

PURPOSE
Approximately 20% of patients with TP53-mutant myelodysplastic syndromes (MDS) achieve complete remission (CR) with hypomethylating agents. Eprenetapopt (APR-246) is a novel, first-in-class, small molecule that restores wild-type p53 functions in TP53-mutant cells.
METHODS
This was a phase Ib/II study to determine the safety, recommended phase II dose, and efficacy of eprenetapopt administered in combination with azacitidine in patients with TP53-mutant MDS or acute myeloid leukemia (AML) with 20%-30% marrow blasts (ClinicalTrials.gov identifier: NCT03072043).
RESULTS
Fifty-five patients (40 MDS, 11 AML, and four MDS/myeloproliferative neoplasms) with at least one TP53 mutation were treated. The overall response rate was 71% with 44% achieving CR. Of patients with MDS, 73% (n = 29) responded with 50% (n = 20) achieving CR and 58% (23/40) a cytogenetic response. The overall response rate and CR rate for patients with AML was 64% (n = 7) and 36% (n = 4), respectively. Patients with only TP53 mutations by next-generation sequencing had higher rates of CR (69% v 25%; P = .006). Responding patients had significant reductions in TP53 variant allele frequency and p53 expression by immunohistochemistry, with 21 (38%) achieving complete molecular remission (variant allele frequency < 5%). Median overall survival was 10.8 months with significant improvement in responding versus nonresponding patients by landmark analysis (14.6 v 7.5 months; P = .0005). Overall, 19/55 (35%) patients underwent allogeneic hematopoietic stem-cell transplant, with a median overall survival of 14.7 months. Adverse events were similar to those reported for azacitidine or eprenetapopt monotherapy, with the most common grade ≥ 3 adverse events being febrile neutropenia (33%), leukopenia (29%), and neutropenia (29%).
CONCLUSION
Combination treatment with eprenetapopt and azacitidine is well-tolerated yielding high rates of clinical response and molecular remissions in patients with TP53-mutant MDS and oligoblastic AML.
INTRODUCTION
TP53 gene mutations are detected in approximately 10%-20% of patients with de novo myelodysplastic syndromes (MDS)1-4 or acute myeloid leukemia (AML)5,6 and 30%-40% of patients with therapy-related disease.7 Treatment outcomes for patients with TP53 mutations are poor with available therapies.8-12 Hypomethylating agents (HMAs), such as azacitidine and decitabine, yield statistically similar complete remission (CR) rates of approximately 15%-20% in patients with either TP53-mutant or wild-type MDS.12-14 However, remissions in TP53-mutant patients are brief with a median overall survival (OS) ranging from 5 to 12 months,8,9,12,13,15-17 reflecting the significant unmet medical need for novel, targeted therapies for patients with TP53-mutant MDS and AML. The TP53 mutation burden (ie, high variant allele frequency [VAF], concurrent complex karyotype, and/or multihit state) is integrally linked to inferior survival with median OS of 6-8 months.8-10,12 Increased HMA duration, such as 10-day decitabine, initially suggested improved outcomes in a limited number of patients with TP53-mutant myeloid malignancies (patients with MDS; n = 9),18 but there was no improvement in CR or OS in a prospective randomized study comparing 10-day versus 5-day decitabine in AML, and this treatment schedule has not been widely adopted.19,20
CONTEXT
Key Objective
To evaluate if it is possible to improve outcomes for patients with TP53-mutant myelodysplastic syndromes (MDS) and oligoblastic acute myeloid leukemia, who have a complete remission rate of < 20% with standard-of-care azacitidine therapy.
Knowledge Generated
Eprenetapopt (APR-246) is a first-in-class mutant p53 reactivator and the combination of azacitidine and eprenetapopt was well-tolerated in patients with TP53-mutant MDS or acute myeloid leukemia, with an adverse event profile similar to azacitidine and eprenetapopt monotherapies. Azacitidine and eprenetapopt resulted in a 71% overall response rate and 44% complete remission rate in the intention-to-treat population (50% for patients with MDS) with a median overall survival of 10.8 months, comparing favorably with single-agent azacitidine.
Relevance
Together, these data support the ongoing pivotal phase III, multicenter, randomized study of eprenetapopt in combination with azacitidine versus azacitidine alone in patients with TP53-mutant MDS (ClinicalTrials.gov identifier: NCT03745716).
Eprenetapopt (APR-246) is a novel, first-in-class, small molecule that selectively induces apoptosis in TP53-mutant cancer cells. Eprenetapopt is a prodrug that, under physiological conditions, is spontaneously converted to methylene quinuclidinone, a Michael acceptor that covalently binds to cysteine residues in mutant p53, leading to thermodynamic stabilization of the p53 protein and shifting the equilibrium toward a functional conformation.21 In addition, eprenetapopt increases oxidative stress by depletion of glutathione and inhibition of thioredoxin reductase, leading to accumulation of reactive oxygen species and further promoting tumor cell death.22,23 Eprenetapopt has synergistic cytotoxicity when combined with azacitidine in TP53-mutant MDS and AML cell lines, primary patient specimens, and in vivo models.24 In a phase I study including patients with AML, eprenetapopt monotherapy demonstrated clinical activity with corresponding activation of p53-dependent pathways.25,26 In January 2020, the US Food and Drug Administration granted breakthrough therapy designation for the treatment of patients with TP53-mutant MDS with the combination of eprenetapopt and azacitidine. Here, we report the safety and efficacy findings from the phase Ib/II trial evaluating combined treatment with eprenetapopt and azacitidine in patients with TP53-mutant myeloid malignancies.
METHODS
Study Design
This was a phase Ib/II open-label, multicenter, dose-escalation and dose-expansion study. The initial dose-escalation phase employed a standard 3 + 3 design to evaluate eprenetapopt in combination with azacitidine. Dose-limiting toxicities (DLTs) were evaluated to establish the recommended phase II dose (RP2D). Eprenetapopt was administered as a 6-hour intravenous infusion on days 1-4 of each 28-day cycle. Azacitidine was administered at the standard dose of 75 mg/m2 (seven consecutive days, days 4-10 or 2 + 5 [ie, days 4-5 and 8-12]) as a subcutaneous injection or an intravenous infusion (Data Supplement, online only). In the dose-escalation phase, 12 patients initially received eprenetapopt monotherapy (lead-in phase) over the same schedule on days −14 to −11 (prior to the start of combination therapy) at three dose levels (50, 75, and 100 mg/kg/d lean body mass [LBM]). As no DLTs occurred in the phase Ib study, eprenetapopt was subsequently given at a fixed dose of 4,500 mg/patient/d in phase II (100 mg/kg/d LBM equivalent on the basis of population pharmacokinetic modeling). The phase II portion of the study used a Simon's two-stage minimax design (Data Supplement). Disease assessments were undertaken after the lead-in phase (day −10) and then every three cycles. The study Protocol (online only) was approved by the institutional review boards of the participating sites. Written informed consent was provided by all the patients before screening and enrollment. The study was conducted in accordance with the principles of the Declaration of Helsinki and Good Clinical Practice guidelines.
