

Abstract
The Sodium Glucose Co-Transporter-2 inhibitor, empagliflozin (EMPA), reduces mortality and hospitalisation for heart failure following myocardial infarction irrespective of diabetes status. While the findings suggest an inherent cardioprotective capacity, the mechanism remains unknown. We studied infarct size (IS) ex-vivo in isolated hearts exposed to global IR injury and in-vivo in rats subjected to regional myocardial ischemia reperfusion (IR) injury, in whom we followed left ventricular dysfunction for 28 days. We compared rats that were given EMPA orally for 7 days before, EMPA 1.5 h before IR injury and at onset of reperfusion and continued orally during the follow-up period. We used echocardiography, high resolution respirometry, microdialysis and plasma levels of β-hydroxybutyrate to assess myocardial performance, mitochondrial respiration and intermediary metabolism, respectively. Pretreatment with EMPA for 7 days reduced IS in-vivo (65 ± 7% vs. 46 ± 8%, p < 0.0001 while administration 1.5 h before IR, at onset of reperfusion or ex-vivo did not. EMPA alleviated LV dysfunction irrespective of the reduction in IS. EMPA improved mitochondrial respiration and modulated myocardial interstitial metabolism while the concentration of β-hydroxybutyric acid was only transiently increased without any association with IS reduction. EMPA reduces infarct size and yields cardioprotection in non-diabetic rats with ischemic LV dysfunction by an indirect, delayed intrinsic mechanism that also improves systolic function beyond infarct size reduction. The mechanism involves enhanced mitochondrial respiratory capacity and modulated myocardial metabolism but not hyperketonemia.
Subject terms: Heart failure, Myocardial infarction, Experimental models of disease, Cardiology
Introduction
Although mortality from acute MI has declined over the last 25 years1,2, long-term mortality, i.e. 1 year and beyond, and post-MI heart failure is still significant1,3. The disarrays that may lead to deterioration of left ventricular (LV) contractile function are determined by the extent of myocardial damage caused by MI4. The underlying mechanisms are multifactorial and involve not only complex remodeling at the organ level but also neurohormonal changes to compensate for the lack of contractile function5.
Recent groundbreaking clinical studies have shown that treatment with Sodium Glucose Co-Transporter 2 (SGLT2) inhibitors markedly reduced cardiovascular mortality and hospitalization in patients with HF6–9. The reduction was independent of diabetic status8,9. While originally intended as a glucose-lowering alternative to metformin for patients with type II diabetes mellitus, contemporary discoveries indicate that SGLT2 inhibitors have a class effect on HF in individuals surviving an acute MI, whereas the incidence of recurrent MI is unchanged. The mechanisms underlying these observations are unknown.
Supporting the clinical findings, a recent experimental study demonstrated that the SGLT2 inhibitor, canagliflozin, has a cardioprotective capability by reducing infarct size in an in vivo rat model of ischemia reperfusion (IR) independently not only of circulating glucose but also circulating ketone levels10 that are known to be increased by SGLT2 inhibitor treatment11. Despite absent infarct size reduction in an in vitro rat heart model, another SGLT2 inhibitor, empagliflozin (EMPA), improves post-MI mitochondrial respiration and increases inner membrane permeability in non-diabetic rats12. The ischemic myocardium is characterized by suppressed mitochondrial oxidative phosphorylation, which may be further deranged by reperfusion13. We hypothesized that cardioprotection by SGLT2 inhibitors is associated with improved mitochondrial respiration. Consequently, the aim of the present study was to investigate the effect of EMPA on IS and cardiac and mitochondrial function after IR induced chronic ventricular dysfunction and after acute IR injury in an in vivo rat heart model.
Materials and methods
Animals and design
Male Sprague Dawley rats (250–300 g, Taconic, Ry, Denmark) were kept for acclimatization at a constant temperature of 23 °C, with a 12 h light–dark cycle and with unlimited access to food and water.
Our study included three experimental series:
Impact of EMPA (30 mg/kg) on infarct size in vivo and ex vivo was investigated to determine if the cardioprotection by EMPA, if any, was dependent on a whole-body system or could be induced directly on the heart. We compared rats receiving chronic oral EMPA treatment daily for 7 days before MI (EMPA-Chronic) to rats receiving an acute oral administration 1.5 h before MI (EMPA-Acute) or at the onset of reperfusion (EMPA-Post). Placebo animals were matched in dose and time of administration of vehicle (Supplementary Fig. S1a).
Impact of EMPA on post-MI LV dysfunction after 28 days evaluated by echocardiography and high resolution respirometry for mitochondrial respiratory capacity. In this series we compared EMPA-Chronic, EMPA-Post, a sham group receiving EMPA (SHAM) and corresponding placebo animals. In all groups in this experimental series, the administration of EMPA (30 mg/kg) continued consecutively for 28 days after myocardial infarction (Supplementary Fig. S1b).
Impact of EMPA on mitochondrial respiratory capacity measured by high resolution respirometry and myocardial intermediary metabolism measured by microdialysis during early reperfusion (30 min) was investigated to delineate the metabolic changes caused by EMPA treatment. In this series we compared EMPA-Chronic, a matched placebo group (PLACEBO-Chronic), EMPA-Acute and a sham group (SHAM) (Supplementary Fig. S1c).
Times of administration were chosen for several reasons. To test the most acute effect of EMPA we chose to administer EMPA 1.5 h before intervention, since the drug reaches maximum bioavailability after 1 h14. To test any effect requiring longer time and repeated administrations we chose to administer EMPA repeatedly for 7 days before intervention. Also, levels of β-OHB were measured at the different time points to determine the relation to any potential cardioprotection.
Exclusion criteria included death during procedure, death during follow up, complications to oral gavage, no sign of myocardial infarction evaluated during procedure; no paling or hypokinesia, no sign of living mitochondria during high resolution respirometry, increase of > 10% in the oxygen consumption rate after addition of cytochrome C during high resolution respirometry, coronary flow rate > 20 ml/min in the isolated perfused heart model indicating unspecific leakage, incorrect microdialysis probe placement and leaking probe membrane.
