

Abstract
IL-33 is upregulated in ulcerative colitis and has a protective role in chemically-induced acute murine colitis. We aimed to determine whether IL-33 influences Il10−/− chronic colitis and its cellular source in health and during colitis. Il10−/−Il33−/− and Il10−/−Il33+/+ littermates developed colitis of similar severity. Colon Il33 was induced in WT and Il10−/− mice exposed to DSS, but not in unchallenged Il10−/− mice with colitis. Il33-citrine reporter mice showed that Il33-citrine colocalized with α-smooth muscle actin+ myofibroblasts and vimentin+ fibroblasts in WT mice. Citrine+CD74+CD90hi inflammatory fibroblasts were increased with DSS treatment. IL-1β induced Il33 expression in colon myofibroblasts, but colon Il33 expression did not differ between DSS-treated WT and Il1r1−/− mice. In conclusion, deficiency of IL-33 does not alter the severity of chronic colitis in Il10−/− mice. Induction of Il33 upon DSS exposure in WT and Il10−/− mice, but not in unchallenged Il10−/− mice, suggests epithelial injury induces colon IL-33. Fibroblasts are the primary colonic source of IL-33 and IL-33-expressing CD90hiCD74+ fibroblasts are increased during DSS-induced colitis. IL-1β induces Il33 in colon myofibroblasts in vitro, but signaling through the IL-1R1 is not necessary for induction of IL-33 in DSS-induced colitis.
Subject terms: Cytokines, Ulcerative colitis
Introduction
Inflammatory bowel disease (IBD), including ulcerative colitis (UC) and Crohn’s disease, is a group of disorders of chronic intestinal inflammation and progressive bowel damage caused by a complex interplay of genetic, microbial and environmental factors1. Cytokines are central to IBD pathogenesis, with roles for both driving and controlling the mucosal inflammation2. Advances in our understanding of cytokine biology have led to the development of transformative therapies for IBD. Still, 40 and 70% of UC patients will not achieve mucosal healing with anti-TNF therapy and anti-IL12/23 therapy, respectively, and 70% of available anti-cytokine biologic drugs frequently fail to heal the mucosa3,4. Defining new mechanisms for intestinal inflammation is a clear need for developing new effective therapies in IBD.
IL-33, a member of the IL-1 superfamily, is increased in patients with UC and a polymorphism has been associated with UC and an extensive colitis phenotype5–11. IL-33 is often described as an alarmin released by epithelial and endothelial cells in the setting of injury, but it is also expressed by innate immune cells12–14. In the intestine, previous studies have shown that IL-33 is expressed in subepithelial fibroblasts, but further phenotyping has not been done15,16. Our lab has previously identified a protective role for IL-33 in oxazolone colitis, through protecting epithelial cells, particularly goblet cells17, which is mediated by ILC2-derived IL-1318. Other studies have also shown that IL-33 has a protective role by inducing epithelial-derived miR-320 that promotes epithelial repair and the resolution of inflammation19 and decreasing the pathogenic Th17 response in chronic DSS colitis20,21. Of the studies done on chronic models of colitis, only the role of exogenous IL-33 was explored22,23, and exogenous IL-33 is known to effect intestinal physiology through goblet cell hypertrophy and hyperplasia5. However, studies have not examined the role of endogenous IL-33 in a chronic model of colitis.
The role for endogenous IL-33 in chronic colitis as well as regulation of IL-33 expression remains uncertain. In our study, we examine differences in the role of IL-33 in chronic vs. acute colitis. Interestingly, IL-33 is strongly induced during DSS-induced colitis, but not during IL-10–/– chronic colitis. Although a protective role has been shown for IL-33 in other models of colitis, we could find no differences in colitis development or severity in Il10−/− compared to Il10−/−Il33−/− mice. Using IL-33-citrine reporter mice (Il33Cit/+), we show that vimentin+CD90+ fibroblasts are the source of IL-33 at baseline and this population increases in DSS-induced colitis. Although IL-1β induced IL-33 expression in vitro, using Il1r1−/− mice, we showed this signaling pathway is not required for Il33 induction during DSS-induced colitis. Interestingly, CD90+CD74+ inflammatory fibroblasts express IL-33 and are increased in DSS-induced colitis, indicating this population is likely responsible for the increases in Il33 in acute colitis induced by epithelial damage.
Results
IL-33 deletion does not alter course of Il10−/− colitis
To determine the role of IL-33 in a spontaneous chronic model of colitis, we crossed Il10−/− mice to Il33−/− to generate Il10−/− Il33−/− mice. Il10−/−Il33−/− and Il10−/−Il33+/+ littermates developed colitis of similar severity beginning at approximately 12 weeks of age. There were no differences in histopathologic severity between the two strains (Fig. 1A). Tissue Ifng and Il17a, two T cell cytokines known to be involved in disease, were examined in the two strains and both were induced similarly in Il10−/−Il33−/− and Il10−/−Il33+/+ compared to WT mice (Fig. 1B). We examined weight loss and rectal prolapse occurrence in both strains of mice through 30 weeks of age and found no differences (Fig. 1C). Histopathological scores were also not different at 30 weeks of age (Fig. 1D). Collectively this data does not support a role for IL-33 in regulating spontaneous chronic colitis in Il10−/− mice.
Figure 1.

IL-33 deletion did not alter severity of Il10−/− colitis. (A) Representative H&E sections from 12-week old Il10−/−Il33+/+ and Il10−/−Il33−/− mice and histological scoring. (B) RT PCR analysis at 12 weeks old. (C) Weight change and prolapse occurrence up to 30 weeks old. (D) Histopathological scores of 30-week old Il10−/−Il33+/+ and Il10−/−Il33−/− mice. N = 7–16/group.
