

Abstract
Persistent high levels of proinflammatory and Th1 responses contribute to cerebral malaria (CM). Suppression of inflammatory responses and promotion of Th2 responses prevent pathogenesis. IL-4 commonly promotes Th2 responses and inhibits inflammatory and Th1 responses. Therefore, IL-4 is widely considered as a beneficial cytokine via its Th2-promoting role that is predicted to provide protection against severe malaria by inhibiting inflammatory responses. However, IL-4 may also induce inflammatory responses, as the result of IL-4 action depends on the timing and levels of its production and the tissue environment in which it is produced. Recently, we showed that dendritic cells (DCs) produce IL-4 early during malaria infection in response to a parasite protein and that this IL-4 response may contribute to severe malaria. However, the mechanism by which IL-4 produced by DCs contributing to lethal malaria is unknown. Using Plasmodium berghei ANKA-infected C57BL/6 mice, a CM model, we show here that mice lacking IL-4Rα only in CD8α+ DCs are protected against CM pathogenesis and survive, whereas WT mice develop CM and die. Compared with WT mice, mice lacking IL-4Rα in CD11c+ or CD8α+ DCs showed reduced inflammatory responses leading to decreased Th1 and cytotoxic CD8+ T cell responses, lower infiltration of CD8+ T cells to the brain, and negligible brain pathology. The novel results presented here reveal a paradoxical role of IL-4Rα signaling in CM pathogenesis that promotes CD8α+ DC-mediated inflammatory responses that generate damaging Th1 and cytotoxic CD8+ T cell responses.
Keywords: IL-4Rα, inflammatory cytokines, cytotoxic T cells, infiltration to brain, endothelial damage, cerebral malaria
Abbreviations: BBB, blood–brain barrier; CM, cerebral malaria; ECM, experimental CM; DC, dendritic cell; IFN-I, type I interferon; IRBC, infected red blood cell
Malaria caused by the protozoan parasites of the Plasmodium genus, especially by Plasmodium falciparum, manifests in several organ-related fatal pathologies, including cerebral malaria (CM), acute lung and liver injury, and kidney damage (1). Of these pathologies, CM is the most devastating (2). Proinflammatory cytokines play key roles in the development of protective immunity to malaria, but their persistent elevated levels subsequently contribute to severe malaria (3). Dendritic cells (DCs) play key roles in malaria by producing early proinflammatory responses and inducing adaptive immune responses through antigen presentation (4, 5). After malaria infection, DCs of the host are the major early responders that produce proinflammatory cytokines such as type I interferon (IFN-I), TNF-α, IL-6, and IL-12 (3, 4, 5). The combined action of DC presenting antigens and cytokine signaling, particularly that of IL-12, activates NK and T cells, lead to Th1 development, IFN-γ production, and CD8+ T cell cytotoxicity (6). Normally, as infection progresses, DCs gradually produce lower levels of proinflammatory cytokines, concomitantly increasing the production of anti-inflammatory IL-10, leading to Th2 development and IL-4 production (4, 5). Collectively, these responses result in protective immunity to malaria, reducing the risk of pathogenesis. However, unbalanced pro-/anti-inflammatory and Th1/Th2 responses contribute to severe pathologies (5).
During the liver stage of malaria infection, IL-4/IL-4R signaling plays important roles in the development of protective effector and memory CD8+ T cell responses (7). In the blood stage of malaria infection, IL-4 and IL-4R gene polymorphisms associate with increased incidences of CM, and severe anemia and illnesses (8, 9). However, how IL-4/IL-4Rα signaling contributes to severe malaria illnesses remains not understood. Although IL-4 is primarily a master Th2 effector, it induces opposing effects depending on timing and levels of its production in the prevailing immune and tissue microenvironments (10). Gaining insight into the mechanisms by which IL-4Rα signaling contributes to lethal malaria is likely to be useful in developing effective vaccine and/or therapeutics.
Studies have shown that DCs treated with IL-4 produce IL-12p70, which is inflammatory and induces Th1 and cytotoxic CD8+ T cell responses (11, 12), and that IL-4 can promote protective Th1 response against pathogenic infections (13, 14). Recently, we showed that, splenic CD8α− DCs, but not CD8α+ DCs, in PbA-infected mice produce IL-4 (15). In addition, DCs treated in vitro with P. falciparum–infected red blood cells (IRBCs) or whole parasite proteins produce IL-4 and that IL-4 production may contribute to severe malaria (15). However, it is not known how IL-4Rα signaling induced by endogenous IL-4 contributes to lethal malaria. Based on the above observations (11, 12, 13, 14) and the findings that IL-4 produced in infected humans associates with severe malaria (8, 9), we hypothesized that IL-4/IL-4Rα signaling contributes to fatal malaria pathogenesis by upregulating inflammatory responses by DCs. We tested this hypothesis in Plasmodium berghei ANKA (PbA)-infected C57BL/6 mice, an experimental CM (ECM) model that exhibits neurological conditions similar to those of human CM. The results show, for the first time, that mice lacking IL-4Rα signaling in CD8α+ DCs resist the development of CM through downregulation of proinflammatory and Th1 responses, leading to reduction in the cytotoxicity of CD8+ T cells and reduced infiltration of these cells into the brains.