Experimental models
In vivo rat model
Rats were anaesthetized in an induction chamber with 8% sevoflurane (Sevorane, AbbVIE A/S, Copenhagen, Denmark) mixed with atmospheric air (flow: 0.8 L/min). Upon induction of anaesthesia, the rats were intubated and connected to a mechanical ventilator (Ugo Basile 7025 rodent ventilator, Comerio, Varese, Italy) with an adjusted flow of 0.6 L/min with 3.5% sevoflurane. A temperature probe (UNO, Zevenaar, Holland) was inserted, and body temperature was kept at a constant 37 °C ± 1 °C. Prior to the procedure the rats were injected subcutaneously with buprenorfin (Temgesic, Reckitt Benckiser Pharmaceuticals Limited, Slough, England) (0.045 mg/mL) to ensure peri-, and post-operational analgesia. A left sided thoracotomy, via the fourth and fifth rib, followed by pericardiotomy gave access to the myocardium. The left anterior descending coronary artery (LAD) was located and ligated approximately 2 mm distally to the junction between the pulmonary conus and the left atrial appendage using a 4-0 silk suture (Sofsilk, Covidien, Dublin, Ireland)14. Myocardial paling and hypokinesia distally to the ligature confirmed ceased blood flow and ischemia. After 30 min of ischemia, reperfusion of the myocardium was ensured by removing the ligature and confirmed by visualization of hyperaemia and enlargement of the LAD. In animals used for infarct size evaluation, the ligature was only loosened but remained in the chest during reperfusion, to ensure exact delineation of the area at risk (AAR). After reperfusion the chest was closed. Animals in the long-term survival group were given buprenorfin (7.4 μg/ml) in the drinking water for three days following MI.
Preparation of the isolated perfused heart
The hearts were isolated and perfused ex vivo as previously described15. In brief, rats were anaesthetised by a subcutaneous injection of Dormicum (midazolam (0.5 mg kg−1 body weight); Matrix Pharmaceuticals, Herlev, Denmark) mixed with Hypnorm (fentanyl citrate (0.158 mg kg−1 body weight) and fluanisone (0.5 mg Kg−1 body weight)). The rats were tracheotomized and connected to a mechanical ventilator and ventilated with atmospheric air (Ugo Basile 7025 rodent ventilator, Comerio, Varese, Italy). A thoracotomy and laparotomy were performed and a bolus of 500 IU heparin (Leo Pharma, Ballerup, Denmark) was administered through the femoral vein. Subsequently, the ascending aorta was cannulated and retrogradely perfused at a constant pressure of 80 mmHg with an oxygenated (95% O2 and 5% CO2) Krebs Henseleit buffer: composition in mM: NaCl 118.5, KCl 4.7, NaHCO3 25.0, glucosemonohydrate 11.0, MgSO4⋅7 H2O 1.2, CaCl2 2.4 and KH2PO4 1.2. The heart was excised and transferred under continuous perfusion to an isolated perfused heart system (IH-SR type844/1; HSE, March-Huhstettem, Germany) with a constant temperature of 37 °C. An intraventricular balloon (size 7, HSE, March-Hugstetten, Germany) was inserted in the LV through the left atrial appendage and the volume adjusted to a LV end-diastolic pressure of 4–8 mmHg to simulate preload. Data was digitally converted (DT9804; Data Translation. Marlboro, MA, USA) and stored using Notocord Hem software (version 2.0, Notocord systems, Croissy sur Seine, France).
Infarct size
In the in vivo model, the ligature around LAD was tightened again after 2 h of reperfusion, and a 4% Evans Blue (Sigma-Aldrich, St. Louis, MO, USA) solution was injected in vena cava inferior, to outline the myocardial area at risk (AAR), as previously described15.
The hearts were excised, frozen at − 80 °C and sliced into 5, 2 mm thick slices, guided by a rat heart slicer matrix (Zivic Instruments, Pittsburgh, PA, USA). The slices were then incubated for 3 min with 1% 2,3,5-triphenyltetrazoliumchloride (TTC) (Sigma-Aldrich, St. Louis, MO, USA) to delineate areas of infarction and after 24 h of storage in 4% formaldehyde buffer (VWR International, Leuven, Belgium), AAR and IS were assessed using ImageJ software (NIH, Bethesda, Maryland, USA).
In the isolated perfused heart model, the same protocol was followed without Evans Blue staining, since the hearts were subjected to global no flow ischemia. IS/AAR, AAR/LV and IS/LV were calculated and adjusted to the wet weight of the individual slices.
Echocardiography
Transthoracic echocardiography was performed at baseline, day 1 and day 27 after MI, as previously described15. Transthoracic echocardiography was performed with a Vevo 2100 high-frequency ultrasound system (Visual Sonic, Toronto, ON, Canada) with a 21 MHz rat probe. Animals were lightly sedated (3% sevoflurane, atmospheric air), fixated in a supine position to a heating pad and connected to ECG electrodes and a rectal thermal probe. Movement of the transducer was facilitated by a mechanical setup to eliminate movement disturbances. Two-dimensional and M-mode images were obtained.
Left ventricular volumes in end diastole (LV Vold) and end systole (LV Vols) were calculated using the bullet method (5/6 × LV Vold/LV Vols area x LV length). Left ventricular ejection fraction (EF) was calculated using LV Vold and LV Vols by the formula:
All images were analyzed using the Vevo 2100 software.
Mitochondrial respiratory capacity
We analyzed the mitochondrial respiratory capacity with non-fatty acid and fatty acid substrates using high-resolution respirometry (Oxygrah-2 k; Oroboros Instruments, Innsbruck, Austria) as described previously16. A mid-papillary biopsy of LV myocardial tissue was prepared by manual dissection of fiber bundles (~ 1.5 m), 30 min after MI in series 3 and 28 days after MI in series 2. In series 3 fibers were dissected at mid papillary level central in the area at risk, to ensure evaluation of mitochondria exposed to IR. In series 2 fibers were dissected from the remote myocardium, since fibrotic areas are not suited for analysis.
Fiber bundles were permeabilized in cold BIOPS solution mixed with saponin (50 μg mL−1) by gentle agitation for 30 min16. After permeabilization the fiber bundles were rinsed twice for 10 min by agitation in a cold respiration medium, MiR05 (in mmol L−1: 110 sucrose, 60 K-lactobionate, 0.5 EGTA, 0,1% BSA, 3 mgCl2, 20 taurine, 10 KH2PO4 and 20 Hepes; pH 7.1).
The fibre bundles were added to the chambers in the Oxygraph-2 k respirometer, filled with 2 mL MiR05. Substrates and inhibitors were added in the following order for non-fatty acid linked oxidation: (1) Glutamate (G) (10 mmol L−1) + Malate (M) (2 mmol L−1), (2) ADP (5 mmol L−1), (3) Cytochrome c (10 μmol L−1), (4) Succinate (S) (10 mmol L−1), (5) Oligomycin (complex V inhibitor) (2 μg mL−1), (6) Rotenone (complex I inhibitor) (0.5 μmol L−1) + Antimycin A (complex III inhibitor) (2.5 mmol L−1).
