

Abstract
Intravenous infusion of high dose (>10 g) vitamin C (IVC) is a common alternative cancer therapy. IVC results in millimolar levels of circulating ascorbate, which is proposed to generate cytotoxic quantities of H2O2 in the presence of transition metal ions. In this study we report on the in vitro and in vivo effects of millimolar ascorbate on erythrocytes. Addition of ascorbate to whole blood increased erythrocyte intracellular ascorbate approximately 35-fold. Within 10 min of ascorbate addition, we detected increased oxidation of erythrocyte peroxiredoxin 2 (Prx2), a major thiol antioxidant protein and a sensitive marker of H2O2 production. Up to 50% of Prx2 was present in the oxidised form after 60 min. The presence of extracellular catalase, removal of plasma or the addition of a metal chelator did not prevent ascorbate-induced Prx2 oxidation, suggesting that the H2O2 responsible for Prx2 oxidation was generated within the erythrocyte. Ascorbate is known to increase the rate of haemoglobin autoxidation and H2O2 production. Through spectral monitoring of oxidised haemoglobin we estimated a generation rate of 15 μM H2O2/min inside erythrocytes. We also investigated changes in erythrocyte ascorbate concentration and Prx2 oxidation following IVC infusion in a cohort of patients with cancer. Plasma ascorbate levels ranged from 7.8 to 35 mM immediately post infusion, while erythrocyte ascorbate levels reached 1.5–3.4 mM 4 h after beginning infusion. Transient oxidation of erythrocyte Prx2 was observed. We conclude that erythrocytes accumulate ascorbate during IVC infusion, providing a significant reservoir of ascorbate, and this ascorbate increases H2O2 generation within the cells. The consequence of increased erythrocyte Prx2 oxidation warrants further investigation.
Keywords: Ascorbate, Haemoglobin, Hydrogen peroxide, Peroxiredoxin, Thiol oxidation, Erythrocytes
Graphical abstract
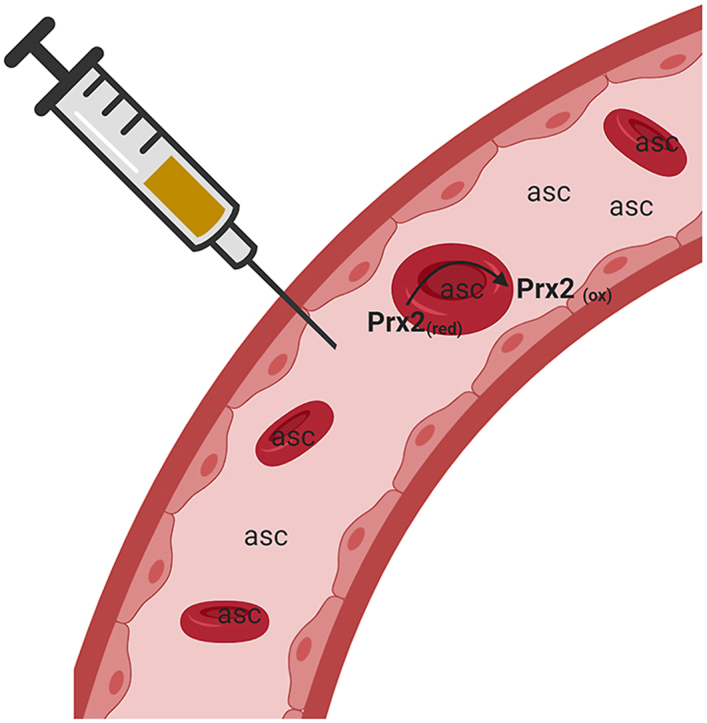
1. Introduction
For many decades patients with cancer have been receiving high-dose intravenous vitamin C infusions as an adjunctive or alternative therapy. Whilst plasma ascorbate concentrations following oral intake rarely exceed 200 μM [1], intravenous administration can result in hundred-fold higher levels [[2], [3], [4], [5]]. Ascorbate is cleared by the kidneys with a half-life of approximately 90 min, therefore normal plasma concentrations are restored within hours of intravenous administration [2,3]. In the presence of oxygen and transition metal ions, ascorbate oxidation can result in the generation of hydrogen peroxide (H2O2). This reaction becomes significant when ascorbate concentrations are in the millimolar range and hence IVC is thought to promote oxidative stress in cancer cells [[6], [7], [8], [9], [10]].
Erythrocytes constitute ~40–50% of the blood volume, with levels in males generally higher than in females. We were therefore interested to investigate the impact of high concentrations of plasma ascorbate on these cells. Haemoglobin (Hb) is the predominant protein in erythrocytes, with the reduced form acting as an oxygen carrier (oxyhaemoglobin, Hb2+O2). Electron transfer from the haem to bound oxygen can occur (Hb3+O2−), resulting in the slow release of superoxide (O2−) [11]. Ascorbate (AscH−) accelerates the rate of autoxidation by donating an additional electron to the Hb3+O2− complex, generating H2O2 [12] (Eq (1)). The methaemoglobin (Hb3+) produced during autoxidation is either reduced back to oxyhaemoglobin by methaemoglobin reductase, or can be further oxidised to unstable products that denature and form Heinz bodies [13].