Patients
Patients age ≥ 18 years with an Eastern Cooperative Oncology Group performance status of 0-2 and adequate renal and hepatic function and who had HMA-naive MDS, MDS/myeloproliferative neoplasm (MDS/MPN) overlap syndrome, chronic myelomonocytic leukemia, or oligoblastic AML (20%-30% blasts) and were classified as higher risk as defined by the revised International Prognostic Scoring System (ie, intermediate, high, or very high) were eligible for inclusion. All patients were required to have a TP53 mutation identified on the basis of local next-generation sequencing (NGS) testing.
Study Assessments
The primary end point of the phase II study was CR as defined by the 2006 International Working Group criteria.27 Key predefined secondary end points included overall response rate (ORR), duration of response, and OS. In addition, p53 reactivation (lead-in phase only), TP53 clonal suppression (measurable residual disease [MRD]) via high-sensitivity NGS analyses, and biomarkers of response including p53 protein expression and presence of co-occurring non-TP53 mutations were evaluated (Data Supplement).
Statistical Analysis
The intention-to-treat (ITT) and safety populations included all patients who received at least one dose of eprenetapopt. All patients who completed at least one treatment cycle of eprenetapopt and azacitidine and had at least one post-treatment clinical response assessment were included in the response-evaluable population. Duration of response was calculated from the date of first response to the date of progression or death and was not censored at the date of allogeneic hematopoietic stem-cell transplant (HSCT). OS was calculated from the date of initiation of therapy to death or last follow-up and was evaluated using the Kaplan-Meier method. Log-rank test was used to compare the survival curves between the patient groups of interest. Landmark analyses were used for survival comparisons in responding versus nonresponding patients. The Cox proportional hazards model was used to evaluate the impact of HSCT as a time-dependent covariate. Fisher's exact and paired t-tests were used for comparative analyses. Biomarker and subgroup analyses were performed as exploratory end points (Data Supplement). A P value < .05 was considered statistically significant.
RESULTS
Baseline Patient Characteristics and Disposition
Patients were enrolled between May 2017 and February 2019, and the data cutoff was November 15, 2019, at which point all patients had been enrolled for ≥ 6 months. A total of 55 patients received eprenetapopt in combination with azacitidine (12 in the dose-escalation phase [including six patients treated at RP2D] and 43 patients in the phase II dose-expansion phase). Baseline demographics and clinical characteristics are shown in Table 1 and a heat map containing individual patient molecular and cytogenetic characteristics is provided in Figure 1A. Most patients had significant adverse molecular and genetic features (89% complex karyotype and/or multihit [ie, > 1 TP53 mutation or deletion 17p/-17]). TP53 was the dominant clone, that is, only mutation or mutation with highest VAF, in 89% of patients. The median number of TP53 mutations was one (range, 1-3), with 17 (31%) patients having > 1 TP53 mutation, and median VAF in peripheral blood was 21%. Fifty-three (96%) patients had at least one mutation in the DNA-binding domain, including 48 (87%) with a missense mutation in the DNA-binding domain. A lollipop plot of all variants is shown in Figure 1B. Co-occurring somatic mutations were found in 38% of patients, of which TET2 (11%) and DNMT3A (9%) were the most common (Data Supplement).
TABLE 1.
Baseline Characteristics in Intention-to-Treat Population

FIG 1.
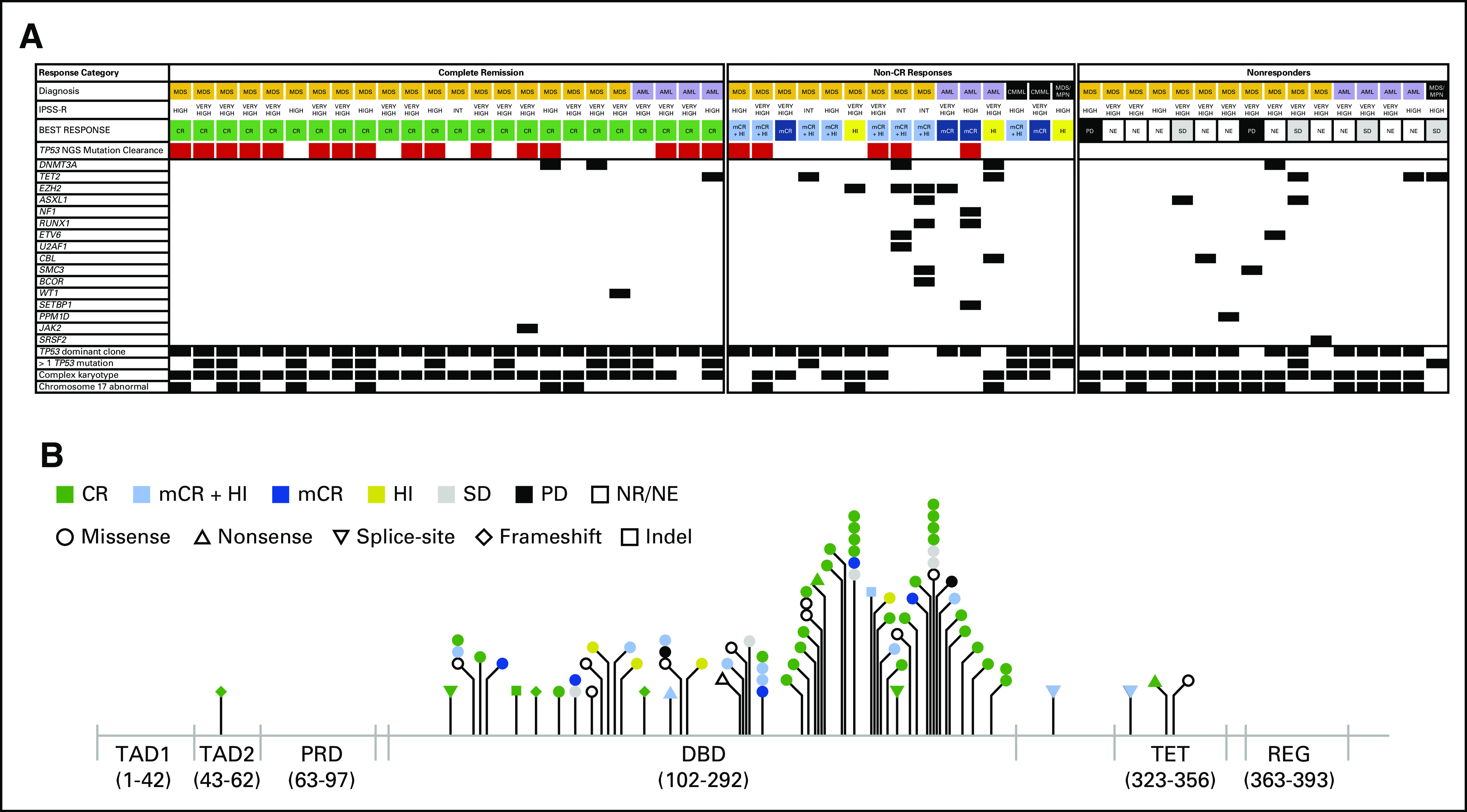
(A) Heat map of individual patient baseline risk characteristics including molecular and genetic features and best response to treatment, and (B) matchstick plot of TP53 mutation types and locations by best response. AML, acute myeloid leukemia; CMML, chronic myelomonocytic leukemia; CR, complete remission; DBD, DNA-binding domain; HI, hematologic improvement; INT, intermediate; IPSS-R, revised International Prognostic Scoring System; mCR, marrow complete remission; MDS, myelodysplastic syndromes; MDS/MPN, myelodysplastic syndromes/myeloproliferative neoplasms; NE, not evaluable; NR, not reported; ORR, overall response rate; PD, progressive disease; PRD, proline-rich domain; REG, C-terminal regulatory domain; SD, stable disease; TAD, transactivation domain; TET, tetramerization domain.
