

Abstract
Despite the increasing prevalence of fatty liver diseases worldwide, the molecular mechanism underlying their pathogenesis remains poorly defined. This study examines the expression and significance of tumor necrosis factor (TNF) receptor-associated factor 6 (TRAF6) in the high-fat diet (HFD)-induced mouse obesity model and the oleic acid/palmitic acid (OA/PA)-induced cell model. After developing these models, we measured the expressions of TRAF6, enhancer of the zeste homolog 2 (EZH2), and peroxisome proliferator activated receptor alpha (PPARα). The expression of TRAF6, EZH2, and PPARα was manipulated to investigate their roles in cholesterol accumulation through evaluating the plasma levels of total cholesterol (TC), high-density lipoprotein cholesterol (HDL-C), and low-density lipoprotein cholesterol (LDL-C). Co-immunoprecipitation (coIP) assay was used to determine the interaction between TRAF6 and EZH2 and chromatin immunoprecipitation (ChIP) assay to detect the enrichment of EZH2 and H3K27me3 in microRNA-429 (miR-429) promoter. We found that HFD resulted in elevated TRAF6 and miR-429 in fatty liver and reduced EZH2 and PPARα. TRAF6 mediated the ubiquitination of EZH2 and increased miR-429 expression, and miR-429 targeted PPARα. TRAF6 increased cholesterol accumulation in liver cells in vitro via the EZH2/miR-429/PPARα axis. Collectively, HFD upregulates TRAF6 and ubiquitinates EZH2 to promote the miR-429-dependent inhibition of PPARα, leading to cholesterol accumulation in liver and the occurrence of fatty liver.
Keywords: TRAF6, EZH2, microRNA-429, PPARα, ubiquitination, high-fat diet, cholesterol, fatty liver
Graphical Abstract
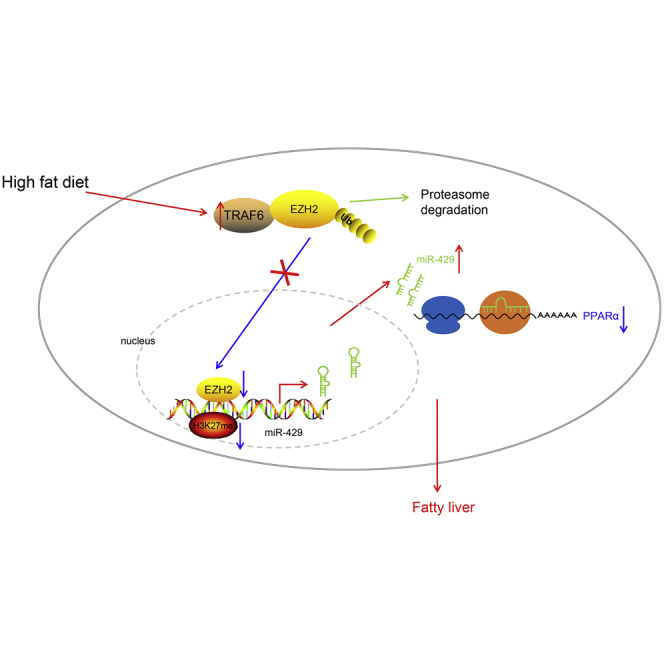
This paper proposes that a HFD enhanced TRAF6 expression in the liver and TRAF6 increased cholesterol accumulation in liver cells via the EZH2/miR-429/PPARα axis. Collectively, HFD upregulates TRAF6 and ubiquitinates EZH2 to promote miR-429-dependent inhibition of PPARα, thereby promoting cholesterol accumulation in the liver and the occurrence of fatty liver.
Introduction
The liver has a wide array of pivotal metabolic functions such as lipid metabolism, which may be impaired in the context of obesity, as well as fatty liver disease.1 Chronic liver disease is a disorder significantly contributing to morbidity and mortality across the globe, which frequently develops from chronic inflammation, eventually leading to advanced liver disease accompanied by severe complications.2 Fatty liver (also known as hepatic steatosis) is widely recognized to arise when lipid accumulation exceeds the rate of lipid disposal.3 Multiple causes can contribute to the occurrence of fatty liver, such as alcoholism, chemotherapy, nonalcoholic fatty liver disease, abnormal metabolism, and toxic injury, as well as infections.4 Disturbance of bile acid synthesis along with deregulated cholesterol efflux leads to cholesterol accumulation in the liver, and the dysfunction of liver lipid metabolism consequently results in progression to fatty liver from lipid deposition in the liver via the steatosis process.5 The biochemical mechanisms underlying an elevation in hepatic cholesterol accumulation should be elaborated to develop new strategies to prevent fat deposition in the liver.6
Signaling through the receptors in the tumor necrosis factor receptor (TNFR) family is a critical player in the secondary complexes and lipid rafts, which determine the metabolic fate of cells.7 Specifically, membrane microdomains that are enriched with cholesterol are positively related to the expression of TNF receptor associated factor 6 (TRAF6), which is a E3 ubiquitin-protein ligase.8 Moreover, TRAF6 expression is stimulated in response to diet-induced obesity, corresponding to boosted secretion of pro-inflammatory cytokines.9 Previous evidence has suggested that enhancement of TRAF6 expression could augment ubiquitination of enhancer of the zeste homolog 2 (EZH2) leading to enhanced degradation, which results in a net reduction in EZH2 expression.10 EZH2 is as a key element of polycomb repressive complex 2, which can catalyze H3K27me3 and thus trigger transcriptional inhibition of target genes.11 Furthermore, EZH2 has been documented to repress the expression of microRNA-429 (miR-429), which is a member of the miR-200 family (miR-200b, miR-200a, and miR-429).12 Importantly, there is upregulation of miR-200 family members including miR-429 in the high-fat-diet (HFD)-fed rodent model, which may underlie the molecular pathogenesis of diet-induced nonalcoholic fatty liver disease.13 EZH2 is a specific methyltransferase, which interacts with the activated peroxisome proliferator activated receptor (PPAR).14 Specifically, the artificial promotion of PPARα expression in nonalcoholic fatty liver disease has been highlighted to restore lipid metabolism and to diminish hepatocyte apoptosis.15 On the basis of the aforementioned literature, we propose the hypothesis that TRAF6, EZH2, miR-429, and PPARα interact in the molecular pathogenesis of fatty liver. Hence, we aimed to test this hypothesis by determining the ability of TRAF6 to influence the progression of fatty liver in relation with the EZH2-miR-429/PPARα axis.
Results
TRAF6 showed high expression in fatty liver caused by HFD
First, we set out to explore the potential regulatory effect of TRAF6 on the development of fatty liver disease, by first validating fatty liver degeneration of HFD-model mice. The results showed that, compared with chow-fed mice, the HFD-fed mice had significant increases in body weight, liver weight, and liver index (Figure 1A, p < 0.01). Moreover, the plasma contents of serum triglyceride (TG), total cholesterol (TC), and low-density lipoprotein cholesterol (LDL-C) increased, while the ratio of high-density lipoprotein cholesterol (HDL-C)/LDL-C decreased in the HFD-fed mice compared to chow-fed mice (p < 0.001); there was no significant difference in content of HDL-C (p > 0.05) between the chow-fed mice and the HFD-fed mice.
Figure 1.

TRAF6 shows high expression in fatty liver caused by HFD
(A) The statistical plot for body weight, liver weight, and liver index in chow-fed mice and HFD-fed mice. (B) The scatterplot for FPG, FINS, and HOMA-IR in chow-fed mice and HFD-fed mice. (C) H&E staining for liver tissues in chow-fed mice and HFD-fed mice (400×). (D) Oil red O staining for fatty liver tissues in chow-fed mice and HFD-fed mice (200×). (E) The scatterplot for the contents of TG and TC in liver tissues of chow-fed and HFD-fed mice. (F) Histogram representing TNF-α levels in serum of chow-fed mice and HFD-fed mice. (G) Western blot analysis of CPT1 protein expression in liver tissues of chow-fed and HFD-fed mice and its statistical diagram. (H) Filipin staining for the fatty liver cell model (200×) and the quantitative histogram. (I) Nile red staining for the fatty liver cell model (200×) and the statistical histogram. (J) Histogram of FFA content in liver cells after OA/PA induction. (K) The histogram for the content of TG and TC in the fatty liver cell model. (L) The expression of TRAF6 in fatty liver tissues of chow-fed and HFD-fed mice as detected by immunohistochemistry (400×). (M) qRT-PCR determination of TRAF6 expression in fatty liver tissues of chow-fed mice and HFD-fed mice and the statistical histogram. (N) The histogram for TRAF6 expression in the fatty liver cell model as detected by qRT-PCR. (O) The protein expression of TRAF6 in the fatty liver cell model as detected by western blot analysis. These data were measurement data, expressed as mean ± standard deviation. Data between two groups were compared utilizing independent sample t test, while data among multiple groups at variant time points were analyzed through repeated-measures ANOVA, followed by Bonferroni post hoc test. Compared with chow or control, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001. In chow-fed mice and HFD-fed mice, each n = 10. In hepatocytes used as control and induced with OA/PA, each n = 3.
Liver function related indexes revealed that, relative to the chow-fed mice, the expression of alanine aminotransferase (ALT) and aspartate aminotransferase (AST) was markedly increased in the serum of HFD-fed mice (Table 1, p < 0.001). Moreover, fasting plasma glucose (FPG), fasting insulin (FINS), and homeostasis model assessment-insulin resistance (HOMA-IR) were significantly higher in the HFD-fed mice than in the chow-fed mice (Figure 1B, p < 0.001). Hematoxylin and eosin (H&E) staining results displayed that, compared with the chow-fed mice, HFD-fed mice showed fatty degeneration, ballooning degeneration, and mixed inflammatory cell infiltration in lobules of the liver tissues (Figure 1C). Oil red O staining showed a notable increase in neutral fat content in the liver tissues of HFD-fed mice compared to the chow-fed mice (Figure 1D, p < 0.001). Additionally, the contents of TG and TC in liver tissues were significantly higher in the HFD-fed mice than in the chow-fed mice (Figure 1E, p < 0.001). These results confirmed successful construction of the fatty liver model induced by HFD. Next, we found significantly increased levels of TNF-α in serum of HFD-fed mice compared to chow-fed control mice (Figure 1F, p < 0.001). To detect changes in enzymes associated with fatty acid oxidation in primary hepatocytes, we further examined the level of CPT1, which is critical in this process; the CPT1 level was significantly lower in liver tissues of HFD-fed mice than in chow-fed mice (Figure 1G, p < 0.001).
