

Simple group testing designs to improve SARS-CoV-2 surveillance in resource-constrained settings are identified using modeling and experimental data.
Batch testing for SARS-CoV-2
Frequent and accurate RT-PCR–based testing is essential for preventing and managing SARS-CoV-2 infection; however, active infection surveillance is still often limited by time or resources. Cleary et al. demonstrate that considering population-level viral prevalence and individual viral loads allows for efficiency gains upon pooled testing with minimal loss of sensitivity, both theoretically and as validated in vitro using human swab and sputum samples. Barak et al. show that pooled testing of 133,816 hospital-collected patient nasopharyngeal samples eliminated three quarters of testing reactions with only a minor reduction in sensitivity, demonstrating the efficacy of the approach in the field. Both studies suggest that considered pooling of individual samples before testing could reliably increase SARS-CoV-2 testing throughput.
Abstract
Virological testing is central to severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) containment, but many settings face severe limitations on testing. Group testing offers a way to increase throughput by testing pools of combined samples; however, most proposed designs have not yet addressed key concerns over sensitivity loss and implementation feasibility. Here, we combined a mathematical model of epidemic spread and empirically derived viral kinetics for SARS-CoV-2 infections to identify pooling designs that are robust to changes in prevalence and to ratify sensitivity losses against the time course of individual infections. We show that prevalence can be accurately estimated across a broad range, from 0.02 to 20%, using only a few dozen pooled tests and using up to 400 times fewer tests than would be needed for individual identification. We then exhaustively evaluated the ability of different pooling designs to maximize the number of detected infections under various resource constraints, finding that simple pooling designs can identify up to 20 times as many true positives as individual testing with a given budget. Crucially, we confirmed that our theoretical results can be translated into practice using pooled human nasopharyngeal specimens by accurately estimating a 1% prevalence among 2304 samples using only 48 tests and through pooled sample identification in a panel of 960 samples. Our results show that accounting for variation in sampled viral loads provides a nuanced picture of how pooling affects sensitivity to detect infections. Using simple, practical group testing designs can vastly increase surveillance capabilities in resource-limited settings.
INTRODUCTION
The ongoing pandemic of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), a novel coronavirus, has caused more than 83 million reported cases of coronavirus disease 2019 (COVID-19) and 1.8 million reported deaths between December 2019 and January 2021 (1). Although wide-spread virological testing is essential to inform disease status and where outbreak mitigation measures should be targeted or lifted, sufficient testing of populations with meaningful coverage has proven difficult (2–7). Disruptions in the global supply chains for testing reagents and supplies, as well as on-the-ground limitations in testing throughput and financial support, restrict the usefulness of testing—both for identifying infected individuals and to measure community prevalence and epidemic trajectory. Although these issues have been at the fore in even the highest-income countries, the situation is even more dire in low-income regions of the world. Cost barriers alone mean that it is often simply not practical to prioritize community testing in any useful way, with the limited testing that exists necessarily reserved for the health care setting. These limitations urge new, more efficient, approaches to testing to be developed and adopted both for individual diagnostics and to enable public health epidemic control and containment efforts.
Group or pooled testing offers a way to increase efficiency by combining samples into a group or pool and testing a small number of pools rather than all samples individually (8–10). For classifying individual samples, including for diagnostic testing, the principle is simple: If a pool tests negative, then all of the constituent samples are assumed negative. If a pool tests positive, then the constituent samples are putatively positive and must be tested again individually or in minipools (Fig. 1A). Further efficiency gains are possible through combinatorial pooling, where instead of testing every sample in every positive pool, each sample is instead represented across multiple pools and potential positives are identified on the basis of the pattern of pooled results (Fig. 1B) (9, 10).
Fig. 1. Group testing designs for sample identification or prevalence estimation.

In group testing, multiple samples are pooled, and tests are run on one or more pools. The results of these tests can be used for identification of positive samples (A and B) or to estimate prevalence (C). (A) In the simplest design for sample identification, samples are partitioned into nonoverlapping pools. In stage 1 of testing, a negative result (pool 2) indicates that each sample in that pool was negative, whereas a positive result (pool 1) indicates that at least one sample in the pool was positive. These putatively positive samples are subsequently individually tested in stage 2 to identify positive results. (B) In a combinatorial design, samples are included in multiple pools as shown in stage 1. All samples that were included in negative pools are identified as negative, and the remaining putatively positive samples that were not included in any negative test are tested individually in stage 2. (C) In prevalence estimation, samples are partitioned into pools. The pool measurement depends on the number and viral load of positive samples as well as the dilution factor. The (quantitative) results from each pool can be used to estimate the fraction of samples that would have tested positive, had they been tested individually.
Simple pooling designs can also be used to assess prevalence without individual specimen identification (Fig. 1C). It has already been shown that the frequency of positive pools can allow estimation of the overall prevalence (11). However, we ask here whether prevalence estimates can be honed by considering quantitative viral loads measured in each positive pool, rather than simply using binary (positive/negative) results, where the viral RNA load measurement from a pool is proportional to the sum of the diluted viral loads from each positive sample in the pool. An extreme, albeit less precise, example of this is the quantitation of viral loads in wastewater as a metric for whole community prevalence (12).
Although the literature on theoretically optimized pooling designs for COVID-19 testing has grown rapidly, formal incorporation of biological variation (such as viral loads) or general position along the epidemic curve has received little attention (13–16). Crucially, test sensitivity is not a fixed value but depends on viral load, which can vary by many orders of magnitudes across individuals and over the course of an infection, with implications for appropriate intervention and the interpretation of a viral load measurement from a sample pool (17–19). Further, the distribution of viral loads in surveillance testing is also sensitive to the course in the epidemic (growth versus decay) that will, thus, also affect the measured test sensitivity (20).
Here, we comprehensively evaluated designs for pooled testing of SARS-CoV-2 while accounting for epidemic dynamics and variation in viral loads arising from viral kinetics and extraneous features such as sampling variation. We demonstrate efficient, logistically feasible pooling designs for individual identification (diagnostics) and prevalence estimation (population surveillance). To do this, we used realistic simulated viral load data at the individual level over time, representing the entire time course of an epidemic to generate synthetic data that reflect the true distribution of viral loads in the population at any given time of the epidemic. We then used these data to derive optimal pooling strategies for different use cases and resource constraints in silico. In particular, we show how evaluating viral loads provides substantial efficiency gains in prevalence estimates, enabling robust public health surveillance where it was previously out of reach. Last, we demonstrate the approach using discarded de-identified human nasopharyngeal swabs initially collected for diagnostic and surveillance purposes.
RESULTS
Modeling a synthetic population to assess pooling designs
To identify optimal pooling strategies for distinct scenarios, we required realistic estimates of viral loads across epidemic trajectories. We developed a population-level mathematical model of SARS-CoV-2 transmission that incorporates empirically measured within-host virus kinetics and used the model to simulate population-level viral load distributions representing real data sampled from population surveillance, either using pharyngeal swab or sputum samples (Fig. 2). These simulations generated a synthetic, realistic epidemic with a peak daily incidence of 19.5 infections per 1000 people and peak daily prevalence of RNA positivity (viral load greater than 100 virus RNA copies/ml) of 265 per 1000 (Fig. 2D). We used these simulation data to evaluate optimal group testing strategies at different points along the epidemic curve for diagnostic and public health surveillance, where the true viral loads in the population are known fully.
Fig. 2. Viral kinetics model fits, simulated infection dynamics, and population-wide viral load kinetics.
