

Abstract
As part of the US Food and Drug Administration (FDA)’s Prescription Drug User Fee Act (PDUFA) VI commitments, the Center for Biologics Evaluation and Research (CBER) and Center for Drug Evaluation and Research (CDER) are conducting a model‐informed drug development (MIDD) pilot program. Sponsor(s) who apply and are selected will be granted meetings that aim to facilitate the application of MIDD approaches throughout the product development lifecycle and the regulatory process. Due to their complex mechanisms of action and limited clinical experience, cell and gene therapies have the potential to benefit from the application of MIDD methods, which may facilitate their safety and efficacy evaluations. Leveraging data that are generated from all stages of drug development into appropriate modeling and simulation techniques that inform decisions remains challenging. Additional discussions regarding the application of quantitative modeling approaches to drug development decisions, such as through the MIDD pilot program, may be crucial for both the sponsor(s) and regulatory review teams. Here, we share some perspectives on the opportunities and challenges for utilizing MIDD approaches for product review, which we hope will encourage investigators to publish their experiences and application of MIDD in gene therapy product development.
INTRODUCTION
Advances in scientific methods have led to the development of new approaches, such as modeling and simulation to facilitate medical product discovery as well as safety and efficacy evaluation. Model‐informed drug development (MIDD) is the science of developing quantitative models from preclinical and clinical data sources to help inform decision making throughout the drug development process. The MIDD pilot program was established as a performance commitment under the sixth iteration of the Prescription Drug and User Fee Act (PDUFA VI). Through this pilot program, the US Food and Drug Administration (FDA) accepts meeting requests for 2 to 4 sponsors quarterly each fiscal year to discuss the potential of quantitative approaches (such as exposure‐based, biological, or statistical models) to inform product development and regulatory decisions. 1 The FDA has recently provided perspectives on MIDD approaches, 2 , 3 as have industry MIDD practitioners. 4 In general, there is a shared outlook that MIDD can provide insight and supportive evidence for product safety and efficacy, and that best practices will continue to evolve and mature. When applied in the appropriate context, successful MIDD methods may improve clinical trial efficiency, increase the probability of regulatory success, and optimize drug dosing/therapeutic individualization in the absence of dedicated trials. 1 Although there are a number of regulatory aspects MIDD can help facilitate, the FDA has identified three priority areas for the pilot program: dose selection/estimation, clinical trial simulation, and predictive/mechanistic safety evaluation (Figure 1 ). Indeed, the utility of the guidance documents for exposure‐response modeling approaches can be appreciated by the increasing trend in the application of pharmacokinetic‐pharmacodynamic (PK‐PD) methods to inform product approval and labeling over the last decade. 5
FIGURE 1.
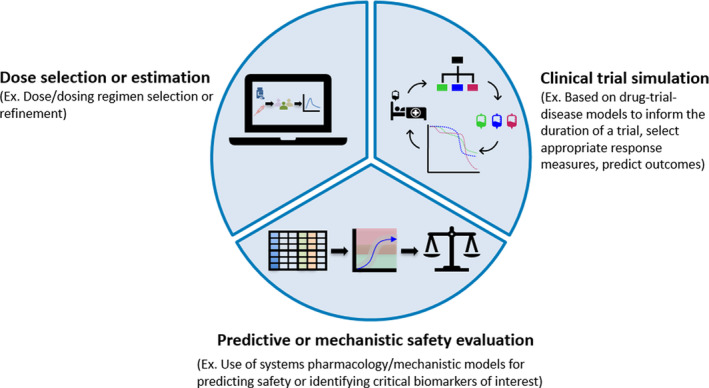
Priority areas for the model‐informed drug development (MIDD) pilot program. As described in the Federal Register Notice, 1 the initial MIDD priority areas include dose selection or estimation, clinical trial simulation, and predictive or mechanistic safety evaluation
Gene therapies (GTs) are a class of products regulated by Center for Biologics Evaluation and Research (CBER) and exhibit unique features compared to traditional small molecules or protein therapies. GTs have a complex life cycle starting from their manufacturing to clinical application, are often administered as a single dose, have unique PK‐PD properties, and have multifaceted mechanisms of action (MOA). These properties pose unique benefits and risks to patients and require additional considerations for the manufacturing, dose selection, and clinical trial design to establish the safety and efficacy of the product. 6 , 7 , 8 GT products are often studied in small clinical trials, which lead to difficulties in assessing the overall benefit‐risk profile of the treatment and selecting a safe and effective dose. As implied in their name, GTs are often composed of many cellular and chemical components, giving them an inherently larger material mass and structural complexity compared to those of small molecules or protein therapies. This makes it increasingly difficult to predict the likelihood of an immunogenic response to the therapy, as well as the epitopes responsible for immune‐mediated adverse events. These factors highlight an opportunity where a comprehensive and robust approach for integrating relevant data sources into a quantitative model may assist the safety and efficacy evaluation of GT products.