| Hb2+O2 ↔ Hb3+O2− + AscH− + 2H+ → Hb3+ + H2O2 + AscH.- | (1) |
Erythrocytes have high levels of superoxide dismutase to protect against O2−, and catalase, glutathione peroxidase and peroxiredoxin 2 (Prx2) to reduce H2O2 to water [14,15]. Erythrocyte Prx2 reacts with H2O2 (Eq (2)) with a rate constant of 1 × 108 M−1s−1 [18], and a slow rate of reduction results in accumulation of the oxidised form, detectable as a disulfide–linked intermolecular dimer. It is difficult to determine the contribution of the different erythrocyte peroxidases in removing intracellular H2O2 [47], but accumulation of oxidised Prx2 indicates either an increase in H2O2 levels or impaired reductive mechanisms. We have observed increased Prx2 oxidation in erythrocytes treated with as little as 0.5 μM H2O2 in vitro [14], upon storage of whole blood for transfusion [19], and upon activation of neighbouring neutrophils, both in in vitro and in vivo models [16]. The redox status of erythrocyte Prx2 has not been assessed following exposure to high plasma ascorbate concentrations.
| Prx2(reduced) + H2O2 + 2H+ → Prx2(oxidised) + 2H2O | (2) |
A number of studies have shown that the ascorbate concentration in erythrocytes is very similar to that observed in plasma [[20], [21], [22], [23], [24]]. Mature erythrocytes do not express the sodium-dependent vitamin C transporters SVCT1 and SVCT2 that actively transport vitamin C into many different cell types [24]. Instead, they are thought to be reliant on glucose transporters (GLUTs) for obtaining vitamin C from circulation [25,26]. In this situation, the oxidised form of ascorbate, dehydroascorbate (DHA), enters cells by facilitated diffusion through GLUT1, GLUT3 and GLUT4 [[27], [28], [29]], and is reduced by glutathione-dependent mechanisms that use NADPH derived from the hexose monophosphate shunt [30,31]. The reducing power of NADPH is also required for the thioredoxin and glutathione-based systems responsible for turnover of oxidised Prx2 [15].
In this study we measured the accumulation of ascorbate in human erythrocytes following exposure to the plasma concentrations observed following high-dose IVC treatment. Alongside, we monitored the redox status of erythrocyte Prx2 following exposure to high-dose vitamin C, both in vitro and in patients receiving IVC.
2. Materials and methods
2.1. In vitro experiments
Peripheral blood was collected into lithium heparin vacutainers from healthy volunteers at the University of Otago, Christchurch. Ethical approval was granted by the Southern Health and Disability Ethics Committee (URA/06/12/083/AM05) and all donors provided written informed consent. Whole blood, or isolated erythrocytes resuspended in Hanks’ balanced salt solution at the original blood volume, were treated with the stated doses of ascorbate at 37 °C with rotation. Aliquots were removed at indicated time points and processed for ascorbate analysis, Prx2 oxidation and haemoglobin analysis.
Some in vitro experiments were conducted in a hypoxia chamber to allow manipulation of the oxygen concentration. Whole blood (1 mL) was incubated in a 12-well dish and placed in a H35 Hypoxystation (Don Whitley Scientific Limited, Shipley, UK) at 2% oxygen for 1 h to equilibrate, prior to the addition of the stated doses of ascorbate for 1 h. The control was performed in a regular incubator (21% oxygen) for the same time (i.e. 2 h total).
2.2. Patient samples
Non-fasting blood samples were collected from patients receiving IVC infusions at a general practice in Christchurch, New Zealand. The study was approved by the Northern A Health and Disability Ethics Committee (18/NTA/5), and the 15 participants recruited provided written informed consent. The cohort included 7 females, 8 males; median age 64 y; age range 42–76 y. The participants had a range of diagnoses: multiple myeloma (1), metastatic mesothelioma (2), metastatic breast (4), non-small cell squamous lung carcinoma (2), cervical carcinoma (1), adenocarcinoma metastatic bowel carcinoma (2), prostatic carcinoma (2) and cholangiocarcinoma (1).
The participants received IVC infusions at the doses indicated in the figures, ranging from 25 to 100 g IVC (Ascor L 500®, McGuff Pharmaceuticals, Santa Ana, CA, USA). The participants received different doses, based on the recommendation of the clinician. Vitamin C was administered in 250 mL or 500 mL water bags with a 30–120 min infusion time depending on the dose. The body weights of eleven participants were available to determine calculation of the dose as grams of ascorbate administered per kg body weight.
Baseline and post-infusion blood samples were drawn from the cannulated port used for vitamin C infusion after flushing with saline. For later time points (4 and 24 h), blood samples were collected by venepuncture. Samples for ascorbate analysis were collected into lithium heparin vacutainers (BD Biosciences, Franklin Lakes, NJ, USA). For erythrocyte Prx2 oxidation analysis, N-ethylmaleimide (NEM; Sigma-Aldrich, St. Louis, MO, USA) sufficient to give a final concentration of 100 mM was added to the lithium heparin vacutainers prior to blood collection, thereby avoiding any post-collection oxidation of the reduced Prx2 monomer [32]. The blood samples for ascorbate analysis were placed on ice until processing, while those for Prx2 analysis were incubated at ambient temperature for 15 min prior to dilution in sample buffer as described below.
