

A photoactivated fluorescent voltage sensor protein combines high sensitivity and optical sectioning in tissue.
Abstract
Photoactivated genetically encoded voltage indicators (GEVIs) have the potential to enable optically sectioned voltage imaging at the intersection of a photoactivation beam and an imaging beam. We developed a pooled high-throughput screen to identify archaerhodopsin mutants with enhanced photoactivation. After screening ~105 cells, we identified a novel GEVI, NovArch, whose one-photon near-infrared fluorescence is reversibly enhanced by weak one-photon blue or two-photon near-infrared excitation. Because the photoactivation leads to fluorescent signals catalytically rather than stoichiometrically, high fluorescence signals, optical sectioning, and high time resolution are achieved simultaneously at modest blue or two-photon laser power. We demonstrate applications of the combined molecular and optical tools to optical mapping of membrane voltage in distal dendrites in acute mouse brain slices and in spontaneously active neurons in vivo.
INTRODUCTION
Improved tools for voltage imaging in tissue would be a powerful enabling capability across broad domains of neuroscience (1–3). In many neurons, electrical dynamics can be substantially different in distal dendrites compared to the soma. Dendritic voltage is thought to be critical for neuronal computation (4). Despite some recent progress in dendritic voltage imaging with voltage-sensitive dyes (5–7), measurement of dendritic voltage in intact tissue with genetically encoded reporters remains a technical challenge due to the small size of distal dendrites and, hence, the small in-focus signals relative to fluorescence background from surrounding tissue. For somatic recordings in vivo, signals from out-of-focus cells can confound inference of subthreshold dynamics (8, 9), so optically sectioned voltage imaging is also important for quantitative measurements.
Recent advances in fluorescent genetically encoded voltage indicators (GEVIs) have enabled one-photon (1P) fluorescence voltage imaging in near-surface neurons in mice in vivo (10–13) and simultaneous voltage imaging and optogenetic stimulation in vivo with near-infrared archaerhodopsin (Arch)–derived GEVIs (8, 11, 13). Using a random-access two-photon (2P) imaging system, Chamberland and co-workers (14) mapped back-propagating action potentials (bAPs) in organotypic slice cultures, but these measurements required averaging of 20 to 30 trials and used a GEVI whose kinetics substantially distorted the waveforms. In our previous work, we 50,000 recorded near-soma bAPs in the mouse hippocampus in vivo with 1P imaging of a fast Arch-derived GEVI but also had to average ~90 spikes (11). The GEVI ASAP3 has been combined with 2P microscopy for single-trial recordings of near-soma dendrites in layer 5 pyramidal neurons in live mice (15), and the GEVI Voltron has been used for 1P single-trial recordings of specialized dendritic structures in Drosophila (10). However, existing combinations of GEVIs and microscopes have not simultaneously achieved sufficiently high voltage sensitivity, background rejection, and time resolution for single-trial mapping of dendritic voltage in distal dendrites, and despite recent advances in 2P voltage imaging (15, 16), high-resolution optically sectioned measurements in vivo remain challenging.
Scanning 2P microscopy has revolutionized Ca2+ imaging in vivo (17), so a natural impulse is to apply the same technique to voltage imaging. It is technically challenging to scan a single point focus with sufficient speed to sample membrane voltage from multiple regions simultaneously at the kilohertz rates needed for voltage imaging while also maintaining the submicron pointing relative to the brain tissue needed to intersect the focus with the cell membranes (15, 16, 18–20). Microbial rhodopsin-derived GEVIs, which have the greatest speed and sensitivity, have not been compatible with 2P voltage imaging (21). Quantitative estimates by us (21) and others (22) suggest that large-area 2P voltage imaging in intact tissue will be technically difficult with any GEVI.
We explored whether the complex photocycles of microbial rhodopsins could open the door to nontraditional optically sectioned voltage imaging techniques. Microbial rhodopsins have multiple conformational states, which interconvert through light- and/or voltage-dependent processes (23). One can further engineer or evolve these proteins to have specific photocycle topologies connecting fluorescent states and voltage-sensitive states. We previously engineered these multistate dynamics to develop microbial rhodopsin reporters for absolute voltage (24), light-gated voltage integrators, and a light-gated voltage sample and hold (25).
The V59A mutation in the Arch-derived GEVI QuasAr3 introduced an unusual property: The fluorescence under red 1P excitation was reversibly enhanced about twofold by weak blue illumination. This photoactivated mutant, which we termed paQuasAr3, provided a route to partial optical sectioning in tissue (11). Intersecting a red and blue beam in the focal plane enhanced the signal-to-background ratio (SBR). The 1P photoactivation of paQuasAr3 was beneficial for voltage imaging of near-surface neurons in vivo but was insufficient to enable optically sectioned imaging at greater depth or in distal dendrites.
Here, we evolve paQuasAr3 into a variant, NovArch, with enhanced photoactivation contrast under conditions useful for imaging in tissue. To apply NovArch in tissue, we combined either 1P or 2P photoactivation with 1P structured illumination epifluorescence imaging. Together, the photoactivation and wide-field imaging gave good optical sectioning and high signal-to-noise ratio (SNR) voltage recordings. We imaged bAPs with high sensitivity in dendrites in acute mouse brain slices and demonstrated optically sectioned recordings of spontaneous activity in hippocampal neurons in vivo.
RESULTS
2P reversibly photoactivatable GEVI for optical sectioning
We began by verifying and extending the prior measurements (11) of photoactivation in paQuasAr3. In human embryonic kidney (HEK) 293T cells expressing paQuasAr3, red illumination (λ = 640 nm, 90 W/cm2) induced weak near-infrared fluorescence (FR). Addition of much dimmer blue light (λ = 488 nm, 12 W/cm2) on top of the red illumination sharply increased the fluorescence (Fig. 1A). The fluorescence from simultaneous red and blue illumination (FR,B) was, on average, more than twice the sum of the fluorescence from red-only (FR) and blue-only (FB) illumination (Fig. 1B) (FB may partially have come from Citrine fluorescence leakage through the emission filter), confirming that the blue illumination enhanced the overall brightness of the protein. We quantified the nonadditive fluorescence relative to red illumination alone by (FR,B-FB-FR)/FR = ∆Fblue/FR and found ∆Fblue/FR = 1.3 ± 0.5 (mean ± SD, n = 95 cells; Figs. 1B and 3D), consistent with the earlier report (11). The parent protein, QuasAr3, was previously reported to show only very slight (~1%) photoactivation under blue light illumination (11).
Fig. 1. Photoactivation of paQuasAr3.
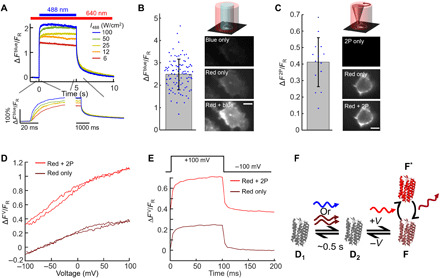
(A) Fluorescence response of paQuasAr3 to blue (488 nm) illumination superposed on steady red illumination (640 nm, 90 W/cm2). Representative recording from a single cell. Inset: Magnified views of the fluorescence dynamics when the blue light was applied and removed. (B) In HEK cells expressing paQuasAr3, blue light (488 nm, 12 W/cm2) enhanced red-excited (640 nm, 90 W/cm2) near-infrared fluorescence (n = 95 cells). Right: Images of HEK cell under the three illumination conditions, shown with the same brightness and contrast scales. Scale bars, 10 μm. (C) Same experiment as (B), with the blue light replaced by 3-mW focused 900-nm laser light scanned along the perimeter of the cell (2P) (n = 12 cells). Error bars in (B) and (C) indicate SDs. (D) Voltage response of paQuasAr3 without (brown) and with (red) 2P activation. Fluorescence changes for both traces are measured relative to, and normalized by, fluorescence with red-only illumination at −70 mV (FR). (E) Voltage step response of paQuasAr3 without (brown) and with (red) 2P activation. Fluorescence changes for both traces are measured relative to, and normalized by, fluorescence with red-only illumination at −100 mV (FR). Data in (D) and (E) are representative recordings from n = 4 cells. (F) Model photocycle for photoactivation and voltage sensitivity in paQuasAr3. States D1 and D2 are nonfluorescent. State F can be excited by red light and emits in the near infrared.
Fig. 3. NovArch is a photoactivated GEVI.
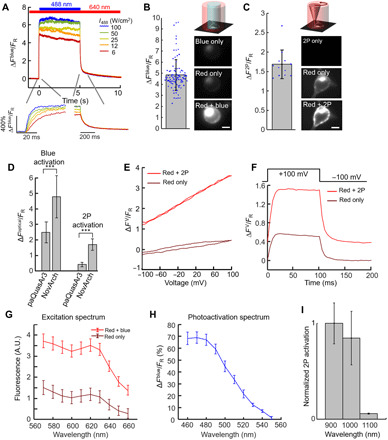
(A) Representative fluorescence response of NovArch to blue and red (90 W/cm2) illumination. Inset: Fluorescence turn-on and turn-off dynamics. (B) In HEK cells expressing NovArch, blue light (12 W/cm2) significantly enhanced red-excited (90 W/cm2) fluorescence (n = 95 cells). Right: HEK cell under the three illumination conditions. Scale bars, 10 μm. (C) Same as (B), with blue light replaced by 3-mW focused 900-nm pulsed laser scanned along the perimeter of the cell (2P) (n = 12 cells). (D) Comparison of fluorescence enhancements in paQuasAr3 and NovArch when photoactivated with blue light (12 W/cm2; P = 3 × 10−5, two-tailed t test) or with 2P illumination (3 mW, P = 8 × 10−12, two-tailed t test). Data from Figs. 1 (B and C) and 3 (B and C), means ± SD. (E) Voltage response of NovArch. Fluorescence changes are measured relative to, and normalized by, fluorescence with red-only illumination at −70 mV (FR). See also fig. S1C. (F) Voltage step response of NovArch. Fluorescence changes are measured relative to, and normalized by, fluorescence with red-only illumination at −100 mV (FR). See also fig. S1D. Data in (D) and (E) are representative recordings from n = 4 cells. (G) Fluorescence excitation spectrum of NovArch without (brown) and with (red) blue light photoactivation. A.U., arbitrary unit. (H) Single-photon photoactivation spectrum of NovArch. (I) 2P photoactivation spectrum of NovArch. Error bars in (G) to (I) represent SDs of successive measurements on a single sample.
We then tested whether the blue activation photon could be replaced by two infrared photons coherently absorbed from a femtosecond laser pulse. Figure 1C shows that 2P excitation (λ = 900 nm, 3 mW) scanned along the periphery of a HEK cell expressing paQuasAr3 reversibly enhanced the red-excited near-infrared emission, while 2P illumination alone did not induce detectable near-infrared fluorescence. We defined (FR,2P-F2P-FR)/FR = ∆F2P/FR and found ∆F2P/FR = 0.4 ± 0.1 (mean ± SD, n = 14 cells, P900nm = 3 mW; Figs. 1C and 3D). We ascribe the smaller 2P enhancement compared to the 1P blue light enhancement to the fact that the 2P scan pattern only covered a portion of the cell membrane. QuasAr3 did not show detectable enhanced fluorescence under 2P infrared excitation (fig. S1A).
