

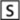
Abstract
The severe acute respiratory syndrome coronavirus (SARS-CoV, or SCV), which caused a world-wide epidemic in 2002 and 2003, binds to a receptor, angiotensin-converting enzyme 2 (ACE2), through the receptor-binding domain (RBD) of its envelope (spike, S) glycoprotein. The RBD is very immunogenic; it is a major SCV neutralization determinant and can elicit potent neutralizing antibodies capable of out-competing ACE2. However, the structural basis of RBD immunogenicity, RBD-mediated neutralization, and the role of RBD in entry steps following its binding to ACE2 have not been elucidated. By mimicking immune responses with the use of RBD as an antigen to screen a large human antibody library derived from healthy volunteers, we identified a novel potent cross-reactive SCV-neutralizing monoclonal antibody, m396, which competes with ACE2 for binding to RBD, and determined the crystal structure of the RBD-antibody complex at 2.3-Ä resolution. The antibody-bound RBD structure is completely defined, revealing two previously unresolved segments (residues 376–381 and 503–512) and a new disulfide bond (between residues 378 and 511). Interestingly, the overall structure of the m396-bound RBD is not significantly different from that of the ACE2-bound RBD. The antibody epitope is dominated by a 10-residue-long protruding β6–β7 loop with two putative ACE2-binding hotspot residues (Ile-489 and Tyr-491). These results provide a structural rationale for the function of a major determinant of SCV immunogenicity and neutralization, the development of SCV therapeutics based on the antibody paratope and epitope, and a retrovaccinology approach for the design of anti-SCV vaccines. The available structural information indicates that the SCV entry may not be mediated by ACE2-induced conformational changes in the RBD but may involve other conformational changes or/and yet to be identified coreceptors.
The severe acute respiratory syndrome coronavirus (SARS-CoV, or SCV)4 infected more than 8000 humans with a fatality rate of ∼10% (1, 2, 3, 4). Although there have been no recent outbreaks, the need to develop potent therapeutics and vaccines against a re-emerging SCV or a related virus remains of high priority. The amazingly rapid pace of SARS research for the last few years has resulted in a wealth of information for the virus, especially about its interactions with the host leading to disease and immune responses, which could also be helpful for the development of strategies to cope with other viral pathogens including influenza and HIV.
Entry of viruses into animal cells is initiated by binding to cell-surface-associated receptors and can be prevented by neutralizing antibodies (nAbs) targeting the virus receptor-binding site (5, 6). In the case of SCV entry, the spike (S) glycoprotein (7, 8) binds to a receptor, angiotensin-converting enzyme 2 (ACE2) (9), through the receptor-binding site of its receptor-binding domain (RBD) (7, 10, 11). The RBD is an attractive target for neutralizing antibodies that could prevent SCV entry by blocking the attachment of ACE2 (12, 13, 14, 15, 16, 17, 18). To understand the structural mechanisms underlying SCV immunogenicity and neutralization and help in the design of vaccines capable of eliciting predetermined highly effective neutralizing antibodies, we used a retrovaccinology (19) approach based on the combination of phage display and x-ray crystallography.
The SCV is a member of the genus Coronavirus, which belongs to the Coronaviridae family of the order Nidovirales, which also includes families Arteriviridae and Roniviridae. Not only SCV but also other nidoviruses can infect humans and animals, resulting in a variety of severe diseases. The infection is initiated by the attachment of virus envelope glycoproteins (Envs) to receptors, which can be blocked by nAbs. The structural mechanisms of receptor recognition and neutralization by antibodies against any nidovirus were not previously known. Recently, the crystal structure of the SCV S RBD in complex with ACE2 was reported at 2.9-Ä resolution (20). However, the structures of a receptor-free RBD and its complexes with nAbs are not known. Thus, fundamental questions related to the mechanism of SCV (and any other nidovirus) entry and neutralization, such as conformational changes induced by the binding of either the receptor or the nAbs, remain unanswered. To date, only a few structures of viral Envs complexed with nAbs are available, including Envs from influenza, picornaviruses, HIV, and West Nile virus (5, 21); these structures have played an essential role in elucidating the mechanisms of viral neutralization. Here, we describe the identification of a potent cross-reactive SCV-neutralizing human monoclonal antibody, m396, and report the crystal structure of the antibody-antigen complex (Fab m396-SCV RBD) at 2.3-Ä resolution (Supplemental Table S1). The structure reveals a major neutralization determinant and its relationship with receptor recognition, providing structural insights into the mechanism of SCV entry and neutralization.
EXPERIMENTAL PROCEDURES
Expression and Purification of the RBD—A fragment containing residues 317–518 from the S glycoprotein was cloned into pSecTag2B (Invitrogen) using BamHI and EcoRI restriction sites as previously described (7, 22). The insert was further cloned into pAcGP67-A using the forward primer 5′-ACT GTC TAG ATG GTA CCG AGC TCG GAT CC-3′ (XbaI) and the reverse primer 5′-CAG TAG ATC TCG AGG CTG ATC AGC G-3′ (BglII). The pAcGP67-S was co-transfected with BaculoGold linearized baculovirus DNA into SF9 cells. High titer recombinant baculovirus stock was prepared by multiple amplifications. The protein was expressed in SF9 cells, cultured in serum-free HyQ-SFX-insect medium (HyClone), and purified from conditioned medium with a HiTrap nickel-chelating column. The eluted monomeric protein was concentrated, further purified with a Superdex 75 10/300GL column equilibrated with phosphate-buffered saline plus 0.2 m NaCl, and concentrated to 5–10 mg/ml in phosphate-buffered saline plus 0.2 m NaCl.