Although clinical applications of GTs are still limited, there are over 800 active investigational new drug applications under review for GTs at the FDA. 9 These drug development efforts continue to increase the amount of data available to investigate GT products from a quantitative perspective, such as estimating parameters for informing and validating MIDD applications in the regulatory setting. The accumulating data may not only help describe the population‐level effects of the therapies, but also assist in personalizing doses for specific patient populations. Traditional quantitative methods used to evaluate the safety and efficacy of small molecule drugs (i.e., pharmacometric modeling and simulation approaches) are difficult to apply to GTs, as typical PK‐PD model structures and parameter paradigms may not be directly applicable to these complex products. Common clinical pharmacology concepts, such as product plasma clearance and the impact of intrinsic and extrinsic factors (such as age, body weight, comedications, etc.), are difficult to determine accurately with small clinical trials and the uncertainty surrounding in vivo processing of GTs. Despite these inherent difficulties, efforts are underway to explore and implement MIDD approaches to describe GTs in vivo and their PK‐PD profile. 10 , 11 , 12 , 13 Below, we will describe a few GT products and references from the literature as to how MIDD might be applied for these therapies.
Chimeric Antigen Receptor T cells
Chimeric antigen receptor T cell (CAR T cell) therapies are genetically modified T cells that are engineered to recognize specific cell surface antigens. The complex manufacturing process of autologous CAR T cells generally entails the collection of patient leukapheresis material, ex vivo delivery of the CAR transgene, expansion to meet the required dose, and administration of the modified T cells back into the patient. 14 CAR T cell MOA includes recognition of specific cell surface antigens (e.g., proteins such as CD19 on B cells), leading to the production of cytokines and subsequent destruction of the target cell. CAR T cells’ efficacy (in particular anti‐CD19 CAR T cells) has been demonstrated in the clinic, but complex safety risks also exist. The pro‐inflammatory molecules produced by the activated CAR T cell can activate neighboring immune cells, promote cytokine release syndrome (CRS), and lead to severe adverse events. 15 Estimating the optimal dose of CAR T cells by balancing these efficacy and safety concerns provides an intriguing opportunity for MIDD methods development.
Unlike traditional small molecule dose‐response relationships, CAR T cells undergo expansion after administration leading to a maximal therapeutic cell number several orders of magnitude larger than the administered dose. 12 These therapies also exhibit atypical clearance, as a certain proportion of CAR T cells persist as memory cells for months to years after the initial dose. The effects of these two properties on the PK profile of CAR T cells were recently explored with a mixed‐effects model. 13 Additionally, the investigators looked at the potential covariate effects from concomitant administration of anti‐inflammatory drugs (corticosteroids and tocilizumab). These efforts demonstrate an application of MIDD to better understand the safety and efficacy of CAR T cell therapies and helped facilitate FDA approval of three CD19‐directed CAR T cell products: tisagenlecleucel 16 and axicabtagene ciloleucel 17 in 2017, and brexucabtagene autoleucel 18 in 2020. During the regulatory review of tisagenlecleucel, the FDA review team conducted an exploratory model‐based analysis to address several regulatory questions. 16 An MIDD approach was explored to investigate the relationship between CAR T cells kinetics versus the CRS risk. The analysis indicated that a higher CAR T cell expansion rate is associated with higher probability of CRS onset. Additionally, an MIDD approach was used to characterize the impact of comedication with corticosteroid on efficacy outcomes. The regression analysis on overall remission rate, Kaplan‐Meier analysis on duration of response, and population PK model‐based analysis showed no significant impacts of corticosteroid on CAR T cells expansion and efficacy outcome. However, it was noted that the clinical trial data has some limitation for unbiased estimates of the impact of corticosteroid use, as the data for steroid exposed versus unexposed was unbalanced, as well as other confounding factors. 16 Further research into the modeling of the CAR T cell MOA is needed to improve the dose recommendations for specific patient populations, and also assist in the prediction of efficacy and safety outcomes in future CAR T cell studies.
