

Abstract
Cardiac fibrosis is a significant component of pathological heart remodeling, yet it is not directly targeted by existing drugs. Systems pharmacology approaches have the potential to provide mechanistic frameworks with which to predict and understand how drugs modulate biological systems. Here, we combine network modeling of the fibroblast signaling network with 36 unique drug‐target interactions from DrugBank to predict drugs that modulate fibroblast phenotype and fibrosis. Galunisertib was predicted to decrease collagen and α‐SMA expression, which we validated in human cardiac fibroblasts. In vivo fibrosis data from the literature validated predictions for 10 drugs. Further, the model was used to identify network mechanisms by which these drugs work. Arsenic trioxide was predicted to induce fibrosis by AP1‐driven TGFβ expression and MMP2‐driven TGFβ activation. Entresto (valsartan/sacubitril) was predicted to suppress fibrosis by valsartan suppression of ERK signaling and sacubitril enhancement of PKG activity, both of which decreased Smad3 activity. Overall, this study provides a framework for integrating drug‐target mechanisms with logic‐based network models, which can drive further studies both in cardiac fibrosis and other conditions.
STUDY HIGHLIGHTS.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
Current drugs do not adequately act against cardiac fibrosis, and some drugs exacerbate it.
WHAT QUESTION DID THIS STUDY ADDRESS?
Can a systems pharmacology model accurately simulate the effects of drugs that act on the fibroblast signaling network?
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
The model predicts mechanisms by 36 drugs affect fibrosis, including more detailed mechanistic insights into Entresto and arsenic trioxide.
HOW MIGHT THIS CHANGE DRUG DISCOVERY, DEVELOPMENT, AND/OR THERAPEUTICS?
This study provides a computational platform for testing new drugs against cardiac fibrosis.
INTRODUCTION
The process for bringing a drug to US Food and Drug Administration (FDA) approval is long and expensive, often taking 8–12 years, with a conservative success rate of 1 in 5000. 1 Even after approval, ~ 30% of new drugs are subject to a postmarket safety event (e.g., withdrawal, black box warning, and safety communication), and the median time for these events to occur is 4.2 years after approval. 2 Therefore, there is a need for methods to quickly and safely identify useful therapeutics early in the discovery process. In silico systems pharmacology has been recognized as one such method to identify drugs. Structural modeling, 3 , 4 , 5 , 6 metabolic network modeling, 7 , 8 and unbiased machine learning approaches that leverage large proteomic or expression datasets 9 , 10 , 11 have all been used to filter through long lists of chemicals to identify putative therapeutics with a higher likelihood of being useful against cancer or microbial infections. There is great potential for systems pharmacology approaches in development of treatments against heart failure and cardiac injury. 12 , 13 , 14 Further, mechanistic models of cardiac signaling can make detailed predictions about how and why specific drugs are effective, thereby facilitating rational drug development. 15
Drugs to prevent or treat fibrosis are needed, as fibrosis is currently considered an end point of organ damage due to a lack of treatment options. 6 , 16 Cardiac fibrosis—an increase in collagen and cross‐linking—is associated with diastolic dysfunction, which can improve if fibrosis is limited. 17 , 18 , 19 Myocardial infarction (MI) is an inciting event that can lead to cardiac fibrosis, and pathologic fibrosis post‐MI is associated with worsening heart failure. 20 , 21 , 22 Given the importance of cardiac fibroblasts in post‐MI remodeling, there are surprisingly no drugs on the market that specifically target cardiac fibroblasts’ post‐MI behavior related to degradation and deposition of collagen. Current post‐MI treatments include administration of drugs, such as ACE inhibitors, 23 , 24 angiotensin receptor blockers, and β‐blockers, 25 , 26 whose impacts on collagen are still unclear.
In this study, we used a systems biology approach to screen for drugs effective against heart failure in the post‐MI environment. We build on our previous literature‐validated model of cardiac fibroblast signaling, 27 which has predicted network mechanisms underlying fibroblast phenotype in vitro and during post‐MI wound healing. 28 In this study, we create a framework for integrating this fibroblast network model with the drug‐target DrugBank database 19 to perform an in silico screen for drugs that are profibrotic or antifibrotic. We validate the predicted drug effects on fibrosis with in vivo studies from the literature, and we further validate the predicted effect of Galunisertib on fibroblast phenotype in vitro. Finally, we use the model to predict pathways by which arsenic trioxide (ATO) is profibrotic and Entresto is antifibrotic.
METHODS
Cardiac fibroblast signaling model
Network model formulation and simulation
We used a previously published computational model of cardiac fibroblast signaling. 27 , 28 Node activity (y) is simulated by logic‐based differential equations 29 with Hill formalism and default parameters for the weight (w), Hill coefficient (n), and half‐maximal effective concentration (EC50). Each node has y init of 0, y max of 1, and time constant, τ, that is scaled based on the type of reaction: 6 min for signaling, 1 h for transcription, and 10 h for translation reactions. Using this methodology, the equation governing the activity of TGFβ mRNA due to activation by the transcription factor AP1 (AP1 → TGFβ mRNA) would be modeled by Equation 1 where f(AP1) is the activity of AP1, calculated using the normalized Hill equation, , where “β” and “K” are parameters used to constrain the function such that f(0) = 0, f(EC50) = 0.5 and f(1) = 1, “x” is the normalized activity of AP1, “w” is the weight of the appropriate reaction, and “n” is the Hill coefficient, which is set to 1.4 in our model. Multiple nodes in the model are combined in reactions using “AND” and “OR” logic gates. Overall, this network model is composed of 106 nodes, with 9 paracrine inputs and 1 mechanical stretch input. Outputs include collagen mRNA, EDAFN, α‐SMA, and MMP9 activity.
| (1) |
Static drug simulations
Baseline paracrine input weights were set to 0.25, with the exception of input mechanical stimulus, which was set to 0.85 to mimic in vitro stiffness of tissue culture plastic. We conducted static simulations by increasing the weights of appropriate inputs from 0.25 to 0.6 and running the simulations to steady‐state. The 106 ordinary differential equations are solved to steady‐state in MATLAB using ode15s.