Table 1.
Comparison of lipid metabolism related indexes in chow-fed and HFD-fed mice
| Group | TG (mM) | TC (mM) | HDL-C (mM) | LDL-C (mM) | HDL-C/LDL-C | ALT (U/L) | AST (U/L) |
|---|---|---|---|---|---|---|---|
| chow | 1.04 ± 0.08 | 2.96 ± 0.31 | 2.95 ± 0.36 | 0.27 ± 0.03 | 11.15 ± 2.23 | 80.52 ± 13.39 | 93.80 ± 14.21 |
| HFD | 1.42 ± 0.15∗∗∗ | 6.04 ± 0.42∗∗∗ | 3.26 ± 0.32 | 0.82 ± 0.11∗∗∗ | 4.03 ± 0.58∗∗∗ | 130.48 ± 16.57∗∗∗ | 150.15 ± 20.46∗∗∗ |
∗∗∗p < 0.001 versus chow-fed mice. Measurement data were presented as the mean ± standard deviation, and data comparison between two groups was conducted using independent sample t test. n = 10. HFD, high-fat diet; TG, triglyceride; TC, total cholesterol; HDL-C, high-density lipoprotein cholesterol; LDL-C, low-density lipoprotein cholesterol; ALT, alanine aminotransferase; AST, aspartate aminotransferase; PPARα, peroxisome proliferator activated receptor alpha; sh-NC, short hairpin RNA-negative control.
Subsequently, we verified the cell model of fatty liver induced by oleic acid/palmitic acid (OA/PA) in primary human and mouse hepatocytes. Filipin staining showed that, following induction with OA/PA, there was a notable increase in free cholesterol in the medium (Figure 1H; Figure S1A, p < 0.001). Nile red staining results indicated that the accumulated lipids and lipid droplets in cells were markedly increased after OA/PA induction (Figure 1I; Figure S1B, p < 0.05). The content of free fatty acid (FFA), TG, and TC in hepatocytes also showed a marked elevation following induction with OA/PA. These results confirmed the successful construction of the OA/PA-induced cell model of fatty liver (Figures 1J and 1K; Figures S1C and S1D, p < 0.01).
The results from immunohistochemistry and qRT-PCR illustrated that the expression of TRAF6 was markedly higher in the HFD-fed mice than in the chow-fed mice (Figures 1L and 1M, p < 0.01). Based on the results from qRT-PCR and western blot analysis, the expression of TRAF6 was notably increased following induction with OA/PA (Figures 1N and 1O; Figures S1E and S1F, p < 0.001). These results suggest that TRAF6 is highly expressed in HFD-induced fatty liver cells.
Silencing of TRAF6 inhibited the cholesterol accumulation in hepatocytes
To further verify the effect of TRAF6 on the cholesterol accumulation in hepatocytes, we first used qRT-PCR and western blot analysis to detect the silencing efficiency of TRAF6, which showed significantly reduced TRAF6 expression after treatment with short hairpin (sh)-TRAF6-1 or sh-TRAF6-2 (Figures 2A and 2B, p < 0.001). To test whether silencing TRAF6 affected the expression of other isoforms of TRAF6, we detected the mRNA and protein expression of TRAF1-5 and TRAF7 after silencing TRAF6 by qRT-PCR and western blot. The results showed no significant difference in the expression of TRAF1-5 and TRAF7 after sh-TRAF6-1 or sh-TRAF6-2 treatment, compared to shRNA-negative control (sh-NC) treatment (Figure S2). Next, we explored the effect of TRAF6 on cholesterol accumulation in NCTC1469 and AML-12 cells. The results of filipin staining showed that the free cholesterol level was notably reduced upon silencing of TRAF6 (Figure 2C; Figures S7A, S3A, and S3E, p < 0.01). Nile red staining results demonstrated significantly lesser accumulation of lipids and lipid droplets in cells after silencing TRAF6 (Figure 2D; Figures S8A, S3B, and S3F, p < 0.05). Moreover, the level of FFA in hepatocytes increased noticeably (Figure 2E; Figures S3C and S3G, p < 0.001), while the levels of TC and TG in hepatocytes displayed a marked decline upon silencing of TRAF6 (Figure 2F; Figures S3D and S3H, p < 0.01). These results indicate that the silencing of TRAF6 is capable of inhibiting the cholesterol accumulation in hepatocytes.
Figure 2.

Silencing of TRAF6 inhibits the cholesterol accumulation in hepatocytes
(A) The silencing efficiency of TRAF6 as detected by qRT-PCR. (B) The silencing efficiency of TRAF6 as detected by western blot analysis. (C) Filipin staining for hepatocytes treated with sh-NC or sh-TRAF6 and the quantitative histogram. (D) Nile red staining results and the statistical histogram. (E) Histogram of free FFA content in hepatocytes treated with sh-NC or sh-TRAF6. (F) The histogram for the contents of TG and TC in hepatocytes treated with sh-NC or sh-TRAF6. These data were measurement data, expressed as mean ± standard deviation. Data among multiple groups were compared using one-way ANOVA followed by a Tukey’s test. ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001 compared with sh-NC. The cell experiment was repeated three times. In chow-fed mice and HFD-fed mice, each n = 10. In hepatocytes used as control and induced with OA/PA, each n = 3.
Cholesterol metabolism entails four main processes, namely endogenous synthesis, exogenous uptake, efflux, and esterification. Next, we used qRT-PCR and western blot to examine the effects of TRAF6 on the mRNA and protein expressions of sterol regulatory element-binding protein 2 (SREBP2), a key regulator of cholesterol synthesis, and HMGCR and SM, the two rate-limiting enzymes in cholesterol biosynthesis, the cholesterol absorption regulator NPC1L1, the cholesterol efflux regulator ABC subfamily A member 1 (ABCA1), the ABC subfamily G (ABCG) members 1, 5, and 8, and the cholesterol esterification regulators ACAT1 and ACAT2. The results showed that the mRNA and protein expression of SREBP2, HMGCR, SM, ABCA1, ABCG1, ABCG5, and ABCG8 were significantly decreased after silencing TRAF6, while the mRNA and protein expression of NPC1L1, ACAT1, and ACAT2 were significantly increased (Figures S4A and S4B). The above results indicated that TRAF6 affected the homeostasis of cholesterol.
TRAF6 enhanced ubiquitination of EZH2
Next, we explored the possible interaction between TRAF6 and EZH2. Immunohistochemistry showed notably lower EZH2 expression in HFD-fed mice than in chow-fed mice (Figure 3A). The results from qRT-PCR and western blot analysis revealed that EZH2 expression was markedly lowered in cells after OA/PA induction (Figures 3B and 3C, p < 0.01). As shown by western blot analysis, the protein expression of EZH2 was significantly increased by silencing of TRAF6 (p < 0.001), while no significant difference was found after the addition of MG132 (Figure 3D, p > 0.05), and EZH2 mRNA expression was unchanged after silencing of TRAF6 (Figure S5A, p > 0.05). The results from co-immunoprecipitation (coIP) illustrated that TRAF6 interacted with EZH2 (Figure 3E). In vivo ubiquitination assays showed that silencing of TRAF6 caused a significant decrease in the level of EZH2 ubiquitination (Figure 3F). These results support the proposition that TRAF6 promotes the ubiquitination of EZH2.
Figure 3.

TRAF6 mediates ubiquitination of EZH2
(A) EZH2 expression in liver tissues of HFD-fed mice and chow-fed mice as detected by immunohistochemistry. (B) EZH2 expression after OA/PA induction as detected by qRT-PCR. (C) EZH2 expression after OA/PA induction as detected by western blot analysis. (D) The histogram for TRAF6 and EZH2 expression as detected by western blot analysis. (E) The interaction of TRAF6 with EZH2 as detected by coIP. (F) The level of EZH2 ubiquitination as detected by in vivo ubiquitination assay. These data were measurement data, expressed as mean ± standard deviation. Data between two groups were compared utilizing independent sample t test, while data among multiple groups were compared with the use of one-way ANOVA, followed by a Tukey’s test. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 versus control or sh-NC. The cell experiment was repeated three times. In tissue experiment, n = 10.
EZH2 inhibited miR-429 expression through methylation
Subsequently, we investigated whether the high expression of miR-429 in fatty liver was attributable to the decrease of EZH2 expression. Therefore, qRT-PCR was first used to detect the expression of miR-429 and EZH2 in the mouse and cell models. The results revealed that the expression of miR-429 was significantly higher in HFD-fed mice than in chow-fed mice, while the expression of EZH2 was significantly lower (Figure 4A, p < 0.001). The expression of miR-429 showed a remarkable increase, while the expression of EZH2 exhibited opposite changes following OA/PA induction (Figure 4B, p < 0.01). qRT-PCR and western blot analysis were performed to detect the efficiency of overexpressing EZH2, which displayed a marked increase in the expression of EZH2 upon overexpression (oe) of EZH2 (Figures 4C and 4D, p < 0.001). The expression of miR-429 was detected by qRT-PCR, the results of which showed that it was markedly diminished by oe of EZH2 (Figure 4E, p < 0.01). Chromatin immunoprecipitation (ChIP) assay results demonstrated that oe of EZH2 also notably increased the enrichment of EZH2 and H3K27me3 (Figure 4F, p < 0.05). Afterward, to determine whether EZH2 level could prevent OA/PA-induced accumulation of cholesterol/lipid, we overexpressed EZH2 in model fatty liver cells and detected measured TC and TG levels. Oe of EZH2 markedly reduced TC and TG levels (Figure 4G, p < 0.05). Taken together, the above results elaborate that EZH2 binds to miR-429 promoter to inhibit miR-429 expression by upregulating H3K27me3, thereby reducing accumulated TC in hepatocytes.
Figure 4.