Il33 is induced in DSS colitis but not Il10−/− colitis
To better understand the lack of an effect of Il33 deficiency on colitis in Il10−/− mice, we examined colon Il33 expression in WT and Il10−/− mice at 12 and 30 weeks of age. Il33 was not increased in Il10−/− mice with chronic colitis compared to WT mice (Fig. 2A). Others have demonstrated a protective role for IL-33 in DSS-induced colitis19,24,25. In contrast to the chronic Il10-deficient model, we observed a sevenfold increase in colon Il33 expression in acute colitis induced by epithelial injury from DSS compared to untreated WT mice (Fig. 2B), indicating that factors specific to DSS colitis may be regulating IL-33. To determine whether IL-10 is required for induction of IL-33 during colitis, we exposed Il10−/− mice to DSS. DSS exposure induced colon Il33 expression significantly compared to untreated mice (Fig. 2B), suggesting that IL-10 is not required for Il33 induction during colitis.
Figure 2.

IL-33 was induced in acute DSS colitis but not chronic Il10−/− colitis. Il33 expression assessed by real-time RT-PCR in the distal colon of (A) 12-week- and 30-week-old mice WT and Il10−/− mice or (B) 12-week old WT and Il10−/− mice treated with DSS for 5 days and harvested on day 7. (C) Il1b and Tnf expression in Il10−/− and WT mice with or without DSS treatment. N = 5–8/group **p < 0.01, ****p < 0.0001.
To begin to understand what difference might be leading to differences in regulation of IL-33 in the two models of murine colitis, we examined tissue RNA expression for two cytokines that have previously been shown to induce IL-33, TNF and IL1-β16. Colon Tnf expression was 4.6 fold higher, and, strikingly, Il1b expression was 1419-fold higher in DSS-treated WT mice compared to 12-week old IL-10KO mice with colitis (Fig. 2C).
Colon fibroblasts are the main source of Il33
IL-33 citrine reporter mice (Il33Cit/+) were used to investigate the cellular source of IL-33 in unchallenged WT mice. This reporter uses citrine fluorescence as a surrogate for IL-33 mRNA expression with the GFP-derived Citrine gene inserted directly downstream of the ATG start codon of Il3314. Immunofluorescence microscopy colocalized citrine expression in the proximal colon with alpha smooth muscle actin (α-SMA) and vimentin in unchallenged Il33Cit/+ mice, suggesting IL-33 is expressed by colon myofibroblasts (Fig. 3A,B). To confirm this, flow cytometry was performed for epithelial, hematopoietic and fibroblast markers on the lamina propria fraction. In the lamina propria, citrine+ cells were EpCam−, CD45−, and > 85% vimentin+, consistent with fibroblasts (Fig. 3C).
Figure 3.

Fibroblasts were the primary source of IL-33 in the healthy mouse colon. Representative immunofluorescence photomicrographs of proximal colon from Il33Cit/+ reporter mice co-stained with (A) α-SMA for myofibroblasts and (B) vimentin for total fibroblasts. (C) Flow cytometric analysis of colon lamina propria cells that express citrine in healthy Il33Cit/+ mice. Shaded histogram is fluorescence minus one control. (D) Flow cytometry for fibroblast markers in citrine+ cells. Representative flow plots from 3 independent experiments.
Fibroblasts are a complex population of many different subsets. As single cell studies have demonstrated, stromal cells can be subdivided into as many as six subsets26. To further phenotype these cells, we also examined expression of CD90, CD140a and CD49a (Fig. 3D). CD90 is reported to be expressed on fibroblasts located in close proximity to stem cells and support organoid growth in vitro27. CD49a is α1 integrin and is involved in fibroblast proliferation and adhesion to collagen28. CD140a is PDGFRA and is considered a pan-fibroblast marker and CD140a+ cells are important for maintaining the intestinal stem cell niche29. The IL-33-expressing population of fibroblasts was heterogeneous, with CD90hi, CD90mid and CD90− cell populations. The citrine+CD90hi fibroblast population expressed CD49a, but the majority of these cells were CD140a− (Fig. 3D).
IL-1β induced Il33 expression in colon fibroblasts
Since we observed substantial differences in the induction of Il1b between colitis induced by DSS and IL-10-deficiency, we sought to determine whether IL-1β regulates Il33 expression in colon myofibroblasts, as has been reported by others15. The WEHI-YH2 colon myofibroblast cell line30 was stimulated with IL-1β for 18 h and Il33 expression was analyzed. IL-1β increased Il33 in WEHI cells compared to control (Fig. 4A). To examine primary cells, citrine+ cells were sorted from control and DSS-treated mice. Although Il33 was not increased in unstimulated citrine+ cells from DSS-treated mice compared to those from control mice (Fig. 4B), ex vivo stimulation with IL-1β increased Il33 expression in citrine+ cells from DSS-treated mice (Fig. 4C), indicating that IL-1β could play a role in the regulation of IL-33 in the colon during colitis.
Figure 4.

IL-1β induced Il33 expression in colon fibroblasts in vitro. Il33 expression assessed by real-time RT-PCR in (A) IL-1β treated and untreated colon myofibroblast WEHI-YH2 cells from 3 independent experiments and (B) Citrine+ cells sorted from Control and DSS-treated Il33Cit/+ mice. (C) Citrine+ cells sorted from DSS-treated Il33Cit/+ mice stimulated with IL-1β, and (D) colon tissue from WT and Il1r1−/− mice given DSS. (E) Representative H&E staining and (F) histopathologic scores of WT and Il1r1−/− mice treated with DSS. (G) Representative images of vimentin immunofluorescent staining. N = 5–10 mice/group, *p < 0.05, **p < 0.01.