Results
IL-4Rα signaling in cDCs or CD8α+ DCs contributes to ECM and mortality
To test the hypothesis that IL-4/IL-4Rα signaling in DCs contributes to CM, we conditionally deleted IL-4Rα in CD11c+ DCs, CD8α+ DCs, or T cells by crossing IL-4Rαfl/fl mice, respectively, with CD11cCre (16), Clec9ACre (17), and LckCre mice (18), which express Cre recombinase under the control of CD11c, Clec9A, and Lck promoter, respectively. The mice deficient in IL-4Rα in classical DCs (cDCs, CD11c+ DCs) (CD11cCre.IL-4Rαfl/fl mice), CD8α+ DCs (Clec9ACre.IL-4Rαfl/fl mice), or T cells (LckCre.IL-4Rαfl/fl mice) were selected by genotyping (Fig. S1). We analyzed IL-4Rα expression in splenic DCs of wild-type (WT), IL-4Rα−/−, CD11cCre.IL-4Rαfl/fl, and Clec9ACre.IL-4Rαfl/fl mice by flow cytometry. Both cDCs of WT mice and CD8α− DCs of Clec9ACre.IL-4Rαfl/fl mice expressed IL-4Rα at normal levels. However, there was no significant expression of IL-4Rα by cDCs in CD11cCre.IL-4Rαfl/fl mice, CD8α+DCs in Clec9ACre.IL-4Rαfl/fl mice, or both cDCs and CD8α+ DCs in IL-4Rα−/− mice that lack IL-4Rα globally (Fig. 1). These results indicate that Cre-lox recombination deleted IL-4Rα in cDCs of CD11cCre.IL-4Rαfl/fl mice and CD8α+DCs of Clec9ACre.IL-4Rαfl/fl mice.
Figure 1.

Flow cytometry analysis of IL-4Rα expression in DCs of the conditional IL-4Rα knockout mice. Spleen cells of uninfected WT and IL-4Rα−/−, CD11cCre.IL-4Rαfl/fl, and Clec9ACre.IL-4Rαfl/fl mice (n = 4–5) were stained with dye-conjugated antibodies against surface markers and anti-mouse IL-4Rα antibody. A, shows gating of CD8α+ and CD8α− DCs. B, histogram showing the IL-4Rα expression of a representative mouse from each group on gated CD8α+ and CD8α− DCs. C, shows plot of mean of IL-4Rα expression. Error bars indicate GeoMFI ±SD. ∗∗p < 0.01, ∗∗∗p < 0.001.
The conditional IL-4Rα knockout mice were infected with PbA and monitored for survival and parasitemia. Cohoused WT mice, CD11cCre mice, littermate WT (Cre−.IL-4Rαfl/fl) mice obtained from either CD11cCre-lox or Clec9ACre-lox cross, and IL-4Rα−/− mice were used as controls. All cohoused WT, CD11cCre, and Cre−.IL-4Rαfl/fl mice developed severe symptoms of CM at 6 to 8 days postinfection (dpi), exhibiting limb paralysis, deviation of the head, ataxia, lack of sensory and motor control, and died between 8 and 11 dpi. Also, ∼90% IL-4Rα−/− mice and LckCre.IL-4Rαfl/fl mice developed severe CM conditions and died between 9 and 11 dpi (Fig. 2A). The remaining ∼10% mice developed moderate CM conditions, but still died at 16 dpi. In contrast, ∼85% infected CD11cCre.IL-4Rαfl/fl mice and ∼65% infected Clec9ACre.IL-4Rαfl/fl mice were protected against pathogenesis of CM and survived until 25 and 28 dpi, respectively, and died between 26 and 29 dpi (Fig. 2A). There were no significant differences in parasitemia in all mouse groups, including control mice (Fig. 2B). In the mice that survived CM conditions, parasitemia steadily increased after 14 dpi, reaching ∼50% by 24 dpi and then all died. Together, these results indicate that IL-4Rα signaling in CD8α+ DCs, but not in other cell types, including T cells, contributes to ECM.
Figure 2.
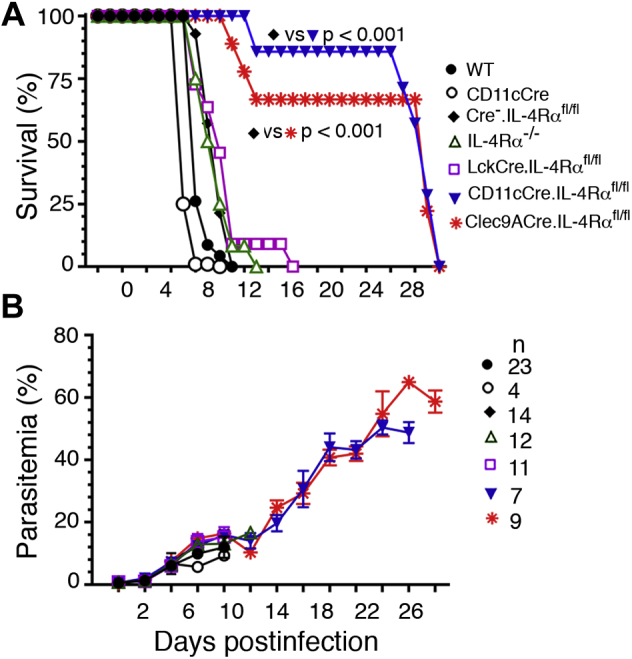
IL-4Rα signaling in cDCs or CD8α+DCs contributes to the development of ECM and mortality.A and B, mice were infected with 2 × 105 PbA IRBCs. Shown are survival (A) and parasitemia (B) in CD11cCre.IL-4Rαfl/fl mice, Clec9ACre.IL-4Rαfl/fl mice, and LckCre.IL-4Rαfl/fl mice. Cohoused WT, CD11cCre, and IL-4Rα−/− mice, and the littermate WT (Cre−.IL-4Rαfl/fl) mice obtained by crossing CD11cCre or Clec9ACre mice with IL-4Rαfl/fl mice were used as controls. Error bars indicate mean values of data ±SD.