EZH2-mediated methylation inhibits miR-429 expression
(A) miR-429 and EZH2 expression in liver tissues of HFD-fed mice and chow-fed mice as detected by qRT-PCR. (B) miR-429 and EZH2 expression after OA/PA induction as detected by qRT-PCR. (C) The efficiency of EZH2 oe as detected by qRT-PCR. (D) The efficiency of EZH2 oe as detected by western blot analysis. (E) The expression of miR-429 in the presence of oe-NC or oe-EZH2. (F) The enrichment of EZH2 and H3K27me3 at the promoter of miR-429. (G) Histogram of TG and TC levels in the cells in the presence of oe-NC or oe-EZH2. These data were measurement data, expressed as mean ± standard deviation. Data between two groups were compared utilizing independent sample t test. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 versus chow, control or oe-NC. The cell experiment was repeated three times. In tissue experiment, n = 10.
TRAF6 promoted the expression of miR-429 and the cholesterol accumulation in hepatocytes through ubiquitination of EZH2
We further verified that TRAF6 could increase the expression of miR-429 and the cholesterol accumulation in hepatocytes via ubiquitination of EZH2. First, qRT-PCR and western blot analysis were used to detect the efficiency of EZH2 silencing. The results showed that the expression of EZH2 was significantly lowered upon treatments either with sh-EZH2-1 or sh-EZH2-2 (Figures 5A and 5B, p < 0.001). Next, we found that silencing EZH2 did not affect the expression of other isoforms of proteins (Figures S5B and S5C). Because the silencing efficiency of sh-EZH2-1 was higher than that of sh-EZH2-2, the subsequent experiments were carried out using sh-EZH2-1. As shown in western blot analysis, the expression of TRAF6 and EZH2 had a marked decline in response to silencing of TRAF6 (p < 0.001), whereas the expression of EZH2 had a notable increase (p < 0.001), while that of TRAF6 remained unchanged in response to silencing of both TRAF6 and EZH2 (Figure 5C, p > 0.05). Moreover, qRT-PCR revealed that the expression of miR-429 was markedly reduced by silencing of TRAF6 but increased by silencing of both TRAF6 and EZH2 (Figure 5D, p < 0.001). We next used qRT-PCR to confirm further that silencing TRAF6 could inhibit miR-429 expression in HFD-fed mice; results showed that depletion of TRAF6 markedly decreased miR-429 expression in HFD-fed mice (Figure S6, p < 0.001). Overall, TRAF6 could inhibit methylated modification of miR-429 promoter via ubiquitination of EZH2, thereby contributing to promoted net miR-429 expression.
Figure 5.

TRAF6 promotes the expression of miR-429 and cholesterol accumulation in hepatocytes through ubiquitination of EZH2
(A) The silencing efficiency of EZH2 as detected by qRT-PCR. (B) The silencing efficiency of EZH2 as detected by western blot analysis. (C) The expression of TRAF6 and EZH2 after different treatment as detected by western blot analysis. (D) The expression of miR-429 after different treatment as detected by qRT-PCR. (E) The expression of TRAF6 and EZH2 after different treatment as detected by western blot analysis. (F) The expression of miR-429 after different treatment as detected by qRT-PCR. (G) Filipin staining for hepatocytes after different treatment. (H) Nile red staining for hepatocytes. (I) Statistical histogram of FFA content in the cells after different treatment. (J) The histogram for the content of TG and TC in hepatocytes after different treatment. These data were measurement data, expressed as mean ± standard deviation. Data among multiple groups were compared with the use of one-way ANOVA, followed by Tukey’s test. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 versus sh-NC or sh-NC + mimic-NC. #p < 0.05, ##p < 0.01, ###p < 0.001 versus sh-TRAF6 + sh-NC or sh-TRAF6 + mimic-NC. The cell experiment was repeated three times.
To further verify the effect of TRAF6 on the cholesterol accumulation in hepatocytes, we first used western blot analysis to determine the expression of TRAF6 following the different treatments. The results showed that TRAF6 expression was markedly reduced after silencing TRAF6 or combined treatment of silencing TRAF6 and overexpressing miR-429 (Figure 5E, p < 0.001). In addition, the expression of miR-429 was significantly reduced in the presence of silenced TRAF6 (p < 0.001), but this expression could be rescued after treatment with a combination of silenced TRAF6 and overexpressed miR-429 (Figure 5F, p < 0.001). The results from filipin staining displayed notably lower free cholesterol upon silencing TRAF6 (p < 0.001), which could be rescued upon combined silencing of TRAF6 and miR-429 oe (Figure 5G; Figure S7B, p < 0.001). The results of Nile red assay showed that silencing of TRAF6 noticeably downregulated levels of free cholesterol and reduced lipids and lipid droplets accumulated in the cells (p < 0.05), whereas upregulation of miR-429 increased the free cholesterol level (Figure 5H; Figure S8B, p < 0.01). Silencing TRAF6 significantly enhanced the FFA level (p < 0.001), which could be reversed by oe of miR-429 (Figure 5I, p < 0.01). Additionally, there were marked declines in TC and TG after silencing of TRAF6, while TRAF6 knockdown in combination with miR-429 oe reversed this effect (Figure 5J, p < 0.01). These results demonstrate that TRAF6 could lead to elevated miR-429 expression and increased cholesterol accumulation in hepatocytes through ubiquitination of EZH2.
miR-429 targeted and downregulated PPARα expression
Next, we used the online analysis website http://mirdb.org/ to explore the targeting relationship between miR-429 and PPARα, which predicted that miR-429 had a targeted binding site with PPARα mRNA 3′ UTR (Figure 6A). Immunohistochemistry showed that PPARα expression in HFD-fed mice was significantly lower than that in chow-fed mice (Figure 6B). The results of qRT-PCR and western blot revealed that PPARα expression was notably reduced after induction with OA/PA (Figures 6C and 6D, p < 0.01). The opposite effects on expressions of miR-429 and PPARα suggest that miR-429 may indeed target and inhibit PPARα expression. Next, qRT-PCR displayed that the expression of miR-429 in HEK293T cells was significantly increased in the presence of miR-429 mimic (Figure 6E, p < 0.01). Further, as demonstrated by dual luciferase reporter gene assay in HEK293T cells, fluorescence activity of miR-429 was notably reduced by co-transfection of miR-429 mimic and PPARα wild-type (WT; p < 0.001) but remained statistically unaffected upon co-transfection of miR-429 mimic and PPARα mutant (MUT; Figure 6F, p > 0.05). qRT-PCR in HEK293T cells showed that the expression of miR-429 was significantly increased by miR-429 mimic but reduced by miR-429 inhibitor (Figure 6G, p < 0.01). The results of qRT-PCR and western blot also revealed that the expression of PPARα was notably decreased by miR-429 mimic but increased by miR-429 inhibitor treatment (Figures 6H and 6I, p < 0.001). These results suggest that miR-429 can target PPARα to inhibit its expression.
Figure 6.

miR-429 targets and downregulates PPARα expression
(A) The targeted binding site between miR-429 and PPARα mRNA 3′ UTR. (B) PPARα expression in HFD-fed mice and chow-fed mice as detected by immunohistochemistry. (C) PPARα expression after induction with OA/PA as detected by qRT-PCR. (D) PPARα expression after induction with OA/PA as detected by western blot analysis. (E) The efficiency of miR-429 oe as detected by qRT-PCR. (F) The targeting relationship between miR-429 and PPARα as verified by dual luciferase reporter gene assay. (G) The overexpressing and silencing efficiency of miR-429 as detected by qRT-PCR. (H) The expression of PPARα after different treatment as detected by qRT-PCR. (I) The expression of PPARα after different treatment as detected by western blot analysis. These data were measurement data, expressed as mean ± standard deviation. Data among multiple groups were compared with the use of one-way ANOVA, followed by a Tukey’s test. ∗∗p < 0.01, ∗∗∗p < 0.001 versus control or mimic-NC. ##p < 0.01, ###p < 0.001 versus inhibitor-NC. In tissue experiment, n = 10. The cell experiment was repeated three times.
miR-429 promoted cholesterol accumulation in hepatocytes by downregulating PPARα expression
Furthermore, we investigated whether miR-429 could promote cholesterol accumulation in hepatocytes by inhibiting the expression of PPARα. First, the efficiency of PPARα silencing was detected using qRT-PCR and western blot analysis. Results showed that the expression of PPARα was significantly inhibited by sh-PPARα-1 or sh-PPARα-2 (Figures 7A and 7B, p < 0.001). qRT-PCR demonstrated a marked decline in the expression of miR-429 upon either inhibition of miR-429 or inhibition of miR-429 in conjunction with silenced PPARα-1 or PPARα-2 (Figure 7C, p < 0.001). In addition, western blot analysis showed that PPARα expression was markedly increased by inhibition of miR-429 (p < 0.001), while this effect was reversed after combined inhibition of miR-429 and PPARα-1 or PPARα-2 (Figure 7D, p < 0.001).
Figure 7.

miR-429 promotes cholesterol accumulation in hepatocytes by downregulating PPARα expression
(A) The silencing efficiency of PPARα as detected by qRT-PCR. (B) The silencing efficiency of PPARα as detected by western blot analysis. (C) The expression of miR-429 after different treatment as detected by qRT-PCR. (D) The expression of PPARα after different treatment as detected by qRT-PCR. (E) Filipin staining for hepatocytes after different treatment. (F) Nile red staining. (G) Statistical histogram of FFA content in the cells after different treatment. (H) The histogram for the content of TG and TC in hepatocytes after different treatment. These data were measurement data, expressed as mean ± standard deviation. Data among multiple groups were compared with the use of one-way ANOVA, followed by a Tukey’s test. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 versus sh-NC or inhibitor-NC + sh-NC. #p < 0.05, ##p < 0.01, ###p < 0.001 versus miR-429 inhibitor + sh-NC. The cell experiment was repeated three times.
From the filipin staining results, we found that inhibition of miR-429 led to markedly reduced free cholesterol levels, which were rescued upon inhibition of both miR-429 and PPARα-1 or PPARα-2 (Figure 7E; Figure S7C, p < 0.001). The Nile red staining results showed that inhibition of miR-429 significantly decreased the accumulated lipid and lipid droplets in the cells, but that silencing of PPARα-1 or PPARα-2 reversed this trend (Figure 7F; Figure S8C, p < 0.05). Furthermore, depletion of miR-429 significantly increased FFA levels (p < 0.001), which could be reversed by silencing PPARα-1 or PPARα-2 (Figure 7G, p < 0.01). Moreover, TC and TG were notably decreased by inhibition of miR-429 (p < 0.05) but increased upon combined inhibition of miR-429 and PPARα (Figure 7H, p < 0.01). Collectively, miR-429 could inhibit PPARα expression and promote cholesterol accumulation in hepatocytes.