Simulation of in vivo fibroblast dynamics after myocardial infarction
To more accurately replicate in vivo conditions post‐MI, we incorporated time‐varying input curves for the nine paracrine inputs in the model using post‐MI data in the literature. The development of these idealized paracrine signaling curves was described previously. 28 Briefly, we first used post‐MI in vivo data from rats to inform idealized, time‐dependent levels of each paracrine input to the model. Specifically, experimental time course data were used to define individual time‐dependent input curves for nine paracrine signals: IL1, IL6, tumor necrosis factor α, angiotensin II (AngII), endothelin‐1 (ET1), TGFβ, norepinephrine (NE), platelet‐derived growth factor, brain natriuretic peptide (corresponding node in the model is natriuretic peptide [NP]). To mimic the in vivo, post‐MI environment, these simulations were conducted with mechanical stimulus set to 0.6. We then used the signaling network described above to predict fibroblast phenotype over time post‐MI. These simulations were linked to a tissue‐level model of fibrosis development that predicts the percent collagen area fraction over time post‐MI. 28 The tissue level model takes as inputs the collagen 1 mRNA and time‐dependent functions for fibroblast proliferation and MMP activity. Simulations with the dynamic paracrine inputs accurately predicted collagen expression dynamics and fibrosis development, as shown in our previous publication. 28 Post‐MI time course of this model is shown in Figure S5.
Drug simulations
Integration with DrugBank and identification of drug information
The simulated drugs were identified from DrugBank version 5.0.3, a web database that lists pharmacokinetic information on over 8000 FDA approved, small molecule, biotech, experimental, nutraceutical, withdrawn, and investigational drugs. 30 , 31 , 32 DrugBank data were integrated with the fibroblast model by identifying drugs from DrugBank that targeted nodes in the model, and then using a Python script to parse DrugBank to extract information about drug agonism (agonist or antagonist). Drug binding properties (competitive or noncompetitive) were manually curated from PubMed (Figure 1). In total, we identified 121 drugs with targets in our model, yielding 132 total and 36 unique drug‐target interactions (Table S1), where a unique interaction comprises of a unique drug, binding, agonism, and target(s) relationship. For example, one unique interaction set includes 25 drugs that are all competitive BAR antagonists, whereas another unique interaction set is for Amiodarone, a noncompetitive BAR antagonist.
FIGURE 1.
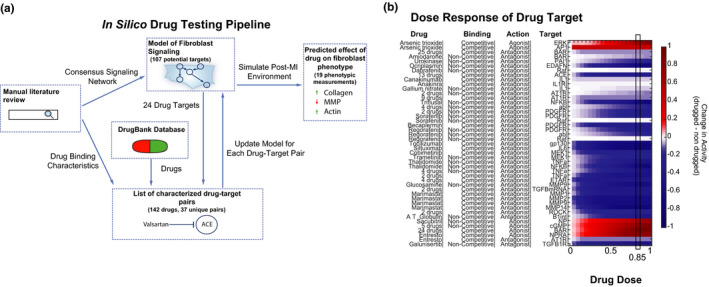
Overview of in silico drug testing method. (a) Schematic of drug testing pipeline showing the use of both the fibroblast signaling network model and DrugBank repository to systematically test drug effects on cardiac fibroblasts. (b) A range of drug doses was simulated with 85% activity highlighted as having a strong effect on the target for nearly all drugs. Note that the heatmap rows have been expanded to only include one target on each row
Model of drug action based on binding mechanisms
Competitive‐binding drugs were simulated by shifting the activity of nodes upstream of the target by the drug dose, “d.” Note the change in shifting direction depends on whether the drug target is part of an activation or inhibition reaction. As observed biologically, the competitive simulations do not change the maximum activation of the reaction, regardless of drug dose. Noncompetitive drugs were simulated by altering the activity of the drug target directly. As seen biologically, the noncompetitive simulations do not change the EC50 of the drugged reaction, regardless of drug dose. Unless specified otherwise, drugs were simulated at 85% of saturating dose. Equations summarizing how these drugs were simulated are shown in Table S2.
Replicating galunisertib experiments in rat cardiac fibroblasts
Control simulations were run with baseline paracrine input weights set to 0.25 and input mechanical stimulus set to 0.85 to mimic in vitro stiffness. The “+TGFβ” simulations were run by increasing the TGFβ input weight from 0.25 to 0.6 and running the simulations to steady‐state.
Literature validation of simulated drug effects
The following four search terms were used to find post‐MI, in vivo cardiac fibrosis validation papers: “DRUG NAME” + (1) “cardiac fibrosis post myocardial infarction,” (2) “cardiac collagen post myocardial infarction,” (3) “cardiac fibrosis post heart attack,” and (4) “cardiac collagen post heart attack.”
Determining mechanistic maps
We used the network structure as well as a series of knockdown screens to determine a simple mechanistic map that shows how ATO and Entresto are predicted to affect collagen expression. Post‐MI simulations described above were performed under no drug, ATO, or Entresto conditions. For each condition, knockdown of each node was individually simulated by setting the ymax of that node to 0, and the change in collagen I mRNA, as compared with no knockdown, was recorded at the 7 day time point. A summary of these knockdown screens is shown in Figure S3. If knockout of a node under a drug condition was predicted to reverse the drug’s effect on collagen mRNA activity, the network structure was used to confirm a connection between the drug’s target and the proposed mechanistic node. The knockdown screen under the control condition was used to exclude nodes that have a universal effect on collagen mRNA that is unrelated to the mechanism of the drug.
In vitro validation with human cardiac fibroblasts
The 96‐well CellBind plates (Corning 3340) were pretreated with 20 µg/ml of Fibronectin (Millipore Sigma, F1141) for 1 h, then rinsed and dried. HCFs (PromoCell, C‐12375) were seeded at a density of 5000 cells per well. After 24 h of plating, the fibroblasts were serum starved for 24 h. Fibroblasts were then treated with DMSO (Life Technologies, D12345), 20 ng/ml TGFβ (Cell Signaling Technology, 8915LC), 5 µM Galunisertib (SelleckChem, LY2157299), or TGFβ + Galunisertib. After 4 days, cells were fixed and stained with collagen primary antibody (Abcam, ab34710), α‐SMA preconjugated antibody (Millipore Sigma, C6198), Phalloidin (Santa Cruz Biotechnology, sc‐363797), and DAPI.