Selection, Expression, and Purification of the High Affinity RBD-specific Fab m396 and Its Conversion to IgG1—A naïve human Fab phage display library (a total of ∼1010 members) was constructed from peripheral blood B cells of 10 healthy donors5 and used for selection of Fabs against purified, soluble, monomeric RBD, conjugated to magnetic beads (Dynabeads M-270 Epoxy, Dynal Inc., New Hyde Park, NY) following a previously described procedure (23). Briefly, amplified libraries of 1012 phage-displayed Fabs were incubated with 5, 3, and 1 μg of RBD in a 500-μl volume for 2 h at room temperature during the first, second, an third rounds of biopanning, respectively. After the third round of biopanning, 95 clones were randomly picked from the infected TG1 cells, and phage enzyme-linked immunosorbent assay was used to identify clones of phage displaying Fabs with high binding affinity. Eight clones that bound to the RBD with A 450 > 1.0 were selected for further characterization. The VH and VL domains (VH and VL denote the variable domains of heavy and light chains, respectively) of these clones were sequenced. The sequences were identical for all selected clones, and the selected Fab was designated as m396. The Fab used for crystallization was purified with a HiTrap nickel-chelating column followed by a Superdex 75 10/300GL column, using phosphate-buffered saline buffer containing 0.2 m NaCl, and concentrated to 10–20 mg/ml. For its conversion to IgG1, the Fab heavy and light chains were amplified and re-cloned in the pDR12 vector (provided by D. Burton, Scripps Research Institute, La Jolla, CA) with the Fc gene fragment replaced with cDNA sequence instead of genomic DNA.
Affinity Determination by Surface Plasmon Resonance—Interactions between m396 and SCV RBD were analyzed by surface plasmon resonance technology using a BIAcore 1000 instrument (Amersham Biosciences). The SCV RBD was covalently immobilized onto a sensor chip (CM5) using carbodiimide coupling chemistry. A control reference surface was prepared for nonspecific binding and refractive index changes. For analysis of the kinetics of interactions, various concentrations of Fab or IgG m396 were injected at a flow rate of 30 μl/min using running buffer containing 150 mm NaCl, 3 mm EDTA, and 0.005% P-20 (pH 7.4). The association and dissociation phase data were fitted simultaneously to a 1:1 Langmuir global model by using the nonlinear data analysis program BIAevaluation 4.1. All the experiments were performed at 25 °C.
Crystallization and Structure Determination—The SCV RBD-Fab m396 complex was formed by mixing individual components in a 1:1 molar ratio and incubating overnight at 4 °C. Crystals were obtained within 2–3 weeks by sitting-drop vapor diffusion technique. The reservoir solution was composed of 15% v/v glycerol, 20% polyethylene glycol 6000, 100 mm MES sodium at pH 6.5; crystals formed only in the drops with a 1:2 ratio for the protein and the reservoir solutions. The crystals of Fab m396 were grown with the sitting-drop vapor diffusion technique within 2 weeks. The reservoir solution was composed of 20% v/v glycerol, 16% v/v ethylene glycol, 20% w/v polyethylene glycol 6000, and 100 mm NaCl in 30 mm Tris-HCl (pH 8.5). Data sets up to 2.3-Ä resolution were collected at cryogenic temperature (100 K) for both the RBD-Fab complex and the unliganded Fab, each from a single crystal, at the Southeast Regional Collaborative Access Team beamline facility 22-ID of the Advanced Photon Source, Argonne National Laboratory. Data processing was carried out with the HKL2000 program suite (24). The structure was solved by molecular replacement with PHASER (25), using the SCV RBD from the receptor complex (PDB code 2AJF) and four individual domains of Fab (VH,VL,CH, and CL)(CH and CL refer to the constant domains of heavy and light chains, respectively) from three different antibody structures (PDB codes: 1ZA6 for CH and CL, 1RZG for VH, and 1W72 for VL) as search models. The RBM (residues 430–490) of SCV RBD and most of the CDRs (complementarity-determining regions) of Fab models, which were not included in the search models, were built on the basis of difference electron density. The complex was refined with CNS (26) at 2.3-Ä resolution. A total of 298 water molecules, a phosphate ion, and one N-linked glucosamine moiety at Asn-330 were added at the final stage of the refinement. The final R and R free values were 19.8 and 26.1, respectively. The unliganded Fab m396 structure was solved by molecular replacement with AMoRe (27), using the constant domains of Fab m396 from the complex structure as the search model. The difference electron density map revealed the location of variable domains. The structure was refined using CNS (26), and a total of 176 water molecules was added at the final stage of the refinement. The final R and R free values were 22.8 and 27.7, respectively. The O program (28) was used for model building for both structures. Data collection, processing, and refinement statistics are summarized in Supplemental Table S1.