The median age of the cohort was 66 years, and 53% were female. The majority of patients had MDS (73%) and 93% had high or very high revised International Prognostic Scoring System. Most patients (69%) were transfusion-dependent at baseline. No patients progressed during the first two cycles of treatment. The disposition of all patients is shown in the Data Supplement. At the time of the data cutoff date, three (5%) patients were continuing study treatment and 22 (40%) had discontinued study treatment to proceed to HSCT. The median time to HSCT was 5.6 months (range, 1.7-9.7 months). Ten (18%) patients were in post-transplant follow-up at the time of data cutoff, and 13 (24%) discontinued treatment because of progressive disease. The rates of discontinuation were similar for patients with MDS and AML. The median treatment duration was 5.2 months (range, 0.4-16.8 months).
Safety
In the dose-escalation phase, no DLTs were observed in 12 patients across the three eprenetapopt dose levels (six were treated with eprenetapopt 100 mg/kg/d LBM) during the eprenetapopt monotherapy phase or in combination with azacitidine. During the eprenetapopt monotherapy lead-in, adverse events (AEs) of any grade that were reported in ≥ 3 patients included nausea (n = 4), peripheral sensory neuropathy (n = 4), and dizziness (n = 3) (Data Supplement). In addition, grade 3 and 4 AEs are listed in the Data Supplement, and AEs assessed related to eprenetapopt treatment (most were grade 1 or 2) are listed in the Data Supplement. All neurologic AEs were fully reversible upon treatment with prochlorperazine and/or temporary eprenetapopt treatment interruption. The RP2D of eprenetapopt with azacitidine was 100 mg/kg/d LBM, equivalent to 4,500 mg/d fixed dosing by population pharmacokinetics.
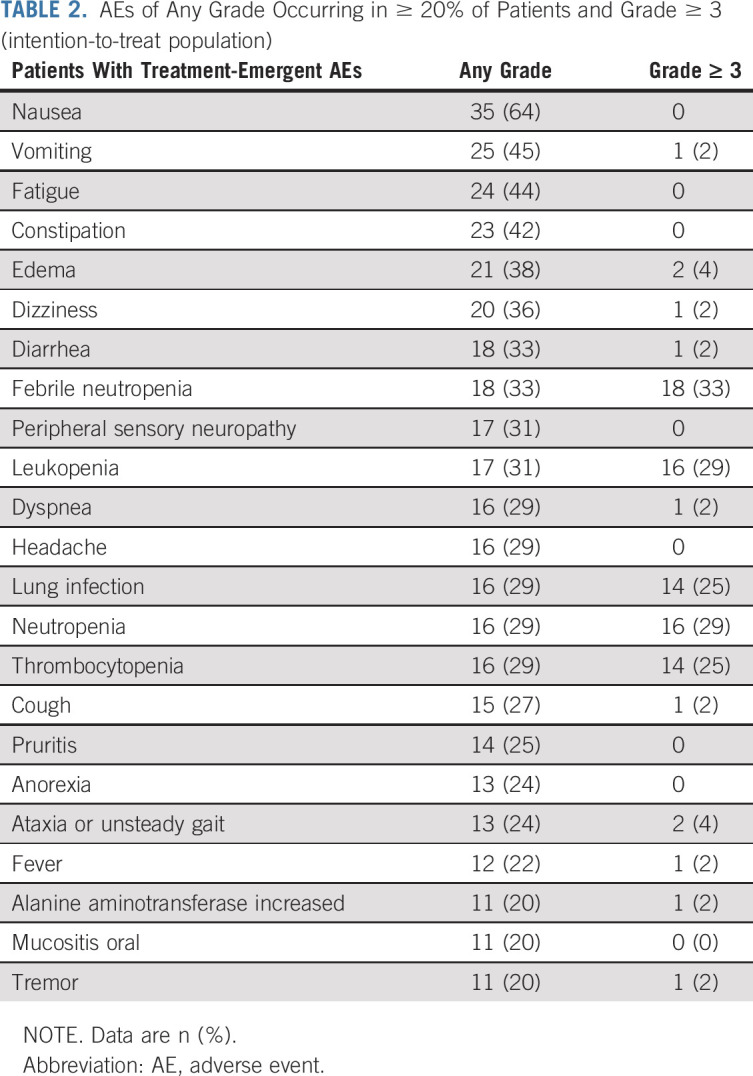
The most commonly reported AEs (> 20%) of any grade in patients receiving eprenetapopt and azacitidine are summarized in Table 2. Of these, dizziness (36%), peripheral sensory neuropathy (31%), ataxia (24%), and tremor (20%) were the most frequently reported neurologic events, occurring during the infusion phase and resolving thereafter (Data Supplement). Among the 15 (27%) patients with recurrent neurologic AEs, the initial occurrence was reported in cycle 1, and 14 of 15 patients had a recurrence of the same neurologic AE in subsequent cycles. No recurrent neurologic AEs worsened to grade ≥ 3. The most commonly reported grade ≥ 3 AEs were febrile neutropenia (33%), leukopenia (29%), neutropenia (29%), thrombocytopenia (25%), and lung infection (25%) with < 5% of grade ≥ 3 AEs considered possibly related to eprenetapopt treatment. Three patients had eprenetapopt dose reductions for nausea: two by one dose level and one by two dose levels. No patients had dose reductions because of neurologic AEs. Three patients (5%) discontinued treatment because of AEs: grade 4 sepsis (1), baseline grade 3 creatinine elevation that did not resolve (1), and pneumonia requiring > 28-day dose delay (1), none were treatment-related. The most common serious AEs were febrile neutropenia (25%), lung infection (20%), respiratory failure (7%), and sepsis (7%). By day 30, one (2%) patient had died because of pneumonia and by day 60, two additional patients (5%) had died because of pneumonia and sepsis, respectively, none were treatment-related.
TABLE 2.
AEs of Any Grade Occurring in ≥ 20% of Patients and Grade ≥ 3 (intention-to-treat population)
Efficacy
ITT population.
Fifty-five patients received combination therapy (ITT cohort); of these, 40 patients had MDS. The ORR was 71%, and the CR rate was 44% (Table 3). By disease subtype, the ORR was 73% for patients with MDS and 64% for those with AML, and the CR rate was 50% for patients with MDS and 36% for those with AML. The median times to ORR and CR were 2.1 months (95% CI, 0.1 to 5.4) and 3.1 months (95% CI, 2.5 to 6.1), respectively. Of the 39 patients who responded, 35 (90%) experienced hematologic improvement by International Working Group criteria. For the 12 patients treated in the dose-escalation phase, after 4 days of eprenetapopt monotherapy, one patient achieved a marrow complete remission (mCR; bone marrow blasts decrease from 18.5% to 0.5% with partial cytogenetic response) and ultimately CR following eprenetapopt and azacitidine therapy.
TABLE 3.
Hematological Responses in All Patients and by Disease Type (intention-to-treat population)

Response-evaluable population.