TRAF6 promoted cholesterol accumulation in hepatocytes through the EZH2/miR-429/PPARα axis
We next validated that TRAF6 could promote cholesterol accumulation in hepatocytes through the EZH2/miR-429/PPARα axis. We first performed western blot analysis and qRT-PCR to respectively detect the expressions of TRAF6, EZH2, and PPARα protein and the expression of miR-429 following each treatment. The results displayed that the expressions of TRAF6 and miR-429 were significantly reduced upon silencing of TRAF6 (p < 0.001), accompanied by notably increased expression of EZH2 and PPARα (p < 0.01). However, further treatment with co-silencing of TRAF6 and PPARα failed to alter the expression of TRAF6, miR-429, and EZH2 (p > 0.05) but notably decreased the expression of PPARα (Figures 8A and 8B, p < 0.01).
Figure 8.

TRAF6 promotes cholesterol accumulation in hepatocytes through the EZH2/miR-429/PPARα axis
(A) The expression of TRAF6, EZH2, and PPARα after different treatment as detected by western blot analysis. (B) The expression of miR-429 after different treatment as detected by qRT-PCR. (C) Filipin staining for hepatocytes after different treatment. (D) Nile red staining after different treatment. (E) Statistical histogram of FFA in the cells after different treatment. (E) The histogram for FFA levels in the cells. (F) The histogram for the content of TG and TC in hepatocytes after different treatment. These data were measurement data, expressed as mean ± standard deviation. Data among multiple groups were compared with the use of one-way ANOVA, followed by a Tukey’s test. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 versus sh-NC. #p < 0.05, ##p < 0.01, ###p < 0.001 versus sh-TRAF6 + sh-NC. The cell experiment was repeated three times.
As displayed by filipin staining, the free cholesterol showed a decline in the presence of silenced TRAF6 (p < 0.001), which could be reversed upon silencing of both TRAF6 and PPARα (Figure 8C; Figure S7D, p < 0.001). The Nile red staining results displayed that TRAF6 knockout markedly decreased the accumulated lipid and lipid droplets, which could be counteracted by depletion of silencing of PPARα (Figure 8D; Figure S8D, p < 0.05). Treatment of the cells with sh-TRAF6 significantly increased FFA expression (p < 0.01), which could be rescued by treatment of sh-PPARα (Figure 8E, p < 0.01). Moreover, silenced TRAF6 markedly diminished TC and TG levels (p < 0.01), which were increased by silencing of both TRAF6 and PPARα (Figure 8F, p < 0.01). The aforementioned results suggest that TRAF6 could regulate the EZH2/miR-429/PPARα axis, thereby promoting the cholesterol accumulation in hepatocytes.
TRAF6 promoted the occurrence of fatty liver through the EZH2/miR-429/PPARα axis
Finally, we verified the aforementioned findings in vitro using the in vivo mouse fatty liver model upon injection of adenovirus through a tail vein. Western blot analysis and qRT-PCR revealed that compared with chow-fed mice, HFD-fed mice exhibited significantly increased TRAF6 and miR-429 expression (p < 0.05) and markedly decreased EZH2 and PPARα protein expression (p < 0.05); the expressions of TRAF6 and miR-429 were reduced upon silencing of TRAF6 (p < 0.001), accompanied by an increase in the EZH2 and PPARα levels (p < 0.001). There were no significant effects on the expressions of TRAF6, miR-429, and EZH2 in response to silencing of TRAF6 or upon co-silencing of TRAF6 and PPARα (p > 0.05), but co-silencing of TRAF6 and PPARα caused decreased PPARα expression (p < 0.001). Additionally, there was no significant difference in the above indicators between mice treated with sh-NC and HFD-fed mice (Figures 9A and 9B, p > 0.05).
Figure 9.

TRAF6 promotes the occurrence of fatty liver through the EZH2/miR-429/PPARα axis
(A) The expression of TRAF6, EZH2, and PPARα after different treatment as detected by western blot analysis. (B) The expression of miR-429 after different treatment as detected by qRT-PCR. (C) The statistical plot for body weight, liver weight, and liver index in mice with different treatment. (D) H&E staining for liver tissues in mice with different treatment (200×). (E) Oil red O staining for fatty liver tissues in mice with different treatment (200×). (F) The scatterplot for the content of TG and TC in liver tissues of chow-fed mice and HFD-fed mice. These data were measurement data, expressed as mean ± standard deviation. Data among multiple groups were compared with the use of one-way ANOVA, followed by a Tukey’s test. ∗p < 0.05 versus chow-fed mice. ###p < 0.001 versus sh-NC. $$$p < 0.001 versus sh-TRAF6 + sh-NC. n = 10.
In the next experiments, we found that body weight, liver weight, and liver index were decreased significantly by silencing of TRAF6 in the fatty liver mice (p < 0.001), all of which were rectified by silencing both TRAF6 and PPARα (p < 0.001); there were no significant differences in the above indicators between mice treated with sh-NC and those fed with HFD (Figure 9C, p > 0.05). Moreover, silencing of TRAF6 notably reduced the serum contents of TG, TC, and LDL-C (p < 0.001) and elevated the HDL-C/LDL-C ratio (p < 0.001) without changing the content of HDL-C (p > 0.05); opposite effects were found after silencing of both TRAF6 and PPARα, except that the TG and HDL-C content were unchanged (p < 0.001). The liver function markers serum ALT and AST expression were notably decreased upon silencing of TRAF6 (p < 0.001) but were increased upon silencing of both TRAF6 and PPARα (p < 0.001); there was no significant difference in the serum ALT and AST expression between mice treated with sh-NC and HFD-fed mice (Table 2, p > 0.05).
Table 2.
Comparison of lipid metabolism related indexes in mice with different treatments
| Group | TG (mM) | TC (mM) | HDL-C (mM) | LDL-C (mM) | HDL-C/LDL-C | ALT (U/L) | AST (U/L) |
|---|---|---|---|---|---|---|---|
| HFD | 1.30 ± 0.08 | 6.05 ± 0.12 | 3.29 ± 0.25 | 0.84 ± 0.13 | 4.01 ± 0.77 | 132.58 ± 14.51 | 155.39 ± 11.27 |
| sh-NC | 1.33 ± 0.19 | 6.10 ± 0.28 | 3.21 ± 0.29 | 0.85 ± 0.16 | 3.90 ± 0.76 | 127.25 ± 22.16 | 152.94 ± 18.32 |
| sh-TRAF6 + sh-NC | 1.02 ± 0.04∗∗∗ | 3.18 ± 0.23∗∗∗ | 3.09 ± 0.18 | 0.35 ± 0.06∗∗∗ | 9.12 ± 2.01∗∗∗ | 75.19 ± 9.37∗∗∗ | 85.63 ± 9.81∗∗∗ |
| sh-TRAF6 + sh-PPARα | 1.32 ± 0.21### | 5.95 ± 0.34### | 3.12 ± 0.24 | 0.79 ± 0.10### | 3.99 ± 0.48### | 125.16 ± 13.28### | 147.39 ± 12.02### |
∗∗∗p < 0.001 versus chow-fed mice. ###p < 0.001 versus sh-TRAF6 + sh-NC. Measurement data were expressed in the form of mean ± standard deviation. Data between two groups were compared utilizing independent sample t test. n = 10.
H&E and oil red O staining demonstrated that silencing of TRAF6 contributed to fatty liver degeneration, ballooning degeneration, and decreased infiltration of mixed inflammatory cells in lobules in the liver tissues, along with decreased neutral fat deposition. However, these changes were counteracted after simultaneous silencing of TRAF6 and PPARα; there was no significant difference in the above indicators between mice treated with sh-NC and HFD-fed mice (Figures 9D and 9E, p > 0.05). Additionally, there was a significant decline in the content of TG and TC upon silencing of TRAF6 (p < 0.001), but these markers were elevated after co-silencing of TRAF6 plus PPARα (p < 0.001); there was no significant difference in the content of TG and TC between mice treated with sh-NC and HFD-fed mice (Figure 9F, p < 0.001). Taken together, TRAF6 could promote fatty liver development by regulating the EZH2/miR-429/PPARα axis.
Discussion
The development of fatty liver carries an increasing risk of cardiovascular diseases, type 2 diabetes, and advanced liver diseases.16 Emerging advances in understanding the molecular mechanism behind fatty liver diseases have identified multiple therapeutic biomarkers that ultimately may help in the development of preventative methods or new treatment options.17,18 Of note, aberrantly expressed E3 ubiquitin ligases may exert beneficial or detrimental functions in fatty liver by regulating liver-related miRNAs and interaction with mRNAs.19,20 Additionally, TRAF6, an E3 ubiquitin ligase, has been documented to modulate ubiquitination of pertinent targets and subsequently alter liver function.21 Based on the present results, it is clear that a HFD upregulates the TRAF6-mediated ubiquitination of EZH2, which promotes miR-429-mediated inhibition of PPARα, leading to cholesterol accumulation in the liver and development of fatty liver.