RESULTS
Previously, we identified several S glycoprotein fragments, containing the RBD, which is a major SCV neutralization determinant (12, 13, 14, 15, 16, 17, 18), and residues critical for the binding of SCV to its receptor ACE2 (7, 22). One of these fragments, containing residues 317–518, was cloned into a baculovirus vector, expressed in insect cells, and purified. This fragment was used as a selecting antigen for panning of a large (∼1010 different antibodies) human antibody Fab library, which we constructed from the B lymphocytes of 10 healthy volunteers. Recently, this library was also used for the selection of potent nAbs against Hendra and Nipah viruses (23). The Fab with the strongest binding to the RBD, m396, was converted to full antibody (IgG1), expressed, and purified. We measured the binding rate constants and affinities of the Fab and the IgG1 m396 to SCV RBD in a BIAcore assay (Supplemental Fig. S1). With two independent experiments, we determined K on = 3.0 (±0.3) and 4 (±3) × 105 m –1 s–1, K off = 6.1 (±0.6) × 10–3 s–1 and2(±1) × 10–5 s–1, and KD = 20 (±0) nm and 4.6 (±0.9) pm, for the Fab and the IgG1 m369, respectively. The high apparent affinity (avidity) observed for IgG1 m369 is due to the effective multivalency of the surface-associated antigen binding to the bivalent IgG1. Further, we found that the antibody potently inhibited 1) cell fusion and pseudovirus entry mediated by the SCV (Tor2 isolate) S glycoprotein with an IC50 of 0.6 and 0.01 μg/ml, respectively, 2) SCV entry mediated by the S glycoprotein from the GD03T0013 isolate, which is not neutralizable by other known human monoclonal antibodies, including 80R (29, 30) and S3.1 (31, 32), and 3) live virus from Urbani and Tor2 isolates with an IC50 of 0.1 and 1 μg/ml, respectively.6
The structure of the SCV RBD-Fab m396 complex is depicted in Fig. 1 . The overall RBD structure in the complex with Fab m396 is similar to that in the complex with ACE2 (20). The root mean square deviation between the common Cα positions in the two RBD structures is 1.3 Ä. However, the new RBD structure reveals a total of 16 amino acid residues localized in two segments (residues 376–381 and 503–512 are shown in brown) that were missing in the RBD·ACE2 complex. The antibody-bound RBD structure also reveals a new disulfide bond between residues 378 and 511 (Fig. 2 ), which was not observed in the RBD·ACE2 complex. As shown in Fig. 1, the RBD consists of a core, which includes a five-stranded anti-parallel β-sheet (β1–β4 and β7), and a long extended loop, which contains a two-stranded anti-parallel β-sheet (β5–β6) in the middle. The complete RBD structure contains eight cysteines that form four disulfide bridges, three in the core and one in the extended loop.
FIGURE 1.

Overall structure of the SCV RBD in complex with the neutralizing antibody Fab m396. The SCV RBD is in green, and the prominent neutralizing site comprising residues 482 through 491 (β6–β7 loop) is in red. The side chains of two important residues, Ile-489 and Tyr-491, of the loop are shown as sticks. A portion of the structure shown in brown constitutes the 16 amino acid residues that were not observed in the RBD·ACE2 structure (20). The light and heavy chains of the Fab are shown in cyan and yellow, respectively, with labeled CDRs, H1, H2, H3, and L3, which make contacts with the RBD.
FIGURE 2.
Comparison of the RBD·Fab and the RBD·ACE2 structures. The newly identified two segments of RBD in the Fab complex are denoted by blue and pink colors. a, sequence and secondary structure assignment of RBD. b, structural alignment between RBD structures based on Cα positions. RBD structures from the Fab and ACE2 complexes are shown in green and cyan, respectively. Blue and pink segments defined in the RBD-Fab structure revealed the fourth disulfide bond within the RBD (between residues Cys-378 and Cys-511), which is shown as a stick model. c and d, stereoviews showing the 2Fo – Fc electron density maps contoured at 1.0 σ level along the segments 375–382 and 501–512.
Fab m396 mainly recognizes the 10-residue (482–491) β6–β7 loop that prominently protrudes from the RBD surface (Fig. 1) and contacts four of the CDRs of Fab, H1, H2, H3, and L3. The four CDRs form a shallow cleft on the surface of the antibody-variable regions, providing a deep binding pocket into which the β6–β7 loop fits tightly. The tip of this loop is a type I β-turn (Gly-Ile-Gly-Tyr, residues 488–491) and is deeply buried in the antibody-combining site, which is a feature most commonly observed in antibody·peptide complexes (33). The same feature involving the recognition of a similar sequence motif (Gly-Pro-Gly-Arg, residues 312–315) has been recently noted at the tip of the gp120 V3 structure (34), which is a major neutralizing determinant of HIV. Most residues in the β6–β7 loop interact with Fab m396 at the binding pocket, and particularly, residues Ile-489 and Tyr-491 from the β-turn penetrate into the deep pocket on the surface of the antibody-combining site. Fifteen residues from the RBD and the Fab participate in the formation of the RBD-antibody interface as defined by a contact distance of 3.5 Ä between the two molecules. The shape correlation statistical parameter (S c) (35), a measure of geometric fit between two juxtaposed surfaces (maximum value, 1.0), calculated for the RBD-antibody interface is 0.66, indicating a high degree of shape complementarity. A total surface area of 1760 Ä2 is buried at the interface of the complex with nearly equal contributions from the two molecules (870 Ä2 from the RBD and 890 Ä2 from the antibody) as determined with a 1.4-Ä probe. The antibody-binding β6–β7 loop alone accounts for 63% of the RBD-antibody interface, which indicates the dominant role of the loop residues in the binding of the two molecules. The heavy chain CDRs contributes 66% of the total surface of the antibody-combining site. The size of the binding interface is close to the average of other antigen·antibody complexes (36). The intermolecular interactions include van der Waals contacts and direct or water-mediated hydrogen bonds (Supplemental Table S2). The details of the buried surface area at the interface between the antibody and the SCV RBD are given in Supplemental Table S3.