To examine the role of IL-1β in vivo, we treated WT and Il1r1−/− mice deficient for the IL-1β receptor with DSS and examined colon Il33 expression. We found no difference in Il33 induction between WT and Il1r1−/− mice treated with DSS (Fig. 4D). WT and Il1r1−/− mice treated with DSS exhibited similar histopathologic severity (Fig. 4E,F). Although Il33 expression was induced with DSS in WT and Il1r1−/− mice, expression did not correlate with histopathologic severity in either strain (WT, Spearman r 0.61, p = 0.16; Il1r1−/−, Spearman r 0.03, p > 0.99). Furthermore, immunofluorescence staining for vimentin indicated a similar number of fibroblasts between WT and Il1r1−/− mice treated with DSS (Fig. 4G). These data indicate that IL1R1 signaling is not required for the induction of Il33 in the colon during DSS-induced colitis in vivo.
IL-33-citrine+ fibroblasts are increased during DSS, but not IL-10-deficient colitis
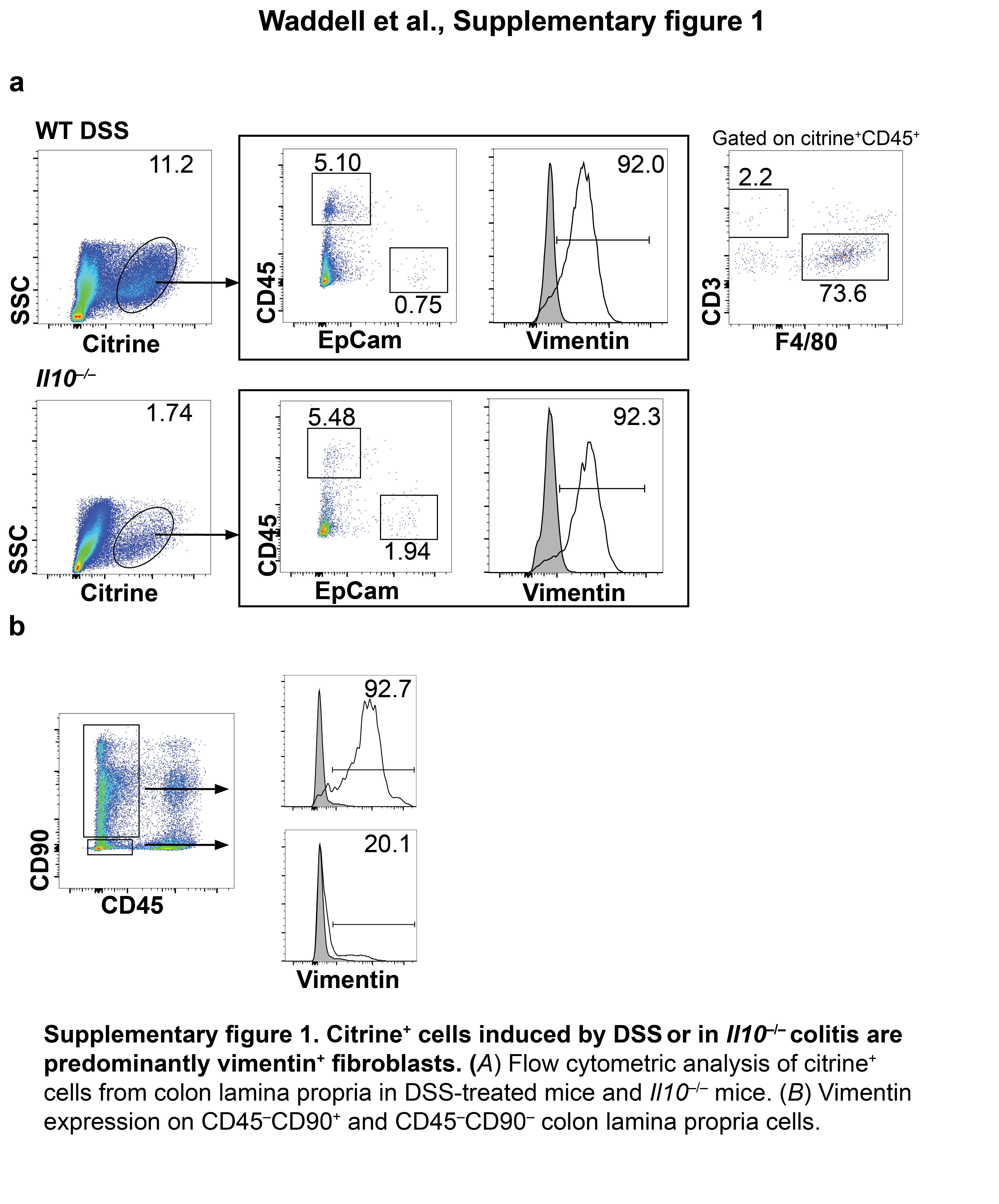
Since Il33 expression increases in DSS-induced colitis, we examined IL-33-citrine+ cells in DSS-exposed and unchallenged Il33Cit/+mice by flow cytometry. Over 90 percent of Citrine+ cells in DSS-induced colitis and IL10−/− colitis were vimentin+ fibroblasts. Approximately 5% were CD45+ leukocytes, most of which were F4/80+ macrophages (Supplementary Fig. 1). We went on to demonstrate that CD45−CD90+ cells correspond to vimentin+ cells, allowing us to use surface markers to identify fibroblasts (Supplementary Fig. 1). Citrine+ cells were significantly increased during DSS-induced colitis compared to unchallenged control mice (Fig. 5A). This increase in citrine+ cells was due to a generalized increase in CD45−CD90+ fibroblasts in the colon (Fig. 5B), but not due to a higher percentage of fibroblasts expressing IL-33 (Fig. 5C), indicating that both IL-33+ and IL-33− fibroblasts are increasing during DSS-induced colitis. Furthermore, citrine mean fluorescence intensity (MFI) was not increased in CD45−CD90+ fibroblasts from DSS-treated mice compared to untreated mice, indicating that transcription of Il33 is not increased on a per cell basis (Fig. 5C). To examine the source of IL-33 in Il10−/− mice, we generated Il10−/−Il33Cit/+mice. In contrast to the DSS model, during colitis in 12-week or 30-week old Il10−/−Il33Cit/+ mice, neither citrine+ cells nor CD45−CD90+ fibroblasts were increased compared to littermate Il10+/-Il33Cit/+ mice (Fig. 5D,E). Immunofluorescence in DSS-treated Il33Cit/+mice (Fig. 5F,G) and Il10−/−Il33Cit/+ mice (Fig. 5H,I) demonstrated vimentin+citrine+ cells in areas of inflammation. Interestingly, these cells are α-SMA−, indicating these cells are different from the α-SMA+citrine+ cells seen in untreated mice (Fig. 3). These data support that increased Il33 expression in DSS-induced colitis may be explained by an expanded population of Il33-expressing colon mucosal fibroblasts, proportional to an overall expansion of fibroblasts, which is not observed in Il10−/− colitis.
