

Abstract
Despite tremendous success of chimeric antigen receptor (CAR) T cell therapy in clinical oncology, the dose‐exposure‐response relationship of CAR‐T cells in patients is poorly understood. Moreover, the key drug‐specific and system‐specific determinants leading to favorable clinical outcomes are also unknown. Here we have developed a multiscale mechanistic pharmacokinetic (PK)‐pharmacodynamic (PD) model for anti‐B‐cell maturation antigen (BCMA) CAR‐T cell therapy (bb2121) to characterize (i) in vitro target cell killing in multiple BCMA expressing tumor cell lines at varying effector to target cell ratios, (ii) preclinical in vivo tumor growth inhibition and blood CAR‐T cell expansion in xenograft mice, and (iii) clinical PK and PD biomarkers in patients with multiple myeloma. Our translational PK‐PD relationship was able to effectively describe the commonly observed multiphasic CAR‐T cell PK profile in the clinic, consisting of the rapid distribution, expansion, contraction, and persistent phases, and accounted for the categorical individual responses in multiple myeloma to effectively calculate progression‐free survival rates. Preclinical and clinical data analysis revealed comparable parameter estimates pertaining to CAR‐T cell functionality and suggested that patient baseline tumor burden could be more sensitive than dose levels toward overall extent of exposure after CAR‐T cell infusion. Virtual patient simulations also suggested a very steep dose‐exposure‐response relationship with CAR‐T cell therapy and indicated the presence of a “threshold” dose, beyond which a flat dose‐response curve could be observed. Our simulations were concordant with multiple clinical observations discussed in this article. Moving forward, this framework could be leveraged a priori to explore multiple infusions and support the preclinical/clinical development of future CAR‐T cell therapies.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
Although a promising platform in immune‐oncology, the drug‐specific and system‐specific determinants governing the observed dose‐exposure‐response relationship of chimeric antigen receptor (CAR)‐T cell therapy in the clinic are poorly understood.
WHAT QUESTION DID THIS STUDY ADDRESS?
Using a multiscale translational pharmacokinetic (PK)‐pharmacodynamic modeling approach, this study integrates key drug‐specific and system‐specific parameters responsible for CAR‐T cell functionality in preclinical (in vitro and in vivo) and clinical settings. These analyses also provide mechanistic insights toward the observed multiphasic CAR‐T cell PK profile as well as the generally observed steep dose‐exposure‐response relationship in the clinic.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
This analysis suggests the presence of a “threshold” CAR‐T dose, beyond which a flat dose‐exposure‐response curve could be observed. In addition, baseline patient tumor burden could be more sensitive than administered dose levels toward the overall extent of CAR‐T cell exposure.
HOW MIGHT THIS CHANGE DRUG DISCOVERY, DEVELOPMENT, AND/OR THERAPEUTICS?
The multiscale modeling framework could be applied in forward translation to support CAR‐T cell dose selection (starting, escalation, and expansion cohorts), simulating multiple infusion regimens and reverse translation to inform optimal drug‐specific characteristics.
INTRODUCTION
Chimeric antigen receptor (CAR)–engineered T cells have recently demonstrated unprecedented efficacy in multiple haematological malignancies and have garnered tremendous recognition in the field of cancer immunotherapy. 1 A typical CAR construct is composed of extracellular single‐chain variable fragment of antibody, a transmembrane domain, and intracellular signalling domain containing CD3ζ linked with 0–2 costimulatory domains. 2 Upon CAR engagement with the tumor‐associated antigen (TAA), a nonclassical immune synapse is formed, which eventually leads to antitumoral effects by releasing perforins and granzymes. With the regulatory approvals of Yescarta (axicabtagene ciloleucel), Kymriah (tisagenlecleucel), and Tecartus (brexucabtagene autoleucel), 3 the clinical landscape of CAR‐T cells is burgeoning with ≥ 500 CAR candidates currently in clinical development. 4 However, the pharmacokinetic (PK)‐pharmacodynamic (PD) characterization of these constructs present many challenges and unique opportunities because of the self‐proliferating and long‐term persistence capabilities in vivo. 5 , 6
CAR‐T cells exhibit a very unique clinical PK profile, which is often discerned by the rapid distribution, expansion, contraction, and persistence phases. 7 , 8 Although mathematical models have been developed recently 9 , 10 to empirically describe the slopes associated with multiphasic PK profiles for CAR‐T cells, they have limited capability toward extrapolation to predict the PK and PD behavior of alternative CAR constructs and dose levels. The cellular kinetic behavior of cell therapies is dependent on several (i) drug‐specific attributes, such as CAR‐affinity, CAR‐density, effector cell type (αβ T cell, γδ T cell, natural killer cells), costimulatory domains (CD28, 4‐1BB); (ii) system‐specific attributes such as disease type, tumor accessibility (solid tumor or heme malignancies), and initial tumor burden; as well as (iii) product‐specific attributes such as CD4:CD8 ratios, phenotypic composition, transduction efficiency, in vitro effector doubling time, etc. Therefore, it is imperative to adopt a more mechanistic PK‐PD approach 5 , 6 while quantitatively describing CAR‐T cell functionalities.
In our prior work, 5 we presented a multiscale mechanistic modeling approach that incorporated key drug‐specific (CAR‐affinity, CAR‐density) and system‐specific (e.g., tumor burden) parameters to characterize preclinical CAR‐T cell activity. The model exhibited the capability of characterizing the distribution of CAR‐T cells to pertinent tissues and site of action, 5 where upon interaction with tumor cells, the CAR‐Target engagement drives the simultaneous killing of tumor cells and the expansion of CAR‐T cells. The developed model was able to well characterize the multiphasic CAR‐T cellular kinetic profile, and some of the model simulations suggested a very steep dose‐exposure‐response relationship.
In this article, a similar modeling paradigm has been extended to characterize clinical PK‐PD data sets for CAR‐T cells. A unified mechanism‐based PK‐PD model is developed to characterize the preclinical (in vitro and in vivo) 11 and clinical PK‐PD data sets for anti‐B‐cell maturation antigen (BCMA) (bb2121, Idecabtagene vicleucel 12 ) CAR‐T cells. 8 Later, model simulations were performed to assess the impact of dose and patient tumor burden on cellular kinetics and clinical responses. Virtual patient simulations were also performed to understand and characterize the dose‐response relationship for bb2121 CAR‐T cell therapy in patients with multiple myeloma. Simulations were later compared with recently reported pivotal clinical trial results for bb2121. 12 , 13 The translational PK‐PD modeling framework described in this article can be used toward forward‐translation and reverse‐translation of cell therapies in the future.
METHODS
Preclinical and clinical data sets used for PK‐PD model development
In vitro cytotoxicity experiments
A single timepoint (4‐hour) killing assay of six BCMA + cell lines was evaluated (Table 1). CAR‐T (bb2121) cells derived from three different T cell donors was cocultured with BCMA + cell lines to obtain mean cytolysis at effector to target cell (E:T) ratios ranging from 1:1 to 10:1. 11
Table 1.