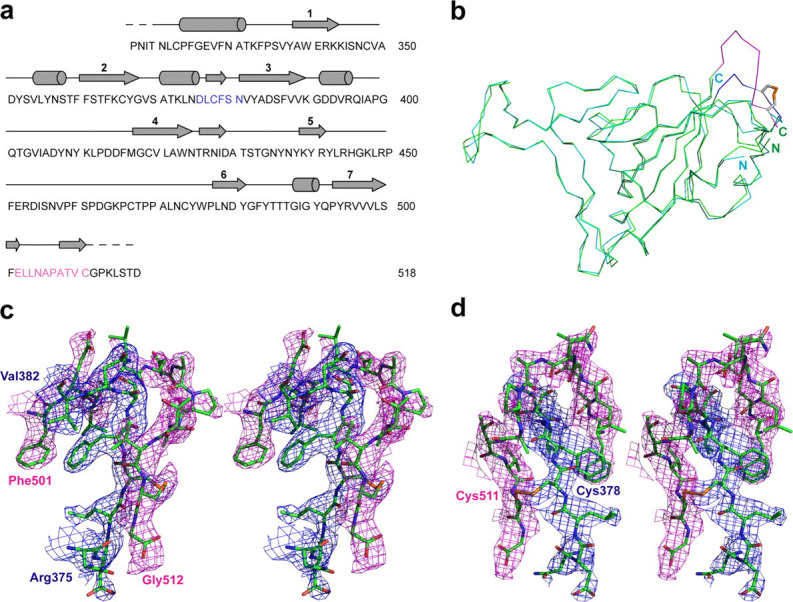
The key interactions in the RBD·antibody complex are mostly between the β6–β7 loop of the RBD and the four CDRs (H1, H2, H3, and L3) of Fab m396. These interactions are clearly defined (Fig. 3 ). H1 makes contacts with hydrophobic residues Tyr-484, Thr-486, and Thr-487; particularly Thr-33 of H1 is in direct contact with the amide nitrogen of Gly-488 in RBD (Fig. 3a). A similar interaction is found in the RBD·ACE2 complex where the amide of Gly-488 is engaged in a hydrogen bond with the carbonyl oxygen of Lys-35 in ACE2. Compared with other CDRs, H2 plays a dominant role in RBD binding; the most conspicuous feature is the burial of Tyr-491 of RBD (122 Ä2) in the shallow cleft rendered by H2, where the amino group of Asn-58 contacts the phenolic oxygen atom of Tyr-491 and the side chain of Thr-52 forms an aromatic on-face hydrogen bond with the π -cloud of Tyr-491 (Fig. 3b). Val-97 is the only residue from H3 that interacts with RBD (Fig. 3c); however, it buries the largest surface area per residue (108 Ä2) among all CDR residues that interact with RBD. The carbonyl oxygen atom of Val-97 forms a strong hydrogen bond with the amino nitrogen of the RBD residue Gln-492 with a distance of 2.7 Ä. Such hydrogen bonds between main-chain and side-chain atoms play an important role in determining the relative orientation of the RBD and the antibody in the complex, and contribute to the specificity of the interactions. The S c parameter calculated for the heavy chain-RBD interaction has a high value of 0.74, which suggests a highly correlated interfacial geometry for the heavy chain-RBD recognition. L3-RBD interaction involves water molecules and additional side chains, including Arg-395, of RBD (Fig. 3d, Supplemental Table S2). Residue Trp-91 of L3 stacks with aromatic residue Ile-489, a major hot spot in the RBD; each of the two residues buries a surface area of ∼100 Ä2 at the interface. The minor binding sites on the RBD include two residues in β2 (Thr-363 and Lys-365), the 310 helix followed by β3 (Lys-390, Gly-391, Asp-392, and Arg-395), and residues Arg-426 and Tyr-436. Apart from the minor contributions of these residues to antibody binding, most of them have significant roles in stabilizing the conformation of the β6–β7 loop. For example, the amide hydrogen atoms of Gly-391 and Asp-392 hydrogen bond to the backbone carbonyls of Gln-492 and Gly-490, respectively; the guanidinium group of Arg-426 forms a hydrogen bond with the carbonyl of Thr-485; and the phenolic oxygen atoms from side chains of Tyr-436 and Tyr-484 are hydrogen-bonded. All these hydrogen bonds help stabilizing the β6–β7 loop conformation in the RBD.
FIGURE 3.