We then quantified the effect of fluorescence sensitization on the voltage response of paQuasAr3. Fluorescence under red light alone showed a voltage sensitivity of ΔFR(V)/FR = 23 ± 6% for a voltage increase from −70 to +30 mV (mean ± SD, n = 4 cells; Fig. 1D). Addition of either 2P or blue illumination increased both the baseline fluorescence (F) and the voltage-induced changes in fluorescence (ΔFV) by the same factor, so when each trace was normalized by its own value at −70 mV, ΔFV/F curves approximately overlapped for all illumination conditions (fig. S1D). Under red-only illumination, the fluorescence responded to a step in membrane voltage with a fast time constant of 1.6 ms (72%) and a slow time constant of 31 ms (28%; Fig. 1E). Neither 2P nor blue illumination affected the kinetics of the voltage response (fig. S1E).
These unusual observations suggested the photocycle model shown in Fig. 1F. Blue illumination shifted an equilibrium between a pair of dark states {D1 ⇌ D2} toward the D2 state. The D2 state, in turn, had a voltage-dependent equilibrium with a fluorescent state, {D2 ⇌ F}. Cessation of blue illumination led to a gradual reversion toward the nonfluorescent D1 state. The blue photon could be replaced by two coherently absorbed near-infrared photons. While the underlying photocycle is likely more complex, this model captures the main features of the data. The key aspect for what follows is that the sensitizing light acts catalytically, rather than stoichiometrically, in governing fluorescence: Absorption of a single blue photon or a pair of infrared photons can lead to many cycles of red excitation and near-infrared fluorescence. The photocycle of Fig. 1F suggested that it might be possible to perform optically sectioned voltage imaging in tissue by intersecting a 1P blue or 2P infrared photoactivation beam with a 1P red imaging beam.
Screening for improved photoactivatable GEVIs
To achieve optically sectioned voltage imaging in tissue, we first had to improve the photoactivation ratio beyond the ∆Fblue/FR = 1.3 observed for 1P activation of paQuasAr3. We used a recently developed screening technique, termed Photostick, to select single cells optically from a pooled library culture, using photoactivation as the selection criterion (Fig. 2A) (26). Starting with paQuasAr3, we made a library of mutants using error-prone polymerase chain reaction (PCR) at a mean mutation rate of 2.8 mutations per gene. This pooled library was introduced into a lentiviral vector. HEK cells were infected with this library at low multiplicity of infection (~10−3) to ensure that few cells were infected with two viral particles. The expressing cells were selected with puromycin and plated at a density of ~50,000/cm2 on coverslip-bottomed dishes.
Fig. 2. Screening of NovArch.
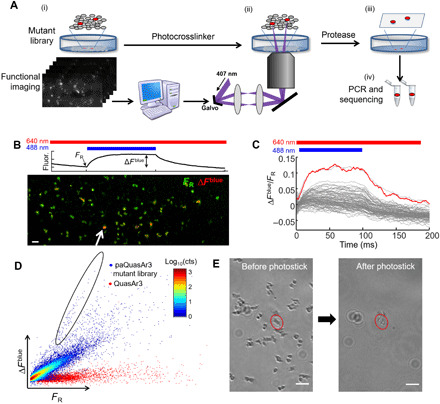
(A) Photostick screening protocol (26). (i) In a pooled library of HEK cells expressing mutants of paQuasAr3, wide-field fluorescence measurements probed blue light sensitization of red-excited fluorescence. Cells with large enhancement were identified via automated image processing. (ii) Focused illumination with 407-nm light cross-linked target cells to the dish. (iii) Nontarget cells were removed via a rinse with protease. (iv) The targeted cells were detached by gentle pipetting, and mutant genes were amplified and sequenced. (B) Top: Illumination protocol and parameters calculated for each cell to quantify photoactivation. Bottom: Example field of view comprising HEK cells expressing mutants of paQuasAr3. Most cells expressed nonfluorescent mutants and were not visible. Green, baseline fluorescence FR under red-only illumination (640 nm, 20 W/cm2); red, increase in fluorescence, ∆Fblue, upon photoactivation with 488-nm light, 0.1 W/cm2. Scale bar, 100 μm. (C) Fluorescence responses, ∆Fblue/FR, for individual cells in the field of view shown in (B). Red line shows response of the cell indicated with white arrow in (B). (D) Scatterplot of photoactivation, ∆Fblue, and baseline brightness, FR, for paQuasAr3 mutants across all fields of view and all dishes (blue dots). Control measurements with QuasAr3 showed no photoactivation (red dots). Selected cells (circled region) had high photoactivation contrast (ratio of blue activation to baseline red fluorescence). cts, counts. (E) Photostick selection of a cell expressing a targeted mutant. Left: Culture dish before photostick. Right: Same field of view after cross-linking the target cell to the dish and removing nontarget cells. Scale bars, 100 μm.
The cells were then imaged on a custom-built ultrawide-field microscope equipped with a low-magnification (2×), high–numerical aperture (NA 0.5) objective (fig. S2) (27), which yielded a field of view 1.2 mm by 3.3 mm, comprising nominally ~2 × 103 cells. Near-infrared fluorescence of each cell was quantified under red-only (FR), blue-only (FB), and simultaneous red and blue (FR,B) illumination. Materials and Methods contain a detailed discussion of the illumination protocol, the calibration of the red and blue illumination profiles, and the image processing steps. Our figure of merit was the blue light–induced nonlinear enhancement in fluorescence, ΔFblue/FR (Fig. 2, B and C). We screened 57 fields of view, corresponding to ~105 cells. Cells with high brightness and large fractional enhancement in fluorescence upon blue illumination were judged to be hits (Fig. 2D).
Hits were isolated via photochemical cross-linking to the dish. A photochemical cross-linker, 4-fluoro-3-nitrophenyl azide (final concentration, 15 μM), was added to the imaging medium. A pair of galvo mirrors directed light from a 405-nm laser (15 mW) sequentially to each selected cell for ~200 ms (Materials and Methods). The cross-linker immobilized the target cells to the dish. Nontarget cells were removed via a wash with trypsin (Fig. 2E). Target cells were then aspirated into ~10 μl of phosphate-buffered saline (PBS) and dispersed for single-cell PCR. The QuasAr mutant gene was amplified from each target, sequenced, and recloned for validation and testing.
The hit with greatest photoactivation contained two additional mutations on the paQuasAr3 background: V209I and I213T (fig. S3). We dubbed paQuasAr3(V209I, I213T) “NovArch” for its rapid increase in brightness (similar to an astronomical nova) upon blue illumination. We cloned additional constructs in which each novel mutation was individually reversed, expressed in HEK cells, and tested the nonlinear photoactivation. None of these constructs had photoactivation as large as NovArch (fig. S4A), demonstrating that both novel mutations were important.
Photophysical characterization of NovArch
We expressed NovArch in HEK cells and measured the near-infrared fluorescence under red illumination (λ = 640 nm, 90 W/cm2) with blue pulses superposed at variable intensity. Under red-only excitation, NovArch had similar brightness to paQuasAr3 (fig. S4). We observed an up to eightfold enhancement in near-infrared fluorescence upon saturating blue illumination (∆Fblue/FR = 7; Fig. 3A), with ~50% maximum enhancement at a blue intensity of Iblue = 3.9 W/cm2 (fig. S1B). For blue illumination intensity compatible with tissue imaging, Iblue = 12 W/cm2, the blue illumination–induced nonlinear enhancement in fluorescence was ∆Fblue/FR = 4.8 ± 0.9 (mean ± SD, n = 95 cells; Fig. 3, B and D).
We compared the brightness in HEK cells of NovArch to Archon1, a recently developed Arch variant that has been reported to have improved brightness (fig. S1C) (28). Under red-only illumination, NovArch was substantially dimmer than Archon1 (230 ± 120 counts per cell versus 450 ± 440 counts, mean ± SD). Under red and blue illumination (Iblue ≈ 26 W/cm2), the NovArch fluorescence increased to (1000 ± 510 counts) more than twice as bright as Archon1. To control for variations in expression level, we divided the near-infrared retinal fluorescence by the fluorescence of an appended Citrine tag. This ratio was 0.2 ± 0.06 for NovArch under red-only illumination, 0.88 ± 0.24 for NovArch under red and blue illumination, and 0.30 ± 0.25 for Archon1. These results establish that without blue light sensitization, NovArch is only ~70% as bright as Archon1, but with blue light sensitization, NovArch is about threefold brighter than Archon1.
Upon onset of blue illumination, NovArch fluorescence rose with single-exponential kinetics and time constant inversely related to illumination intensity: At Iblue = 6 W/cm2, we measured τup = 14 ms, and at Iblue = 100 W/cm2, we measured τup = 4 ms (Fig. 3A, bottom). At the lower blue intensity (6 W/cm2), this time constant implies an absorption cross section for the photoactivation process of σ = 4.8 × 10−18 cm2, corresponding to an extinction coefficient ε = 1270 M−1 cm−1. Upon cessation of the blue illumination, the fluorescence enhancement decayed with double-exponential kinetics, comprising a fast component (τ1 = 69 ± 5 ms, 80 ± 2%) and a slow component (τ2 = 2.7 ± 0.16 s, 20 ± 2%, n = 6 measurements; Fig. 3A). NovArch did not produce detectable photocurrent under red, blue, or combined illumination at holding potentials of either 0 or −60 mV (fig. S4B). Under continuous high-intensity illumination (Iblue = 22 W/cm2 and Ired = 140 W/cm2), NovArch photobleached to 50% of its initial intensity in 650 s (fig. S4C).
We next tested the 2P activation of NovArch. HEK cells expressing NovArch were exposed to wide-field red illumination as above, and illumination from an ultrafast 2P-pulsed laser (λ = 900 nm) was scanned around the periphery of the cell at 500 Hz. The 2P illumination reversibly enhanced the NovArch fluorescence 2.7-fold (Fig. 3C), corresponding to ∆F2P/FR = 1.7 ± 0.4 (mean ± SD, n = 12 cells; Fig. 3D). Upon onset of the 2P illumination, the fluorescence rose with single-exponential kinetics and time constant inversely related to illumination power: At P2P = 2 mW, we measured τup = 160 ms, and at P2P = 12 mW, we measured τup = 40 ms. Upon turning off the 2P illumination, the fluorescence enhancement decayed with double-exponential kinetics, comprising a fast component (τ1 = 169 ± 17 ms, 35 ± 5%) and a slow component (τ2 = 2.5 ± 0.14 s, 65 ± 5%, n = 6 measurements; fig. S4E).
NovArch showed good voltage sensitivity under red-only illumination ( = 41 ± 7% from −70 to +30 mV, n = 4 cells; Fig. 3E) and speed of response (upstroke: 1.2 ms, 76% and 10 ms, 24%; downstroke: 2.7 ms, 76% and 15 ms, 24%; Fig. 3F). As with paQuasAr3, addition of either 2P or blue illumination increased F and ΔFV proportionally, so when each trace was normalized by its own value at −70 mV, the ΔFV/F curves approximately overlapped (fig. S1D). Neither blue nor 2P sensitization affected the kinetics of the voltage response (Fig. 3F and fig. S1E).