The list of parameters, either fixed or estimated, that were used to build the preclinical (in vitro and in vivo) and clinical PK‐PD models for CAR‐T cells
| Parameter name | Description (units) | Estimate (mean/RSE%) | Estimate (/RSE%) | Source | |
|---|---|---|---|---|---|
| Parameters associated with the cell‐level PD model | |||||
|
|
The doubling time of tumor cells (hour) |
Daudi = 24 hours JeKo = 26 hours K562‐bcma = 47 hours NCI‐H929 = 50 hours RPMI‐8226 = 60 hours U266‐B1 = 108 hours |
— | 43 | |
|
|
The doubling time of CAR‐T cells (hour) | 24h | — | 11 | |
|
|
Overall density of TAA on different tumor cell lines (numbers/cell) |
JeKo = 222/cell Daudi = 1173/cell U266‐B1 = 2930/cell NCI‐H929 = 10,000/cell RPMI‐8226 = 12,590/cell K562‐BCMA = 76,942/cell |
— | 11 | |
|
|
Overall density of CARs on CAR‐T cells (numbers/CAR‐T cell) | 15,000/cell | — | Internal data set | |
|
|
The first‐order maximum rate of killing of tumor cells by CAR‐T cells (1/hour) | 0.353 (14) | 0.62 (17) | Estimated | |
|
|
The number of “CAR‐Target complexes per tumor cell” required to achieve 50% of the maximum killing rate (number/cell) | 2.24 (2) | — | Estimated | |
|
|
The Sigmoidicity factor associated with the killing of tumor cells (unitless) | 1.07 (0.2) | — | Estimated | |
|
|
The binding affinity of CAR to TAA (1/Molar/second) | 7.1E4 (Fixed) | — | 44 | |
|
|
The dissociation rate of CAR to TAA (1/second) | 2.39E‐3 (Fixed) | — | 44 | |
|
|
The initial tumor cells (number) | 1E5 (Fixed) | — | 44 | |
| Parameters associated with the preclinical PK‐PD model | |||||
|
|
The first‐order maximum rate of CAR‐T cells expansion (1/day) | 0.9168 (8.47) | — | Estimated | |
|
|
The number of “CAR‐Target complexes per effector cell” required to achieve 50% of the maximum rate of CAR‐T cell expansion (number/cell) | 1.15 (30.9) | — | Estimated | |
|
|
The first‐order conversion rate from effector cells to memory cells (1/day) | — | — | — | |
|
|
The first‐order elimination rate of effector CAR‐T cells (1/day) | 113 (Fixed) | — | Clinical model estimates | |
|
|
The first‐order elimination rate of memory CAR‐T cells (1/day) | — | — | — | |
|
|
The first‐order maximum rate of killing of tumor cells by CAR‐T cells (1/day) | 0.612 (28.2) | — | Estimated | |
|
|
The number of “CAR‐Target complexes per tumor cell” required to achieve 50% of the maximum killing rate (number/cell) | 2.24 (Fixed) | — | In vitro estimates | |
|
|
The first‐order distribution rate from blood compartment to bone marrow compartment (1/day) | 20,304 (20.9) | 0.1 (Fixed) | Estimated | |
|
|
The first‐order redistribution rate from bone marrow compartment to blood compartment (1/day) | 0.3288 (29.5) | 0.1 (Fixed) | Estimated | |
|
|
The first‐order rate of tumor growth (1/day) | 0.0888 (10.9) | 0.1 (Fixed) | Estimated | |
|
|
The volume of blood compartment (mL) | 0.944 (Fixed) | — | 45 | |
|
|
The volume of tumor compartment (mL) | 0.151 (Fixed) | — | 11 | |
|
|
The binding affinity of CAR to TAA (1/Molar/second) | 7.1E4 (Fixed) | — | 44 | |
|
|
The dissociation rate of CAR to TAA (1/second) | 2.39E‐3 (Fixed) | — | 44 | |
|
|
Overall density of CARs on CAR‐T cells (number/CAR‐T cell) | 15,000 (Fixed) | — | Internal data set | |
|
|
Overall density of TAA on different tumor cell lines (number/cell) | 12,590 (Fixed) | — | 44 | |
|
|
The transit time parameter associated with signal transduction of killing signal (hour) | 47.4 (36.7) | — | Estimated | |
| Parameters associated with the clinical PK‐PD model | |||||
|
|
The first‐order maximum rate of CAR‐T cells expansion (1/day) | 1.73 (10) | 0.22 (38) | Estimated | |
|
|
The number of “CAR‐Target complexes per effector cell” required to achieve 50% of the maximum rate of CAR‐T cell expansion (number/cell) | 10 (18) | — | Estimated | |
|
|
The “net” first‐order conversion rate from effector cells to memory cells (1/day) | 0.00002 (66) | 0.00004 (Fixed) | Estimated | |
|
|
The first‐order elimination rate of effector CAR‐T cells (1/day) | 113 (19) | — | Estimated | |
|
|
The first‐order elimination rate of memory CAR‐T cells (1/day) | 0.219 (13) | — | Estimated | |
|
|
The first‐order distribution rate from blood compartment to bone marrow compartment (1/day) | 1.71(11) | — | Estimated | |
|
|
The first‐order redistribution rate from bone marrow compartment to blood compartment (1/day) | 0.176(14) | — | Estimated | |
|
|
The volume of blood compartment (L) | 5 (Fixed) | — | 46 | |
|
|
The volume of bone marrow compartment (L) | 3.65 (Fixed) | — | 46 | |
|
|
Overall density of CARs on CAR‐T cells (number/CAR‐T cell) | 15,000 (Fixed) | — | Internal data set | |
|
|
Overall density of TAA on different tumor cell lines (number/cell) | 12,590 (Fixed) | — | 11 | |
|
|
The binding affinity of CAR to TAA (1/M/s) | 7.1E4 (Fixed) | — | 44 | |
|
|
The dissociation rate of CAR to TAA (1/second) | 2.39E‐3 (Fixed) | — | 44 | |
| TransC | The conversion factor from CAR‐T cells to transgene copies (unitless) | 0.002 (Fixed) | — | 46 | |
|
|
The first‐order rate of tumor growth (1/day) | 0.008 (Fixed) | — | 47 | |
|
|
The first‐order maximum rate of killing of tumor cells by CAR‐T cells (1/day) | 0.343 (21) | 0.50 (37) | Estimated | |
|
|
The number of “CAR‐Target complexes per tumor cell” required to achieve 50% of the maximum killing rate (number/cell) | 2.24 (Fixed) | — | In vitro estimates | |
|
|
The degradation rate of serum M‐protein (1/day) | 0.117 (Fixed) | — | 48, 49 | |
|
|
The production rate of serum M‐protein (picogram/cell/day) | 12.1 (Fixed) | — | 48, 49 | |
|
|
The degradation rate of soluble BCMA (1/day) | 0.7 (Fixed) | — | 50 | |
|
|
The production rate of soluble BCMA (picogram/cell/day) | 0.175 (Fixed) | — | Calculated | |
|
|
The factor associated with the production of M‐protein in response to the tumor change (unitless) | 0.215 (5) | 0.21 (44) | Estimated | |
|
|
The factor associated with the production of sBCMA in response to the tumor change (unitless) | 1 (Fixed) | 1.0 (31) | Estimated | |
BCMA, B‐cell maturation antigen; CAR, chimeric antigen receptor; NSG, NOD scid gamma; PD, pharmacodynamic; PK, pharmacokinetic; RPMI, Roswell Park Memorial Institute; sBCMA, soluble BCMA; TAA, tumor‐associated antigen.
Preclinical xenograft experiments to assess efficacy and expansion of CAR‐T cells
NOD scid gamma mice subcutaneously inoculated with BCMA + RPMI‐8226 tumors were randomized (at 151 mm3 tumor volume) for treatment with vehicle control or ~ 5 million bb2121 CAR‐T cells/mouse. Tumor volumes were measured twice a week, and CD3+ CAR+ T cells were measured (using flow cytometry) to assess PK in blood. 11
Phase 1 clinical trial in patients with relapsed or refractory (r/r) multiple myeloma
In this clinical study (n = 33), 8 anti‐BCMA (bb2121) CAR‐T cells were administered as a single infusion at flat dose levels of 50, 150, 450 and 800 million CAR+ cells per patient. PK was evaluated by measuring median vector transgene copies per µg of genomic DNA. Mean longitudinal data for PD biomarkers, i.e., soluble BCMA and serum M‐protein, was reported as surrogates for antitumor activity. In addition, swimmer plots associated with individual patient categorical response data for 33 patients were also reported based on the International Myeloma Working Group (IMWG) Uniform Response Criteria for Multiple Myeloma.
Mathematical modeling
A sequential mathematical modeling analysis was conducted in which a cell‐level PD model was developed (Figure 1a ) to characterize in vitro functionality of bb2121 in BCMA + cell lines. The developed model was later integrated within an in vivo framework (Figure 1b ) to simultaneously characterize antitumor effects and CAR‐T cell expansion in preclinical and clinical settings. Detailed information on model parameters is described in Table 1 , and the model assumptions and equations are thoroughly described in the [Link] , [Link] .