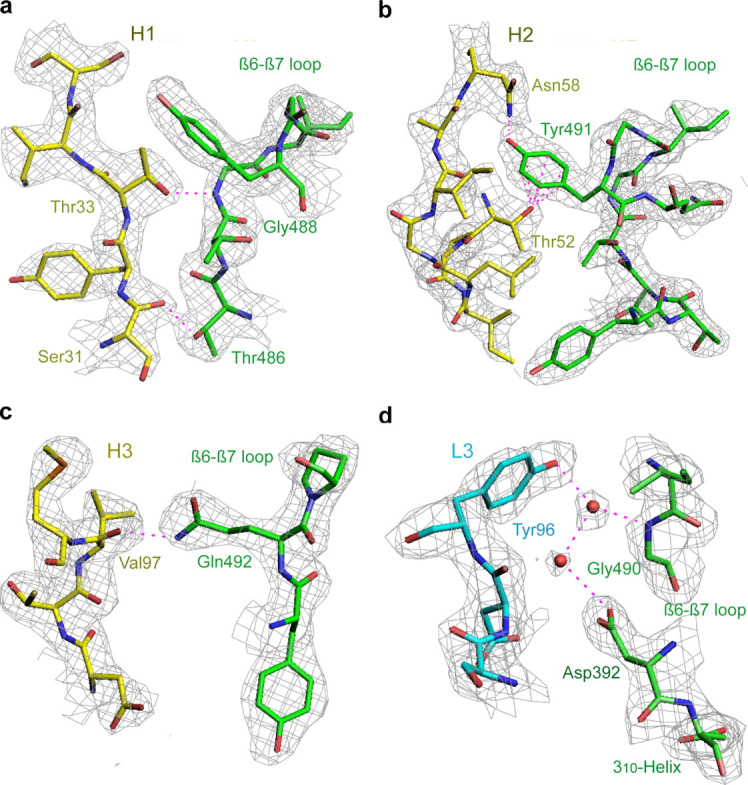
Critical interactions between SCV RBD (green) and Fab m396 (yellow and cyan for heavy- and light-chain CDRs, respectively) depicted with 2Fo – Fc electron density maps contoured at the 1.0 σ level. CDRs H1, H2, and H3 recognize the major neutralizing site, the β6–β7 loop. L3 exclusively contacts minor binding sites with the involvement of bridging water molecules. a, H1 residues Ser-31 and Thr-33 form hydrogen bonds with RBD residues Thr-486 and Thr-488 via backbone-side chain interactions. b, H2 displays a concave surface and contributes to the specific interactions between H2 residues Thr-52 and Asn-58, and RBD residue Tyr-491. c, H3 residue Val-97 contacts the RBD and buries the largest surface area per residue (108 Ä2) among all residues of the antibody combining site. The carbonyl of Val-97 forms a hydrogen bond to the side-chain amide of Gln-492 of RBD. d, L3 is the only light chain CDR that binds to the RBD with two bridging water molecules (pink spheres) involved.
The RBD-antibody interface has two major characteristic features: the high level of complementarity between the interacting surfaces and the anchoring of the putative major hotspot RBD residue Tyr-491 into the antibody combining site. Two putative hotspot residues, Ile-489 and Tyr-491, of the RBD β6–β7 loop form a protruding ridge, shown in yellow on the green RBD surface in Fig. 4a . The antibody binding pocket includes cavities in the shallow cleft mostly formed by the heavy chain, highlighted in green on the yellow antibody surface in Fig. 4b. As shown, the paratope and the epitope structures are highly complementary, which could be a major factor for the high affinity of their interaction. As mentioned earlier, another feature of the RBD-antibody interaction is the insertion of the RBD residue Tyr-491 into the bottom of the binding pocket at the antibody combining site where H2 residues Thr-52 and Asn-58 hold the RBD residue Tyr-491 in place (Fig. 3b). Thus, the preferential recognition of these two H2 residues, which line up the combining-site pocket in the antibody, by Tyr-491 is a unique feature of the SCV RBD-Fab m396 recognition.
FIGURE 4.

Structural features facilitating the binding between the SCV RBD and the Fab m396 include shape complementarity and specific side-chain interactions.a, the protruding structure of RBD (highlighted in yellow) within 3.5 Ä of the antibody surface approximates the antibody epitope on the RBD (green). b, the RBD-interacting area of the antibody (highlighted in green) within 3.5 Ä of the RBD surface approximates the paratope of the antibody (yellow), showing high structural complementarity to the antibody epitope shown in yellow in panel a.
To find out whether the antibody undergoes any conformational changes upon RBD binding, such as rearrangements in the combining site or the elbow angle, we determined the crystal structure of unliganded Fab m396 at 2.3-Ä resolution. There are two similar Fab m396 molecules in the asymmetric unit. Intriguingly, a significant change in the elbow angle between the free and RBD-bound structures of the Fab is observed (Fig. 5 ). The ligand-free Fab, shown in red, has almost a straight elbow angle (173.5°), whereas the RBD-bound Fab, shown in blue, is markedly bent (125.1°). The elbow angle for RBD-bound Fab m396 is the lowest for a human antibody (37). This conformational change supports an early notion that elbow bending may occur in the antibodies upon antigen binding, which could play a role as a signal transfer mechanism (38). However, we found no significant conformational differences in the antibody-combining site between the ligand-free and RBD-bound Fab, except for minor differences in side-chain orientations of residues involved in antigen binding. Therefore, Fab m396 has a structurally stable antibody-combining site.
FIGURE 5.

Stereoview of superimposed structures of RBD-free (red) and RBD-bound Fab m396 (blue) based on the Cα positions in their variable domains, which shows a significant elbow-angle difference between the two conformations of Fab m396. The unliganded Fab has an open or straight elbow angle, whereas the SCV RBD-bound Fab has a closed or highly bent elbow angle.