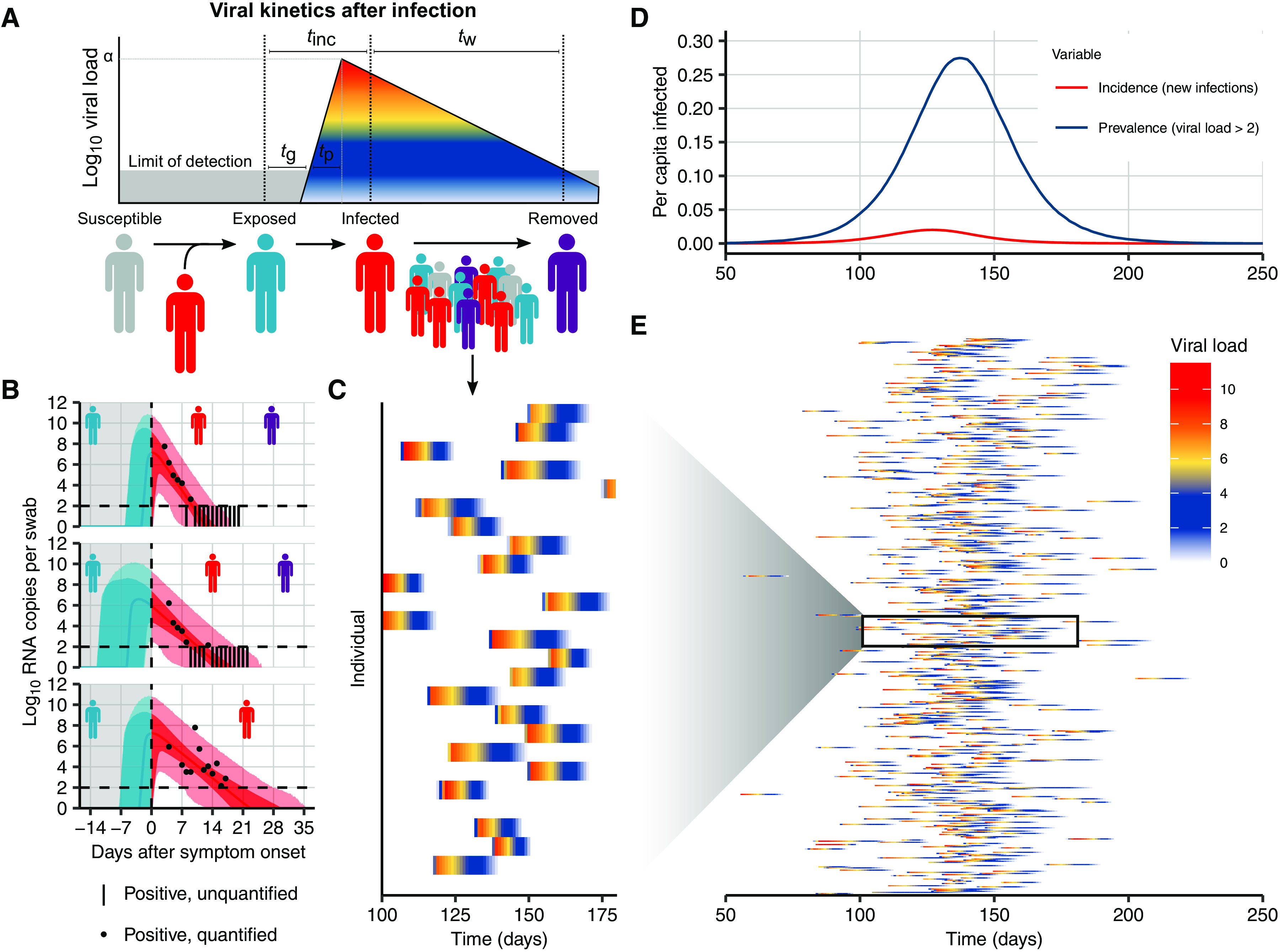
(A) Schematic of the viral kinetics and infection model. Individuals begin susceptible with no viral load (susceptible), acquire the virus from another infectious individual to become exposed but not yet infectious (exposed), experience an increase in viral load to become infectious and possibly develop symptoms (infected), and, last, either recover after viral waning or die (are removed). This process is simulated for many individuals. Symbols shown are tg as the time from infection to viral load crossing the limit of detection, tp as the time from first detectable viral load to peak, tinc as the time from infection to symptom onset, and tw as the time from symptom onset to loss of detectable viral load. (B) Model fits to time-varying viral loads in swab samples. The black dots show observed log10 RNA copies per swab, black bars show positive but unquantified swab samples, solid lines show posterior median estimates, dark shaded regions show 95% credible intervals (CIs) on model-predicted latent viral loads, and light shaded regions show 95% CI on simulated viral loads with added observation noise. The blue region shows viral loads before symptom onset, and the red region shows time after symptom onset. The horizontal dashed line shows the limit of detection. (C and E) Twenty-five and 500 simulated viral loads over time, respectively. The heatmap shows the viral load in each individual over time. The distribution of viral loads reflects the increase and subsequent decline of prevalence. We simulated from inferred distributions for the viral load parameters, thereby propagating substantial individual-level variability through the simulations. Marginal distributions of observed viral loads during the different epidemic phases are shown in fig. S4. (D) Simulated infection incidence and prevalence of virologically positive individuals from the SEIR model. Incidence was defined as the number of new infections per day divided by the population size. Prevalence was defined as the number of individuals with a viral load of >100 (log10 viral load >2) in the population divided by the population size on a given day.
Improved testing efficiency for estimating prevalence
We developed a statistical method to estimate prevalence of SARS-CoV-2 based on cycle threshold (Ct) values measured from pooled samples, potentially using far fewer tests than would be required to assess prevalence based on number of positive samples identified. We used our synthetic viral load data to assess inferential accuracy under a range of sample availabilities and pooling designs. Because RNA extraction and polymerase chain reaction (PCR) efficiency can vary from laboratory to laboratory depending on the methods used and within laboratory from batch to batch, we introduced substantial variability in our simulations in the conversion from viral load to Ct value to capture the multiple levels of uncertainty. In practice, this uncertainty can be refined by focusing on a single assay within a single laboratory.
Across the spectrum of simulated pools and tests, we found that simple pooling allows accurate estimates of prevalence across at least four orders of magnitude, ranging from 0.02 to 20%, with up to 400 times efficiency gains (in other words, 400 times fewer tests) for prevalence estimation than would be needed without pooling (Fig. 3). For example, in a population prevalence study that collects ~2000 samples, we accurately estimated infection prevalences as low as 0.05% using only 24 quantitative PCR (qPCR) tests overall (24 pools of 96 samples each; Fig. 3A and fig. S1). Because the distribution of Ct values may differ depending on the sample type (sputum versus swab), the instrument, and the phase of the epidemic (growth versus decline; fig. S2), the method should be calibrated in practice to viral load data (Ct values) specific to the laboratory and instrument as well as the population under investigation.
Fig. 3. Estimating prevalence from a small number of pooled tests.
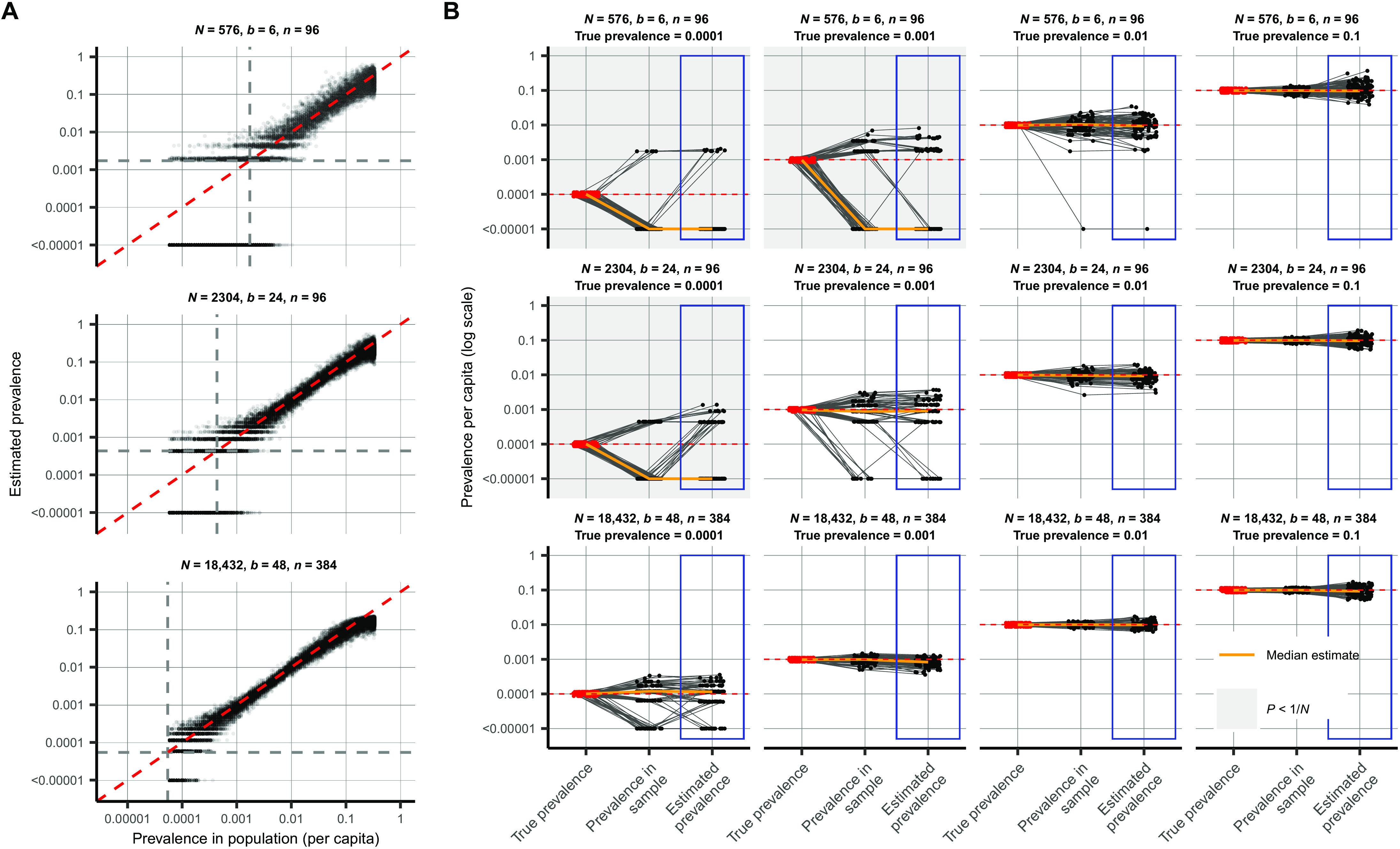
In prevalence estimation, a total of N individuals are sampled and partitioned into b pools (with n = N/b samples per pool). The true prevalence in the entire population varies over time with epidemic spread. Population prevalences shown here are during the epidemic growth phase. (A) Estimated prevalence (y axis) and true population prevalence (x axis) using 100 independent trials sampling N individuals at each day of the epidemic. Each facet shows a different pooling design (additional pooling designs shown in fig. S1). Dashed gray lines, one divided by the sample size, N. (B) For a given true prevalence (top label, red points and horizontal dashed red line), estimation error is introduced both through binomial sampling of positive samples (prevalence in sample) and inference on the sampled viral loads (estimated prevalence, blue boxes). Each set of three connected dots shows one simulation, with points slightly jittered on the x axis for visibility. Horizontal lines indicate accurate inference. The orange line shows the median across 100 simulations. Each panel shows the results from a single pooling design at the specified true prevalence. Sampling variation is a bigger contributor to error at low prevalence and low sample sizes. When prevalence is less than one divided by N (gray-shaded panels), inference is less accurate due to the high probability of sampling only negative individuals or inclusion of false positives.