Oncolytic viral therapies
Oncolytic viral therapies (OVs) are another class of GTs that are biologically engineered to selectively target tumor cells and produce lytic factors to promote cell death. 19 Tumor cells act as incubators for viral particle production, thereby increasing the concentration of the OVs proximal to the tumor microenvironment. In addition to typical clearance pathways, many factors can influence the OV titer in patients, such as neutralizing antibodies generated by the host, the efficiency of virus uptake by tumor cells, and the time‐varying tumor burden, all of which make traditional PK‐PD analysis difficult. Efforts to describe the PK of OVs include using differential equations to model the dynamic interactions between OVs and tumor cells have been proposed. 10 , 11 These models have provided a mathematical perspective of OV biodistribution beyond the site of injection, the predicted viral titer over time at varying doses, and the optimal times for re‐administration of OVs. Although these MIDD approaches are exploratory and based on simplified concepts, they provide a necessary step by incorporating the available knowledge about OVs into a quantitative drug evaluation approach. The initial models may serve as a foundation to further iterate and develop into more comprehensive MIDD approaches for related therapies.
Adeno‐associated virus
Adeno‐associated viruses (AAVs) are a class of GTs that can deliver an exogenous copy of a gene to the patient whose endogenous gene produces a nonfunctional form of the gene. Briefly, AAVs can package a corrected cDNA copy of the defective gene in a viral capsid. The corrected gene is often placed under a tissue‐specific promotor to limit off‐target production of the gene. 20 The therapeutic MOA depends on the AAV tropism, encoded gene, disease context, and the affected biological pathway(s). Examples of AAVs currently being explored in the clinic include the delivery of modified Factor VIII and Factor IX genes for hemophilia A and B, respectively, 21 , 22 , 23 , 24 and delivery of the lipoprotein lipase gene (LPL) for the treatment of associated metabolic diseases. 25 There are a growing number of AAV clinical trials, and two recently FDA approved products. The first approved product was voretigene neparvovec‐rzyl, which is injected into the retina and delivers a copy of RPE65 (a retinoid isomerohydrolase), whose defect causes type 2 Leber congenital amaurosis. 26 The second was onasemnogene abeparvovec‐xioi, an AAV that delivers a copy of survival of motor neuron 1 gene (SMN1), whose aberrant function is associated with spinal muscular atrophy (SMA), a debilitating and often fatal muscle weakness. 27
Although clinical progress has been made, uncertainties remain regarding the short‐term and long‐term effects of AAVs. Immune tolerance to the AAV serotype and each transgene remains poorly understood, but may limit therapeutic longevity. 20 Gene transduction and in vivo expression levels have historically not been a concern for toxicity due to relatively low levels of transgene production. However, recent improvements in engineered AAV vectors as well as gain‐of‐function and modified gene variants have led to supra‐physiologic levels of transgene activity. 21 Predicting transgene expression, protein and enzyme activity levels, and the potential for immune responses or other adverse events will be critical to help ensure GT products remain within a therapeutic range acceptable for efficacy and safety. Although efforts to describe in vivo processing of AAV vectors have been considered decades before the currently available data, 28 predicting the above‐mentioned features of AAVs continue to be a challenging topic for MIDD method developments. Improving on the initial modeling efforts may prove valuable for AAV product development and remains of great interest to the research community. Graphical schematics proposing the theoretical parameters influencing an MIDD approach can aid in taking the first steps to building the quantitative model framework for AAV therapies (Figure 2 ). The development of any valid MIDD approach requires the understanding of multiple kinetic processes, such as clearance of the AAV vector, fate of the transduced cells, and clearance of the expressed transgene (i.e., often the protein of interest). To our knowledge, much of the published preclinical and clinical data, including those referenced in this review, have not been fully leveraged to mechanistically and quantitatively elucidate the impact on downstream pathways that may influence the in vivo processing of AAV therapies. The mechanistic and quantitative insights gained from such an MIDD approach may assist in defining the safety and efficacy of the product, as well as optimize dosing.
FIGURE 2.