To determine the optimal wavelengths for NovArch imaging and activation, we measured the fluorescence excitation and photoactivation spectra. We expressed NovArch in HEK cells, centrifuged the cells to form a dense pellet, and performed spectroscopy in a homebuilt microscope system. Using light from a tunable supercontinuum laser (90 μW per band), we measured the direct fluorescence excitation spectrum, which peaked at 620 nm, validating the choice of 640-nm light for fluorescence excitation (Materials and Methods). We combined the tunable supercontinuum beam with a blue beam (488 nm, 1 mW) and measured the fluorescence excitation spectrum of the photoactivated state. This spectrum had a similar shape, but higher amplitude, compared to the unactivated state (Fig. 3G), confirming that the fluorescence was dominated by a single state whose population was modulated by the blue illumination, as in the photocycle model (Fig. 1F). We then combined the tunable supercontinuum beam with a red (λ = 635 nm, 1 mW) beam and measured the photoactivation action spectrum, which peaked at 470 nm (Fig. 3H), validating the choice of 488 nm as the activation wavelength. 2P activation was more efficient at λ = 900 nm than at 1000 or 1100 nm (Fig 3I). 2P photoactivation showed saturation behavior, with ~50% maximum activation at a 2P laser power of 1.8 mW (fig. S4D).
We tested NovArch expression and its ability to report action potentials in cultured rat hippocampal neurons. NovArch showed excellent trafficking in the soma and throughout the dendritic arbor (Fig. 4A). Under wide-field red illumination (110 W/cm2), action potentials were detected in single-trial recordings with an amplitude of ΔFV/FR = 28 ± 5% (mean ± SD, n = 6 neurons; Fig. 4B). The fluorescence recordings showed close correspondence to simultaneous manual patch-clamp recordings (Fig. 4C). Addition of 2P illumination (900 nm, 3 mW) scanned around the cell periphery led to a 2.8-fold increase in both the baseline fluorescence and the spike amplitudes, i.e., ΔF2P/FR = 1.8 ± 0.4 (mean ± SD, n = 6; Fig. 4B), and preserved the correspondence between optically and electrically recorded waveforms (Fig. 4C). Compared to the baseline fluorescence in the absence of 2P activation, FR, the 2P-activated spikes had an amplitude of ΔFV,2P/FR = 79 ± 11% (mean ± SD, n = 6). To confirm that the 2P enhancement was not a simple consequence of additional fluorescence excitation by the 2P beam, we repeated the experiment on neurons expressing QuasAr3. Here, addition of 2P illumination did not affect the fluorescence signal (Fig. 4D).
Fig. 4. 2P-activated voltage imaging in cultured rat hippocampal neurons.
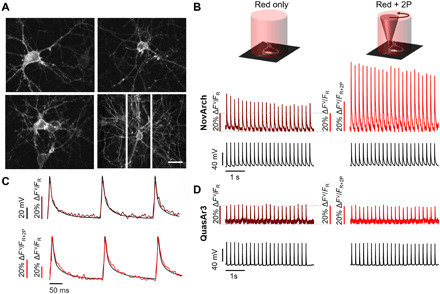
(A) Fluorescence of Citrine in NovArch-Citrine showed good membrane trafficking. Scale bar, 50 μm. (B) Simultaneous voltage and fluorescence recordings of action potentials evoked through current clamp stimulation via patch pipette. Application of 2P illumination scanned around the cell periphery enhanced the NovArch fluorescence. Vertical scale bars show fluorescence normalized by either red-only illumination (FR) or red +2P illumination (FR+2P). The increased amplitude of the spikes under 2P illumination reflects an increase in overall brightness of the reporter (see, e.g., Fig. 3E). (C) The fluorescence recordings show close correspondence to simultaneous manual patch-clamp recordings with (top) red-only illumination or (bottom) red + 2P illumination. (D) QuasAr3 also resolved action potentials but did not show 2P photoactivation. Data are representative recordings from n = 6 neurons expressing NovArch and n = 6 neurons expressing QuasAr3.
NovArch voltage imaging in tissue
We next sought to apply NovArch in tissue using sparsely expressed Cre-dependent constructs for paQuasAr3-Citrine and NovArch-Citrine, delivered via adeno-associated virus 2/9 (AAV2/9) (Materials and Methods). The primary obstacles to voltage imaging in tissue are scattering of excitation and emission light and the high background from out-of-focus fluorescence. We explored several optical approaches to maximize the SBR for NovArch in tissue.
Voltage-sensitive fluorescence comes only from the cell membranes, a two-dimensional manifold embedded in a three-dimensional medium. Any light that enters a tissue and misses the cell membrane contributes solely to background and not to useful signal. Thus, one should focus the illumination on the cell membrane only.
Figure 5 shows the impact of successively more precise optical targeting of excitation in acute brain slice. In each case, we first acquired a conventional 2P fluorescence image of the appended Citrine tag to provide ground truth on cell morphology and to guide the targeted illumination (Fig. 5A). For measurements of near-infrared GEVI fluorescence, we collected images on a back-illuminated electron multiplying charge-coupled device (EMCCD) camera (Andor Ixon3 860).
Fig. 5. Targeted illumination combined with 2P photoactivation enhances GEVI imaging in tissue.

(A) Scanned 2P images of Citrine fluorescence in acute brain slices expressing the indicated GEVI fused to Citrine. Top: A near-surface cell (depth, 20 μm). Bottom: Cells at greater depth (120 μm). (B) Wide-field illumination with red light produced a low-contrast image of fluorescence in a near-surface cell expressing paQuasAr3. No contrast was attained at a depth of 120 μm. (C) Restricting illumination to the soma improved SBR for near-surface cells but did not enable imaging of deeper cells. (D) Scanning a focus along the equatorial periphery of the cell improved SBR for near-surface cells and began to reveal outlines of cells at depth. (E) Coaligned 2P photoactivation and 1P fluorescence excitation along the equatorial periphery revealed the outline of a NovArch-expressing cell at a depth of 120 μm. (F) 2P enhancements calculated from the difference between the images in (E) and (D). (G) Quantification of image contrast along cross sections of the images shown in the corresponding colored boxes in (B) to (E). Scale bars, 10 μm. N.D., no data.
Wide-field illumination of a cell 20 μm below the surface produced a low-contrast image, with an SBR of only 0.19 (Fig. 5B). Wide-field illumination was clearly ill suited to voltage imaging in tissue because many photons missed the target cell altogether yet still contributed to background fluorescence. Closing a field aperture to restrict illumination just to the soma of the targeted cell improved the SBR more than eightfold to 1.55 (Fig. 5C).
Not all parts of the cell membrane contributed equally to the signal. Geometrical projection effects caused the edges of the cell to appear about threefold brighter than the middle, so excitation was about threefold more efficient when targeted to the edges. We used a pair of fast galvo mirrors to scan a red (640 nm) laser focus around the periphery. This “semiconfocal” approach (focal excitation and wide-field detection) further improved the SBR to 3.5, a 19-fold improvement over wide-field illumination (Fig. 5D). This enhancement is applicable to all GEVIs and does not require the photoactivation property of NovArch.
We then sought to image cells deeper in the slice. At a depth of 120 μm, wide-field illumination produced no detectable contrast. Cell-localized excitation produced a faint blur in the cell location. Scanning focal excitation and wide-field detection revealed a faint outline of the cell shape. We then coaligned the 2P laser focus (900 nm, 6.8 mW) with the 1P laser focus (637 nm, 1.7 mW; fig. S5) and jointly scanned the pair of lasers around the cell periphery. For paQuasAr3, this approach did not produce detectable enhancement, presumably because of the small photoactivation ΔF2P/FR for paQuasAr3. For NovArch, the 2P enhancement clearly revealed the outline of the cell (Fig. 5E). The 2P enhancement of the NovArch signal closely matched the conventional 2P image of the Citrine expression marker (Fig. 5F). These results demonstrate that 2P-activated 1P NovArch imaging can resolve single cells at depths where conventional 1P excitation wide-field or confocal approaches fail (Fig 5G).
We next applied 2P-enhanced NovArch imaging to study voltage signals in acute slice. Using 2P Citrine images to define the cell periphery, we coscanned 2P (900 nm, 6.8 mW) and 1P (637 nm, 1.7 mW) foci around cells at depths between 45 and 220 μm (Fig. 6, A and B, and fig. S6). The frequency of the galvo cycles was locked to the frame rate of the camera, both at 500 Hz. Fluorescence signals were low-pass filtered to an effective frame rate of 370 Hz. 2P sensitization enhanced the spike amplitude by a factor of 3.5 ± 0.5 (mean ± SEM, n = 14 neurons between 40 and 70 μm into the slice; Fig. 6C). The 2P-sensitized SNR was 96 ± 12 (mean ± SEM, n = 14 neurons between 40 and 70 μm into the slice). The SNR of single spikes diminished approximately exponentially with depth. The decay length was ~150 μm, both without and with 2P sensitization, and prefactors were 39 (without 2P sensitization) and 128 (with 2P sensitization; n = 28 neurons; Fig. 6C). With red-only semiconfocal illumination, single-trial action potentials were resolved to a depth of 100 μm. With 2P sensitization, single-trial action potentials were clearly resolved at a depth of 160 μm and were resolved with marginal SNR at 220 μm.
Fig. 6. Depth-resolved voltage imaging and all-optical electrophysiology in acute brain slice.
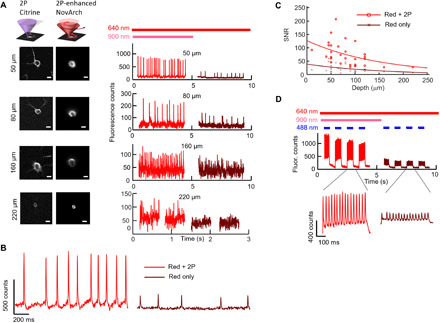
(A) Left: 2P images of Citrine acquired through conventional raster scanning and 2P-enhanced 1P fluorescence images acquired by scanning both foci along the cell periphery and acquiring fluorescence images on a camera. Scale bars, 10 μm. Right: Using 2P Citrine images to define the cell periphery, 2P and 1P foci were coaligned and scanned around neurons at different depths. The presence of 2P sensitization robustly enhanced fluorescence measurements of action potential amplitude. (B) Optical recording from a cell at a depth of 50 μm. (C) Depth-dependent SNR for single-trial AP detection without and with 2P sensitization. The lines represent fits to an exponential decay function. Paired measurements on n = 29 neurons. (D) Simultaneous optogenetic stimulation and 2P-enhanced NovArch imaging in tissue. Top: Pulses of blue light–excited repetitive spiking. 2P illumination–enhanced spike amplitude and SNR. Bottom: Magnified views of the optically induced spiking with and without 2P enhancement.
Combining NovArch imaging with optogenetic stimulation
A key merit of near-infrared GEVIs is that they are, in principle, compatible with optogenetic stimulation for all-optical electrophysiology. We thus sought to combine NovArch imaging with optogenetic stimulation of a blue-shifted channelrhodopsin, CheRiff. The complex photophysics of NovArch raised the possibility of two types of optical cross-talk: (i) The 2P beam used to sensitize NovArch could also activate CheRiff, leading to spurious activation of the target cell, and (ii) the blue light used to activate CheRiff could further sensitize NovArch, leading to spurious signals in the NovArch fluorescence channel.
We coexpressed NovArch and a blue-shifted channelrhodopsin, CheRiff, in acute slices (Materials and Methods) and used wide-field blue stimulation (488 nm, 60 mW/cm2) to activate the CheRiff. Figure 6D shows that tonic blue CheRiff stimulation induced high-frequency spiking but had only a small effect on the NovArch baseline. 2P illumination enhanced the NovArch fluorescence signal by a factor of 3.5 and did not induce spurious spontaneous activity. Combined optogenetic stimulation and 2P-sensitized NovArch imaging were achieved to a depth of 220 μm (fig. S6).