Common pathological features of CM in humans and mice include infiltration of immune cells to the brain and loss of blood–brain barrier (BBB) integrity, leading to vascular leakage and brain dysfunction (19). To determine the role of IL-4Rα signaling in DCs contributing to ECM clinical conditions, we examined pathological features in the brains of PbA-infected Cre−.IL-4Rαfl/fl, IL-4Rα−/−, CD11cCre.IL-4Rαfl/fl, and Clec9ACre.IL-4Rαfl/fl mice. We assessed BBB damage in infected mice by injecting Evans blue, a dye that binds to serum albumin and normally does not enter the brain. There was negligible entry of albumin–Evans blue complex into the brains of infected CD11cCre.IL-4Rαfl/fl and Clec9ACre.IL-4Rαfl/fl mice (Fig. 3, A and B). By contrast, infected littermate WT (Cre−.IL-4Rαfl/fl) mice had substantially high levels of Evans blue in the brains. These results demonstrated that IL-4Rα signaling in CD8α+ DCs contributes to malaria-induced BBB disruption. H&E-stained brain sections of cohoused infected WT, IL-4Rα−/− and Cre−.IL-4Rαfl/fl mice at 6 dpi showed high levels of immune cell infiltration in blood vessels (Fig. 3, C and D). By contrast, there was no significant infiltration of immune cells in infected CD11cCre.IL-4Rαfl/fl and Clec9ACre.IL-4Rαfl/fl mice (Fig. 3, C and D). Together the above results indicated that IL-4Rα signaling in CD8α+ DCs contributes to ECM.
Figure 3.
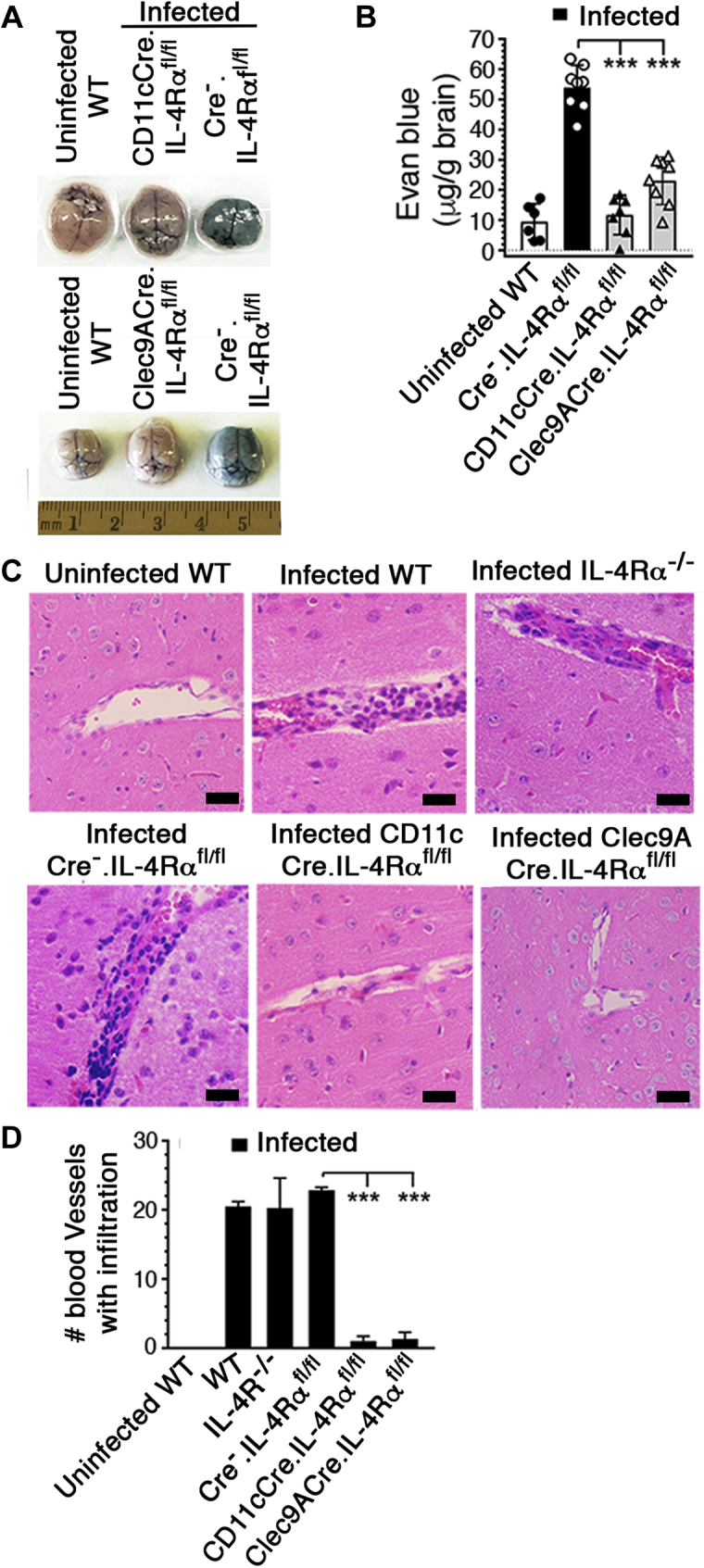
IL-4Rα signaling in cDCs or CD8α+DCs contributes to brain pathology.A–D, mice were infected with 2 × 105 PbA IRBCs. At 6 dpi, Evans blue was injected into mice (n = 6–8/group) via tail vein. After 2 h, brains were harvested and photographed, and the amounts of Evans blue entered into the brains were assessed as outlined under Experimental procedures. A and B, photographs of the brain of a representative mouse in each group; uninfected WT, and infected CD11cCre.IL-4Rαfl/fl, Clec9ACre.IL-4Rαfl/fl, and Cre−.IL-4Rαfl/fl mice (A), and the amounts of Evans blue entered into the brains (B). Mean data ±SD plotted. Panel A, the numbers in the horizontal scale represent cm. C, at 6 dpi, 6-μm brain sections of the indicated mice (n = 3–4/group) were assessed by H&E staining and 40× images of a representative mouse in each group are shown. In each image, the black bar is 10 μm. Images of uninfected CD11cCre.IL-4Rαfl/fl and Clec9ACre.IL-4Rαfl/fl mice were similar to those of uninfected WT mice. D, 20× microscopic views of the brain sections of mice were assessed under light microscopy and the numbers of blood vessels having large numbers of immune cell filtration were counted and mean values plotted. Error bars indicate mean values of data ±SD plotted. ∗∗∗p ≤ 0.001.