Plates were imaged using an Operetta CLS High‐Content Analysis System. These images were then processed using CellProfiler, 33 which used the DAPI signal to identify nuclei and the propagate algorithm to define cell borders. Collagen (or actin or αSMA) intensity per cell was background subtracted. The mean measurement of each well was normalized for comparison across plates such that the mean of the medians of negative control wells (DMSO) on each plate = 0, and mean of the medians of positive control wells (TGF‐β) on each plate = 1.
Statistics
Statistical significance between control and all conditions or drug versus no drug was determined by t‐test with Benjamini‐Hochberg correction for multiple comparisons. Calculations were performed using statsmodels version 0.11.1. 34
RESULTS
Virtual drug screen
We sought to develop a method by which publicly available drug datasets could be used in combination with a mechanistic fibroblast signaling model to screen for drugs that can affect cardiac remodeling (Figure 1a). The previously published fibroblast signaling network 27 , 28 incorporates 10 signaling pathways involved in cardiac wound healing, and the nodes of this network (representing gene products) were used as potential targets. Using the DrugBank database version 5.0.3, we identified 121 drugs that have targets within the fibroblast signaling network (Figure S1). Additionally, we added the combination drug Entresto (sacubitril/valsartan) because both component drugs target the fibroblast signaling network and the drug has recently shown promise in the management of heart failure. The identified drugs targeted all of the major pathways in the fibroblast signaling network (Figure S2). After using published literature to identify the binding characteristics of each drug, we narrowed to 36 unique drug‐target interactions to model in a screen (see Methods and Figure S3).
We initially modeled different doses of each drug and predicted concentration‐response relationship for target activity. A normalized drug dose of 85% was used in all subsequent screens as this resulted in a strong but incomplete action on the targets for all drugs.
Virtual screen for modulation of fibroblast activity in a modeled in vitro context
Static application of individual paracrine stimuli or pairs of paracrine stimuli as would be seen in an in vitro experiment was modeled, and administration of each drug category was simulated. These paracrine single or paired inputs were chosen based on our prior study showing that these paracrine stimuli are representative of different phases of infarct healing. 28 The effect of each unique drug type on the target protein(s) was consistent with the known activity of the drug (agonist vs. antagonist), but the magnitude of the effect was dependent on the paracrine stimulus (Figure S4). As shown in Figure 2 and Figure S6, the effect of each drug on extracellular matrix proteins is also dependent on the signaling context. For example, many drug categories have the strongest effect in the context of a TGFβ + ET1 stimulus. The majority of the drug categories modeled decrease fibrosis‐associated proteins (collagen, EDAFN, αSMA) in the signaling contexts shown. The effect of these drugs on MMPs is highly context‐specific and often unique to the specific MMP. For example, Entresto upregulates MMP1 in the context of TGFβ or TGFβ + ET1 signaling, but it downregulates MMP1 in the context of AngII or AngII + NE signaling. This highlights the context‐specific regulation of MMP1. Further, Entresto negatively regulates MMP2 in the context of TGFβ + ET1 signaling, but it upregulates both MMP1 and MMP9 indicating a specific action toward an antifibrotic MMP profile. This is supported by an Entresto‐driven decrease in collagen expression predicted in that signaling context.
FIGURE 2.
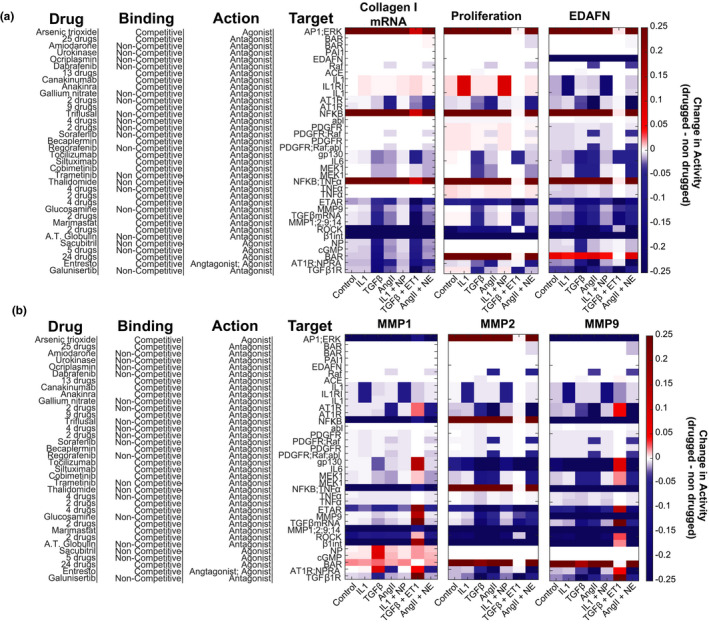
In silico drug screen with static conditions mimicking an in vitro experiment. The predicted change in profibrotic phenotypic outputs (a) or MMPs (b) for each drug is shown under simulated conditions of different sustained paracrine inputs. White on the color scale refers to without drug treatment at the specified condition
We experimentally validated the predicted effect of the ALK5 inhibitor Galunisertib. Our model predicted Galunisertib would decrease collagen expression in both control and TGFβ‐treated conditions, and we validated this prediction in cultured human cardiac fibroblasts, as shown in Figure 3. Additionally, cytoskeletal proteins were downregulated by Galunisertib in all conditions, which is in agreement with model predictions (Figure S3).
FIGURE 3.