We also measured the capacity of mitochondrial fatty acid oxidation using substrates in the following order: (1) Octanoyl-L-carnitine (250 μmol L−1), a medium chain fatty acid, + Malate (2 mmol L−1), (2) ADP (5 mmol L−1), Cytochrome c (10 μmol L−1).
To avoid any O2 limitations to respiration the chambers were hyperoxygenated and all measurements were carried out in duplicate. The integrity of the outer mitochondrial membrane was tested by adding cytochrome c and an increase of > 10% in the oxygen consumption rate led to exclusion. The respiratory rates (O2 consumption rates) are expressed as the O2 flux normalised to the cardiac muscle mass of the permeabilized fibers (pmol s−1 kg−1 wet weight of permeabilized fibers).
GM: complex I respiration without ADP. GM3: Complex I respiration with ADP. GMS3: Complex I + II respiration with ADP (maximal coupled respiration). 4o: LEAK/non-phosphorylating resting respiration. ROX: residual oxygen consumption. Moc: fatty acid respiration without ADP. Moc3: fatty acid respiration with ADP. The respiratory control ratio (RCR) was calculated as state 3 respiration/state 2 respiration and expresses the respiratory coupling efficiency of the electron transport system (ETS) independent of muscle weight and mitochondrial density.
Mitochondrial enzymatic activity
We analyzed citrate synthase (CS) activity in the cardiac tissue by spectrophotometry as described previously16. The results are expressed as μmol min−1 g protein−1.
Ketone body assay
β-hydroxybutyrate (β-OHB) was quantified in rat plasma using hydrophilic interaction liquid chromatography tandem mass spectrometry as previously described17. The blood sample was taken from the left femoral vein immediately prior to excision of the heart.
Myocardial interstitial concentrations of metabolites assessed by microdialysis in vivo
Myocardial interstitial concentrations of metabolites were assessed by microdialysis as described previously18. After placement of the ligature around the LAD in the in vivo model but before inducing ischemia, a microdialysis probe (membrane length 4 mm, cut-off 6 Da; AgnTho’s, Lidingoe, Sweden) was carefully guided by a 26 g needle into the free anterior wall of the LV in the estimated area of infarction. The probe was connected to a microdialysis pump (Univentor Limited, Zejtun, Malta) and perfused at a flow speed of 1μL min-1 with deoxygenated KHB (95%N2 and 5% CO2). Following insertion, a 20-min period of constant flow was allowed for stabilization, to ensure the concentrations of myocardial metabolites reach equilibrium. We measured concentrations of the TCA cycle intermediates; citrate, malate and succinate and ATP degradation products; hypoxanthine, xanthine, adenosine and inosine, during stabilization, ischemia and reperfusion at 10-min intervals and stored samples at − 80 °C. During collection the vials were cooled to approximately 4 °C. Samples were analysed by liquid chromatography and mass spectrometry. The absolute values were corrected to recovery rate as described previously19.
Myocyte cross sectional area
Hematoxylin/eosin (HE) staining was used for visualization of myocyte cross-sectional area.
Briefly, cross sections of the left ventricle (3 µm) were deparaffinized and counterstained with hematoxylin II (Ventana Medical Systems, AZ, USA) 28 days after MI.
Myocyte cross-sectional area was evaluated at 400 × magnification in the HE stained sections. Cells were measured manually by outlining the cell contour. Only cells with a visible nucleus, a clear and intact cell membrane and cells located perpendicular to the plane were measured. Five to seven randomly selected fields in each section were selected for evaluation in the remote myocardium, which was characterized as the area most distally from the infarcted area.
Microscopy was performed on a light microscope (BX50F4, Olympus, Tokyo, Japan) and image analysis was performed using Image J software (NIH, Bethesda, MD, USA).
Drug preparation and administration
EMPA (30 mg/kg) (Empagliflozin, Merck, Darmstadt, Germany)20 was dissolved in a 0.5% hydroxyethylcellulose (Sigma-Aldrich, St. Louis, MO, USA) solution and administered by oral gavage. Placebo animals received vehicle only. The relatively high dose of EMPA was based on pivotal non-clinical safety studies20, a pilot trial in (Supplementary Fig. S2) and since this study serves as a proof-of-concept study exploring the mechanism of action rather than the clinical potential of EMPA.
Statistical analysis
Based on own experience and reports by other groups, a sample size of n = 10 was considered adequate to identify a treatment effect12,15,21. All results are expressed as mean ± SD unless otherwise stated. Comparisons of means between three or more groups were analysed by one-way ANOVA with post-hoc Bonferroni test. Comparison between two groups was done using a student’s t-test. Concentrations of myocardial interstitial metabolites were analysed using two-way ANOVA. All analyses were performed using GraphPad Prism 8.2.0 (Graph Pad Software, CA, USA). P < 0.05 was considered statistically significant.
Ethics approval
All animal handling was in accordance with national guidelines in Denmark and the Guide for the Care and Use of Laboratory Animals37 and all experiments conformed to Danish Law (Act. No. 1306 of 23/11/2007) and the experimental setup was approved by The Animal Experiments Inspectorate, Ministry of Environment and Food of Denmark with the license number: 2018-15-0201-01475. All animal experiments were carried out in accordance with the ARRIVE guidelines.
Results
In the in vivo infarct size study, we included 66 rats. Numbers for the final analysis and those excluded are shown in the flow chart in supplementary figure S1a. In the ex vivo infarct size series, we included 20 animals. Two rats were excluded due to protocol violations. In the post MI LV-dysfunction series, we included 50 rats. The flow chart is shown in supplementary figure S1b. In the mitochondrial respiratory capacity and intermediary metabolism series, we included 50 rats. Supplementary Figure S1c depicts the flow chart and specifies reasons for exclusion of 21 rats.
EMPA treatment for 7 days prior to MI significantly reduced myocardial in vivo IS by 20% points compared to placebo (65 ± 7% vs. 46 ± 8%p < 0.0001) (Fig. 1a). Administration of EMPA 1.5 h before MI yielded no IS reduction in vivo (p > 0.99) (Fig. 1a). AAR did not differ between groups (Fig. 1b). EMPA did not reduce IS ex vivo (63 ± 16% and 53 ± 13%, p = 0.14) or affect hemodynamic performance (Supplementary Fig. S3).
Figure 1.