IL-4Rα signaling in cDCs or CD8α+ DCs contributes to malaria-induced inflammatory responses by spleen cells
As IL-4/IL-4Rα signaling can induce inflammatory responses by cDCs via induction of IL-12p70 production (11, 12), we hypothesized that IL-4Rα signaling in response to malaria early during infection activates DCs to produce inflammatory cytokines, contributing to ECM. Therefore, we analyzed the expression of costimulatory molecules and IL-12p35 that indicates the levels of functional IL-12p70 by splenic DCs in PbA-infected mice. At 4 dpi, compared with cDCs of littermate WT (Cre−.IL-4Rαfl/fl) mice, cDCs of infected CD11cCre.IL-4Rαfl/fl and Clec9ACre.IL-4Rαfl/fl mice were substantially less activated as indicated by lower surface levels of the costimulatory molecule CD86, which is normally upregulated upon DC activation in response to pathogenic infections (Fig. 4A). At 5 dpi, the percentages of IL-12p70-producing CD8α+ DCs in CD11cCre.IL-4Rαfl/fl and Clec9ACre.IL-4Rαfl/fl mice were significantly lower compared with Cre−.IL-4Rαfl/fl mice (Fig. 4B). These results demonstrated that IL-4Rα signaling in CD8α+ DCs promotes the activation and proinflammatory cytokine production by DCs in response to malaria infection.
Figure 4.

IL-4Rα signaling in cDCs or CD8α+DCs contributes to inflammatory responses by DCs and T cells of PbA-infected mice. The indicated mice (n = 8–12/group) were infected with 2 × 105 PbA IRBCs. A–F, spleen cells from the infected and uninfected WT control mice were analyzed by flow cytometry. A and B, the expression of costimulatory molecule CD86 at 4 dpi (A) and the percentages of IL-12p70+ at 5 dpi (B) in CD8α+ DCs and CD8α− DCs. C, gating strategy of CD4+ T and CD8+ T cells. D, the percentages of CD69 in the CD4+ T and CD8+ T cells at 4 dpi. E, the percentages of IL-10+CD4+ T and IFN-γ+CD8+ T cells at 4 dpi. F, the percentages of Tbet+ CD4+ T and granzyme B+CD8+ T cells at 6 dpi. Error bars indicate mean values ±SD. ns, not significant, ∗p ≤ 0.05, ∗∗p ≤ 0.01, ∗∗∗p ≤ 0.001.
Activated DCs produce cytokines that signal to regulate the activation of innate and adaptive immune responses (6). In particular, IL-12 signaling and antigen presentation by DCs activate T cells, promoting Th1 development. At 4 dpi, the activation of both splenic CD4+ T and CD8+ T cells, measured by expression of CD69, was significantly lower in CD11cCre.IL-4Rαfl/fl and Clec9ACre.IL-4Rαfl/fl mice than in Cre−.IL-4Rαfl/fl mice (Fig. 4, C and D). Further, the frequencies of CD4+ T cells expressing the transcription factor Tbet, which indicates differentiation into Th1 cells, were significantly lower in CD11cCre.IL-4Rαfl/fl and Clec9ACre.IL-4Rαfl/fl mice (Fig. 4F). Consistent with these results, the frequency of CD8+ T cells expressing the proinflammatory cytokine IFN-γ was lower and the frequency of CD4+ T cells expressing IL-10 was higher in both infected CD11cCre.IL-4Rαfl/fl and Clec9ACre.IL-4Rαfl/fl mice compared with Cre−.IL-4Rαfl/fl mice (Fig. 4E). Importantly, the frequency of granzyme B-expressing CD8+ T cells, which are known to migrate to the brains of PbA-infected mice and damage the BBB (19), was lower in infected CD11cCre.IL-4Rαfl/fl and Clec9ACre.IL-4Rαfl/fl mice than those in Cre−.IL-4Rαfl/fl WT mice (Fig. 4F). Collectively, the above data demonstrated that IL-4Rα signaling in DCs creates an inflammatory state, leading to marked increase in Th1 response and cytolytic function of CD8+ T cells, each of which contribute to ECM. Further, the above results taken together with our observation that global or T-cell-specific deficiency in IL-4Rα signaling results in ECM pathology and mortality indicate that IL-4Rα signaling in CD8α+ DCs contributes to ECM pathogenesis.
The IL-4Rα signaling contributes to infiltration of pathogenic immune cells into the brains of PbA-infected mice
During ECM, T cells enter the brain, creating an inflammatory condition contributing to ECM pathology via CD4+ T cells producing IFN-γ and CD8+ T cells producing granzyme B (20, 21, 22). So, we examined the role of IL-4Rα signaling in T cell infiltration into the brains. At 6 dpi, the numbers of immune cells that infiltrated into the brains of PbA-infected CD11cCre.IL-4Rαfl/fl and Clec9ACre.IL-4Rαfl/fl mice were substantially lower compared with Cre−.IL-4Rαfl/fl mice (Fig. 5, A and B). CXCR3 is known to mediate the infiltration of T cells into the brains of PbA-infected mice (23). The numbers of CXCR3 expressing CD4+ T and CD8+ T cells infiltrated into the brains were also significantly lower in infected CD11cCre.IL-4Rαfl/fl and Clec9ACre.IL-4Rαfl/fl mice than in Cre−.IL-4Rαfl/fl mice (Fig. 5C). Although both CD4+ T and CD8+ T cells move into the brain during ECM, it is granzyme B-producing cytotoxic CD8+ T cells that disrupt BBB integrity by interacting with endothelial cells, contributing to ECM pathology (19, 22). The numbers of granzyme B+CD8+ T cells were significantly lower in CD11cCre.IL-4Rαfl/fl and Clec9ACre.IL-4Rαfl/fl compared with Cre−.IL-4Rαfl/fl WT mice (Fig. 5D). Additionally, the numbers of CD11a/LFA-1-expressing CD4+ T and CD8+ T cells were significantly lower in CD11cCre.IL-4Rαfl/fl and Clec9ACre.IL-4Rαfl/fl mice compared with Cre−.IL-4Rαfl/fl control mice (Fig. 5E). In addition to its role as a marker of T cell activation, LFA-1 functions as a receptor for the adherence of CD8+ T cells to endothelia by binding to ICAM-1 (24). The LFA-1-mediated ICAM-1-dependent interaction of granzyme B-expressing CD8+ T cells with endothelial cells results in the disruption of BBB and ECM pathology (25). Together these results indicated that IL-4Rα signaling in CD8α+ DCs promotes infiltration of T cells into the brains and increases the cytotoxicity in CD8+ T cells, contributing to ECM pathology and mortality.