Individual responses for the 45 patients in the response-evaluable cohort are shown in Figure 2A. The ORR for the overall cohort was 87%: 88% each for patients with MDS (n = 29) and AML (n = 7) (Data Supplement). The overall CR rate was 53%: 61% for patients with MDS and 50% for patients with AML. Cytogenetic responses were observed in 59% of patients (18% partial and 41% complete), with responses highest in the MDS population (70%; 24% partial and 46% complete). The median durations of ORR and CR were 8.0 and 7.3 months, respectively, with no difference between MDS and AML.
FIG 2.
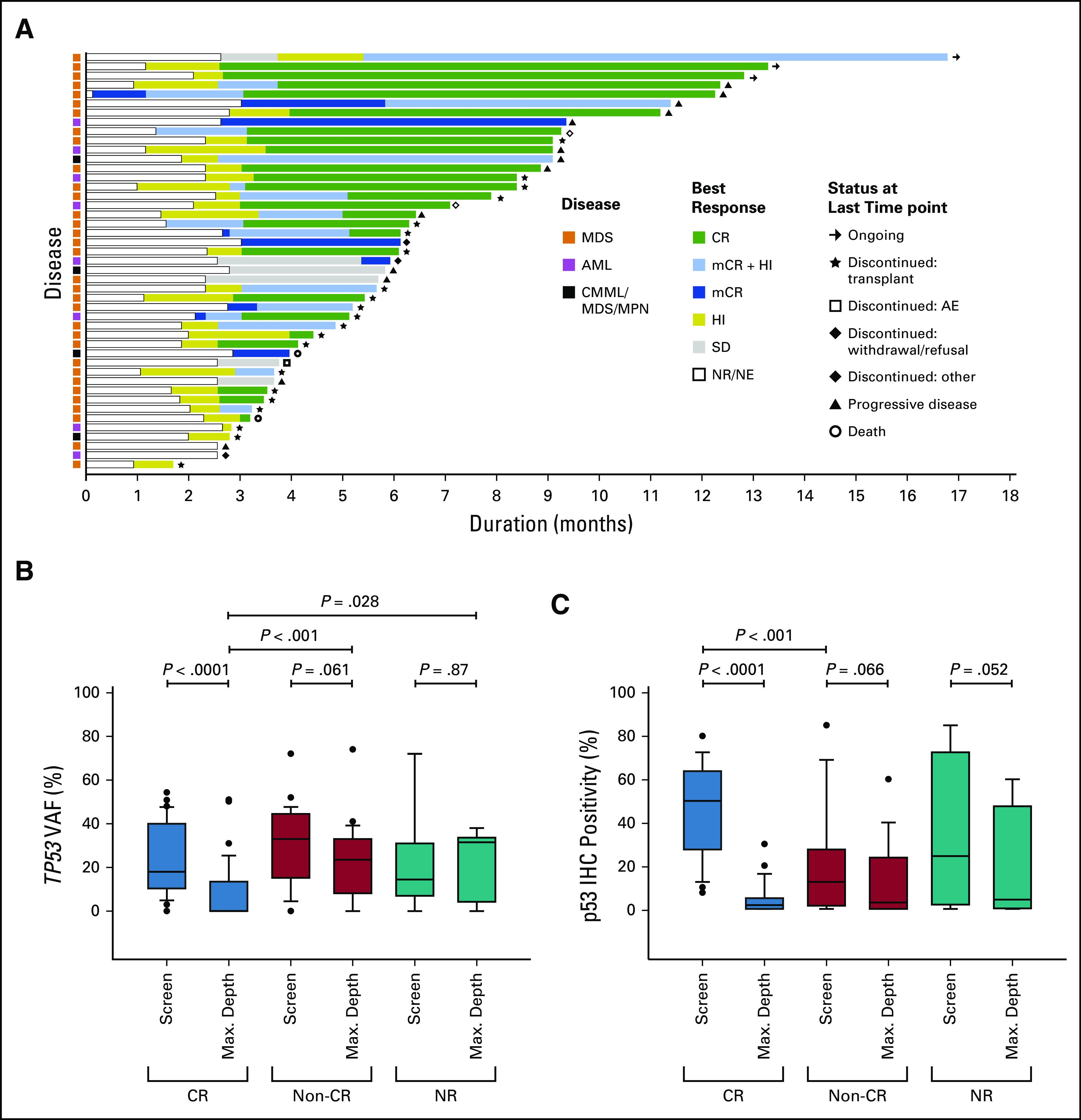
(A) Treatment response and duration for all 45 response-evaluable patients, (B) TP53 variant allele frequency by best response, and (C) serial p53 IHC positivity by best response. AE, adverse event; AML, acute myeloid leukemia; CMML, chronic myelomonocytic leukemia; CR, complete remission; HI, hematologic improvement; IHC, immunohistochemistry; mCR, marrow complete remission; MDS, myelodysplastic syndromes; MDS/MPN, myelodysplastic syndromes/myeloproliferative neoplasms; NE, not evaluable; NR, not reported; SD, stable disease; VAF, variant allele frequency.
Survival
At a median follow-up time of 10.5 months, the median OS in the ITT population was 10.8 months (95% CI, 8.1 to 13.4; Fig 3A), which was similar between the MDS (10.4 months; 95% CI, 7.6 to 13.3) and AML disease cohorts (10.8 months; 95% CI, 5.1 to 16.5). A landmark analysis of responders and nonresponders at 4 months demonstrated significantly longer median OS in responding patients (14.6 months; 95% CI, 12.4 to 16.8) versus 7.5 months in nonresponding patients (95% CI, 6.1 to 8.7 months; hazard ratio, 0.23; P = .0005; Fig 3B). Patients achieving CR and non-CR responses had significantly improved OS compared with nonresponding patients in landmark analysis at 4 months (P = .001; Fig 3C), although the median OS between CR and non-CR responding patients was not significantly different (12.8 [95% CI, 10.8 to 14.7] v 14.7 [95% CI, 8.4 to 21.1] months; P = .63). The median OS for patients who were bridged to HSCT was 14.7 months (95% CI, 8.6 to 20.9). HSCT was modeled as a time-dependent covariate in the Cox proportional hazards model, which showed that HSCT was not significantly associated with survival (hazard ratio, 1.01; P = .98). However, patients receiving ≥ 4 cycles of combination therapy prior to HSCT had significantly improved OS compared with those with < 4 cycles (16.1 months; 95% CI, 10.4 to not reported [NR] v 9.3 months; 95% CI, 8 to NR months, respectively; P = .01; Data Supplement). The median OS was not reached for patients with TP53 VAF clearance to < 5% (n = 6) prior to HSCT versus 14.7 months (95% CI, 10.1 to NR) in patients without clearance (n = 12), although this was not statistically significant in this limited data set (P = .56; Data Supplement).
FIG 3.

Kaplan-Meier curves for OS (A) in intention-to-treat population, (B) in patients with response versus no response by landmark analysis at 4 months, and (C) in patients with CR and non-CR response versus no response by landmark analysis at 4 months. CR, complete remission; OS, overall survival.