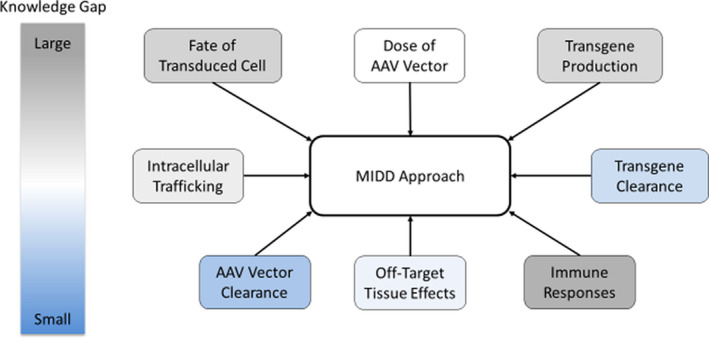
An example of a web diagram showing possible topics to include in a model‐informed drug development (MIDD) approach for adeno‐associated virus (AAV)‐based gene therapies. Topic areas are color‐coded according to the knowledge gap gradient shown on the left, with gray and blue representing topic areas with larger and smaller knowledge gaps, respectively. For example, AAV vector concentration has been measured over time in a number of tissues (colored light blue), whereas details regarding drivers of immune responses are poorly understood (colored gray)
PERSPECTIVES/CONCLUSION
As quantitative methods evolve and are increasingly applied in the development of biologic therapies, many opportunities remain to further refine MIDD approaches to new and innovative products. GTs have profound potential, such as replacing absent or mutated genes and restoring normal physiology after a single dose, but also carry unique and complex risks. The increasing clinical experience and data being generated on GTs can inform the development of MIDD methods to assess the benefits and risks of these therapies. Quantitative models may help provide insight on safety and efficacy to inform innovation, policy, and ultimately benefit the patient. As the use of MIDD by regulators and industry expands, standards and best practices must be developed to establish when, where, and what methods are appropriate in disparate settings. Further discussions between stakeholders to establish applied examples will assist the field in developing a robust scientific MIDD approach for each disease‐GT product context. We hope that this review will bring additional context to the FDA MIDD pilot program and encourage other stakeholders to share their experience on the application of quantitative methods for GT products.
Conflict of Interest
The authors declared no competing interests for this work.
Disclaimer
The opinions expressed in this manuscript are those of the authors and should not be interpreted as the position of the US Food and Drug Administration.
Funding information
No funding was received for this work.
References
- 1. U.S. Food and Drug Administration . Pilot Meetings Program for Model‐Informed Drug Development Approaches; 2018. <https://www.federalregister.gov/documents/2018/04/17/2018‐08010/pilot‐meetings‐program‐for‐model‐informed‐drug‐development‐approaches>. Accessed May 7, 2019.
- 2. Madabushi R, Wang Y, Zineh I. A holistic and integrative approach for advancing model‐informed drug development. CPT Pharmacometrics Syst Pharmacol. 2019;8(1):9–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wang Y, Zhu H, Madabushi R, Liu Q, Huang SM, Zineh I. Model‐informed drug development: current us regulatory practice and future considerations. Clin Pharmacol Ther. 2019;105(4):899–911. [DOI] [PubMed] [Google Scholar]
- 4. Marshall S, Madabushi R, Manolis E, et al. Model‐informed drug discovery and development: current industry good practice and regulatory expectations and future perspectives. CPT Pharmacometrics Syst Pharmacol. 2019;8(2):87–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tegenge MA, Mahmood I, Forshee R. Clinical pharmacology review of plasma‐derived and recombinant protein products: CBER experience and perspectives on model‐informed drug development. Haemophilia. 2019;25:e240–e246. [DOI] [PubMed] [Google Scholar]
- 6. FDA . Draft Guidance for Industry: Chemistry, Manufacturing, and Control Information for Human Gene Therapy Investigational New Drug Applications; 2018. <https://www.FDAgov/regulatory‐information/search‐fda‐guidance‐documents/chemistry‐manufacturing‐and‐control‐cmc‐information‐human‐gene‐therapy‐investigational‐new‐drug>. Accessed May 7, 2019.
- 7. FDA . Guidance for Industry: Considerations for the Design of Early‐Phase Clinical Trials of Cellular and Gene Therapy Products; 2017. <https://www.FDAgov/regulatory‐information/search‐fda‐guidance‐documents/considerations‐design‐early‐phase‐clinical‐trials‐cellular‐and‐gene‐therapy‐products>. Accessed May 7, 2019.
- 8. FDA . Draft Guidance for Industry: Human Gene Therapy for Rare Diseases; 2018. <https://www.FDAgov/regulatory‐information/search‐fda‐guidance‐documents/human‐gene‐therapy‐rare‐diseases>. Accessed May 7, 2019.
- 9. FDA . Statement from FDA Commissioner Scott Gottlieb, M.D. and Peter Marks, M.D., Ph.D. Director of the Center for Biologics Evaluation and Research on new policies to advance development of safe and effective cell and gene therapies; 2017. <https://www.fda.gov/news‐events/press‐announcements/statement‐fda‐commissioner‐scott‐gottlieb‐md‐and‐peter‐marks‐md‐phd‐director‐center‐biologics>. Accessed May 7, 2019.