Figure 5.

IL-4Rα signaling in cDCs or CD8α+DCs contributes to infiltration of immune cells, including pathogenic CD8+T cells, to the brain.A–D, the indicated mice (n = 6–7/group) were infected with 2 × 105 PbA IRBCs. At 6 dpi, immune cells in the brains were isolated, stained with dye-conjugated antibodies, and analyzed by flow cytometry. A, cell gating strategy. B–E, the numbers of immune cells infiltrated into the brains; total immune cells (B), CXCR3+ CD4+ T and CD8+ T cells (C), granzyme B-expressing CD8+ T cells (D), and CD11a/LFA-1+ CD4+ T and CD8+ T cells. (E). Error bars indicate mean values ±SD. ∗p ≤ 0.05, ∗∗p ≤ 0.01, ∗∗∗p ≤ 0.001.
Discussion
The novel finding of this study is that IL-4Rα signaling in CD8α+ DCs induces inflammatory responses to malaria, promoting Th1 development and cytotoxic lytic function of CD8+ T cells. Subsequently, CXCR3+CD8+ T cells infiltrate into brains and damage BBB via LFA-I-mediated ICAM-1-dependent interaction with endothelial cells, contributing to ECM and mortality (19, 22, 24, 26). Our findings agree with the reports that treatment with IL-4 at the initial stage of DC maturation inhibits the IL-10 response and promotes the production of IL-12p70 by DCs, leading to Th1 and cytotoxic CD8+ T cell responses (11, 12, 13, 14). Our results also agree with reports that IL-4 imparts Th1 responses and resistance to Leishmania major (14) and that IL-4 is required for the protective Th1 responses to Candida albicans (13). The ability of a cytokine to exert opposing effects, depending on its source, concentration, and tissue environment, is not uncommon. For example, IFN-γ treatment at the initiation of autoimmune encephalomyelitis in mice exacerbated the disease, whereas treatment during effector phase reduced the disease severity (27). Also, IFN-I, a well-known pleiotropic cytokine (28), plays a dual role in malaria (29). In the blood stage malaria, early IFN-I production promotes cytotoxic CD8+ T cell response contributing to ECM, whereas increased production of IFN-α/β by DCs inhibits proinflammatory and Th1 responses, promoting Th2 response as infection progresses to the acute stage (29). Thus, our results demonstrate that IL-4/IL-4Rα signaling in CD8α+ DCs plays an important role in the pathogenesis of ECM by promoting inflammatory responses by DCs and Th1 and cytotoxic CD8+ T cell responses. We recently reported that IL-4 treatment early during malaria infections protects mice from ECM (15). The findings reported here resemble the previous findings by others regarding the role of IFN-I and IFNα/βR in ECM pathology; mice treated with relatively high doses of INF-α as well as IFNα/βR−/− mice do not develop ECM (30, 31).
Our conclusion that IL-4/IL-4Rα signaling in CD8α+ DCs contributes to ECM is further supported by the report that conditional ablation of CD8α+ DCs results in significantly lower expression of granzyme B by CD8+ T cells and markedly reduced infiltration of these cells to the brain, providing protection against ECM (32). Our conclusion also agrees with the report that cDCs are important mediators of CM development (33). Further, in the liver stage malaria, IL-4/IL-4R signaling is critical for the development of antigen-dependent effector CD8+ T cell response (7), an important protective cellular immunity that kills infected hepatocytes, although in the blood stage the effector CD8+ T cells are pathogenic, contributing to CM. Collectively, our results highlight the key, but complex, role of IL-4/IL-4Rα signaling in ECM pathogenesis, and the results have important implication for the development of treatment strategies for severe malaria.
Experimental procedures
Mice, malaria infection, and assessment of parasitemia and clinical symptoms
Mice with a cell-specific deficiency in IL-4Rα were generated from C57BL/6 IL-4Rαfl/fl mice. Originally, IL-4Rαfl/fl mice were generated in BALB/c background by targeting ES cells (34), and these mice were backcrossed with C57BL/6 mice for >10 generations in Dr Ajay Chawla’s lab (35). Dr Chawla provided C57BL/6 IL-4Rαfl/fl mice. Dr Finkelman, University of Cincinnati College of Medicine, provided C57BL/6 IL-4Rα−/− mice. We generated mice having CD11c+ DC-, CD8α+ DC-, and T-cell-specific deficiency in IL-4Rα by crossing C57BL/6 IL-4Rαfl/fl mice, respectively, with CD11cCre, Clec9ACre, or LckCre mice (16, 17, 18). In all experiments, gender- and age-matched mice were used. PbA parasite strain was obtained from MR4/BEI Resources Manassas, VA. WT C57BL/6 mice, infected with frozen stocks of PbA, were used as donor mice for infecting experimental mice. Mice were injected intraperitoneally (ip) with ∼2 × 105 IRBCs from donor mice. Parasitemia was determined by examining Giemsa-stained thin blood smears under the light microscopy. Mice were monitored daily and assessed for the following neurological conditions: ataxia, limb paralysis, seizures, coma, and lack of sensory and motor functions.
Ethics statement
The husbandry and care of mice were in accordance with the recommendations in the National Institutes of Health Guide for the Care and Use of Laboratory Animals. The Institutional Animal Care and Use Committee of The Pennsylvania State University College of Medicine, Hershey, approved the use of mice for experiments described in this study.