Biomarkers
Evaluation of response biomarkers showed that patients with an isolated TP53 mutation, that is, absence of a mutation in any other gene by NGS, had a higher rate of CR than those with co-occurring mutations (69% v 25%; P = .006). Additionally, p53 protein accumulation, defined as ≥ 10% p53 immunohistochemistry (IHC)+ bone marrow (BM) mononuclear cells at baseline, was associated with CR (66% v 13%; P = .01). NGS for TP53 mutations using a VAF cutoff of 5% showed that 21 (38%) patients achieved NGS negativity. TP53 VAF clearance was significantly associated with CR (P < .0001; Fig 2B) with a trend for clearance in patients with non-CR responses (P = .061). There was no change in TP53 VAF clearance in patients without a response (P = .87). Additionally, patients who achieved a CR had significantly lower TP53 VAF (maximum reduction at disease assessment) than those who achieved a non-CR response (P < .001). The median MRD VAF at maximum clearance was 0.63% (range, 0%-5%) with 2 (4%) patients having undetectable MRD. Similarly, p53 protein clearance on the basis of IHC was significantly associated with CR (P < .0001; Fig 2C) with mild reductions in non-CR responders (P = .066) and nonresponding patients (P = .052) (Data Supplement).
DISCUSSION
The presence of a TP53 gene mutation in patients with MDS or AML confers a poor prognosis, highlighting the urgent need for novel, effective therapy in this molecularly defined subgroup. Patients enrolled to this study comprised very high-risk patients with TP53-mutant myeloid malignancies, the majority of whom (91%) had more than one TP53 mutation, complex cytogenetics, and/or abnormalities in chromosome 17, thus representing a molecular subset with the poorest of outcomes.8-10,12,28 In this study, treatment with eprenetapopt and azacitidine yielded a high rate of CR (44%, 50% in MDS), which is substantially higher than previously reported for single-agent HMAs in patients with MDS, including those with TP53 mutations.9,13,14,29,30 In addition, the majority (90%) of responding patients experienced hematologic improvement with a high degree of cytogenetic clearance (59%) and molecular remission (47% with TP53 VAF < 5%; 4% undetectable MRD), which is notable given recent data supporting improved OS in patients with TP53 VAF clearance to < 5%, which is rare in patients treated with azacitidine monotherapy.16,31
Predefined biomarker analyses identified subgroups with higher rates of response to treatment. Consistent with the mechanism by which eprenetapopt restores wild-type function to the misfolded mutant p53 protein, patients with insufficient p53 levels on baseline BM biopsies (< 10% p53 IHC+ BM mononuclear cells) had significantly lower rates of CR than those who were p53-positive (13% v 66%, respectively). Notably, there was only one patient on study who had an isolated nonsense or frameshift mutation where an absence of p53 protein was confirmed by IHC, and this patient did not respond to treatment. Patients with mutant TP53 without other somatic gene mutations had the highest CR rates (69% v 25%). Notably, 100% of CR patients had TP53 in the dominant clone.
The median OS was 10.8 months in the ITT cohort. Median OS was significantly longer in responding patients than nonresponders by landmark analysis (14.6 v 7.5 months; P = .0005). These findings compare favourably to historical outcomes with HMA treatment, including azacitidine or low-dose cytarabine administered with venetoclax where median OS was 7.2 and 3.7 months, respectively.32,33 Combination of azacitidine with other novel agents such as magrolimab and pevonedistat are in development for TP53-mutant MDS or AML34,35 and if warranted on the basis of promising data could be studied in novel combinatorial approaches to further improve outcomes in this patient population.
Historically, the proportion of patients with MDS who proceed to HSCT is < 10%.36 Among the 19 (35%) patients who proceeded to HSCT in our study, the median OS of 14.7 months was encouraging. As patients experienced a deepening of cytogenetic and molecular responses over time, a longer duration of azacitidine and eprenetapopt prior to HSCT may be important to reduce the risk for relapse. Indeed, our study showed that median OS was significantly longer in patients who received at least four cycles of treatment.
The combination of eprenetapopt and azacitidine was well-tolerated and displayed a similar safety profile to that reported for eprenetapopt and azacitidine monotherapies. The majority of grade ≥ 3 AEs were hematologic with similar frequencies to those reported in a phase III study of azacitidine.14 Furthermore, AEs with eprenetapopt monotherapy were transient and infusion-related neurologic events, which were generally grade ≤ 2 and similar to those reported previously.25 Importantly, no treatment-related AEs led to permanent discontinuation of eprenetapopt. Moreover, the mortality rate with the combination was very low (one death within 30 days), which may relate to the more rapid time to response with the eprenetapopt combination.
In conclusion, eprenetapopt in combination with azacitidine is well-tolerated and yields high remission rates in patients with TP53-mutant MDS and AML. Consistent with these data, results from a phase II study of eprenetapopt and azacitidine by the Groupe Francophone des Myélodysplasies (ClinicalTrials.gov identifier: NCT03588078) showed comparable response rates.37 Together, these data support the ongoing pivotal phase III, multicenter, randomized study of eprenetapopt in combination with azacitidine versus azacitidine alone in patients with TP53-mutant MDS (ClinicalTrials.gov identifier: NCT03745716).
ACKNOWLEDGMENT
We thank the participating patients and their families, the nurses, research coordinators and the study management team, and Juliet Fawcett, PhD, for editorial assistance (supported by Aprea Therapeutics).
David A. Sallman
Consulting or Advisory Role: Celyad, Agios, Abbvie, Aprea AB, Bristol-Myers Squibb, Gilead Sciences, Intellia Therapeutics, Kite Pharma, Magenta Therapeutics, Novartis, Syndax
Speakers' Bureau: Agios, Incyte, Bristol-Myers Squibb
Research Funding: Celgene, Jazz Pharmaceuticals
Patents, Royalties, Other Intellectual Property: Intellectual Property Patent for LB-100 in MDS
Amy E. DeZern
Consulting or Advisory Role: Celgene, Novartis, Gilead Sciences
Research Funding: Celgene, Astex Pharmaceuticals
Travel, Accommodations, Expenses: Abbvie
Guillermo Garcia-Manero
Honoraria: Celgene, Astex Pharmaceuticals, Acceleron Pharma, Helssin, Abbvie