Figure 5.

Increased colon mucosal fibroblasts in colitis induced by DSS but not IL-10-deficiency. Comparisons of percent of colon lamina propria cells that are (A) citrine+ ll33-expressing cells or (B) CD45−CD90+ fibroblasts, and (C) percent of lamina propria citrine+ ll33-expressing fibroblasts and citrine MFI in unchallenged and DSS-treated Il33Cit/+ mice. Comparisons of percent of colon lamina propria cells that are (D) citrine+ ll33-expressing cells or (E) CD45−CD90+ fibroblasts in 12- and 30-week old WT and Il10−/− mice. Representative immunofluorescent images from DSS-treated Il33Cit/+mice (F) and (G) and Il10−/− Il33Cit/+mice (H) and (I) N = 5–8 mice/group **p < 0.01, ****p < 0.0001.
Recently single cell RNA-seq approaches have brought attention to an expanded population of inflammatory fibroblasts as a key signaling hub in UC26,31. To further phenotype the IL-33+ fibroblasts in DSS-induced colitis, we examined the inflammatory marker CD74, which has been shown to be expressed on inflammatory fibroblasts26. Inflammatory citrine+ CD90hiCD74+ fibroblasts were markedly increased in DSS-treated compared to unchallenged Il33Cit/+mice (Fig. 6A,B). Furthermore, > 90% of CD90hiCD74+ fibroblast are citrine+ while only 33–41% of CD90lowCD74− fibroblasts are citrine+ (Fig. 6C,D) demonstrating that CD90hiCD74+ fibroblasts have a greater capacity for making IL-33.
Figure 6.

Citrine+ fibroblasts induced by DSS are CD90hiCD74+ inflammatory fibroblasts. (A) Flow cytometric analysis and (B) quantification of CD90hiCD74+ cells gated on citrine+CD45−CD90+ fibroblasts in the colon lamina propria at baseline and during DSS-induced colitis. (C) Flow cytometric analysis and (D) quantification of citrine+ cells gated on CD45−CD90+ fibroblasts to show which fibroblast population is mainly expressing IL-33-citrine during DSS. N = 8–9 mice/group ****p < 0.0001.
Discussion
In the present study, we demonstrate that mucosal fibroblasts are the main cellular source of IL-33 in the mouse colon. During acute colitis induced with DSS, we show there is an expansion of IL-33-producing inflammatory fibroblasts similar to those reported in human UC. Surprisingly, we found that neither Il33 expression, nor IL-33 producing fibroblasts were increased during the development of chronic colitis in Il10-deficient mice, although Il33 expression could be induced in Il10−/− mice by epithelial injury with DSS. Accordingly, using double knockout mice, we demonstrated that IL-33 does not play a significant role in the development or progression of chronic colitis in Il10−/− mice. We found that colon mucosal Il1b expression was induced to a much higher degree in DSS-induced compared to Il10−/− colitis, and that IL-1β induced Il33 from colon fibroblasts in vivo, however IL1R signaling was not required for induction of Il33 expression during DSS.
Many studies have explored the role of IL-33 in acute murine models of colitis or recovery from colitis, but there is a paucity of studies in models of chronic colitis. The role of exogenous IL-33 has been examined in chronic colitis22,23, but supra-physiologic levels of IL-33 have profound effects on intestinal morphology through induction of Th2 cytokines5. To determine the role of endogenous IL-33 in chronic colitis, we bred Il10−/−/Il33−/− mice and compared them to littermate Il10−/−/Il33+/+ mice. Mice spontaneously began to develop colitis around 8 weeks old and some mice were followed for up to 30 weeks. We expected to find a protective role for IL-33 in chronic colitis due to its protective role in other models of colitis, including our previous study in oxazolone colitis17,19,22,24,25. Some studies have shown a protective role for IL-33 by acting directly on the intestinal epithelium17,19. However, other studies have shown that IL-33 acts on ST2+ regulatory T cells to induce their proliferation and suppressive function32,33. Since IL-10 from regulatory T cells is known to be important in preventing development of intestinal inflammation34, the lack of functional IL-10-producing regulatory T cells could have prevented IL-33 from ameliorating colitis in this model. On the other hand, this potential explanation does not explain the finding of unchanged colon Il33 expression during colitis in Il10−/− mice. The lack of increased Il33 expression Il10−/− mice with colitis stands in stark contrast to the increased Il33 expression consistently observed in human UC7–10,17. It may be that the degree of epithelial injury in chronic Il10−/− colitis is insufficient to induce tissue IL-33 given that colon tissue Il33 expression could be induced with DSS treatment in Il10−/− mice.