FIGURE 1.
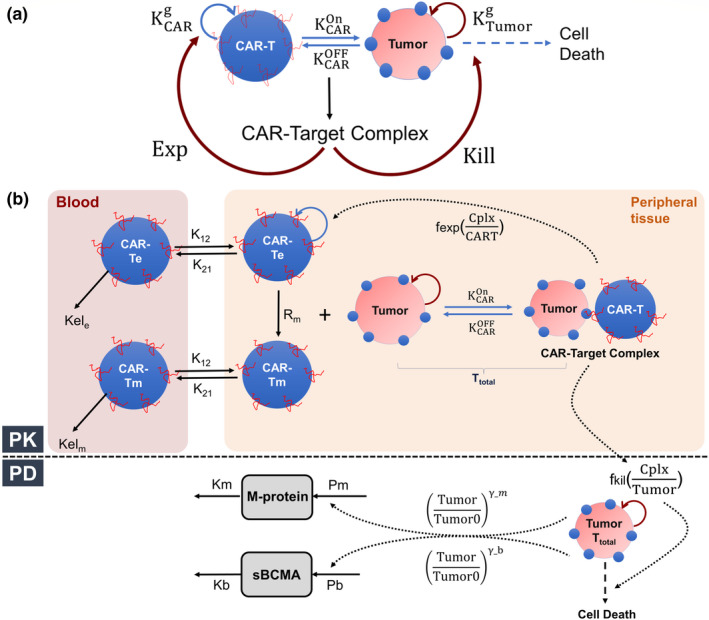
Schematics for Multiscale PK‐PD Model for CAR‐T Cell Therapy (a) A schematic diagram of a cell‐level pharmacodynamic model for chimeric antigen receptor (CAR) T cell activity: Dynamic populations of CAR‐T cells and tumor cells were assumed in an in vitro coculture system, which are proliferating with their respective first‐order growth rates. Upon binding and interaction between CAR‐T cell and tumor cell population, there is the formation of “CAR‐Target complexes,” which simultaneously mediate the killing of tumor cells and the antigen‐mediated expansion of CAR‐T cells. (b) A schematic diagram of a mechanism‐based pharmacokinetic (PK)‐pharmacodynamic (PD) model for CAR‐T cells: The PK model is compartmentalized into blood and peripheral tissues (site of action). In the blood compartment, effector CAR‐T cell (CAR‐Te) and memory CAR‐T cell (CAR‐Tm) were assumed to exhibit first‐order elimination rates (via and ) and distribution rates (via and ) to the peripheral tissues. Within the peripheral tissue (solid tumor (preclinical xenograft) and bone marrow (multiple myeloma patients)), effector CAR‐T cells differentiate into memory CAR‐T phenotypes using a “net” first‐order rate of conversion (Rm), and both cell types could redistribute to the blood compartment. At the site of action, total CAR‐T cells interact with tumor cells and form “CAR‐Target complexes (Cplx),” which drive the simultaneous expansion of effector CAR‐T cells (fexp) and the killing of the tumor cells (fkill). In the PD model, the turnover of two disease‐associated biomarkers, i.e., serum M‐protein and soluble BCMA (sBCMA), were described using a zero‐order production (from tumor) rate and first‐order degradation rate. The fractional change in the tumor burden (c.f. baseline) over time is assumed to impact the production rate of these two biomarkers in a nonlinear manner with a coefficient factor (). The model equations, assumptions and parametrization has been discussed in supplementary text. EXP, expansion.
RESULTS
Model‐based characterization of in vitro functional activity of bb2121
Figure 2a describes the simultaneous model‐fitted cytolysis percentage profiles overlaid with observed data (Figure 2a 1–2a6) from six BCMA + cell lines after coculture with bb2121 CAR‐T cells at different E:T ratios. Our previously proposed in vitro modeling framework accounts for simultaneous CAR‐T cell expansion and tumor cell depletion. However, because of the lack of an in vitro expansion data set of bb2121 CAR‐T cells within the underlying experiment, the activated bb2121 CAR‐T cell doubling time () parameter was fixed (Table 1 ) to known values. IN addition, while performing data analysis, the system‐specific parameters such as CAR‐affinity, BCMA receptor densities, and CAR densities were fixed to the known values (Table 1 ), whereas the parameters associated with the efficacy of CAR‐T cells, i.e., maximum killing rate () and killing potency () were estimated. The estimated revealed that ~ 2.24 CAR‐Target complexes per tumor cells were required to achieve 50% of the maximum killing rate (). The extent of the cytolysis percentage was not correlated with the antigen densities on six cell lines. Hence, ~ 60% interindividual variability (IIV) was estimated on the mean maximum killing rate () parameter. In the subsequent modeling analysis, the cell‐level in vitro potency estimates () were fixed while characterizing in vivo CAR‐T cell–induced tumor growth inhibition (TGI).
FIGURE 2.
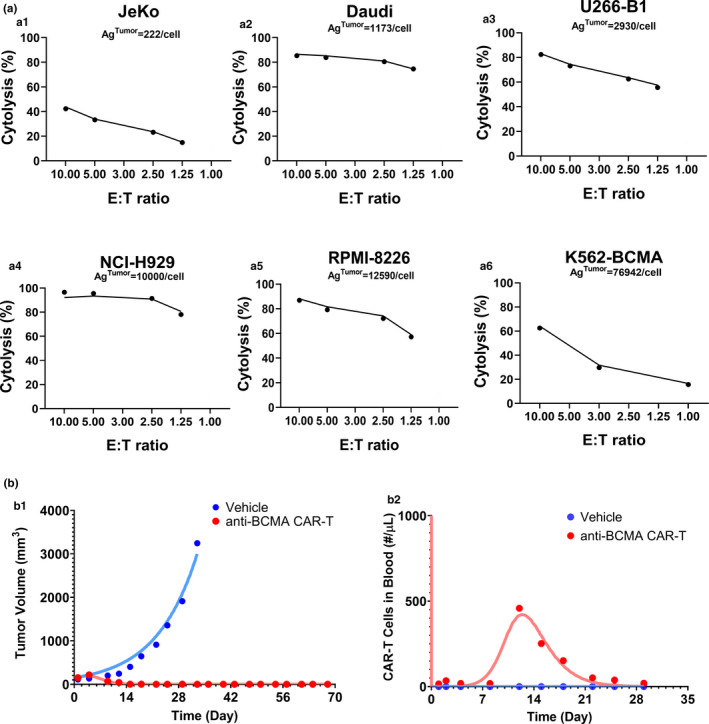
Observed and model‐fitted profiles for the preclinical data sets for anti B‐cell maturation antigen (BCMA) chimeric antigen receptor (CAR) T cells (bb2121). (a) Tumor cell killing: Observed (dots) and model‐generated (solid lines) curves of viability of BCMA‐expressing tumor cell lines with varying antigen‐densities (Ag‐tumor: 222–76,942 receptors/cell) upon incubation with CAR‐T cells with varying effector to target (E:T) ratios for 4 hours. (b) Tumor growth inhibition and CAR‐T cell expansion: Observed (dots) and model‐generated (solid lines) profiles of vehicle (blue) or 5 × 106 BB2121 CAR‐T cells (red) induced (b1) tumor growth inhibition and (b2) blood CAR‐T cell expansion in NOD scid gamma mice inoculated with RPMI‐8226 tumors
Model‐based characterization of in vivo expansion and bb2121 CAR‐T cell–induced TGI in preclinical xenograft mice
Figure 2b describes the simultaneous model‐fitted profiles overlaid with the observed data for bb2121 induced TGI (Figure 2 b1) and bb2121 CAR‐T cell expansion in blood (Figure 2 b2) in a RPMI‐8226 tumor‐bearing xenograft mouse model using the PK‐PD model described in Figure 1b . While performing the preclinical modeling analysis, the memory differentiation of CAR‐T cells at the site of action (as described in Figure 1b ) was neglected due to the lack of observed data at later timepoints. Overall, the model was able to characterize the integrated CAR‐T cell PK and TGI data set, where all of the known drug‐specific and system‐specific parameters were fixed (as listed in Table 1 ), and the parameters associated with CAR‐T cell expansion and CAR‐T cell–induced tumor killing were estimated.
