., Lopes M., Groth A., New histone supply regulates replication fork speed and PCNA unloading. J. Cell Biol. 204, 29–43 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang Z., Shibahara K., Stillman B., PCNA connects DNA replication to epigenetic inheritance in yeast. Nature 408, 221–225 (2000). [DOI] [PubMed] [Google Scholar]
- 17.Choe K. N., Moldovan G. L., Forging ahead through darkness: PCNA, still the principal conductor at the replication fork. Mol. Cell 65, 380–392 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Johnson C., Gali V. K., Takahashi T. S., Kubota T., PCNA retention on DNA into G2/M phase causes genome instability in cells lacking Elg1. Cell Rep. 16, 684–695 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tan Z., Wortman M., Dillehay K. L., Seibel W. L., Evelyn C. R., Smith S. J., Malkas L. H., Zheng Y., Lu S., Dong Z., Small-molecule targeting of proliferating cell nuclear antigen chromatin association inhibits tumor cell growth. Mol. Pharmacol. 81, 811–819 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Densham R. M., Garvin A. J., Stone H. R., Strachan J., Baldock R. A., Daza-Martin M., Fletcher A., Blair-Reid S., Beesley J., Johal B., Pearl L. H., Neely R., Keep N. H., Watts F. Z., Morris J. R., Human BRCA1-BARD1 ubiquitin ligase activity counteracts chromatin barriers to DNA resection. Nat. Struct. Mol. Biol. 23, 647–655 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rowbotham S. P., Barki L., Neves-Costa A., Santos F., Dean W., Hawkes N., Choudhary P., Will W. R., Webster J., Oxley D., Green C. M., Varga-Weisz P., Mermoud J. E., Maintenance of silent chromatin through replication requires SWI/SNF-like chromatin remodeler SMARCAD1. Mol. Cell 42, 285–296 (2011). [DOI] [PubMed] [Google Scholar]
- 22.Mermoud J. E., Rowbotham S. P., Varga-Weisz P. D., Keeping chromatin quiet: How nucleosome remodeling restores heterochromatin after replication. Cell Cycle 10, 4017–4025 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Costelloe T., Louge R., Tomimatsu N., Mukherjee B., Martini E., Khadaroo B., Dubois K., Wiegant W. W., Thierry A., Burma S., van Attikum H., Llorente B., The yeast Fun30 and human SMARCAD1 chromatin remodellers promote DNA end resection. Nature 489, 581–584 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Toledo L. I., Altmeyer M., Rask M. B., Lukas C., Larsen D. H., Povlsen L. K., Bekker-Jensen S., Mailand N., Bartek J., Lukas J., ATR prohibits replication catastrophe by preventing global exhaustion of RPA. Cell 155, 1088–1103 (2013). [DOI] [PubMed] [Google Scholar]
- 25.Chakraborty S., Pandita R. K., Hambarde S., Mattoo A. R., Charaka V., Ahmed K. M., Iyer S. P., Hunt C. R., Pandita T. K., SMARCAD1 phosphorylation and ubiquitination are required for resection during DNA double-strand break repair. iScience 2, 123–135 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pierce A. J., Johnson R. D., Thompson L. H., Jasin M., XRCC3 promotes homology-directed repair of DNA damage in mammalian cells. Genes Dev. 13, 2633–2638 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lange S. S., Takata K., Wood R. D., DNA polymerases and cancer. Nat. Rev. Cancer 11, 96–110 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Follonier C., Oehler J., Herrador R., Lopes M., Friedreich’s ataxia-associated GAA repeats induce replication-fork reversal and unusual molecular junctions. Nat. Struct. Mol. Biol. 20, 486–494 (2013). [DOI] [PubMed] [Google Scholar]
- 29.Neelsen K. J., Lopes M., Replication fork reversal in eukaryotes: From dead end to dynamic response. Nat. Rev. Mol. Cell Biol. 16, 207–220 (2015). [DOI] [PubMed] [Google Scholar]
- 30.Ray Chaudhuri A., Hashimoto Y., Herrador R., Neelsen K. J., Fachinetti D., Bermejo R., Cocito A., Costanzo V., Lopes M., Topoisomerase I poisoning results in PARP-mediated replication fork reversal. Nat. Struct. Mol. Biol. 19, 417–423 (2012). [DOI] [PubMed] [Google Scholar]