Myocardial infarct size following ischemia reperfusion in vivo. (a) Histological evaluation of infarct size with 2,3,5-triphenyltetrazolium chloride (TTC) staining, 2 h after reperfusion in PLACEBO-Chronic (n = 10); EMPA-Chronic (n = 10), PLACEBO-Acute (n = 7), EMPA-Acute (n = 8), PLACEBO-Post (n = 7) and EMPA-Post (n = 6) (b) The ratio of AAR/LV indicating the similarity of AAR between groups. AAR area at risk, IS infarct size, LV left ventricle. Mean ± SD. Statistical significance is shown as * p < 0.05, ** p < 0.01, **** p < 0.0001.
LV EF was similar in all groups at baseline. LV EF was reduced after MI at day 1 and day 27 compared to baseline in the placebo (p < 0.0001), EMPA-Chronic (p = 0.0005) and EMPA-Post (p < 0.0001) groups (Fig. 3a), whereas there was no reduction in the sham groups between baseline and day 27 (p = 0.34). EMPA (EMPA-Chronic and EMPA-Post) significantly improved LV EF at day 1 and at 27 after MI compared to placebo (50 ± 5 and 49 ± 11 vs. 41 ± 9%, p < 0.05) (Fig. 2a).
Figure 3.

Mitochondrial respiratory capacity in early reperfusion. (a) Summarized data of mitochondrial respiration in each respiratory state. (b) RCR with complex I linked substrates. (c) RCR with complex I + II linked substrates. (d) Enzymatic activity of CS. SHAM (n = 4), PLACEBO-Chronic (n = 8), EMPA-Acute (n = 9), EMPA-Chronic (n = 10). GM Complex I respiration with glutamate and malate and ADP, GM3 Complex I respiration with glutamate, malate and ADP, GMS3 Complex I + II respirations with glutamate, malate, succinate and ADP, 4o LEAK/non-phosphorylating respirations with oligomycin, ROX residual oxygen consumption evaluated after adding rotenone and antimycin A, Moc fatty acid respiration with malate and octanoyl-L-carnitine without ADP, Moc3 fatty acid respiration with malate, octanyol-L-carnitine and ADP. RCR respiratory control ratio, CS citrate synthase. Data are mean ± SD. Statistical significance is shown as * p < 0.05, ** p < 0.01, *** p < 0.001 vs. PLACEBO-Chronic.
Figure 2.

Effects of EMPA on left ventricular function. Effects of EMPA on (a) left ventricular ejection fraction, left ventricular volumes in (b) end systole and (c) end diastole were assessed by echocardiography in PLACEBO-Post (n = 7), EMPA-Chronic (n = 6), EMPA-Post (n = 8), PLACEBO SHAM (n = 7) and EMPA SHAM (n = 7). EF ejection fraction, LV Vols left ventricular volume in the systole, LV Vold left ventricular volume in the diastole. Mean ± SD. Statistical significance is shown as * p < 0.05, *** p < 0.001.
Neither LV Vols nor LV Vold differed between groups at baseline (Fig. 2b,c). Animals treated with EMPA (EMPA-Chronic and EMPA-Post) had significantly improved LV Vols at day 27 after MI compared to placebo (368 ± 63 and 322 ± 59 vs. 427 ± 71 , p < 0.05) (Fig. 2b). We observed no difference in LV Vold between the EMPA treated and placebo groups at day 27 after MI (Fig. 2c).
Compared to placebo EMPA-Chronic and EMPA-Acute increased non-ADP stimulated complex I respiration (31.6 ± 3.7 and 29.4 ± 6.3 vs. 21.7 ± 8.3 mol O2 s−1 mg−1, p < 0.05) (Fig. 3a) as well as ADP stimulated complex I respiration (127.1 ± 35,16 and 106.1 ± 36,86 vs. 56.4 ± 36,01 mol O2 s−1 mg−1, p < 0.05) with glucose linked substrates 30 min after MI. Accordingly, the RCR increased (4.0 ± 0.99 and 3.6 ± 0.97 vs. 2.2 ± 0.34, p < 0.05) (Fig. 3b).
Stimulation of complex I + II yielded an enhanced response in both EMPA-Chronic and EMPA-Acute compared to placebo (231.7 ± 59.04 and 238.1 ± 65.77 vs. 156.6 ± 40.25 mol O2 s-1 mg-1, p < 0.05). State 4o respiration (non-phosphorylating respiration) and residual oxygen consumption (ROX) were similar between all groups.
Respiration with fatty acid substrates 30 min after MI was similar between all groups in complex I (non-ADP stimulated) linked respiration (Fig. 3a). In contrast, EMPA-Chronic increased complex I + II (ADP stimulated) respiration compared to placebo (99.2 ± 47.3 vs 40.8 ± 40.3 mol O2 s−1 mg−1, p < 0.05) (Fig. 3a). This was reflected in an increased RCR by EMPA-Chronic (2.7 ± 0.6 vs. 1.7 ± 0.7, p < 0.05) (Fig. 3c).
At 28 days after MI EMPA-Chronic increased ADP stimulated complex I + II respiration with glucose linked substrates, but the increment was not statistically significantly different from placebo (296.3 ± 128.9 vs. 183.9 ± 81. mol O2 s−1 mg−1, p = 0.17), (Fig. 4a).
Figure 4.

Mitochondrial respiratory capacity at day 28 after reperfusion. (a) Summarized data of mitochondrial respiration in each respiratory state. (b) Mitochondrial respiratory capacity at day 28 after sham operation. (c) Enzymatic activity of CS. PLACEBO-Chronic (n = 7), EMPA-Chronic (n = 6), EMPA-Post (n = 7), EMPA SHAM (n = 7), PLACEBO SHAM (n = 7). GM State 2 respirations with glutamate and malate, GM3 State 3 respirations with glutamate and malate, GMS3 State 3 respirations with glutamate, malate, and succinate, 4o State 4 respirations with oligomycin, ROX residual oxygen consumption evaluated after adding rotenone and antimycin A, Moc state 2 respiration with malate and octanoyl-L-carnitine, Moc3 state 3 respiration with malate and octanyol-L-carnitine, RCR respiratory control ratio, CS citrate synthase. Data are mean ± SD. Statistical significance is shown as * p < 0.05, ** p < 0.01, *** p < 0.001 vs. PLACEBO-Chronic. † p < 0.05, †† p < 0.01 vs. PLACEBO SHAM.
Respiration with fatty acid linked substrates demonstrated an increase of complex I respiration (non-ADP stimulated) by EMPA-Chronic and EMPA-Post compared to placebo (46.6 ± 5.4 and 44.5 ± 8.3 vs. 33.2 ± 8.2 mol O2 s−1 mg−1, p < 0.05) (Fig. 4a).
SHAM animals were evaluated after 28 days of exposure to either EMPA or vehicle.