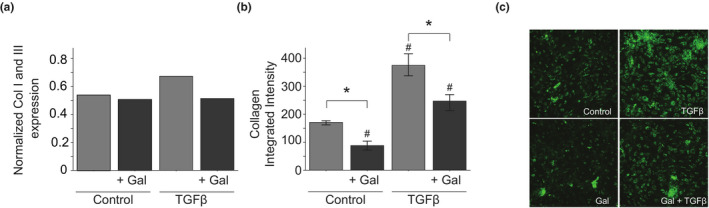
Experimental validation of predicted effect of galunisertib on collagen expression by cardiac fibroblasts. (a) Predicted collagen expression in control or TGFβ stimulated network model with and without galunisertib simulation. (b) Collagen production by cultured human cardiac fibroblasts under control or TGFβ‐treated conditions with and without galunisertib with representative images (c). N = 9 wells, error bar indicates SEM, * p < 0.05 comparing no drug vs. drug, # p < 0.05 comparing to control
Virtual screen for modulation of fibroblast activity during post‐MI wound healing
We next modeled the effects of these drugs within a dynamic post‐MI signaling environment, which was previously described. 28 For collagen, proliferation, and EDAFN, the predicted effect of each drug at the different phases of infarct healing (Figure 4a) was similar to the predicted effect shown in the static paracrine contexts that are representative of those phases (Figure 2). The strongest effect of most drugs is predicted to occur at day 7, when collagen expression is highest. This highlights the phase‐specificity of drugs, such as Glucosamine or Sacubitril. However, in the dynamic post‐MI model, drugs, such as TGFB mRNA inhibitors that upregulated some MMPs in some contexts, are now primarily downregulating all MMPs. More broadly the in vitro, static simulations had more upregulation events compared with the in vivo, dynamic simulations. For example, Triflusal, an NF‐κB antagonist, upregulates collagen in all in vitro contexts, but not at any post‐MI time point in the in vivo simulation. Another such example to note is the effect of IL1 and IL1R1 inhibition on proliferation. Inhibition of these nodes is predicted to increase proliferation in vitro, but decrease it in vivo. This behavior could perhaps be explained by an absence of additional cytokine inputs that would better fully mimic the in vivo environment. Additionally, we used the dynamic model to predict percent collagen area fraction post‐MI. We found that ATO has a strong profibrotic effect and is predicted to dramatically increase the collagen area fraction in a short time post‐MI. In contrast, Entresto is predicted to decrease the collagen area fraction post‐MI. Post‐MI time courses are shown for untreated, ATO, and Entresto treatments in Figure S5.
FIGURE 4.
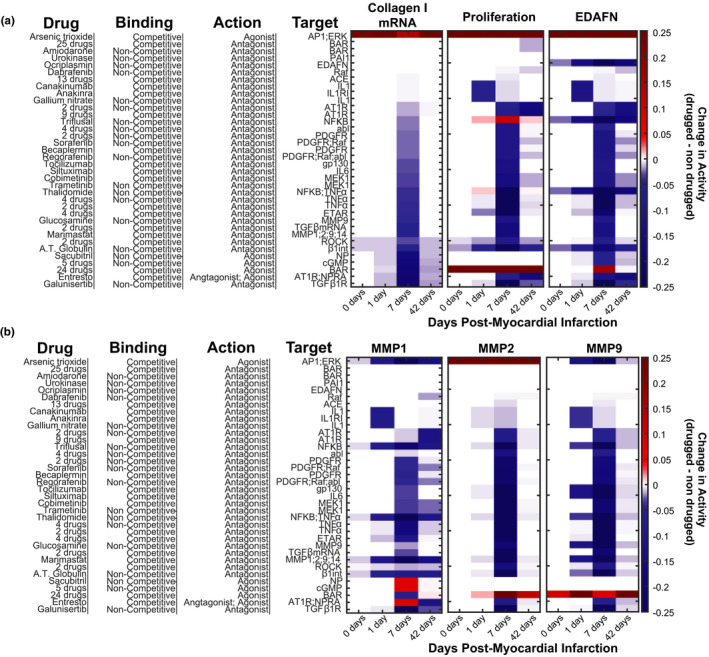
In silico drug screen with dynamic paracrine signaling mimicking the post‐myocardial infarction (MI) environment. The predicted change in profibrotic phenotypic outputs (a) or MMPs (b) for each drug is shown for specific days post‐MI representative of the different phases of wound healing (inflammatory = 1 day, reparative = 7 day, and mature = 42 day). White on the color scale refers to without drug treatment at the specified timepoint
We validated many of the predictions from our in silico screen against previously published in vivo post‐MI data (Table 1), and found that our predictions aligned with experimental data for 10 out of the 14 drug‐target interactions that have generated consistent results in the literature. Further, the model predicted antifibrotic roles for BAR antagonists and ETAR antagonists, which is consistent with some but not all of the literature.
TABLE 1.
Validation of model predictions against in vivo literature data
| Drug category | Predicted | Measured | PMID |
|---|---|---|---|
| Arsenic trioxide: competitive AP1, ERK agonist | ↑ | ↑ | 22853924 |
| 25 drugs: competitive BAR antagonist | − | − / ↓ | 10898446 |
| Amiodarone: noncompetitive BAR antagonist | − | − | 27652141 |
| Urokinase: noncompetitive PAI1 antagonist | − | ↑ | 15297377, 20380835 |
| 13 drugs: competitive ACE antagonist | ↓ | ↓ | 11851355, 9593063, 10993857, 9330127, 10898446 |
| 9 drugs: competitive AT1R antagonist | ↓ | ↓ | 25823960, 22128836, 28656296, 14516412, 9349385, 28656296, 23429590, 23727946 |
| 2 drugs: noncompetitive PDGFR antagonist | ↓ | ↓ | 17161265 |
| Sorafenib: noncompetitive PDGFR, Raf antagonist | ↓ | − | 24718482 |
| Cobimetinib: competitive MEK1 antagonist | ↓ | ↓ | 27936014 |
| Thalidomide: noncompetitive NF‐κB, TNFa antagonist | ↓ | ↓ | 16549389 |
| 2 drugs; competitive TNFa antagonist | ↓ | ↓ | 15949474 |
| 4 drugs: competitive ETAR antagonist | ↓ | ↓ / ↑ | 12738614, 12061394 / 11179039 |
| Marimastat: competitive MMP1, MMP2, MMP9, MMP14 antagonist | ↓ | ↑ | 12658202 |
| 24 drugs: competitive BAR agonist | ↓ | ↑ | 31615408, 2527639, 28549109 |
| Entresto: competitive AT1R antagonist, competitive NPRA agonist | ↓ | ↓ | 25362207 |
| Isosorbide Dinitrate: NPRA agonist | ↓ | ↓ | 28810603 |
Abbreviation: PDGFR, platelet‐derived growth factor receptor.
Mechanistic and context‐dependent effects of specific drugs
We sought to predict the mechanisms by which two drugs of interest, ATO and Entresto, affect fibroblast phenotype post‐MI. Therefore, we performed simulated knockdown screens in the post‐MI context with both drugs (Figure S6). We further combined this information with the network topology of the manually curated fibroblast signaling network topology to identify mechanisms by which they affect fibroblast phenotype.
