

ABSTRACT
At the junction between the glycolysis and the tricarboxylic acid cycle—as well as various other metabolic pathways—lies the phosphoenolpyruvate (PEP)-pyruvate-oxaloacetate node (PPO-node). These three metabolites form the core of a network involving at least eleven different types of enzymes, each with numerous subtypes. Obviously, no single organism maintains each of these eleven enzymes; instead, different organisms possess different subsets in their PPO-node, which results in a remarkable degree of variation, despite connecting such deeply conserved metabolic pathways as the glycolysis and the tricarboxylic acid cycle. The PPO-node enzymes play a crucial role in cellular energetics, with most of them involved in (de)phosphorylation of nucleotide phosphates, while those responsible for malate conversion are important redox enzymes. Variations in PPO-node therefore reflect the different energetic niches that organisms can occupy. In this review, we give an overview of the biochemistry of these eleven PPO-node enzymes. We attempt to highlight the variation that exists, both in PPO-node compositions, as well as in the roles that the enzymes can have within those different settings, through various recent discoveries in both bacteria and archaea that reveal deviations from canonical functions.
Keywords: phosphoenolpyruvate, pyruvate, oxaloacetate, PPO-node, central metabolism, enzyme biochemistry
Three metabolites, phosphoenolpyruvate, pyruvate and oxaloacetate together form the PPO-node, and are interconnected by a subset of 11 different enzymes; the current knowledge regarding these enzymes at the heart of metabolism is presented.
INTRODUCTION
Different organisms can have many differences in their metabolism, yet they universally share twelve precursor metabolites that form the basis of all biomass on earth (Fig. 1) (Noor et al. 2010). Although significant variation exists, the set of reactions connecting those twelve precursor metabolites are generally well conserved and will be referred to as the core metabolism. The majority of the twelve precursor metabolites are part of the glycolysis, which is a largely linear sequence of reactions. The three precursor metabolites at the end of the glycolysis, phosphoenolpyruvate (PEP), pyruvate and oxaloacetate form a node with often multiple reactions connecting them. This PEP-pyruvate-oxaloacetate node (PPO-node) lies at the junction between the glycolysis and the TCA cycle, as well as many other metabolic pathways, and as such lies at the heart of the metabolism.
Figure 1.

Structural formulas of the 12 universal precursor metabolites and the topology of the core metabolism that connects them. The PPO-node is highlighted in red. G6P, d-glucose-6-phosphate; F6P, d-fructose-6-phosphate; R5P, d-ribose-5-phosphate; E4P, d-erythrose-4-phosphate; GAP, d-glyceraldehyde-3-phosphate; 3PG, glycerate-3-phosphate; PEP, phosphoenolpyruvate; PYR, pyruvate; OAA, oxaloacetate; AcCoA, acetyl-CoA; AKG, α-ketoglutarate; SuccCoA, succinyl-CoA.
PEP has the highest-energy phosphate bond of all known natural organo-phosphates (Bowman et al. 1988), and serves as a precursor for aromatic amino acids (Herrmann and Weaver 1999). Pyruvate is a precursor for alanine, valine, leucine, isoleucine and lysine,1 and is also important as the first entry point for many fermentation pathways that re-oxidize NAD(P)H (Müller 2008). Oxaloacetate is part of the tricarboxylic acid cycle, where it accepts acetyl-CoA or is reduced to malate. Oxaloacetate also functions as a precursor for aspartate, which in turn is used for the biosynthesis of many other amino acids, as well as nucleotides (Park and Lee 2010).
Yet, despite forming the heart of metabolism, the PPO-node is also the part of the core metabolism that seems to vary the most between different organisms, as many possible biochemical reactions are known to exist in the PPO-node, summarized in Fig. 2. This is in stark contrast with the lower part, or trunk of glycolysis, directly leading towards the PPO-node (starting with glyceraldehyde-3-phosphate), which is one of the oldest and most conserved metabolic pathways that exist2 (Ronimus and Morgan 2003).
Figure 2.
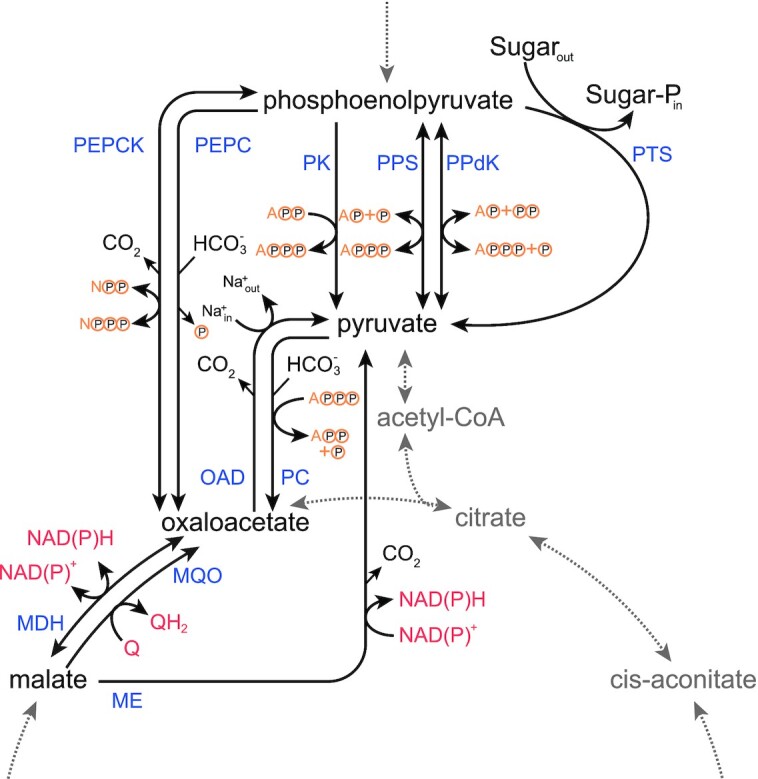
Overview of all known reactions in the PPO-node. MDH: Malate dehydrogenase, ME: Malic enzyme, MQO: Malate:quinone oxidoreductase, OAD: Oxaloacetate decarboxylase, PC: Pyruvate carboxylase, PEPC: Phosphoenolpyruvate carboxylase, PEPCK: Phosphoenolpyruvate carboxykinase, PK: Pyruvate kinase, PPdK: Pyruvate phosphate dikinase, PPS: Phosphoenolpyruvate synthase, PTS: Phosphotransferase system. The yellow circles represent phosphate (groups).
The conversion of PEP to pyruvate—typically considered the last step of the glycolysis—is also the step where the glycolytic energy is harvested, in the form of ATP (equivalents). This conversion can either occur directly, through pyruvate kinase, pyruvate phosphate dikinase,3 the phosphotransferase system, or even phosphoenolpyruvate synthase; or can occur via the intermediate formation of oxaloacetate, with or without malate as intermediate as well (Fig. 2). And not only do each of these different routes have different enzymes associated with (with different properties), each route also has a different set of cofactors that are used (i.e. red and yellow elements in Fig. 2). Variations in the PPO-node are therefore profoundly linked to cellular energetics and the constraints on them.
Anaerobic and extremophilic microorganisms most often show deviations from the ‘typical’ central metabolism found in most model organisms, as a consequence of the way in which the different constraints that dictate cellular metabolism are prioritized (differently) in ‘extreme’ environments.4 Research into the PPO-node enzymes of anaerobic- and extremophilic bacteria and archaea will therefore be featured extensively in this review, where we aim to give a concise view of what is currently known about the biochemical reactions that form the PPO-node, putting emphasis on the variations that exist.
THE DIFFERENT ENZYMES OF THE PPO-NODE5
Pyruvate kinase (EC 2.7.1.40)
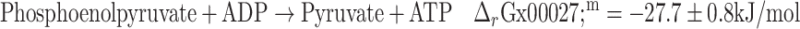 |
Pyruvate kinase (PK) catalyzes the transphosphorylation from phosphoenolpyruvate (PEP) to ADP, forming pyruvate and ATP, which is typically the final, and a rate determining step of the glycolysis. The reaction proceeds in two steps. First, the phosphoryl-group is transferred from PEP to ADP, and then the enolate that is formed is converted into pyruvate via the addition of a proton (Kumar and Barth 2010). It has to be noted, however, that the specificity for ADP is very low, as many other NDPs can function as an acceptor (Weber 1969; Abbe and Yamada 1982; Sakai, Suzuki and Imahori3 1986). The reaction is typically considered irreversible, however, recent findings suggest that the ΔrG of PK is not as negative as was previously thought (Park et al. 2016). PK has an absolute requirement for divalent cations, in particular Mg2+ (Enriqueta Muñoz and Ponce 2003). Phylogenetically, PKs can be clustered in two distinct groups, K+-dependent (cluster I) and K+-independent (cluster II) enzymes. These two clusters do not correlate with the three domains of life (Schramm et al. 2000; Johnsen, Hansen and Schönheit 2003; Oria-Hernández, Riveros-Rosas and Ramírez-Sílva 2006; Guerrero-Mendiola et al. 2017), although PKs are ubiquitously present in all three, with only a few exceptions that will be discussed below (Enriqueta Muñoz and Ponce 2003). Members of the gammaproteobacteria, including Escherichia coli, Yersinia enterocolitica and Salmonella typhimurium, are unique in that they possess isozymes of both the K+-dependent and the K+-independent clusters (Guerrero-Mendiola et al. 2017). For Y. enterocolitica it was shown that both PykF (cluster I) and PykA (cluster II) form homotetramers, which is the most common quaternary structure for PKs (Enriqueta Muñoz and Ponce 2003), although monomers (Knowles, Dennis and Plaxton 1989), homodimers (Pawluk, Scopes and Griffiths-Smith 1986), heterotetramers (Plaxton 1989), heterohexamers (Plaxton, Smith and Knowles 2002), and homodecamers (Lin, Turpin and Plaxton 1989) are also known to exist.
Pyruvate kinase is responsible for one of the three rate determining steps, and one of the two ATP-generating steps of glycolysis. Consequently, its regulation is complex and features many layers; especially its allosteric regulation is exceptionally intricate, and is different between the two phylogenetic clusters (Valentini et al. 2000). K+-dependent PKs are stimulated by bi-phosphorylated sugars, such as fructose-1,6-bisphosphate (Jurica et al. 1998) or fructose-2,6-bisphosphate (Schaftingen, Opperdoes and Hers 1985; Ernest et al. 1994), whereas K+-independent PKs are generally allosterically regulated by AMP and mono-phosphorylated sugars, such as glucose-6-phosphate and ribose-5-phosphate (Yamada and Carlsson 1975; Malcovati and Valentini 1982; Guerrero-Mendiola et al. 2017). Several PKs have been described to deviate from this typical allosteric control. The PK from the hyperthermophilic archaeon Pyrobaculum aerophilum was found to respond to 3-phosphoglycerate, rather than to any of the previously mentioned effectors (Solomons et al. 2013), and the PKs from the parasite Cryptosporidium parvum and other hyperthermophilic archaea have no known effectors at all (Denton et al. 1996; Schramm et al. 2000; Johnsen, Hansen and Schönheit 2003; Cook et al. 2012). In Corynebacterium glutamicum, PK is subject to serine-phosphorylation. Whether the PK activity is controlled via this post-translational modification is not known (Bendt et al. 2003).
Besides the kinase activity, PKs also have an intrinsic and conserved oxaloacetate-decarboxylase activity. And although this decarboxylase activity is rather low and likely the result of a non-mutable requirement in the active site, a potential biological role cannot be ruled out (Zhong et al. 2014).