Complex I (ADP stimulated) respiration (193.8 ± 25.2 vs. 148.8 ± 27.8 mol O2 s−1 mg−1, p = 0.008) as well as complex I + II (ADP stimulated) respiration (370.3 ± 64.0 vs. 277.4 ± 67.2 mol O2 s−1 mg−1, p = 0.02) were increased by EMPA compared to placebo (Fig. 4b).
Enzymatic activities of CS did not differ between either of the experimental series (Figs. 3d, 4c).
At the beginning of stabilization, the myocardial interstitial concentration of the tricarboxylic acid (TCA) cycle intermediates citrate, succinate and malate were similar in all groups (Fig. 5), but the concentration of glutamate was significantly higher in EMPA-Chronic compared to placebo and remained so during stabilization. EMPA-Acute increased the myocardial citrate level and EMPA-Chronic increased the succinate level before the induction of ischemia compared to EMPA-Chronic and placebo respectively. During ischemia, the interstitial citrate concentration was significantly higher in the EMPA-Acute group compared to EMPA-Chronic and remained continuously increased during reperfusion. The interstitial concentrations of succinate, malate and glutamate were significantly increased by EMPA-Chronic compared to placebo during ischemia and also in early reperfusion.
Figure 5.

Myocardial interstitial concentrations of metabolites. The myocardial interstitial concentration of TCA cycle intermediates (citrate, succinate and malate) and purine metabolites (adenosine, hypoxanthine, inosine and xanthine) during stabilization, ischemia and reperfusion. Data are mean ± SEM. Statistical significance is shown as * p < 0.05 EMPA-Acute vs. EMPA-Chronic, † p < 0.05 EMPA-Chronic vs. PLACEBO-Chronic.
At the end of stabilization, the interstitial concentrations of purine metabolites were similar in all study groups. Concentrations of adenosine and inosine were significantly elevated in early ischemia compared with EMPA-Acute. During early reperfusion, the interstitial concentrations of adenosine, inosine, hypoxanthine and xanthine were significantly increased in the EMPA-Chronic group compared to EMPA-Acute.
The interstitial concentration of lactate was significantly elevated during ischemia in EMPA-Chronic compared to EMPA-Acute but not compared to placebo.
EMPA increased circulating β-OHB acid levels after 1.5 h of its administration. After 7 days of its administration, the level of β-OHB acid was normalized (Fig. 6).
Figure 6.
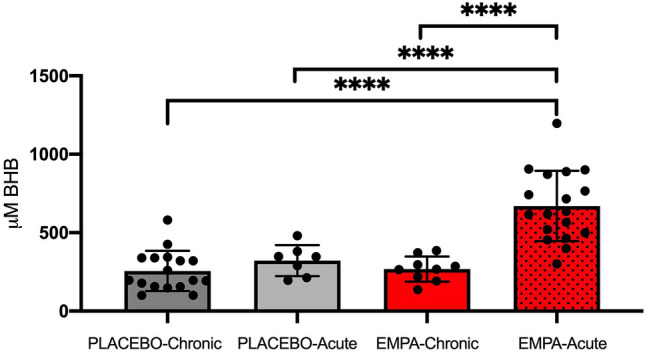
Plasma levels of β-hydroxybutyrate in early reperfusion. β-hydroxybutyrate levels measured from a venous blood sample after 2 h of reperfusion in. PLACEBO (n = 17), PLACEBO-Acute (n = 1.5), EMPA-Chronic (n = 18), EMPA1.5 (n = 9). BHB: β-hydroxybutyrate. Data are mean ± SD. Statistical significance is shown as **** p < 0.0001.
At 28 days after MI, myocyte cross sectional area was slightly increased in the EMPA-Post group compared to placebo but otherwise similar across the study groups (ANOVA, p < 0.05) (Fig. 7).
Figure 7.

Myocyte cross sectional area. Histological analysis of Myocyte cross sectional area. PLACEBO-Post (n = 7), EMPA-Chronic (n = 7–8), EMPA-Post (n = 7), PLACEBO SHAM (n = 6–7) and EMPA SHAM (n = 8). Mean ± SD. Statistical significance is shown as * p < 0.05, **p < 0.01 vs. PLACEBO-Post.
Discussion
The present study demonstrates that continuous treatment with EMPA for 7 days prior to MI reduces IS in an in vivo rat heart model, while a single administration 1.5 h before MI does not. IS reduction is associated with a subsequent improvement of LV function at 28 days post MI. The most notable finding is that administration of EMPA in a post MI period of 28 days improves LV function regardless of time of therapy initiation and hence independently of IS reduction in the in vivo rat heart. The hemodynamic effect of SGLT2 inhibition was associated with an improvement in mitochondrial respiration that was documented in healthy, sham-operated rats. The cardioprotective effect and the effect on mitochondrial respiration seem to be independent of circulating β-OHB levels. Our results confirm that EMPA yields no cardioprotection in the isolated heart. EMPA did not affect mortality rate (Fig. S1).
The mechanisms underlying the cardioprotective effect of treatment with EMPA is of importance in the light of the recent demonstration of a beneficial cardiovascular effect of SGLT2 inhibitors. Notably, the beneficial effects were obtained not only by patients with8,22 but also in patients without diabetes8,9.
In accordance with previous studies, we found that EMPA administered for one week prior to MI reduced IS in non-diabetic rat hearts23–25. Administration of EMPA as a single dose shortly before MI did not offer the same effect23. Even though acute intravenous administration of dapagliflozin and canagliflozin is protective26,27, this observation may indicate that the main mechanism of EMPA does not work by a primary and direct modulation of the myocardium. This assumption is supported by the fact that administration of SGLT2 inhibitors directly into the perfusate in an ex vivo, Langendorff setting leaves cardioprotection unobtainable10,12. We confirmed these findings in a Langendorff setting that eliminated any effect of circulating substances (e.g. metabolic substrates such as β-OHB), since the initial washout with high coronary flow rates rapidly dilutes substances, which are carried over from the body. Hence, the mediating cardioprotective signal appears to be dependent on a whole-body system.
EMPA improves contractility in hypoxic cardiomyocytes25. We extended these findings by demonstrating that EMPA enhanced EF irrespective of the time of treatment onset in rats with post infarction compromised LV function. Our findings demonstrated that IS reduction alone is not responsible for the effect. EMPA improved end systolic volume, while end diastolic volume remained unchanged, reflecting increased contractility. In accordance with our findings, canagliflozin increased stroke volume and myocardial efficiency without altering myocardial substrate utilization, i.e. uptake of glucose, fatty acids or ketones, in otherwise healthy swine subjected to IR injury28 and alleviated post-ischemic systolic and diastolic function in non-diabetic male rats27. Together, these findings suggest that SGLT2 inhibitors have an inherent beneficial modulating class effect that reduces the cardiac derangements following MI.