Estimation error arises in two stages: sample collection effects and as part of the inference method (Fig. 3B). Error from sample collection became less important with increasing numbers of positive samples, which occurred with increasing population prevalence or by increasing the total number of tested samples (Fig. 3B and fig. S2). At very low prevalence, small sample sizes (N) risk missing positives together or becoming biased by false positives. We found that accuracy in prevalence estimation was greatest when population prevalence is greater than 1/N and that when this condition was met, partitioning samples into more pools always improved accuracy (fig. S2). In summary, very accurate estimates of prevalence can be attained using only a small fraction of the tests that would be needed in the absence of pooling.
Pooled testing for individual identification
We next analyzed effectiveness of group testing for identifying individual sample results at different points along the epidemic curve with the aim of identifying simple, efficient pooling strategies that are robust across a range of prevalences (Fig. 1, A and B). Using the simulated viral load data, we evaluated a large array of pooling designs in silico (table S1). On the basis of our models of viral kinetics and given a PCR limit of detection (LOD) of 100 viral copies/ml, we first estimated a baseline sensitivity of conventional (nonpooled) PCR testing of 85% during the epidemic growth phase: 15% of the time, we sampled an infected individual with a viral load greater than 1 but below the LOD of 100 viral copies/ml, (Fig. 4A), which largely agrees with reported estimates (21, 22). This estimate reflects sampling during the latent period of the virus (after infection but before substantial viral growth) or in the relatively long duration of low viral titers during viral clearance.
Fig. 4. Group testing for sample identification.
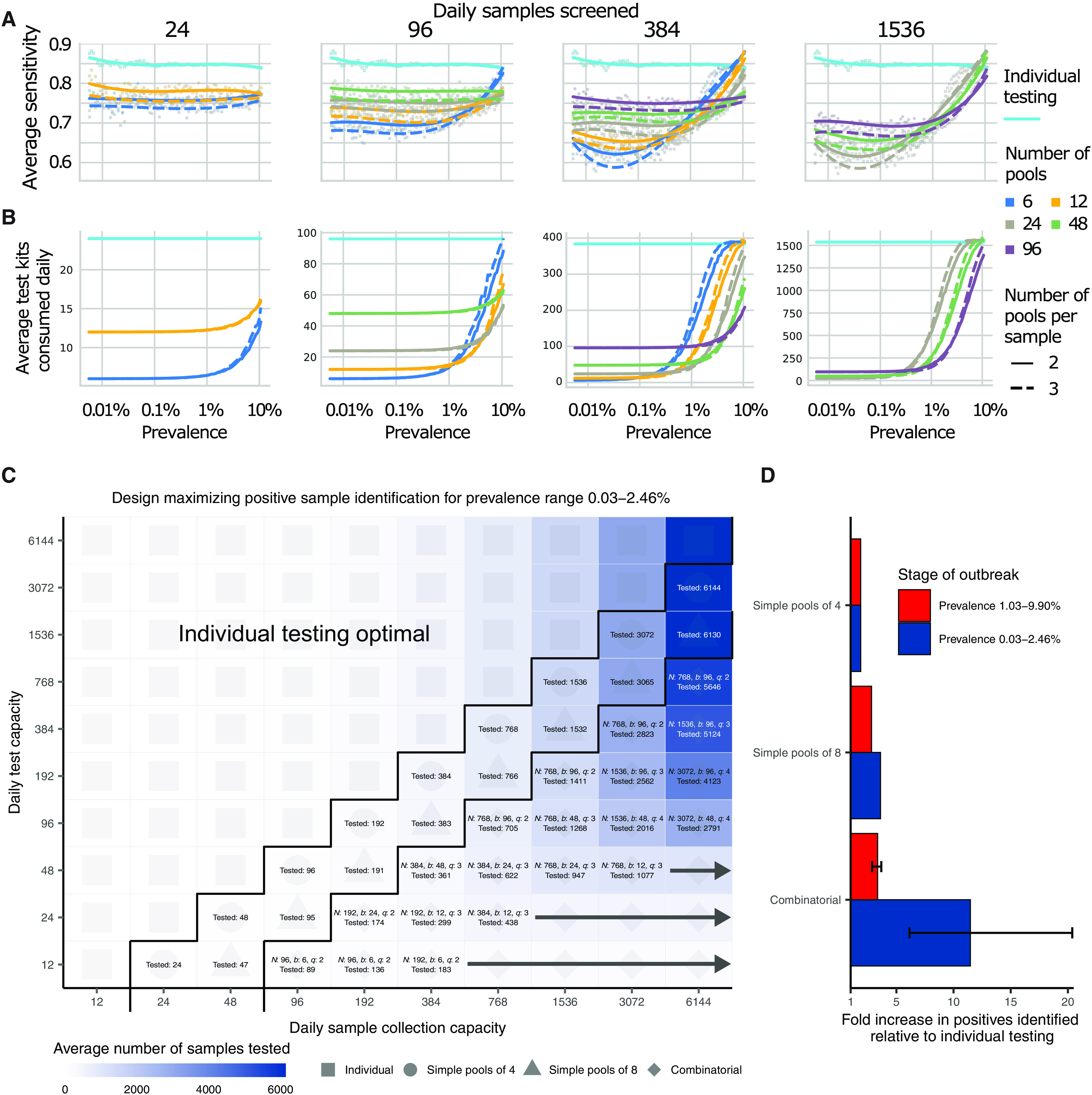
We evaluated a variety of group testing designs for sample identification (table S1) based on sensitivity (A), efficiency (B), total number of positive samples identified (C), and the fold increase in positive samples identified relative to individual testing (D). (A and B) The average sensitivity (A) (y axis, individual points and spline) and average number of tests needed to identify individual positive samples (B) (y axis) using different pooling designs (individual lines) were measured over days 20 to 110 in our simulated population, with results plotted against prevalence (x axis, log scale). Results show the average of 200,000 trials, with individuals selected at random on each day in each trial. Pooling designs are separated by the number of samples tested on a daily basis (individual panels), the number of pools (color), and the number of pools into which each sample is split (dashed versus solid line). Solid teal line indicates results for individual testing. Note that the average number of test kits consumed increases with prevalence due to a greater number of positive pools requiring confirmatory testing. (C) Every design was evaluated under constraints on the maximum number of samples collected (columns) and average number of reactions that can be run on a daily basis (rows) over days 40 to 90. Text in each box indicates the optimal design for a given set of constraints [number of samples per batch (N), number of pools (b), number of pools into which each sample is split (q), and average number of total samples screened per day]. Color indicates the average number of samples screened on a daily basis using the optimal design. Arrows indicate that the same pooling design is optimal at higher sample collection capacities due to testing constraints. (D) Fold increase in the number of positive samples identified relative to individual testing with the same resource constraints. Error bar shows range among optimal designs.
Sensitivity of pooled tests, relative to individual testing, is affected by the dilution factor of pooling and by the population prevalence, with lower prevalence resulting in generally lower sensitivity as positives are diluted into many negatives (Fig. 4A). The decrease in sensitivity is roughly linear with the log of the dilution factor used, which largely depends on the number and size of the pools and, for combinatorial pooling, the number of pools that each sample is placed into (fig. S3, A to C).
There is a less intuitive relationship between sensitivity and prevalence as it changes over the course of the epidemic. Early in an epidemic, there is an initial dip in sensitivity for both individual and pooled testing (Fig. 4A). Early on during exponential growth of an outbreak, in a random sample of infected individuals, a relatively greater fraction of positives will be sampled early in their infection and thus closer to their peak viral load. Later on, there is an increasing mixture of newly infected with individuals at the tail end of their infections and thus with lower viral loads at the time of sampling. We found that, as a result, sensitivity of pooled testing increases at peak prevalence because samples with lower viral loads, which would otherwise be missed because of dilution, are more likely to be “rescued” by coexisting in the same pool with high viral load samples and thus ultimately get individually retested (at their undiluted or less diluted concentration) during the validation stage. During epidemic decline, fewer new infections arose over time in our simulated data, and therefore, a randomly selected infected individual was more likely to be sampled during the recovery phase of their infection, when viral loads are lower (fig. S4D). Overall sensitivity is therefore lower during epidemic decline, because more infected individuals have viral loads below the LOD. During epidemic growth (up to day 108 in the simulation), we found that overall sensitivity of reverse transcription PCR (RT-PCR) for individual testing was 85%, whereas during epidemic decline (from day 168 onward), it was 60% (fig. S5A). Mean sensitivity of RT-PCR for individual testing was ~75% across the whole epidemic. We note that, in practice, sensitivity is likely higher than estimated here, because individuals are not sampled entirely at random but instead tend to be enriched with symptomatic people sampled nearer to peak viral loads. Together, these results describe how sensitivity is affected by the combination of epidemic dynamics, viral kinetics, and pooling design when individuals are sampled randomly from the population.