The absence of cross-talk in this system can be explained by the differing photophysical properties of NovArch and CheRiff. To maximize the fluorescence SNR, the 2P sensitization and 1P imaging beams were confined to the brightest part of the cell, the equatorial periphery. While the 2P beam likely excited CheRiff molecules in its path, the cumulative conductance of these molecules was too low to induce spiking. Because of the low unit conductance of channelrhodopsins, 2P optogenetic stimulation has remained technically challenging and is best achieved either by spiral scanning (29) or by advanced beam shaping approaches that blanket the cell membrane (30, 31). The localized scan pattern that favored voltage imaging disfavored 2P CheRiff stimulation.
The absence of cross-talk from blue light into the NovArch fluorescence channel can be explained by the differing sensitivities of CheRiff and NovArch. CheRiff has 50% activation at a blue intensity of 20 mW/cm2 (32), while NovArch has ~50% of maximum sensitization at a blue intensity of 3.9 W/cm2 (fig. S1B). At the blue illumination used in these experiments, 60 mW/cm2, the blue light–induced change in NovArch fluorescence is predicted to be <10%. Thus, despite the spectral overlap between CheRiff activation and NovArch sensitization, blue light optogenetic stimulation at intensities between 20 and 500 mW/cm2 can activate CheRiff, with little spurious sensitization of NovArch.
Mapping bAPs in tissue
We explored the capability to map dendritic voltage associated with bAPs in acute slices. We expressed NovArch sparsely in vivo and then prepared acute cortical slices. NovArch trafficked well to the membrane throughout the dendritic arbor (fig. S7). We identified NovArch-expressing L5 pyramidal cells, filled the cells with Alexa Fluor 594 via a patch pipette, and mapped the shape of the dendritic tree (Fig. 7A). The patch-clamp recordings were used to establish somatic ground truth but were not necessary for the voltage imaging experiments. Through careful alignment of the physical slicing angle with the dendritic axis, the primary apical dendrites remained within ~50 μm of the slice surface along their length. Since this distance was less than the optical scattering length for blue light, we found it most convenient to sensitize the NovArch with 1P blue illumination rather than 2P illumination. We used a digital micromirror device to co-illuminate selected patches of dendrite with localized red and blue illumination (see Fig. 8A). The blue-induced enhancement was critical for resolving small dendritic signals above the background. We then used a patch pipette at the soma to induce bAPs and mapped their propagation along the apical dendrites.
Fig. 7. Voltage imaging of dendritic bAPs in acute brain slice.
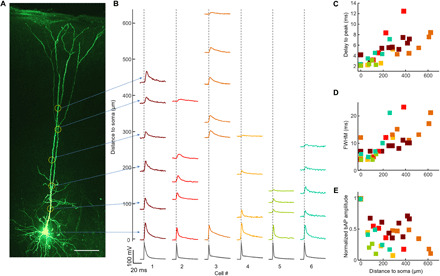
(A) L5 pyramidal neuron-expressing NovArch-Citrine filled with Alexa Fluor 594 via a patch pipette. The image shows Alexa Fluor 594 fluorescence. Scale bar, 100 μm. (B) Blue light–enhanced fluorescence recordings of action potentials at the soma and bAPs in the dendrites to a distance 625 μm from the soma; n = 6 neurons. Spikes were induced via current injection at the soma. Fluorescence recordings represent a spike-triggered average over 10 spikes, with timing aligned to the action potential peak recorded at the soma. For each cell, fluorescence recordings were normalized by the spike height at the soma. bAP (C) delay to spike peak, (D) full width at half maximum (FWHM), and (E) amplitude as a function of distance from the soma. Colors signify data from corresponding cells in (B).
Fig. 8. NovArch recordings in vivo.
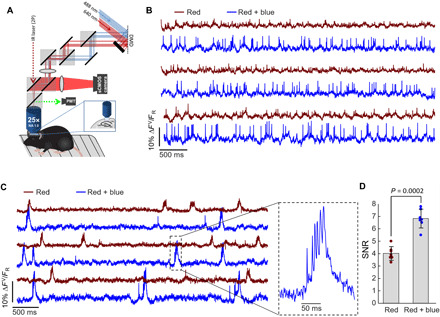
(A) Instrument for 2P imaging and overlapping red and blue structured illumination. The red and blue light were directed to adjacent halves of the digital micromirror device (DMD), and then, the two images were combined with dichroic mirrors and projected onto the brain. (B) Fluorescence recordings from a single oriens interneuron alternately illuminated with red-only and red + blue light. The vertical scale indicates fluorescence relative to red-only illumination, so the difference in amplitude reflects the blue light–enhanced brightness. Traces have been corrected for photobleaching but not otherwise filtered. (C) Fluorescence recordings from a single CA1 pyramidal cell alternately illuminated with red-only and red + blue light. The cell shows both simple spikes and complex spikes. (D) Comparison of SNR in paired recordings with red-only or red + blue illumination (n = 6 cells, two mice). IR, infrared; sCMOS, scientific complementary metal-oxide semiconductor.
bAP waveforms were clearly resolved at the single-trial level throughout the dendritic arbor (fig. S8). We mapped bAPs at 65 locations over n = 6 neurons to distances up to 625 μm from the soma (Fig. 7B). Spike-triggered averages, triggered from the somatic patch-clamp recording, showed distance-dependent changes in the spike waveform. The delay to peak increased with distance with a slope of 68 ± 16 mm/s (95% confidence bounds on linear regression; Fig. 7C) in approximate concordance with published dual patch-clamp measurements (33). The optically recorded spike full width at half maximum also increased with distance, from a mean of 5.9 ± 1.5 ms between 50 and 100 μm to 15 ± 4 ms between 500 and 625 μm (mean ± SD, n = 6 neurons in both cases; Fig. 7D), also consistent with prior patch-clamp recordings that also showed about threefold broadening over this range (34). While fluorescence measurement is not a reliable reporter of absolute voltage, we found that the relative amplitude of the bAP, measured as ΔF/F was only 16 ± 11% as big between 500 and 625 μm as at the soma (mean ± SD, n = 6 neurons; Fig. 7E). Prior dual patch-clamp measurements had reported a substantially smaller decay in amplitude, only ~50%, over a comparable distance (33, 34). We speculate that this discrepancy may be due to a bias in patch-clamp measurements toward larger-diameter dendritic branches. Although our analysis sought to subtract background fluorescence, increased relative contribution of residual background may also have artificially decreased ΔF/F in our measurements on very thin dendrites.
Recording neural activity in vivo
To test NovArch in vivo, we constructed a soma-localized variant by making a C-terminal fusion to the KV2.1 trafficking motif (11, 35, 36). We used AAV to express Cre-on soma-localized NovArch in the CA1 region of the hippocampus of adult mice using coinjected hSyn-Cre to control the sparsity of expression. We aspirated the cortex above CA1 and installed a glass window. Recordings were performed in awake mice at a 1-kHz bandwidth using a custom microscope (11) that combined 2P illumination for imaging or photoactivation with digital micromirror device–patterned red (~5 mW per cell) and blue (~200 μW per cell) illumination (Fig. 8A). Previous numerical simulations showed that optical heating is minimal under these conditions (11).
2P images of the appended Citrine tag showed good membrane localization and expression predominantly localized to the soma and proximal dendrites (fig. S9A). We identified neuron cell bodies using 2P microscopy and then made recordings successively with red-only, red + spiral scan 2P, and red + blue illumination targeted to the cell bodies (fig. S9B). Fluorescence waveforms had sufficient SNR to detect spikes in 11 of 15 recordings. The combination of red + blue gave better SNR compared to red + 2P, so subsequent experiments were performed with red + blue illumination. We performed single-cell recordings alternating red-only and red + blue illumination. These recordings clearly resolved spikes and subthreshold events in oriens interneurons (Fig. 8B) and pyramidal cells (Fig. 8C). Addition of blue light significantly improved the SNR of the spikes compared to red-only illumination from 4.0 ± 0.5 (red only) to 6.8 ± 0.7 (red + blue) (mean ± SD, n = 6 neurons, two mice, P = 0.0002, two-tailed paired t test).
DISCUSSION
By combining protein engineering, detailed spectroscopy, and optical design, we developed a new tool for high-sensitivity voltage imaging in tissue. The NovArch protein functions as a reversibly photoactivated GEVI, enabling 1P or 2P optical “highlighting” of cells or subcellular regions of interest. Key to maximizing the benefits of NovArch was an optical design that localized the fluorescence excitation to the peripheral cell membrane. This approach maximized the ratio of signal fluorescence to input light, achieving a 19-fold enhancement in SBR compared to wide-field excitation. 2P sensitization of NovArch further enhanced signal levels by an additional factor of 3.5, while 1P sensitization enhanced signal levels by a factor of 6 to 7.
Neuroscientists can now choose among several GEVIs that have recently been used for in vivo imaging. Villette and co-workers (15) used the green fluorescent protein–based GEVI ASAP3 and a specialized 2P optical system to record from individual neurons as deep as 420 μm in the cortex. ASAP3 showed high sensitivity in the subthreshold regime (−100 to 0 mV) but little sensitivity above 0 mV, and the ~6-ms response time distorted spike waveforms. Even with fast acousto-optic 2P scanning, no more than three cells could be recorded simultaneously.
In principle, 1P voltage imaging offers better SNR, faster time resolution, and wider field of view but at the expense of decreased depth penetration and increased contamination from out-of-focus signals. The fractional voltage sensitivities (ΔF/F per 100 mV), expression levels, and speeds of the opsin-based 1P GEVIs QuasAr3 (11), paQuasAr3 (11), Archon1 (13), and NovArch are all similar. The reported depths for single-cell recordings in vivo are as follows: paQuasAr3, 130 μm; Voltron, 195 μm [250 μm asserted in (10)]; Archon1, 150 μm using disk-shaped illumination and 230 μm using holographically targeted illumination (8); and NovArch, 220 μm (this work). The SNR of hippocampal recordings in vivo with paQuasAr3 was modestly higher than with NovArch (13.2 ± 6, n = 10 neurons versus 6.8 ± 0.8, n = 7 neurons), but substantial animal-to-animal variations make such comparisons difficult.
The high photoactivation contrast of NovArch made this reporter particularly well suited for recordings from fine processes, such as dendrites, in intact tissue where background can be high relative to signals. In this context, the photoactivation was critical for lifting the signal above the background. NovArch is the only GEVI to demonstrate single-trial recordings of bAPs in distal dendrites in intact tissue, although high-resolution voltage maps have previously been demonstrated with injected voltage-sensitive dyes in acute slices (5, 6) and recently in cerebellar Purkinje neurons in vivo (7).
Although 2P sensitization of NovArch, in principle, offers better background rejection than 1P sensitization in tissue, for near-surface imaging experiments, e.g., of dendrites, light scattering is not a concern. In this case, 1P sensitization was more effective because of its larger overall enhancement in brightness. For in vivo soma-targeted recordings, the broader spatial profile of the 1P sensitization also provided lower susceptibility to motion artifacts. Recently developed motion compensation schemes (37) might improve 2P targeting of membranes in vivo, and recent signal extraction algorithms can help mitigate the cross-talk from out-of-focus sources (9).