DISCUSSION
The major findings of this study are the identification of a novel, potent SCV-neutralizing human monoclonal antibody, the elucidation of the structural mechanism of its function, and the determination of a complete high resolution structure of the SCV RBD. The comparison between the RBD·ACE2 and the RBD·m396 structures provided important clues for the mechanism of SCV entry and the molecular basis of antibody-mediated neutralization. The antibody and the receptor occupy a common region on the surface of RBD, consisting of the β6–β7 loop (Thr-484, Thr-486, Thr-487, Gly-488, and Tyr-491) and Arg-426. Therefore, these residues are critical for the binding of RBD to both the antibody and the receptor. Fig. 6a shows a superimposed stereoview of the RBD complexes with antibody (yellow) and with receptor (cyan). Fig. 6 (b and c) illustrates the binding regions of the antibody and the receptor, respectively (red on the yellow and cyan surface, respectively). It is seen that the neutralizing determinants are located contiguously in one major segment of the β6–β7 loop, whereas the receptor ACE2 have determinants over most of the extended loop appearing on the top of RBD. These observations demonstrate that the antibody neutralizes SCV by competing for the same set of critical residues in the β6–β7 loop of RBD and therefore blocking the receptor binding site. Recently, bats were reported as a reservoir of SARS-like coronaviruses (39, 40). The sequences of human and civet isolates differ greatly from those of bat isolates, although they are phylogenetically related. Notably, residues Arg-426, Ile-489, and Tyr-491 of SCV RBD, which are involved in the antibody binding, are conserved among the bat isolates, indicating a potential neutralizing activity of m396, although this possibility should be evaluated experimentally.
FIGURE 6.

Comparison of the RBD·Fab and the RBD·ACE2 complexes: lack of induced conformational changes of RBD and overlapping but not identical binding sites of Fab and ACE2.a, stereoview of the superposition of the RBD-antibody (in yellow) and RBD-receptor (in cyan) complexes. The superposition was based on the alignment of the Cα positions of RBD in the two complexes. The heavy chain of the antibody significantly overlaps with the receptor. A common binding region, the β6–β7 loop of RBD, is shared by the antibody and the receptor. The residues in the common binding region of RBD may contribute to the orientation of the antibody and the receptor to the spike on viral surface, the high affinity of the antibody binding, and the ACE2 binding specificities that lead to SARS infection of humans and cross-species transmission. b, structural footprints of the antibody and c, the receptor on the SCV RBD shown as red batches on the RBD surface. The circled area on the RBD surface represents the common binding site for the antibody and the receptor.
Fab m396 potently neutralizes virus pseudotyped with the S glycoprotein from the 2003/2004 Guangdong index patient (GD03T0013 isolate).6 Compared with the middle/late phase 2002/2003 isolate Tor2, five residues in the RBD were mutated in the late isolate GD03T0013, including Lys-344 → Arg, Phe-360 → Ser, Leu-472 → Pro, Asp-480 → Gly, and Thr-487 → Ser. Among the five residues, only Thr-487 contacts the antibody-combining site as observed in the RBD·m396 structure. However, it is only involved in minor van der Waals interactions with Fab, and therefore the Thr-487 → Ser mutation does not significantly affect the neutralizing activity of the antibody.6 Our analysis of 86 available RBD sequences of human SCV isolates revealed that 35 sequences contain the Thr-487 → Ser mutation. Thus, being able to neutralize all existing SCV isolates, m396 appears to be a broadly cross-reactive antibody and may have clinical potential for future outbreaks.
Unexpectedly, we found that the RBD structure in the complex with antibody is similar to that in the complex with ACE2 (Figs. 2 and 6a). The unliganded RBD structure is unknown. However, it is very unlikely that binding of ACE2 and of m396 could induce exactly the same conformational changes that would lead to virtually the same RBD structure, especially in light of the overlapping but different binding sites of ACE2 and m396 as well as the molecular specificities of their binding as discussed above. However, although the overall structure of the RBD is preserved, differences are observed in the conformations of the side chains that are involved in ligand binding. Also, it remains to be seen whether conformational changes occur in other segments of an intact S glycoprotein upon ligand binding. The 16 amino acid residues in the two segments (residues 376–381 and 503–512), which were not observed in the RBD·ACE2 complex (20), are located on the surface of RBD, opposite to the neutralization and receptor-binding sites, and also could undergo and/or mediate conformational changes. One can therefore only hypothesize that the SCV entry is not through an ACE2-activating mechanism that causes conformational changes directly in the RBD, although it is possible that membrane-associated ACE2 could induce conformational changes in other regions of the S glycoprotein, e.g. by hinge motions or changes in the conformation of the oligomeric S glycoprotein through multivalent binding to ACE2. Another possibility is that the ACE2 function is to bind specifically the S glycoprotein followed by binding to co-receptor(s) that can induce conformational changes activating the fusogenic machinery of the S glycoprotein.
Our findings have implications for the development of vaccines and therapeutics against SCV and for understanding the mechanisms of antibody-mediated virus neutralization and virus entry. The newly identified antibody, m396, itself may have therapeutic potential, and we plan to test it in animal models. Based on its structure, one could design other therapeutic modalities. The structure of the antibody epitope could be used for the design of vaccine immunogens that are likely to elicit m396 or m396-like antibodies (a retrovaccinology approach (19)). Especially attractive is the potential use of the main neutralizing determinant, the β6–β7 loop, and constraint peptides based on its sequence as vaccine immunogens. Its protruding nature, absence of carbohydrates in close vicinity, exposure, and easy access by antibodies suggest a critical role in neutralization mechanisms, and it is likely that it also binds other antibodies in addition to the receptor ACE2 and antibody m396. Finally, the finding that the overall RBD structure in the complex with the antibody is virtually the same as the one in the complex with the receptor should stimulate further research to resolve the fundamental question of what is the activation mechanism that operates during the SCV entry into cells, and whether there are other SCV receptors or co-receptors.