Prior evidence provided by Gallot et al.22 has identified high expression of TRAF6 in response to a HFD and highlighted that ablation of TRAF6 in adipocytes caused a relative decline in the body weight gain triggered by a HFD. Present results are consistent with these findings in showing that a HFD resulted in upregulated expression of TRAF6 in the mouse model of fatty liver disease. Moreover, others have shown that hydrogenated fat intake augments the expression of TRAF6 in white adipose tissue.23 Furthermore, our present observations have revealed that shRNA-mediated silencing of TRAF6 impeded cholesterol accumulation in liver cells. The repression of TRAF6 was previously revealed to abrogate nuclear factor κB (NF-κB) transcriptional activity and the secretion of pro-inflammatory cytokines.24 In the presence of HFD-induced liver inflammation and fibrosis, enhanced hepatocyte TRAF6 expression resulted in pro-inflammatory and profibrogenic reactions, suggesting a supporting role of TRAF6 in nonalcoholic steatohepatitis.25 Accordingly, the downregulation of TRAF6 signaling might cause an appreciable decline in the excessive inflammation induced by a HFD in obesity.26
We extended our mechanistic findings to determine how TRAF6 affected the progression of fatty liver disease. The experimental data unveiled that TRAF6 accelerated the ubiquitination of methyltransferase EZH2 and diminished its net expression levels. There is prior evidence that EZH2 is responsible for adipogenesis through repressing the Wnt genes during adipogenesis.27 EZH2 plays an essential role in the maintenance of both the proliferative and self-renewal capacities of hepatic stem/progenitor cells.28 In a rat model of nonalcoholic fatty liver disease, the aberrant downregulation of EZH2 was associated with lipid accumulation and pro-inflammatory reactions, which accelerated the hepatic steatosis and inflammation.29
Additionally, we found in the present study that EZH2 bound to the miR-429 promoter and repressed miR-429 expression by promoting H3K27me3. This finding corroborates that of a previous study, which demonstrated that TRAF6 facilitated ubiquitination and degradation of EZH2 to diminish EZH2 and H3K27me3 levels, while the silencing of TRAF6 reduced the EZH2 ubiquitination resulting an elevation in EZH2 expression.10 A recent study highlighted that activated EZH2 depleted the levels of miR-200 family members (miR-200b, miR-200a, and miR-429).30 The study of Wang et al.12 also illuminated that EZH2 restricted miR-429 expression and that ablation of EZH2 could restore the miR-429 expression in vitro and in vivo. Hu et al.31 suggested that leptin-triggered hepatic lipid metabolism and increased liver weight were positively associated with elevated miR-429. Further evidence has revealed that artificial ablation of miR-200c could diminish the expression of genes related to hepatic proinflammation and fibrosis in HFD-induced nonalcoholic steatohepatitis.32 Moreover, our present mechanistic investigation suggested that miR-429 targeted and negatively regulated PPARα expression. PPARα regulates reverse cholesterol transport by directly mediating its downstream genes, which are the main structure proteins of HDL-C and apolipoproteinA-I,33 and also indirectly regulates ABCA1 through LXRα to promote reverse cholesterol transport.34 Previous evidence has also shown that PPARα is involved in bile acid synthesis to promote cholesterol excretion35 and can regulate the key enzyme CYP7A1 in cholesterol metabolism to affect cholesterol homeostasis.36 Consistent with those findings, PPARα is reported to be responsible for maintaining lipid, cholesterol, and bile acid homeostasis through mediation of genes participating in bile acid synthesis such as CYP7A1 and CYP27A1.37 Others have unmasked that a deficiency of PPARα expression caused significant exacerbation of fatty liver in HFD-fed mice, indicating a potential role of PPARα as a therapeutic biomarker for nonalcoholic steatohepatitis.38 Emerging evidence has identified the involvement of fatty acid metabolism with PPARα target genes and shown that PPARα activation could alleviate steatosis, inflammation, and fibrosis in the liver of animal models of nonalcoholic fatty liver disease.39 Partly in line with our study, Li and other researchers40 found that lipid oxidative stress in HFD-induced nonalcoholic fatty liver disease mice promotes an interaction between apoptosis signal-regulating kinase 1 (ASK1) and TNF receptor factors including TRAF1, TRAF2, and TRAF6, leading to ASK1 deubiquitination and thus increasing protein stability of ASK1. Further, ASK1 could facilitate the occurrence of fatty liver by activating the MKK3/6-p38-MKK4/7-JNK signal cascade. In line with these results, we have substantiated that TRAF6 promoted cholesterol accumulation in liver cells in vitro and fatty liver development in vivo through the EZH2/miR-429/PPARα axis.
Conclusions
Taken together, our findings indicate that the TRAF6-mediated EZH2/miR-429/PPARα axis is likely to prove critical for a better understanding of the development of fatty liver and may guide the development of targeted therapies. TRAF6 upregulated the ubiquitination of EZH2 and promoted miR-429-mediated inhibition of PPARα, leading to cholesterol accumulation in the liver and the occurrence of fatty liver (Figure 10). Although TRAF6-based therapeutics are in their infancy, the present findings are encouraging in suggesting that TRAF6 is indeed a potential therapeutic target and holds some promise for clinical translation for the treatment of fatty liver diseases. We note that we used a glucose concentration of 2 g/L in the Williams’ medium, which is much lower than that in Dulbecco’s modified Eagle’s medium (DMEM). In the next step of this project, we shall undertake experiments with normal glucose concentrations.
Figure 10.

HFD increases the expression of TRAF6
TRAF6 reduces the expression of EZH2 by enhancing the ubiquitination of EZH2, which inhibits H3K27me3 to promote miR-429 expression. miR-429 inhibits the expression of PPARα in the cytoplasm, leading to the development of fatty liver.
Materials and methods
Ethics statement
The study was approved by the medical ethics committee of The Fifth Affiliated Hospital, Southern Medical University. All animal experiments were performed strictly in accordance with recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health.
Animal model establishment
60 male 57BL/6J mice (8 weeks old), purchased from the experimental animal center of Southern Medical University (Guangzhou, P.R. China), were reared with free access to food and water, with alternative 12-h light/dark cycles at 22°C with 70% relative humidity. 10 of these mice were fed a chow diet (conventional laboratory diet: 10% kcal fat, Slacom, Buenos Aires, Argentina, #M01), and 50 mice were fed a HFD (60% kcal fat diet, D12492, Research Diets, New Brunswick, NJ, USA) for 12 consecutive weeks. At the 10th week of feeding, 40 HFD-fed mice were randomly assigned into four groups with 10 mice in each group; one group was not injected with adenovirus, but the other three groups were injected with the corresponding adenovirus (1 × 109 plaque-forming units [PFUs], sh-NC, sh-TRAF6 + sh-NC, sh-TRAF6 + sh-PPARα) via tail vein through Invivofectamine 2.0 (Invitrogen, Carlsbad, CA, USA) that has high transfection efficiency in the liver in vivo. After adenovirus injections for 2 weeks, mice were fasted for 1 night, whereupon their blood was collected and FPG was measured utilizing a blood glucose meter (ACCU-CHEK Performa, Roche Diagnostics GmbH, Mannheim, Germany). Serum was collected through centrifugation at 4°C and 3,000 rpm for 15 min and used for assays of TC, HDL-C, LDL-C, ALT, AST, and FINS. Next, liver tissues were collected, weighed, and stored at −80°C for further use. The weight of mice was measured every week, and the liver index was calculated as the ratio of liver weight to body weight. Based on the relationship between FPG and FINS, HOMA-IR was calculated as follows: HOMA-IR = FPG (mmol/L) × FINS (mU/L)/22.5. All adenoviruses for mouse infections were purchased from Shanghai Sangon Biotechnology (Shanghai, P.R. China), and primer sequence synthesis, plasmid construction, and virus purification were also completed by that company. The experimental steps were carried out according to the instructions.
Cell treatment
C57BL/6J male mice (aged 8 weeks old) were anesthetized with 1% pentobarbital sodium (P3761, Sigma-Aldrich Chemical Company, St. Louis, MO, USA). Next, the portal vein was dissected to expose the inferior vena cava, followed by heart catheterization. Hank’s balanced salt solution (14170112, GIBCO, Carlsbad, CA, USA) containing 0.2 g/L ethylenediamine tetraacetic acid (EDTA) was infused into the mice at a rate of 14 mL/min at 37°C for 5 min, followed by infusion of 0.1% type IV collagenase (17104019, GIBCO, Carlsbad, CA, USA) at a rate of 7 mL/min for 5 min. The hepatocytes were dispersed in DMEM (10569044, GIBCO, Carlsbad, CA, USA) supplemented with 2% fetal bovine serum (FBS; 10099141, GIBCO, Carlsbad, CA, USA), 100 μL/mL penicillin, and 100 μg/mL streptomycin (15070063, GIBCO, Carlsbad, CA, USA), as well as 4.5 g/L glucose. After the hepatocytes were filtered through a 40 μm filter (Falcon; Becton Dickinson, Franklin Lakes, NJ, USA), the cell suspension was centrifuged twice for 5 min each time. After trypan blue dye exclusion, the survival rate of the isolated hepatocytes was about 85%–95%. Hepatocytes were seeded in a 6-well plate (Corning Glass Works, Corning, NY, USA) at a density of 8 × 105 cells/well and then cultured in a 37°C incubator with 5% CO2 along with Williams’ medium E (W4125, Sigma-Aldrich Chemical Company, St. Louis, MO, USA) that contained 0.11 g/L sodium pyruvate, 1× ITS-X (51500056, GIBCO, Carlsbad, CA, USA), 100 U/mL penicillin, 100 μg/mL streptomycin, 10% FBS, and 2 g/L glucose.
To construct a nonalcoholic fatty liver cell model, we dissolved OA (O1008, Sigma-Aldrich Chemical Company, St. Louis, MO, USA) and PA (292125, Sigma-Aldrich Chemical Company, St. Louis, MO, USA) in 0.1 M NaOH at 70°C and then stored them at −20°C. On the day before use, OA/PA working solution was prepared with 1% serum-free bovine serum albumin (BSA; B2064, Sigma-Aldrich Chemical Company, St. Louis, MO, USA). After 24 h of adenovirus infection, hepatocytes used for cell model establishment were treated with 1.32 mM OA + 0.66 mM PA for 48 h, while hepatocytes used as control were cultured under the same condition except with addition of 1.32 mM OA + 0.66 mM PA. Before the OA/PA treatment, 10 different MOI adenoviruses were used to infect hepatocytes for 24 h. The adenoviruses included sh-NC, sh-TRAF6-1 (sh-TRAF6), sh-TRAF6-2, oe-NC, oe-EZH2, sh-EZH2-1 (sh-EZH2), sh-EZH2-2, sh-TRAF6 + sh-NC, sh-TRAF6 + sh-EZH2, sh-NC + mimic-NC, sh-TRAF6 + mimic-NC, sh-TRAF6 + miR-429 mimic, mimic-NC, miR-429 mimic, inhibitor-NC, miR-429 inhibitor, sh-PPARα-1 (sh-PPARα), sh-PPARα-2, inhibitor-NC + sh-NC, miR-429 inhibitor + sh-NC, miR-429 inhibitor + sh-PPARα, and sh-TRAF6 + sh-PPARα. All adenoviruses for cell infection were purchased from Shanghai Sangon Biotechnology (Shanghai, P.R. China), and synthesis of the primer sequences, plasmid construction, and virus purification were completed by that company. The experimental steps were carried out in strict accordance with the instructions.