2.3. Analysis of plasma and erythrocyte ascorbate
Whole blood was centrifuged at 1000×g for 15 min at 4 °C with no brake. The plasma was removed and an aliquot added to an equal volume of ice-cold 0.54 M perchloric acid with 100 μM diethylenetriaminepentaacetic acid (DTPA). After 5 min on ice, the extracts were centrifuged and the acidified plasma supernatants stored at −80 °C until analysis.
Red blood cell ascorbate concentrations were determined using an adaptation of the method of Levine and co-workers [21]. Remaining plasma and the buffy coat layer were removed from the centrifuged blood and discarded. The packed erythrocytes were washed with a 10-fold excess of ice-cold PBS containing 500 μM DTPA, and 150 μL aliquots stored at −80 °C until HPLC analysis. To measure the ascorbate content of the cells, the samples were quickly defrosted and a four times volume of ice-cold milliQ water containing 500 μM DTPA added to lyse the cells. The samples were vortexed thoroughly, and incubated on ice for 2 min. A 200 μL aliquot was applied to a centrifugal filter unit (Amicon Ultra 0.5 mL, 10K Ultracel®, Millipore, Burlington, Massachusetts, USA) and the lysate centrifuged at 14000×g for 20 min at 4 °C to remove haemoglobin. An equal volume of ice-cold 0.54 M perchloric acid containing 100 μM DTPA was immediately added to the ultrafiltrate and the samples vortexed and centrifuged. Samples were incubated with 32 mM of the reducing agent tris(2-carboxyethyl)phosphine hydrochloride (TCEP) for 3 h on ice prior to analysis to reduce any DHA present in the sample [33]. The haemoglobin content of the packed cells was determined using Drabkin's reagent [34] to correct for any differences in the packed cell content between samples [35].
The ascorbate content of the plasma and erythrocyte samples was analysed by reverse-phase HPLC with coulometric electrochemical detection as described previously [36]. A standard curve of sodium ascorbate was prepared daily in 77 mM perchloric acid containing 100 μM DTPA and the ascorbate content of the samples calculated by linear regression after taking into account any dilutions. Total ascorbate levels are reported. Dehydroascorbate, determined from the difference between with and without TCEP reduction, was found to constitute only a small fraction of the total levels.
2.4. Detection of erythrocyte Prx2 oxidation by gel electrophoresis
The blood samples collected into NEM for Prx2 analysis were further diluted 1:500 into non-reducing sample buffer (10% glycerol, 2% SDS, 62.5 mM Tris pH 6.8) and the samples stored at −80 °C. Prx2 oxidation was analysed by running the extracts on non-reducing 12% SDS-polyacrylamide gels (Bio-Rad, Hercules, CA, USA) then transferring to polyvinylidene difluoride membrane for Western blotting [32]. Membranes were probed with an anti-Prx2 antibody (1:10000; catalogue number R8656; Sigma-Aldrich, St. Louis, MO, USA) and the relative band density of the monomer and dimer were quantified using the Alliance Q9 advanced chemiluminescence imager and its associated software (Uvitec, Cambridge, England, UK).
2.5. Measurement of erythrocyte methaemoglobin and oxyhaemoglobin
Aliquots of erythrocytes were lysed in 5 mM sodium phosphate buffer, pH 7.4 with gentle mixing. Samples were centrifuged and the absorbance of the haemolysate determined at 577 nm, 630 nm and 700 nm [13]. The A700 value was subtracted from the A577 and A630 measurements to control for turbidity. The following calculations were then used to determine concentrations of oxyhaemoglobin and methaemoglobin [13]:[Oxyhaemoglobin] = 66A577 – 80A630, [Methaemoglobin] = 279A630 – 3A577
2.6. Statistical analyses
Statistical analyses were carried out using GraphPad Prism version 8.0 (La Jolla, CA, USA). Data are presented as the mean ± SEM as stated, and analysis of data was performed using two-tailed Student's t-test with the p value for statistical significance set at 0.05. Correlations were tested using Spearman's rank order correlation.
3. Results
3.1. Ascorbate accumulates in erythrocytes and promotes Prx2 oxidation
Freshly collected whole blood was incubated with varying concentrations of ascorbate to mimic levels reported following intravenous infusion [[1], [2], [3], [4]]. At initial collection, the intracellular erythrocyte ascorbate concentration was 0.043 ± 0.005 mM (mean, SEM, n = 3). This increased to 1.51 ± 0.03 mM 60 min after the addition of 20 mM ascorbate.
The redox state of erythrocyte Prx2 was assessed by western blotting. Increased oxidation, manifest as the presence of the oxidised dimeric form on non-reducing SDS-PAGE, was observed with addition of increasing concentrations of ascorbate (Fig. 1A and B). Formation of the oxidised dimer was time dependent, with oxidation apparent at 10 min and reaching 40-50% by 60 min (Fig. 1C).
Fig. 1.