We found that, on average, most false negatives arose in our simulations from individuals sampled 7 days or more after their peak viral loads or around 7 days after what is normally considered symptom onset: These samples accounted for ~75% of false negatives in swab samples collected during epidemic growth and ~96% in swab samples during epidemic decline. This reflects that the majority of time spent in the PCR positive state is usually postinfectious and the asymmetry highlights differences in prevalence of more recent versus older infections during epidemic growth versus decay. Only ~3% of false-negative swab samples arose from individuals tested during the first week after peak viral load during epidemic growth, and only ~1% during epidemic decline (peak titers usually coincide with symptom onset, as reflected in our simulation); thus, most false negatives were from individuals with the least risk of onward transmission (fig. S3, D and E) (19).
The lower sensitivity of dilutional pooled testing is counterbalanced by gains in efficiency. When prevalence is low, efficiency is roughly the number of samples divided by the number of pools, as there are rarely putative positives to test individually. However, the number of validation tests required will increase as prevalence increases, and designs that are initially more efficient will lose efficiency (Fig. 4B). In general, we found that, at very low population prevalence, the use of fewer pools each with larger numbers of specimens offers relative efficiency gains compared to larger numbers of smaller pools, as most pools will test negative. However, as prevalence increases, testing a greater number of smaller pools pays off as more validations will be performed on fewer samples overall (Fig. 4B). For combinatorial designs with a given number of total samples and pools, splitting each sample across fewer pools resulted in modest efficiency gains (dashed versus solid lines in Fig. 4B).
To address realistic resource constraints, we integrated our analyses of sensitivity and efficiency with limits on daily sample collection and testing capacity to maximize the number of positive individuals identified. We analyzed the total number of samples screened and the fold increase in the number of positive samples identified relative to individual testing for a wide array of pooling designs evaluated over a period of 50 days during epidemic spread (days 40 to 90 where point prevalence reaches ~2.5%; Fig. 4, C and D). Because prevalence changes over time, the number of validation tests may vary each day despite constant pooling strategies. Thus, tests saved on days requiring fewer validation tests can be stored for days where more validation tests are required.
Across all resource constraints considered, we found that effectiveness ranged from 1 (when testing every sample individually is optimal) to 20 (that is, identifying 20× more positive samples on a daily basis compared with individual testing within the same budget; Fig. 4D). As expected, when capacity to collect samples exceeds capacity to test, group testing becomes increasingly effective. Simple pooling designs were most effective when samples are in moderate excess of testing capacity (2 to 8×), whereas increasingly complex combinatorial designs became the most effective when the number of samples to be tested greatly exceeded testing capacity. In addition, when prevalence was higher (sample prevalence from 1.03 to 9.90%), the optimal pooling designs shifted toward combinatorial pooling, and the overall effectiveness decreased—but still remained up to 4× more effective than individual testing (fig. S6). Our results were qualitatively unchanged when evaluating the effectiveness of pooling sputum samples, and the optimal pooling designs under each set of sample constraints were either the same or very similar (for example, using the same number of samples and pools, but a different number of pools per sample; fig. S7). Furthermore, we evaluated the same strategies during a 50-day window of epidemic decline (days 190 to 250) and found that similar pooling strategies were optimally effective, despite lower overall sensitivity as described above (fig. S5).
Pooled testing in a sustained, multiwave epidemic
Modeling the time evolution of viral load distributions and prevalence in sustained or multiple epidemic phases is important for understanding realistic performance of pooled testing for SARS-CoV-2 and other respiratory viral diseases. Building on our results above, we next simulated an epidemic with an initial wave, followed by a decline phase and subsequently another growth phase (fig. S8A).
Using this simulated epidemic, we first assessed how pooled testing for prevalence estimation would be affected by multiple waves. Because the results for a single epidemic peak (fig. S2) demonstrated that it is best to match training data for calibrating viral load distributions with the same phase in which the distributions will be used to estimate prevalence (that is, to match training data from growth phase with application during growth phase and decline with decline), we asked whether viral loads observed during a first wave of epidemic growth are appropriate training data for prevalence estimation during a second growth phase. We found that models using either training phase had low, nearly identical, degrees of error when predicting prevalence in the second growth phase (fig. S8B).
We next evaluated pooled testing for sample identification under the two-wave epidemic model. Our results above demonstrated a difference in sensitivity between growth and decline in a single-wave epidemic, driven by a shift in the viral load distribution sampled on any given day, which is, in turn, driven by viral kinetics and a shifting bias in the time relative to infection at which individuals are sampled (20). In an epidemic model with two waves, there is a dip in sensitivity over the transition from the first growth to decline phase, followed by a rise in sensitivity during the second growth phase (fig. S9A). The reduction in sensitivity for pooled testing compared to individual testing is relatively consistent across epidemic time for any given pooling design, demonstrating robustness to more complicated epidemic dynamics. Moreover, the changes in sensitivity are quantitative reflections of growth rate, so that the lower growth rate of the second growth phase results in a lower sensitivity than the first phase, due to more samples being from individuals with lower viral loads (fig. S9A).
The model also allowed us to assess any impacts on sample identification resulting from different epidemic dynamics in each period, for example, during initial epidemic growth with a high basic reproductive number (2.5) and a large susceptible population, compared with a second growth phase with smaller basic reproductive number (1.5) and a smaller susceptible population (due to past infections). We found that pooled testing for sample identification remained advantageous throughout the modeled time series, with very similar optimal pooling strategies under the same array of resource constraints considered above (fig. S9, B and C). Together, these results demonstrate generalizability to sustained or multiwave epidemics and robustness to changes in viral load distributions from different epidemic growth rates.
Pilot and validation experiments
We validated our pooling strategies using anonymized clinical nasopharyngeal swab specimens. To evaluate simple pooling across a range of inputs, we diluted 5 nasopharyngeal clinical swab samples with viral loads of 89,000, 12,300, 1280, 140, and 11 viral RNA copies/ml, respectively, into 23 negative nasopharyngeal swab samples (pools of 24). The results matched the simulated sampling results: The first three pools were all positive, the fourth was inconclusive (negative on N1 and positive on N2), and the remaining pool was negative (Fig. 5A and table S2). These results are as expected because the Emergency Use Authorization (EUA)–approved assay used has an LOD of ~100 virus copies/ml, such that the last two specimens fell below the LOD given a dilution factor of 24 (0.46 and 5.8 virus copies/ml once pooled).
Fig. 5. Experimental validation of simple and combinatorial pooling.

(A) Five pools (columns of matrix), each consisting of 24 nasopharyngeal swab samples (rows of matrix: 23 negative samples per pool and 1 positive, with viral load indicated on right) were tested by viral extraction and RT-qPCR. Pooled results indicated as negative (blue), inconclusive (yellow), or positive (red). (B) Six combinatorial pools (columns) of 48 samples (rows: 47 negative and 1 positive with viral load of 12,300) were tested as above. Pools 1, 2, and 4 tested positive. Arrows indicate two samples that were in pools 1, 2, and 4: sample 32 (negative) and sample 48 (positive). (C) Previously tested de-identified samples were pooled using a combinatorial design with 96 samples, 6 pools, and 2 pools per sample. Thirty positive samples were randomly distributed across 10 batches of the design. Viral RNA extraction and RT-qPCR were performed on each pool, with the results used to identify potentially positive samples. (D) Samples were pooled according to a simple design (48 pools with 48 samples per pool). Twenty-four positive samples were randomly distributed among the pools (establishing 1% prevalence). The pooled test results were used with an MLE procedure to estimate prevalence (0.87%), and bootstrapping was used to estimate a 95% confidence interval (0.52 to 1.37%).
We next tested combinatorial pooling, first using only a modest pooling design. We split 48 samples, including one positive sample with a viral load of 12,300, into six pools with each sample spread across three different pools. The method correctly identified the three pools containing the positive specimen (Fig. 5B and table S2). One negative sample was included in the same three pools as the positive sample; thus, eight total tests (six pools and two validations) were needed to accurately identify the status of all 48 samples, a 6× efficiency gain, which matched our expectations from the simulations.
We next performed two larger validation studies. To validate combinatorial pooling, we used anonymized samples representing 930 negative and 30 distinct positive specimens (3.1% prevalence), split across 10 batches of 96 specimens each (table S3). For each batch of 96, we split the specimens into six pools and spread each specimen across two pools (Fig. 5C and see table S4 for sample allocation and table S6 for pooled test results). For this combinatorial pooling design and prevalence, our simulations suggested that we would expect to identify ~26 of 30 known positives (~87%) and would see a 2.81× efficiency gain, using only 35% of the number of tests compared to no pooling. We identified 24 of the 30 known positives (80%) and required 35% fewer tests (341 versus 960, a 2.8× efficiency gain). Regarding the slightly reduced sensitivity, we note that positive samples and negative samples were accumulated over a longer period of time by the Broad’s testing facility, aggregated into separate plates, then delivered to our laboratory for pooling. Many of these samples went through multiple freeze-thaw cycles, likely leading to degradation of some viral RNA, which may have contributed to the reduced sensitivity that we observed relative to predicted levels.