The “CAR‐Target complexes per tumor cell” based potency parameter () was fixed to the value estimated from the in vitro model fitting (Figure 2a , Table 1 ). In addition, a brief delay was observed between the formation of the CAR‐Target complexes to the initiation of TGI, which was estimated as the transit time of 7.84 days. The model‐estimated CAR‐T cell expansion parameters revealed that ~ 1.15 CAR‐Target complexes per CAR‐T cells were required () to achieve 50% of the maximum expansion rate (), which was estimated to be 0.92 1/day (~18‐hour half‐life). Estimation of the distributional rate constants (, ) between the blood and tumor compartments revealed a very rapid distribution of CAR‐T cells to the peripheral tissues on systemic administration.
Model‐based characterization of in vivo expansion and bb2121 CAR‐T cell–induced TGI in patients with r/r multiple myeloma
Figure 3 describes the simultaneous model‐fitted profiles overlaid with the mean observed data (with standard deviations) for bb2121 blood PK (Figure 3a ), change in soluble BCMA levels compared with baseline (Figure 3b ), and change in serum M‐protein levels compared with baseline (Figure 3c ) at four different dose levels ranging from 50–800 million CAR‐T cells/patient. The model was able to capture the overall dose‐dependent multiphasic PK profile of bb2121, highlighted by the rapid distribution, expansion, contraction, and persistence phases.
FIGURE 3.
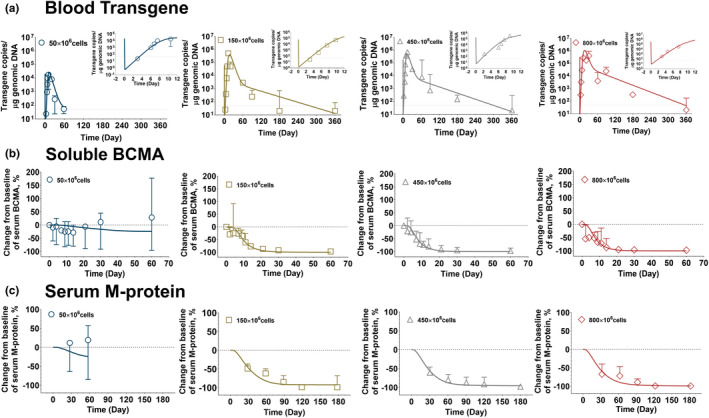
Observed and model fitted profiles for clinical pharmacokinetic (PK)‐pharmacodynamic (PD) profiles for anti‐B‐cell maturation antigen (BCMA) chimeric antigen receptor (CAR) T cell therapy (bb2121). (a) Blood transgene level: Observed mean data (symbols), standard deviation (error bars), and model‐generated (solid lines) profiles of CAR transgene copies/g genomic DNA over time. (b) soluble BCMA: Observed mean data (symbols), standard deviation (error bars), and model‐generated (solid lines) profiles of percentage of change from baseline of soluble BCMA concentration over time. (c) Serum M‐protein: Observed mean data (symbols), standard deviation (error bars), and model‐generated (solid lines) profiles of percentage of change from baseline of M‐protein concentration over time following 50 × 106, 150 × 106, 450 × 106, and 800 × 106 doses of CAR‐T infusion in patients with relapsed/refractory multiple myeloma.
It was observed that the lowest dose of 50 million CAR‐T cell resulted in lower CAR‐T peak blood concentration (Cmax) (Figure 3a ) and poor persistence in comparison to the rest of clinically investigated dose levels. 8 The insert between within each plot (Figure 3a ) shows the model characterization of the rapid expansion phase of bb2121, within the time frame of 12 days. The model estimated CAR‐T cell expansion parameters revealed that ~ 10 CAR‐Target complexes per CAR‐T cells () were required to achieve 50% of the maximum CAR‐T cell expansion rate (), which was estimated to be 1.73 1/day (~9.16‐hour half‐life). The estimated values for and were < 2‐fold and ~ 10‐fold higher than the corresponding preclinical in vivo expansion parameter estimates. The model estimation also revealed that the blood elimination rate constant for CAR‐T cells with effector phenotype () was much higher (shorter half‐life) than that for the persistent memory phenotypic () population. Global sensitivity analysis (Figure S1 ) revealed the relative sensitivities of PK parameters at different phases of blood PK, for example, the elimination parameter associated with the effector pool () was more sensitive in the expansion phase, whereas the memory differentiation parameter () and elimination parameter for memory pool () was more sensitive in the persistent phases. Estimation of the distributional rate constants (, ) between the blood and bone marrow compartments also revealed a very rapid distribution of CAR‐T cells to peripheral tissues on systemic administration in concordance with our preclinical in vivo observations.
Figure 3b,c describes the decrease in two biomarkers, i.e., soluble BCMA (sBCMA) and serum M‐protein levels, which serve as surrogates to the rate and extent of CAR‐T cell–induced tumor cell depletion (Figure 1b ). The extent of observed decrease in both biomarkers directly correlated with the observed blood PK of CAR‐T cells, where lower Cmax and poor persistence in the 50 million CAR‐T dosing cohort (Figure 3 , in blue) led to the limited decrease in mean sBCMA and serum M‐protein profiles compared with baseline levels. While characterizing the data set, the production and degradation rates of the two biomarkers were obtained from literature sources as described in Table 1 . The model estimated maximum rate constant of CAR‐T induced tumor cell depletion rate () was estimated to be 0.343 1/day (2‐day half‐life), which was < 2‐fold higher than the preclinical estimate (0.612 1/day), and the killing potency () was fixed to the in vitro estimated value. Unlike preclinical TGI studies, the direct tumor cell depletion data set was unavailable in the clinical settings and hence no delay (transit time) was assumed between the formation of “CAR‐Target complexes” and the induction of tumor cell depletion.
Impact of CAR‐T dose level and patient tumor burden on clinical PK and PD end points
Figure 4 describes contour plots where the impact of patient initial tumor burden and CAR‐T dose level was evaluated on bb2121 exposure (Cmax, time to reach maximum concentration (Tmax)) and PD biomarkers (sBCMA and serum‐M protein) end points. The model simulations suggested that the observed Cmax after CAR‐T cell expansion in blood is more sensitive to initial patient tumor burden in comparison to the CAR‐T dose level (Figure 4a ). This observation suggests a steep dose‐exposure relationship, where beyond a “threshold” dose, the overall Cmax after CAR‐T cell administration saturates. Simulations also revealed that increasing tumor burden and CAR‐T cell doses led to a saturable increase in the overall rate of expansion (lower Tmax; Figure 4b ). In the current case study, this saturation in the Tmax occurs within a range of 2–3 weeks.
FIGURE 4.
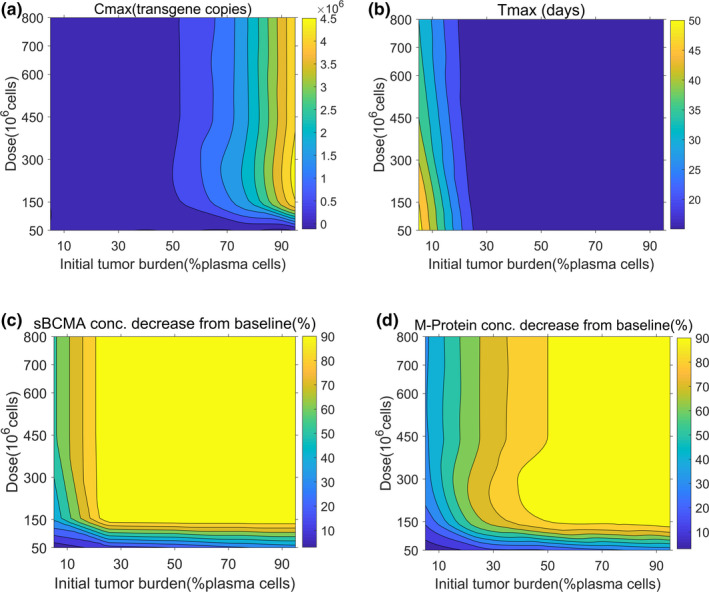
Contour plots using developed clinical pharmacokinetic‐pharmacodynamic model of chimeric antigen receptor (CAR) T cell therapy to evaluate the impact of CAR‐T dose and initial tumor burden on (a) peak blood concentration (Cmax), (b) time to reach maximum concentration (Tmax), (c) soluble BCMA (sBCMA) concentration decrease from baseline (%), and (d) serum M‐protein concentration decrease from baseline (%). Model simulations were performed after single intravenous administration of anti‐BCMA (bb2121) CAR‐T cells to patients with multiple myeloma at a dose range from 50 to 800 × 106 CAR‐T cells and a tumor burden from 5% to 95% of plasma cells.