Oxidation of Prx2 in erythrocytes incubated with millimolar concentrations of ascorbate. (A, B) Ascorbate (10–30 mM) was added to whole blood for 60 min and Prx2 oxidation assessed by quantification of the oxidised (ox) and reduced (red) Prx2 bands following non-reducing gel electrophoresis. (C) Time-course of erythrocyte Prx2 oxidation with whole blood and 20 mM ascorbate. (D) Whole blood treated with 20 mM ascorbate under 21% and 2% oxygen conditions. (E) Whole blood was incubated with ascorbate for 60 min in the presence or absence of added catalase (10 μg/mL). (F) Ascorbate was added to whole blood or erythrocytes suspended in Hanks balanced salt solution (60 min treatment). (G) Whole blood was incubated with ascorbate for 60 min in the presence or absence of 100 μM DTPA. For all experiments data is from 3 to 5 donors/experiment. A representative blot is shown for A. Error bars for all graphs show SEM, *p < 0.05, by paired t-test.
Incubating erythrocytes with ascorbate in 2% oxygen lowered the extent of Prx2 oxidation, consistent with a role for H2O2 production (Fig. 1D). However, addition of 10 μg/mL of catalase to whole blood had no effect (Fig. 1E), suggesting that Prx2 oxidation was driven by intracellular H2O2. Consistent with this conclusion, plasma constituents were not required as ascorbate-dependent oxidation still occurred in isolated erythrocytes (Fig. 1F), and the extracellular iron chelator DTPA had little effect on Prx2 oxidation (Fig. 1G).
3.2. In vitro incubation of ascorbate with isolated erythrocytes caused haemoglobin oxidation
Since ascorbate appeared to drive Prx2 oxidation within erythrocytes, we tested the ability of high-dose vitamin C to oxidise oxyhaemoglobin to methaemoglobin. Methaemoglobin reductases convert methaemoglobin back to oxyhaemoglobin, using NADH/NADPH. The removal of glucose slows the reductive restoration of oxyhaemoglobin, allowing methaemoglobin to accumulate and the rate of oxidation to be measured. In the absence of glucose, we measured a significant increase in methaemoglobin formation upon incubation of isolated erythrocytes with 20 mM ascorbate (Fig. 2). There was a significant lag phase of approximately 3 h, reflecting the need to deplete endogenous cellular glucose, and then approximately 15% of the oxyhaemoglobin was converted to methaemoglobin (Fig. 2). Since erythrocytes contain 20 mM haemoglobin, we estimate the ascorbate is resulting in the production of approximately 15 μM/min H2O2 within erythrocytes (10% oxidation in final 120 min = 16.7 μM/min H2O2).
Fig. 2.

Ascorbate causes haemoglobin oxidation. Isolated erythrocytes were resuspended in phosphate buffered saline containing 1 mM calcium chloride and 0.5 mM magnesium chloride, either with or without 5.5 mM glucose prior to treatment with 20 mM ascorbate. Error bars show SEM; n = 2. The baseline oxyhaemoglobin concentration of the extract was 74 μM.
3.3. Ascorbate accumulation in erythrocytes following IVC
Ascorbate pharmacokinetics was investigated in five individuals who received IVC infusions. Blood was drawn just prior to and immediately post vitamin C infusion, as well as 4 and 24 h after the infusion began. Plasma and erythrocyte ascorbate concentrations were measured (Fig. 3). Three of the five participants had received IVC infusions 48 h prior to the monitored infusion. The mean ± SEM pre-infusion plasma ascorbate concentration of the five individuals was 79 ± 10 μM. In contrast, the pre-infusion erythrocyte ascorbate concentration of the five participants was 268 ± 100 μM. The three individuals who had IVC infusions 48 h prior to the monitored infusion had substantially higher starting erythrocyte ascorbate concentrations than those who had not had recent infusions (Table 1).
Fig. 3.

Time-course of plasma and erythrocyte ascorbate concentrations after IVC infusion. (A) Plasma and (B) erythrocyte ascorbate concentrations over the 24 h following an IVC infusion in 5 individuals. Participant 2 received a 100 g dose, all others received 50 g.
Table 1.
Baseline pre-infusion ascorbate concentrations.
| Participant number | 1 | 2 | 3 | 4 | 5 |
|---|---|---|---|---|---|
| IVC dose infused 48 h prior (g) | none | 100 | 50 | 25 | none |
| Plasma ascorbate baseline (mM) | 0.077 | 0.098 | 0.042 | 0.083 | 0.095 |
| Erythrocyte ascorbate baseline (mM) | 0.070 | 0.605 | 0.381 | 0.189 | 0.097 |
| IVC dose received on day (g) | 50 | 100 | 50 | 50 | 50 |
| Plasma ascorbate post-infusion (mM) | 18.176 | 18.651 | 12.811 | 14.703 | 34.577 |
| Erythrocyte ascorbate post-infusion (mM) | 1.100 | 2.867 | 0.944 | 1.009 | 1.121 |
| Plasma ascorbate 4 h (mM) | 2.697 | 7.327 | 1.833 | 2.510 | 3.640 |
| Erythrocyte ascorbate 4 h (mM) | 2.152 | 3.425 | 1.516 | 2.048 | 1.980 |
| Plasma ascorbate 24 h (mM) | 0.116 | 0.238 | 0.086 | 0.167 | 0.153 |
| Erythrocyte ascorbate 24 h (mM) | 1.094 | 1.685 | 0.597 | 0.932 | 1.284 |
Plasma ascorbate increased sharply to 20 ± 4 mM immediately post-infusion and erythrocyte ascorbate levels peaked 4 h after infusion at 2.2 ± 0.3 mM (Fig. 3). Plasma levels remained elevated and within the millimolar range for up to 4 h (Table 1) and we noted that erythrocyte levels continued to increase over this time (Fig. 3). After 24 h, the ascorbate concentration had decreased from the post-infusion peak in both plasma and erythrocytes, but was still above pre-infusion concentrations at 152 μM and 1118 μM, respectively (Fig. 3). This result, and the observation that erythrocyte ascorbate concentrations remained elevated in those participants who had undertaken prior ascorbate infusions, suggests that erythrocyte levels may be elevated above baseline for at least 48 h post IVC.