Since previous studies have shown a protective role for IL-33 in DSS-induced colitis, we compared Il33 induction in Il10−/− chronic colitis and DSS-induced acute colitis. We found that Il33 is significantly increased in DSS-induced colitis, but not in Il10−/− colitis. However, Il33 can be induced in Il10−/− mice by DSS, suggesting that epithelial damage triggers colon IL-33 expression and that IL-33 induction is not limited by IL-10 deficiency itself. We sought to determine what molecular differences between DSS and Il10−/− colitis could lead to this difference in Il33 induction. We firstly found that Il1b was induced over 1000-fold more in DSS compared to Il10−/− colitis. Furthermore, IL-1β has been shown to induce Il33 in dermal and ileal fibroblasts15,16. Although we demonstrated that IL-1β induced Il33 expression in both a colon myofibroblast cell line and Il33-expressing citrine+ cells isolated from DSS-treated Il33Cit/+ mice in vitro, we did not observe any effect of loss of IL-1β signaling through its receptor on DSS-induced mucosal inflammation or Il33 expression in vivo. Furthermore, our data in vivo suggest that an increase in the number of Il33-expressing fibroblasts, not fibroblast-intrinsic Il33 expression, explains the overall increase in ll33 in the colon. Therefore, although IL-1β may, in part, regulate colon fibroblast Il33 expression, IL-1β signaling is not required for induction of Il33 after epithelial injury with DSS. Further research is warranted to determine the factors that stimulate recruitment, differentiation, or proliferation of IL-33-producing colon fibroblasts by epithelial injury and or mucosal inflammation.
Others have demonstrated that intestinal IL-33 is also regulated by bacterial infection and colonization. Similar to the observed induction of IL-33 in DSS-induced colitis, colon IL-33 and IL-33 receptor are upregulated in acute infectious colitis with Citrobacter rodentium. In this infectious model, IL-33 drives mucosal inflammation and impairs bacterial clearance through enhancing epithelial permeability and limiting the induction of Th17 cells35. Stable colonization of mice with the Crohn’s disease-associated pathobiont adherent invasive E. coli, permitted only after colitis induced by Salmonella or DSS, also results in marked upregulation of IL-33 and the IL-33 receptor, which contributes to intestinal fibrosis in this model36. The sources of IL-33 have not been fully elucidated in these microbe-induced models.
Previous studies have shown that in the intestine, myofibroblasts and epithelial cells express IL-33 at baseline8,15,16,37. Using IL-33-citrine reporter mice14, we showed that the main cellular source of IL-33 in unchallenged mice and during acute colitis with DSS is CD90+vimentin+ fibroblasts. Immunofluorescent microscopy revealed that a subset of these cells were myofibroblasts expressing α-SMA. Further heterogeneity in this population is evidenced by populations of Il33-expressing citrine+ cells that are CD90hiCD49a+CD140a− and CD90midCD49a−CD140a+/−. Recent single cell RNA-seq studies of stromal cells in the intestine have shown diverse populations of fibroblasts in the colon of patients with UC26,31. Following DSS treatment, we observed an increase in citrine+ cells in DSS-treated mice compared to control mice, confirming an increase in Il33-expressing cells is leading to the increase in Il33 during DSS.
We sought to determine which IL-33-citrine+ fibroblast population was increasing during DSS-induced colitis. One study found that IL-33-expressing colon fibroblasts also expressed the major histocompatibility complex class II invariant chain CD74 and these cells were enriched in active UC26. We found that CD90hiCD74+ cells expressing IL-33 were increased in DSS-treated mice compared to control mice. CD90 has previously been shown to be a marker for intestinal stromal cells that express IL-33 in response to Salmonella typhimurium and flagellin27. Furthermore, CD90+ fibroblasts have been shown to more strongly support epithelial growth compared to CD90− fibroblasts using co-cultures with colonoids, resulting in significantly more colonoid budding and growth in co-cultures with CD90+ fibroblasts38. These studies support a protective, immunoregulatory role for this fibroblast population during colitis.
In conclusion, the primary source of IL-33 in the mouse colon, at baseline and during acute colitis induced by epithelial injury, is mucosal fibroblasts. The induction of tissue Il33 expression after epithelial injury with DSS is related to an increase in Il33-expressing CD90hiCD74+ inflammatory fibroblasts. Il33-expressing fibroblasts are not induced during spontaneous chronic colitis in Il10−/− mice and IL-33 deficiency does not affect colitis development or severity in Il10−/− mice. IL-1β induces Il33 expression from colon fibroblasts in vitro, but signaling through IL1R1 is dispensable for induction of Il33 expression in colon tissue after epithelial injury with DSS in vivo. This study uncovers important cell and molecular differences between animal models of colitis and human UC with regard to cytokine expression and stromal cell populations. Future study of the action of IL-33-producing fibroblasts during colitis may uncover new pathways to translate for UC treatment.
Materials and methods
Mice and in vivo treatment
Il10−/− (Jackson Labs strain 002251), Il1r1−/− (Jackson Labs strain 003245), Il33−/−, Il33-citrine reporter (Il33Cit/+) and WT mice, all on the C57BL/6 background, were bred at CCHMC under specific-pathogen-free conditions and maintained on a standard laboratory chow diet in a half-day light cycle exposure and temperature-controlled environment. The generation of the Il33−/− and Il33Cit/+ mice were previously described14,39. Il10−/− and Il33−/− strains were crossed to generate littermate Il10−/−Il33−/− and Il10−/−Il33+/+ mice. Il33Cit/+ mice were crossed to Il10−/− mice to generate Il10−/−Il33Cit/+. Mice in this facility tested positive for Helicobacter spp. Male and female age- and sex-matched mice were used and were age 6–12 weeks at the start of the experiments. The study was carried out following recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The CCHMC Institutional Animal Care and Use Committee approved the protocol. This study was carried out in accordance to ARRIVE guidelines.