Expectedly, the mitochondrial complex I and complex I + II linked oxidative phosphorylation (OXPHOS) capacity was impaired by IR injury during early reperfusion after MI. EMPA-Chronic significantly improved mitochondrial function. EMPA-Chronic also improved mitochondrial fatty acid oxidation. The main electron entry sites in the ETS after β-oxidization of fatty acids are complexes I and II via NADH and electron transfer flavoprotein via FADH229. Hence, the increased respiration by EMPA might reflect an overall improvement in the electron flow through complex I and II, as reflected in the respiratory coupling efficiency (RCR) and driven by an increase in state 3 respiration. Reduction of the oxidative capacity of fatty acids plays an important role in the development of HF after IR injury30,31. This reduction provides an early indication of deranged cardiac mitochondrial performance in HF30. EMPA-Chronic improved both complex I and fatty acid respiration.
Because we measured mitochondrial respiration under unloaded resting conditions in the absence of EMPA or its potential mediator, the preserved mitochondrial function may favour a mechanism underlying the improved cardiac function rather than a consequence and that the modulation has already happened at the organ level in the intact body. Supporting this, we observed a significantly improved mitochondrial respiratory capacity in sham animals treated with EMPA compared with sham animals treated with placebo. Thus, our findings seem to represent a specific effect of EMPA on mitochondrial respiratory capacity. We have previously demonstrated a similar effect in an in vitro model12.
ATP degradation end products (i.e. adenosine, inosine, hypoxanthine and xanthine) were elevated during ischemia19. In accordance with an increased ATP-turnover from the elevated respiratory capacity, in particular during reperfusion when the respiratory chain is recovering32, we found an amplified increase in the ATP degradation products in the EMPA-Chronic group. The myocardial interstitial concentrations of TCA cycle intermediates succinate, malate and glutamate were significantly elevated in the EMPA-Chronic compared to placebo during ischemia.
In our experimental rat model, we found that levels of β-OHB increased only transiently after administration of EMPA. Conversely, in the EMPA-Chronic group, circulating β-OHB levels were normalized at the time of exposure to IR with no apparent association with the observed IS reduction. Since we were unable to associate elevated β-OHB levels in plasma with IS reduction and increased mitochondrial respiration, it seems unlikely that the cardioprotective effect should be coupled to increased β-OHB metabolism.
Some limitation must be acknowledged. We used healthy young male animals without comorbidities and without previous exposure to pharmacological treatment. Furthermore, we used a supra-therapeutic dose of EMPA to investigate the cardioprotective properties and mitochondrial modulatory effects on MI and post-MI HF. During several of the study procedures, the animals underwent anesthesia with volatile anaesthetics, which has been shown to be cardioprotective33. However, we found no differences in length of anesthesia or doses of sevoflurane between our study groups.
We found no differences in the number of mitochondria, measured by citrate synthase activity. A biomarker such as citrate synthase may not be an optimal marker for mitochondrial content across varying pathological conditions such as IR34. However, the same constraint relates to all markers that seem to have similar validity as citrate synthase35. To circumvent the limitation, we calculated the respiratory control ratio, as a reliable measure of respiratory coupling efficiency, independently from the number of mitochondria. Mitochondrial respiration was measured 30 min after reperfusion, whereas infarct size was evaluated after 2 h. The damaging process of reperfusion may extend beyond 30 min and thus mitochondrial respiration may still be significant as part of the cardioprotective mechanism, as shown in the chronic experimental series. However, the time discrepancy may explain that we found no IS reduction in EMPA-Acute animals, while mitochondrial respiratory capacity was similar to that observed in the EMPA-Chronic group. Similarly, it might explain that concentrations in intermediary metabolites were different in EMPA-Acute and EMPA-Chronic, while we found no difference in mitochondrial respiration.
The microdialysis samples were collected in 10-min spans to ensure sufficient material for analysis. This may limit the interpretation as a result of low temporal resolution compared to the rapid changes that occur during early reperfusion. Catheter implantation and surgical procedures may per se affect levels of myocardial interstitial metabolites36 and may challenge the interpretation of the physiology in our specific experimental setup. The influence was observed initially and was similar in all study groups, whereas no increase was observed in the sham group during ischemia. Hence, we considered the differences between the study groups during ischemia and reperfusion valid. Furthermore, microdialysis is not the most accurate method to estimate exact concentrations in tissues, but we chose the continuous measurement approach from the same animal to assess dynamic changes.
We did not investigate causes of mortality during the experiments, specifically we did not monitor arrhythmicity, which might have provided information about differences in causes of mortality between groups.
Conclusion
EMPA yields cardioprotection against acute IR injury in vivo but not ex vivo, indicating dependency of an intact body and an indirect effect by a delayed intrinsic cardioprotective mechanism. The protection persists in the failing rat heart by restoring systolic function and the effect is present irrespective of infarct size reduction. The cardioprotective effect is associated with enhanced mitochondrial respiratory capacity. Our data support a beneficial effect of EMPA in non-diabetic individuals with post infarction left ventricular dysfunction.
Supplementary Information
Acknowledgements
We sincerely thank Casper Carlsen Elkjær for excellent technical assistance.
Abbreviations
- AAR
Area at risk
- ADP
Adenosine diphosphate
- ATP
Adenosine triphosphate
- ANOVA
Analysis of variance
- β-OHB
β-Hydroxybutyrate
- CS
Citrate synthase
- EF
Ejection fraction
- ETS
Electron transport system
- HF
Heart failure
- IR
Ischemia reperfusion
- IS
Infarct size
- LAD
Left anterior descending coronary artery
- LV
Left ventricle
- MI
Myocardial infarction
- mPTP
Mitochondrial permeability transition pore
- RCR
Respiratory control ratio
- SD
Standard deviation
- SGLT2
Sodium glucose Co-Transporter 2
- TTC
2,3,5-Triphenyltetrazoliumchloride
- TCA
Tricarboxylic acid cycle
- Vold
End diastolic volume
- Vols
End systolic volume
Author contributions
J.M.S., T.R.L., N.R.J. and H.E.B. participated in the experimental design. J.M.S., M.V.H. and J.H. participated in the experimental work. J.M.S., T.R.L, N.R.J., F.K. and H.E.B. conducted the data analysis. J.M.S., T.R.L., N.R.J., and H.E.B. wrote the manuscript. All authors declare that they have sufficiently contributed to the manuscript to substantiate the authorship. The manuscript is currently neither published nor under review anywhere else. All authors have approved the manuscript at hand.