As mentioned, only a few (sugar fermenting) organisms are known that do not possess a PK. These include fermentative protists, such as Entamoeba histolytica and Tritrichomonas foetus (Hrdý, Mertens and Van Schaftingen 1993; Mertens 1993; Saavedra-Lira, Ramirez-Silva and Perez-Montfort 1998), and the thermophilic bacterium Clostridium thermocellum, the best cellulose degrader that is currently known (Zhou et al. 2013; Olson et al. 2016). For the latter, it was recently proven that conversion of PEP to pyruvate proceeds via the ‘malate shunt’ and via pyruvate phosphate dikinase, as shown in Fig. 3 (Olson et al. 2016). The malate shunt, running via PEP carboxykinase, malate dehydrogenase, and malic enzyme accounts for a third of the flux to pyruvate, with the remainder going via pyruvate phosphate dikinase. Interestingly, the malate shunt could only be disrupted after introduction of a heterologous PK (Deng et al. 2013), whereas pyruvate phosphate dikinase could be knocked-out in the wild-type strain, redirecting all flux via the malate shunt (Olson et al. 2016). In fermentative protists, the function of PK is replaced by pyruvate phosphate dikinase as well (Saavedra et al. 2019). Flux through the malate shunt has not been described in those cases, but it is possible that this pathway also plays a role in fermentative protists. Overall, the malate shunt is an efficient mechanism for the transhydrogenation of NADH to NADPH, considering that NADPH generation through the pentose-phosphate pathway results in loss of substrate in the form of CO2 and that (bifurcating) transhydrogenases require high-energy electron donors (i.e. ferredoxin or flavodoxin) (Wang et al. 2010).
Figure 3.
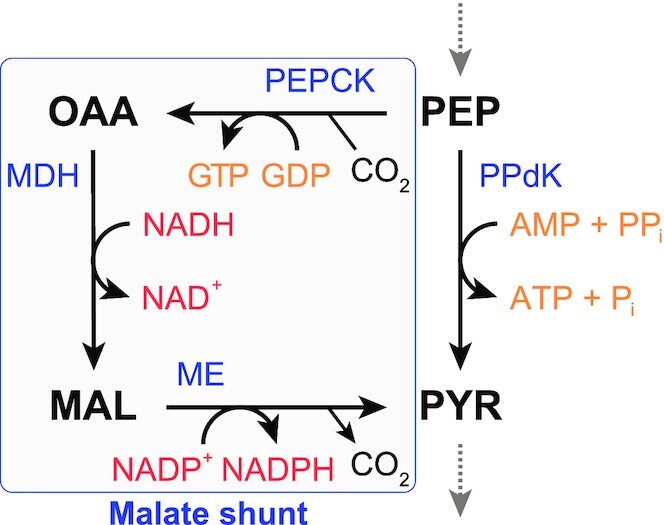
Conversion of PEP to pyruvate through the malate shunt in the PPO-node of C. thermocellum. The malate shunt results in the transhydrogenation of NADH to NADPH. MAL: malate, MDH: malate dehydrogenase, ME: malic enzume, OAA: oxaloacetate, PEP: phosphoenolpyruvate, PEPCK: PEP carboxykinase, PPdK: pyruvate phosphate dikinase, PYR: pyruvate.
Almost paradoxically, in C. glutamicum PK was also found to be important for growth on certain gluconeogenic substrates, including acetate and citrate, which are first converted to PEP via PEP carboxykinase (Netzer et al. 2004). PK is then required for the formation of pyruvate-derived biomass constituents.
Pyruvate phosphate dikinase (EC 2.7.9.1)
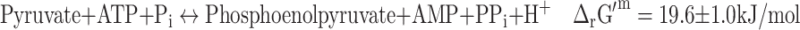 |
Pyruvate phosphate dikinase (PPdK) catalyzes the reversible conversion of PEP to pyruvate via three partial reactions: First, PPdK combines with ATP to form AMP and the enzyme-diphosphate, then pyrophosphate is formed via the combination of orthophosphate and a phosphate group from the enzyme-diphosphate. Finally, the other phosphate bound to PPdK is transferred to pyruvate, forming PEP (Evans and Wood 1968). The mechanism involves one of the largest single protein-domain movements known so far (Minges, Höppner and Groth 2017), and is believed to proceed via an alternate binding change mechanism, akin to ATP synthase (Ciupka and Gohlke 2017).
Most of what we know about PPdK comes from studies with plants and parasitic protists, but PPdKs are also present in archaea and in bacteria, from which they are in fact believed to originate (Slamovits and Keeling 2006). In C4 plants, PPdK is responsible for the first step of carbon-fixation starting with pyruvate, and can comprise up to 10% of soluble protein in C4 leaves (Chastain 2010). It is no surprise then that PPdK has mostly been studied in plants, and is therefore typically considered a gluconeogenic enzyme, which actually is surprising, in view of the ΔrG′m. Furthermore, for several bacteria, including Microbispora rosea and Acetobacter xylinum PPdK was shown to be important during gluconeogenesis as well (Benziman and Eizen 1971; Eisaki et al. 1999). However, the thermodynamics certainly do not restrict it from working in the glycolytic direction. In fact, for a multitude of protists (Deramchia et al. 2014; González-Marcano et al. 2016; Rivero et al. 2016) and bacteria (Reeves 1968; Bielen et al. 2010; Olson et al. 2016), and the archaeon Thermoproteus tenax (Tjaden et al. 2006), PPdK has been demonstrated to function as a glycolytic enzyme, which is likely also its original function. In C3 plants, PPdK is ubiquitously present in the plant tissue, where its role is currently not well understood, since those plants do not use PPdK for carbon fixation (Chastain et al. 2011; Hyskova and Ryslava 2016).
In bacteria (Ernst, Budde and Chollet 1986; Ciupka and Gohlke 2017) and archaea (Tjaden et al. 2006), PPdK appears to be active in homodimeric form, whereas for C4 plants it typically functions optimally as a homotetramer (Shirahashi, Hayakawa and Sugiyama 1978; Ohta, Ishida and Usami 2004). For protists, both dimeric (Hiltpold, Thomas and Köhler 1999) and tetrameric (Saavedra-Lira, Ramirez-Silva and Perez-Montfort 1998) forms have been described. Free Mg2+ is required for the formation of PPdK tetramers in plants and thus its activity, whereas inactivation occurs at low temperatures due to dissociation into dimers and monomers. For the dimeric PPdK, neither Mg2+ nor low temperature affects the stability of the quaternary structure, but Mg2+ is still required for its catalytic activity (Hiltpold, Thomas and Köhler 1999). Other effectors described to stimulate PPdK activity in plants, protists and bacteria are the monovalent cations NH4+ and K+, whereas PPdKs are not subject to regulation by metabolites. Furthermore, pH significantly influences the direction that is favoured, with low pH favouring the glycolytic (proton consuming) direction, and vice versa (Chastain 2010). The PPdK of the archaeon T. tenax, on the contrary, is not affected by any monovalent cations. Instead, ATP was found to act as an potent competitive inhibitor with AMP (Tjaden et al. 2006).
In C4 plants, PPdK activity is tightly regulated via inactivation and activation by the PPdK regulatory protein (PDRP), respectively through ADP-dependent phosphorylation and Pi-dependent dephosphorylation of a threonine residue in the active site. This regulation is imposed by light intensity, and is believed to be dictated by the adenylate energy charge, which is low under dark conditions (Edwards et al. 1985; Chastain 2010). A homolog of the PDRP (DUF 299) is also present in all bacteria harbouring PPdK, and was shown in Listeria monocytogenes to activate and inactivate PPdK via an identical mechanism as described for plant PPdK (Tolentino, Chastain and Burnell 2013). Bacteria harbouring PDRP without PPdK also exist, in which case PDRP likely controls the activity of PEP synthase, as was shown in E. coli (Burnell 2010). The DUF299 protein is absent in archaea and protists (Burnell 2010; Chastain et al. 2011), although inactivation via phosphorylation of the threonine residue still occurs in the PPdK from Trypanosoma cruzi. Furthermore, T. cruzi PPdK is irreversibly inactivated via proteolytic cleavage from 100 to 75 kDa, after which the protein associates with the glycosomal membrane (González-Marcano et al. 2014). Nevertheless, the full extent of the regulation via posttranslational modification of PPdK remains unknown, as multiple phosphorylation sites have been discovered more recently in plants, of which not all are phosphorylated by PDRP, as well as an acetylation site near the N-terminus, whose function is unknown (Chen et al. 2014).
Phosphoenolpyruvate synthase (EC 2.7.9.2)
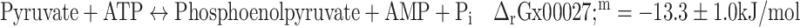 |
Phosphoenolpyruvate synthase (PPS), or sometimes called pyruvate, water dikinase resembles pyruvate phosphate dikinase, both catalytically and structurally (Nguyen and Saier 1995). Like pyruvate phosphate dikinase, the reaction proceeds via three partial reactions, where first the enzyme-diphosphate is formed. The first phosphate group is then transferred to water, rather than orthophosphate, expending a high-energy phosphate bond. Finally, the second phosphate is transferred to pyruvate (Cooper and Kornberg 1967), forming PEP. As a consequence of the energy that is dissipated at the cost of the high-energy phosphate bond, the reaction strongly favours the gluconeogenic direction, to PEP. Nonetheless, in the extreme case of Thermococcus kodakarensis, but probably also in related Thermococcus/Pyrococcus species, PPS is responsible for the glycolytic conversion of PEP to pyruvate (Imanaka et al. 2006). This makes sense in view of the fact that Thermococcus/Pyrococcus species have an ADP-dependent glucokinase and 6-phosphofructokinase (Kengen et al. 1994)—forming AMP. This supply of AMP fits well with ATP synthesis from AMP by PPS, as shown in Fig. 4.
Figure 4.
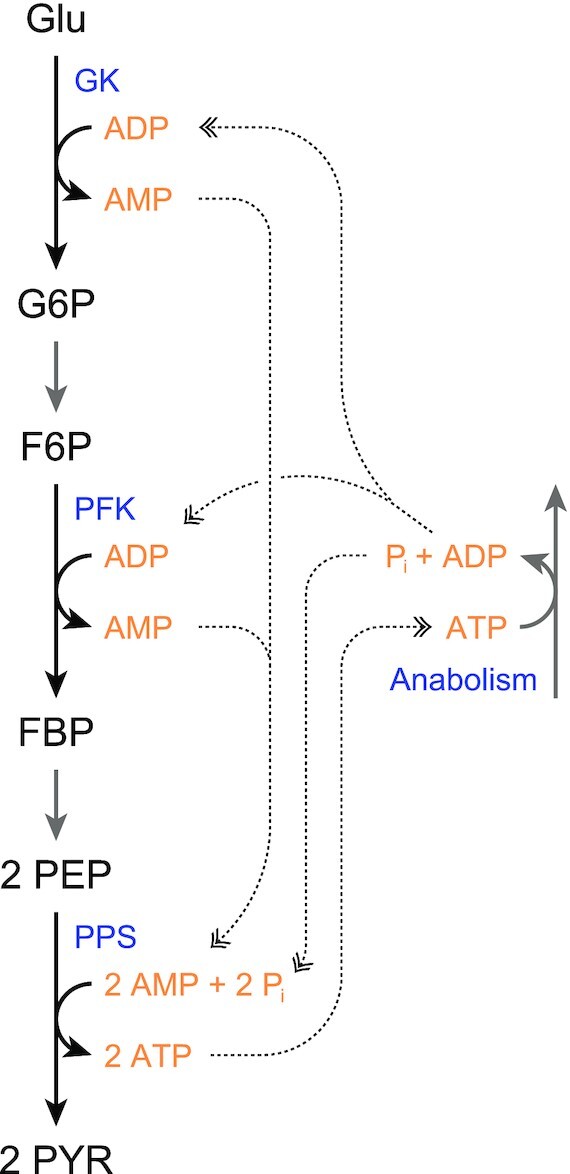
Cycling of ATP/ADP/AMP in the metabolism of T. kodakarensis, which relies on PPS for glycolysis, as well as ADP-dependent GK and PFK. F6P, fructose 6-phosphate; FBP, fructose 1,6-bisphosphate; G6P, glucose 6-phosphate; GK, glucokinase; Glu, glucose; PEP, phosphoenolpyruvate; PPS, PEP synthase; PFK, 6-phosphofructokinase; PYR, pyruvate.