The percentage of decline of two PD biomarkers, i.e., soluble BCMA and serum M‐protein, was also predicted to be sensitive to both dose and initial tumor burden values in a nonlinear saturable manner. At lower initial tumor burden values and lower CAR‐T dose levels (Figure 4c,d ), the decrease in sBCMA and serum M‐protein increases as a function of dose and tumor burden; however, this effect saturates after a threshold. The relative sensitivity of these PD biomarkers to initial tumor burden was also dependent on their relative degradation half‐lives (,), where in sBCMA has a faster turnover rate in comparison to serum‐M protein.
Model fittings for individual patient response data and calculation of progression‐free survival (PFS)
Figure 5 describes the model‐fitting results to the individual patient response data categorically scored based on percentage changes in serum‐M protein measurements according to the IMWG Uniform Response Criteria for Multiple Myeloma. 14 Figure 5a describes the individual model fits for representative subjects, whereas Figure S2 describes the model fittings for all 33 subjects. The error bars in Figures a1–4 5 represent the bandwidth of each categorical bin based on the response criteria. The model could simultaneously characterize individual patient responses, and the calculated PFS rates (dashed lines) were concordant with the reported values (solid lines) as shown in Figure 5b . While performing this modeling step, the IIV in the PD parameters was estimated (as shown in Table 1 ) while fixing the mean estimates for parameters from the previous step.
FIGURE 5.
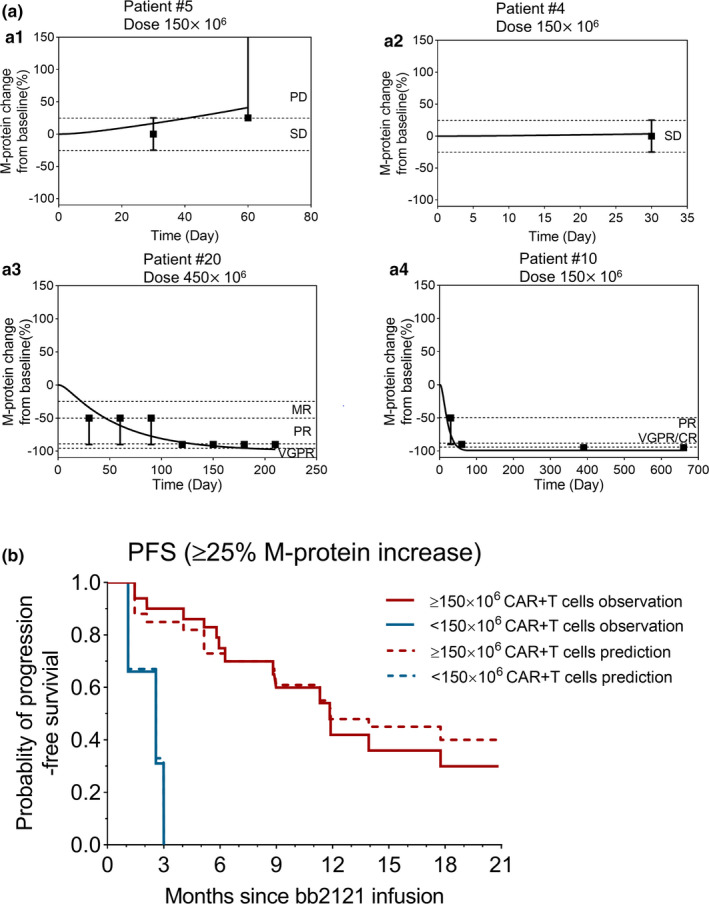
Observed and model‐fitted profiles of individual responses and progression‐free survival (PFS) in the bb2121 clinical trial. (a) Four representative patient profiles with different responses based on M‐protein change over time: a describes the observed response criteria (symbols), upper or lower bound (error bars), and model‐generated (lines) profiles of patients who developed progressive disease (PD; a1), retained stable disease (SD; a2), exhibited tumor regression to very good partial response (VGPR; a3), and had complete response (CR; a4), respectively. (b) PFS: The observed (solid lines) and model‐generated (dashed lines) profiles of PFS over time for the total 33 patients involved in the BB2121 clinical trial categorized into lower dose (<150 × 106) and higher dose (≥150 × 106) levels. MR, Minor Response; PR, Partial Response
Virtual patient simulations and calculation of patient response rates
Figure 6a describes virtual patient simulations (250/1000) for percentage of change in serum‐M protein after single‐dose administration of CAR‐T cells ranging from 50–800 million cells/patient. The [Link] , [Link] describes the list of parameters and criteria for performing these Monte Carlo simulations, whereas Figure S3 describes all 1000 profiles at each dose level. The overall intensity of simulated longitudinal change in serum‐M protein profiles in 1000 virtual patients describes the extent of variability in response. The simulations revealed that the 50 million dose level exhibited responses in fewer patients, with an overall higher probability of relapses. The subsequent dose level of 150 million exhibited a higher rate of response with fewer cases of relapse. The two higher dose levels (i.e., 450 and 800 million) exhibited a similar rate (c.f. 150 million), but marginally higher depth of response (Figure S3 ). In fact, the dose level of 450 million was determined to be most efficacious with subsequent pivotal clinical studies of idecabtagene vicleucel. 12 , 13
FIGURE 6.
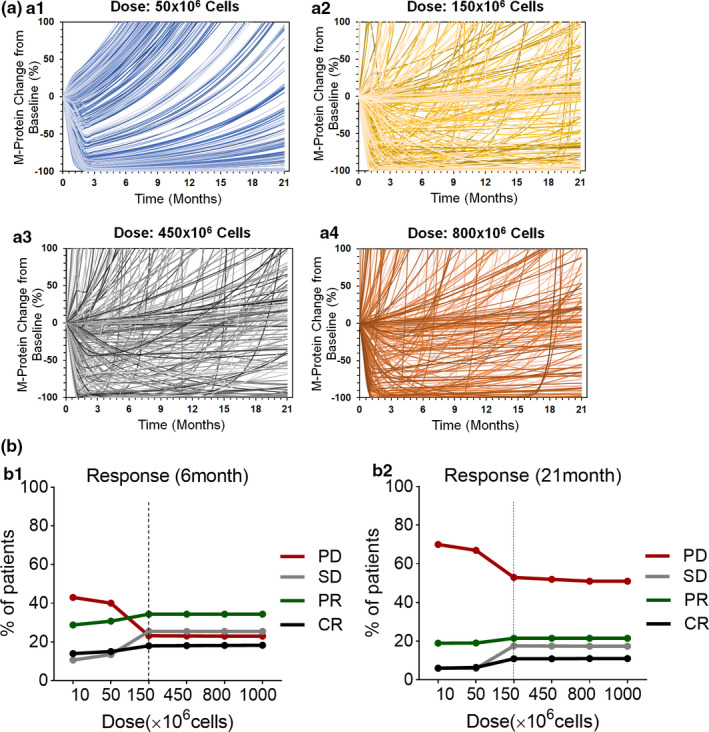
Virtual patient simulations using a developed chimeric antigen receptor (CAR) T pharmacokinetic‐pharmacodynamic model to predict the dose‐response relationship. (a) Spider plots for the first 250 (of 1000) virtual patients describing the percentage change in serum M‐protein over time at dose levels of 50 × 106 (a1), 150 × 106 (a2), 450 × 106 (a3), 800 × 106 (a4) CAR + T cells. (b) The response rates of progressive disease (PD), stable disease (SD), partial response (PR) (combined minor response, partial response, and very good partial response), and complete response (CR) at 6 months and 21 months after the administration of 10 to 1000 million CAR‐T cells.