3.4. IVC causes erythrocyte Prx2 oxidation
To seek evidence of H2O2 generation in blood following IVC administration, oxidation of erythrocyte Prx2 was used as an indicator. Blood was obtained from the 15 individuals immediately pre and post IVC infusion. Prx2 oxidation increased significantly in response to IVC administration regardless of infusion dose, and the extent of oxidation in these individuals correlated with the ascorbate dose received (Fig. 4A–C).
Fig. 4.

Oxidation of Prx2 in erythrocytes before and after IVC infusion. (A) Plasma ascorbate concentration and (B) erythrocyte Prx2 oxidation before and immediately after IVC infusion at increasing doses (n = 15, error bars show mean ± SEM, *p < 0.05 by paired t-test). (C) Significant correlation between dose of ascorbate and erythrocyte Prx2 oxidation (n = 11). (D) Erythrocyte Prx2 oxidation over the 24 h following an IVC infusion in five individuals. Prx2 samples were collected at the same time as the ascorbate samples shown in Fig. 3. See Table 1 for IVC dose. For D, a significant difference was observed between pre-infusion and immediately post-infusion Prx-2 oxidation (*p < 0.05, paired t-test). Prx2 oxidation at four and 24 h were not different from 0 time.
We also monitored the dynamics of Prx2 oxidation following IVC infusion. Prx2 oxidation peaked immediately post-infusion, and returned to basal levels 4 and 24 h later (Fig. 4D).
4. Discussion
In this study we show that ascorbate accumulates in the erythrocytes of people administered IVC, reaching millimolar levels, and that this increase is associated with the oxidation of erythrocyte Prx2. Accumulation of ascorbate into erythrocytes could have a significant impact on plasma pharmacokinetics, as these cells constitute up to half the volume of blood and could therefore comprise a significant ascorbate reservoir. Slow release from erythrocytes could potentially sustain elevated plasma levels and result in higher baseline levels. There is already substantial evidence that erythrocytes are involved in ascorbate recycling in vivo and the maintenance of plasma ascorbate [26], possibly via transmembrane electron transfer to reduce any plasma ascorbyl radicals [[37], [38], [39]]. Our data indicated that even after 24–48 h post infusion, plasma ascorbate levels were elevated above the initial baseline. In our recent study with colorectal patients receiving IVC for four consecutive days, we noted retention of ascorbate in erythrocytes and a steady increase in baseline plasma levels over the four days [35]. However, we are unable to distinguish whether increases in erythrocyte ascorbate are contributing to the subsequently elevated plasma ascorbate, as compared to plasma increases caused by tissue saturation.
The consequences of Prx2 oxidation, which is involved in the removal of H2O2 [15] and is a marker of oxidative stress [17], are not so clear. There was no evidence of the haemolysis that can occur in erythrocytes at high levels of oxidative stress. Catalase and glutathione peroxidase will still remove intracellular H2O2, and Prx2 reductive pathways may keep pace without significant impact on erythrocyte function. However, the reducing equivalents required to turnover oxidised Prx2 and oxidised ascorbate may impact the antioxidant capacity of erythrocytes in vivo. Oxidation also alters the oligomeric conformation of Prx2 and its association with other cellular constituents [15,40]. Further investigations into the consequences of erythrocyte Prx2 oxidation are warranted.
There are a number of proposed activities for supra-physiological concentrations of ascorbate, including the ability to act as a pro-oxidant to generate H2O2 through autoxidation facilitated by the presence of redox-active metal ions [10,30,41]. This is best characterised in vitro, where ascorbate toxicity to cancer cell lines is mediated by H2O2 generation through the contribution of catalytic metals in the cell culture media [[7], [8], [9],42,43]. There is limited evidence for such a pathway operating in vivo, where catalytic metal ions will be much less abundant [4,10,30]. An earlier study in patients with pancreatic cancer showed that markers of oxidative stress, including plasma F2-isoprostanes and erythrocyte glutathione (GSH) and glutathione disulphide (GSSG), either decreased or did not change after IVC infusion [4]. While EPR evidence has shown the formation of the ascorbyl radical intermediate in blood during high-dose IVC infusion of animal models [44], only very low concentrations were observed. The same authors have been unable to demonstrate H2O2 generation in blood after IVC despite showing its formation in the extracellular fluid, with erythrocytes proposed to scavenge any H2O2 produced in circulation [44].