Cardiac fibrosis is a known side effect of ATO, which is used clinically as a chemotherapeutic. In model simulations, ATO enhanced collagen, AP1, TGFβ, and MMP2 activity. As shown in Figure 5, the fibrosis induced by ATO could be partially blocked by either knockdown of TGFβ or by knockdown of MMP2. TGFβ mRNA and MMP2 both affected TGFβ expression and downstream collagen expression. Therefore, the profibrotic effect of ATO is likely due to a feed‐forward circuit in which ATO stimulates two separate mechanisms that converge to enhance TGFβ activity and collagen expression. Additionally, cotreatment of ATO with Marimastat (a pan‐MMP inhibitor) is predicted to partially rescue the fibroblast phenotype and block the increase in collagen expression.
FIGURE 5.
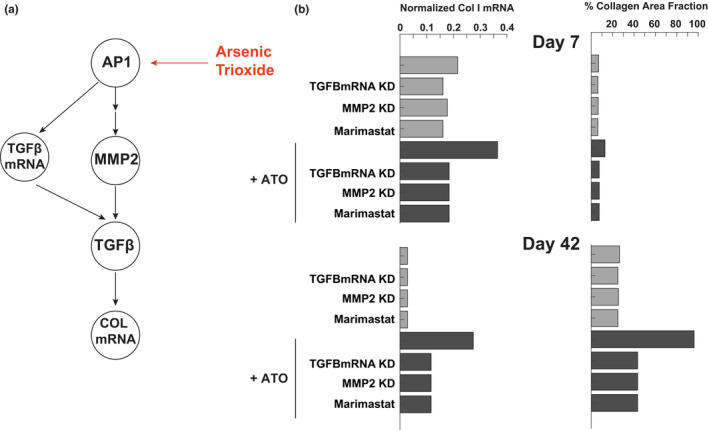
Mechanism of profibrotic effect of arsenic trioxide (ATO). (a) Mechanistic map of major effectors of ATO in the fibroblast signaling network. (b) Predicted effect of knockdown of effectors of ATO activity on collagen mRNA and collagen area fraction at day 7 and day 42 of post‐myocardial infarction simulation
The combination drug valsartan/sacubitril (commercially known as Entresto) is currently used to improve cardiac function in patients with heart failure. The model predicts that the valsartan component reduces ERK activity, which is an indirect activator of smad3, whereas sacubitril activates PKG, which inhibits smad3 activity (Figure 6a). As shown in Figure 6, these two mechanisms work together to reduce collagen expression. The antifibrotic effects of valsartan/sacubitril were predicted to be partly reversed by either ERK overexpression or PKG knockdown. In contrast, the effect of valsartan/sacubitril on collagen expression could be fully mimicked by smad3 knockdown, indicating it as a key integration point for the ERK and PKG effects.
FIGURE 6.
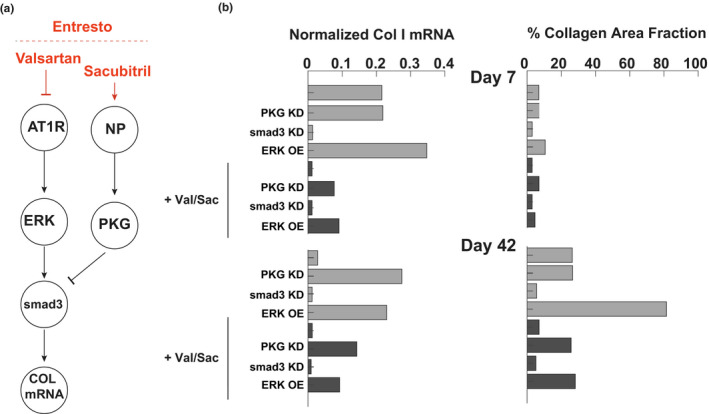
Mechanism of antifibrotic effect of valsartan/sacubitril (Entresto). (a) Mechanistic map of major effectors of val/sac in the fibroblast signaling network. (b) Predicted effect of knockdown of effectors of val/sac activity on collagen mRNA and collagen area fraction at day 7 and day 42 of post‐myocardial infarction simulation
DISCUSSION
Fibroblasts are highly plastic cells that respond to injury, but overactive fibroblast activity can result in fibrosis. Although there are medications that have an antifibrotic effect clinically (e.g., RAAS inhibitors), some patients will still develop fibrosis on these medications, indicating a need for more therapeutic options. 23 Therefore, in this study, we developed a method for screening drugs that could increase the risk of fibrosis or decrease post‐MI fibrosis with a mechanistic model of cardiac fibroblast signaling. Using the database from DrugBank, we identified 36 unique drug‐target interactions. The computational model of fibroblast signaling was used to predict each drug sets’ effect on fibroblast phenotype in paracrine signaling contexts representative of in vitro treatment or post‐MI wound healing. We validated our predictions against published literature, and found that our predictions aligned with the bulk of experimental data. Our screen correctly identified a known profibrotic drug, ATO, and predicted 35 putative antifibrotic drug sets, including combination valsartan‐sacubitril (Entresto). We used the network structure and knockout screens to map the network mechanism by which ATO and Entresto affect collagen expression.