To further elucidate the marginal differences in responses at various dose levels, categorical binning of simulated continuous profiles was performed at 6 months and 21 months post–CAR‐T infusion based on the IMWG Uniform Response Criteria for Multiple Myeloma. 14 Dose‐response curves were later derived (Figure 6b ) from Monte Carlo simulations, highlighting the percentage of subjects achieving progressive disease (PD), stable disease (SD), partial response (PR), and complete response (CR) compared with baseline at the end of 6 and 21 months. It was observed that the fraction of patients with PD increases by the end of 21 months in comparison to 6 months due to the reduction in CAR‐T exposures at later timepoints and hence relapse in the overall tumor burden. In addition, dose‐dependent (i) decrease in PD and (ii) increases in SD, PR, and CR were predicted until a threshold dose level (150 × 106, dashed vertical line), beyond which a nearly flat dose‐response curve was observed with no substantial improvement in exposure or efficacy with dose escalation.
DISCUSSION
The field of cancer immunotherapy has been revolutionized with the emergence of CAR‐T cells. This “living” therapeutic modality possesses the potential for in vivo expansion, effective tumor cell killing, and long‐term persistence. 15 Although a promising new platform in oncology, the drug‐specific and system‐specific determinants influencing the PK and PD of these agents are poorly understood. 16 Consequently, there are no established paradigms to facilitate preclinical to clinical translation of CAR‐T cell therapies and to a priori predict safe and efficacious first‐in‐human dose levels. Moreover, due to the limited clinical experience with CAR‐T cell therapy, the impact of dose escalation and baseline tumor burden on patient responses is still unclear. We believe that the utility of mechanism‐based PK‐PD models can be paramount in understanding the underlying dose‐exposure‐response relationship of CAR‐T cell therapy and facilitate the discovery and development of these agents. 5 , 6 , 9 , 17
Here we have adopted a stepwise modeling approach that uses CAR‐T cell in vitro and in vivo functional activity data sets from different drug development phases using anti‐BCMA (bb2121) CAR‐T cells 8 as a case study. The first step toward this goal was to develop a cell‐level PD model (Figure 1a ). The model simultaneously characterized the bb2121 CAR‐T cell mediated killing of BCMA‐expressing tumor cell lines (Figure 2a ). Such model‐based characterization of in vitro killing potential of CAR‐T cells can help determine the cell‐level potency parameter for CAR‐T cells (, which could later be translated toward the development of in vivo (both preclinical and clinical) mathematical relationships. 5 It is imperative that when designing such functional in vitro experiments in discovery settings, patient representative cell lines, CAR‐T cells derived from multiple donors (healthy and diseased), relevant matrices (media vs. whole blood) and adequate E:T ratios should be used to characterize cytolysis, CAR‐T cell expansion, and the release of relevant cytokines. 18 , 19
The second step involved the development of a mechanism‐based PK‐PD model to characterize in vivo TGI and CAR‐T cell expansion. A unified PK‐PD model (Figure 1b ) was used to adequately characterize both preclinical and clinical data sets for anti‐BCMA (bb2121) CAR‐T cells. Upon single infusion of ~ 5 million bb2121 CAR‐T cells in RPMI‐8226 xenograft mice, it was observed that CAR‐T cells rapidly extravasate from the blood compartment to the site of action (solid tumor) and other peripheral tissues, followed by their expansion, which is later reflected in the blood pool with a delay (Figure 2b ). When designing such in vivo xenograft studies, a range of dose levels should be explored to characterize dose dependency on TGI, blood/tumor CAR‐T cell expansion, and the release of relevant cytokines. If feasible, the impact of donor‐to‐donor variability on in vivo CAR‐T cell function should also be assessed. 11 , 20 Based on our sequential modeling analysis, there was ~ 10‐fold difference in the expansion potency () parameter estimate for bb2121 between preclinical xenograft studies and phase I clinical studies. However, mechanistic vigilance is required while directly translating this parameter estimate to predict the clinical expansion rate because it is dependent on donor‐to‐donor variability in the autologous CAR‐T cell product, CAR‐T cell fitness, and overall manufacturing process differences in the CAR‐T cell product in preclinical and clinical settings. In addition, the host environment differences between immunocompromised mice and patients with r/r multiple myeloma along with the accessibility of the tumor site among two species 21 should also be taken into consideration.
In the third step of our modeling analysis, the proposed PK‐PD model (Figure 1b ) was used to characterize the mean PK and two PD biomarkers (sBCMA, serum M‐protein) data sets in patients with r/r multiple myeloma (Figure 3 ). The model was able to effectively capture the multiphasic PK profile of bb2121 CAR‐T cells. The mean estimates revealed a very rapid extravasation of CAR‐T cells from the blood compartment () in comparison to their circulation back to the systemic circulation (). 5 This observation was consistent with the known reports highlighting that only a fraction (~5%) of overall CAR‐T cells reside in the peripheral circulation. 9 The data analysis also revealed the differential elimination rates ( vs. ) of two CAR‐T cell phenotypes, i.e., effector vs. memory cells (Table 1 ). This observation is also consistent with the expected half‐lives of these two phenotypes, where effector phenotypes are known to be short lived compared with memory phenotypes. The shorter half‐life of the effector pool could be explained by mechanisms such as activation‐induced cell death, 22 exhaustion, 23 and loss of antigen stimulus as well as antibody and/or complement‐driven lysis due to immunogenicity. 24 , 25 , 26
Although quiescent (memory) T cells (Figure 2b ) could also differentiate back into short‐lived, rapidly proliferating effector T cells (CAR‐Te) upon CAR‐Target engagement, 27 a “net” unidirectional first‐order rate constant of conversion (Rm) from short‐lived effector to long‐lived memory CAR‐T cells was estimated. Due to the unavailability of measurements for different CAR‐T cell phenotypes (e.g., naïve/stem cell memory (Tscm), central memory (Tcm), effector memory (Tem) and effector (Te)) in the infused cell product or during the duration of the study, it was assumed that 100% of the infused CAR‐T cells were of the effector phenotype, which gradually differentiate (rm) into the memory phenotype at the site of action (bone marrow). However, with the availability of flow‐based measurements pertaining to different subsets (CD4/CD8) and phenotypes of CAR‐T cells in the peripheral blood and tumor site, the developed base model could be evolved to mathematically characterize distinct kinetics, efficacy, and expansion capabilities of different CAR‐T subsets/phenotypes moving forward.
The impact of CAR‐T dose and initial patient disease burden on CAR‐T cell expansion and percentage decrease in serum sBCMA and M‐protein levels was assessed by simulations shown in Figure 4 . It was observed that the patient tumor burden could be more sensitive to apparent Cmax and Tmax after expansion in comparison to the administered doses of CAR‐T cells. These results were not only consistent with our prior preclinical PK‐PD modeling 5 but also a similar observation has been observed in several clinical trials, including anti‐CD19, 7 , 28 anti‐BCMA, 29 and Tmunity anti‐prostate specific membrane antigen (PSMA)/transforming growth factor beta dominant negative (TGFβdn) 20 CAR‐T cell clinical studies. 30 Within the Tmunity anti‐PSMA/TGFβdn clinical trial in patients with metastatic castration‐resistant prostate cancer, 30 it was observed that a ~ 10‐fold increase in CAR‐T doses does not result in a proportional increase to overall extent of exposure (Cmax); however, the rate to maximum exposure is increased (decrease in Tmax). Similarly, in an anti‐BCMA CAR‐T cells clinical study in patients with multiple myeloma, 29 a 10‐fold increase in CAR‐T dose levels among different cohorts (with or without lymphodepletion) led to no significant differences in Cmax. Furthermore, in anti‐CD19 CAR‐T clinical trials in patients with B cell acute lymphoblastic leukemia, 7 it was also observed that patients with higher bone marrow acute myeloid leukemia blast counts led to higher expansion (increased Cmax) compared with patients with lower tumor burdens. These clinical observations provide further validity of adopting a systems approach while developing translational PK‐PD relationships for CAR‐T cells. 31 Some other factors that could impact the cellular kinetics and efficacy of CAR‐T cells include (i) product characteristics such as CAR‐T cell subsets (CD4/CD8 ratios 27 , 32 , 33 ), phenotypes (Tscm, Tcm, Tem, and Te 28 , 34 ), and exhaustion markers (PD‐1, LAG‐3, TIM‐3 28 , 34 ) in patient apheresate and preinfusion products; (ii) presence of immunosuppressive immune cells and soluble factors 35 ; (iii) presence or absence of lymphodepletion 28 , 29 , 36 , 37 ; and (iv) resistance mechanisms leading to antigen escape. 29 These factors are further discussed in the [Link] , [Link] .