PPSs are present in all three domains of life. In eukaryotes, however, the majority of PPSs are annotated in a specific class of fungi (leotiomyceta), without any functional evidence available. Structural data regarding PEP synthase is limited to one resolved crystal structure, from Neisseria meningitidis. In the hyperthermophilic archaeon Staphylothermus marinus, PPS forms a large homomultimeric complex, composed of 24 subunits, assembled in a rather unusual octahedral arrangement. For Pyrococcus furiosus, PPS was found to exist in a large and a smaller homomultimeric complex, of which the smaller octamer was found to be the active form (Hutchins, Holden and Adams 2001). PPS from T. tenax also exists as a large homomultimer (Tjaden et al. 2006), so it is likely a general feature of PPSs from hyperthermophilic archaea, as PPS from E. coli is active as a homodimer (Narindrasorasak and Bridger 1977). Unfortunately, no other PPSs have been characterized in such detail.
All characterized PPSs require divalent cations for activity. Furthermore, P. furiosus PPS contains one Ca-atom per subunit, and its activity is stimulated by K+ and NH4+ (Hutchins, Holden and Adams 2001). The optimal pH of the PEP-forming reaction was 9.0, while the optimal pH of the pyruvate formation reaction was 7.5 (Hutchins, Holden and Adams 2001). Contrary to PPS from P. furiosus, and also Methanothermobacter thermoautotrophicus, activity of PPS from T. tenax is not stimulated by the monovalent cations (Tjaden et al. 2006). T. tenax PPS is strongly inhibited by 2-oxoglutarate, and AMP and ADP, competitively with pyruvate and ATP, respectively, in the PEP-forming direction (Tjaden et al. 2006). For E. coli PPS, Mg2+ and Mn2+ were required for activation, but higher concentrations led to decreased activity (Berman and Cohn 1970). As was already mentioned, it was shown that PPdK regulatory protein (PDRP) is a regulator for PPS activity in E. coli, analogous to its regulation of pyruvate phosphate dikinase (Burnell 2010). Nothing is known regarding post-translational modification in archaeal PPSs.
In T. tenax, PPS is downregulated during heterotrophic growth and upregulated during autotrophic growth, indicative of a gluconeogenic role (Tjaden et al. 2006), whereas in T. kodakarensis, transcript levels of PPS increased under glycolytic conditions when compared with cells grown on pyruvate or amino acids, indicating a glycolytic role, which was confirmed by the knock-out of PPS (Imanaka et al. 2006). T. kodakarensis also harbours a pyruvate kinase, but this appeared to be less essential for glycolysis than PPS (Imanaka et al. 2006). In line with this, PPS from P. furiosus is also considered to function in the pyruvate-forming direction, and was shown to be upregulated by maltose (Sakuraba et al. 1999, 2001; Sakuraba and Ohshima 2002). In most organisms, however, PPS functions as a gluconeogenic enzyme. Knock-outs in E. coli and S. typhimurium showed that it is essential for growth on C3 substrates, such as pyruvate (Smyer and Jeter 1989). In M. thermoautotrophicus, like for T. tenax, PPS plays a role in autotrophic growth (Eyzaguirre, Jansen and Fuchs 1982).
Phosphoenolpyruvate carboxylase (EC 4.1.1.31)
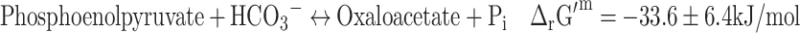 |
Phosphoenolpyruvate carboxylase (PEPC) catalyzes the irreversible bicarbonate fixation on PEP, yielding oxaloacetate and orthophosphate. This reaction likely proceeds via the following three steps: First, carboxyphosphate and enolate are formed by the transfer of the phosphate group. The carboxyphosphate is then cleaved into hydrogen phosphate and the—compared to bicarbonate—more reactive CO2. Finally, the CO2 is fixed on the enolate, forming oxaloacetate (Kai, Matsumura and Izui 2003). This reaction is highly exergonic, and essentially irreversible.
PEPC is present in plants, bacteria and archaea, but not in fungi and animals. The archaeal-type PEPC, which is also present is several bacteria, differs strongly from its counterpart found in plants and bacteria (Ettema et al. 2004; Patel, Kraszewski and Mukhopadhyay 2004). As with pyruvate phosphate dikinase, PEPC plays a crucial role in C4 photosynthesis, and consequently, a lot of what we know about PEPC comes from plants. Fortunately, significant effort has gone into investigating both bacterial and archaeal PEPCs as well. The most obvious difference between the PEPC from bacteria and plants (bpPEPC) compared to the archaeal-type PEPC (atPEPC), is the size; the atPEPC, with monomer sizes ranging from 55 to 60 kDa, is a lot smaller than the bpPEPC, which ranges from 90 to 110 kDa (Ettema et al. 2004). Nevertheless, bpPEPC and atPEPC both exist as homotetramers, with two sets of dimers being somewhat loosely associated with each other (Izui et al. 2004; Dharmarajan et al. 2011). In plants and algae, a second class of PEPCs has been discovered that forms a hetero-octameric complex, consisting of four typical ‘class-1’ plant-type monomers, and four monomers related to bacterial PEPC, typically referred to as bacterial-type PEPC (BTPC) to contrast them with the plant-type PEPCs (PTPC) (O'Leary et al. 2009). Thus, bpPEPCs encompass three separate phylogenetic groups, bacterial PEPCs, BTPCs, and PTPCs (O'Leary, Park and Plaxton 2011). Together with the atPEPCs, four main PEPC types can therefore be distinguished, summarized in Fig. 5.
Figure 5.
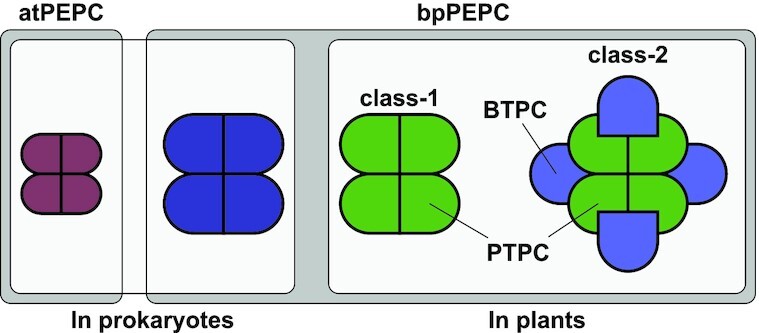
Overview of the different PEPC complexes and the typically used nomenclature. atPEPC, archaeal-type PEPCs; bpPEPC, bacterial and plant PEPC. Most PEPCs are tetrameric, except for the class-2 PEPCs in plants, which is a hetero-octameric combination of the plant-type PEPCs (PTPC; in green) and bacterial-type PEPC (BTPC; in light-blue). Although PTPC and BTPC are both bpPEPCs, BTPCs are more homologous to those found in Bacteria than the others found in plants (i.e. class-1/PTPC).
Many different allosteric effectors for PEPC are known, and listing all known effectors, and the variations of allosteric control in different organisms is beyond the scope of this review. Generally, class-1 PEPCs in plants are inhibited by malate and activated by glucose-6-phosphate. Aspartate and glutamate are important feedback effectors in tissues active in nitrogen assimilation and/or transamination reactions (O'Leary, Park and Plaxton 2011). In bacteria, PEPC is typically activated by fructose 1,6-bisphosphate and acetyl-CoA, and inhibited by aspartate and malate (Chen et al. 2002). Individually, the effects of those allosteric regulators are rather modest. However, for E. coli it was shown that at physiological concentrations, the combination of these effectors turned fructose 1,6-bisphosphate into an ultrasensitive allosteric regulator for PEPC. Upon fructose 1,6-bisphosphate depletion resulting from glucose removal, PEPC is very rapidly virtually switched off, which is important for the build-up of PEP, required for future glucose import via the phosphotransferase system (Xu et al. 2012). In cyanobacteria and plants, the allosteric control is determined by a single amino acid residue (Takeya, Hirai and Osanai 2017). The hetero-octameric class-2 PEPCs from plants are found to have significantly lower sensitivity for allosteric control compared to class-1 PEPCs, which is believed to allow them to bypass PEPC inhibition under specific conditions that would inhibit class-1 PEPCs (O'Leary et al. 2009). It was also shown that class-2 PEPC expression correlates with the formation of storage organelles in pollen, so its function might also be related to the formation of storage molecules (Igawa et al. 2010). The atPEPCs have no known allosteric activators, and residues related to the binding of inhibitors in bpPEPCs are absent (Matsumura, Izui and Mizuguchi 2006; Dharmarajan et al. 2011). Inhibition of atPEPC by aspartate was found to be competitive with PEP, rather than allosteric (Dharmarajan et al. 2011).
In plants, PEPC functionality is also controlled via phosphorylation, by PEPC protein kinase, and via mono-ubiquitination (Aldous et al. 2014; Ruiz-Ballesta et al. 2016; Ying et al. 2017). In bacteria, much less is known regarding PEPC control via post-translational modification. Recently, however, it was shown that in C. glutamicum, (possibly non-enzymatic) PEPC acetylation of lysine-653 decreased PEPC activity, which was activated again via enzymatic deacetylation.
Phosphoenolpyruvate carboxykinase (EC 4.1.1.49) (EC 4.1.1.32) (EC 4.1.1.38)
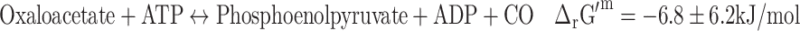 |
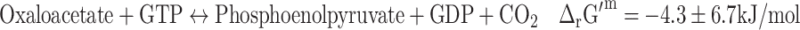 |
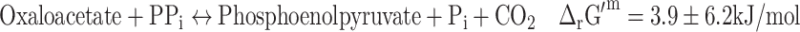 |
Phosphoenolpyruvate carboxykinase (PEPCK) catalyzes the reversible decarboxylation and phosphorylation between oxaloacetate and PEP. Enzymes exist that can use either ATP, GTP or PPi as phosphate donor (EC 4.1.1.49, 4.1.1.32 and 4.1.1.38, respectively). The enzymes using PPi are not evolutionary related to the enzymes using either ATP or GTP, which do share an evolutionary origin, despite having relatively low sequence homology (Aich and Delbaere 2007; Chiba et al. 2015). PPi-PEPCK will be discussed further down, and for now, PEPCK will refer to the ATP- and GTP-dependent enzymes, which are believed to have a conserved mechanism of catalysis, despite the low sequence identity (Matte et al. 1997).
In contrast to phosphoenolpyruvate carboxylase, PEPCK uses CO2 rather than HCO3−, and has a specific binding site for CO2, which is uncommon for (de)carboxylating enzymes (Cotelesage et al. 2007). PEPCK requires both Mg2+ and Mn2+ for maximal activity, where Mg2+ is associated with the nucleotide and Mn2+ with the active site of the enzyme (Goldie and Sanwal 1980; Machová et al. 2015). The reaction is believed to proceed in a stepwise manner, with oxaloacetate binding first to the Mn2+-enzyme complex followed by binding of the Mg2+-nucleotide complex. Catalysis then proceeds by closing of a Ω–loop lid-domain, during which a stabilized enolate intermediate is formed after decarboxylation of oxaloacetate, allowing for phosphoryl transfer from the nucleotide, rather than the energetically more favourable protonation (Carlson and Holyoak 2009; Cui et al. 2017).
PEPCK is present in all domains of life. Mammalian PEPCKs are all GTP-dependent, and all plant PEPCKs are ATP-dependent. Archaeal PEPCKs are mostly GTP-dependent, and bacterial and other eukaryotic PEPCKs can be both ATP- and GTP-dependent (Aich and Delbaere 2007). No clear explanation exists for the use of either ATP or GTP. Except for bacterial ATP-PEPCKs, which are monomeric, most other ATP-PEPCKs are multimeric, with two, four, or six subunits (Matte et al. 1997). Conversely, most known GTP-PEPCKs are monomeric, with the exception of the sole characterized archaeal GTP-PEPCK, which forms a homotetramer (Fukuda et al. 2004).