Consulting or Advisory Role: Celgene, Astex Pharmaceuticals, Acceleron Pharma, Jazz Pharmaceuticals, Bristol-Myers Squibb, Helsinn Therapeutics
Research Funding: Celgene, Astex Pharmaceuticals, Amphivena, Helsinn Therapeutics, Novartis, Abbvie, Bristol-Myers Squibb, Onconova Therapeutics, H3 Biomedicine, Merck
David P. Steensma
Stock and Other Ownership Interests: Arrowhead Pharmaceuticals, Sage Therapeutics
Honoraria: Daiichi Sankyo, Summer Road, Stemline Therapeutics, Celgene, Astex Pharmaceuticals
Consulting or Advisory Role: Pfizer, Janssen Oncology, Agios, Onconova Therapeutics, Geron, Astex Pharmaceuticals
Research Funding: Aprea AB, Celgene/BMS, H3 Biomedicine
Gail J. Roboz
Consulting or Advisory Role: Amphivena, Janssen, Amgen, Astex Pharmaceuticals, Celgene, Genoptix, MedImmune, Novartis, Pfizer, Abbvie, Argenx, Array BioPharma, Bayer, Celltrion, Jazz Pharmaceuticals, Orsenix, Genentech/Roche, Sandoz, Actinium Pharmaceuticals, Astellas Pharma, Eisai, Bayer, Daiichi Sankyo, MEI Pharma, Otsuka, Takeda, Roche, Agios, Trovagene, GlaxoSmithKline, Bristol-Myers Squibb, Helsinn Therapeutics, Mesoblast
Research Funding: Abbvie, Agios, Astex Pharmaceuticals, Celgene, CTI, Karyopharm Therapeutics, MedImmune, MEI Pharma, Moffitt, Novartis, Onconova Therapeutics, Pfizer, Sunesis Pharmaceuticals, Tensha Therapeutics, Cellectis, Janssen, Amphivena
Travel, Accommodations, Expenses: Amphivena, Astex Pharmaceuticals, Janssen, Pfizer, Array BioPharma, Novartis, Abbvie, Jazz Pharmaceuticals, Celgene, Celltrion, Roche/Genentech, Sandoz, Bayer, Clovis Oncology, Amgen, Sunesis Pharmaceuticals, Eisai, Agios
Mikkael A. Sekeres
Consulting or Advisory Role: Celgene, Millennium, Syros Pharmaceuticals
Research Funding: Takeda, Pfizer, Bristol-Myers Squibb
Thomas Cluzeau
Consulting or Advisory Role: Abbvie, Agios, Bristol-Myers Squibb, Jazz Pharmaceuticals, Novartis, Roche, Takeda, Syros Pharmaceuticals
Speakers' Bureau: Novartis, Amgen, Sanofi, Astellas Pharma
Travel, Accommodations, Expenses: Bristol-Myers Squibb, Novartis, Pfizer, Sanofi
Kendra L. Sweet
Leadership: Immtech
Consulting or Advisory Role: Astellas Pharma, Bristol-Myers Squibb, Novartis, Takeda, Stemline Therapeutics
Research Funding: Incyte
Kathy L. McGraw
Research Funding: Genentech, Celgene
Eric Padron
Honoraria: Stemline Therapeutics, Blueprint Medicines
Speakers' Bureau: Novartis, Taiho Pharmaceutical
Research Funding: Incyte, Bristol-Myers Squibb, Kura Oncology
Greg Korbel
Employment: Aprea Therapeutics, Inc
Leadership: Aprea Therapeutics, Inc
Stock and Other Ownership Interests: Aprea Therapeutics, Inc
Eyal C. Attar
Employment: Agios, Aprea AB
Leadership: Aprea AB
Stock and Other Ownership Interests: Agios, Aprea AB
Consulting or Advisory Role: Teladoc
Travel, Accommodations, Expenses: Aprea AB, Agios
Hagop M. Kantarjian
Honoraria: Abbvie, Amgen, ARIAD, Bristol-Myers Squibb, Immunogen, Orsenix, Pfizer, Agios, Takeda, Actinium Pharmaceuticals
Research Funding: Pfizer, Amgen, Bristol-Myers Squibb, Novartis, ARIAD, Astex Pharmaceuticals, Abbvie, Agios, Cyclacel, Immunogen, Jazz Pharmaceuticals
Jeffrey E. Lancet
Stock and Other Ownership Interests: Arvinas
Consulting or Advisory Role: Jazz Pharmaceuticals, Astellas Pharma, Abbvie, Agios, BerGenBio, Daiichi Sankyo, ElevateBio
Research Funding: Pfizer
Pierre Fenaux
Honoraria: Celgene
Research Funding: Celgene
Alan F. List
Honoraria: Celgene, Aileron Therapeutics, Cellular Biomedicine Group
Consulting or Advisory Role: Celgene, Cellular Biomedicine Group, Aileron Therapeutics, Acceleron Pharma, International Personalized Cancer Center, Precision Biosciences, CTI BioPharma Corp, Prelude Therapeutics
Research Funding: Celgene
Travel, Accommodations, Expenses: Celgene, Cellular Biomedicine Group
Other Relationship: Thousand Talents Award
Rami S. Komrokji
Stock and Other Ownership Interests: Abbvie
Consulting or Advisory Role: Novartis, Incyte, Bristol-Myers Squibb, Jazz Pharmaceuticals, Abbvie, Geron, Acceleron Pharma
Speakers' Bureau: Jazz Pharmaceuticals, Bristol-Myers Squibb, Agios
Travel, Accommodations, Expenses: Incyte, Jazz Pharmaceuticals, Bristol-Myers Squibb, Agios
No other potential conflicts of interest were reported.
PRIOR PRESENTATION
Presented in part at the 60th American Society of Hematology (ASH) Annual Meeting, San Diego, CA, November 30-December 4, 2018, and the 61st ASH Annual Meeting, Orlando, FL, December 7-10, 2019.
SUPPORT
Supported by MDS Foundation Young Investigator Grant, the Early Career Award of the Dresner Foundation (DAS), and the Edward P. Evans MDS Clinical Research Consortium. This work has also been supported in part by the Flow Cytometry and Molecular Genomics Core Facilities at the H. Lee Moffitt Cancer Center & Research Institute, an NCI-designated Comprehensive Cancer Center (P30-CA076292).
CLINICAL TRIAL INFORMATION
A.F.L. and R.S.K. contributed equally to this work.
DATA SHARING STATEMENT
Data and clinical protocols are available from the corresponding author David Sallman (david.sallman@moffit.org). Details of the study are also available at www.clinicaltrials.gov/ct2/show/NCT03072043.
AUTHOR CONTRIBUTIONS
Conception and design: David A. Sallman, Guillermo Garcia-Manero, David P. Steensma, Mikkael A. Sekeres, Thomas Cluzeau, Pierre Fenaux, Alan F. List, Rami S. Komrokji
Financial support: David A. Sallman
Administrative support: David A. Sallman, Lisa Nardelli
Provision of study materials or patients: David A. Sallman, Amy E. DeZern, Guillermo Garcia-Manero, David P. Steensma, Gail J. Roboz, Ling Zhang, Eric Padron, Hagop M. Kantarjian, Alan F. List, Rami S. Komrokji
Collection and assembly of data: David A. Sallman, Amy E. DeZern, Guillermo Garcia-Manero, David P. Steensma, Gail J. Roboz, Mikkael A. Sekeres, Kendra L. Sweet, Amy McLemore, Kathy L. McGraw, John Puskas, Ling Zhang, Lisa Nardelli, Najla H. Al Ali, Eric Padron, Eyal C. Attar
Data analysis and interpretation: David A. Sallman, Amy E. DeZern, Guillermo Garcia-Manero, David P. Steensma, Gail J. Roboz, Mikkael A. Sekeres, Thomas Cluzeau, Kendra L. Sweet, Amy McLemore, Ling Zhang, Jiqiang Yao, Qianxing Mo, Greg Korbel, Eyal C. Attar, Hagop M. Kantarjian, Jeffrey E. Lancet, Pierre Fenaux, Alan F. List, Rami S. Komrokji
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Eprenetapopt (APR-246) and Azacitidine in TP53-Mutant Myelodysplastic Syndromes
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated unless otherwise noted. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/jco/authors/author-center.
Open Payments is a public database containing information reported by companies about payments made to US-licensed physicians (Open Payments).