Acknowledgments
We thank the Advanced Biomedical Computing Center, NCI-Frederick for computing facilities. X-ray diffraction data were collected at the Southeast Regional Collaborative Access Team 22-ID beamline of the Advanced Photon Source, Argonne National Laboratory. Supporting institutions may be found at www.ser-cat.org/members.html.
Footnotes
The abbreviations used are: SCV, severe acute respiratory syndrome coronavirus (or SARS-CoV); mAb, monoclonal antibody; ACE2, angiotensin-converting enzyme 2; RBD, receptor-binding domain; Env, virus envelope glycoprotein; HIV, human immunodeficiency virus; MES, 4-morpholineethanesulfonic acid; CDR, complementarity-determining region; S glycoprotein; spike glycoprotein.
Z. Y. Zhu and D. S. Dimitrov, manuscript in preparation.
Zhu, Z., Prabakaran, P., Gan, J., Chakraborti, S., He, Y., Choudhry, V., Feng, Y., Xiao, X., Wang, L., Ji, X., Jiang, S., and Dimitrov, D. S., manuscript to be submitted.
References
- 1.Ksiazek T.G., Erdman D., Goldsmith C.S., Zaki S.R., Peret T., Emery S., Tong S., Urbani C., Comer J.A., Lim W., Rollin P.E., Dowell S.F., Ling A.E., Humphrey C.D., Shieh W.J., Guarner J., Paddock C.D., Rota P., Fields B., DeRisi J., Yang J.Y., Cox N., Hughes J.M., LeDuc J.W., Bellini W.J., Anderson L.J. N. Engl. J. Med. 2003;348:1953–1966. doi: 10.1056/NEJMoa030781. [DOI] [PubMed] [Google Scholar]
- 2.Peiris J.S., Lai S.T., Poon L.L., Guan Y., Yam L.Y., Lim W., Nicholls J., Yee W.K., Yan W.W., Cheung M.T., Cheng V.C., Chan K.H., Tsang D.N., Yung R.W., Ng T.K., Yuen K.Y. Lancet. 2003;361:1319–1325. doi: 10.1016/S0140-6736(03)13077-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Drosten C., Gunther S., Preiser W., van der W.S., Brodt H.R., Becker S., Rabenau H., Panning M., Kolesnikova L., Fouchier R.A., Berger A., Burguiere A.M., Cinatl J., Eickmann M., Escriou N., Grywna K., Kramme S., Manuguerra J.C., Muller S., Rickerts V., Sturmer M., Vieth S., Klenk H.D., Osterhaus A.D., Schmitz H., Doerr H.W. N. Engl. J. Med. 2003;348:1967–1976. doi: 10.1056/NEJMoa030747. [DOI] [PubMed] [Google Scholar]
- 4.Holmes K.V. N. Engl. J. Med. 2003;348:1948–1951. doi: 10.1056/NEJMp030078. [DOI] [PubMed] [Google Scholar]
- 5.Dimitrov D.S. Nat. Rev. Microbiol. 2004;2:109–122. doi: 10.1038/nrmicro817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Smith A.E., Helenius A. Science. 2004;304:237–242. doi: 10.1126/science.1094823. [DOI] [PubMed] [Google Scholar]
- 7.Xiao X., Chakraborti S., Dimitrov A.S., Gramatikoff K., Dimitrov D.S. Biochem. Biophys. Res. Commun. 2003;312:1159–1164. doi: 10.1016/j.bbrc.2003.11.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xiao X., Dimitrov D.S. Cell Mol. Life Sci. 2004;61:2428–2430. doi: 10.1007/s00018-004-4257-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li W., Moore M.J., Vasilieva N., Sui J., Wong S.K., Berne M.A., Somasundaran M., Sullivan J.L., Luzuriaga K., Greenough T.C., Choe H., Farzan M. Nature. 2003;426:450–454. doi: 10.1038/nature02145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wong S.K., Li W., Moore M.J., Choe H., Farzan M. J. Biol. Chem. 2004;279:3197–3201. doi: 10.1074/jbc.C300520200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Babcock G.J., Esshaki D.J., Thomas W.D., Jr., Ambrosino D.M. J. Virol. 2004;78:4552–4560. doi: 10.1128/JVI.78.9.4552-4560.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.He Y., Zhou Y., Liu S., Kou Z., Li W., Farzan M., Jiang S. Biochem. Biophys. Res. Commun. 2004;324:773–781. doi: 10.1016/j.bbrc.2004.09.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang M.Y., Choudhry V., Xiao X., Dimitrov D.S. Curr. Opin. Mol. Ther. 2005;7:151–156. [PubMed] [Google Scholar]
- 14.Jiang S., He Y., Liu S. Emerg. Infect. Dis. 2005;11:1016–1020. doi: 10.3201/eid1107.050219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.He Y., Lu H., Siddiqui P., Zhou Y., Jiang S. J. Immunol. 2005;174:4908–4915. doi: 10.4049/jimmunol.174.8.4908. [DOI] [PubMed] [Google Scholar]