To further validate our methods for prevalence estimation, we created a large study representing 2304 samples with a (true) positive prevalence of 1%. We aimed to determine how well our methods would work to estimate the true prevalence using 1/48th the number of tests compared to testing samples individually. To do this, we randomly assigned 24 distinct positive samples into 48 pools, with each pool containing 48 samples (table S3; to create the full set of pools, we treated some known negatives as distinct samples across separate pools). By using the measured viral loads detected in each of the pools, our methods estimated a prevalence of 0.87% (compared to the true prevalence of 1%) with a bootstrapped 95% confidence interval of 0.52 to 1.37% (Fig. 5D) and did so using 48× fewer tests than without pooling. This degree of accuracy was in line with our expectations from our simulations. The inference algorithm applied to these data used viral load distributions calibrated from our simulated epidemic, which, in turn, had viral kinetics calibrated to samples collected and tested on another continent, demonstrating robustness of the training procedure.
DISCUSSION
Our results show that group testing for SARS-CoV-2 can be a highly effective tool to increase surveillance coverage and capacity when resources are constrained. For prevalence testing, we found that fewer than 40 tests can be used to accurately infer prevalences across four orders of magnitude, providing large savings on the number of tests required. For individual identification, we determined an array of designs that optimize the rate at which infected individuals were identified under constraints on sample collection and daily test capacities. These results provide pooling designs that maximize the number of positive individuals identified on a daily basis, while accounting for epidemic dynamics, viral kinetics, viral loads measured from pharyngeal swabs or sputum, and practical considerations of laboratory capacity.
Although our experiments suggest that pooling designs may be beneficial for SARS-CoV-2 surveillance and identification of individual specimens, there are substantial logistical challenges to implementing theoretically optimized pooling designs. Large-scale testing without the use of pooling already requires managing thousands of specimens per day, mostly in series. Pooling adds complexity because samples must be tracked across multiple pools and stored for potential retesting. These complexities can be overcome with proper tracking software (including simple spreadsheets) and standard operating procedures in place before pooling begins. Such procedures can mitigate the risk of handling error or specimen mix-up.
In addition, expecting laboratories to regularly adapt their workflow and optimize pool sizes based on prevalence may not be feasible in some settings (8, 15). A potential solution is to follow a simple, fixed protocol that is robust to a range of prevalences. We provide an example spreadsheet (data file S1) guiding a technician receiving 96 labeled samples to create six pools, enter the result of each pool, and be provided a list of putative positives to be retested. Despite these logistical challenges, simple Dorfman pooling has been successfully used in Israel during the COVID-19 pandemic to process ~133,000 samples over a period of variable prevalence, leading to resource savings with expected and acceptable sensitivity loss (23).
For certain viscous sample types (for example saliva or sputum), the process of pooling samples may be challenging. Whereas robotic pooling with small volumes of viscous samples is error prone, manual pipetting can be reliable. Our designs can accommodate both robotic and manual pipetting, although pooling may still be a challenge for viscous samples compared to swabs in viral transport media or similar. In these cases, pretreating samples with proteinase K can additionally make samples easier to pipette (as is performed with SalivaDirect) and increase retention of viral RNA by degrading nucleases (24). Overall, our results are largely applicable to a variety of sampling landscapes in which viral loads are determined. However, individual laboratories will of course need to determine which approach is best suited for their own purposes.
Depending on the purpose of testing and resources available, enhancing efficiency at the expense of sensitivity must be considered. We recommend validation of the selected pooling strategy to identify potential differences in predicted versus observed sensitivity as demonstrated here, which may be unacceptable if the aim is clinical testing rather than overall throughput. However, we found that the most efficient pooling designs were the same when using swab- or sputum-calibrated simulations, suggesting that the ideal strategies were robust to misspecification in overall sensitivity. For prevalence testing, accurate estimates can be obtained using relatively few tests if individual identification is not the aim. For individual testing, although identifying all positive samples that are tested is of course the objective, increasing the number of specimens tested when sacrificing sensitivity may be a crucially important trade-off.
As an example of how to evaluate the allocation of limited resources in the context of reduced sensitivity and enhanced efficiency, consider our experience with a laboratory in The Gambia. Simplifying a bit, the resource constraints being faced were a limited capacity for sample collection (several hundred per day, initially, this has since increased), a limited supply of test kits for the foreseeable future, and an excess of laboratory technician capacity. For the purposes of screening, we assumed that up to 384 samples could be collected and up to 24 test kits consumed per day (on average, because there is a fixed supply over a longer horizon, sample collection and testing for triaging or testing higher-risk individuals were handled separately). With individual testing, effectively 20.4 individuals (24 available tests × 85% sensitivity) would be screened per day. On the other hand, with the pooling strategy identified under these constraints (N = 192, b = 12, and q = 3), one could effectively screen 202.57 individuals per day, on average (192 individuals screened per batch × 68% sensitivity × average of 1.55 batches run per day). Therefore, despite substantially reduced sensitivity, the pooling strategy would identify about 10× as many infected individuals. Given the unused technician capacity, the logistical cost (in our experience, it takes one person ~1.5 hours to pool 192 samples by hand in an unoptimized workflow, although there are other costs to consider) is very likely outweighed by the positive benefits on infection control.
Furthermore, the specimens most likely to be lost due to dilution are those samples with the lowest viral loads already near the LOD. Although there is a chance that the low viral load samples missed are on the upswing of an infection, when identifying the individual would be maximally beneficial, the asymmetric course of viral titers over the full duration of positivity means that most false negatives would arise from failure to detect late-stage, low-titer individuals who are less likely to be infectious (19). Optimal strategies and expectations of sensitivity should also be considered alongside the phase of the epidemic and how samples are collected, as this will dictate the distribution of sampled viral loads. For example, if individuals are under a regular testing regimen and therefore likely to be removed from the tested population before reaching the long tail of low viral loads at the end of infection or are tested because of recent exposure or symptom onset, then viral loads at the time of sampling will typically be higher, leading to higher sensitivity despite dilution effects.
For individual identification, errors may arise when a positive sample is split into multiple pools, but only some of those pools test positive (see Supplementary Materials and Methods for an example and further discussion). For an error tolerance, e, error correction is possible by allowing a putative positive to be in up to e negative pools, where e > 0 will result in not only higher sensitivity but also more putative positives and lower efficiency. The optimal error tolerance can be derived analytically in terms of the independent error rate r (that is, rates of PCR failure not related to low viral load, dilution, or other systemic effects) (25). With a conservative estimate of the rate of independent PCR failure, r, of <5% and samples split into less than five pools, q < 5 (as in all pooling designs we consider), the optimal e is less than 0.5; thus, it is optimal to not correct these errors (e = 0) in all pooling designs considered here. We note that, for alternative pooling designs, particularly those with each sample split into many pools, error correction may increase effectiveness. Other methods that incorporate error correction [such as Pooling-Based Efficient SARS-CoV-2 Testing (P-BEST) (16)] have more complicated pooling designs with samples split into more pools (q = 6 or more). An alternative to individually testing all putative positives would be to simply retest each pool (optionally repooling samples before retesting). Whether to do so would be a choice made by each laboratory, similar to decisions about whether to retest samples with failed controls or inconclusive results. Furthermore, error correction does not address the primary source of error: low viral load, below the LOD and possibly due to dilution, which is a systemic problem affecting all pools with a given sample.
Testing throughput and staffing resources should also be considered. If a testing facility can only run a limited number of tests per day, then it may be preferable to process more samples at a slight cost to sensitivity. Backlogs of individual testing can result in substantial delays in returning individual test results, which can ultimately defeat the purpose of identifying individuals for isolation, potentially further justifying some sensitivity losses (26, 27). Choosing a pooling strategy will therefore depend on target population and availability of resources. For testing in the community or in existing sentinel surveillance populations (for example, antenatal clinics), point prevalence is likely to be low (<0.1 to 3%), which may favor strategies with fewer pools (6, 28–30). Conversely, secondary attack rates in contacts of index cases may vary from <1 to 17% depending on the setting (e.g., casual versus household contacts) (31–33) and may be even higher in some instances (34, 35), favoring more pools. These high-point prevalence subpopulations may represent less efficient use cases for pooled testing, because our results suggest that pooling for individual identification is inefficient once prevalence reaches 10%. However, group testing may still be useful if testing capacity is severely limited, for example, samples from all members of a household could be tested as a pool and quarantined if tested positive, enabling faster turnaround than testing individuals. This approach may be even more efficient if samples can be pooled at the point of collection, requiring no change to laboratory protocols.
Related to throughput, another important consideration is the overall time between sample collection and testing, particularly where individuals are advised to self-isolate until their result is returned. In settings where results are being returned within 24 to 48 hours already and there is no backlog of samples, pooling is unlikely to decrease turnaround time due to the extra time needed to retest some samples. However, in settings where testing capacity is insufficient to meet demand and results are delayed simply due to the queue of samples, pooling will likely decrease turnaround time because the time from sample to processing will be shorter.
Our modeling results have a number of limitations and may be updated as more data become available. First, our simulation results depend on the generalizability of the simulated Ct values, which were based on pharyngeal swab and sputum viral load data from symptomatic patients. Although some features of viral trajectories, such as viral waning, differ between symptomatic and asymptomatic individuals, population-wide data suggest that the range of Ct values does not differ based on symptom status (19, 36). Furthermore, we have assumed a simple hinge function to describe viral kinetics. Different shapes for the viral kinetics trajectory may become apparent as more data become available. Nonetheless, our simulated population distribution of Ct values is comparable to existing data, and we propagated substantial uncertainty in viral kinetics parameters to generate a wide range of viral trajectories. For prevalence estimation, the maximum likelihood estimate (MLE) framework requires training on a distribution of Ct values. Such data can be available based on past tests from a given laboratory, but care should be taken to use a distribution appropriate for the population under consideration. For example, training the virus kinetics model on data skewed toward lower viral loads (as would be observed during the tail end of an epidemic curve) may be inappropriate when the true viral load distribution is skewed higher (as might be the case during the growth phase of an epidemic curve). Nevertheless, we used our simulated distribution of Ct, which was fit to virus kinetics in published reports from distinct laboratories across the world, and we obtained highly accurate results throughout. Thus, despite the limitations just mentioned, this shows that virus kinetics models are quite robust and may not, in practice, require new fitting to the individual laboratory or population. In addition, although we assumed that individuals are sampled from the population at random in our analysis, in practice, samples that are processed together are also typically collected together, which may bias the distribution of positive samples among pools.