We propose that the smaller sensitization induced by 2P versus 1P sensitization is likely due to a geometrical effect. The 2P excitation spot is tightly constrained along the optical axis, whereas even highly focused 1P illumination excites molecules above and below the focal plane. Thus, the 2P sensitization only activated a portion of the molecules contributing to near-infrared fluorescence, while 1P sensitization activated molecules in the same spatial profile as the 1P fluorescence excitation. In addition, it is possible that the photocycle model in Fig. 1F is too simple. Upon turning off the blue or 2P sensitization, the red fluorescence decayed with different rates under prior blue versus 2P sensitization (compare Fig. 3A versus fig. S4E). Since neither sensitization beam was present during this fluorescence decay, this observation suggests that the two modes of sensitization may have acted differently. Further studies will be required to characterize the full NovArch photocycle.
The technical capability to coalign a 2P focus and a red 1P focus is not commonly available in most microscopes and may present a barrier to adoption. While this approach provides the most optimized SNR, we note that substantial enhancements can be obtained simply by crossing a red and blue beam in the sample, provided that each beam is restricted to an approximately cell-sized region. This capability can be achieved in most wide-field epifluorescence microscopes.
It is instructive to compare the effects of photoactivation to other approaches that one might take to improve the SNR of voltage imaging in tissue. The shot noise–limited SNR for optical voltage measurements is
| (1) |
where ΔF is the change in sample fluorescence due to the electrical event of interest, F is the baseline fluorescence of the indicator, and B is the background fluorescence (all measured in photon counts). There are many ways to increase the SNR. Increases in laser power increase ΔF, F, and B proportionally, leading to only square root improvements in SNR. Enhancements to GEVI brightness or expression level also increase ΔF, F, and B proportionally (assuming that background fluorescence is dominated by out-of-focus reporter molecules). Thus, brightness also only helps SNR by its square root.
On the other hand, enhancements to the focal plane signals, ΔF and F, without changing B, can increase SNR linearly with the enhancement in the high background limit (B >> F), which applies for 1P GEVI measurements in tissue. Thus, the 3.5-fold enhancement in focal plane voltage signal achieved through 2P photoactivation is equivalent, from an SNR perspective, to a 14-fold enhancement in overall brightness.
The concept of photoactivation for enhanced signal localization has previously been applied in other domains of fluorescence imaging. Multistep reversible photoexcitation can improve optical sectioning for opsins (23) and other fluorescent proteins (38). At the single-molecule level, this phenomenon enables STORM (Stochastic optical reconstruction microscopy) and PALM (Photoactivation light microscopy) superresolution microscopies. Multiphoton photoconversion of DsRed has been used as an optical “highlighter” (39). A photoactivated Ca2+ indicator has been used for optically targeted measurements in vivo (40). However, in these reporters, the photoactivation was irreversible, so imaging could only be performed on one set of neurons per sample. NovArch has the useful property of being reversibly switchable, allowing for sequential targeting of distinct cells or focal planes.
Spectroscopic studies on homologous mutations in bacteriorhodopsin (BR) provide some insights into the molecular mechanism of photoactivation in NovArch. The BR mutation V49A, analogous to V59A in Arch, has been studied in detail. In BR, this mutation stabilizes the N state, increasing its lifetime from ~5 to ~100 ms (41). In our model of voltage-sensitive fluorescence in wild-type Arch, the fluorescent Q state is reached from the N state (23), so stabilization of this photocycle intermediate would be expected to lead to photoactivation behavior. Several other mutations in BR have also been found to stabilize the N intermediate (42). These might be plausible alternative mutations sites for making photoactivatable GEVIs.
A report on photochromic mutations in BR found that the mutant D85N/V49A had exceptionally high photoswitching efficiency (43). In BR, aspartic acid 85 is the proton acceptor from the Schiff base. NovArch contains a similar mutation at the homologous position, D95Q, in which the glutamine (Q) probably plays a similar role to asparagine (N).
On the basis of the recent crystal structure of Arch 3 (44), the V209I mutation resides in the extracellular loop between the F and G helices, and the I213T mutation resides near the extracellular face of helix G, both far from the retinal chromophore (fig. S3B). The mechanisms by which these residues affect photoactivation is unclear, although their proximity to each other suggests that saturation mutagenesis of the surrounding neighborhood would be a plausible strategy for further enhancing the photoactivation.
MATERIALS AND METHODS
The constructs developed in this project are available on Addgene (Table 1).
Table 1. NovArch constructs available on Addgene.
| Construct | Description | Backbone |
Addgene ID |
| MPC020 (NovArch) | FCK-CKII-NovArch- TS-Citrine_TSx3-ER2 |
FCK | 153193 |
| MPC021 (sNovArch) | FCK-CKII-NovArch- PP-Citrine_KV2.1TS- ER2 (soma targeting) |
FCK | 153194 |
| MPC026 (AAV-NovArch) |
pAAV-CAG-FLEX- NovArch-TS- Citrine_TSx3-ER2 |
CAG-FLEX | 153195 |
| MPC027 (AAV-sNovArch) |
pAAV-CAG-FLEX- NovArch-PP- Citrine_KV2.1TS-ER2 (soma targeting) |
CAG-FLEX | 153196 |
Photostick screening of photoactivatable mutants of paQuasAr3
Mutant library preparation
The starting gene for the mutant library was derived from a QuasAr variant with improved expression and trafficking in vivo compared to the previously published QuasAr2 (11). This variant, called QuasAr3, contained enhanced trafficking sequences, the mutation K171R, and Citrine as a fluorescent expression marker. paQuasAr3 additionally had the V59A mutation.
paQuasAr3-Citrine was initially cloned into a modified FCK lentivirus vector, in which the original calcium/calmodulin-dependent protein kinase II promoter was replaced by a cytomegalovirus (CMV) promoter, and the WPRE (woodchuck hepatitis virus post-transcriptional regulatory element) enhancer sequence was included at the 3′ end. Ampicillin and puromycin resistance genes were also included in the construct. We denote this plasmid as pLenti-CMV-paQuasAr3-Citrine-Puro. This plasmid was then used as the template for error-prone PCR with the following primers, which flanked the paQuasAr3 region: forward, GACACCGACTCTAGAGCGCGGATCCACCATGGTAAGTATC and reverse, TCTCGTAGCAGAACTTGTAGAATTCTTATTCATTCTCATAACAAAAG.
An error-prone PCR reaction was performed as follows: In a 50-μl reaction, we combined 5 μl of ThermoPol Buffer (New England Biolabs), 5 μl of 2 mM deoxynucleotide triphosphate (New England Biolabs), 5 μl of 10 mM 2′-deoxycytidine 5′-triphosphate and 3′-deoxythymidine 5′-triphosphate, 5 μl of 35 mM MgSO4, 1.5 μl of each 20 μM primer, 0.5 μl of the plasmid template, 1 μl of Taq polymerase (5 U), and 25.5 μl of nuclease-free water. The PCR condition comprised 1 cycle of 95°C for 3 min; 35 cycles of the following three steps, 95°C for 25 s, 55°C for 30 s, and 72°C for 1 min; followed by 1 cycle of 72°C for 10 min; and then maintenance at 4°C. The error-prone PCR products were cloned back into the starting plasmid to create pLenti-CMV-NovArchX-Citrine-Puro, where X indicates mutants of NovArch.
Lentivirus production
HEK 293T cells were grown to 80% confluence in a 10-cm dish. Fresh Dulbecco’s modified Eagle’s medium 10 (DMEM10) was exchanged 1 to 2 hours before DNA transfection. A mixture of DNA plasmids [6.2 μg of paQuasAr3 plasmid and two virus packing plasmids: 4 μg of psPAX2 (Addgene, ID 12260) and 1.78 μg of VsVg (Addgene ID 8454)] were added into an Eppendorf tube with Opti-MEM medium (0.6 ml) followed by 36 μl of PEI solution (1 mg/ml) (branched polyethylenimine; average molecular weight, 25,000; Sigma-Aldrich, catalog no. 408727). The mixture was vortexed briefly and incubated at room temperature for 10 min. The DNA/PEI mixture was then added dropwise to the dish of HEK cells and incubated for 4 hours before changing to 10 ml of fresh DMEM10 without antibiotics. After 48 hours, cell culture supernatant was collected and centrifuged at 500g for 5 min to remove cells and debris. The supernatant was further filtered using a 0.45-μm filter and aliquoted for storage at −80°C.
NovArchX stable HEK cell line generation
NovArchX lentivirus was thawed and added at a titer of 0.001 multiplicity of infection in a 10-cm dish of HEK 293T cells grown to 80% confluency. Forty-eight hours after infection, puromycin was added to a final concentration of 2 mg/ml. Cells were selected for 14 days to stabilize the expression of NovArchX. The stably expressing HEK cells were then frozen in liquid nitrogen for later use.
Screening NovArchX followed by Photostick selection
Glass-bottom dishes covalently modified with fibronectin
Glass-bottom dishes (In Vitro Scientific, D35-14-1.5-N) were covalently modified with fibronectin to facilitate subsequent photochemical cross-linking of cells to the dish. Dishes were first cleaned and chemically activated by 3-min treatment in a plasma cleaner with low-pressure ambient air. The glass was aldehyde-functionalized with a 1% solution of 11-(triethoxysilyl) undecanal (Gelest Inc.) in ethanol, which reacted for 1 hour in a nitrogen-purged glove box. Dishes were rinsed twice with ethanol and once with Nanopure water and then cured in a vacuum oven at 65°C for 1 to 2 hours.
Fibronectin (0.1 mg/ml in PBS) was added to the dishes and incubated overnight at 4°C or at 37°C for 2 hours, resulting in a covalent imine bond between the surface and free primary amines on the fibronectin. Dishes were then immersed in 0.1% Tween 20–PBS for 10 min followed by rinsing three times with PBS. Completed dishes could either be seeded with cells directly or desiccated and stored at −80°C.
Fibronectin-coated dishes were seeded with 150,000 HEK cells stably expressing NovArchX mutants for 16 to 24 hours. Before imaging, the culture medium was exchanged for XC buffer [125 mM NaCl, 2.5 mM KCl, 2 mM CaCl2, 1 mM MgCl2, 10 mM HEPES, and 30 mM glucose (pH 7.3)].
Photostick optics
Photostick experiments were performed on a custom-built microscope (fig. S2), modified from (27). Red illumination was provided by six 635-nm diode lasers (Dragon Lasers), each 500 mW, for a total of 3 W. The beams were combined in pairs via polarizing beam splitters and then directed to the sample via a custom-fused silica prism placed between the objective and the sample. The beams were coupled from the prism to the sample via immersion oil, entering the sample at close to the critical angle for total internal reflection at the glass-water interface. Blue illumination was provided by a 488-nm, 300-mW light-emitting diode, mounted above the sample and expanded to illuminate the whole field of view.
Violet light for photocrosslinking was provided by a 407-nm, 200-mW laser (Lilly Electronics). Collimated 407-nm laser light at the back focal plane of the objective was focused at the sample to obtain a 5-μm spot. The position of the 407-nm laser focus was set by a pair of galvo mirrors (Thorlabs, GVS202) located in a conjugate plane.
Imaging was performed with a large dissecting microscope objective (MV PLAPO 2×, 0.5 NA; Olympus). Fluorescence was separated from illumination light using an emission filter (Semrock, no. Em01-R405/488/635). A 0.63× dissecting microscope objective served as the tube lens and projected an image onto a scientific complementary metal-oxide semiconductor camera (Hamamatsu ORCA-Flash 4.0).