DSS-induced colonic injury and histopathologic examination
DSS (Colitis grade, GoJira Chemicals) was administered in the drinking water as a 2% (w/v) solution for up to 7 days to induce acute colitis40. Colons were stained with H&E and examined and histologically scored by light microscopy. DSS-induced colitis was scored by a blinded scorer on a 0–21 scale including: percentage of involved area, amount of follicles, edema, erosion/ulceration, crypt loss and infiltration of immune cells40. Il10−/− colitis was scored by a blinded scorer on a 0–4 scale for each colon segment as previously described41.
Immunofluorescence
For immunofluorescence analysis, frozen sections from Il33Cit/+ mice were fixed in 10% acetone for 15 min, rinsed in PBS, blocked with 3% donkey serum/PBS for 2 h at room temperature as previously described18, and incubated with primary Ab rabbit anti-mouse alpha smooth muscle actin (1:100, ab5694 from AbCam) or Alexa Fluor 594 anti-Vimentin Antibody (1:100, Biolegend, clone W16220A) in 3% normal donkey serum/PBS. For vimentin staining from WT and Il1r1−/− mice, formalin-fixed paraffin-embedded sections were deparaffinized and rehydrated. Antigen retrieval was performed using citrate-based antigen unmasking solution (Vector Laboratories) for 10 min in a pressure cooker. Sections were rinsed in PBS, blocked with 3% donkey serum/PBS for 2 h at room temperature and incubated with primary Ab rabbit anti-Vimentin Antibody (ab92547, AbCam). After an overnight incubation at 4 °C, sections were washed with 0.1% BSA and 0.05% Tween/PBS and incubated with donkey anti-rabbit Alexa Fluor 594 (Invitrogen, Carlsbad, CA) for 2 h at room temperature. Slides were washed in PBS and counterstained with DAPI/Supermount G solution (Southern Biotechnology Associates, Birmingham, AL). Images were acquired using an Olympus BX51 microscope with a DP80 camera (Olympus America Inc., PA, USA) and CellSens Dimension digital imaging software (Olympus Corporation, version 1.18). Images were merged using ImageJ 1.52q (FIJI) software (NIH, https://imagej.nih.gov/ij/).
RNA expression
RNA was isolated from tissue or cells using the RNeasy Mini Kit (Qiagen, Valencia, CA) per the manufacturer’s instructions, and RNA (100 ng) was reverse transcribed using the High-Capacity cDNA Reverse Transcription Kit (ThermoFisher Scientific, Waltham, MA) as previously described18. Real-time RT-PCR was performed with TaqMan Gene Expression Assays for Il33 (Mm00505399), Il1b (Mm00434228_m1), Tnf (Mm00443258), Il17a (Mm01189488), Ifng (Mm01168134) and Gapdh (Mm99999915_g1). All reactions were performed on a StepOnePlus real-time PCR system (ThermoFisher Scientific). Relative mRNA levels were determined using the 2−ΔΔCT method with Gapdh as the reference.
Flow cytometric analysis and cell sorting
Mouse colons were washed in cold CMF-HBSS and placed in CMF-HBSS containing 5 mM EDTA and shaken gently at 37 °C for 30 min to remove epithelial cells. The remaining colon tissue was minced and agitated in RMPI with 2 mg/mL dispase (Gibco), 0.5 mg/mL collagenase I (Gibco) and 0.2 mg/mL DNase (Roche) at 37 °C for 45 min. Tissue was broken up using a 19-gauge needle and filtered through a 100 μM filter. Cells were centrifuged at 350 × g for 20 min and used for flow cytometry. For cell sorting, cells were resuspended in 40%Percoll/RPMI and spun at room temperature for 20 min at 600 × g and the cell pellet was collected. Single-cell suspensions were washed with FACS buffer (PBS/1% FCS) and incubated with combinations of the following Abs: APC-Cy7 anti-CD45 (Clone 30-F11), Pacific Blue anti-CD90 (Clone 30.H12), APC anti-EpCam (Clone G8.8), Alexa Fluor 594 anti-vimentin (Clone W1622A), APC anti-CD140a (Clone APA5), Alexa Fluor 647 anti-CD74 (Clone In1/CD74), PE anti-CD49a (Clone HMα1), PE Cy7 anti-F4/80 (Clone BM8) and PerCP Cy5.5 anti-CD3 (Clone 17A2) (Biolegend, San Diego, CA). Cells were then analyzed with an LSR Fortessa cytometer (BD Biosciences, San Jose, CA). Citrine+ cells were sorted with a Sony SH800S supported by NIH S10OD023410.
In vitro and ex vivo colon fibroblast stimulation
WEHI-YH2 cells, a colon myofibroblast cell line30, or citrine+ cells sorted from DSS-treated mice were stimulated in vitro with 20 ng/mL IL-1β (Biolegend, San Diego, CA) for 18 h and then cells were harvested for RNA.
Statistical analysis
For all data from experiments with three or more groups, non-parametric Kruskal–Wallis test was performed followed by two-stage step-up method of Benjamini, Krieger, and Yekutieli for false discovery rate. Data from experiments with two groups was analyzed using the non-parametric Mann–Whitney test. Rates of prolapse were compared using the Log-rank (Mantel-Cox) test. Spearman correlation was used for correlation analyses. Individual data points and mean ± SE are plotted on all graphs. The analysis was performed on Prism software (version 8.0.1, GraphPad Software, La Jolla, CA, www.graphpad.com). All authors had access to the study data and had reviewed and approved the final manuscript.