Mechanisms of profibrotic and antifibrotic drugs
TGFβ is known to be a strong profibrotic regulator of fibrosis, but it is not likely to be a good pharmacologic target due to its critical role in regulating integral cellular processes. 35 , 36 Activation of the TGFβ pathway is a strong self‐inducing fibrotic stimulus. ATO is one of many cancer drugs with cardiotoxicity—namely QT prolongation, which is associated with increased fibrosis. 37 , 38 Our model predicts ATO activates the TGFβ pathway via AP1, which is a hub of fibroblast signaling 27 and increases both expression and activation of TGFβ, that induces a strong positive feedback loop. ATO has been shown to upregulate TGFβ and MMPs in cardiac fibroblasts, and it increases cardiac fibrosis in vivo in guinea pigs and in humans. 38 , 39 The results of our model prediction here raise the possibility that an MMP inhibitor, such as marimastat or glucosamine, could protect the heart from ATO‐induced fibrosis. In fact, MMP inhibition has been shown to decrease TAC‐induced fibrosis in mice. 40 , 41
Entresto (sacubitril‐valsartan) has an indirect effect on the TGFB pathway via both ERK inhibition and PKG activation, which decreases smad3 complex activation without fully eliminating cardiac fibrosis. Simulated knockdown screens and the structure of the signaling network indicate the NP pathway is the major effector of this drug, as ERK overexpression was predicted to only partially rescue collagen expression. Entresto has been shown to decrease TAC‐induced cardiac fibrosis in rats, 42 , 43 and it has been shown to decrease profibrotic biomarkers in patients with heart failure. 44
These results cannot necessarily be generalized to modulation of fibrosis in any organ. For example, ATO has been shown to have an antifibrotic effect in lung fibroblasts and in the lungs of bleomycin‐treated mice, 45 which highlights the organ‐specificity of fibrosis and the focus of this study on cardiac fibroblasts. Further studies that specifically characterize fibroblasts in other organs could use the same method described here to screen for drugs likely to result in damage to that organ.
Developing a method for mechanistic in silico drug screening
Due to the expense and time necessary to bring a new therapeutic into clinical use, there is a need for in silico or ex vivo screening processes that can filter putative targets earlier in drug development. 1 , 46 , 47 Fibrosis in any organ is an indicator of end‐stage organ failure due to the lack of useful therapeutic options. There have been efforts to perform broad searches for organ‐specific anti‐fibrotic therapies. For example, FIBROTARGETS is a consortium focused on rational design of antifibrotic drugs based on clinical biomarkers and high‐throughput in vitro screening supplemented with protein structural modeling. 6 , 16 Although that study is a promising approach to find antifibrotic drugs, our method offers a tool for in silico screening against well‐studied biological signaling networks. This allows for mechanistic predictions of how a drug affects fibroblast phenotype that can be easily translated into experimental validation. Indeed, we identified 30 putative antifibrotic therapies among our list of 121 of drugs in DrugBank’s dataset of over 8000 drugs, all of which could be tested experimentally and, if successful, repurposed into clinical use post‐MI at a much lower cost than testing the entire list.
One benefit to our approach is that, due to the use of default parameters, this model can incorporate data as it becomes available, such as more detailed signaling connections, half‐maximal inhibitory concentration (IC50) for all drugs or expression levels of each gene modeling in the signaling network. In this way, the model can be continuously improved for better predictions or adapted to specific testing beyond the broad screen we showed here (e.g., creating models based on patient expression data).
Another strength of this platform is its ability to mechanistically simulate drug combinations. Six of the modeled drugs exhibit polypharmacology, or have multiple targets within this network. The model predicted that Entresto had actions distinct from its components sacubitril or valsartan alone. The effect of Entresto was closer to sacubitril for suppressing collagen, and closer to valsartan for suppressing proliferation and EDAFN. More systematic simulations of drug combinations may be warranted to optimize fibrosis.
Additionally, this approach could be applied to other models or drug datasets. DrugBank has datasets of investigational or experimental drugs and academic or corporate investigators have proprietary drug datasets, all of which could be used separately or together in the method outlined here. There are large scale, logic‐based models of cardiomyocytes, 48 , 49 hepatocytes, 50 and inflammatory cells, 51 , 52 among others. 53 This pipeline could be applied to these models as a cell‐specific screen.
Limitations and future directions
The main limitation of this approach is the limited data available to incorporate in the fibroblast model. Although we outlined major, well‐studied pathways involved in cardiac fibroblast signaling in the signaling network, 27 further pathways or more specific interactions could be outlined. Additionally, data such as cardiac bioavailability and IC50 were not available for a majority of the drugs involved in the screen. Therefore, we limited our characterization of each drug to the reported binding characteristics. However, the methodologies presented here could easily incorporate such data as it becomes available.
The dynamic paracrine signaling profile that defines the post‐MI simulations described here allows for phase‐specific modeling, 28 which could predict effective timing of post‐MI therapeutics. The timing of administration of a therapeutic to target post‐MI remodeling is likely important, 54 , 55 , 56 but designing such an onerous experimental investigation could benefit from prior in silico testing. This model is uniquely suited to answer such a question. Other interesting future directions include modeling feedback with immune cells and cardiomyocytes, integration with pharmacokinetics to model dosing regimens, simulation of drug combinations, and retrospective analysis of failed clinical trials. Multiscale extensions of this model including immune cells and mechanics may be needed to predict transitions between fibrosis, transient wound healing, and cardiac rupture. It may also be possible to use network states as biomarkers of specific cell phenotypes or disease progression.
In order to apply the findings from this study to clinical practice, the putative antifibrotic drugs would need to be tested experimentally and clinically. We do not expect this model to predict drug effects with 100% accuracy, but rather intend the model to be used as a tool for narrowing from a large list of drugs to one drug that is experimentally feasible. Overall, this study provides a framework with which to integrate drug‐target interaction mechanisms with large‐scale signaling networks, which is needed both for fibrosis and other syndromes.
Funding information
This study was supported by grants from the National Institutes of Health (HL137755, HL137100, HL127944, and OD021723).
CONFLICTS OF INTEREST
The authors declared no competing interests for this work.
AUTHOR CONTRIBUTIONS
Angela C. Zeigler, Anirudha S. Chandrabhatla, and Jeffrey J. Saucerman wrote the manuscript. Angela C. Zeigler, Anirudha S. Chandrabhatla, Jeffrey W. Holmes, and Jeffrey J. Saucerman designed the research. Angela C. Zeigler, Anirudha S. Chandrabhatla, Steven L. Christiansen, and Anders R. Nelson performed the research. Angela C. Zeigler, Anirudha S. Chandrabhatla, and Anders R. Nelson analyzed the data.
Supporting information
Supplementary Material
Supplementary Material
Supplementary Material
Angela C. Zeigler and Anirudha S. Chandrabhatla equally contributed to this study.