Pooling samples for qPCR testing is widespread in infectious disease surveillance, and our approach could be easily adapted to investigate pooled testing strategies in silico for other disease systems (37–41). The viral kinetics model could be replaced with any model for the within-host process of interest (for example, an antibody response), and the epidemic model could be reparameterized for particular settings (such as a vector-borne with seasonal transmission). In general, modeling the key mechanisms behind the biological and epidemiological processes that give rise to measured quantities such as viral load provides a more nuanced picture of sensitivity and efficiency than treating test characteristics as fixed values.
We have shown that simple designs that are straightforward to implement have the potential to greatly improve testing throughput across the time course of the pandemic. These principles likely also hold for pooling of sera for antibody testing, which remains an avenue for future work. There are logistical challenges and additional costs associated with pooling that we do not consider deeply here, and it will therefore be up to laboratories and policy makers to decide where these designs are feasible. Substantial coordination will therefore be necessary to make group testing practical, but investing in these efforts could enable community screening where it is currently infeasible and provide epidemiological insights that are urgently needed.
MATERIALS AND METHODS
Study design
The aim of this study was to understand the potential effectiveness of pooled testing for SARS-CoV-2 infection identification and prevalence estimation in a population where prevalence and viral loads evolve over time. The study included both simulation-based experiments where the effectiveness of different pooling strategies was evaluated against known inputs and laboratory experiments using previously tested nasopharyngeal swab specimens to validate our proposed pooling designs.
In the modeling experiments, we simulated a SARS-CoV-2 outbreak in a large population roughly the size of a small U.S. state (12,500,000 individuals), ensuring that there were at least 100 unique infected individuals early in the epidemic at prevalence as low as 0.001%. For each tested pooling design and day of the outbreak, we randomly sampled individuals from the simulated population to generate in silico sample pools with known viral loads. We repeated this sampling process across independent trials until 2500 positive individuals had been sampled across all trials, or until 200,000 trials had been run.
In the laboratory validation trials of our pooling designs, human nasopharyngeal swab specimens with previously determined positive or negative status were randomly assigned a sample ID within a validation experiment, for example, 1 of 2304 sample IDs in the large prevalence study or 1 of 960 IDs in the large identification study, without regard for their positivity status. Once IDs were assigned, samples were distributed to pools according to prespecified pooling designs. Samples with predetermined inconclusive status were not included. The study followed all relevant ethical guidelines for the use of discarded patient specimens. An exempt protocol for the study was approved by the Broad Institute Office of Research and Sponsored Programs (ORSP).
Simulation model of infection dynamics and viral load kinetics
We developed a population-level mathematical model of SARS-CoV-2 transmission that incorporates realistic within-host virus kinetics. Full details are provided in Supplementary Materials and Methods, but we provide an overview here. First, we fit a viral kinetics model to published longitudinally collected viral load data from pharyngeal swab and sputum samples using a Bayesian hierarchical model that captures the variation of peak viral loads, delays from infection to peak, and virus decline rates across infected individuals (Fig. 2, A and B, and fig. S10) (18). By incorporating estimated biological variation in virus kinetics, this model allows random draws each representing distinct within-host virus trajectories. We then simulated infection prevalence during a SARS-CoV-2 outbreak using a deterministic Susceptible-Exposed-Infected-Removed (SEIR) model with parameters reflecting the natural history of SARS-CoV-2 (Fig. 2D). For each simulated infection, we generated longitudinal virus titers over time by drawing from the distribution of fitted virus kinetic curves, using distributions derived using either pharyngeal swab or sputum data (fig. S4). All estimated and assumed model parameters are shown in table S5, with model fits shown in fig. S10. Posterior estimates and Markov chain Monte Carlo trace plots are shown in figs. S11 and S12. We accounted for measurement variation by transforming viral loads into Ct values under a range of Ct calibration curves, simulating false positives with 1% probability, and simulating sampling variation. We assumed an LOD of 100 RNA copies/ml.
Estimating prevalence from pooled test results
We adapted a statistical (maximum likelihood) framework initially developed to estimate HIV prevalence with pooled antibody tests to estimate prevalence of SARS-CoV-2 using pooled samples (42, 43). The framework accounts for the distribution of viral loads (and uncertainty around them) measured in pools containing a mixture of negative and potentially positive samples. By measuring viral loads from multiple such pools, it is possible to estimate the prevalence of positive samples without individual testing.
We evaluated prevalence estimation under a range of sample availabilities (N total samples; N = 288 to ~18,000) and pooling designs. We varied the pool size of combined specimens (n samples per pool; n = 48, 96, 192, or 384) and the number of pools (b = 6, 12, 24, or 48). For each combination, we estimated the point prevalence from pooled tests on random samples of individuals drawn during epidemic growth (days 20 to 120) and decline (days 155 to 300). Because the data were realistic but simulated, we used ground truth prevalence in the population and, separately, in the specific set of samples collected from the overall population to assess accuracy of our estimates (see for example Fig. 3B). We calculated estimates for 100 entirely distinct epidemic simulations.
Pooled tests for individual sample identification
Using the same simulated population, we evaluated a range of simple and combinatorial pooling strategies for individual positive sample identification. In simple pooling designs, each sample is placed in one pool, and each pool consists of some prespecified number of samples. If a pool tests positive, then all samples that were placed in that pool are retested individually (Fig. 1A). For combinatorial pooling, each sample is split into multiple, partially overlapping pools (Fig. 1B) (9, 10). Every sample that was placed in any pool that tested negative is inferred to be negative, and the remaining samples are identified as potential positives. Here, we consider a very simple form of combinatorial testing, where identified potential positive samples are individually tested in a validation stage.
A given pooling design is defined by three parameters: the total number of individuals to be tested (N), the total number of pools to test (b), and the number of pools a given sample is included in (q). For instance, if we have 50 individuals (N) to test, then we might split the 50 samples into four pools (b) of 25 samples each, where each sample is included in two pools. Note that, by definition, in simple pooling designs, each sample is placed in one pool (q = 1).
To identify optimal testing designs under different resource constraints, we systematically analyzed a large array of pooling designs under various sample and test kit availabilities. We evaluated different combinations of between 12 and ~6000 available samples/tests per day. The daily testing capacity shown is the daily average, although we assume that there is some flexibility to use fewer or more tests day to day (in other words, that there is a budget for period of time under evaluation).
For each set of resource constraints, we evaluated designs that split N samples between 1 and 96 distinct pools and with samples included in q = 1 (simple pooling), 2, 3, or 4 (combinatorial pooling) pools (table S1). To ensure robust estimates (especially at low prevalences of less than 1 in 10,000), we repeated each simulated pooling protocol at each time point in the epidemic up to 200,000 times. In each scenario, we calculated the sensitivity to detect positive samples when they existed in the pool; the efficiency, defined as the total number of samples tested divided by the total number of tests used; the total number of identified true positives (total recall); and the effectiveness, defined as the total recall relative to individual testing.
Pilot experiments
For validation experiments of our simulation-based approach, we used fully de-identified, discarded human nasopharyngeal specimens obtained from the Broad Institute of Massachusetts Institute of Technology (MIT) and Harvard. In each experiment, sample aliquots were pooled before RNA extraction and qPCR, and pooled specimens were tested using the EUA-approved SARS-CoV-2 assay performed by the Broad Institute Clinical Laboratory Improvement Amendments (CLIA) laboratory. qPCR Ct values were calibrated to a standard curve of known viral RNA copies (fig. S13). The protocol and specifics of each pooling approach are described in full detail in Supplementary Materials and Methods.
Acknowledgments
We thank B. Gnangnon, R. Dey, X. Lin, E. Dobriban, R. Niehus, and H. Shakerchi for useful discussions. Funding: This work was supported by the Merkin Institute Fellowship at the Broad Institute of MIT and Harvard (to B.C.), by the National Institute of General Medical Sciences (#U54GM088558 to J.A.H. and M.J.M.), by an NIH DP5 grant (to M.J.M.), and the Dean’s Fund for Postdoctoral Research of the Wharton School and NSF BIGDATA grant IIS 1837992 (to D.H.). Author contributions: B.C., J.A.H., A.R., and M.J.M. conceived the study. B.C. and J.A.H. performed the simulations and modeling. B.B., M.H., M.C., J.B., and B.S. performed the pooled testing. D.H. and B.C. designed the combinatorial pools used for validation. M.S. and A.K.S. contributed to study design, discussion, and analysis of effectiveness in a resource constrained environment. S.G. contributed to study design of validation experiments. B.C., J.A.H., A.R., and M.J.M. analyzed results and wrote the manuscript with input from all authors. Competing interests: A.R. is a cofounder and equity holder of Celsius Therapeutics, an equity holder in Immunitas, and, until 31 July 2020, was an SAB member of Thermo Fisher Scientific, Syros Pharmaceuticals, Asimov, and Neogene Therapeutics. From 1 August 2020, A.R. is an employee of Genentech, a member of the Roche group. M.J.M. is a medical advisor for Detect. Data and materials availability: Raw data generated in this study are available in tables S2 and S6. Simulated data can be regenerated using the accompanying code. The SEIR model, viral kinetics model, and Markov chain Monte Carlo were implemented in R version 3.6.2. The remainder of the work was performed in Python version 3.7. The code used for the Markov chain Monte Carlo framework is available at 10.5281/zenodo.4521688. All other code and data used are available at 10.5281/zenodo.4521668. This work is licensed under a Creative Commons Attribution 4.0 International (CC BY 4.0) license, which permits unrestricted use, distribution, and reproduction in any medium, provided that the original work is properly cited. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/. This license does not apply to figures/photos/artwork or other content included in the article that is credited to a third party, obtain authorization from the rights holder before using this material.