Genotyping of mice
The primers used for genotyping were: CD11cCre: Forward 5’-ACTTGGCAGCTGT CTCCAAG-3’ and Reverse 5’-GCGAACAT CTTCAGGTTCTG-3’, and internal controls 5’-CAAATG TTGCTTGTCTGGTG-3’ and 5’-GTCAGTCGAGTGCACAGTTT-3’. Clec9ACre_WT; Forward 5’-AAAAGTTCC ACTTCTGGATGATGA-3’ and Reverse 5’-TCACTTACTCCTCCATGC TGACG-3’. Clec9ACre_trasgene: 5’-GGCTCTCTCC CCAGCATCCACA-3’. LckCre. Forward 5’-ATGCCCAAGAAGAAGAGGAAGGT-3’ and Revise: 5’-GAAATCAGTGCGTTCGAA CGCTAGA-3’. The primers for IL-4Rα LoxP were: Forward 5’-CCCTTCCTGGCCCTGA ATTT-3’ and Reverse 5’-GTTTCCTCCTA CCGCTGATT-3’.
Analysis of brain pathology and BBB damage
Brains of mice were fixed with 4% paraformaldehyde, embedded in paraffin, 6-μm sections were taken on glass slides, deparaffinized, and stained with H&E dye. The brain pathology was assessed under the light microscopy. BBB damage was assessed by injecting 200 μl of 0.5% Evans blue in saline through tail vein. After 2 h, mice were euthanized, perfused, the brains were harvested and photographed, as described previously. The brains were weighed and Evans blue was extracted and quantified as reported previously (36).
Antibodies for flow cytometry
The antibodies used were directed against mouse antigens and were purchased from the following vendors: eFluor 450-anti-CD11a (M17/4), AlexaFluor 700 anti-CD11c (N418), purified anti-CD16/32 (2.4G2), PE- and Brilliant Violet (BV)421-anti-I-A/I-E (M5/114.15.2), BV421-anti-CD49b (DX5), BV510-anti-CD86 (GL-1), and PerCP-Cy5.5-anti-Tbet (eBio4B10) antibodies from e-Biosciences. APC- and APC-Cy7-anti-CD3ε (145-2C11), AlexaFluor 700-anti-CD4 (RM4-5), Alexa Fluor 700- and BV605-anti-CD8α (53–6.7), FITC-anti-CD11c (N418), BV510- and PerCP-Cy5.5-anti-CD69 (H1.2F3), PerCP-Cy5.5-anti-I-A/I-E (M5/114.15.2), PE-anti-NK1.1 (PK136), PerCP-Cy5.5-anti-TCRβ (H57-597), PE-anti-IL-10 (JES5-16E3), APC-anti-IFN-γ (XMG1.2) antibodies from BD Biosciences. BV510-anti-CD45 (30-F11), PE-Cy7-anti-CXCR3 (CXCR3-173), and FITC-anti-Granzyme B (QA16A02), and APC-anti-IL-4Rα (I015F8) antibodies were from BioLegend. APC-anti-human/mouse IL-12p35 (clone 25537) antibody was from R&D Systems.
Preparation of spleen cells and analysis of cytokine responses
Spleen cells were prepared, suspended in incomplete Dulbecco's modified Eagle's medium, stained with antibodies against CD69, CD86, and other surface markers of cells followed by intracellular staining of cytokines, Tbet, and granzyme B, as described previously (36). The stained cells were analyzed by flow cytometry using BD 16-color Fortessa instrument. The data were analyzed using FlowJo software version 10 (36).
Analysis of immune cells infiltrated to brains by flow cytometry
Cell suspensions of mouse brains, prepared as described (36), were centrifuged at 300g, and cells were suspended in 40% Percoll in PBS, pH 7.2, centrifuged on 70% Percoll cushion at 1200g (36). The cell layers on the top of 70% Percoll were collected, stained with antibodies against cell markers, CXCR3 and CD11a followed by intracellular staining of cytokines and granzyme B. The stained cells were analyzed by flow cytometry.
Statistical analysis
Statistical analysis was performed using GraphPad Prism version 7.03. Significance of data from experimental groups compared with control groups was determined by nonparametric Kruskal–Wallis test followed by two-stage step-up procedure of Benjamini, Krieger, and Yekutieli to control the false discovery rates (Figs. 2B, 3 and 4) or one-way ANOVA followed by the Newman–Keuls test (Figs. 1, 2D and 5). Survival data were analyzed by log-rank (Mantel–Cox) test. The results are presented as the mean ± SD p values ≤0.05 were considered statistically significant.
Data availability
All experimental data for this article are available upon e-mail request to the following authors: Xianzhu Wu (The Penn State University College of Medicine, xianzhuwu@pennstatehealth.psu.edu) or D. Channe Gowda (The Penn State University College of Medicine, gowda@psu.edu).
Supporting information
This article contains supporting information.
Conflict of interest
The authors declare that they have no conflicts of interest with the contents of this article.
Acknowledgments
We thank Dr Ajay Chawla, University of California, San Francisco, and Dr Finkelman, University of Cincinnati College of Medicine, who respectively provided us IL-4Rαfl/fl mice and IL-4Rα−/− mice in C57BL/6 background. We also thank MR4/BeiResources (Manassas, VA) for providing P. berghei ANKA strain.
Author contributions
D. C. G. and X. W. conceptualization; X. W., D. C. G., C. C. N., and Z. C. C. investigation; D. C. G. and X. W. writing original draft; D. C. G., X. W., C. C. N., F. B., and Z. C. C. reviewing and editing; X. W., D. C. G., and C. C. N. formal analysis; X. W., D. C. G., C. C. N., F. B., and Z. C. C. methodology; F. B. IL-4Rα floxed mouse resource; Z. C. C. Cre mice resource; D. C. G. and C. C. N. funding acquisition; X. W., D. C. G., and C. C. N. data curation; D. C. G. supervision; D. C. G. project administration.