References
- 1. Lipsky MS, Sharp LK. From idea to market: the drug approval process. J Am Board Fam Pract. 2001;14:362‐367. [PubMed] [Google Scholar]
- 2. Downing NS, Shah ND, Aminawung JA et al. Postmarket safety events among novel therapeutics approved by the US Food and Drug Administration between 2001 and 2010. JAMA. 2017;317:1854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sohraby F, Bagheri M, Aliyar M, Aryapour H. In silico drug repurposing of FDA‐approved drugs to predict new inhibitors for drug resistant T315I mutant and wild‐type BCR‐ABL1: a virtual screening and molecular dynamics study. J Mol Graph Model. 2017;74:234‐240. [DOI] [PubMed] [Google Scholar]
- 4. Lavecchia A, Cerchia C. In silico methods to address polypharmacology: current status, applications and future perspectives. Drug Discov Today. 2016;21:288‐298. [DOI] [PubMed] [Google Scholar]
- 5. March‐Vila E, Pinzi L, Sturm N et al. On the integration of in silico drug design methods for drug repurposing. Front Pharmacol. 2017;8:298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gyöngyösi M, Winkler J, Ramos I et al. Myocardial fibrosis: biomedical research from bench to bedside. Eur J Heart Fail. 2017;19:177‐191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Blais EM, Rawls KD, Dougherty BV et al. Reconciled rat and human metabolic networks for comparative toxicogenomics and biomarker predictions. Nat Commun. 2017;8:14250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Folger O, Jerby L, Frezza C, Gottlieb E, Ruppin E, Shlomi T. Predicting selective drug targets in cancer through metabolic networks. Mol Syst Biol. 2011;7:501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hu G, Agarwal P. Human disease‐drug network based on genomic expression profiles. PLoS One. 2009;4:e6536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Guney E, Menche J, Vidal M, Barábasi AL. Network‐based in silico drug efficacy screening. Nat Commun. 2016;7:10331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Vanhaelen Q, Mamoshina P, Aliper AM et al. Design of efficient computational workflows for in silico drug repurposing. Drug Discov Today. 2017;22:210‐222. [DOI] [PubMed] [Google Scholar]
- 12. Amanfu RK, Saucerman JJ. Cardiac models in drug discovery and development: a review. Crit Rev Biomed Eng. 2011;39:379‐395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zeigler AC, Richardson WJ, Holmes JW, Saucerman JJ. Computational modeling of cardiac fibroblasts and fibrosis. J Mol Cell Cardiol. 2016;93:73‐83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Shim JV, Chun B, van Hasselt JGC , Birtwistle MR, Saucerman JJ, Sobie EA. Mechanistic systems modeling to improve understanding and prediction of cardiotoxicity caused by targeted cancer therapeutics. Front Physiol. 2017;8:651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Amanfu RK, Saucerman JJ. Modeling the effects of β1‐adrenergic receptor blockers and polymorphisms on cardiac myocyte Ca2+ handling. Mol Pharmacol. 2014;86:222‐230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ferreira JP, Machu J‐L, Girerd N et al. Rationale of the FIBROTARGETS study designed to identify novel biomarkers of myocardial fibrosis. ESC Heart Fail. 2018;5:139‐148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jalil JE, Doering CW, Janicki JS, Pick R, Shroff SG, Weber KT. Fibrillar collagen and myocardial stiffness in the intact hypertrophied rat left ventricle. Circ Res. 1989;64:1041‐1050. [DOI] [PubMed] [Google Scholar]
- 18. Burlew BS, Weber KT. Cardiac fibrosis as a cause of diastolic dysfunction. Herz. 2002;27:92‐98. [DOI] [PubMed] [Google Scholar]
- 19. López B, Ravassa S, González A et al. Myocardial collagen cross‐linking is associated with heart failure hospitalization in patients with hypertensive heart failure. J Am Coll Cardiol. 2016;67:251‐260. [DOI] [PubMed] [Google Scholar]
- 20. Conrad CH, Brooks WW, Hayes JA et al. Myocardial fibrosis and stiffness with hypertrophy and heart failure in the spontaneously hypertensive rat. Circulation. 1995;91:161‐170. [DOI] [PubMed] [Google Scholar]
- 21. Bulluck H, Rosmini S, Abdel‐Gadir A, et al. Automated extracellular volume fraction mapping provides insights into the pathophysiology of left ventricular remodeling post‐reperfused ST‐elevation myocardial infarction. J Am Heart Assoc Cardiovasc Cerebrovasc Dis. 2016;5:e003555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Beltrami CA, Finato N, Rocci M et al. Structural basis of end‐stage failure in ischemic cardiomyopathy in humans. Circulation. 1994;89:151‐163. [DOI] [PubMed] [Google Scholar]
- 23. Fang L, Murphy AJ, Dart AM. A clinical perspective of anti‐fibrotic therapies for cardiovascular disease. Front Pharmacol. 2017;8:186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Park S, Nguyen NB, Pezhouman A, Ardehali R. Cardiac fibrosis: potential therapeutic targets. Transl Res J Lab Clin Med. 2019;209:121‐137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Roffi M, Patrono C, Collet JP et al. 2015 ESC Guidelines for the management of acute coronary syndromes in patients presenting without persistent ST‐segment elevation: Task Force for the Management of Acute Coronary Syndromes in Patients Presenting without Persistent ST‐Segment Elevation of the European Society of Cardiology (ESC). Eur Heart J. 2016;37:267‐315. [DOI] [PubMed] [Google Scholar]
- 26. Ibanez B, James S, Agewall S et al. 2017 ESC Guidelines for the management of acute myocardial infarction in patients presenting with ST‐segment elevation: The Task Force for the management of acute myocardial infarction in patients presenting with ST‐segment elevation of the European Society of Cardiology (ESC). Eur Heart J. 2018;39:119‐177. [DOI] [PubMed] [Google Scholar]