Screening protocol
Cells were illuminated with 640-nm light (12 s, 20 W/cm2) and with weak 488-nm light (4 s, 0.1 W/cm2) superimposed at intervals to probe photoactivation. Before each experimental run, a control measurement was performed with a dish not containing cells for calibration of background autofluorescence levels and illumination profiles. The fluorescence intensity at each pixel depended on the following eight initially unknown parameters: Ir, illumination profile of red light; Ib, illumination profile of blue light; α, proportionality between red light excitation and near-infrared fluorescence; γ, proportionality between blue light excitation and near-infrared fluorescence; β, nonlinear coupling of blue excitation to red-excited fluorescence; Cdark, dark counts of camera; BrIr, background sample autofluorescence upon red light illumination; and BbIb, background sample autofluorescence upon blue light illumination.
Six fluorescence values were measured at each pixel (Table 2). Two additional pieces of data comprised the total optical powers in the red and blue beams. With these eight measurements, it was possible to solve for the eight variables at each pixel. The map of β as a function of position probed the nonlinear response, and the map of α probed the overall brightness. Movies were analyzed immediately after acquisition to determine whether the measured field of view contained cells with desirable photoactivation properties.
Table 2. Procedure for calibrating fluorescence measurements to detect nonlinear photoactivation.
|
Measurement number |
Cells on dish |
Red on | Blue on | Fluorescence value |
| 1 | − | − | + | BbIb + Cdark |
| 2 | − | + | − | BrIr + Cdark |
| 3 | + | − | − | Cdark |
| 4 | + | − | + | γIb + BbIb + Cdark |
| 5 | + | + | − | αIr + BrIr + Cdark |
| 6 | + | + | + | αIrβIb+γIb + αIr + BbIb + BrIr + Cdark |
Photostick protocol
A photochemical cross-linker phenyl azide derivative, 4-fluoro-3-nitrophenyl azide (15 μM), was applied as the Photostick reagent. Light from a 407-nm laser (15 mW) was scanned over the target cells in a small raster pattern. The illumination time was 6 × 10−2 ms/μm2 or 100 to 200 ms per cell, depending on the cell size. Subsequently, the dish was rinsed with PBS and digested with trypsin for 5 min to detach nonilluminated cells. The remaining adhered cells were detached by gently pipetting with 10 μl of PBS and transferred into a PCR tube. When more than one cell was harvested, additional dilutions were used to ensure that each PCR tube contained at most one cell. Each single cell was then lysed with 2.8 μl of lysis buffer (40 mM dithiothreitol, 2 mM EDTA, and 200 mM KOH) at 65°C for 10 min and neutralized with 1.4 μl of neutralization buffer [400 mM HCl and 600 mM tris (pH 7.5)] before conducting a PCR reaction. Q5 NEB 2× master mix was used for a PCR amplification using the following conditions: one cycle of 95°C for 5 min; 45 cycles of the following three steps: 95°C for 30 s, 58°C for 45 s, and 72°C for 1 min; followed by 1 cycle of 72°C for 10 min; and then 4°C for ∞.
1P, 2P, and combined fluorescence imaging
2P microscopy
We built a beam-scanning 2P microscope optimized for excitation at wavelengths between 950 and 1300 nm and for detection of fluorescence at wavelengths shorter than 775 nm. Illumination was provided by a Spectra-Physics InSight DeepSee, tunable between 680 and 1300 nm, with pulses of ~120 fs at 80-MHz repetition rate. The pulse dispersion was adjusted via an internal, motorized prism pair compressor controlled by Spectra-Physics software.
The beam was steered by a pair of galvo mirrors (Cambridge Technology 6215HM40B, driven by Cambridge Technologies 671215H-1HP MicroMax servo driver on the X axis and Cambridge Technologies 671215H-1 MicroMax servo driver on the Y axis). The beam then passed through an Olympus PL scan lens and an Olympus TLUIR tube lens onto the back aperture of an Olympus water immersion XPLN25XWMP2 objective (NA 1.05). The imaging plane was selected by moving the objective with a Physik Instrumente PIFOC P725 piezoelectric nanomanipulator, driven by a PIFOC E625 driver. Fluorescence emission was separated from back-scattered excitation light via a Semrock 775-nm long-pass dichroic beam splitter (FF775-Di01-25x36). Residual laser light was rejected using a Semrock 790-nm short-pass filter (FF01-790/SP-25).
For direct two-photon imaging, fluorescence was reimaged onto a Hamamatsu H10492-13 PMT (photomultiplier tube). The signal was amplified and low-pass filtered through an USBPGF-S1 low-pass filter (Alligator Technologies) with a typical cutoff of 62.5 kHz for digitization at 125 kHz. Signals were recorded using a National Instruments PCIe-6259 board.
Camera-based imaging
For camera-based imaging of NovArch, optical excitation of CheRiff, and initial screening of the photoactivation effect with blue light, we used 640- and 488-nm Coherent OBIS lasers for wide-field illumination. Beams of both lasers were combined and fed through a Gooch and Housego acousto-optic modulator for temporal control, collimated, and passed through an iris on an x-y stage. The iris was reimaged into the sample via the fluorescence excitation pathway; fluorescence and excitation light were separated using a 664-nm long-pass dichroic (Semrock BLP01-664R-025). The size and position of the iris provide control of the excitation spot. Fluorescence was imaged onto an Andor iXon X3 860 EMCCD for spatially resolved imaging of 1P or 2P signals. This pathway was also used in white light illumination when applying a patch pipette to a cell. All images of NovArch fluorescence in cells represent time-averaged fluorescence. Fluorescence was quantified as mean counts per pixel per frame.
Coscanning of red and 2P focal spots
For the double focus scanning, the beam from a Coherent OBIS 640-nm laser was led through a 1:1 telescope and a pair of steering mirrors and combined with the near-infrared beam from the DeepSee using a FF660-Di02 dichroic mirror. The combined beam was steered along the same pathway used for conventional 2P imaging. Fluorescence was separated from the excitation light using a custom dichroic with a 25-nm transmission band around 640 nm and a long-pass edge at 775 nm (Alluxa). In addition to the 790-nm short-pass filter, a 664-nm long-pass filter (Semrock BLP01-664R-025) was added in the detection path to reject the 640-nm laser light. Fluorescence was resolved on the Andor iXon Camera via the wide-field imaging path. For typical measurements, a field of view would be selected on the camera that would allow 500- to 1000-Hz frame rates.
Alignment of red and 2P PSFs
In the coscanned geometry, the 640-nm 1P and near-infrared 2P PSFs (point-spread functions) must be precisely aligned to maximize photoactivation. Alignment was performed using 100-nm-diameter beads that fluoresced under both excitation beams (0.1 μm, fluorescent blue, green, orange, and dark red; TetraSpeck Microspheres) and happened in three stages.
First, a bead was selected, and each beam was moved in a short scan along each of the three Cartesian axes, while fluorescence was recorded on a PMT. The beam positions were adjusted to maximize the peak fluorescence signal and to localize the peak to the midpoint of each scan.
Second, small volumetric scans of a bead were made, leading to three-dimensional images representing the PSFs of both lasers. The laser positions were adjusted to overlap the PSFs. This step provided a complete but slower image of the offsets and orientations of both PSFs.
Last, the first two steps were repeated at different points in the field of view to assure overlap of the PSFs in all locations. We found no PSF offsets in a range of {Δx, Δy} ≤ ±700 μm from the center of the field of view. We found minor changes in the angle relative to the optical axis of the 640-nm PSF toward the edge of the field of view, indicating residual chromatic aberration in the excitation path, but this did not affect the efficiency of photoactivation.
Photophysical characterization of paQuasAr3 and NovArch
Patch-clamp recordings
We coupled a HEKA EPC 800 patch-clamp amplifier to the setup to provide simultaneous electrophysiological and optical measurements. The pipette was positioned by a Sutter MP-285 micromanipulator. Whole-cell voltage clamp and current clamp signals were filtered at 3 kHz with the internal Bessel filter and digitized with a National Instruments PCIe-6259 board. The electrophysiology and optical measurements were synchronized via custom software written in LabVIEW.
Expression of constructs in HEK293 cells
HEK 293T cells (American Type Culture Collection, CRL-11268) were cultured and transfected as described before (45). Briefly, cells were grown at 37°C, 5% CO2, in DMEM supplemented with 10% fetal bovine serum and penicillin-streptomycin. Cells were tested negative for mycoplasma. Cells were transfected with GEVI constructs under the upstream CMV promoter of the FCK plasmid. Plasmid DNA (200 to 400 ng) was transfected using TransIT 293T (Mirus) following the manufacturer’s instructions and assayed 48 hours later. The day before recording, cells were replated onto Matrigel-coated glass-bottom dishes (In Vitro Scientific) at a density of ∼10,000 cells/cm2.
Simultaneous fluorescence and patch-clamp electrophysiology
All imaging and electrophysiology were performed in extracellular buffer containing 125 mM NaCl, 2.5 mM KCl, 3 mM CaCl2, 1 mM MgCl2, 15 mM Hepes, and 30 mM glucose (pH 7.3) and adjusted to 305 to 310 mosm with sucrose. Simultaneous whole-cell patch-clamp and fluorescence recordings were acquired on the homebuilt, combined 2P and inverted epifluorescence microscope described above. For simultaneous electrophysiology and imaging, filamented glass micropipettes (World Precision Instruments) were pulled to a tip resistance of 5 to 10 megohms and filled with internal solution containing the following: 125 mM potassium gluconate, 8 mM NaCl, 0.6 mM MgCl2, 0.1 mM CaCl2, 1 mM EGTA, 10 mM Hepes, 4 mM Mg–adenosine 5′-triphosphate, and 0.4 mM Na–guanosine 5′-triphosphate (pH 7.3; adjusted to 295 mosm with sucrose). Pipettes were positioned with a Sutter MP-285 manipulator.
For Figs. 4 and 7, action potentials were induced by current injection via patch-clamp pipette. For channelrhodopsin-stimulated activity (Fig. 6), we positioned an iris in the 488-nm beam, imaged the iris into the sample, and closed the iris and positioned it so that it stimulated a single soma. For Fig. 8, the red and blue illumination were patterned on adjacent halves of a digital micromirror device and combined via dichroic beam splitters using the apparatus described in (11).
NovArch fluorescence excitation and activation spectra
We expressed NovArch in HEK 293T cells, centrifuged the cells to form a dense pellet, and performed spectroscopy in a homebuilt microscope system using light from a tunable supercontinuum laser (Fianium SC-450-6). This beam was combined with a Coherent OBIS 640-nm laser to measure the photosensitization spectrum and an Omicron PhoxX 488-nm laser to measure the excitation spectrum. The cell pellet was illuminated through a 60× oil immersion 1.45 NA objective. Fluorescence was separated from excitation light using a 660-nm long-pass dichroic and 664-nm long-pass fluorescence filter. Fluorescence from the cell pellet was imaged onto an Andor IXon X3 860 EMCCD. The same imaging sequence as in the “Screening protocol” section above was used. To measure the excitation spectrum, “red” was varied between 570 and 660 nm, and “blue” was kept at 488 nm; for the photoactivation spectrum, red was kept at 640 nm, and blue was varied between 460 and 550 nm.
Characterization of NovArch in brain slice and in vivo
AAV preparation
NovArch-Citrine (NovArch-TS-Citrine_TSx3-ER2) was created by fusing NovArch with the Citrine fluorescent reporter. TS represents the trafficking sequence (TS) from Kir2.1, and ER2 represents the endoplasmic reticulum export signal FCYENEV (46). The construct was then cloned into an AAV vector to create MPC026 AAV2/9.CAG.FLEX.NovArch-Citrine.WPRE.SVPA for AAV production.