Erythrocyte Prx2 is a sensitive marker for the measurement of peroxides in blood, due primarily to high expression levels of this protein in these cells, and its high reactivity with H2O2 [[15], [16], [17], [18]]. Under normal conditions >95% of erythrocyte Prx2 is present in the reduced form, but low levels of thioredoxin reductase relative to other cell types results in slow turnover of oxidised Prx2 following challenge with increased H2O2 [14]. Our in vitro experiments suggest that the mechanism of H2O2 generation by ascorbate was increased haemoglobin autoxidation. Plasma components were not required, and extracellular catalase and the metal chelator DTPA did not have a major effect on Prx2 oxidation. However, it is interesting to note that Prx2 oxidation returned towards baseline levels in patients 4 h after infusion (Fig. 4D), despite erythrocyte ascorbate remaining high (Fig. 3B). Plasma ascorbate levels were significantly lower at 4 h (Fig. 3A), indicating that the high levels of extracellular ascorbate may also contribute to increased Prx2 oxidation. Rather than invoke extracellular H2O2 production, we propose that impairment of the reducing capacity of erythrocytes as a consequence of transmembrane-dependent reduction of extracellular ascorbyl radicals [[37], [38], [39]] promotes the accumulation of oxidised Prx2.
By monitoring haemoglobin oxidation in the absence of glucose, which limits the reduction of both Prx2 and methaemoglobin, we were able to estimate that 15 μM H2O2/min was generated in erythrocytes containing millimolar ascorbate levels. This represents a significant physiological challenge. By comparison, activated neutrophils generate large amounts of H2O2 to kill pathogens, but when taking into account the high abundance of erythrocytes in blood, the generation rate in the presence of ascorbate is almost twice that expected upon activation of the neutrophils in the same volume of blood (i.e. 1500 cells/μL). This highlights that glucose-dependent reductive mechanisms will be essential for maintaining erythrocyte reductive mechanisms during IVC treatment. It also provides a mechanistic basis for the adverse effects associated with IVC treatment of glucose-6-phosphate dehydrogenase (G6PD) deficient individuals [45]. Indeed, an in vitro study has previously shown that erythrocytes treated with high dose ascorbate undergo haemolysis, which is exacerbated in erythrocytes from individuals with G6PD deficiency [46]. There was also increased flux of glucose through the pentose phosphate pathway in the erythrocytes incubated with ascorbate, as well as an increase in oxidative stress and glutathione depletion, which may be the result of H2O2 generation. The authors also show that co-culturing with erythrocytes protects several cancer cell lines from the cell death induced by pharmacological ascorbate concentrations, implying these cells can help detoxify ascorbate [46].
5. Conclusions
We show that erythrocytes accumulate millimolar levels of ascorbate during IVC infusion that results in the haemoglobin-dependent generation of intracellular H2O2, as monitored by the reversible oxidation of erythrocyte Prx2. The consequences of this oxidation require further investigation.
Author contributions
AP and JP had equal responsibility for the experimental work and writing the manuscript. JC was responsible for providing clinical samples, ES performed plasma ascorbate analyses, MV contributed to data interpretation and editing the manuscript, AC assisted with the design of the clinical study, data interpretation and the manuscript, and MH initiated the project and contributed to experimental design, data interpretation and final editing of the manuscript.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
This study was supported by the Canterbury Medical Research Foundation (15/03). AC was supported by a Health Research Council of New Zealand Sir Charles Hercus Health Research Fellowship. We would also like to thank the study participants for their valuable contribution, and Prof Christine Winterbourn for informative discussions.
Contributor Information
Andree G. Pearson, Email: andree.pearson@otago.ac.nz.
Mark B. Hampton, Email: mark.hampton@otago.ac.nz.
References
- 1.Padayatty S.J. Vitamin C pharmacokinetics: implications for oral and intravenous use. Ann. Intern. Med. 2004;140(7):533–537. doi: 10.7326/0003-4819-140-7-200404060-00010. [DOI] [PubMed] [Google Scholar]
- 2.Stephenson C.M. Phase I clinical trial to evaluate the safety, tolerability, and pharmacokinetics of high-dose intravenous ascorbic acid in patients with advanced cancer. Canc. Chemother. Pharmacol. 2013;72(1):139–146. doi: 10.1007/s00280-013-2179-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nielsen T.K. Elimination of ascorbic acid after high-dose infusion in prostate cancer patients: a pharmacokinetic evaluation. Basic Clin. Pharmacol. Toxicol. 2015;116(4):343–348. doi: 10.1111/bcpt.12323. [DOI] [PubMed] [Google Scholar]