- 31.Zellweger R., Dalcher D., Mutreja K., Berti M., Schmid J. A., Herrador R., Vindigni A., Lopes M., Rad51-mediated replication fork reversal is a global response to genotoxic treatments in human cells. J. Cell Biol. 208, 563–579 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sakato M., O’Donnell M., Hingorani M. M., A central swivel point in the RFC clamp loader controls PCNA opening and loading on DNA. J. Mol. Biol. 416, 163–175 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee K. Y., Fu H., Aladjem M. I., Myung K., ATAD5 regulates the lifespan of DNA replication factories by modulating PCNA level on the chromatin. J. Cell Biol. 200, 31–44 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Park S. H., Kang N., Song E., Wie M., Lee E. A., Hwang S., Lee D., Ra J. S., Park I. B., Park J., Kang S., Park J. H., Hohng S., Lee K. Y., Myung K., ATAD5 promotes replication restart by regulating RAD51 and PCNA in response to replication stress. Nat. Commun. 10, 5718 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sau S., Liefshitz B., Kupiec M., The yeast PCNA unloader Elg1 RFC-like complex plays a role in eliciting the DNA damage checkpoint. MBio 10, e01159-19 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Thangavel S., Berti M., Levikova M., Pinto C., Gomathinayagam S., Vujanovic M., Zellweger R., Moore H., Lee E. H., Hendrickson E. A., Cejka P., Stewart S., Lopes M., Vindigni A., DNA2 drives processing and restart of reversed replication forks in human cells. J. Cell Biol. 208, 545–562 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ait-Saada A., Khorosjutina O., Chen J., Kramarz K., Maksimov V., Svensson J. P., Lambert S., Ekwall K., Chromatin remodeler Fft3 plays a dual role at blocked DNA replication forks. Life Sci. Alliance 2, e201900433 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Al Kubaisy E., Arafat K., De Wever O., Hassan A. H., Attoub S., SMARCAD1 knockdown uncovers its role in breast cancer cell migration, invasion, and metastasis. Expert Opin. Ther. Targets 20, 1035–1043 (2016). [DOI] [PubMed] [Google Scholar]
- 39.Arafat K., al Kubaisy E., Sulaiman S., Karam S. M., al Natour Z., Hassan A. H., Attoub S., SMARCAD1 in breast cancer progression. Cell. Physiol. Biochem. 50, 489–500 (2018). [DOI] [PubMed] [Google Scholar]
- 40.Henneman L., van Miltenburg M. H., Michalak E. M., Braumuller T. M., Jaspers J. E., Drenth A. P., de Korte-Grimmerink R., Gogola E., Szuhai K., Schlicker A., Bin Ali R., Pritchard C., Huijbers I. J., Berns A., Rottenberg S., Jonkers J., Selective resistance to the PARP inhibitor olaparib in a mouse model for BRCA1-deficient metaplastic breast cancer. Proc. Natl. Acad. Sci. U.S.A. 112, 8409–8414 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jaspers J. E., Kersbergen A., Boon U., Sol W., van Deemter L., Zander S. A., Drost R., Wientjens E., Ji J., Aly A., Doroshow J. H., Cranston A., Martin N. M. B., Lau A., O’Connor M. J., Ganesan S., Borst P., Jonkers J., Rottenberg S., Loss of 53BP1 causes PARP inhibitor resistance in Brca1-mutated mouse mammary tumors. Cancer Discov. 3, 68–81 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Barazas M., Gasparini A., Huang Y., Küçükosmanoğlu A., Annunziato S., Bouwman P., Sol W., Kersbergen A., Proost N., de Korte-Grimmerink R., van de Ven M., Jonkers J., Borst G. R., Rottenberg S., Radiosensitivity is an acquired vulnerability of PARPi-resistant BRCA1-deficient tumors. Cancer Res. 79, 452–460 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Duarte A. A., Gogola E., Sachs N., Barazas M., Annunziato S., R de Ruiter J., Velds A., Blatter S., Houthuijzen J. M., van de Ven M., Clevers H., Borst P., Jonkers J., Rottenberg S., BRCA-deficient mouse mammary tumor organoids to study cancer-drug resistance. Nat. Methods 15, 134–140 (2018). [DOI] [PubMed] [Google Scholar]