GTP-PEPCKs are not subject to allosteric control, but for ATP-PEPCKs some allosteric effectors are reported (Fukuda et al. 2004): In E. coli, Ca2+ was shown to act as an allosteric activator (Goldie and Sanwal 1980), and in C4 plants, PEPCK was shown to be inhibited by 3-phosphoglycerate, fructose-6-P, fructose-1,6-bisphosphate and dihydroxyacetone phosphate (Burnell 1986).
In most organisms, the primary function of PEPCK is believed to be gluconeogenesis, in order to convert pyruvate to PEP; first by pyruvate carboxylase, followed by PEPCK. In some C4 plants, PEPCK is used for the localized enrichment of CO2 in the bundle-sheath cells. In contrast to (most) eukaryotes, many prokaryotes possess PPS and/or PPdK for the conversion of pyruvate to PEP; PEPCK (or malic enzyme/oxaloacetate decarboxylase) is required for the growth on TCA-intermediates such as malate and succinate (Liao, Chao and Patnaik 1994). The reaction operates close to equilibrium, and there are also more and more examples where PEPCK is responsible for the conversion of PEP to oxaloacetate fixing CO2. These notably include natural succinic acid producers, which thus are able to couple succinate fermentation to the generation of ATP-equivalents, but also other capnophilic (CO2-loving) bacteria (Laivenieks, Vieille and Zeikus 1997; Schöcke and Weimer 1997; Koendjbiharie, Wiersma and van Kranenburg 2018). In C. thermocellum—not a succinate producer—PEPCK, followed by malate dehydrogenase and malic enzyme, was shown to be involved in the glycolytic conversion of PEP to pyruvate, via the malate shunt (Olson et al. 2016). In M. tuberculosis, Fe2+ inhibits the gluconeogenic direction of PEPCK while it activates the anaplerotic oxaloacetate synthesis (Machová et al. 2015). The human cytosolic PEPCK—considered a gluconeogenic or cataplerotic enzyme (Yang, Kalhan and Hanson 2009)—was recently shown to be converted into an anaplerotic enzyme upon p300-dependent acetylation at high-glucose conditions (Latorre-Muro et al. 2018). Other post-translational control of cytosolic PEPCK in humans include phosphorylation, and ubiquitination.
PPi-PEPCK (or PEP carboxytransphosphorylase; PEPCT) is distributed in limited but diverse lineages of unicellular eukaryotes and bacteria, but not in archaea. Most of these organisms also possess the canonical ATP/GTP-PEPCKs; E. histolytica, however, possesses three PPi-PEPCK genes, but lacks any other PEPCK-types (Chiba et al. 2015; Saavedra et al. 2019). In Propionibacterium shermanii, PPi-PEPCK was found to require Mg2+, Mn2+, Co2+ for activity (Lochmüller, Wood and Davis 1966). Sulfate, malate and PPi were found to be effective inhibitors.
Pyruvate carboxylase (EC 6.4.1.1)
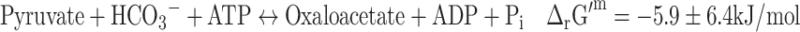 |
Pyruvate carboxylase (PC) catalyzes the irreversible, ATP-dependent carboxylation of pyruvate by HCO3− to oxaloacetate. The enzyme is a biotin-carboxylase, a family of enzymes that includes amongst others, acetyl-CoA carboxylase and urea carboxylase. Biotin-carboxylases contain three main functional domains: The biotin carboxylase (BC) responsible for the ATP-dependent carboxylation of the biotin with HCO3−; the biotin carboxyl carrier protein (BCCP) that has the biotin covalently linked to it and is responsible for the translocation of the carboxylated biotin to the third domain; and the carboxyltransferase (CT), where the CO2 is transferred to the acceptor, which is pyruvate in the case of PC. The BC and BCCP domains are conserved between biotin-carboxylases, whereas the CT domain is different, depending on the substrate.
Two different PC variants are known: The single-polypeptide enzyme and the two-subunit enzyme, in which the BC domain composes the α-chain, and the BCCP and CT domains the β-chain. The former is present in eukaryotes and bacteria and is only active as a tetramer (α4). The two-subunit enzyme is present in archaea and some bacteria and is active as a tetramer of the two subunits (α4β4) (Lai et al. 2006; Tong 2013). PC has an absolute requirement for one divalent cation bound per subunit, typically in the form of Mg2+, although isoforms requiring Zn2+ are also described (Jitrapakdee and Wallace 1999; St Maurice et al. 2007). Furthermore, K+ and larger monovalent cations can stimulate PC activity (Charles and Willer 1984; Scrutton and Taylor 1974). The tetramer is arranged as a dimer of dimers, where the BCCP domains swings between any of the two BC and two CT domains of a dimer, allowing four different translocation routes to take place (Liu et al. 2018).
The α4-type PC is allosterically activated by binding of acetyl-CoA to the BC domain, which specifically activates the translocation of BCCP from the BC domain of its own subunit to the CT domain of the opposing subunit. The α4β4-type PC is insensitive to acetyl-CoA. The degree of activation by acetyl-CoA can vary significantly, depending on the origin (Adina-Zada et al. 2012). A notable exception is the α4-type PC from C. glutamicum, which seems inhibited by acetyl-CoA, as well as ADP and AMP (Peters-Wendisch et al. 1997). Aspartate for microbial PCs and glutamate for vertebrate PCs can inhibit α4-type PCs via a mechanism distinct from that for acetyl-CoA, even though binding of aspartate is mutual exclusive with acetyl-CoA. The α4β4-type PCs are insensitive to aspartate as well. Inhibition by α-ketoglutarate, another dicarboxylate, seems to be more widely distributed, albeit not universal, as it can also modestly inhibit activity of α4β4-type PC from methanogenic archaea (Zeczycki, Maurice and Attwood 2010).
The only known post-translational modification of PC is its biotinylation, which is required for activity (Jitrapakdee and Wallace 1999). For mammals it was recently proposed that biotinylation of the five biotin-dependent carboxylases is a novel regulatory mechanism linked to the circadian clock (He et al. 2016). Evidence for a similar regulatory mechanism in bacteria does not exist. However, the efficiency of biotinylation is influenced by the allosteric effectors acetyl-CoA (positive) and aspartate (negative), acting on PC in Geobacillus stearothermophilus and Saccharomyces cerevisiae (Sundaram, Cazzulo and Kornberg 1971).
PC is present in many bacteria, but whereas for most eukaryotes it is the primary anaplerotic enzyme, in many bacteria it co-occurs with PEP carboxylase (PEPC) (Sauer and Eikmanns 2005). Notably, many enteric bacteria, including E. coli, lack PC and only seem to rely on PEPC for the production of oxaloacetate (Jitrapakdee et al. 2008). In non-photosynthetic eukaryotes, PC is also essential during gluconeogenesis, where the formed oxaloacetate is converted to PEP by PEP carboxykinase. Plants are also known to possess PC, where it might contribute to gluconeogenesis during germination (Jitrapakdee and Wallace 1999).
Oxaloacetate decarboxylase (EC 4.1.1.112)
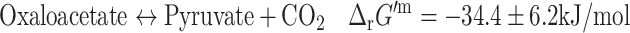 |
Oxaloacetate decarboxylases (OAD) are enzymes that catalyze the irreversible decarboxylation of oxaloacetate to pyruvate. Many enzymes, including malic enzymes, pyruvate kinases, malate dehydrogenases, pyruvate carboxylases and PEP carboxykinases are known to have OAD ‘side-activity’. However, at least four different types of dedicated OADs have been described. We will try to give a concise summary of what is known about these different types, which deserve their own review.
Best known are the Na+-pumping, biotin-dependent OAD and the soluble, divalent cation-dependent OAD. These are the two classes of OADs that are typically acknowledged, and will be described in more detail below. However, in Lactococcus lactis and Enterococcus faecalis malic enzymes have been reported that have lost their ability to decarboxylate malate to pyruvate, and as such can be seen as dedicated OADs (Sender et al. 2004; Espariz et al. 2011). Mg2+ or Mn2+ are required for activity. Fumarate, succinate and NAD(P)H were found to inhibit their OAD activity, while these are positive effectors for many malic enzymes. The ME-related OAD takes part in the citrate fermentation pathway, converting the oxaloacetate formed by citrate lyase (Pudlik and Lolkema 2011). Additionally, a novel OAD from Pseudomonas aeruginosa has been characterized that belongs to the PEP mutase/isocitrate lyase superfamily (Narayanan et al. 2007). The enzyme is homotetrameric, and forms a complex with Mg2+ and oxalate. Orthologs are found in all Pseudomonad genomes, and a few other Gram-negative bacteria. Gene disruption showed that the enzyme is not essential, and the genomic context led the authors to speculate that the OAD helps in the supply of pyruvate or CO2 for a variety of cellular processes (Narayanan et al. 2007).
The Na+-pumping OADs belong to the biotin-dependent enzyme family, like the above described pyruvate carboxylase. Other known biotin-dependent Na+-pumping decarboxylases are methylmalonyl-CoA decarboxylase, glutaconyl-CoA decarboxylase, and malonate decarboxylase. In the biotin-dependent decarboxylases the carboxyl moiety bound to biotin is not transferred to a substrate. Instead, the CO2 is released at the membrane bound β-subunit in a reaction that consumes a periplasmic proton and is coupled to the translocation of up to two Na+ ions from the cytoplasm to the periplasm (Dimroth, Jockel and Schmid 2001; Lietzan and St. Maurice 2014). The Na+ gradient is then used to drive ATP-synthesis, solute uptake, or movement of flagella.
Na+-pumping decarboxylases consist of three different subunits: α, β and γ, of which the latter two are situated in the membrane. The quaternary structure of OAD has for long remained elusive, but for Vibrio cholerae the complex has now been shown to contain the α, β and γ subunits in a 4:2:2 ratio (Balsera, Buey and Li 2011; Inoue and Li 2015). More recently, the β and γ subunits of S .typhimurium OAD were found to form a β3γ3 hetero-hexamer, which likely binds up to 6 α subunits through a more dynamic association (Xu et al. 2020). Both the α and the γ subunits contain a Zn+ ion (Studer et al. 2007). The Na+-pumping OAD is found in both bacteria and archaea, but occurs mainly in proteobacteria. It is known to be important for citrate/tartrate fermentation, as are the other biotin-dependent Na+-pumping decarboxylases for the fermentation of other dicarboxylic acids (Dimroth and Schink 1998; Buckel 2001). Whether the Na+-pumping OAD is exclusively used in citrate/tartrate fermenting bacteria is not clear. To our understanding, there seems to be no clear reason why it should not be able to take part in other types of energy metabolism. At least one archaeal Na+-pumping OAD was (partly) characterized, from Archaeoglobus fulgidus, which does not ferment citrate, suggesting indeed that the role of Na+-pumping OADs is not restricted to citrate fermentation (Dahinden et al. 2004). In E. faecalis a variant of the Na+-pumping OAD had been described that has an additional subunit that allows activity in a soluble—non membrane associated—form (Repizo et al. 2013).
The soluble divalent cation-dependant OAD had been found and characterized in a range of different microorganisms already, until in 2010 the encoding gene was finally identified in C. glutamicum (Klaffl and Eikmanns 2010). The soluble OAD from C. glutamicum is reported to be homotetrameric as well as homodimeric (Jetten and Sinskey 1995; Klaffl and Eikmanns 2010), whereas the one from Pseudomonas stutzeri is monomeric (Labrou and Clonis 1999). Activity was absolutely dependent on Mn2+ or Mg2+, NAD+ was found to stimulate activity, and acetate and various dicarboxylic acids were found to act as inhibitors. ADP and ATP are both reported as inhibitors and activators (Benziman et al. 1978; Ng, Wong and Hamilton 1982; Jetten and Sinskey 1995; Labrou and Clonis 1999; Klaffl and Eikmanns 2010). The function of the soluble OAD is not quite clear. C. glutamicum did not appear to be affected by the deletion of the soluble OAD. Based on its relatively high Km value for oxaloacetate, typically present in low concentrations, the authors suggested that the soluble OAD might function as an overflow mechanism (Klaffl and Eikmanns 2010). It has also been suggested that it might be required for the supply of pyruvate during growth on TCA-cycle intermediates (Benziman et al. 1978). Recently, a homologous OAD was identified in eukaryotes as well, potentially involved in maintaining an optimal oxaloacetate pool (Pircher et al. 2015; Etemad et al. 2019).