- 27. Zeigler AC, Richardson WJ, Holmes JW, Saucerman JJ. A computational model of cardiac fibroblast signaling predicts context‐dependent drivers of myofibroblast differentiation. J Mol Cell Cardiol. 2016;94:72‐81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zeigler AC, Nelson AR, Chandrabhatla AS, et al. Computational model predicts paracrine and intracellular drivers of fibroblast phenotype after myocardial infarction. Matrix Biol. 2020;91‐92:136‐151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kraeutler MJ, Soltis AR, Saucerman JJ. Modeling cardiac β‐adrenergic signaling with normalized‐Hill differential equations: comparison with a biochemical model. BMC Syst Biol. 2010;4:157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wishart DS, Knox C, Guo AC et al. DrugBank: a comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res. 2006;34:D668‐D672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wishart DS, Feunang YD, Guo AC et al. DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Res. 2018;46:D1074‐D1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Law V, Knox C, Djoumbou Y, et al. DrugBank 4.0: shedding new light on drug metabolism. Nucleic Acids Res. 2014;42:D1091‐D1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Carpenter AE, Jones TR, Lamprecht MR, et al. CellProfiler: image analysis software for identifying and quantifying cell phenotypes. Genome Biol. 2006;7:R100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Seabold S, Perktold J. “statsmodels: Econometric and statistical modeling with python.” Proceedings of the 9th Python in Science Conference. 2010. [Google Scholar]
- 35. Anderton MJ, Mellor HR, Bell A, et al. Induction of heart valve lesions by small‐molecule ALK5 inhibitors. Toxicol Pathol. 2011;39:916‐924. [DOI] [PubMed] [Google Scholar]
- 36. Herbertz S, Sawyer JS, Stauber AJ et al. Clinical development of galunisertib (LY2157299 monohydrate), a small molecule inhibitor of transforming growth factor‐beta signaling pathway. Drug Des Devel Ther. 2015;9:4479‐4499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Chang H‐M, Moudgil R, Scarabelli T, Okwuosa TM, Yeh ETH. Cardiovascular complications of cancer therapy: best practices in diagnosis, prevention, and management: part 1. J Am Coll Cardiol. 2017;70:2536‐2551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Li C, Qu X, Xu W et al. Arsenic trioxide induces cardiac fibroblast apoptosis in vitro and in vivo by up‐regulating TGF‐β1 expression. Toxicol Lett. 2013;219:223‐230. [DOI] [PubMed] [Google Scholar]
- 39. Chu W, Li C, Qu X et al. Arsenic‐induced interstitial myocardial fibrosis reveals a new insight into drug‐induced long QT syndrome. Cardiovasc Res. 2012;96:90‐98. [DOI] [PubMed] [Google Scholar]
- 40. Matsusaka H, Ide T, Matsushima S et al. Targeted deletion of matrix metalloproteinase 2 ameliorates myocardial remodeling in mice with chronic pressure overload. Hypertension. 2006;47:711‐717. [DOI] [PubMed] [Google Scholar]
- 41. Heymans S, Lupu F, Terclavers S et al. Loss or inhibition of uPA or MMP‐9 attenuates LV remodeling and dysfunction after acute pressure overload in mice. Am J Pathol. 2005;166:15‐25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lu H‐I, Tong M‐S, Chen K‐H, et al. Entresto therapy effectively protects heart and lung against transverse aortic constriction induced cardiopulmonary syndrome injury in rat. Am J Transl Res. 2018;10:2290‐2305. [PMC free article] [PubMed] [Google Scholar]
- 43. Burke RM, Lighthouse JK, Mickelsen DM, Small EM. Sacubitril/valsartan decreases cardiac fibrosis in left ventricle pressure overload by restoring PKG signaling in cardiac fibroblasts. Circ Heart Fail. 2019;12:e005565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zile MR, O’Meara E, Claggett B et al. Effects of sacubitril/valsartan on biomarkers of extracellular matrix regulation in patients with HFrEF. J Am Coll Cardiol. 2019;73:795‐806. [DOI] [PubMed] [Google Scholar]
- 45. Luo F, Zhuang Y, Sides MD, et al. Arsenic trioxide inhibits transforming growth factor‐β1‐induced fibroblast to myofibroblast differentiation in vitro and bleomycin induced lung fibrosis in vivo. Respir Res. 2014;15:51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Morrato EH, Smith MY. Integrating risk minimization planning throughout the clinical development and commercialization lifecycle: an opinion on how drug development could be improved. Ther Clin Risk Manag. 2015;11:339‐348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Rouse R, Kruhlak N, Weaver J, Burkhart K, Patel V, Strauss DG. Translating new science into the drug review process: the US FDA’s Division of Applied Regulatory Science. Ther Innov Regul Sci 2018;52:244‐255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ryall KA, Holland DO, Delaney KA, Kraeutler MJ, Parker AJ, Saucerman JJ. Network reconstruction and systems analysis of cardiac myocyte hypertrophy signaling. J Biol Chem. 2012;287:42259‐42268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Frank DU, Sutcliffe MD, Saucerman JJ. Network‐based predictions of in vivo cardiac hypertrophy. J Mol Cell Cardiol. 2018;121:180‐189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Saez‐Rodriguez J, Alexopoulos LG, Zhang M, Morris MK, Lauffenburger DA, Sorger PK. Comparing signaling networks between normal and transformed hepatocytes using discrete logical models. Cancer Res. 2011;71:5400‐5411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Saez‐Rodriguez J, Simeoni L, Lindquist JA, et al. A logical model provides insights into T cell receptor signaling. PLoS Comput Biol. 2007;3:e163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Niarakis A, Bounab Y, Grieco L et al. Computational modeling of the main signaling pathways involved in mast cell activation. Curr Top Microbiol Immunol. 2014;382:69‐93. [DOI] [PubMed] [Google Scholar]
- 53. Morris MK, Saez‐Rodriguez J, Sorger PK, Lauffenburger DA. Logic‐based models for the analysis of cell signaling networks. Biochemistry. 2010;49:3216‐3224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Garcia RA, Go KV, Villarreal FJ. Effects of timed administration of doxycycline or methylprednisolone on post‐myocardial infarction inflammation and left ventricular remodeling in the rat heart. Mol Cell Biochem. 2007;300:159‐169. [DOI] [PubMed] [Google Scholar]
- 55. Adamopoulos C, Ahmed A, Fay R, et al. Timing of eplerenone initiation and outcomes in patients with heart failure after acute myocardial infarction complicated by left ventricular systolic dysfunction: insights from the EPHESUS trial. Eur J Heart Fail. 2009;11:1099‐1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Yarbrough WM, Mukherjee R, Escobar GP, et al. Selective targeting and timing of matrix metalloproteinase inhibition in post‐myocardial infarction remodeling. Circulation. 2003;108:1753‐1759. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material
Supplementary Material
Supplementary Material