- 44.Tacconi E. M., Lai X., Folio C., Porru M., Zonderland G., Badie S., Michl J., Sechi I., Rogier M., Matía García V., Batra A. S., Rueda O. M., Bouwman P., Jonkers J., Ryan A., Reina-San-Martin B., Hui J., Tang N., Bruna A., Biroccio A., Tarsounas M., BRCA1 and BRCA2 tumor suppressors protect against endogenous acetaldehyde toxicity. EMBO Mol. Med. 9, 1398–1414 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ding D., Bergmaier P., Sachs P., Klangwart M., Rückert T., Bartels N., Demmers J., Dekker M., Poot R. A., Mermoud J. E., The CUE1 domain of the SNF2-like chromatin remodeler SMARCAD1 mediates its association with KRAB-associated protein 1 (KAP1) and KAP1 target genes. J. Biol. Chem. 293, 2711–2724 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hashimoto Y., Ray Chaudhuri A., Lopes M., Costanzo V., Rad51 protects nascent DNA from Mre11-dependent degradation and promotes continuous DNA synthesis. Nat. Struct. Mol. Biol. 17, 1305–1311 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bantele S. C. S., Pfander B., Nucleosome remodeling by Fun30SMARCAD1 in the DNA damage response. Front. Mol. Biosci. 6, 78 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Taneja N., Grewal S. I. S., Shushing histone turnover: It’s FUN protecting epigenome-genome. Cell Cycle 16, 1731–1732 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bai G., Kermi C., Stoy H., Schiltz C. J., Bacal J., Zaino A. M., Hadden M. K., Eichman B. F., Lopes M., Cimprich K. A., HLTF promotes fork reversal, limiting replication stress resistance and preventing multiple mechanisms of unrestrained DNA synthesis. Mol. Cell 78, 1237–1251.e7 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Quinet A., Tirman S., Jackson J., Šviković S., Lemaçon D., Carvajal-Maldonado D., González-Acosta D., Vessoni A. T., Cybulla E., Wood M., Tavis S., Batista L. F. Z., Méndez J., Sale J. E., Vindigni A., PRIMPOL-mediated adaptive response suppresses replication fork reversal in BRCA-deficient cells. Mol. Cell 77, 461–474.e9 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Berti M., Cortez D., Lopes M., The plasticity of DNA replication forks in response to clinically relevant genotoxic stress. Nat. Rev. Mol. Cell Biol. 21, 633–651 (2020). [DOI] [PubMed] [Google Scholar]
- 52.Fradet-Turcotte A., Canny M. D., Escribano-Díaz C., Orthwein A., Leung C. C. Y., Huang H., Landry M. C., Kitevski-LeBlanc J., Noordermeer S. M., Sicheri F., Durocher D., 53BP1 is a reader of the DNA-damage-induced H2A Lys 15 ubiquitin mark. Nature 499, 50–54 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Awad S., Ryan D., Prochasson P., Owen-Hughes T., Hassan A. H., The Snf2 homolog Fun30 acts as a homodimeric ATP-dependent chromatin-remodeling enzyme. J. Biol. Chem. 285, 9477–9484 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Byeon B., Wang W., Barski A., Ranallo R. T., Bao K., Schones D. E., Zhao K., Wu C., Wu W. H., The ATP-dependent chromatin remodeling enzyme Fun30 represses transcription by sliding promoter-proximal nucleosomes. J. Biol. Chem. 288, 23182–23193 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Navarro C., Lyu J., Katsori A. M., Caridha R., Elsasser S. J., An embryonic stem cell-specific heterochromatin state promotes core histone exchange in the absence of DNA accessibility. Nat. Commun. 11, 5095 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bunting S. F., Callén E., Kozak M. L., Kim J. M., Wong N., López-Contreras A. J., Ludwig T., Baer R., Faryabi R. B., Malhowski A., Chen H. T., Fernandez-Capetillo O., D’Andrea A., Nussenzweig A., BRCA1 functions independently of homologous recombination in DNA interstrand crosslink repair. Mol. Cell 46, 125–135 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wienholz F., Zhou D., Turkyilmaz Y., Schwertman P., Tresini M., Pines A., van Toorn M., Bezstarosti K., Demmers J. A. A., Marteijn J. A., FACT subunit Spt16 controls UVSSA recruitment to lesion-stalled RNA Pol II and stimulates TC-NER. Nucleic Acids Res. 47, 4011–4025 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Smal I., Loog M., Niessen W., Meijering E., Quantitative comparison of spot detection methods in fluorescence microscopy. IEEE Trans. Med. Imag. 29, 282–301 (2010). [DOI] [PubMed] [Google Scholar]
- 59.Bray N. L., Pimentel H., Melsted P., Pachter L., Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 34, 525–527 (2016). [DOI] [PubMed] [Google Scholar]
- 60.The Cancer Genome Atlas Research Network , Genome Atlas Research, Integrated genomic analyses of ovarian carcinoma. Nature 474, 609–615 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/19/eabe7804/DC1