Funding
This study was supported with grants from The Novo Nordisk Foundation: NNF17OC0029602 and A.P. Møller Fonden.
Data availability
All data included in this article is displayed or can be displayed upon request.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-021-89149-9.
References
- 1.Schmidt M, Jacobsen JB, Lash TL, Botker HE, Sorensen HT. 25 year trends in first time hospitalisation for acute myocardial infarction, subsequent short and long term mortality, and the prognostic impact of sex and comorbidity: A Danish nationwide cohort study. BMJ. 2012;344:e356. doi: 10.1136/bmj.e356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Szummer K, Wallentin L, Lindhagen L, Alfredsson J, Erlinge D, Held C, et al. Improved outcomes in patients with ST-elevation myocardial infarction during the last 20 years are related to implementation of evidence-based treatments: Experiences from the SWEDEHEART registry 1995–2014. Eur. Heart J. 2017;38(41):3056–3065. doi: 10.1093/eurheartj/ehx515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen J, Hsieh AF, Dharmarajan K, Masoudi FA, Krumholz HM. National trends in heart failure hospitalization after acute myocardial infarction for Medicare beneficiaries: 1998–2010. Circulation. 2013;128(24):2577–2584. doi: 10.1161/CIRCULATIONAHA.113.003668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stone GW, Selker HP, Thiele H, Patel MR, Udelson JE, Ohman EM, et al. Relationship between infarct size and outcomes following primary PCI: Patient-level analysis from 10 randomized trials. J. Am. Coll. Cardiol. 2016;67(14):1674–1683. doi: 10.1016/j.jacc.2016.01.069. [DOI] [PubMed] [Google Scholar]
- 5.Shih H, Lee B, Lee RJ, Boyle AJ. The aging heart and post-infarction left ventricular remodeling. J. Am. Coll. Cardiol. 2011;57(1):9–17. doi: 10.1016/j.jacc.2010.08.623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fitchett D, Zinman B, Wanner C, Lachin JM, Hantel S, Salsali A, et al. Heart failure outcomes with empagliflozin in patients with type 2 diabetes at high cardiovascular risk: results of the EMPA-REG OUTCOME(R) trial. Eur. Heart J. 2016;37(19):1526–1534. doi: 10.1093/eurheartj/ehv728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Neal B, Perkovic V, Mahaffey KW, de Zeeuw D, Fulcher G, Erondu N, et al. Canagliflozin and cardiovascular and renal events in type 2 diabetes. N. Engl. J. Med. 2017;377(7):644–657. doi: 10.1056/NEJMoa1611925. [DOI] [PubMed] [Google Scholar]
- 8.McMurray JJV, Solomon SD, Inzucchi SE, Kober L, Kosiborod MN, Martinez FA, et al. Dapagliflozin in patients with heart failure and reduced ejection fraction. N. Engl. J. Med. 2019;381(21):1995–2008. doi: 10.1056/NEJMoa1911303. [DOI] [PubMed] [Google Scholar]
- 9.Packer M, Anker SD, Butler J, Filippatos G, Pocock SJ, Carson P, et al. Cardiovascular and renal outcomes with empagliflozin in heart failure. N. Engl. J. Med. 2020;383(15):1413–1424. doi: 10.1056/NEJMoa2022190. [DOI] [PubMed] [Google Scholar]
- 10.Lim VG, Bell RM, Arjun S, Kolatsi-Joannou M, Long DA, Yellon DM. SGLT2 Inhibitor, canagliflozin, attenuates myocardial infarction in the diabetic and nondiabetic heart. JACC Basic Transl. Sci. 2019;4(1):15–26. doi: 10.1016/j.jacbts.2018.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ferrannini E, Mark M, Mayoux E. CV Protection in the EMPA-REG OUTCOME trial: A "Thrifty Substrate" hypothesis. Diabetes Care. 2016;39(7):1108–1114. doi: 10.2337/dc16-0330. [DOI] [PubMed] [Google Scholar]
- 12.Jespersen NR, Lassen TR, Hjortbak MV, Nb S, He B. Sodium glucose transporter 2 (SGLT2) inhibition does not protect the myocardium from acute ischemic reperfusion injury but modulates post-ischemic mitochondrial function. Cardiovasc. Pharm. Open Access. 2017;6:210. doi: 10.4172/2329-6607.1000210. [DOI] [Google Scholar]
- 13.Halestrap AP, Clarke SJ, Javadov SA. Mitochondrial permeability transition pore opening during myocardial reperfusion–a target for cardioprotection. Cardiovasc. Res. 2004;61(3):372–385. doi: 10.1016/S0008-6363(03)00533-9. [DOI] [PubMed] [Google Scholar]
- 14.(CHMP) CfMPfHU. Assessment report, Jardiance, International non-proprietary name: empagliflozin. Procedure No. EMEA/H/C/002677/0000.: European Medicines Agency; 2014 [Available from: https://www.ema.europa.eu/en/documents/assessment-report/jardiance-epar-public-assessment-report_en.pdf.