- 4.Welsh J.L. Pharmacological ascorbate with gemcitabine for the control of metastatic and node-positive pancreatic cancer (PACMAN): results from a phase I clinical trial. Canc. Chemother. Pharmacol. 2013;71(3):765–775. doi: 10.1007/s00280-013-2070-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lykkesfeldt J., Tveden-Nyborg P. The pharmacokinetics of vitamin C. Nutrients. 2019;11(10) doi: 10.3390/nu11102412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Buettner G.R., Jurkiewicz B.A. Catalytic metals, ascorbate and free radicals: combinations to avoid. Radiat. Res. 1996;145(5):532–541. [PubMed] [Google Scholar]
- 7.Chen Q. Pharmacologic ascorbic acid concentrations selectively kill cancer cells: action as a pro-drug to deliver hydrogen peroxide to tissues. Proc. Natl. Acad. Sci. U. S. A. 2005;102(38):13604–13609. doi: 10.1073/pnas.0506390102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen Q. Pharmacologic doses of ascorbate act as a prooxidant and decrease growth of aggressive tumor xenografts in mice. Proc. Natl. Acad. Sci. U. S. A. 2008;105(32):11105–11109. doi: 10.1073/pnas.0804226105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Michels A.J., Frei B. Myths, artifacts, and fatal flaws: identifying limitations and opportunities in vitamin C research. Nutrients. 2013;5(12):5161–5192. doi: 10.3390/nu5125161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vissers M.C.M., Das A.B. Potential mechanisms of action for vitamin C in cancer: reviewing the evidence. Front. Physiol. 2018;9:809. doi: 10.3389/fphys.2018.00809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Misra H.P., Fridovich I. The generation of superoxide radical during the autoxidation of hemoglobin. J. Biol. Chem. 1972;247(21):6960–6962. [PubMed] [Google Scholar]
- 12.Lemberg R., Legge J.W., Lockwood W.H. Coupled oxidation of ascorbic acid and haemoglobin. Insect Biochem. J. 1939;33(5):754–758. doi: 10.1042/bj0330754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Winterbourn C.C. Oxidative reactions of hemoglobin. Methods Enzymol. 1990;186:265–272. doi: 10.1016/0076-6879(90)86118-f. [DOI] [PubMed] [Google Scholar]
- 14.Low F.M. Peroxiredoxin 2 functions as a noncatalytic scavenger of low-level hydrogen peroxide in the erythrocyte. Blood. 2007;109(6):2611–2617. doi: 10.1182/blood-2006-09-048728. [DOI] [PubMed] [Google Scholar]
- 15.Low F.M., Hampton M.B., Winterbourn C.C. Peroxiredoxin 2 and peroxide metabolism in the erythrocyte. Antioxidants Redox Signal. 2008;10(9):1621–1630. doi: 10.1089/ars.2008.2081. [DOI] [PubMed] [Google Scholar]
- 16.Bayer S.B. Neutrophil-mediated oxidation of erythrocyte peroxiredoxin 2 as a potential marker of oxidative stress in inflammation. Faseb. J. 2013;27(8):3315–3322. doi: 10.1096/fj.13-227298. [DOI] [PubMed] [Google Scholar]
- 17.Poynton R.A., Hampton M.B. Peroxiredoxins as biomarkers of oxidative stress. Biochim. Biophys. Acta. 2014;1840(2):906–912. doi: 10.1016/j.bbagen.2013.08.001. [DOI] [PubMed] [Google Scholar]
- 18.Peskin A.V. The high reactivity of peroxiredoxin 2 with H(2)O(2) is not reflected in its reaction with other oxidants and thiol reagents. J. Biol. Chem. 2007;282(16):11885–11892. doi: 10.1074/jbc.M700339200. [DOI] [PubMed] [Google Scholar]
- 19.Bayer S.B., Hampton M.B., Winterbourn C.C. Accumulation of oxidized peroxiredoxin 2 in red blood cells and its prevention. Transfusion. 2015;55(8):1909–1918. doi: 10.1111/trf.13039. [DOI] [PubMed] [Google Scholar]
- 20.Williamson D., Winterbourn C.C. Effect of oral administration of ascorbate on acetylphenylhydrazine-induced Heinz body formation. Br. J. Haematol. 1980;46(2):319–321. doi: 10.1111/j.1365-2141.1980.tb05974.x. [DOI] [PubMed] [Google Scholar]
- 21.Li H. Vitamin C in mouse and human red blood cells: an HPLC assay. Anal. Biochem. 2012;426(2):109–117. doi: 10.1016/j.ab.2012.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Evans R.M., Currie L., Campbell A. The distribution of ascorbic acid between various cellular components of blood, in normal individuals, and its relation to the plasma concentration. Br. J. Nutr. 1982;47(3):473–482. doi: 10.1079/bjn19820059. [DOI] [PubMed] [Google Scholar]
- 23.Pullar J.M. Erythrocyte ascorbate is a potential indicator of steady-state plasma ascorbate concentrations in healthy non-fasting individuals. Nutrients. 2020;12(2) doi: 10.3390/nu12020418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.May J.M. Maturational loss of the vitamin C transporter in erythrocytes. Biochem. Biophys. Res. Commun. 2007;360(1):295–298. doi: 10.1016/j.bbrc.2007.06.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sage J.M., Carruthers A. Human erythrocytes transport dehydroascorbic acid and sugars using the same transporter complex. Am. J. Physiol. Cell Physiol. 2014;306(10):C910–C917. doi: 10.1152/ajpcell.00044.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tu H. Chemical transport knockout for oxidized vitamin C, dehydroascorbic acid, reveals its functions in vivo. EBioMedicine. 2017;23:125–135. doi: 10.1016/j.ebiom.2017.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vera J.C. Mammalian facilitative hexose transporters mediate the transport of dehydroascorbic acid. Nature. 1993;364(6432):79–82. doi: 10.1038/364079a0. [DOI] [PubMed] [Google Scholar]