David A. Sallman
Consulting or Advisory Role: Celyad, Agios, Abbvie, Aprea AB, Bristol-Myers Squibb, Gilead Sciences, Intellia Therapeutics, Kite Pharma, Magenta Therapeutics, Novartis, Syndax
Speakers' Bureau: Agios, Incyte, Bristol-Myers Squibb
Research Funding: Celgene, Jazz Pharmaceuticals
Patents, Royalties, Other Intellectual Property: Intellectual Property Patent for LB-100 in MDS
Amy E. DeZern
Consulting or Advisory Role: Celgene, Novartis, Gilead Sciences
Research Funding: Celgene, Astex Pharmaceuticals
Travel, Accommodations, Expenses: Abbvie
Guillermo Garcia-Manero
Honoraria: Celgene, Astex Pharmaceuticals, Acceleron Pharma, Helssin, Abbvie
Consulting or Advisory Role: Celgene, Astex Pharmaceuticals, Acceleron Pharma, Jazz Pharmaceuticals, Bristol-Myers Squibb, Helsinn Therapeutics
Research Funding: Celgene, Astex Pharmaceuticals, Amphivena, Helsinn Therapeutics, Novartis, Abbvie, Bristol-Myers Squibb, Onconova Therapeutics, H3 Biomedicine, Merck
David P. Steensma
Stock and Other Ownership Interests: Arrowhead Pharmaceuticals, Sage Therapeutics
Honoraria: Daiichi Sankyo, Summer Road, Stemline Therapeutics, Celgene, Astex Pharmaceuticals
Consulting or Advisory Role: Pfizer, Janssen Oncology, Agios, Onconova Therapeutics, Geron, Astex Pharmaceuticals
Research Funding: Aprea AB, Celgene/BMS, H3 Biomedicine
Gail J. Roboz
Consulting or Advisory Role: Amphivena, Janssen, Amgen, Astex Pharmaceuticals, Celgene, Genoptix, MedImmune, Novartis, Pfizer, Abbvie, Argenx, Array BioPharma, Bayer, Celltrion, Jazz Pharmaceuticals, Orsenix, Genentech/Roche, Sandoz, Actinium Pharmaceuticals, Astellas Pharma, Eisai, Bayer, Daiichi Sankyo, MEI Pharma, Otsuka, Takeda, Roche, Agios, Trovagene, GlaxoSmithKline, Bristol-Myers Squibb, Helsinn Therapeutics, Mesoblast
Research Funding: Abbvie, Agios, Astex Pharmaceuticals, Celgene, CTI, Karyopharm Therapeutics, MedImmune, MEI Pharma, Moffitt, Novartis, Onconova Therapeutics, Pfizer, Sunesis Pharmaceuticals, Tensha Therapeutics, Cellectis, Janssen, Amphivena
Travel, Accommodations, Expenses: Amphivena, Astex Pharmaceuticals, Janssen, Pfizer, Array BioPharma, Novartis, Abbvie, Jazz Pharmaceuticals, Celgene, Celltrion, Roche/Genentech, Sandoz, Bayer, Clovis Oncology, Amgen, Sunesis Pharmaceuticals, Eisai, Agios
Mikkael A. Sekeres
Consulting or Advisory Role: Celgene, Millennium, Syros Pharmaceuticals
Research Funding: Takeda, Pfizer, Bristol-Myers Squibb
Thomas Cluzeau
Consulting or Advisory Role: Abbvie, Agios, Bristol-Myers Squibb, Jazz Pharmaceuticals, Novartis, Roche, Takeda, Syros Pharmaceuticals
Speakers' Bureau: Novartis, Amgen, Sanofi, Astellas Pharma
Travel, Accommodations, Expenses: Bristol-Myers Squibb, Novartis, Pfizer, Sanofi
Kendra L. Sweet
Leadership: Immtech
Consulting or Advisory Role: Astellas Pharma, Bristol-Myers Squibb, Novartis, Takeda, Stemline Therapeutics
Research Funding: Incyte
Kathy L. McGraw
Research Funding: Genentech, Celgene
Eric Padron
Honoraria: Stemline Therapeutics, Blueprint Medicines
Speakers' Bureau: Novartis, Taiho Pharmaceutical
Research Funding: Incyte, Bristol-Myers Squibb, Kura Oncology
Greg Korbel
Employment: Aprea Therapeutics, Inc
Leadership: Aprea Therapeutics, Inc
Stock and Other Ownership Interests: Aprea Therapeutics, Inc
Eyal C. Attar
Employment: Agios, Aprea AB
Leadership: Aprea AB
Stock and Other Ownership Interests: Agios, Aprea AB
Consulting or Advisory Role: Teladoc
Travel, Accommodations, Expenses: Aprea AB, Agios
Hagop M. Kantarjian
Honoraria: Abbvie, Amgen, ARIAD, Bristol-Myers Squibb, Immunogen, Orsenix, Pfizer, Agios, Takeda, Actinium Pharmaceuticals
Research Funding: Pfizer, Amgen, Bristol-Myers Squibb, Novartis, ARIAD, Astex Pharmaceuticals, Abbvie, Agios, Cyclacel, Immunogen, Jazz Pharmaceuticals
Jeffrey E. Lancet
Stock and Other Ownership Interests: Arvinas
Consulting or Advisory Role: Jazz Pharmaceuticals, Astellas Pharma, Abbvie, Agios, BerGenBio, Daiichi Sankyo, ElevateBio
Research Funding: Pfizer
Pierre Fenaux
Honoraria: Celgene
Research Funding: Celgene
Alan F. List
Honoraria: Celgene, Aileron Therapeutics, Cellular Biomedicine Group
Consulting or Advisory Role: Celgene, Cellular Biomedicine Group, Aileron Therapeutics, Acceleron Pharma, International Personalized Cancer Center, Precision Biosciences, CTI BioPharma Corp, Prelude Therapeutics
Research Funding: Celgene
Travel, Accommodations, Expenses: Celgene, Cellular Biomedicine Group
Other Relationship: Thousand Talents Award
Rami S. Komrokji
Stock and Other Ownership Interests: Abbvie
Consulting or Advisory Role: Novartis, Incyte, Bristol-Myers Squibb, Jazz Pharmaceuticals, Abbvie, Geron, Acceleron Pharma
Speakers' Bureau: Jazz Pharmaceuticals, Bristol-Myers Squibb, Agios
Travel, Accommodations, Expenses: Incyte, Jazz Pharmaceuticals, Bristol-Myers Squibb, Agios
No other potential conflicts of interest were reported.