Soma-localized NovArch-Citrine (NovArch-PP-Citrine_KV2.1TS-ER2) was created by fusing NovArch with the Citrine fluorescent reporter through a two–amino acid linker (PP) and with a KV2.1 TS to the C terminus of NovArch (11). ER2 represents the endoplasmic reticulum export signal FCYENEV (11, 46). The construct was then cloned into an AAV vector to create MPC027 AAV2/9.CAG.FLEX.sNovArch-Citrine.WPRE.SVPA for AAV production. AAVs were produced in the Gene Transfer Vector Core at Massachusetts Eye and Ear Infirmary and Schepens Eye Research Institute, Harvard Medical School.
Viral injections
All animal experiments were approved by the Harvard University Institutional Animal Care and Use Committee. C57BL/6 mouse pups were cryo-anesthetized at postnatal day 0 (P0) to P1 (47). Pups were immobilized dorsal side up, facing away from the experimenter under a stereotaxic microscope. Injections were made using home-pulled micropipettes (Sutter P1000 pipette puller), mounted in a microinjection pump (World Precision Instruments Nanoliter 2010) controlled by a microsyringe pump controller (World Precision Instruments Micro4). The micropipette was positioned using a stereotaxic instrument (Stoelting Digital Mouse Stereotaxic Instrument). Pups were injected in the right hemisphere, 1 mm lateral, 1 mm anterior, and 0 to 2 mm ventral from lambda with a virus mixture containing CKII(0.4)-Cre [8.3 × 109 GC (genome copies)/ml], hSyn.CheRiff-CFP (1.5 × 1012 GC/ml), and CAG.FLEX.NovArch-Citrine (5.8 × 1012 GC/ml). Forty nanoliters was injected at each site. Typically, nine sites were injected per pup. Pups were placed back in their home cage once they were awake.
Brain slice preparation
Acute brain slices were prepared from P15 to P38 male and female mice. The mice were deeply anesthetized by intraperitoneal injection of ketamine (90 mg/kg) and xylazine (10 mg/kg) and then perfused with carbogen (95% O2, 5% CO2)–saturated ice-cold slicing solution with the following composition: 110 mM choline chloride, 2.5 mM KCl, 1.25 mM NaH2PO4, 25 mM NaHCO3, 25 mM glucose, 0.5 mM CaCl2, 7 mM MgCl2, 11.6 mM Na-ascorbate, and 3.1 mM Na-pyruvate. Mice were then decapitated, and the brains were removed into ice-cold slicing solution and then rapidly blocked for coronal sectioning at a thickness of 300 μm on a vibratome (Leica VT1200S). Slices were then incubated for 45 min at 34°C in a carbogenated artificial cerebrospinal fluid (ACSF) with the following composition: 127 mM NaCl, 2.5 mM KCl, 1.25 mM NaH2PO4, 25 mM NaHCO3, 25 mM glucose, 2 mM CaCl2, and 1 mM MgCl2. The pH was maintained at 7.4 under constant bubbling with carbogen, and slices could be used for 4 to 6 hours after harvest.
Brain slice imaging
Brain slice imaging was performed on a homebuilt inverted microscope (see below). The slice was immobilized in a Warner Instruments RC-27LD flow chamber using a slice anchor (harp). ACSF, perfused with carbogen as above, was flowed through the chamber at a rate of 1.5 ml/min. Recordings were performed at 23°C.
Virus injection and cranial window surgery
Ten- to 18-week-old C57BL/6 mice (male and female) were deeply anesthetized with 2% isoflurane and maintained with ~1% isoflurane throughout the surgery. The skull was exposed and thoroughly dried, and a 3-mm round craniotomy was opened using a biopsy punch (Miltex). To target CA1, the craniotomy center was 1.8 mm lateral and 2.0 mm caudal of bregma. Virus was then injected in one to three locations in the center of the craniotomy.
For single-cell measurements with NovArch, sparse expression was achieved by mixing AAV encoding Cre-on soma-localized NovArch (CAG-FLEX-NovArch-PP-Citrine_KV2.1TS-ER2; final titer, 2 × 1013 GC/ml) with hSyn-Cre virus (final titer, 2 × 1010 to 1 × 1011 GC/ml). Injections were made using home-pulled micropipettes (Sutter P1000 pipette puller), mounted in a microinjection pump (World Precision Instruments Nanoliter 2010) controlled by a microsyringe pump controller (World Precision Instruments Micro4). The micropipette was positioned using a stereotaxic instrument (Stoelting Digital Mouse Stereotaxic Instrument). For CA1 expression, injections were made −1.5 to −1 mm from the dura, at 0.1-mm increments (40 nl per depth, 5 nl/s). Brain surface was kept moist with saline throughout the injection.
The procedure for imaging in CA1 followed Dombeck and co-workers (48). Briefly, a cannula was prepared before the surgery and comprised a 1.5-mm segment of a thin-walled stainless steel tube (outer diameter, 3 mm; MicroGroup). A no. 1 round cover glass (diameter, 3 mm; Harvard Apparatus) was cemented to one end of the tube using ultraviolet (UV)–curable adhesive (Norland Products) and cured for at least 5 min on a standard laboratory UV table. Following hippocampal virus injection, we removed the dura and then slowly aspirated the cortex while continuously irrigating with saline until bleeding stopped. After exposure of the external capsule, a small region of the capsule in the center was gently removed, exposing the CA1 surface. To reduce brain motion, in some of the animals, a small amount of Kwik-Sil (World Precision Instruments) was applied to the surface of the brain. The cannula was then inserted and cemented to the skull with dental cement (C&B-Metabond). After the cannula cured, a titanium headplate [similar to (49)] was glued around the cannula and any exposed skull was covered with dental cement. Animals were returned to their home cage for recovery and treated for 3 days with carprofen (5 mg/kg) and buprenorphine (0.1 mg/kg) twice a day. To avoid damage to the implant, mice were housed in separate cages.
In vivo imaging
Head-fixed animals were imaged while resting on a stationary platform. Before imaging sessions started, mice were habituated to head restraint. To adjust mice to imaging conditions, at least one training session took place under the microscope with the objective on top of the cranial window. Awake animals were imaged 3 weeks to 3 months from virus injection without any detectable effects on cell health.
Analysis of electrophysiology and voltage imaging data
Data were recorded with a 1000-Hz (Figs. 4, 7, and 8) or 500-Hz (Fig. 6) camera frame rate. To convert the movie into a data trace, we averaged the pixels in the frame weighed by their response to 2P activation. Data in Figs. 4 and 6 were Fourier filtered with a 370-Hz bandwidth to remove camera-related electronic noise. For SNR calculations, we first corrected the traces for photobleaching and photoactivation artifacts. We calculated spike amplitude as the difference between the peak value of the spikes and the value of the baseline in a window around the spike; window size was adapted depending on the spiking frequency. To estimate the noise, we sought to distinguish shot noise from true subthreshold dynamics. A recording interval without spikes was high-pass filtered with a 20-ms sliding window. The shot noise was calculated from the SD of the high-pass–filtered signal. SNR was determined by the ratio of spike amplitude to shot noise. Automated spike finding was performed by detecting events that were at least 5σ above baseline.
Statistical methods
Statistical tests were used to determine whether illumination led to significant changes in GEVI fluorescence and to compare sensitization levels between GEVIs. For measurements of photo-enhancement, each cell was measured before and after photo-enhancement. This approach largely eliminated the effects of biological variation (e.g., from variations in expression level) and also eliminated the effects of variation in red illumination intensity. Significance was tested via a paired-sample Student’s t test. Probabilities of the null hypothesis P < 0.05 were judged to be statistically significant.
Acknowledgments
We thank K. Williams for technical assistance with molecular biology and M. Lee for technical assistance with cell culture, animal husbandry, and acute slice preparation. We thank S. Farhi, S. Lou, and L. Fan for help with mouse injections and acute slice preparation. Funding: This work was supported by the Howard Hughes Medical Institute and U.S. NIH grant 1-R01-MH117042. D.B. acknowledges support by a Rubicon grant from the Netherlands Organisation for Scientific Research (NWO). M.-P.C. was supported by a Gordon and Betty Moore Foundation Life Sciences Research Foundation Postdoctoral Fellowship GBMF2550.05. Y.A. was supported by a long-term fellowship of the Human Frontier Science Program. Author contributions: M.-P.C. created the Photostick screening technology and performed the screening experiments to create NovArch, with input on screening protocol from D.B. D.B. designed the targeted illumination scheme and built it with the help of W.B. Y.A. conducted the proof-of-concept experiments on paQuasAr3 together with D.B. and performed the NovArch in vivo measurements. W.B. performed the spectroscopy experiments on paQuasAr3 and NovArch under the supervision of D.B. G.T.-S. performed the patch-clamp experiments in slices followed by dendritic voltage imaging. H.T., F.P.B., and B.G. performed the photophysical characterizations. D.B. performed all other experiments described in the paper with input from M.-P.C., W.B., S.K., and Y.A. D.B., M.-P.C., W.B., and A.E.C. analyzed the data. D.B., M.P.C., and A.E.C. wrote the paper with input from all authors. A.E.C. supervised all aspects of the project. Competing interests: A.E.C. is a founder of Q-State Biosciences and is on the Scientific Advisory Board. All authors declare that they have no other competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/19/eabe3216/DC1
REFERENCES AND NOTES
- 1.Antic S. D., Empson R. M., Knöpfel T., Voltage imaging to understand connections and functions of neuronal circuits. J. Neurophysiol. 116, 135–152 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lin M. Z., Schnitzer M. J., Genetically encoded indicators of neuronal activity. Nat. Neurosci. 19, 1142–1153 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Xu Y., Zou P., Cohen A. E., Voltage imaging with genetically encoded indicators. Curr. Opin. Chem. Biol. 39, 1–10 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Häusser M., Mel B., Dendrites: Bug or feature? Curr. Opin. Neurobiol. 13, 372–383 (2003). [DOI] [PubMed] [Google Scholar]
- 5.Popovic M., Gao X., Zecevic D., Voltage-sensitive dye recording from axons, dendrites and dendritic spines of individual neurons in brain slices. J. Vis. Exp. 69, e4261 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tanese D., Weng J. Y., Zampini V., de Sars V., Canepari M., Rozsa B., Emiliani V., Zecevic D., Imaging membrane potential changes from dendritic spines using computer-generated holography. Neurophotonics 4, 031211 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Roome C. J., Kuhn B., Simultaneous dendritic voltage and calcium imaging and somatic recording from Purkinje neurons in awake mice. Nat. Commun. 9, 3388 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fan L. Z., Kheifets S., Böhm U. L., Wu H., Piatkevich K. D., Xie M. E., Parot V., Ha Y., Evans K. E., Boyden E. S., Takesian A. E., Cohen A. E., All-optical electrophysiology reveals the role of lateral inhibition in sensory processing in cortical layer 1. Cell 180, 521–535.e18 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xie M. E., Adam Y., Fan L. Z., Böhm U. L., Kinsella I., Zhou D., Rozsa M., Singh A., Svoboda K., Paninski L., Cohen A. E., High-fidelity estimates of spikes and subthreshold waveforms from 1-photon voltage imaging in vivo. Cell Reports 35, 108954 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Abdelfattah A. S., Kawashima T., Singh A., Novak O., Liu H., Shuai Y., Huang Y.-C., Campagnola L., Seeman S. C., Yu J., Zheng J., Grimm J. B., Patel R., Friedrich J., Mensh B. D., Paninski L., Macklin J. J., Murphy G. J., Podgorski K., Lin B.-J., Chen T.-W., Turner G. C., Liu Z., Koyama M., Svoboda K., Ahrens M. B., Lavis L. D., Schreiter E. R., Bright and photostable chemigenetic indicators for extended in vivo voltage imaging. Science 365, 699–704 (2019). [DOI] [PubMed] [Google Scholar]