Funding and additional information
This work was funded in parts by the grant R01 AI41139 from the National Institute of Allergy and Infectious Diseases, NIH, and the grant with the Pennsylvania Department of Health using Tobacco CURE Funds SAP# 4100083097 (D. C. G. and C. C. N.). The Pennsylvania Department of Health specifically disclaims responsibility for any analysis, interpretations, or conclusions. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Edited by Peter Cresswell
Supporting information
References
- 1.de Souza M.C., Pádua T.A., das Graças Henriques M. Multiple organ dysfunction during severe malaria: The role of the inflammatory response. In: Rodriguez-Marales A.J., editor. Current Topics in Malaria. InTech Publishing; Rijeka, Croatia: 2016. [Google Scholar]
- 2.Storm J., Craig A.G. Pathogenesis of cerebral malaria-inflammation and cytoadherence. Front. Cell. Infect. Microbiol. 2014;4:1–8. doi: 10.3389/fcimb.2014.00100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hunt N.H., Grau G.E. Cytokines: Accelerators and brakes in the pathogenesis of cerebral malaria. Trends Immunol. 2003;24:491–499. doi: 10.1016/s1471-4906(03)00229-1. [DOI] [PubMed] [Google Scholar]
- 4.Amorim K.N., Chagas D.C., Sulczewski F.B., Boscardin S.B. Dendritic cells and their multiple roles during malaria infection. J. Immunol. Res. 2016;2016 doi: 10.1155/2016/2926436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Deroost K., Pham T.-T., Opdenakker G., Van den Steen P.E. The immunological balance between host and parasite in malaria. FEMS Microbiol. Rev. 2015;40:208–257. doi: 10.1093/femsre/fuv046. [DOI] [PubMed] [Google Scholar]
- 6.Walsh K.P., Mills K.H. Dendritic cells and other innate determinants of T helper cell polarisation. Trends Immunol. 2013;34:521–530. doi: 10.1016/j.it.2013.07.006. [DOI] [PubMed] [Google Scholar]
- 7.Morrot A., Zavala F. Regulation of the CD8+ T cell responses against Plasmodium liver stages in mice. Int. J. Parasitol. 2004;34:1529–1534. doi: 10.1016/j.ijpara.2004.10.001. [DOI] [PubMed] [Google Scholar]
- 8.Verra F., Luoni G., Calissano C., Troye-Blomberg M., Perlmann P., Perlmann H., Arcà B., Sirima B.S., Konaté A., Coluzzi M., Kwiatkowski D., Modiano D. IL4-589C/T polymorphism and IgE levels in severe malaria. Acta Trop. 2004;90:205–209. doi: 10.1016/j.actatropica.2003.11.014. [DOI] [PubMed] [Google Scholar]
- 9.Cabantous S., Poudiougou B., Oumar A.A., Traore A., Barry A., Vitte J., Bongrand P., Marquet S., Doumbo O., Dessein A.J. Genetic evidence for the aggravation of Plasmodium falciparum malaria by interleukin 4. J. Infect. Dis. 2009;200:1530–1539. doi: 10.1086/644600. [DOI] [PubMed] [Google Scholar]
- 10.Paul W.E. History of interleukin-4. Cytokine. 2015;75:3–7. doi: 10.1016/j.cyto.2015.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yao Y., Li W., Kaplan M.H., Chang C.-H. Interleukin (IL)-4 inhibits IL-10 to promote IL-12 production by dendritic cells. J. Exp. Med. 2005;201:1899–1903. doi: 10.1084/jem.20050324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Guenova E., Volz T., Sauer K., Kaesler S., Müller M.R., Wölbing F., Chen K., Schwärzler C., Brossart P., Röcken M., Biedermann T. IL-4-mediated fine tuning of IL-12p70 production by human DC. Eur. J. Immunol. 2008;38:3138–3149. doi: 10.1002/eji.200838463. [DOI] [PubMed] [Google Scholar]
- 13.Mencacci A., Del Sero G., Cenci E., d'Ostiani C.F., Bacci A., Montagnoli C., Kopf M., Romani L. Endogenous interleukin 4 is required for development of protective CD4+ T helper type 1 cell responses to Candida albicans. J. Exp. Med. 1998;187:307–317. doi: 10.1084/jem.187.3.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Biedermann T., Zimmermann S., Himmelrich H., Gumy A., Egeter O., Sakrauski A.K., Seegmuller I., Voigt H., Lunois P., Levine A.D., Wagner H., Heeg K., Louis J.A., Röcken M. IL-4 instructs Th1 responses and resistance to Leishmania major in susceptible BALB/c mice. Nat. Immunol. 2001;2:1054–1060. doi: 10.1038/ni725. [DOI] [PubMed] [Google Scholar]
- 15.Wu X., Gowda N.M., Kawasawa Y.I., Gowda D.C. A malaria protein factor induces IL-4 production by dendritic cells via PI3K-Akt-NF-κB signaling independent of MyD88/TRIF and promotes Th2 response. J. Biol. Chem. 2018;293:10425–10434. doi: 10.1074/jbc.AC118.001720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Caton M.L., Smith-Raska M.R., Reizis S. Notch-RBP-J signaling controls the homeostasis of CD8- dendritic cells in the spleen. J. Exp. Med. 2007;204:1653–1664. doi: 10.1084/jem.20062648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schraml B.U., Van Blijswijk J., Zelenay S., Whitney P.G., Filby A., Acton S.E., Rogers N.C., Moncaut N., Carvajal J.J., Reis e Sousa C. Genetic tracing via DNGR-1 expression history defines dendritic cells as a hematopoietic lineage. Cell. 2013;154:843–858. doi: 10.1016/j.cell.2013.07.014. [DOI] [PubMed] [Google Scholar]