- 15.Lassen TR, Nielsen JM, Johnsen J, Ringgaard S, Botker HE, Kristiansen SB. Effect of paroxetine on left ventricular remodeling in an in vivo rat model of myocardial infarction. Basic Res. Cardiol. 2017;112(3):26. doi: 10.1007/s00395-017-0614-5. [DOI] [PubMed] [Google Scholar]
- 16.Christiansen LB, Dela F, Koch J, Hansen CN, Leifsson PS, Yokota T. Impaired cardiac mitochondrial oxidative phosphorylation and enhanced mitochondrial oxidative stress in feline hypertrophic cardiomyopathy. Am. J. Physiol. Heart Circ. Physiol. 2015;308(10):H1237–H1247. doi: 10.1152/ajpheart.00727.2014. [DOI] [PubMed] [Google Scholar]
- 17.Sorensen LK, Rittig NF, Holmquist EF, Jorgensen KA, Jorgensen JO, Moller N, et al. Simultaneous determination of beta-hydroxybutyrate and beta-hydroxy-beta-methylbutyrate in human whole blood using hydrophilic interaction liquid chromatography electrospray tandem mass spectrometry. Clin. Biochem. 2013;46(18):1877–1883. doi: 10.1016/j.clinbiochem.2013.08.011. [DOI] [PubMed] [Google Scholar]
- 18.Kavianipour M, Wikstrom G, Ronquist G, Waldenstrom A. Validity of the microdialysis technique for experimental in vivo studies of myocardial energy metabolism. Acta Physiol. Scand. 2003;179(1):61–65. doi: 10.1046/j.1365-201X.2003.01145.x. [DOI] [PubMed] [Google Scholar]
- 19.Birkler RI, Stottrup NB, Hermannson S, Nielsen TT, Gregersen N, Botker HE, et al. A UPLC-MS/MS application for profiling of intermediary energy metabolites in microdialysis samples–a method for high-throughput. J. Pharm. Biomed. Anal. 2010;53(4):983–990. doi: 10.1016/j.jpba.2010.06.005. [DOI] [PubMed] [Google Scholar]
- 20.Bogdanffy MS, Stachlewitz RF, van Tongeren S, Knight B, Sharp DE, Ku W, et al. Nonclinical safety of the sodium-glucose cotransporter 2 inhibitor empagliflozin. Int. J. Toxicol. 2014;33(6):436–449. doi: 10.1177/1091581814551648. [DOI] [PubMed] [Google Scholar]
- 21.Jespersen NR, Yokota T, Stottrup NB, Bergdahl A, Paelestik KB, Povlsen JA, et al. Pre-ischaemic mitochondrial substrate constraint by inhibition of malate-aspartate shuttle preserves mitochondrial function after ischaemia-reperfusion. J. Physiol. 2017;595(12):3765–3780. doi: 10.1113/JP273408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Inzucchi SE, Kosiborod M, Fitchett D, Wanner C, Hehnke U, Kaspers S, et al. Improvement in cardiovascular outcomes with empagliflozin is independent of glycemic control. Circulation. 2018;138(17):1904–1907. doi: 10.1161/CIRCULATIONAHA.118.035759. [DOI] [PubMed] [Google Scholar]
- 23.Nikolaou PE, Efentakis P, Abu Qourah F, Femmino S, Makridakis M, Kanaki Z, et al. Chronic empagliflozin treatment reduces myocardial infarct size in nondiabetic mice through STAT-3-mediated protection on microvascular endothelial cells and reduction of oxidative stress. Antioxid. Redox Signal. 2021 doi: 10.1089/ars.2019.7923. [DOI] [PubMed] [Google Scholar]
- 24.Andreadou I, Efentakis P, Balafas E, Togliatto G, Davos CH, Varela A, et al. Empagliflozin limits myocardial infarction in vivo and cell death in vitro: Role of STAT3, mitochondria, and redox aspects. Front. Physiol. 2017;8:1077. doi: 10.3389/fphys.2017.01077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lu Q, Liu J, Li X, Sun X, Zhang J, Ren D, et al. Empagliflozin attenuates ischemia and reperfusion injury through LKB1/AMPK signaling pathway. Mol. Cell Endocrinol. 2020;501:110642. doi: 10.1016/j.mce.2019.110642. [DOI] [PubMed] [Google Scholar]
- 26.Lahnwong S, Palee S, Apaijai N, Sriwichaiin S, Kerdphoo S, Jaiwongkam T, et al. Acute dapagliflozin administration exerts cardioprotective effects in rats with cardiac ischemia/reperfusion injury. Cardiovasc. Diabetol. 2020;19(1):91. doi: 10.1186/s12933-020-01066-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sayour AA, Korkmaz-Icoz S, Loganathan S, Ruppert M, Sayour VN, Olah A, et al. Acute canagliflozin treatment protects against in vivo myocardial ischemia-reperfusion injury in non-diabetic male rats and enhances endothelium-dependent vasorelaxation. J. Transl. Med. 2019;17(1):127. doi: 10.1186/s12967-019-1881-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Baker HE, Kiel AM, Luebbe ST, Simon BR, Earl CC, Regmi A, et al. Inhibition of sodium–glucose cotransporter-2 preserves cardiac function during regional myocardial ischemia independent of alterations in myocardial substrate utilization. Basic Res. Cardiol. 2019 doi: 10.1007/s00395-019-0733-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gnaiger E. Capacity of oxidative phosphorylation in human skeletal muscle: New perspectives of mitochondrial physiology. Int. J. Biochem. Cell Biol. 2009;41(10):1837–1845. doi: 10.1016/j.biocel.2009.03.013. [DOI] [PubMed] [Google Scholar]
- 30.Lemieux H, Semsroth S, Antretter H, Hofer D, Gnaiger E. Mitochondrial respiratory control and early defects of oxidative phosphorylation in the failing human heart. Int. J. Biochem. Cell Biol. 2011;43(12):1729–1738. doi: 10.1016/j.biocel.2011.08.008. [DOI] [PubMed] [Google Scholar]
- 31.Lou PH, Zhang L, Lucchinetti E, Heck M, Affolter A, Gandhi M, et al. Infarct-remodelled hearts with limited oxidative capacity boost fatty acid oxidation after conditioning against ischaemia/reperfusion injury. Cardiovasc. Res. 2013;97(2):251–261. doi: 10.1093/cvr/cvs323. [DOI] [PubMed] [Google Scholar]
- 32.Lopaschuk GD. Metabolic changes in the acutely ischemic heart. Heart Metabol. 2016;70:32–35. [Google Scholar]
- 33.Cason BA, Gamperl AK, Slocum RE, Hickey RF. Anesthetic-induced preconditioning: Previous administration of isoflurane decreases myocardial infarct size in rabbits. Anesthesiology. 1997;87(5):1182–1190. doi: 10.1097/00000542-199711000-00023. [DOI] [PubMed] [Google Scholar]
- 34.Groennebaek T, Nielsen J, Jespersen NR, Botker HE, de Paoli FV, Miller BF, et al. Utilization of biomarkers as predictors of skeletal muscle mitochondrial content after physiological intervention and in clinical settings. Am. J. Physiol. Endocrinol. Metab. 2020;318(6):E886–E889. doi: 10.1152/ajpendo.00101.2020. [DOI] [PubMed] [Google Scholar]
- 35.Larsen S, Nielsen J, Hansen CN, Nielsen LB, Wibrand F, Stride N, et al. Biomarkers of mitochondrial content in skeletal muscle of healthy young human subjects. J. Physiol. 2012;590(14):3349–3360. doi: 10.1113/jphysiol.2012.230185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kitagawa H, Yamazaki T, Akiyama T, Sugimachi M, Sunagawa K, Mori H. Microdialysis separately monitors myocardial interstitial myoglobin during ischemia and reperfusion. Am. J. Physiol. Heart Circ. Physiol. 2005;289(2):H924–H930. doi: 10.1152/ajpheart.01207.2004. [DOI] [PubMed] [Google Scholar]
- 37.Council FMNR. Guide for the Care and Use of Laboratory Animals. The National Academies Press; 1996. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data included in this article is displayed or can be displayed upon request.