Later, the validated clinical PK‐PD model was leveraged to describe the reported categorical response data for individual patients (n = 33; Figure 5 and Figure S2 ) based on the decrease in serum M‐protein levels. 14 Adopting this methodology not only bolstered our confidence in identifying mean parameter values but also enabled us to estimate intersubject variability around the mean estimates. PFS rates were calculated using this approach, which were later validated using the observed PFS rates, as shown in Figure 5b .
Once validated, the population PK‐PD model was leveraged to do virtual population simulations (n = 1000) at different dose levels (50–800 × 106 CAR‐T cells; Figure 6a ), and categorical dose‐response curves were generated at 6 months (when CAR‐T PK is active) and 21 months (CAR‐T PK is inactive) post–CAR‐T cell infusion based on the IMWG Uniform Response Criteria for Multiple Myeloma. 14 Simulated dose‐response curves suggested a very steep dose‐exposure‐response relationship (Figure 6 ). This observation is in concordance with the clinical behavior of two approved autologous CAR‐T cell products (i.e., Kymriah 38 and Yescarta 39 ), where a flat dose‐response relationship was observed. Our model simulations for anti‐BCMA CAR‐T (bb2121) suggested that beyond a “threshold” CAR‐T cell dose level, dose‐escalation does not result in further improvement in exposure and patient responses. Recently reported clinical observations from the pivotal phase II karMMa trial on bb2121, evaluating three different dose levels (150, 300, and 450 million CAR + T cells) in 128 patients with r/r multiple myeloma also suggested very similar PK behavior across three dose levels and marginal improvement in response rates when escalating from 150 to 450 million dose levels. 12 Not only are these clinical observations somewhat concordant with our model predictions but also when done a priori, they could significantly influence the clinical development of future CAR‐T programs moving forward. Besides robust dose‐efficacy relationships, when selecting recommended phase II dose level(s) for subsequent pivotal studies, dose‐toxicity relationships and manufacturing capacity/costs of CAR‐T cells should also be taken into consideration.
In summary, we have developed a multiscale translational PK‐PD model to effectively characterize preclinical (in vitro and in vivo) and clinical PK‐PD data sets for CAR‐T cell therapy. This integrated PK‐PD framework to describe the cellular kinetics of CAR‐T cells could be leveraged in the future to characterize the two commonly observed toxicities with CAR‐T cell therapies, i.e., cytokine release syndrome 40 and immune effector cell–associated neurological syndrome. 41 , 42 Currently the complexity of the model structure is limited by the availability of clinical data sets. Hence many important components, such as (i) CAR‐T product–related attributes, (ii) integration of multiple bioanalytical measurements, and (iii) presence of a host‐immune system (and its interactions with CAR‐T cells), are currently missing. However, with exponential evolution of the cell‐therapy platform, this modeling and simulation framework can be extended and evolved in the future to explore multiple infusion regimens and facilitate the discovery and development of other cell‐therapy programs.
CONFLICT OF INTEREST
All authors were employees of Johnson and Johnson during the execution and completion of this study.
AUTHOR CONTRIBUTIONS
A.P.S. wrote the manuscript. A.P.S., W.C., X.Z., A.Z., and D.H. designed the research. W.C., X.Z., H.M., T.J.C., and A.P.S. performed the research. W.C., X.Z., T.J.C. A.P.S., and H.M. analyzed the data. All authors contributed new reagents/analytical tools.
Supporting information
Supplementary Material
Supplementary Material
Acknowledgments
The authors acknowledge Weirong Wang (Clinical Pharmacology and Pharmacometrics, Janssen R&D), Gopi Shankar (Biologics Development Sciences), Professor Yanguang (Carter) Cao (University of North Carolina Chapel Hill), and other team members from Discovery and Translational Research at Janssen Biotherapeutics for multiple discussions on the pharmacokinetic‐pharmacodynamic modeling aspects of chimeric antigen receptor T cells. The authors are indebted to the teachings from Dr. Dhaval K. Shah Laboratory at University at Buffalo on “bench to bedside translational modeling,” which has significantly impacted the presented research in this article.
Aman P. Singh and Wenbo Chen contributed equally to this study.
Funding information
This study was funded by the Janssen Biotherapeutics, Janssen R&D, The Pharmaceutical Company of Johnson and Johnson.
REFERENCES
- 1. Labanieh L, Majzner RG, Mackall CL. Programming CAR‐T cells to kill cancer. Nat Biomed Eng. 2018;2:377‐391. [DOI] [PubMed] [Google Scholar]
- 2. Pan C, Liu H, Robins E et al. Next‐generation immuno‐oncology agents: current momentum shifts in cancer immunotherapy. J Hematol Oncol. 2020;13:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. U.S. FDA approves Kite’s Tecartus™, the first and only CAR T treatment for relapsed or refractory mantle cell lymphoma. Gilead Sciences, 2020. [Google Scholar]
- 4. Beacon Targeted Therapies Database. https://data.beacon‐intelligence.com/ (2020).
- 5. Singh AP, Zheng X, Lin‐Schmidt X et al. Development of a quantitative relationship between CAR‐affinity, antigen abundance, tumor cell depletion and CAR‐T cell expansion using a multiscale systems PK‐PD model. MAbs. 2020;12:1688616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hardiansyah D, Ng CM. Quantitative systems pharmacology model of chimeric antigen receptor T‐cell therapy. Clin Transl Sci. 2019;12:343‐349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Awasthi R, Pacaud L, Waldron E et al. Tisagenlecleucel cellular kinetics, dose, and immunogenicity in relation to clinical factors in relapsed/refractory DLBCL. Blood Adv. 2020;4:560‐572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Raje N, Berdeja J, Lin YI et al. Anti‐BCMA CAR T‐cell therapy bb2121 in relapsed or refractory multiple myeloma. N Engl J Med. 2019;380:1726‐1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Stein AM, Grupp SA, Levine JE et al. Tisagenlecleucel model‐based cellular kinetic analysis of chimeric antigen receptor‐T cells. CPT Pharmacometrics Syst Pharmacol. 2019;8:285‐295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Liu C, Ayyar VS, Zheng X et al. Model‐based cellular kinetic analysis of chimeric antigen receptor‐T cells in humans. Clin Pharmacol Ther. 10.1002/cpt.2040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Friedman KM, Garrett TE, Evans JW et al. Effective targeting of multiple B‐cell maturation antigen‐expressing hematological malignances by anti‐B‐cell maturation antigen chimeric antigen receptor T cells. Hum Gene Ther. 2018;29:585‐601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Munshi NC, Anderson LD, Shah N et al. Idecabtagene vicleucel (ide‐cel; bb2121), a BCMA‐targeted CAR T‐cell therapy, in patients with relapsed and refractory multiple myeloma (RRMM): Initial KarMMa results. J Clin Oncol. 2020;38:8503. [Google Scholar]
- 13. Delforge M. KarMMa‐3: a phase 3 study of idecabtagene vicleucel (ide‐cel, bb2121), a BCMA‐Directed CAR T cell therapy vs standard regimens in relapsed and refractory multiple myeloma. 62nd ASH Annual Meeting and Exposition; Virtual conference; December 5, 2020.