Malate dehydrogenase (EC 1.1.1.37) (EC 1.1.1.82)
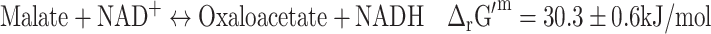 |
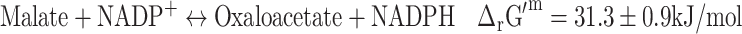 |
Malate dehydrogenase (MDH) catalyzes the reversible oxidation of malate to oxaloacetate using NAD(P)+. Another malate dehydrogenase exists (in bacteria) that uses a quinone instead of NAD(P)+, called malate:quinone oxidoreductase (MQO), which will be reviewed further down. The MDH reaction mechanism proceeds via the binding of NAD(P)+ followed by malate in a hydrophobic pocket, which is then closed by an external loop through a conformational change (Goward and Nicholls 1994; Minárik et al. 2002). The mechanism is identical to that of L-lactate dehydrogenase (LDH), which shares an evolutionary origin with MDHs.
MDHs are ubiquitously present in all domains of life. Two distinctive classes of MDHs exist: dimeric and tetrameric (Madern 2002). The latter is more closely related to LDHs, which also form tetramers, and occurs predominantly in prokaryotes; primarily in Gram-positives, while in archaea they are the only MDH present (Takahashi-Íñiguez et al. 2016). In some databases they can be wrongly annotated as LDH, and a single amino acid substitution has shown to convert an E. coli MDH to LDH. The dimeric MDHs are further split into two distinct phylogenetic groups. Both are present only in bacteria—in particular Gram-negatives (Takahashi-Íñiguez et al. 2016), and eukaryotes. In eukaryotes, this split follows mitochondrial (and glycosomal) MDHs versus cytosolic (and chloroplastic) MDHs (Madern 2002). In eukaryotes all MDHs are NAD+-dependent, except for the chloroplast MDHs, which use NADP+ (Minárik et al. 2002). Bacterial MDHs are predominantly NAD+-dependent; archaeal MDHs can be either NAD+- or NADP+-dependent, or have equal affinities for both (Takahashi-Íñiguez et al. 2016).
The precise dependency on the oligomeric state of MDHs is not known, but a study with monomeric mutants of the (dimeric) E. coli MDH showed a dramatic reduction in activity (Breiter, Resnik and Banaszak 1994), suggesting that at least the dimeric state is essential for activity. MDHs do not depend on any metal ions for activity, but are subject to some allosteric regulation, especially oxaloacetate seems to act as an inhibitor across the full spectrum of MDHs (Takahashi-Íñiguez et al. 2016). Mitochondrial MDHs are also activated by citrate in the oxaloacetate forming direction, while the reverse reaction is inhibited by citrate (Mullinax et al. 1982). NADP+-dependent MDHs of chloroplasts are activated by light, through the reduction of specific disulfide-residues. NADP+ inhibits the kinetics of this light-dependent activation (Scheibe and Jacquot 1983; Kagawa and Bruno 1988). Furthermore, MDHs both from mitochondria and B. subtilis (i.e. tetrameric) have been shown to form multi-enzyme complexes, or metabolons with other TCA-cycle enzymes, thereby regulating their activity and channelling the metabolites (Fahien et al. 1988; Meyer et al. 2011; Bartholomae et al. 2014; Jung and Mack 2018).
MDHs primarily function in the TCA-cycle, converting malate to oxaloacetate; generating NADH that is subsequently used by the respiratory complex. Additionally in eukaryotes, MDH—both the mitochondrial and the cytosolic isoforms, together with aspartate aminotransferase, the malate-α-ketoglutarate antiporter, and the glutamate-aspartate antiporter are responsible for the transport of electrons (i.e. NADH from glycolysis) from the cytosol into the mitochondrial matrix, to be used for oxidative phosphorylation (Lu et al. 2008). This is known as the malate-aspartate shuttle, which is depicted in Fig. 6. Furthermore, MDH is involved in the fermentation pathway to succinic acid via the reductive branch of the TCA-cycle (Thakker et al. 2012), and it has been shown to be involved in the formation of pyruvate from PEP via the so-called ‘malate shunt’. In this shunt, PEP is converted to pyruvate via PEPCK, MDH, and then ME—all part of the PPO-node. The primary function of this shunt is likely the trans-hydrogenation of NADH (oxidized by MDH) to NADPH (reduced by ME) (McKinlay et al. 2007; Taillefer et al. 2015; Olson et al. 2016).
Figure 6.
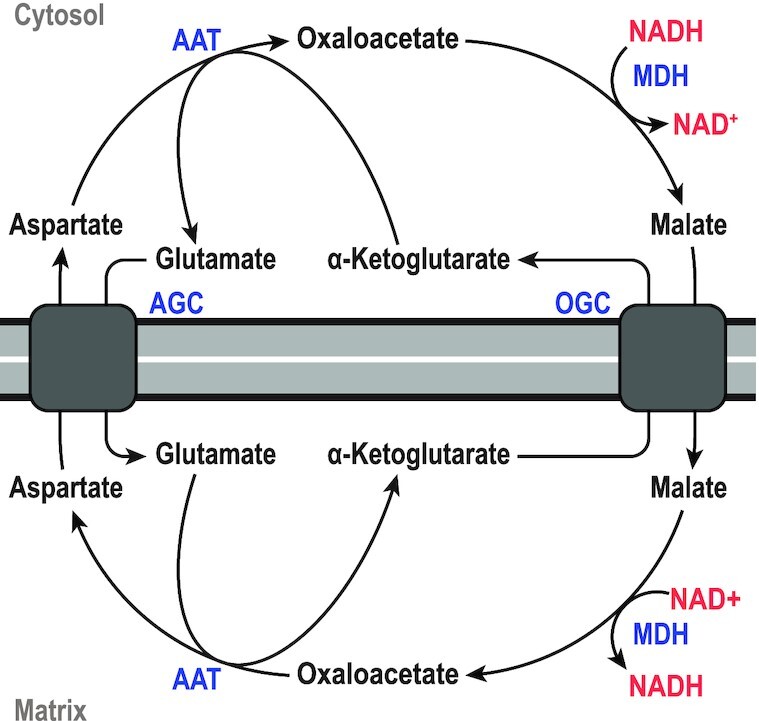
The malate-aspartate shuttle. The shuttle results in the net exchange of NADH with NAD+ from the cytosol into the mitochondrial matrix. AAT, aspartate aminotransferase; AGC, glutamate-aspartate antiporter; MDH, malate dehydrogenase; OGC, malate-α-ketoglutarate antiporter. Figure adapted from (Korla, Vadlakonda and Mitra 2015).
Malate:quinone oxidoreductase (EC 1.1.5.4)
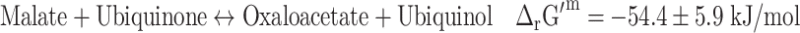 |
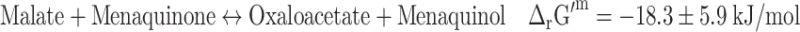 |
Just like MDH, malate:quinone oxidoreductase (MQO) facilitates the oxidation of malate to oxaloacetate, but uses quinone, rather than NAD(P)+, rendering the reaction physiologically irreversible. The enzyme is membrane associated and contains a tightly non-covalently bound prosthetic FAD-group (Molenaar, van der Rest and Petrovic 1998). However, no crystal structure is available at the moment, and details about the precise reaction mechanism are lacking.
MQOs are present mostly in bacteria, with only a handful of archaeal MQOs known, but none characterized. One study found MQOs to be present in 27% of analysed bacterial genomes. Eukaryotic MQOs are almost exclusively limited to Apicomplexa, a large phylum of parasitic protists (Mogi et al. 2009; Kabashima et al. 2013; Marreiros et al. 2016). MQOs can be split in two clear phylogenetic groups, with the second—minor—group being restricted mainly to Apicomplexa, Archaea and ϵ-proteobacteria (Mogi et al. 2009). Some Apicomplexa, Archaea and ϵ-proteobacteria are also known to possess MQOs from the major group, however. It is not clear whether there is a functional difference between the two phylogenetic groups.
Both ubiquinone and menaquinone can function as electron acceptor, however, many bacteria only possess menaquinone, which has a much lower redox potential (−74 mV versus + 113 mV) and therefore potentially allows the (reverse) reduction of oxaloacetate (Kather et al. 2000). Nevertheless, this is still much higher than that of NAD+/NADH (−320 mV) (Molenaar, van der Rest and Petrovic 1998). When both MDH and MQO are present in the same organism—as is very often the case, the simultaneous oxidation of malate (by MDH) and reduction of oxaloacetate (by MQO) can take place. This effectively channels electrons from NADH to quinone, mimicking the reaction of the non-proton pumping type II NADH dehydrogenase (NDH2), an enzyme believed to share a common ancestor with MQO (Mogi et al. 2009). In fact, there is evidence that in C. glutamicum, this is indeed one of several mechanisms for NADH:quinone oxidoreductase activity, and in Mycobacterium smegmatis, NDH2 mutants could be complemented by MDH (Miesel et al. 1998; Nantapong et al. 2004; Cook et al. 2014).
In organisms that contain MQO, but lack MDH, such as Helicobacter pylori, it clearly functions as part of the TCA-cycle (Kather et al. 2000). In Staphylococcus aureus MQO is also important for resistance against nitric oxide—a weapon of the host immune system—and the ensuing virulence (Spahich et al. 2016). In P. aeruginosa and Pseudomonas citronellolis, MQO was found to be essential for growth on acetate and ethanol, through the glyoxylate cycle, but nor for growth on glucose, lactate, succinate or malate (Görisch et al. 2002; Förster-Fromme and Jendrossek 2005). What the precise role of MQO is in organisms that have both MQO and MDH is not fully understood. It could be the observed NADH:quinone oxidoreductase activity as discussed above. Further observations are that in C. glutamicum MQO was the main contributor to the TCA-cycle, compared to MDH (Molenaar et al. 2000), while in E. coli, to opposite was true (van der Rest, Frank and Molenaar 2000).
Malic enzyme (EC 1.1.1.38) (EC 1.1.1.39) (EC 1.1.1.40)
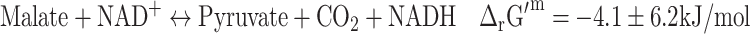 |
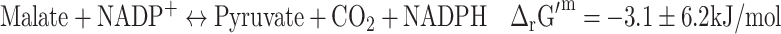 |
Malic enzymes (ME) catalyze the reversible decarboxylation reaction from malate to pyruvate via the reduction of NADP+ or NAD+, proceeding in three steps: First, the dehydrogenation of malate to oxaloacetate, followed by the decarboxylation where enolpyruvate is formed, which is converted to pyruvate via the tautomerization reaction. The catalysis requires a divalent cation bound in the active site (Tao, Yang and Tong 2003). ME variants are classified in various ways. Typically, they are classified based on their cofactor usage (NADP+ versus NAD+) and the ability to decarboxylate oxaloacetate (OAA), in the following three classes: EC1.1.1.38 (NAD+-dependent; OAA decarboxylating), EC1.1.1.39 (NAD+-dependent) and EC 1.1.1.40 (NADP+-dependent; OAA decarboxylating). However, NADP+-dependent MEs that do not decarboxylate OAA are also described, but they do not have their own EC-number (Fukuda et al. 2005; Taillefer et al. 2015; Olson et al. 2016).