REFERENCES
- 1.Papaemmanuil E Gerstung M Malcovati L, et al. : Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood 122:3616-3627, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Haferlach T Nagata Y Grossmann V, et al. : Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia 28:241-247, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Walter MJ Shen D Shao J, et al. : Clonal diversity of recurrently mutated genes in myelodysplastic syndromes. Leukemia 27:1275-1282, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bejar R Stevenson K Abdel-Wahab O, et al. : Clinical effect of point mutations in myelodysplastic syndromes. N Engl J Med 364:2496-2506, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sasaki K Kanagal-Shamanna R Montalban-Bravo G, et al. : Impact of the variant allele frequency of ASXL1, DNMT3A, JAK2, TET2, TP53, and NPM1 on the outcomes of patients with newly diagnosed acute myeloid leukemia. Cancer 126:765-774, 2020 [DOI] [PubMed] [Google Scholar]
- 6.Hunter AM, Sallman DA: Current status and new treatment approaches in TP53 mutated AML. Best Pract Res Clin Haematol 32:134-144, 2019 [DOI] [PubMed] [Google Scholar]
- 7.Singhal D Wee LYA Kutyna MM, et al. : The mutational burden of therapy-related myeloid neoplasms is similar to primary myelodysplastic syndrome but has a distinctive distribution. Leukemia 33:2842-2853, 2019 [DOI] [PubMed] [Google Scholar]
- 8.Sallman DA Komrokji R Vaupel C, et al. : Impact of TP53 mutation variant allele frequency on phenotype and outcomes in myelodysplastic syndromes. Leukemia 30:666-673, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bernard E Nannya Y Hasserjian RP, et al. : Implications of TP53 allelic state for genome stability, clinical presentation and outcomes in myelodysplastic syndromes. Nat Med 26:1549-1556, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Haase D Stevenson KE Neuberg D, et al. : TP53 mutation status divides myelodysplastic syndromes with complex karyotypes into distinct prognostic subgroups. Leukemia 33:1747-1758, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kulasekararaj AG Smith AE Mian SA, et al. : TP53 mutations in myelodysplastic syndrome are strongly correlated with aberrations of chromosome 5, and correlate with adverse prognosis. Br J Haematol 160:660-672, 2013 [DOI] [PubMed] [Google Scholar]
- 12.Montalban-Bravo G Kanagal-Shamanna R Benton CB, et al. : Genomic context and TP53 allele frequency define clinical outcomes in TP53-mutated myelodysplastic syndromes. Blood Adv 4:482-495, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sallman DA Al Ali N Yun S, et al. : Clonal suppression of TP53 mutant MDS and oligoblastic AML with hypomethylating agent therapy improves overall survival. Blood 132:1817, 2018 [Google Scholar]
- 14.Fenaux P Mufti GJ Hellstrom-Lindberg E, et al. : Efficacy of azacitidine compared with that of conventional care regimens in the treatment of higher-risk myelodysplastic syndromes: A randomised, open-label, phase III study. Lancet Oncol 10:223-232, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bally C Adès L Renneville A, et al. : Prognostic value of TP53 gene mutations in myelodysplastic syndromes and acute myeloid leukemia treated with azacitidine. Leuk Res 38:751-755, 2014 [DOI] [PubMed] [Google Scholar]
- 16.Falconi G Fabiani E Piciocchi A, et al. : Somatic mutations as markers of outcome after azacitidine and allogeneic stem cell transplantation in higher-risk myelodysplastic syndromes. Leukemia 33:785-790, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Craddock CF Houlton AE Quek LS, et al. : Outcome of azacitidine therapy in acute myeloid leukemia is not improved by concurrent vorinostat therapy but is predicted by a diagnostic molecular signature. Clin Cancer Res 23:6430, 2017 [DOI] [PubMed] [Google Scholar]
- 18.Welch JS Petti AA Miller CA, et al. : TP53 and decitabine in acute myeloid leukemia and myelodysplastic syndromes. N Engl J Med 375:2023-2036, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Short NJ Kantarjian HM Loghavi S, et al. : Treatment with a 5-day versus a 10-day schedule of decitabine in older patients with newly diagnosed acute myeloid leukaemia: A randomised phase 2 trial. Lancet Haematol 6:e29-e37, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zeidan AM Wang R Wang X, et al. : Clinical outcomes of older patients with AML receiving hypomethylating agents: A large population-based study in the United States. Blood Adv 4:2192-2201, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lambert JM Gorzov P Veprintsev DB, et al. : PRIMA-1 reactivates mutant p53 by covalent binding to the core domain. Cancer Cell 15:376-388, 2009 [DOI] [PubMed] [Google Scholar]
- 22.Bykov VJ Zhang Q Zhang M, et al. : Targeting of mutant p53 and the cellular redox balance by APR-246 as a strategy for efficient cancer therapy. Front Oncol 6:21, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Peng X Zhang MQ Conserva F, et al. : APR-246/PRIMA-1MET inhibits thioredoxin reductase 1 and converts the enzyme to a dedicated NADPH oxidase. Cell Death Dis 4:e881, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Maslah N Salomao N Drevon L, et al. : Synergistic effects of PRIMA-1Met (APR-246) and 5-azacitidine in TP53-mutated myelodysplastic syndromes and acute myeloid leukemia. Haematologica 105:1539, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lehmann S Bykov VJ Ali D, et al. : Targeting p53 in vivo: A first-in-human study with p53-targeting compound APR-246 in refractory hematologic malignancies and prostate cancer. J Clin Oncol 30:3633-3639, 2012 [DOI] [PubMed] [Google Scholar]
- 26.Deneberg S Cherif H Lazarevic V, et al. : An open-label phase I dose-finding study of APR-246 in hematological malignancies. Blood Cancer J 6:e447, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cheson BD Greenberg PL Bennett JM, et al. : Clinical application and proposal for modification of the International Working Group (IWG) response criteria in myelodysplasia. Blood 108:419-425, 2006 [DOI] [PubMed] [Google Scholar]
- 28.Ganster C Schabb R Shirneshan K, et al. : TP53 status and outcome in myelodysplastic syndromes with complex aberrant karyotypes. EHA Libr 294702:EP785, 2020 [Google Scholar]
- 29.Kantarjian H Issa J-PJ Rosenfeld CS, et al. : Decitabine improves patient outcomes in myelodysplastic syndromes. Cancer 106:1794-1803, 2006 [DOI] [PubMed] [Google Scholar]
- 30.Bejar R Stevenson KE Caughey B, et al. : Somatic mutations predict poor outcome in patients with myelodysplastic syndrome after hematopoietic stem-cell transplantation. J Clin Oncol 32:2691-2698, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yun S Geyer SM Komrokji RS, et al. : Prognostic significance of serial molecular annotation in myelodysplastic syndromes (MDS) and secondary acute myeloid leukemia (sAML). Leukemia 10.1038/s41375-020-0997-4[epub ahead of print on July 29, 2020] [DOI] [PMC free article] [PubMed]
- 32.DiNardo CD Pratz K Pullarkat V, et al. : Venetoclax combined with decitabine or azacitidine in treatment-naive, elderly patients with acute myeloid leukemia. Blood 133:7-17, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wei AH Strickland SA Hou J-Z, et al. : Venetoclax combined with low-dose cytarabine for previously untreated patients with acute myeloid leukemia: Results from a phase Ib/II study. J Clin Oncol 37:1277-1284, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Swords RT Coutre S Maris MB, et al. : Pevonedistat, a first-in-class NEDD8-activating enzyme inhibitor, combined with azacitidine in patients with AML. Blood 131:1415-1424, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sallman DA Al Malki M Asch AS, et al. : Tolerability and efficacy of the first-in-class anti-CD47 antibody magrolimab combined with azacitidine in MDS and AML patients: Phase Ib results. J Clin Oncol 38:7507, 2020. (suppl 15) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sekeres MA Schoonen WM Kantarjian H, et al. : Characteristics of US patients with myelodysplastic syndromes: Results of six cross-sectional physician surveys. J Natl Cancer Inst 100:1542-1551, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cluzeau T Sebert M Rahmé R, et al. : Eprenetapopt plus azacitidine in TP53-mutated myelodysplastic syndromes and acute myeloid leukemia: A phase II study by the Groupe Francophone des Myélodysplasies (GFM). J Clin Oncol 39:1575-1583, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data and clinical protocols are available from the corresponding author David Sallman (david.sallman@moffit.org). Details of the study are also available at www.clinicaltrials.gov/ct2/show/NCT03072043.