- 18.Hennet T., Hagen F.K., Tabak L.A., Marth J.D. T-cell-specific deletion of a polypeptide N-acetylgalactosaminyltransferase gene by site-directed recombination. Proc. Natl. Acad. Sci. U. S. A. 1995;92:12070–12074. doi: 10.1073/pnas.92.26.12070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Riggle B.A., Manglani M., Maric D., Johnson K.R., Lee M.-H., Neto O.L.A., Taylor T.E., Seydel K.B., Nath A., Miller L.H. CD8+ T cells target cerebrovasculature in children with cerebral malaria. J. Clin. Invest. 2019;130:1128–1138. doi: 10.1172/JCI133474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Villegas-Mendez A., Greig R., Shaw T.N., de Souza J.B., Findlay E.G., Stumhofer J.S., Hafalla J.C., Blount D.G., Hunter C.A., Riley E.M., Couper K.N. IFN-γ-producing CD4+ T cells promote experimental cerebral malaria by modulating CD8+ T cell accumulation within the brain. J. Immunol. 2012;189:968–979. doi: 10.4049/jimmunol.1200688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ghazanfari N., Mueller S.N., Heath W.R. Cerebral malaria in mouse and man. Front. Immunol. 2018;9:2016. doi: 10.3389/fimmu.2018.02016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Renia L., Grau G.E., Wassmer S.C. CD8+ T cells and human cerebral malaria: A shifting episteme. J. Clin. Invest. 2020;130:1109–1111. doi: 10.1172/JCI135510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Campanella G.S., Tager A.M., El Khoury J.K., Thomas S.Y., Abrazinski T.A., Manice L.A., Colvin R.A., Luster A.D. Chemokine receptor CXCR3 and its ligands. CXCL9 and CXCL10 are required for the development of murine cerebral malaria. Proc. Natl. Acad. Sci. U. S. A. 2008;105:4814–4819. doi: 10.1073/pnas.0801544105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Makgoba M.W., Sanders M.E., Luce G.E.G., Gugel E.A., Dustin M.L., Springer T.A., Shaw S. Functional evidence that intercellular adhesion molecule-1 (ICAM-1) is a ligand for IFA-1-dependent adhesion in T cell-mediated cytotoxicity. Eur. J. Immunol. 1988;18:637–640. doi: 10.1002/eji.1830180423. [DOI] [PubMed] [Google Scholar]
- 25.Swanson P.A., II, Hart G.T., Russo M.V., Nayak D., Yazew T., Peña M., Khan S.M., Janse C.J., Pierce S.K., McGavern D.B. CD8+ T cells induce fatal brainstem pathology during cerebral malaria via luminal antigen-specific engagement of brain vasculature. PLoS Pathog. 2016;12 doi: 10.1371/journal.ppat.1006022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Morris S.C., Heidorn S.M., De'Broski R.H., Perkins C., Hildeman D.A., Khodoun M.V., Finkelman F.D. Endogenously produced IL-4 nonredundantly stimulates CD8+ T cell proliferation. J. Immunol. 2009;182:1429–1438. doi: 10.4049/jimmunol.182.3.1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Naves R., Singh S.P., Cashman K.S., Rowse A.L., Axtell B.C., Steinman L., Mountz J.D., Steele C., De Sarno P., Raman C. The interdependent, overlapping, and differential roles of type I and II IFNs in the pathogenesis of experimental autoimmune encephalomyelitis. J. Immunol. 2013;191:2967–2977. doi: 10.4049/jimmunol.1300419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Trinchieri G. Type I interferon: Friend or foe? J. Exp. Med. 2010;207:2053–2063. doi: 10.1084/jem.20101664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sebina I., Haque A. Effects of type I interferons in malaria. Immunology. 2018;165:176–185. doi: 10.1111/imm.12971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vigário A.M., Belnoue E., Grüner A.C., Mauduit M., Kayibanda M., Deschemin J.-C., Marussig M., Snounou G., Mazier D., Gresser I. Recombinant human IFN-α inhibits cerebral malaria and reduces parasite burden in mice. J. Immunol. 2007;178:6416–6425. doi: 10.4049/jimmunol.178.10.6416. [DOI] [PubMed] [Google Scholar]
- 31.Sharma S., DeOliveira R.B., Kalantari P., Parroche P., Goutagny N., Jiang Z., Chan J., Bartholomeu D.C., Lauw F., Hall J.P., Barber G.N., Gazzinelli R.T., Fitzgerald K.A., Golenbock D.T. Innate immune recognition of an AT-rich stem-loop DNA motif in the Plasmodium falciparum genome. Immunity. 2011;35:194–207. doi: 10.1016/j.immuni.2011.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Piva L., Tetlak P., Claser C., Karjalainen K., Renia L., Ruedl C. Cutting edge: Clec9A+ dendritic cells mediate the development of experimental cerebral malaria. J. Immunol. 2012;189:1128–1132. doi: 10.4049/jimmunol.1201171. [DOI] [PubMed] [Google Scholar]
- 33.deWalick S., Amante F.H., McSweeney K.A., Randall L.M., Stanley A.C., Haque A., Kuns R.D., MacDonald K.P., Hill G.R., Engwerda C.R. Cutting edge: Conventional dendritic cells are the critical APC required for the induction of experimental cerebral malaria. J. Immunol. 2007;178:6033–6037. doi: 10.4049/jimmunol.178.10.6033. [DOI] [PubMed] [Google Scholar]
- 34.Mohrs M., Ledermann B., Kohler G., Dorfmuller A., Gessner A., Brombacher F. Differences between IL-4- and IL-4 receptor alpha-deficient mice in chronic leishmaniasis reveal a protective role for IL-13 receptor signaling. J. Immunol. 1999;162:7302–7308. [PubMed] [Google Scholar]
- 35.Lee M.-W., Odegaard J.I., Mukundan L., Qiu Y., Molofsky A.B., Nussbaum J.C., Yun K., Locksley R.M., Chawla A. Activated type 2 innate lymphoid cells regulate beige fat biogenesis. Cell. 2015;160:74–87. doi: 10.1016/j.cell.2014.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wu X., Dayanand K.K., Thylur R.P., Norbury C.C., Gowda D.C. Small molecule-based inhibition of MEK1/2 proteins dampens inflammatory responses to malaria, reduces parasite load, and mitigates pathogenic outcomes. J. Biol. Chem. 2017;292:13615–13634. doi: 10.1074/jbc.M116.770313. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All experimental data for this article are available upon e-mail request to the following authors: Xianzhu Wu (The Penn State University College of Medicine, xianzhuwu@pennstatehealth.psu.edu) or D. Channe Gowda (The Penn State University College of Medicine, gowda@psu.edu).