Phylogenetically, there are two main groups. The first group is exclusively prokaryotic, with the exception of Trichomonas vaginalis and E. histolytica, and has subunits of about 40–50 kDa, which is smaller than group 2 ME subunits of about 60 kDa. Group 2 MEs are often referred to as the ‘eukaryotic’ MEs, however, they also occur in prokaryotes. NAD+ or NADP+ dependency does not appear to correlate very strongly with the phylogeny (Tronconi, Andreo and Drincovich 2018), explained from the fact that only minor amino acid changes are needed to change the specificity (Hsieh, Chen and Hung 2011). Whether or not OAA decarboxylation correlates better with phylogeny is less clear; the EC1.1.1.39 (NAD+-dependent; non-OAA decarboxylating) ME class is exclusively composed of plant mitochondrial MEs. However, the C. thermocellum and T. kodakarensis MEs, shown to be non-decarboxylating NADP+-MEs belongs to the group 1 MEs (Fukuda et al. 2005; Taillefer et al. 2015). Group 2 MEs can subsequently also be divided in two clades, but these—recently reviewed—details are beyond the scope of this review (Doležal et al. 2004). Group 2 also include the malolactic enzyme from lactic-acid bacteria that catalyzes the reversible decarboxylation reaction from malate to lactate via the reduction of NAD+, and the oxaloacetate decarboxylases described earlier (Tronconi, Andreo and Drincovich 2018).
The group 2 MEs are typically active as homotetramers, with the exception of the plant mitochondrial MEs (EC1.1.1.39), which form heterodimers of two ME paralogs. However, reports of homodimeric, -hexameric, -octameric and -decameric forms are also known (Suye et al. 1992; Tronconi et al. 2008). Group I MEs are also primarily found as tetramers (Kobayashi et al. 1989; Chen et al. 1998). The two characterized archaeal MEs from Sulfolobus solfataricus and T. kodakarensis, however, are both dimeric (Bartolucci et al. 1987; Fukuda et al. 2005), as well as the ones from the eukaryotes T. vaginalis and E. histolytica, the latter of which is also closely related to the archaeal MEs (Doležal et al. 2004). Many type I MEs are described that have a molecular mass of around 80–90 kDa, due to a fused C-terminal domain homologous to phosphate acetyltransferase (PTA). The PTA domain is not essential for ME activity, nor does it show any PTA activity (Mitsch et al. 1998). Instead, it was found that deletion of the PTA domain from the E. coli enzyme abolished allosteric inhibition by acetyl-CoA and activation by glutamate, aspartate, and glucose-6-P, indicating that the PTA domain is important for allosteric control (Bologna, Andreo and Drincovich 2007). Rhizobium meliloti, a Gram-negative nitrogen fixing bacterium contains two group 1 MEs, both with PTA domains, with divergent allosteric regulation, of which only one is sensitive to acetyl-CoA (Voegele, Mitsch and Finan 1999). E. coli contains ME isoforms of both groups. Both are inhibited by fumarate and oxaloacetate, and activated by aspartate. Interestingly, acetyl-phosphate inhibits the group 2 ME while it activates the group 1 ME, independently of the PTA domain (Bologna, Andreo and Drincovich 2007). Many other allosteric regulators for MEs are known, including NH4+ and ATP (Kawai et al. 1996; Hsieh, Chen and Hung 2011). The complex allosteric regulation, often varying between paralogs, illustrates the different roles that can be played by ME and PPO-node enzymes in general.
Generally, NAD+-dependent MEs are used in the catabolism of malate, and NADP+-dependent MEs are used for NADPH generation(Sauer and Eikmanns 2005). Although both have been shown to be required for acetyl-CoA generation via pyruvate, primarily NADP+-dependent MEs are thought to be responsible for gluconeogenesis (Voegele, Mitsch and Finan 1999; Sauer and Eikmanns 2005; Bologna, Andreo and Drincovich 2007). Only one example is known of a wildtype bacterium that uses a NADP+-dependent ME to fix CO2 (Matula, McDonald and Martin 1969).
Phosphotransferase system
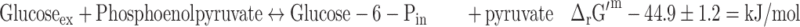 |
The phosphotransferase system (PTS) is a system consisting of several proteins that is involved in active uptake of sugars, driven by the transfer of the phosphate group from PEP to the imported sugar. As such, it is simultaneously responsible for sugar phosphorylation and the conversion of PEP to pyruvate. The mechanism proceeds as follows: First, the phosphate group of PEP is transferred to Enzyme I (EI), after which the phosphate group is transferred sequentially from EI to histidine protein (HPr), then to Enzyme II A (EIIA), and finally to Enzyme II B (EIIB). EIIB then phosphorylates the sugar as it is imported through the cell membrane via the transmembrane Enzyme II C (EIIC). Each of these steps is reversible (Deutscher, Francke and Postma 2006). The complete sequence of steps is shown in Fig. 7. The sugar phosphorylation increases the polarity of the sugar, decreasing the rate at which it can leak out of the cell again (Bar-Even et al. 2012), while simultaneously keeping a low intracellular concentration of the non-phosphorylated sugar, maintaining a high driving force. The EII proteins are specific for the sugar they transport, and many different EII proteins can be present in parallel in one cell that are all connected to a common PEP/EI/HPr pathway. For example, E. coli contains at least 15 different EII complexes (Deutscher, Francke and Postma 2006).
Figure 7.
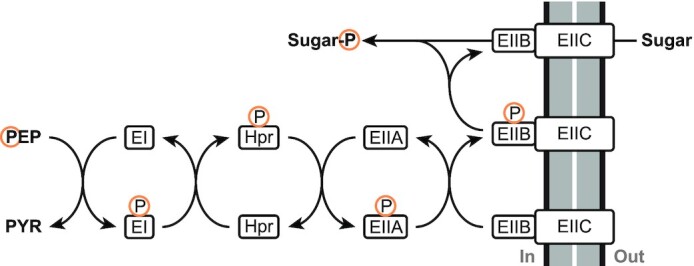
Sequence of reactions in the phosphotransferase system.
The PTS is almost exclusively present in bacteria, and only recently it has been identified and characterized in archaea (Pickl, Johnsen and Schonheit 2012; Cai et al. 2014); the PTS proteins are completely absent from eukaryotes. A comparative genomics analysis in 2005 found that 57% of studied bacteria possessed genes for all required PTS constituents, while 22% did not possess any (Barabote and Saier 2005). The remaining 21% contained soluble ‘PTS’ proteins, which are also known to possess regulatory functions, which will be discussed briefly below. Complete systems have been found in almost all bacterial kingdoms, and have been gained and lost with high frequency.
Seven different PTS types are distinguished, of which the substrate-recognizing EIIC proteins have evolved from at least four independent sources, giving rise to the Glc-Fru-Lac superfamily, the Asc-Gat superfamily, the Man family and the Dha family. Overall, the different EII components form a true mosaic, derived from many ancestral sources with a complex phylogeny between the different PTS types. EI exhibits both sequence and structural similarity with PPdK (Oberholzer et al. 2005; Deutscher, Francke and Postma 2006). EIIA, B and C can either exist as separate proteins, or may be fused in various combinations (Barabote and Saier 2005). The individual components of the PTS each form functional dimers (or oligomers), including the transmembrane EIIC, but there is no evidence for the existence of larger complexes or metabolons between EI, HPr and EII (Deutscher, Francke and Postma 2006; Patel et al. 2006; McCoy, Levin and Zhou 2015).
As mentioned before, the PTS also possesses regulatory functions, which are related to carbon, nitrogen and phosphate utilizations, as well as virulence in some pathogens, chemotaxis and potassium transport (Galinier and Deutscher 2017). By and large, the signal for these processes is provided by the phosphorylation state of the PTS components, which is dictated by the metabolic state of the cell, and the availability of its substrates (Deutscher et al. 2014). The signal is transduced via phosphorylation of non-PTS proteins by PTS components or via protein-protein interactions. In enteric bacteria, EIIA of the glucose PTS plays a central role in regulating carbon metabolism, in particular for catabolite repression and the uptake of sugars (Somavanshi, Ghosh and Sourjik 2016), making it a common target for metabolic engineering.
Although PTSs are only known to facilitate active solute import and not the export, EI was found recently to facilitate a significant (gluconeogenic) reverse flux from pyruvate to PEP in E. coli—both in wild type and in a PPS knock-out mutant during gluconeogenic as well as glycolytic growth (Long et al. 2017). The responsible phosphate donor is not known, but it suggests that the PTS might have a more central metabolic role (in the PPO-node) beyond sugar uptake.
PHYLOGENETIC DISTRIBUTION OF PPO-NODE ENZYMES
No single organism harbours enzymes to catalyse all 11 reactions described above. Instead, they have different subsets of those, illustrated by the global overview presented in Fig. 8. From the overview it is already evident that most of the PPO-node enzymes are distributed over both Bacteria and Archaea. Although OADs, MQOs and PTSs are rare in Archaea. And with the exception of the PTS, Eukaryotes—absent from the figure—also possess enzymes for all of the PPO-node reactions; albeit occurrence of PPS and MQO is very restricted. The ubiquity and especially the pervasiveness of many of the PPO-node enzymes amongst prokaryotes suggest that they are evolutionary old and that some may have been present already in the last universal common ancestor, LUCA. Conversely, the existence of multiple enzymes/routes connecting PEP, pyruvate and oxaloacetate (as well as malate) could also be indicative of convergent evolution, in which case they emerged in parallel after LUCA (and were then distributed through horizontal gene transfer, to account for their pervasiveness). The study into which genes were present in LUCA is still a contentious, but mostly nebulous subject. A phylogenetic study from 2016 nevertheless gives the first small insight into LUCA's physiology and habitat (Weiss et al. 2016). LUCA's metabolism likely relied on H2 dependent CO2 fixation through the Wood-Ljungdahl pathway, which would indicate that the PPO-node primarily had a gluconeogenic role, to generate the precursor metabolites upwards from acetyl-CoA (Fig. 1). What the composition of that PPO-node would have been—if even a real node—is still an open question.
Figure 8.
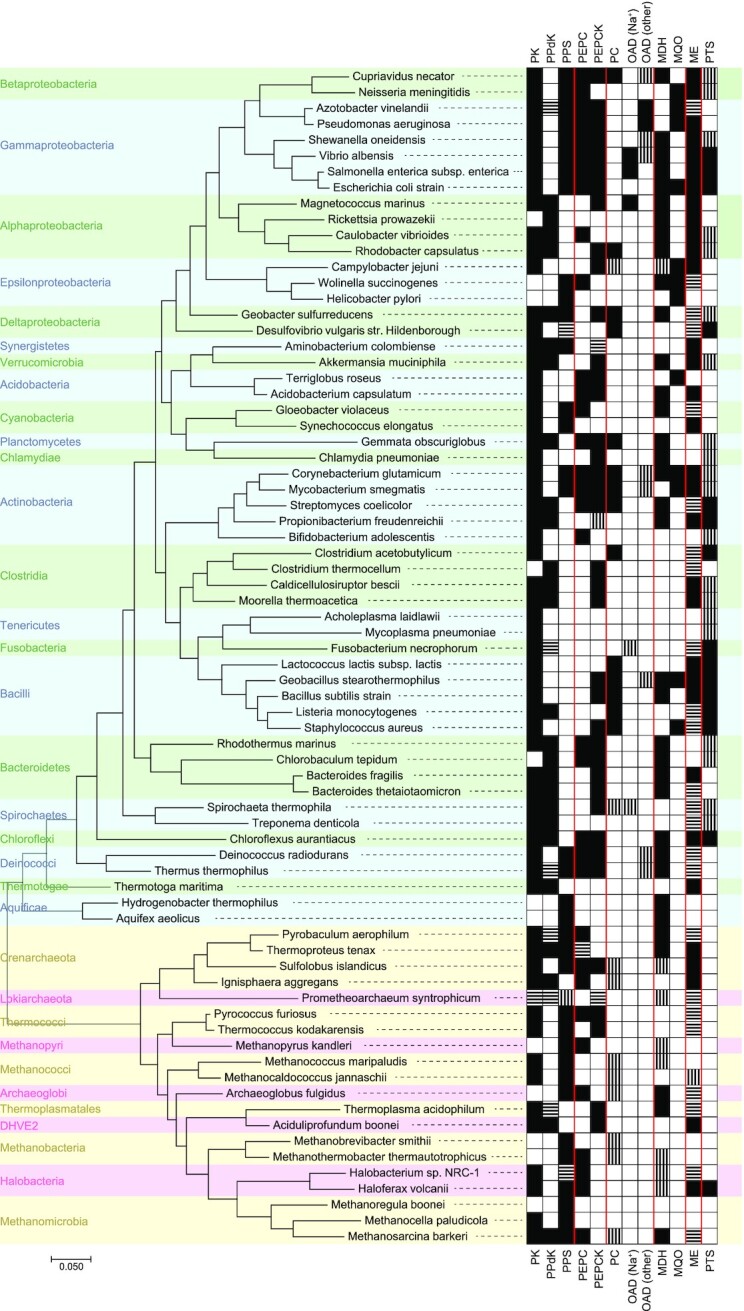
Different subsets of PPO-node enzymes as present in selected (cultivated) prokaryotes, spread-out through the tree of life. Presence of an enzyme is based on data from the UniProt database, with vertical stripes indicating presence of a corresponding enzyme commission (EC) number in the given organism, and horizontal stripes the presence of corresponding InterPro accession numbers; fully black indicates the presence of both. The results are a conservative estimation, as not all enzymes have (unique) InterPro accession numbers (i.e. a described protein families) or annotated EC numbers. The PPi-dependent PEPCK, the soluble OAD and the ME-related OAD do not have InterPro protein families described for them. Conversely, for multi-subunit proteins (i.e. Na+-pumping OAD, and PTS-systems) the EC numbers are the same for the different subunits, and can therefore yield an overestimation. Finally, some MDHs are known to be annotated as LDHs, and might also not be picked up (Taillefer et al. 2015). The phylogenetic tree is the result of the Maximum Likelihood algorithm, based on 16s rRNA sequences that were aligned using MUSCLE. A list of the selected EC numbers and InterPro accession numbers can be found in the supplementary data.