SUPPLEMENTARY MATERIAL
stm.sciencemag.org/cgi/content/full/scitranslmed.abf1568/DC1
Fig. S1. True prevalence against maximum likelihood estimates.
Fig. S2. Prevalence estimation can depend on training and application period.
Fig. S3. Sensitivity of sample identification relative to dilution factor and time since peak viral load.
Fig. S4. Simulated viral loads.
Fig. S5. Group testing for sample identification during epidemic decline.
Fig. S6. Effectiveness of optimal testing design under resource constraints at high prevalence.
Fig. S7. Effectiveness of optimal testing design under resource constraints using sputum data.
Fig. S8. Evaluation of pooled testing in a sustained, multiwave epidemic.
Fig. S9. Evaluation of pooled testing for sample identification in the multiwave epidemic shown in fig. S8A.
Fig. S10. Model fits to swab viral loads.
Fig. S11. Posterior distributions of estimated parameters fitted to swab and sputum data.
Fig. S12. Markov chain Monte Carlo trace plots from fitting to swab and sputum data.
Fig. S13. qPCR calibration curve using standard viral RNA copies.
Table S1. List of all group test designs for sample identification.
Table S2. Ct values from qPCR on pooled samples with variable viral load.
Table S3. Positive sample distribution within validation pools.
Table S4. Pool design for combinatorial test with 96 samples.
Table S5. Description of all parameters used in the viral kinetics and transmission models.
Table S6. RT-qPCR results for pooling validations.
Data file S1. Ninety-six–sample pooling template (Excel).
REFERENCES AND NOTES
- 1.World Health Organization, “Weekly epidemiological update - 5 January 2021” (World Health Organization, 2021); www.who.int/publications/m/item/weekly-epidemiological-update---5-january-2021.
- 2.World Health Organization, “Critical preparedness, readiness and response actions for COVID-19” (World Health Organization, 2020); www.who.int/publications/i/item/critical-preparedness-readiness-and-response-actions-for-covid-19.
- 3.Cheng M. P., Papenburg J., Desjardins M., Kanjilal S., Quach C., Libman M., Dittrich S., Yansouni C. P., Diagnostic testing for severe acute respiratory syndrome–Related coronavirus-2. Ann. Intern. Med. 172, 726–734 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Korean Society of Infectious Diseases; Korean Society of Pediatric Infectious Diseases; Korean Society of Epidemiology; Korean Society for Antimicrobial Therapy; Korean Society for Healthcare-associated Infection Control and Prevention; Korea Centers for Disease Control and Prevention , Report on the epidemiological features of coronavirus disease 2019 (COVID-19) outbreak in the Republic of Korea from January 19 to March 2, 2020. J. Korean Med. Sci. 35, e112 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lee V. J., Chiew C. J., Khong W. X., Interrupting transmission of COVID-19: Lessons from containment efforts in Singapore. J. Travel Med. 27, taaa039 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gudbjartsson D. F., Helgason A., Jonsson H., Magnusson O. T., Melsted P., Norddahl G. L., Saemundsdottir J., Sigurdsson A., Sulem P., Agustsdottir A. B., Eiriksdottir B., Fridriksdottir R., Gardarsdottir E. E., Georgsson G., Gretarsdottir O. S., Gudmundsson K. R., Gunnarsdottir T. R., Gylfason A., Holm H., Jensson B. O., Jonasdottir A., Jonsson F., Josefsdottir K. S., Kristjansson T., Magnusdottir D. N., le Roux L., Sigmundsdottir G., Sveinbjornsson G., Sveinsdottir K. E., Sveinsdottir M., Thorarensen E. A., Thorbjornsson B., Löve A., Masson G., Jonsdottir I., Möller A. D., Gudnason T., Kristinsson K. G., Thorsteinsdottir U., Stefansson K., Spread of SARS-CoV-2 in the Icelandic population. N. Engl. J. Med. 382, 2302–2315 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Grassly N. C., Pons-Salort M., Parker E. P. K., White P. J., Ferguson N. M.; Imperial College COVID-19 Response Team , Comparison of molecular testing strategies for COVID-19 control: A mathematical modelling study. Lancet Infect. Dis. 2020, 1381–1389 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dorfman R., The detection of defective members of large populations. Ann. Math. Stat. 14, 436–440 (1943). [Google Scholar]
- 9.D. Du, F. K. Hwang, F. Hwang, Combinatorial Group Testing and Its Applications (World Scientific, 2000). [Google Scholar]
- 10.E. Porat, A. Rothschild, (n.d.). Explicit Non-adaptive Combinatorial Group Testing Schemes, in Automata, Languages and Programming (Vol. 5125, Lecture Notes in Computer Science, Berlin, Heidelberg: Springer Berlin Heidelberg), pp. 748–759. [Google Scholar]
- 11.European Centre for Disease Prevention, “Methodology for estimating point prevalence of SARS-CoV-2 infection by pooled RT-PCR testing” (European Centre for Disease Prevention, 2020); www.ecdc.europa.eu/en/publications-data/methodology-estimating-point-prevalence-sars-cov-2-infection-pooled-rt-pcr.
- 12.Wu F., Zhang J., Xiao A., Gu X., Lee W. L., Armas F., Kauffman K., Hanage W., Matus M., Ghaeli N., Endo N., Duvallet C., Poyet M., Moniz K., Washburne A. D., Erickson T. B., Chai P. R., Thompson J., Alm E. J., SARS-CoV-2 titers in wastewater are higher than expected from clinically confirmed cases. mSystems 5, e00614-20 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hogan C. A., Sahoo M. K., Pinsky B. A., Sample pooling as a strategy to detect community transmission of SARS-CoV-2. JAMA 323, 1967 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bilder C. R., Iwen P. C., Abdalhamid B., Tebbs J. M., McMahan C. S., Tests in short supply? Try group testing. Significance 17, 15–16 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ben-Ami R., Klochendler A., Seidel M., Sido T., Gurel-Gurevich O., Yassour M., Meshorer E., Benedek G., Fogel I., Oiknine-Djian E., Gertler A., Rotstein Z., Lavi B., Dor Y., Wolf D. G., Salton M., Drier Y.; Hebrew University-Hadassah COVID-19 Diagnosis Team , Large-scale implementation of pooled RNA extraction and RT-PCR for SARS-CoV-2 detection. Clin. Microbiol. Infect. 26, 1248–1253 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shental N., Levy S., Wuvshet V., Skorniakov S., Shalem B., Ottolenghi A., Greenshpan Y., Steinberg R., Edri A., Gillis R., Goldhirsh M., Moscovici K., Sachren S., Friedman L. M., Nesher L., Shemer-Avni Y., Porgador A., Hertz T., Efficient high-throughput SARS-CoV-2 testing to detect asymptomatic carriers. Sci. Adv. 6, eabc5961 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zou L., Ruan F., Huang M., Liang L., Huang H., Hong Z., Yu J., Kang M., Song Y., Xia J., Guo Q., Song T., He J., Yen H. L., Peiris M., Wu J., SARS-CoV-2 viral load in upper respiratory specimens of infected patients. N. Engl. J. Med. 382, 1177–1179 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wölfel R., Corman V. M., Guggemos W., Seilmaier M., Zange S., Müller M. A., Niemeyer D., Jones T. C., Vollmar P., Rothe C., Hoelscher M., Bleicker T., Brünink S., Schneider J., Ehmann R., Zwirglmaier K., Drosten C., Wendtner C., Virological assessment of hospitalized patients with COVID-2019. Nature 581, 465–469 (2020). [DOI] [PubMed] [Google Scholar]
- 19.Cevik M., Tate M., Lloyd O., Maraolo A. E., Schafers J., Ho A., SARS-CoV-2, SARS-CoV, and MERS-CoV viral load dynamics, duration of viral shedding, and infectiousness: A systematic review and meta-analysis. Lancet Microbe 2, e13–e22 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.J. A. Hay, L. Kennedy-Shaffer, S. Kanjilal, M. Lipsitch, M. J. Mina, Estimating epidemiologic dynamics from single cross-sectional viral load distributions. medRxiv 2020.10.08.20204222 [Preprint]. 13 October 2020. 10.1101/2020.10.08.20204222. [DOI] [PMC free article] [PubMed]
- 21.T. C. Williams, E. Wastnedge, G. McAllister, R. Bhatia, K. Cuschieri, K. Kefala, F. J. Hamilton, I. Johannessen, I. F. Laurenson, J. Shepherd, A. Stewart, D. Waters, H. Wise, K. Templeton, Sensitivity of RT-PCR testing of upper respiratory tract samples for SARS-CoV-2 in hospitalised patients: A retrospective cohort study. medRxiv 2020.06.19.20135756 [Preprint]. 20 June 2020. 10.1101/2020.06.19.20135756. [DOI] [PMC free article] [PubMed]