HEK293T cells, human normal hepatocyte HL-7702, mouse normal hepatocyte NCTC 1469, and AML-12 purchased from American Type Culture Collection (ATCC, VA, USA), were cultured in high-glucose DMEM (10569044, GIBCO, Carlsbad, CA, USA) supplemented with 10% FBS (10099141, GIBCO, Carlsbad, CA, USA) and 1% penicillin streptomycin (15070063, GIBCO, Carlsbad, CA, USA) and incubated in a cell incubator at 37°C with 5% CO2.
Enzyme-linked immunosorbent assay (ELISA)
Serum of mice in each group was collected, and TNF-α expression was measured according to the instructions of the TNF-α ELISA kit (MTA00B, R&D Systems, USA). In brief, the absorbance (A) values of each well at 450 nm were measured using a multimode microplate reader (Synergy 2, BioTek, USA), and optical density of each well was measured and normalized to the blank control. Regression equation of the standard curve was calculated by taking the concentration of the standard substance as x-coordinate and the value of A as y-coordinate. Then, the value of sample A was substituted into the equation to calculate the concentration of the target protein.
Determination of metabolites
After transfection, liver cells or tissues of mice were collected in phosphate-buffered saline (PBS) and sonicated. TC and FFA were extracted with chloroform/isopropanol/NP-40 (7:11:0.1 v/v/v) or chloroform-Triton X-100 (1% Triton X-100). Then, the organic phase (substrate) was collected for vacuum drying followed by 10 min-high-speed revolution in a microcentrifuge to remove trace amounts of chloroform. The extracts were dissolved again for further experimentation.
TC (A111-1-1), FFA (A042-1), HDL-C (A112-1-1), and LDL-C (A113-1-1) in serum, liver homogenate, and hepatocytes were determined, followed by detection of ALT (C009-3-1), AST (c010-3-1), and FINS (H203) in serum. All the detection kits were purchased from NanJing JianCheng Bioengineering Institute (Nanjing, P.R. China).
H&E, oil red O, and filipin staining
Liver tissues were fixed overnight in 4% paraformaldehyde solution, dehydrated in 20% sucrose water, and then frozen and embedded for examination by optical coherence tomography. Afterward, the tissues were sectioned, stained with oil red O and filipin, paraffin-embedded, and then stained with H&E to detect the pathological changes of liver tissues. In brief, the complete sections were dried at room temperature, fixed at room temperature for 30 s, stained with hematoxylin (at 60°C) for 60 s, and immersed in 1% hydrochloric acid alcohol differentiation solution. Following eosin staining for 3 min, the sections were dehydrated with 70%, 80%, and 95% ethanol and then anhydrous ethanol for 5 min each and then cleared 3 times with xylene for 5 min each. Finally, the sections were sealed with gum and observed and photographed under a microscope (BX63, Olympus, Tokyo, Japan).
Oil red O staining was performed to detect lipid accumulation in hepatocytes. In brief, the treated hepatocytes were washed with PBS, fixed with 4% paraformaldehyde for 5 min, and then incubated in 60% isopropanol solution containing 0.5% oil red O for 30 min. Finally, the hepatocytes were observed and photographed under a microscope (BX63, Olympus, Tokyo, Japan).
Free cholesterol in liver tissues and hepatocytes was detected by filipin staining. The samples were fixed in 4% paraformaldehyde for 30 min. Fresh filipin solution prepared with a filipin fluorescence staining kit (GMS80059.1 v.A, Genemed Biotechnologies, South San Francisco, CA, USA) was applied to stain the samples for 30 min. Phenylenediamine/glycerin was added to the samples, which were then fixed with a coverslip. The experimental steps were carried out according to the instructions of the kit. Image-Pro software was used to quantify the fluorescence intensity.
Nile red assay
The accumulated lipids and lipid droplets in cells were detected with the Nile red assay. Nile red powder (N1142, Thermo Fisher Scientific, Rockford, IL, USA) was dissolved into 1 mM in dimethyl sulfoxide (D2650, Sigma-Aldrich Chemical Company, St. Louis MO, USA) and stored at −20°C for later use. Liver cells after different treatments were cultured using medium added with 1 μM Nile red dye (diluted from 1 mM stock solution) in the dark for 10 min followed by two PBS washes. Then, the cells were fixed with 4% paraformaldehyde at room temperature for 30 min, washed twice with PBS, and photographed under a microscope (BX63, Olympus Optical, Tokyo, Japan).
qRT-PCR
Trizol (16096020, Thermo Fisher Scientific, USA) was utilized to extract total RNA from tissues or cells. miRNA was synthesized using a miRNA first strand complementary DNA (cDNA) synthesis kit (B532451, Shanghai Sangon Biotechnology, Shanghai, P.R. China) and mRNA using a mRNA first strand scDNA synthesis kit (D7168L, Beyotime Biotechnology, Shanghai, P.R. China) following the manufacturer’s instructions. qRT-PCR was carried out with a qRT-PCR kit (Q511-02, Vazyme Biotechnology, Nanjing, P.R. China) in strict accordance with the instructions. The internal inference for miR-429 was U6, while that for TRAF6, EZH2, and PPARα was β-actin. A miRNA reverse transcription kit provided U6 primers and general downstream primers. Primer sequences for TRAF6, EZH2, PPARα, and β-actin were designed and provided by Shanghai Sangon Biotechnology (Shanghai, P.R. China; Table 3). The 2–ΔΔCt method was applied to calculate the fold changes in gene expression.
Table 3.
Primer sequences for qRT-PCR
| Target gene | Primer sequence (5′-3′) |
|---|---|
| TRAF6 | F: AAAGCGAGAGATTCTTTCCCTG |
| R: ACTGGGGACAATTCACTAGAGC | |
| TRAF1 | F: GCCACCTCTATCCACCAGA R: ACAATGCCTGTCCGTGCAAA |
| TRAF2 | F: CTGTCCCTCTTCTTTGTGG R: CATTAAGGTCACCTTCTGGT |
| TRAF3 | F: AGTCAGGTTCCGATGATCG R: GACAAGTGTGCACTCAACTC |
| TRAF4 | F: CTACGTCATCTACAGGGCC R: CAAGTGTGCAGGTAGATCAC |
| TRAF5 | F: CAATGGCTTATTCAGAAGAGCA R: CCAAGGAAATGGAGTTGCC |
| TRAF7 | F: ATGAGCTCAGGCAAGAGTG R: CGTTTCCATTCTGGTCCCT |
| EZH1 | F: GATGGAAAGCAGGATGACAG R: GTTCTCGATACCTTCAATAGCA |
| EZH2 | F: AGTGACTTGGATTTTCCAGCAC |
| R: AATTCTGTTGTAAGGGCGACC | |
| miR-429 | TAATACTGTCTGGTAATGCCGT |
| PPARα | F: AACATCGAGTGTCGAATATGTGG |
| R: AGCCGAATAGTTCGCCGAAAG | |
| β-actin | F: GGCTGTATTCCCCTCCATCG |
| R: CCAGTTGGTAACAATGCCATGT |
miR, microRNA; TRAF6, TNF receptor associated factor 6; EZH2, enhancer of the zeste homolog 2; F, forward; R, reverse.
Western blot analysis
The radioimmunoprecipitation assay (RIPA) lysate containing phenylmethanesulfonyl fluoride (PMSF) (P0013B, Beyotime Biotechnology, Shanghai, P.R. China) was applied to lyse tissues and cells, with the total protein extracted according to the instructions provided on the kit (P0028, Beyotime Biotechnology, Shanghai, P.R. China). After that, 8%–12% sodium dodecyl sulfate (SDS) gels were prepared. After electrophoretic separation, the proteins were transferred from the gel to a polyvinylidene fluoride membrane (1620177, Bio-Rad Laboratories, Hercules, CA, USA), followed by blockade with 5% skim milk or 5% BSA at room temperature for 1 h. Next, the membrane was incubated overnight at 4°C with primary rabbit anti-mouse to β-actin (ab8227, 1:5,000, Abcam, UK), TRAF6 (ab33915, 1:1,000, Abcam, UK), TRAF1 (4710, 1:1,000, Cell Signaling Technology, P.R. China), TRAF2 (ab126758, 1:1,000, Abcam, UK), TRAF3 (ab239357, 1:1,000, Abcam, UK), TRAF4 (ab108991, 1:1,000, Abcam, UK), TRAF5 (41658, 1:1,000, Cell Signaling Technology, P.R. China), TRAF7 (11780-1-AP, 1:1,000, Proteintech, USA), EZH1 (ab189833, 1:1,000, Abcam, UK), PPARα (ab215270, 1:1,000, Abcam, UK), SREBP2 (ab30682, 1:1,000, Abcam, UK), HMGCR (ab174830, 1:1,000, Abcam), SM (12544-1-AP, 1:1,000, Proteintech, USA), NPC1L1 (NB400-128, 1:1,000, Novus Biologicals, UK), ABCA1 (ab66217, 1:1,000, Abcam, USA), ABCG1 (ab52617, 1:1,000, Abcam, UK), ABCG5 (27722-1-AP, 1:1,000, Proteintech, USA), ABCG8 (ab223056, 1:1,000, Abcam, UK), ACAT1 (ab168342, 1:1,000, Abcam, UK), and ACAT12 (ab131215, 1:1,000, Abcam, UK), as well as mouse anti-mouse EZH2 (3147, 1:1,000, Cell Signaling Technology, China) and CPT1 (ab128568, 1:1000, Abcam, UK). The next day, the membranes were incubated at room temperature for 1 h with horseradish peroxidase (HRP)-labeled secondary goat anti-rabbit to immunoglobulin G (IgG; ab6721, 1:5,000) or rabbit anti-mouse to IgG (ab6728, 1:5,000). All the aforementioned antibodies were purchased from Abcam (Cambridge, MA, USA), except for EZH2 (Cell Signaling Technology, Beverly, MA, USA). The membrane was immersed in enhanced chemiluminescence (ECL) reaction solution (1705062, Bio-Rad Laboratories, Hercules, CA, USA) at room temperature for 1 min. The excess liquid was removed, and the membrane was covered with plastic wrap. Exposure imaging was performed on the protein bands using the Image Quant LAS 4000C gel imager (GE Company, NY, USA). β-actin was used as the internal reference for the total protein, and the ratio of gray value of target band to that of internal reference band was considered to be the relative protein expression.