- 16.He Y., Zhu Q., Liu S., Zhou Y., Yang B., Li J., Jiang S. Virology. 2005;334:74–82. doi: 10.1016/j.virol.2005.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen Z., Zhang L., Qin C., Ba L., Yi C.E., Zhang F., Wei Q., He T., Yu W., Yu J., Gao H., Tu X., Gettie A., Farzan M., Yuen K.Y., Ho D.D. J. Virol. 2005;79:2678–2688. doi: 10.1128/JVI.79.5.2678-2688.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yi C.E., Ba L., Zhang L., Ho D.D., Chen Z. J. Virol. 2005;79:11638–11646. doi: 10.1128/JVI.79.18.11638-11646.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Burton D.R. Nat. Rev. Immunol. 2002;2:706–713. doi: 10.1038/nri891. [DOI] [PubMed] [Google Scholar]
- 20.Li F., Li W., Farzan M., Harrison S.C. Science. 2005;309:1864–1868. doi: 10.1126/science.1116480. [DOI] [PubMed] [Google Scholar]
- 21.Nybakken G.E., Oliphant T., Johnson S., Burke S., Diamond M.S., Fremont D.H. Nature. 2005;437:764–769. doi: 10.1038/nature03956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chakraborti S., Prabakaran P., Xiao X., Dimitrov D.S. Virol. J. 2005;2:73. doi: 10.1186/1743-422X-2-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhu Z., Dimitrov A.S., Bossart K.N., Crameri G., Bishop K.A., Choudhry V., Mungall B.A., Feng Y.R., Choudhary A., Zhang M.Y., Feng Y., Wang L.F., Xiao X., Eaton B.T., Broder C.C., Dimitrov D.S. J. Virol. 2006;80:891–899. doi: 10.1128/JVI.80.2.891-899.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Otwinowski Z., Minor W. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 25.Storoni L.C., McCoy A.J., Read R.J. Acta Crystallogr. Sect. D Biol. Crystallogr. 2004;60:432–438. doi: 10.1107/S0907444903028956. [DOI] [PubMed] [Google Scholar]
- 26.Brunger A.T., Adams P.D., Rice L.M. Structure. 1997;5:325–336. doi: 10.1016/s0969-2126(97)00190-1. [DOI] [PubMed] [Google Scholar]
- 27.Navaza J. Acta Crystallogr. Sect. A. 1994;50:157–163. [Google Scholar]
- 28.Jones T.A., Kjeldgaard M. Methods Enzymol. 1997;277:173–208. doi: 10.1016/s0076-6879(97)77012-5. [DOI] [PubMed] [Google Scholar]
- 29.Sui J., Li W., Murakami A., Tamin A., Matthews L.J., Wong S.K., Moore M.J., Tallarico A.S., Olurinde M., Choe H., Anderson L.J., Bellini W.J., Farzan M., Marasco W.A. Proc. Natl. Acad. Sci. U. S. A. 2004;101:2536–2541. doi: 10.1073/pnas.0307140101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sui J., Li W., Roberts A., Matthews L.J., Murakami A., Vogel L., Wong S.K., Subbarao K., Farzan M., Marasco W.A. J. Virol. 2005;79:5900–5906. doi: 10.1128/JVI.79.10.5900-5906.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Traggiai E., Becker S., Subbarao K., Kolesnikova L., Uematsu Y., Gismondo M.R., Murphy B.R., Rappuoli R., Lanzavecchia A. Nat. Med. 2004;10:871–875. doi: 10.1038/nm1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yang Z.Y., Werner H.C., Kong W.P., Leung K., Traggiai E., Lanzavecchia A., Nabel G.J. Proc. Natl. Acad. Sci. U. S. A. 2005;102:797–801. doi: 10.1073/pnas.0409065102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wilson I.A., Stanfield R.L. Curr. Opin. Struct. Biol. 1994;4:857–867. doi: 10.1016/0959-440x(94)90267-4. [DOI] [PubMed] [Google Scholar]
- 34.Huang C.C., Tang M., Zhang M.Y., Majeed S., Montabana E., Stanfield R.L., Dimitrov D.S., Korber B., Sodroski J., Wilson I.A., Wyatt R., Kwong P.D. Science. 2005;310:1025–1028. doi: 10.1126/science.1118398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lawrence M.C., Colman P.M. J. Mol. Biol. 1993;234:946–950. doi: 10.1006/jmbi.1993.1648. [DOI] [PubMed] [Google Scholar]
- 36.Davies D.R., Cohen G.H. Proc. Natl. Acad. Sci. U. S. A. 1996;93:7–12. doi: 10.1073/pnas.93.1.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stanfield R.L., Zemla A., Wilson I.A., Rupp B. J. Mol. Biol. 2006;357:1566–1574. doi: 10.1016/j.jmb.2006.01.023. [DOI] [PubMed] [Google Scholar]
- 38.Huber R. Science. 1986;233:702–703. doi: 10.1126/science.2426777. [DOI] [PubMed] [Google Scholar]
- 39.Li W., Shi Z., Yu M., Ren W., Smith C., Epstein J.H., Wang H., Crameri G., Hu Z., Zhang H., Zhang J., McEachern J., Field H., Daszak P., Eaton B.T., Zhang S., Wang L.F. Science. 2005;310:676–679. doi: 10.1126/science.1118391. [DOI] [PubMed] [Google Scholar]
- 40.Lau S.K., Woo P.C., Li K.S., Huang Y., Tsoi H.W., Wong B.H., Wong S.S., Leung S.Y., Chan K.H., Yuen K.Y. Proc. Natl. Acad. Sci. U. S. A. 2005;102:14040–14045. doi: 10.1073/pnas.0506735102. [DOI] [PMC free article] [PubMed] [Google Scholar]