- 22.Arevalo-Rodriguez I., Buitrago-Garcia D., Simancas-Racines D., Zambrano-Achig P., del Campo R., Ciapponi A., Sued O., Martinez-Garcia L., Rutjes A., Low N., Perez-Molina J., Zamora J., False-negative results of initial RT-PCR assays for COVID-19: A systematic review. PLOS ONE 15, e0242958 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Barak N., Ben-Ami R., Sido T., Perri A., Shtoyer A., Rivkin M., Licht T., Peretz A., Magenheim J., Fogel I., Livneh A., Daitch Y., Oiknine-Djian E., Benedek G., Dor Y., Wolf D. G., Yassour M.; Hebrew University-Hadassah COVID-19 Diagnosis Team , Lessons from applied large-scale pooling of 133,816 SARS-CoV-2 RT-PCR tests. Sci. Transl. Med. 13, eabf2823 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vogels C. B., Watkins A. E., Harden C. A., Brackney D. E., Shafer J., Wang J., Caraballo C., Kalinich C. C., Ott I. M., Fauver J. R., Kudo E., SalivaDirect: A simplified and flexible platform to enhance SARS-COV-2 testing capacity. Med. 2, 263.e6–280.e6 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cheraghchi M., Hormati A., Karbasi A., Vetterli M., Group testing with probabilistic tests: Theory, design and application. IEEE Trans. Inf. Theory 57, 7057–7067 (2011). [Google Scholar]
- 26.Larremore D. B., Wilder B., Lester E., Shehata S., Burke J. M., Hay J. A., Tambe M., Mina M. J., Parker R., Test sensitivity is secondary to frequency and turnaround time for COVID-19 surveillance. medRxiv 10.1101/2020.06.22.20136309 , (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Paltiel A. D., Zheng A., Walensky R. P., Assessment of SARS-CoV-2 screening strategies to permit the safe reopening of college campuses in the United States. JAMA Netw. Open 3, e2016818 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.REACT Study Investigators, Riley S., Ainslie K., Eales O., Jeffrey B., Walters C., Atchison C., Diggle P., Ashby D., Donnelly C., Cooke G., Barclay W., Ward H., Taylor G., Darzi A., Elliott P., Community prevalence of SARS-CoV-2 virus in England during May 2020: REACT study. medRxiv 10.1101/2020.07.10.20150524 , (2020). [Google Scholar]
- 29.K. B. Pouwels, T. House, J. V. Robotham, P. Birrell, A. B. Gelman, N. Bowers, I. Boreham, H. Thomas, J. Lewis, I. Bell, J. I. Bell, J. Newton, J. Farrar, I. Diamond, P. Benton, S. Walker; the COVID-19 Infection Survey Team, Community prevalence of SARS-CoV-2 in England: Results from the ONS Coronavirus Infection Survey Pilot. medRxiv 2020.07.06.20147348 [Preprint]. 7 July 2020. 10.1101/2020.07.06.20147348. [DOI]
- 30.Lavezzo E., Franchin E., Ciavarella C., Cuomo-Dannenburg G., Barzon L., Del Vecchio C., Rossi L., Manganelli R., Loregian A., Navarin N., Abate D., Sciro M., Merigliano S., De Canale E., Vanuzzo M. C., Besutti V., Saluzzo F., Onelia F., Pacenti M., Parisi S., Carretta G., Donato D., Flor L., Cocchio S., Masi G., Sperduti A., Cattarino L., Salvador R., Nicoletti M., Caldart F., Castelli G., Nieddu E., Labella B., Fava L., Drigo M., Gaythorpe K. A. M.; Imperial College COVID-19 Response Team, Brazzale A. R., Toppo S., Trevisan M., Baldo V., Donnelly C. A., Ferguson N. M., Dorigatti I., Crisanti A., Suppression of a SARS-CoV-2 outbreak in the Italian municipality of Vo’. Nature 584, 425–429 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jing Q.-L., Liu M.-J., Zhang Z.-B., Fang L.-Q., Yuan J., Zhang A.-R., Dean N. E., Luo L., Ma M.-M., Longini I., Kenah E., Lu Y., Ma Y., Jalali N., Yang Z.-C., Yang Y., Household secondary attack rate of COVID-19 and associated determinants in Guangzhou, China: A retrospective cohort study. Lancet Infect. Dis. 2020, 1141–1150 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li W., Zhang B., Lu J., Liu S., Chang Z., Peng C., Liu X., Zhang P., Ling Y., Tao K., Chen J., Characteristics of household transmission of COVID-19. Clin. Infect. Dis. 71, 1943–1946 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Huang Y.-T., Tu Y.-K., Lai P.-C., Estimation of the secondary attack rate of COVID-19 using proportional meta-analysis of nationwide contact tracing data in Taiwan. J. Microbiol. Immunol. Infect. 54, 89–92 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu Y., Eggo R. M., Kucharski A. J., Secondary attack rate and superspreading events for SARS-CoV-2. Lancet 395, e47 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hamner L., Dubbel P., Capron I., Ross A., Jordan A., Lee J., Lynn J., Ball A., Narwal S., Russell S., Patrick D., Leibrand H., High SARS-CoV-2 attack rate following exposure at a choir practice - Skagit County, Washington, March 2020. MMWR Morb. Mortal. Wkly Rep. 69, 606–610 (2020). [DOI] [PubMed] [Google Scholar]
- 36.N. J. Lennon, R. P. Bhattacharyya, M. J. Mina, H. L. Rehm, D. T. Hung, S. Smole, A. Woolley, E. S. Lander, S. B. Gabriel, Comparison of viral levels in individuals with or without symptoms at time of COVID-19 testing among 32,480 residents and staff of nursing homes and assisted living facilities in Massachusetts. medRxiv 2020.07.20.20157792 [Preprint]. 26 July 2020. 10.1101/2020.07.20.20157792. [DOI]
- 37.Papaiakovou M., Wright J., Pilotte N., Chooneea D., Schär F., Truscott J. E., Dunn J. C., Gardiner I., Walson J. L., Williams S. A., Littlewood D. T. J., Pooling as a strategy for the timely diagnosis of soil-transmitted helminths in stool: Value and reproducibility. Parasit. Vectors 12, 443 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hsiang M. S., Hwang J., Kunene S., Drakeley C., Kandula D., Novotny J., Parizo J., Jensen T., Tong M., Kemere J., Dlamini S., Moonen B., Angov E., Dutta S., Ockenhouse C., Dorsey G., Greenhouse B., Surveillance for malaria elimination in Swaziland: A national cross-sectional study using pooled PCR and serology. PLOS ONE 7, e29550 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Taylor S. M., Juliano J. J., Trottman P. A., Griffin J. B., Landis S. H., Kitsa P., Tshefu A. K., Meshnick S. R., High-throughput pooling and real-time PCR-based strategy for malaria detection. J. Clin. Microbiol. 48, 512–519 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Preiser W., Van Zyl G. U., Pooled testing: A tool to increase efficiency of infant HIV diagnosis and virological monitoring. Afr. J. Lab. Med. 9, 1035 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Van T. T., Miller J., Warshauer D. M., Reisdorf E., Jernigan D., Humes R., Shult P. A., Pooling nasopharyngeal/throat swab specimens to increase testing capacity for influenza viruses by PCR. J. Clin. Microbiol. 50, 891–896 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zenios S. A., Wein L. M., Pooled testing for HIV prevalence estimation: Exploiting the dilution effect. Stat. Med. 17, 1447–1467 (1998). [DOI] [PubMed] [Google Scholar]
- 43.Wein L. M., Zenios S. A., Pooled testing for HIV screening: Capturing the dilution effect. Oper. Res. 44, 543–569 (1996). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
stm.sciencemag.org/cgi/content/full/scitranslmed.abf1568/DC1
Fig. S1. True prevalence against maximum likelihood estimates.
Fig. S2. Prevalence estimation can depend on training and application period.
Fig. S3. Sensitivity of sample identification relative to dilution factor and time since peak viral load.
Fig. S4. Simulated viral loads.
Fig. S5. Group testing for sample identification during epidemic decline.
Fig. S6. Effectiveness of optimal testing design under resource constraints at high prevalence.
Fig. S7. Effectiveness of optimal testing design under resource constraints using sputum data.
Fig. S8. Evaluation of pooled testing in a sustained, multiwave epidemic.
Fig. S9. Evaluation of pooled testing for sample identification in the multiwave epidemic shown in fig. S8A.
Fig. S10. Model fits to swab viral loads.
Fig. S11. Posterior distributions of estimated parameters fitted to swab and sputum data.
Fig. S12. Markov chain Monte Carlo trace plots from fitting to swab and sputum data.
Fig. S13. qPCR calibration curve using standard viral RNA copies.
Table S1. List of all group test designs for sample identification.
Table S2. Ct values from qPCR on pooled samples with variable viral load.
Table S3. Positive sample distribution within validation pools.
Table S4. Pool design for combinatorial test with 96 samples.
Table S5. Description of all parameters used in the viral kinetics and transmission models.
Table S6. RT-qPCR results for pooling validations.
Data file S1. Ninety-six–sample pooling template (Excel).