- 28.Rumsey S.C. Glucose transporter isoforms GLUT1 and GLUT3 transport dehydroascorbic acid. J. Biol. Chem. 1997;272(30):18982–18989. doi: 10.1074/jbc.272.30.18982. [DOI] [PubMed] [Google Scholar]
- 29.Washko P.W., Wang Y., Levine M. Ascorbic acid recycling in human neutrophils. J. Biol. Chem. 1993;268(21):15531–15535. [PubMed] [Google Scholar]
- 30.Du J., Cullen J.J., Buettner G.R. Ascorbic acid: chemistry, biology and the treatment of cancer. Biochim. Biophys. Acta. 2012;1826(2):443–457. doi: 10.1016/j.bbcan.2012.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mendiratta S., Qu Z.C., May J.M. Erythrocyte ascorbate recycling: antioxidant effects in blood. Free Radic. Biol. Med. 1998;24(5):789–797. doi: 10.1016/s0891-5849(97)00351-1. [DOI] [PubMed] [Google Scholar]
- 32.Cox A.G., Winterbourn C.C., Hampton M.B. Measuring the redox state of cellular peroxiredoxins by immunoblotting. Methods Enzymol. 2010;474:51–66. doi: 10.1016/S0076-6879(10)74004-0. [DOI] [PubMed] [Google Scholar]
- 33.Lykkesfeldt J. Determination of ascorbic acid and dehydroascorbic acid in biological samples by high-performance liquid chromatography using subtraction methods: reliable reduction with tris[2-carboxyethyl] phosphine hydrochloride. Anal. Biochem. 2000;282(1):89–93. doi: 10.1006/abio.2000.4592. [DOI] [PubMed] [Google Scholar]
- 34.Beutler E. Grune and Stratton; New York, USA: 1984. Red Cell Metabolism: A Manul of Biochemical Methods. [Google Scholar]
- 35.Dachs G.U. Vitamin C administration by intravenous infusion increases tumour ascorbate content in patients with colon cancer: a clinical intervention study. Frontiers in Oncology. 2021;10:600715. doi: 10.3389/fonc.2020.600715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pullar J.M., Bayer S., Carr A.C. Appropriate handling, processing and analysis of blood samples is essential to avoid oxidation of vitamin C to dehydroascorbic acid. Antioxidants (Basel) 2018;7(2) doi: 10.3390/antiox7020029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Su D. Human erythrocyte membranes contain a cytochrome b561 that may be involved in extracellular ascorbate recycling. J. Biol. Chem. 2006;281(52):39852–39859. doi: 10.1074/jbc.M606543200. [DOI] [PubMed] [Google Scholar]
- 38.May J.M., Qu Z., Cobb C.E. Extracellular reduction of the ascorbate free radical by human erythrocytes. Biochem. Biophys. Res. Commun. 2000;267(1):118–123. doi: 10.1006/bbrc.1999.1906. [DOI] [PubMed] [Google Scholar]
- 39.VanDuijn M.M. Erythrocytes reduce extracellular ascorbate free radicals using intracellular ascorbate as an electron donor. J. Biol. Chem. 2000;275(36):27720–27725. doi: 10.1074/jbc.M910281199. [DOI] [PubMed] [Google Scholar]
- 40.Bayer S.B. Interactions between peroxiredoxin 2, hemichrome and the erythrocyte membrane. Free Radic. Res. 2016;50(12):1329–1339. doi: 10.1080/10715762.2016.1241995. [DOI] [PubMed] [Google Scholar]
- 41.Carr A.C., Cook J. Intravenous vitamin C for cancer therapy - identifying the current gaps in our knowledge. Front. Physiol. 2018;9:1182. doi: 10.3389/fphys.2018.01182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sestili P. Hydrogen peroxide mediates the killing of U937 tumor cells elicited by pharmacologically attainable concentrations of ascorbic acid: cell death prevention by extracellular catalase or catalase from cocultured erythrocytes or fibroblasts. J. Pharmacol. Exp. Therapeut. 1996;277(3):1719–1725. [PubMed] [Google Scholar]
- 43.Clement M.V. The in vitro cytotoxicity of ascorbate depends on the culture medium used to perform the assay and involves hydrogen peroxide. Antioxidants Redox Signal. 2001;3(1):157–163. doi: 10.1089/152308601750100687. [DOI] [PubMed] [Google Scholar]
- 44.Chen Q. Ascorbate in pharmacologic concentrations selectively generates ascorbate radical and hydrogen peroxide in extracellular fluid in vivo. Proc. Natl. Acad. Sci. U. S. A. 2007;104(21):8749–8754. doi: 10.1073/pnas.0702854104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Udomratn T. Effects of ascorbic acid on glucose-6-phosphate dehydrogenase-deficient erythrocytes: studies in an animal model. Blood. 1977;49(3):471–475. [PubMed] [Google Scholar]
- 46.Zhang Z.Z. Glutathione depletion, pentose phosphate pathway activation, and hemolysis in erythrocytes protecting cancer cells from vitamin C-induced oxidative stress. J. Biol. Chem. 2016;291(44):22861–22867. doi: 10.1074/jbc.C116.748848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Orrico F., Moller M.N., Cassina A. Kinetic and stoichiometric constraints determine the pathway of H2O2 consumption by red blood cells. Free Rad. Biol. Med. 2018;121:231–239. doi: 10.1016/j.freeradbiomed.2018.05.006. [DOI] [PubMed] [Google Scholar]