Immunohistochemistry
The paraffin-embedded liver tissue sections were baked at 60°C for 20 min and immersed in xylene solution (twice, 15 min each). Following treatment with anhydrous alcohol hydration (twice, 5 min each), the sections were successively hydrated in 70% and 95% alcohol for 10 min each. Each section was then immersed with 3% H2O2 for 10 min at room temperature to block endogenous peroxidase. Following addition of citric acid buffer, the sections were boiled in a microwave oven for 3 min, treated with antigen repair solution, and allowed to stand at room temperature for 10 min. Following blockade with normal goat serum blocking solution (Shanghai Sangon Biotechnology, Shanghai, P.R. China) at room temperature for 20 min, the sections were incubated overnight at 4°C with diluted primary rabbit anti-mouse antibodies to TRAF6 (ab33915, 1:1,000), EZH2 (5246, 1:300, Cell Signaling Technology, Beverly, MA, USA), and PPARα (ab215270, 1:1,000). The following day, the sections were incubated with goat anti-rabbit to IgG (ab6721, 1:5,000) for 30 min, and then stored in a 37°C incubator for 30 min along with streptavidin biotin-peroxidase complex (Vector Laboratories, Burlingame, CA, USA). A diaminobenzidine color developing kit (Sigma-Aldrich Chemical Company, St. Louis, MO, USA) was used for color development for 6 min. After 30 s of staining in hematoxylin, the sections were dehydrated with 70%, 80%, 90%, and 95% ethanol and then anhydrous ethanol for 2 min, respectively. Finally, the sections were cleared with xylene (twice, 5 min each) and observed under an upright microscope (BX63, Olympus, Tokyo, Japan).
coIP
The interaction between endogenous TRAF6 and EZH2 protein was detected by coIP. The cells were lysed with Pierce IP buffer (1% Triton X-100, 150 mM NaCl, 1 mM EDTA, and 25 mM Tris HCl pH 7.5), followed by addition of protease and phosphatase inhibitors. The lysate was incubated overnight at 4°C with rabbit anti-mouse to EZH2 (5246, 1:300, Cell Signaling Technology, Beverly, MA, USA) and rabbit antibody to IgG (ab172730, 1:100, Abcam, Cambridge, MA, USA). The following day, the lysate was supplemented with protein G beads Dynabeads (Thermo Fisher Scientific, Rockford, IL, USA), followed by slow rotation at 4°C for 8 h, and then western blot analysis.
In vivo ubiquitination assay
For the determination of endogenous EZH2-ubiquitin (Ub), 10 μM MG132 (HY-13259, MedChemExpress, Monmouth Junction, NJ, USA) was added to cells infected with virus, followed by incubation for 6 h. Cells were lysed in RIPA buffer containing 1% SDS and treated with ultrasound. IP reaction was carried out; EZH2 (5246, 1:300, Cell Signaling Technology, Beverly, MA, USA) was co-incubated overnight with diluted cell lysate at 4°C. The following day, protein G beads were added to the cells for incubation of 8 h at 4°C. Following three rinses in IP buffer, rabbit anti-mouse to Ub (ab7780, 1:1,000, Abcam, Cambridge, MA, USA) was used in western blot analysis to detect the ubiquitination level of EZH2.
ChIP
ChIP was conducted using a ChIP kit (P2078, Beyotime Biotechnology, Shanghai, P.R. China) according to the instructions of the kit. Rabbit antibody against IgG (ab172730, 1:100, Abcam, Cambridge, MA, USA) was added to the NC group, rabbit anti-mouse against EZH2 (5246, 1:200, CST, Beverly, MA, USA) to the anti-EZH2 group, and rabbit anti-mouse against (ab192985, 1:100, Abcam, Cambridge, MA, USA) to the anti-trimethylation of histone 3 lysine 27 (H3K27me3) group. Finally, the precipitated miR-429 promoter was analyzed utilizing qRT-PCR. The miR-429 ChIP primer was F: 5′-GCTGTGGGTCTGTGGGGTCT-3′; R: 5′- TTTGGAGCAATGAAGGGACC-3′.
Dual luciferase reporter gene assay
Through the online analysis website (http://mirdb.org/), we predicted the targeting binding site between miR-429 and PPARα mRNA 3′ UTR. WT and MUT PPARα mRNA 3′ UTR gene fragments (PPARα WT and PPARα MUT) binding to miR-429 were synthesized and respectively introduced into a pGL3-basic vector (E1751, Promega, Madison, WI, USA). After restriction endonuclease digestion, T4 DNA ligase (M0204S, New England Biolabs, Beverly, MA, USA) was used to insert the target fragments into the pGL3 vector. The Renilla luciferase and constructed luciferase reporter plasmids were co-transfected into HEK293T cells with mimic-NC and miR-429 mimic, respectively. The cells were collected and lysed after 48 h of transfection in a 37°C incubator with 5% CO2 and saturated humidity. The luciferase activity was detected on the Glomax 20/20 Luminometer detector (E5311, Promega, Madison, WI, USA) using a dual luciferase reporter gene assay kit (E1910, Promega, Madison, WI, USA). With Renilla luciferase activity as the internal reference, the relative luciferase activity (Ratio) was regarded as the ratio of luciferase activity (RLU1) to luciferase activity (RLU2): Ratio = RLU1/RLU2. All vectors were constructed by Shanghai Sangon Biotechnology (Shanghai, P.R. China).
Statistical analysis
The SPSS 21.0 (IBM, Armonk, NY, USA) was employed for data analysis. Measurement data were expressed in the form of mean ± standard deviation. Data between two groups were compared utilizing independent sample t test, while data among multiple groups were compared with the use of one-way analysis of variance (ANOVA), followed by a Tukey’s test. Data among multiple groups at variant time points were analyzed through repeated-measures ANOVA or two-way ANOVA, followed by Bonferroni post hoc test. A value of p <0.05 was regarded as a statistically significant difference.
Acknowledgments
We would like express our sincere appreciation to the reviewers for critical comments on this article. Funding: This study is supported by Science and Technology Innovation Project of Guangdong Province (Basic and Applied Basic Research Foundation of Natural Science Foundation of Guangdong Province) (No. 2021A1515011040).
Author contributions
Z.Z., H.W., and B.P. designed the experiment; Z.Z., H.W., and J.W. performed experiments; B.P., J.W., and F.Z. analyzed the data; and Z.Z., H.W., and F.Z. wrote the paper. All authors have read and approved the final submitted manuscript.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtn.2021.01.026.
Supplemental information
References
- 1.Peng Q., Chen B., Wang H., Zhu Y., Wu J., Luo Y., Zuo G., Luo J., Zhou L., Shi Q. Bone morphogenetic protein 4 (BMP4) alleviates hepatic steatosis by increasing hepatic lipid turnover and inhibiting the mTORC1 signaling axis in hepatocytes. Aging (Albany NY) 2019;11:11520–11540. doi: 10.18632/aging.102552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Karanjia R.N., Crossey M.M., Cox I.J., Fye H.K., Njie R., Goldin R.D., Taylor-Robinson S.D. Hepatic steatosis and fibrosis: Non-invasive assessment. World J. Gastroenterol. 2016;22:9880–9897. doi: 10.3748/wjg.v22.i45.9880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Xi Y., Li H. Role of farnesoid X receptor in hepatic steatosis in nonalcoholic fatty liver disease. Biomed. Pharmacother. 2020;121:109609. doi: 10.1016/j.biopha.2019.109609. [DOI] [PubMed] [Google Scholar]
- 4.Idilman I.S., Ozdeniz I., Karcaaltincaba M. Hepatic steatosis: Etiology, patterns, and quantification. Semin. Ultrasound CT MR. 2016;37:501–510. doi: 10.1053/j.sult.2016.08.003. [DOI] [PubMed] [Google Scholar]
- 5.Chen Y., Chen Y., Zhao L., Chen Y., Mei M., Li Q., Huang A., Varghese Z., Moorhead J.F., Ruan X.Z. Inflammatory stress exacerbates hepatic cholesterol accumulation via disrupting cellular cholesterol export. J. Gastroenterol. Hepatol. 2012;27:974–984. doi: 10.1111/j.1440-1746.2011.06986.x. [DOI] [PubMed] [Google Scholar]
- 6.Ferramosca A., Zara V. Modulation of hepatic steatosis by dietary fatty acids. World J. Gastroenterol. 2014;20:1746–1755. doi: 10.3748/wjg.v20.i7.1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Muppidi J.R., Tschopp J., Siegel R.M. Life and death decisions: secondary complexes and lipid rafts in TNF receptor family signal transduction. Immunity. 2004;21:461–465. doi: 10.1016/j.immuni.2004.10.001. [DOI] [PubMed] [Google Scholar]
- 8.Ha H., Kwak H.B., Le S.W., Kim H.H., Lee Z.H. Lipid rafts are important for the association of RANK and TRAF6. Exp. Mol. Med. 2003;35:279–284. doi: 10.1038/emm.2003.38. [DOI] [PubMed] [Google Scholar]
- 9.Aslani M.R., Keyhanmanesh R., Khamaneh A.M., Abbasi M.M., Fallahi M., Alipour M.R. Tracheal overexpression of IL-1β, IRAK-1 and TRAF-6 mRNA in obese-asthmatic male Wistar rats. Iran. J. Basic Med. Sci. 2016;19:350–357. [PMC free article] [PubMed] [Google Scholar]