Supplementary Information
{kind=link}
Acknowledgements
We thank Andrew N. J. McKenzie for generously providing the Il33-citrine reporter mice, Susume Nakae and Hirohisa Saito for sharing the Il33-deficient mice, and Antony Burgess for providing the WEHI-YH2 cells used in these studies. All flow cytometric data were acquired using equipment maintained by the Research Flow Cytometry Core in the Division of Rheumatology at Cincinnati Children’s Hospital Medical Center.
Author contributions
A.W., J.V. and S.F. acquired data. M.R. and A.W. designed the experiments, analyzed and interpreted data and wrote the manuscript. M.R. supervised the study. All authors reviewed the manuscript.
Funding
Research reported in this publication was supported by the National Institute of Diabetes and Digestive and Kidney Diseases of the National Institutes of Health under award R01DK117119 to Michael J. Rosen. This project was supported in part by NIH P30 DK078392 for the Research Flow Cytometry Core and Integrated Morphology Core of the Digestive Disease Research Center in Cincinnati. This work was also supported by a Crohn’s & Colitis Foundation Research Fellowship Award to Amanda Waddell and a Trustee Award from the Cincinnati Children’s Research Foundation to Michael J. Rosen.
Data availability
Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-021-89119-1.
References
- 1.de Souza HSP, Fiocchi C, Iliopoulos D. The IBD interactome: An integrated view of aetiology, pathogenesis and therapy. Nat. Rev. Gastroenterol. Hepatol. 2017;14:739–749. doi: 10.1038/nrgastro.2017.110. [DOI] [PubMed] [Google Scholar]
- 2.Neurath MF. Cytokines in Inflammatory Bowel Disease. Nature Reviews Immunology. Nature Publishing Group; 2014. pp. 329–342. [DOI] [PubMed] [Google Scholar]
- 3.Rutgeerts P, et al. Infliximab for induction and maintenance therapy for ulcerative colitis. N. Engl. J. Med. 2005;353:2462–2476. doi: 10.1056/NEJMoa050516. [DOI] [PubMed] [Google Scholar]
- 4.Sands BE, et al. Ustekinumab as induction and maintenance therapy for ulcerative colitis. N. Engl. J. Med. 2019;381:1201–1214. doi: 10.1056/NEJMoa1900750. [DOI] [PubMed] [Google Scholar]
- 5.Schmitz J, et al. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity. 2005;23:479–490. doi: 10.1016/j.immuni.2005.09.015. [DOI] [PubMed] [Google Scholar]
- 6.Kakkar R, Lee RT. The IL-33/ST2 pathway: Therapeutic target and novel biomarker. Nat. Rev. Drug Discovery. 2008;7:827–840. doi: 10.1038/nrd2660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Beltran CJ, et al. Characterization of the novel ST2/IL-33 system in patients with inflammatory bowel disease. Inflamm. Bowel Dis. 2010;16:1097–1107. doi: 10.1002/ibd.21175. [DOI] [PubMed] [Google Scholar]
- 8.Kobori A, et al. Interleukin-33 expression is specifically enhanced in inflamed mucosa of ulcerative colitis. J. Gastroenterol. 2010;45:999–1007. doi: 10.1007/s00535-010-0245-1. [DOI] [PubMed] [Google Scholar]
- 9.Pastorelli L, et al. Epithelial-derived IL-33 and its receptor ST2 are dysregulated in ulcerative colitis and in experimental Th1/Th2 driven enteritis. Proc. Natl. Acad. Sci. USA. 2010;107:8017–8022. doi: 10.1073/pnas.0912678107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Seidelin JB, et al. IL-33 is upregulated in colonocytes of ulcerative colitis. Immunol. Lett. 2010;128:80–85. doi: 10.1016/j.imlet.2009.11.001. [DOI] [PubMed] [Google Scholar]
- 11.Latiano A, et al. Associations between genetic polymorphisms in IL-33, IL1R1 and risk for inflammatory bowel disease. PLoS ONE. 2013;8:e62144. doi: 10.1371/journal.pone.0062144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pei C, et al. Emerging role of interleukin-33 in autoimmune diseases. Immunology. 2014;141:9–17. doi: 10.1111/imm.12174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pichery M, et al. Endogenous IL-33 is highly expressed in mouse epithelial barrier tissues, lymphoid organs, brain, embryos, and inflamed tissues: In situ analysis using a novel Il-33-LacZ gene trap reporter strain. J. Immunol. 2012;188:3488–3495. doi: 10.4049/jimmunol.1101977. [DOI] [PubMed] [Google Scholar]
- 14.Hardman CS, Panova V, McKenzie ANJ. IL-33 citrine reporter mice reveal the temporal and spatial expression of IL-33 during allergic lung inflammation. Eur. J. Immunol. 2012;42:488–498. doi: 10.1002/eji.201242863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sponheim J, et al. Inflammatory bowel disease-associated interleukin-33 is preferentially expressed in ulceration-associated myofibroblasts. Am. J. Pathol. 2010;177:2804–2815. doi: 10.2353/ajpath.2010.100378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mahapatro M, et al. Programming of intestinal epithelial differentiation by IL-33 derived from pericryptal fibroblasts in response to systemic infection. Cell Rep. 2016;15:1743–1756. doi: 10.1016/j.celrep.2016.04.049. [DOI] [PubMed] [Google Scholar]