Perhaps the most striking from Fig. 8 is how limited the correlation seems to be between the organisms’ phylogeny and the PPO node composition, despite its place at the heart of the core metabolism (Fig. 1). It is illustrative of the evolutionary flexibility of metabolism in general and suggests that the PPO-node composition is strongly dictated by the organism's niche, together with its evolutionary history.
CONCLUDING REMARKS
Many of the PPO-node enzymes are often referred to as either glycolytic, gluconeogenic, anaplerotic or cataplerotic. Although those are typical roles they can fulfil, such generalizations do not do justice to the varying and dynamic roles certain PPO-node enzymes play in different metabolisms, of which several examples have been discussed in this review. The most extreme example in this regard is PPS, which—thermodynamically—highly favours the gluconeogenic conversion of pyruvate to PEP, and yet has been shown to function as a glycolytic enzyme in several Archaea, despite the simultaneous presence of a PK in the genome, and of course the existence of PPdK, which is even more ambivalent with respect to its role in glycolysis and gluconeogenesis. Similarly, the role of PEPCK can be either glycolytic, gluconeogenic, anaplerotic, or cataplerotic. Also, the quintessentially-glycolytic PK is required for growth on gluconeogenic substrates in certain cases. And finally, their variability and dynamicity are further exemplified by the complex allosteric control that many PPO-node enzymes are subjected to, which often vary (or even work oppositely) for different homologs and paralogs.
So, not only can PPO-node enzymes have different roles in different organisms, but also within one organism can they operate in opposing directions, or seemingly contradicting roles. We therefore recommend that using the labels glycolytic, gluconeogenic, anaplerotic and cataplerotic to define PPO-node enzymes should be avoided, as it might well only be a partial description; but also to prevent being put on the wrong track while trying to understand an organism's metabolism.
Supplementary Material
Footnotes
Two pathways exist for de novo lysine biosynthesis: the diaminopimelate (DAP) pathway and α–aminoadipate (AAA) pathway. Only the former uses pyruvate as precursor, and is found in prokaryotes and plants. The AAA pathway occurs primarily in yeasts and fungi, but some hyperthermophilic prokaryotes also possess a variant of the AAA pathway (Bowman et al. 1988).
The only two variations known—both occurring in Archaea—are glyceraldehyde-3-phosphate:ferredoxin oxidoreductase (GAPOR) replacing glyceraldehyde-3-phosphate dehydrogenase (GAPDH), and the non-phosphorylating glyceraldehyde-3-phosphate dehydrogenase (GAPN), which also replaces phosphoglycerate kinase (PGK), abolishing the accompanying ATP generation (Brasen et al. 2014).
Pyruvate phosphate dikinase is normally written as pyruvate, phosphate dikinase, using a comma. However, to improve readability, we have written it without a comma throughout this review.
An example of such a different prioritization can be found in some extreme thermophiles, which do not phosphorylate certain glycolytic metabolites. At very high temperatures those become unstable and can undergo spontaneous dephosphorylation (Bar-Even et al. 2012; Brasen et al. 2014). The consequence is that non-phosphorylated compounds are more prone to membrane leakage (due to their increased hydrophobicity), but at higher temperature this constraint seemingly weighs less than loss in energy resulting from spontaneous dephosphorylation. Other alternative prioritizations could relate to the trade-off between rate and yield of energy conservation; toxicity of metabolic intermediates; the costs for the required enzyme machinery; requirement for metals, cofactors & coenzymes, or ecologically rare/limiting elements; etc.
Values for the Gibbs free energy of the reaction (ΔrG) are obtained from eQuilibrator (Flamholz et al. 2012). The ‘m’ in ΔrG′m indicates that in the calculations 1 mM was used as concentration for all the reactants, at a pH of 7.0 and ionic strength of 1.0 M. ΔrG is defined such that at negative values, the net reaction proceeds in the forward direction. However, it is good to keep in mind that ΔrG′m does not reflect the true ΔrG, which is ultimately determined by the actual concentrations of the reactants; it essentially reflects to which direction (and extend) the equilibrium leans. A good example is glyceraldehyde 3-phosphate dehydrogenase, which has a ΔrG′m of 25.9 kJ/mol in the glycolytic direction, yet it still proceeds (because the concentrations of the substrates are many times higher than that of the products). The often-large error of ΔrG′m is a result of the fact that for only ∼10% of the metabolic reactions in E. coli and human cell metabolic models, experimental equilibrium data are available and that for the remainder the group contribution method is used. This method can cover the majority (∼80%) of relevant biochemical reactions, but is less accurate since it relies on a simplifying assumption that the contributions of groups are additive. Thus, the average estimation error attributed to group contribution is about 9–10 kJ/mol (Noor et al. 2013).
Contributor Information
Jeroen G Koendjbiharie, Laboratory of Microbiology, Wageningen University, Stippeneng4, 6708 WE Wageningen, The Netherlands.
Richard van Kranenburg, Laboratory of Microbiology, Wageningen University, Stippeneng4, 6708 WE Wageningen, The Netherlands; Corbion, Arkelsedijk 46, 4206 AC Gorinchem, The Netherlands.
Servé W M Kengen, Laboratory of Microbiology, Wageningen University, Stippeneng4, 6708 WE Wageningen, The Netherlands.
Conflicts of interest
None declared.
REFERENCES
- Abbe K, Yamada T. Purification and properties of pyruvate kinase from Streptococcus mutans. J Bacteriol. 1982;149:299–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adina-Zada A, Zeczycki TN, St Maurice Met al. . Allosteric regulation of the biotin-dependent enzyme pyruvate carboxylase by acetyl-CoA. Biochem Soc Trans. 2012;40:567–72. [DOI] [PubMed] [Google Scholar]
- Aich S, Delbaere LTJ. Phylogenetic study of the evolution of PEP-carboxykinase. Evol Bioinform Online. 2007;3:333–40. [PMC free article] [PubMed] [Google Scholar]
- Aldous SH, Weise SE, Sharkey TDet al. . Evolution of the Phosphoenolpyruvate Carboxylase Protein Kinase Family in C3 and C4 Flaveria spp. Plant Physiol. 2014;165:1076–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balsera M, Buey RM, Li X-D. Quaternary structure of the oxaloacetate decarboxylase membrane complex and mechanistic relationships to pyruvate carboxylases. J Biol Chem. 2011;286:9457–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bar-Even A, Flamholz A, Noor Eet al. . Rethinking glycolysis: on the biochemical logic of metabolic pathways. Nat Chem Biol. 2012;8:509–17. [DOI] [PubMed] [Google Scholar]
- Barabote RD, Saier MH. Comparative genomic analyses of the bacterial phosphotransferase system. Microbiol Mol Biol Rev. 2005;69:608–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartholomae M, Meyer FM, Commichau FMet al. . Complex formation between malate dehydrogenase and isocitrate dehydrogenase from Bacillus subtilis is regulated by tricarboxylic acid cycle metabolites. FEBS J. 2014;281:1132–43. [DOI] [PubMed] [Google Scholar]
- Bartolucci S, Rella R, Guagliardi Aet al. . Malic enzyme from archaebacterium Sulfolobus solfataricus. Purification, structure, and kinetic properties. J Biol Chem. 1987;262:7725–31. [PubMed] [Google Scholar]
- Bendt AK, Burkovski A, Schaffer Set al. . Towards a phosphoproteome map ofCorynebacterium glutamicum. Proteomics. 2003;3:1637–46. [DOI] [PubMed] [Google Scholar]
- Benziman M, Eizen N. Pyruvate-phosphate dikinase and the control of gluconeogenesis in Acetobacter xylinum. J Biol Chem. 1971;246:67–61. [PubMed] [Google Scholar]
- Benziman M, Russo A, Hochman Set al. . Purification and regulatory properties of the oxaloacetate decarboxylase of Acetobacter xylinum. J Bacteriol. 1978;134:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman KM, Cohn M. Phosphoenolpyruvate synthetase of Escherichia coli. Purification, some properties, and the role of divalent metal ions. J Biol Chem. 1970;245:5309–18. [PubMed] [Google Scholar]
- Bielen AAM, Willquist K, Engman Jet al. . Pyrophosphate as a central energy carrier in the hydrogen-producing extremely thermophilic Caldicellulosiruptor saccharolyticus. FEMS Microbiol Lett. 2010;307:48–54. [DOI] [PubMed] [Google Scholar]
- Bologna FP, Andreo CS, Drincovich MF. Escherichia coli malic enzymes: two isoforms with substantial differences in kinetic properties, metabolic regulation, and structure. J Bacteriol. 2007;189:5937–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowman E, McQueney M, Barry RJet al. . Catalysis and thermodynamics of the phosphoenolpyruvate/phosphonopyruvate rearrangement. entry into the phosphonate class of naturally occurring organophosphorus compounds. J Am Chem Soc. 1988;110:5575–6. [Google Scholar]
- Brasen C, Esser D, Rauch Bet al. . Carbohydrate metabolism in Archaea: current insights into unusual enzymes and pathways and their regulation. Microbiol Mol Biol Rev. 2014;78:89–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breiter DR, Resnik E, Banaszak LJ. Engineering the quaternary structure of an enzyme: Construction and analysis of a monomeric form of malate dehydrogenase from Escherichia coli. Protein Sci. 1994;3:2023–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckel W. Sodium ion-translocating decarboxylases. Biochim Biophys Acta - Bioenerg. 2001;1505:15–27. [DOI] [PubMed] [Google Scholar]
- Burnell J. Purification and properties of phosphoenolpyruvate carboxykinase from C4 plants. Funct Plant Biol. 1986;13:577. [Google Scholar]
- Burnell JN. Cloning and characterization of Escherichia coli DUF299: a bifunctional ADP-dependent kinase - Pi-dependent pyrophosphorylase from bacteria. BMC Biochem. 2010;11:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai L, Cai S, Zhao Det al. . Analysis of the transcriptional regulator glpr, promoter elements, and posttranscriptional processing involved in fructose-induced activation of the phosphoenolpyruvate-dependent sugar phosphotransferase system in Haloferax mediterranei. Appl Environ Microbiol. 2014;80:1430–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson GM, Holyoak T. Structural insights into the mechanism of phosphoenolpyruvate carboxykinase catalysis. J Biol Chem. 2009;284:27037–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charles AM, Willer DW. Pyruvate carboxylase from Thiobacillus novellus: properties and possible function. Can J Microbiol. 1984;30:535–39. [Google Scholar]