- 14. Kumar S, Paiva B, Anderson KC et al. International Myeloma Working Group consensus criteria for response and minimal residual disease assessment in multiple myeloma. Lancet Oncol. 2016;17:e328‐e346. [DOI] [PubMed] [Google Scholar]
- 15. Feinberg D, Paul B, Kang Y. The promise of chimeric antigen receptor (CAR) T cell therapy in multiple myeloma. Cell Immunol. 2019;345:103964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Till B. Mechanisms of treatment failure in chimeric antigen receptor T cell therapy (National Institutes of Health, Bethesda, MD, 2019). https://grantome.com/grant/NIH/R01‐CA230520‐01A1
- 17. Khot A, Matsueda S, Thomas VA, Koya RC, Shah DK. Measurement and quantitative characterization of whole‐body pharmacokinetics of exogenously administered T cells in mice. J Pharmacol Exp Ther. 2019;368:503‐513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nacasaki Silvestre R, Moco PD, Picanco‐Castro V. Determination of cytotoxic potential of CAR‐T cells in co‐cultivation assays. Methods Mol Biol. 2020;2086:213‐222. [DOI] [PubMed] [Google Scholar]
- 19. Caruso HG, Hurton LV, Najjar A et al. Tuning sensitivity of CAR to EGFR density limits recognition of normal tissue while maintaining potent antitumor activity. Cancer Res. 2015;75:3505‐3518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kloss CC, Lee J, Zhang A et al. Dominant‐negative TGF‐beta receptor enhances PSMA‐targeted human CAR T cell proliferation and augments prostate cancer eradication. Mol Ther. 2018;26:1855‐1866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Paiva B, Mateos MV, Sanchez‐Abarca LI et al. Immune status of high‐risk smoldering multiple myeloma patients and its therapeutic modulation under LenDex: a longitudinal analysis. Blood. 2016;127:1151‐1162. [DOI] [PubMed] [Google Scholar]
- 22. Arakaki R, Yamada A, Kudo Y, Hayashi Y, Ishimaru N. Mechanism of activation‐induced cell death of T cells and regulation of FasL expression. Crit Rev Immunol. 2014;34:301‐314. [DOI] [PubMed] [Google Scholar]
- 23. Cheng J, Zhao L, Zhang Y et al. Understanding the mechanisms of resistance to CAR T‐cell therapy in malignancies. Front Oncol. 2019;9:1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Dooms H, Abbas AK. Life and death in effector T cells. Nat Immunol. 2002;3:797‐798. [DOI] [PubMed] [Google Scholar]
- 25. Gordon S, Pluddemann A. Macrophage clearance of apoptotic cells: a critical assessment. Front Immunol. 2018;9:127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Green DR, Oguin TH, Martinez J. The clearance of dying cells: table for two. Cell Death Differ. 2016;23:915‐926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mahnke YD, Brodie TM, Sallusto F, Roederer M, Lugli E. The who's who of T‐cell differentiation: human memory T‐cell subsets. Eur J Immunol. 2013;43:2797‐2809. [DOI] [PubMed] [Google Scholar]
- 28. Turtle CJ, Hanafi LA, Berger C et al. CD19 CAR‐T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J Clin Invest. 2016;126:2123‐2138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Cohen AD, Garfall AL, Stadtmauer EA et al. B cell maturation antigen‐specific CAR T cells are clinically active in multiple myeloma. J Clin Invest. 2019;129:2210‐2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Narayan V, Gladney W, Plesa G et al. A phase I clinical trial of PSMA‐directed/TGFβ‐insensitive CAR‐T cells in metastatic castration‐resistant prostate cancer. J Clin Oncol. 2019;37(7 suppl):TPS347. [Google Scholar]
- 31. Shah DK, Haddish‐Berhane N, Betts A. Bench to bedside translation of antibody drug conjugates using a multiscale mechanistic PK/PD model: a case study with brentuximab‐vedotin. J Pharmacokinet Pharmacodyn. 2012;39:643‐659. [DOI] [PubMed] [Google Scholar]
- 32. Benmebarek MR, Karches CH, Cadilha BL et al. Killing mechanisms of chimeric antigen receptor (CAR) T cells. Int J Mol Sci. 2019;20:1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yang Y, Kohler ME, Chien CD et al. TCR engagement negatively affects CD8 but not CD4 CAR T cell expansion and leukemic clearance. Sci Transl Med. 2017;9:eaag1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Finney OC, Brakke H, Rawlings‐Rhea S et al. CD19 CAR T cell product and disease attributes predict leukemia remission durability. J Clin Invest. 2019;129:2123‐2132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Rodriguez‐Garcia A, Palazon A, Noguera‐Ortega E, Powell DJ Jr, Guedan S. CAR‐T cells hit the tumor microenvironment: strategies to overcome tumor escape. Front Immunol. 2020;11:1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Haas AR, Tanyi JL, O’Hara MH et al. Phase I study of lentiviral‐transduced chimeric antigen receptor‐modified T cells recognizing mesothelin in advanced solid cancers. Mol Ther. 2019;27:1919‐1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gauthier J, Bezerra ED, Hirayama AV et al. Factors associated with outcomes after a second CD19‐targeted CAR T‐cell infusion for refractory B cell malignancies. Blood. 2020;137:323‐335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mueller KT, Waldron E, Grupp SA et al. Clinical pharmacology of tisagenlecleucel in B‐cell acute lymphoblastic leukemia. Clin Cancer Res. 2018;24:6175‐6184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lee DW, Kochenderfer JN, Stetler‐Stevenson M et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose‐escalation trial. Lancet. 2015;385:517‐528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lee DW, Kochenderfer JN, Stetler‐Stevenson M et al. Mitigating the risk of cytokine release syndrome in a Phase I trial of CD20/CD3 bispecific antibody mosunetuzumab in NHL: impact of translational system modeling. NPJ Syst Biol Appl. 2020;6:28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Santomasso B, Bachier C, Westin J, Rezvani K, Shpall EJ. The other side of CAR T‐cell therapy: cytokine release syndrome, neurologic toxicity, and financial burden. Am Soc Clin Oncol Educ Book. 2019;39:433‐444. [DOI] [PubMed] [Google Scholar]
- 42. Bloomingdale P, Mager DE. Machine learning models for the prediction of chemotherapy‐induced peripheral neuropathy. Pharm Res. 2019;36:35. [DOI] [PubMed] [Google Scholar]
- 43. ExPASy Bioinformatics Resource Portal. https://www.expasy.org/vg/index/Cell (2020).
- 44. Bu DX, Singh R, Choi E et al. Pre‐clinical validation of B cell maturation antigen (BCMA) as a target for T cell immunotherapy of multiple myeloma. Oncotarget. 2018;9:25764‐25780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Shah DK, Betts AM. Towards a platform PBPK model to characterize the plasma and tissue disposition of monoclonal antibodies in preclinical species and human. J Pharmacokinet Pharmacodyn. 2012;39:67‐86. [DOI] [PubMed] [Google Scholar]
- 46. Kalos M, Levine BL, Porter DL et al. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Transl Med. 2011;3:95ra73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hokanson JA, Brown BW, Thompson JR, Drewinko B, Alexanian R. Tumor growth patterns in multiple myeloma. Cancer. 1977;39:1077‐1084. [DOI] [PubMed] [Google Scholar]
- 48. Dingli D, Pacheco JM, Dispenzieri A et al. Serum M‐spike and transplant outcome in patients with multiple myeloma. Cancer Sci. 2007;98:1035‐1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Dingli D, Pacheco JM, Dispenzieri A et al. In vivo and in silico studies on single versus multiple transplants for multiple myeloma. Cancer Sci. 2007;98:734‐739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ghermezi M, Li M, Vardanyan S et al. Serum B‐cell maturation antigen: a novel biomarker to predict outcomes for multiple myeloma patients. Haematologica. 2017;102:785‐795. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material
Supplementary Material