- 17.Waddell A, et al. IL-33 signaling protects from murine oxazolone colitis by supporting intestinal epithelial function. Inflamm. Bowel Dis. 2015;21:2737–2746. doi: 10.1097/MIB.0000000000000532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Waddell A, Vallance JE, Hummel A, Alenghat T, Rosen MJ. IL-33 induces murine intestinal goblet cell differentiation indirectly via innate lymphoid cell IL-13 secretion. J. Immunol. 2019;202:598–607. doi: 10.4049/jimmunol.1800292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lopetuso LR, et al. IL-33 promotes recovery from acute colitis by inducing miR-320 to stimulate epithelial restitution and repair. Proc. Natl. Acad. Sci. USA. 2018;115:E9362–E9370. doi: 10.1073/pnas.1803613115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhu J, et al. IL-33 alleviates DSS-induced chronic colitis in C57BL/6 mice colon lamina propria by suppressing Th17 cell response as well as Th1 cell response. Int. Immunopharmacol. 2015;29:846–853. doi: 10.1016/j.intimp.2015.08.032. [DOI] [PubMed] [Google Scholar]
- 21.Zhu JF, et al. IL-33 protects mice against DSS-induced chronic colitis by increasing both regulatory B cell and regulatory T cell responses as well as decreasing Th17 cell response. J. Immunol. Res. 2018;2018:1827901. doi: 10.1155/2018/1827901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Grobeta P, Doser K, Falk W, Obermeier F, Hofmann C. IL-33 attenuates development and perpetuation of chronic intestinal inflammation. Inflamm. Bowel Dis. 2012;18:1900–1909. doi: 10.1002/ibd.22900. [DOI] [PubMed] [Google Scholar]
- 23.Sattler S, et al. IL-10-producing regulatory B cells induced by IL-33 (Breg(IL-33)) effectively attenuate mucosal inflammatory responses in the gut. J. Autoimmun. 2014;50:107–122. doi: 10.1016/j.jaut.2014.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Seo DH, et al. Interleukin-33 regulates intestinal inflammation by modulating macrophages in inflammatory bowel disease. Sci. Rep. 2017;7:851. doi: 10.1038/s41598-017-00840-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Monticelli LA, et al. IL-33 promotes an innate immune pathway of intestinal tissue protection dependent on amphiregulin-EGFR interactions. Proc. Natl. Acad. Sci. USA. 2015;112:10762–10767. doi: 10.1073/pnas.1509070112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kinchen J, et al. Structural remodeling of the human colonic mesenchyme in inflammatory bowel disease. Cell. 2018;175:372–386.e317. doi: 10.1016/j.cell.2018.08.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Karpus ON, et al. Colonic CD90+ crypt fibroblasts secrete semaphorins to support epithelial growth. Cell Rep. 2019;26:3698–3708.e3695. doi: 10.1016/j.celrep.2019.02.101. [DOI] [PubMed] [Google Scholar]
- 28.Boudjadi S, et al. Involvement of the integrin alpha1beta1 in the progression of colorectal cancer. Cancers (Basel) 2017;9:96. doi: 10.3390/cancers9080096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Greicius G, et al. PDGFRalpha(+) pericryptal stromal cells are the critical source of Wnts and RSPO3 for murine intestinal stem cells in vivo. Proc. Natl. Acad. Sci. USA. 2018;115:E3173–E3181. doi: 10.1073/pnas.1713510115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hirokawa Y, Yip KH, Tan CW, Burgess AW. Colonic myofibroblast cell line stimulates colonoid formation. Am. J. Physiol. Gastrointest. Liver Physiol. 2014;306:G547–556. doi: 10.1152/ajpgi.00267.2013. [DOI] [PubMed] [Google Scholar]
- 31.Smillie CS, et al. Intra- and inter-cellular rewiring of the human colon during ulcerative colitis. Cell. 2019;178:714–730.e722. doi: 10.1016/j.cell.2019.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Duan L, et al. Interleukin-33 ameliorates experimental colitis through promoting Th2/Foxp3(+) regulatory T-cell responses in mice. Mol. Med. 2012;18:753–761. doi: 10.2119/molmed.2011.00428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schiering C, et al. The alarmin IL-33 promotes regulatory T-cell function in the intestine. Nature. 2014;513:564–568. doi: 10.1038/nature13577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rubtsov YP, et al. Regulatory T cell-derived interleukin-10 limits inflammation at environmental interfaces. Immunity. 2008;28:546–558. doi: 10.1016/j.immuni.2008.02.017. [DOI] [PubMed] [Google Scholar]
- 35.Palmieri, V. et al. Interleukin-33 signaling exacerbates experimental infectious colitis by enhancing gut permeability and inhibiting protective Th17 immunity. Mucosal Immunol. (2021). Mar 2. Online ahead of print. [DOI] [PMC free article] [PubMed]
- 36.Imai J, et al. Flagellin-mediated activation of IL-33-ST2 signaling by a pathobiont promotes intestinal fibrosis. Mucosal Immunol. 2019;12:632–643. doi: 10.1038/s41385-019-0138-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Maywald RL, et al. IL-33 activates tumor stroma to promote intestinal polyposis. Proc. Natl. Acad. Sci. USA. 2015;112:E2487–2496. doi: 10.1073/pnas.1422445112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Owens BM, et al. CD90(+) stromal cells are non-professional innate immune effectors of the human colonic mucosa. Front. Immunol. 2013;4:307. doi: 10.3389/fimmu.2013.00307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Oboki K, et al. IL-33 is a crucial amplifier of innate rather than acquired immunity. Proc. Natl. Acad. Sci. USA. 2010;107:18581–18586. doi: 10.1073/pnas.1003059107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ahrens R, et al. Intestinal macrophage/epithelial cell-derived CCL11/eotaxin-1 mediates eosinophil recruitment and function in pediatric ulcerative colitis. J. Immunol. 2008;181:7390–7399. doi: 10.4049/jimmunol.181.10.7390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Berg DJ, et al. Rapid development of colitis in NSAID-treated IL-10-deficient mice. Gastroenterology. 2002;123:1527–1542. doi: 10.1053/gast.2002.1231527. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.