- 10. Mok W, Stylianopoulos T, Boucher Y, Jain RK. Mathematical modeling of herpes simplex virus distribution in solid tumors: implications for cancer gene therapy. Clin Cancer Res. 2009;15(7):2352–2360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Titze MI, Frank J, Ehrhardt M, Smola S, Graf N, Lehr T. A generic viral dynamic model to systematically characterize the interaction between oncolytic virus kinetics and tumor growth. Eur J Pharm Sci. 2017;97:38–46. [DOI] [PubMed] [Google Scholar]
- 12. Mueller KT, Waldron E, Grupp SA, et al. Clinical pharmacology of tisagenlecleucel in B‐cell acute lymphoblastic leukemia. Clin Cancer Res. 2018;24(24):6175–6184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Stein AM, Grupp SA, Levine JE, et al. Tisagenlecleucel model‐based cellular kinetic analysis of chimeric antigen receptor‐T cells. CPT Pharmacometrics Syst Pharmacol. 2019;8(5):285–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. June CH, O'Connor RS, Kawalekar OU, Ghassemi S, Milone MC. CAR T cell immunotherapy for human cancer. Science. 2018;359(6382):1361–1365. [DOI] [PubMed] [Google Scholar]
- 15. Grigor EJM, Fergusson D, Kekre N, et al. Risks and benefits of chimeric antigen receptor T‐cell (CAR‐T) therapy in cancer: a systematic review and meta‐analysis. Transfus Med Rev. 2019;33(2):98–110. [DOI] [PubMed] [Google Scholar]
- 16. FDA . Clinical Pharmacology BLA Review – Kymriah; 2017. <https://www.fda.gov/vaccines‐blood‐biologics/cellular‐gene‐therapy‐products/kymriah‐tisagenlecleucel>. Accessed May 7, 2019.
- 17. FDA . Clinical Pharmacology Review: Yescarta; 2017. <https://www.fda.gov/vaccines‐blood‐biologics/cellular‐gene‐therapy‐products/yescarta‐axicabtagene‐ciloleucel>. Accessed May 7, 2019.
- 18. FDA . Clinical Pharmacology BLA Review – Tecartus; 2020. <https://www.fda.gov/vaccines‐blood‐biologics/cellular‐gene‐therapy‐products/tecartus‐brexucabtagene‐autoleucel>.
- 19. Lawler SE, Speranza MC, Cho CF, Chiocca EA. Oncolytic viruses in cancer treatment: a review. JAMA Oncol. 2017;3(6):841–849. [DOI] [PubMed] [Google Scholar]
- 20. Wang D, Tai PWL, Gao G. Adeno‐associated virus vector as a platform for gene therapy delivery. Nat Rev Drug Discov. 2019;18(5):358–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. George LA, Sullivan SK, Giermasz A, et al. Hemophilia B gene therapy with a high‐specific‐activity factor IX variant. N Engl J Med. 2017;377(23):2215–2227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Miesbach W, Meijer K, Coppens M, et al. Gene therapy with adeno‐associated virus vector 5‐human factor IX in adults with hemophilia B. Blood. 2018;131(9):1022–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Nathwani AC, Reiss UM, Tuddenham EG, et al. Long‐term safety and efficacy of factor IX gene therapy in hemophilia B. N Engl J Med. 2014;371(21):1994–2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rangarajan S, Walsh L, Lester W, et al. AAV5‐factor VIII gene transfer in severe hemophilia A. N Engl J Med. 2017;377(26):2519–2530. [DOI] [PubMed] [Google Scholar]
- 25. Gaudet D, Methot J, Dery S, et al. Efficacy and long‐term safety of alipogene tiparvovec (AAV1‐LPLS447X) gene therapy for lipoprotein lipase deficiency: an open‐label trial. Gene Ther. 2013;20(4):361–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Product label for voretigene neparvovec‐rzyl; 2017. <https://www.fda.gov/media/109487/download>. Accessed May 7, 2019.
- 27. Product Label for onasemnogene abeparvovec‐xioi ; 2019. <https://www.fda.gov/media/126109/download> Accessed May 7, 2019.
- 28. Shapiro Ledley T, Ledley FD. Pharmacokinetic considerations in somatic gene therapy. Adv Drug Deliv Rev. 1998;30(1–3):133–150. [DOI] [PubMed] [Google Scholar]