- 10.Lu W., Liu S., Li B., Xie Y., Izban M.G., Ballard B.R., Sathyanarayana S.A., Adunyah S.E., Matusik R.J., Chen Z. SKP2 loss destabilizes EZH2 by promoting TRAF6-mediated ubiquitination to suppress prostate cancer. Oncogene. 2017;36:1364–1373. doi: 10.1038/onc.2016.300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Margueron R., Reinberg D. The Polycomb complex PRC2 and its mark in life. Nature. 2011;469:343–349. doi: 10.1038/nature09784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang Y., Guo W., Li Z., Wu Y., Jing C., Ren Y., Zhao M., Kong L., Zhang C., Dong J. Role of the EZH2/miR-200 axis in STAT3-mediated OSCC invasion. Int. J. Oncol. 2018;52:1149–1164. doi: 10.3892/ijo.2018.4293. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 13.Alisi A., Da Sacco L., Bruscalupi G., Piemonte F., Panera N., De Vito R., Leoni S., Bottazzo G.F., Masotti A., Nobili V. Mirnome analysis reveals novel molecular determinants in the pathogenesis of diet-induced nonalcoholic fatty liver disease. Lab. Invest. 2011;91:283–293. doi: 10.1038/labinvest.2010.166. [DOI] [PubMed] [Google Scholar]
- 14.Hu K., Liu L., Qian H., Zhou T., Li Y., Yu J., Tan B. [Alpinetin promotes the binding of PPAR and methyltransferase] Xibao Yu Fenzi Mianyixue Zazhi. 2017;33:1610–1614. [PubMed] [Google Scholar]
- 15.Wu Z., Zhang Y., Gong X., Cheng G., Pu S., Cai S. The preventive effect of phenolic-rich extracts from Chinese sumac fruits against nonalcoholic fatty liver disease in rats induced by a high-fat diet. Food Funct. 2020;11:799–812. doi: 10.1039/c9fo02262g. [DOI] [PubMed] [Google Scholar]
- 16.Kotronen A., Yki-Järvinen H. Fatty liver: a novel component of the metabolic syndrome. Arterioscler. Thromb. Vasc. Biol. 2008;28:27–38. doi: 10.1161/ATVBAHA.107.147538. [DOI] [PubMed] [Google Scholar]
- 17.Samuel V.T., Liu Z.X., Qu X., Elder B.D., Bilz S., Befroy D., Romanelli A.J., Shulman G.I. Mechanism of hepatic insulin resistance in non-alcoholic fatty liver disease. J. Biol. Chem. 2004;279:32345–32353. doi: 10.1074/jbc.M313478200. [DOI] [PubMed] [Google Scholar]
- 18.Lieber C.S. Alcoholic fatty liver: its pathogenesis and mechanism of progression to inflammation and fibrosis. Alcohol. 2004;34:9–19. doi: 10.1016/j.alcohol.2004.07.008. [DOI] [PubMed] [Google Scholar]
- 19.Ghosh M., Niyogi S., Bhattacharyya M., Adak M., Nayak D.K., Chakrabarti S., Chakrabarti P. Ubiquitin ligase cop1 controls hepatic fat metabolism by targeting atgl for degradation. Diabetes. 2016;65:3561–3572. doi: 10.2337/db16-0506. [DOI] [PubMed] [Google Scholar]
- 20.Zhang T., Kho D.H., Wang Y., Harazono Y., Nakajima K., Xie Y., Raz A. Gp78, an E3 ubiquitin ligase acts as a gatekeeper suppressing nonalcoholic steatohepatitis (NASH) and liver cancer. PLoS ONE. 2015;10:e0118448. doi: 10.1371/journal.pone.0118448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cheng K.K., Lam K.S., Wang Y., Wu D., Zhang M., Wang B., Li X., Hoo R.L., Huang Z., Sweeney G., Xu A. TRAF6-mediated ubiquitination of APPL1 enhances hepatic actions of insulin by promoting the membrane translocation of Akt. Biochem. J. 2013;455:207–216. doi: 10.1042/BJ20130760. [DOI] [PubMed] [Google Scholar]
- 22.Gallot Y.S., McMillan J.D., Xiong G., Bohnert K.R., Straughn A.R., Hill B.G., Kumar A. Distinct roles of TRAF6 and TAK1 in the regulation of adipocyte survival, thermogenesis program, and high-fat diet-induced obesity. Oncotarget. 2017;8:112565–112583. doi: 10.18632/oncotarget.22575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.de Oliveira J.L., Oyama L.M., Hachul A.C., Biz C., Ribeiro E.B., Oller do Nascimento C.M., Pisani L.P. Hydrogenated fat intake during pregnancy and lactation caused increase in TRAF-6 and reduced AdipoR1 in white adipose tissue, but not in muscle of 21 days old offspring rats. Lipids Health Dis. 2011;10:22. doi: 10.1186/1476-511X-10-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zheng C.Z., Shu Y.B., Luo Y.L., Luo J. The role of miR-146a in modulating TRAF6-induced inflammation during lupus nephritis. Eur. Rev. Med. Pharmacol. Sci. 2017;21:1041–1048. [PubMed] [Google Scholar]
- 25.Wang Y., Wen H., Fu J., Cai L., Li P.L., Zhao C.L., Dong Z.F., Ma J.P., Wang X., Tian H. Hepatocyte tnf receptor-associated factor 6 aggravates hepatic inflammation and fibrosis by promoting lysine 6-linked polyubiquitination of apoptosis signal-regulating kinase 1. Hepatology. 2020;71:93–111. doi: 10.1002/hep.30822. [DOI] [PubMed] [Google Scholar]
- 26.Chatzigeorgiou A., Seijkens T., Zarzycka B., Engel D., Poggi M., van den Berg S., van den Berg S., Soehnlein O., Winkels H., Beckers L. Blocking CD40-TRAF6 signaling is a therapeutic target in obesity-associated insulin resistance. Proc. Natl. Acad. Sci. USA. 2014;111:2686–2691. doi: 10.1073/pnas.1400419111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang L., Jin Q., Lee J.E., Su I.H., Ge K. Histone H3K27 methyltransferase Ezh2 represses Wnt genes to facilitate adipogenesis. Proc. Natl. Acad. Sci. USA. 2010;107:7317–7322. doi: 10.1073/pnas.1000031107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Aoki R., Chiba T., Miyagi S., Negishi M., Konuma T., Taniguchi H., Ogawa M., Yokosuka O., Iwama A. The polycomb group gene product Ezh2 regulates proliferation and differentiation of murine hepatic stem/progenitor cells. J. Hepatol. 2010;52:854–863. doi: 10.1016/j.jhep.2010.01.027. [DOI] [PubMed] [Google Scholar]
- 29.Vella S., Gnani D., Crudele A., Ceccarelli S., De Stefanis C., Gaspari S., Nobili V., Locatelli F., Marquez V.E., Rota R., Alisi A. EZH2 down-regulation exacerbates lipid accumulation and inflammation in in vitro and in vivo NAFLD. Int. J. Mol. Sci. 2013;14:24154–24168. doi: 10.3390/ijms141224154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ning X., Shi Z., Liu X., Zhang A., Han L., Jiang K., Kang C., Zhang Q. DNMT1 and EZH2 mediated methylation silences the microRNA-200b/a/429 gene and promotes tumor progression. Cancer Lett. 2015;359:198–205. doi: 10.1016/j.canlet.2015.01.005. [DOI] [PubMed] [Google Scholar]
- 31.Hu Y., Zhang R., Zhang Y., Li J., Grossmann R., Zhao R. In ovo leptin administration affects hepatic lipid metabolism and microRNA expression in newly hatched broiler chickens. J. Anim. Sci. Biotechnol. 2012;3:16. doi: 10.1186/2049-1891-3-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tran M., Lee S.M., Shin D.J., Wang L. Loss of miR-141/200c ameliorates hepatic steatosis and inflammation by reprogramming multiple signaling pathways in NASH. JCI Insight. 2017;2:e96094. doi: 10.1172/jci.insight.96094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tayyeb J.Z., Popeijus H.E., Mensink R.P., Konings M.C.J.M., Mulders K.H.R., Plat J. Amoxicillin modulates apoa-i transcription and secretion, predominantly via pparalpha transactivation inhibition. Int. J. Mol. Sci. 2019;20:5967. doi: 10.3390/ijms20235967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.He X.W., Yu D., Li W.L., Zheng Z., Lv C.L., Li C., Liu P., Xu C.Q., Hu X.F., Jin X.P. Anti-atherosclerotic potential of baicalin mediated by promoting cholesterol efflux from macrophages via the PPARγ-LXRα-ABCA1/ABCG1 pathway. Biomed. Pharmacother. 2016;83:257–264. doi: 10.1016/j.biopha.2016.06.046. [DOI] [PubMed] [Google Scholar]
- 35.Kanemitsu T., Tsurudome Y., Kusunose N., Oda M., Matsunaga N., Koyanagi S., Ohdo S. Periodic variation in bile acids controls circadian changes in uric acid via regulation of xanthine oxidase by the orphan nuclear receptor PPARα. J. Biol. Chem. 2017;292:21397–21406. doi: 10.1074/jbc.M117.791285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Marrapodi M., Chiang J.Y. Peroxisome proliferator-activated receptor alpha (PPARalpha) and agonist inhibit cholesterol 7alpha-hydroxylase gene (CYP7A1) transcription. J. Lipid Res. 2000;41:514–520. [PubMed] [Google Scholar]
- 37.Ghonem N.S., Assis D.N., Boyer J.L. Fibrates and cholestasis. Hepatology. 2015;62:635–643. doi: 10.1002/hep.27744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Abdelmegeed M.A., Yoo S.H., Henderson L.E., Gonzalez F.J., Woodcroft K.J., Song B.J. PPARalpha expression protects male mice from high fat-induced nonalcoholic fatty liver. J. Nutr. 2011;141:603–610. doi: 10.3945/jn.110.135210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pawlak M., Lefebvre P., Staels B. Molecular mechanism of PPARα action and its impact on lipid metabolism, inflammation and fibrosis in non-alcoholic fatty liver disease. J. Hepatol. 2015;62:720–733. doi: 10.1016/j.jhep.2014.10.039. [DOI] [PubMed] [Google Scholar]
- 40.Li D.J., Tong J., Li Y.H., Meng H.B., Ji Q.X., Zhang G.Y., Zhu J.H., Zhang W.J., Zeng F.Y., Huang G. Melatonin safeguards against fatty liver by antagonizing TRAFs-mediated ASK1 deubiquitination and stabilization in a β-arrestin-1 dependent manner. J. Pineal Res. 2019;67:e12611. doi: 10.1111/jpi.12611. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.