- 11.Adam Y., Kim J. J., Lou S., Zhao Y., Xie M. E., Brinks D., Wu H., Mostajo-Radji M. A., Kheifets S., Parot V., Chettih S., Williams K. J., Gmeiner B., Farhi S. L., Madisen L., Buchanan E. K., Kinsella I., Zhou D., Paninski L., Harvey C. D., Zeng H., Arlotta P., Campbell R. E., Cohen A. E., Voltage imaging and optogenetics reveal behaviour-dependent changes in hippocampal dynamics. Nature 569, 413–417 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gong Y., Huang C., Li J. Z., Grewe B. F., Zhang Y., Eismann S., Schnitzer M. J., High-speed recording of neural spikes in awake mice and flies with a fluorescent voltage sensor. Science 350, 1361–1366 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Piatkevich K. D., Bensussen S., Tseng H.-a., Shroff S. N., Lopez-Huerta V. G., Park D., Jung E. E., Shemesh O. A., Straub C., Gritton H. J., Romano M. F., Costa E., Sabatini B. L., Fu Z., Boyden E. S., Han X., Population imaging of neural activity in awake behaving mice. Nature 574, 413–417 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chamberland S., Yang H. H., Pan M. M., Evans S. W., Guan S., Chavarha M., Yang Y., Salesse C., Wu H., Wu J. C., Clandinin T. R., Toth K., Lin M. Z., St-Pierre F., Fast two-photon imaging of subcellular voltage dynamics in neuronal tissue with genetically encoded indicators. eLife 6, e25690 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Villette V., Chavarha M., Dimov I. K., Bradley J., Pradhan L., Mathieu B., Evans S. W., Chamberland S., Shi D., Yang R., Kim B. B., Ayon A., Jalil A., St-Pierre F., Schnitzer M. J., Bi G., Toth K., Ding J., Dieudonné S., Lin M. Z., Ultrafast two-photon imaging of a high-gain voltage indicator in awake behaving mice. Cell 179, 1590–1608.e23 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kulkarni R. U., Vandenberghe M., Thunemann M., James F., Andreassen O. A., Djurovic S., Devor A., Miller E. W., In vivo two-photon voltage imaging with sulfonated rhodamine dyes. ACS Cent. Sci. 4, 1371–1378 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Weisenburger S., Tejera F., Demas J., Chen B., Manley J., Sparks F. T., Martínez Traub F., Daigle T., Zeng H., Losonczy A., Vaziri A., Volumetric Ca2+ imaging in the mouse brain using hybrid multiplexed sculpted light microscopy. Cell 177, 1050–1066.e14 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kazemipour A., Novak O., Flickinger D., Marvin J. S., Abdelfattah A. S., King J., Borden P. M., Kim J. J., Al-Abdullatif S. H., Deal P. E., Miller E. W., Schreiter E. R., Druckmann S., Svoboda K., Looger L. L., Podgorski K., Kilohertz frame-rate two-photon tomography. Nat. Methods 16, 778–786 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu J., Liang Y., Chen S., Hsu C. L., Chavarha M., Evans S. W., Shi D., Lin M. Z., Tsia K. K., Ji N., Kilohertz two-photon fluorescence microscopy imaging of neural activity in vivo. Nat. Methods 17, 287–290 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang T., Hernandez O., Chrapkiewicz R., Shai A., Wagner M. J., Zhang Y., Wu C.-H., Li J. Z., Inoue M., Gong Y., Ahanonu B., Zeng H., Bito H., Schnitzer M. J., Kilohertz two-photon brain imaging in awake mice. Nat. Methods 16, 1119–1122 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brinks D., Klein A. J., Cohen A. E., Two-photon lifetime imaging of voltage indicating proteins as a probe of absolute membrane voltage. Biophys. J. 109, 914–921 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Peron S., Chen T.-W., Svoboda K., Comprehensive imaging of cortical networks. Curr. Opin. Neurobiol. 32, 115–123 (2015). [DOI] [PubMed] [Google Scholar]
- 23.Maclaurin D., Venkatachalam V., Lee H., Cohen A. E., Mechanism of voltage-sensitive fluorescence in a microbial rhodopsin. Proc. Natl. Acad. Sci. U.S.A. 110, 5939–5944 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hou J. H., Venkatachalam V., Cohen A. E., Temporal dynamics of microbial rhodopsin fluorescence reports absolute membrane voltage. Biophys. J. 106, 639–648 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Venkatachalam V., Brinks D., Maclaurin D., Hochbaum D., Kralj J., Cohen A. E., Flash memory: Photochemical imprinting of neuronal action potentials onto a microbial rhodopsin. J. Am. Chem. Soc. 136, 2529–2537 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chien M.-P., Werley C. A., Farhi S. L., Cohen A. E., Photostick: A method for selective isolation of target cells from culture. Chem. Sci. 6, 1701–1705 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Werley C. A., Chien M.-P., Cohen A. E., Ultrawidefield microscope for high-speed fluorescence imaging and targeted optogenetic stimulation. Biomed. Opt. Express 8, 5794–5813 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Piatkevich K. D., Jung E. E., Straub C., Linghu C., Park D., Suk H.-J., Hochbaum D. R., Goodwin D., Pnevmatikakis E., Pak N., Kawashima T., Yang C.-T., Rhoades J. L., Shemesh O., Asano S., Yoon Y.-G., Freifeld L., Saulnier J. L., Riegler C., Engert F., Hughes T., Drobizhev M., Szabo B., Ahrens M. B., Flavell S. W., Sabatini B. L., Boyden E. S., A robotic multidimensional directed evolution approach applied to fluorescent voltage reporters. Nat. Chem. Biol. 14, 352–360 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rickgauer J. P., Tank D. W., Two-photon excitation of channelrhodopsin-2 at saturation. Proc. Natl. Acad. Sci. U.S.A. 106, 15025–15030 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Andrasfalvy B. K., Zemelman B. V., Tang J., Vaziri A., Two-photon single-cell optogenetic control of neuronal activity by sculpted light. Proc. Natl. Acad. Sci. U.S.A. 107, 11981–11986 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ronzitti E., Ventalon C., Canepari M., Forget B. C., Papagiakoumou E., Emiliani V., Recent advances in patterned photostimulation for optogenetics. J. Opt. 19, 113001 (2017). [Google Scholar]
- 32.Zhang H., Reichert E., Cohen A. E., Optical electrophysiology for probing function and pharmacology of voltage-gated ion channels. eLife 5, e15202 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stuart G. J., Sakmann B., Active propagation of somatic action potentials into neocortical pyramidal cell dendrites. Nature 367, 69–72 (1994). [DOI] [PubMed] [Google Scholar]
- 34.Stuart G., Schiller J., Sakmann B., Action potential initiation and propagation in rat neocortical pyramidal neurons. J. Physiol. 505, 617–632 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Baker C. A., Elyada Y. M., Parra A., Bolton M. M., Cellular resolution circuit mapping with temporal-focused excitation of soma-targeted channelrhodopsin. eLife 5, e14193 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wu C., Ivanova E., Zhang Y., Pan Z.-H., rAAV-mediated subcellular targeting of optogenetic tools in retinal ganglion cells in vivo. PLOS ONE 8, e66332 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Griffiths V. A., Valera A. M., Lau J. Y. N., Roš H., Younts T. J., Marin B., Baragli C., Coyle D., Evans G. J., Konstantinou G., Koimtzis T., Nadella K. M. N. S., Punde S. A., Kirkby P. A., Bianco I. H., Silver R. A., Real-time 3D movement correction for two-photon imaging in behaving animals. Nat. Methods 17, 741–748 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ingaramo M., York A. G., Andrade E. J., Rainey K., Patterson G. H., Two-photon-like microscopy with orders-of-magnitude lower illumination intensity via two-step fluorescence. Nat. Commun. 6, 8184 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Marchant J. S., Stutzmann G. E., Leissring M. A., LaFerla F. M., Parker I., Multiphoton-evoked color change of DsRed as an optical highlighter for cellular and subcellular labeling. Nat. Biotechnol. 19, 645–649 (2001). [DOI] [PubMed] [Google Scholar]
- 40.Berlin S., Carroll E. C., Newman Z. L., Okada H. O., Quinn C. M., Kallman B., Rockwell N. C., Martin S. S., Lagarias J. C., Isacoff E. Y., Photoactivatable genetically encoded calcium indicators for targeted neuronal imaging. Nat. Methods 12, 852–858 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schobert B., Brown L. S., Lanyi J. K., Crystallographic structures of the M and N intermediates of bacteriorhodopsin: Assembly of a hydrogen-bonded chain of water molecules between Asp-96 and the retinal Schiff base. J. Mol. Biol. 330, 553–570 (2003). [DOI] [PubMed] [Google Scholar]
- 42.Dioumaev A. K., Brown L. S., Needleman R., Lanyi J. K., Coupling of the reisomerization of the retinal, proton uptake, and reprotonation of Asp-96 in the N photointermediate of bacteriorhodopsin. Biochemistry 40, 11308–11317 (2001). [DOI] [PubMed] [Google Scholar]
- 43.Millerd J. E., Rohrbacher A., Brock N. J., Chau C.-K., Smith P., Needleman R., Improved sensitivity in blue-membrane bacteriorhodopsin films. Opt. Lett. 24, 1355–1357 (1999). [DOI] [PubMed] [Google Scholar]
- 44.I. Moraes, P. J. Judge, D. Axford, J. F. Bada Juarez, J. Vinals, A. Watts, Room temperature structure of Archaerhodopsin-3 via LCP extruder using synchrotron radiation. 10.2210/pdb6GUY/pdb (2019). [DOI]
- 45.Hochbaum D. R., Zhao Y., Farhi S. L., Klapoetke N., Werley C. A., Kapoor V., Zou P., Kralj J. M., Maclaurin D., Smedemark-Margulies N., Saulnier J. L., Boulting G. L., Straub C., Cho Y. K., Melkonian M., Wong G. K.-S., Harrison D. J., Murthy V. N., Sabatini B. L., Boyden E. S., Campbell R. E., Cohen A. E., All-optical electrophysiology in mammalian neurons using engineered microbial rhodopsins. Nat. Methods 11, 825–833 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gradinaru V., Zhang F., Ramakrishnan C., Mattis J., Prakash R., Diester I., Goshen I., Thompson K. R., Deisseroth K., Molecular and cellular approaches for diversifying and extending optogenetics. Cell 141, 154–165 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Phifer C. B., Terry L. M., Use of hypothermia for general anesthesia in preweanling rodents. Physiol. Behav. 38, 887–890 (1986). [DOI] [PubMed] [Google Scholar]
- 48.Dombeck D. A., Harvey C. D., Tian L., Looger L. L., Tank D. W., Functional imaging of hippocampal place cells at cellular resolution during virtual navigation. Nat. Neurosci. 13, 1433–1440 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Goldey G. J., Roumis D. K., Glickfeld L. L., Kerlin A. M., Reid R. C., Bonin V., Schafer D. P., Andermann M. L., Removable cranial windows for long-term imaging in awake mice. Nat. Protoc. 9, 2515–2538 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/19/eabe3216/DC1