- Chastain CJ, Failing CJ, Manandhar Let al. . Functional evolution of C4 pyruvate, orthophosphate dikinase. J Exp Bot. 2011;62:3083–91. [DOI] [PubMed] [Google Scholar]
- Chastain CJ. Chapter 15 structure, function, and post-translational regulation of C4 pyruvate orthophosphate dikinase. Dordrecht: Springer, 2010, 301–15. [Google Scholar]
- Chen F, Okabe Y, Osano Ket al. . Purification and characterization of an NAD-malic enzyme from Bradyrhizobium japonicum A1017. Appl Environ Microbiol. 1998;64:4073–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Omiya T, Hata Set al. . Molecular characterization of a phosphoenolpyruvate carboxylase from a thermophilic cyanobacterium, Synechococcus vulcanus with unusual allosteric properties. Plant Cell Physiol. 2002;43:159–69. [DOI] [PubMed] [Google Scholar]
- Chen Y-B, Lu T-C, Wang H-Xet al. . Posttranslational modification of maize chloroplast pyruvate orthophosphate dikinase reveals the precise regulatory mechanism of its enzymatic activity. Plant Physiol. 2014;165:534–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiba Y, Kamikawa R, Nakada-Tsukui Ket al. . Discovery of PPi-type phosphoenolpyruvate carboxykinase genes in eukaryotes and bacteria. J Biol Chem. 2015;290:23960–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciupka D, Gohlke H. On the potential alternate binding change mechanism in a dimeric structure of Pyruvate Phosphate Dikinase. Sci Rep. 2017;7:8020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook GM, Hards K, Vilchèze Cet al. . Energetics of respiration and oxidative phosphorylation in Mycobacteria. Microbiol Spectr. 2014;2, DOI: 10.1128/microbiolspec.MGM2-0015-2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook WJ, Senkovich O, Aleem Ket al. . Crystal structure of Cryptosporidium parvum pyruvate kinase. PLoS One. 2012;7:e46875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper RA, Kornberg HL. The mechanism of the phosphoenolpyruvate synthase reaction. Biochim Biophys Acta - Gen Subj. 1967;141:211–3. [DOI] [PubMed] [Google Scholar]
- Cotelesage JJH, Puttick J, Goldie Het al. . How does an enzyme recognize CO2? Int J Biochem Cell Biol. 2007;39:1204–10. [DOI] [PubMed] [Google Scholar]
- Cui DS, Broom A, Mcleod MJet al. . Asymmetric anchoring is required for efficient Ω-loop opening and closing in cytosolic phosphoenolpyruvate carboxykinase. Biochemistry. 2017;56:2106–15. [DOI] [PubMed] [Google Scholar]
- Dahinden P, Pos KM, Taralczak Met al. . Oxaloacetate decarboxylase of Archaeoglobus fulgidus: cloning of genes and expression in Escherichia coli. Arch Microbiol. 2004;182:414–20. [DOI] [PubMed] [Google Scholar]
- Deng Y, Olson DG, Zhou Jet al. . Redirecting carbon flux through exogenous pyruvate kinase to achieve high ethanol yields in Clostridium thermocellum. Metab Eng. 2013;15:151–8. [DOI] [PubMed] [Google Scholar]
- Denton H, Brown SM, Roberts CWet al. . Comparison of the phosphofructokinase and pyruvate kinase activities of Cryptosporidium parvum, Eimeria tenella and Toxoplasma gondii. Mol Biochem Parasitol. 1996;76:23–9. [DOI] [PubMed] [Google Scholar]
- Deramchia K, Morand P, Biran Met al. . Contribution of pyruvate phosphate dikinase in the maintenance of the glycosomal ATP/ADP balance in the Trypanosoma brucei procyclic form. J Biol Chem. 2014;289:17365–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deutscher J, Aké FMD, Derkaoui Met al. . The bacterial phosphoenolpyruvate:carbohydrate phosphotransferase system: regulation by protein phosphorylation and phosphorylation-dependent protein-protein interactions. Microbiol Mol Biol Rev. 2014;78:231–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deutscher J, Francke C, Postma PW. How phosphotransferase system-related protein phosphorylation regulates carbohydrate metabolism in bacteria. Microbiol Mol Biol Rev. 2006;70:939–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dharmarajan L, Kraszewski JL, Mukhopadhyay Bet al. . Structure of an archaeal-type phosphoenolpyruvate carboxylase sensitive to inhibition by aspartate. Proteins Struct Funct Bioinforma. 2011;79:1820–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimroth P, Jockel P, Schmid M. Coupling mechanism of the oxaloacetate decarboxylase Na+ pump. Biochim Biophys Acta - Bioenerg. 2001;1505:1–14. [DOI] [PubMed] [Google Scholar]
- Dimroth P, Schink B. Energy Conservation in the Decarboxylation of Dicarboxylic Acids by Fermenting Bacteria. Arch Microbiol. 1998;170:69–77. [DOI] [PubMed] [Google Scholar]
- Doležal P, Vaňáčová Š, Tachezy Jet al. . Malic enzymes of Trichomonas vaginalis: two enzyme families, two distinct origins. Gene. 2004;329:81–92. [DOI] [PubMed] [Google Scholar]
- Edwards GE, Nakamoto H, Burnell JNet al. . Pyruvate,Pi Dikinase and NADP-Malate Dehydrogenase in C4 photosynthesis: properties and mechanism of light/dark regulation. Annu Rev Plant Physiol. 1985;36:255–86. [Google Scholar]
- Eisaki N, Tatsumi H, Murakami Set al. . Pyruvate phosphate dikinase from a thermophilic actinomyces Microbispora rosea subsp. aerata: purification, characterization and molecular cloning of the gene. Biochim Biophys Acta - Protein Struct Mol Enzymol. 1999;1431:363–73. [DOI] [PubMed] [Google Scholar]
- Enriqueta Muñoz M, Ponce E. Pyruvate kinase: current status of regulatory and functional properties. Comp Biochem Physiol Part B Biochem Mol Biol. 2003;135:197–218. [DOI] [PubMed] [Google Scholar]
- Ernest I, Callens M, Opperdoes FRet al. . Pyruvate kinase of Leishmania mexicana mexicana Cloning and analysis of the gene, overexpression in Escherichia coli and characterization of the enzyme. Mol Biochem Parasitol. 1994;64:43–54. [DOI] [PubMed] [Google Scholar]
- Ernst SM, Budde RJ, Chollet R. Partial purification and characterization of pyruvate, orthophosphate dikinase from Rhodospirillum rubrum. J Bacteriol. 1986;165:483–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espariz M, Repizo G, Blancato Vet al. . Identification of malic and soluble oxaloacetate decarboxylase enzymes in Enterococcus faecalis. FEBS J. 2011;278:2140–51. [DOI] [PubMed] [Google Scholar]
- Etemad S, Petit M, Weiss AKHet al. . Oxaloacetate decarboxylase FAHD1 – a new regulator of mitochondrial function and senescence. Mech Ageing Dev. 2019;177:22–9. [DOI] [PubMed] [Google Scholar]
- Ettema TJG, Makarova KS, Jellema GLet al. . Identification and functional verification of archaeal-type phosphoenolpyruvate carboxylase, a missing link in archaeal central carbohydrate metabolism. J Bacteriol. 2004;186:7754–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans HJ, Wood HG. The mechanism of the pyruvate, phosphate dikinase reaction. Proc Natl Acad Sci USA. 1968;61:1448–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eyzaguirre J, Jansen K, Fuchs G. Phosphoenolpyruvate synthetase in Methanobacterium thermoautotrophicum. Arch Microbiol. 1982;132:67–74. [Google Scholar]
- Fahien LA, Kmiotek EH, MacDonald MJet al. . Regulation of malate dehydrogenase activity by glutamate, citrate, alpha-ketoglutarate, and multienzyme interaction. J Biol Chem. 1988;263:10687–97. [PubMed] [Google Scholar]
- Flamholz A, Noor E, Bar-Even Aet al. . eQuilibrator–the biochemical thermodynamics calculator. Nucleic Acids Res. 2012;40:D770–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda W, Fukui T, Atomi Het al. . First characterization of an archaeal GTP-dependent phosphoenolpyruvate carboxykinase from the hyperthermophilic archaeon Thermococcus kodakaraensis KOD1. J Bacteriol. 2004;186:4620–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda W, Ismail YS, Fukui Tet al. . Characterization of an archaeal malic enzyme from the hyperthermophilic archaeon Thermococcus kodakaraensis KOD1. Archaea. 2005;1:293–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Förster-Fromme K, Jendrossek D. Malate:quinone oxidoreductase (MqoB) is required for growth on acetate and linear terpenes in Pseudomonas citronellolis. FEMS Microbiol Lett. 2005;246:25–31. [DOI] [PubMed] [Google Scholar]
- Galinier A, Deutscher J. Sophisticated regulation of transcriptional factors by the bacterial phosphoenolpyruvate: sugar phosphotransferase system. J Mol Biol. 2017;429:773–89. [DOI] [PubMed] [Google Scholar]
- Goldie AH, Sanwal BD. Allosteric control by calcium and mechanism of desensitization of phosphoenolpyruvate carboxykinase of Escherichia coli. J Biol Chem. 1980;255:1399–405. [PubMed] [Google Scholar]
- González-Marcano E, Acosta H, Mijares Aet al. . Kinetic and molecular characterization of the pyruvate phosphate dikinase from Trypanosoma cruzi. Exp Parasitol. 2016;165:81–7. [DOI] [PubMed] [Google Scholar]
- González-Marcano E, Mijares A, Quiñones Wet al. . Post-translational modification of the pyruvate phosphate dikinase from Trypanosoma cruzi. Parasitol Int. 2014;63:80–6. [DOI] [PubMed] [Google Scholar]
- Goward CR, Nicholls DJ. Malate dehydrogenase: A model for structure, evolution, and catalysis. Protein Sci. 1994;3:1883–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerrero-Mendiola C, García-Trejo JJ, Encalada Ret al. . The contribution of two isozymes to the pyruvate kinase activity of Vibrio cholerae: One K+-dependent constitutively active and another K+-independent with essential allosteric activation. Guerrero-Hernandez A (ed.). PLoS One. 2017;12:e0178673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Görisch H, Jeoung J-H, Rückert Aet al. . Malate:quinone oxidoreductase is essential for growth on ethanol or acetate in Pseudomonas aeruginosa. Microbiology. 2002;148:3839–47. [DOI] [PubMed] [Google Scholar]
- He L, Hamm JA, Reddy Aet al. . Biotinylation: a novel posttranslational modification linking cell autonomous circadian clocks with metabolism. Am J Physiol Heart Circ Physiol. 2016;310:H1520–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrmann KM, Weaver LM. The shikimate pathway. Annu Rev Plant Physiol Plant Mol Biol. 1999;50:473–503. [DOI] [PubMed] [Google Scholar]
- Hiltpold A, Thomas RM, Köhler P. Purification and characterization of recombinant pyruvate phosphate dikinase from Giardia. Mol Biochem Parasitol. 1999;104:157–69. [DOI] [PubMed] [Google Scholar]
- Hrdý I, Mertens E, Van Schaftingen E. Identification, purification and separation of different isozymes of NADP-specific malic enzyme from Tritrichomonas foetus. Mol Biochem Parasitol. 1993;57:253–60. [DOI] [PubMed] [Google Scholar]
- Hsieh J-Y, Chen M-C, Hung H-C. Determinants of nucleotide-binding selectivity of malic enzymeUversky VN (ed). PLoS One. 2011;6:e25312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchins AM, Holden JF, Adams MW. Phosphoenolpyruvate synthetase from the hyperthermophilic archaeon Pyrococcus furiosus. J Bacteriol. 2001;183:709–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyskova V, Ryslava H. Unusual Properties and Functions of Plant Pyruvate, Orthophospate Dikinase. Biochem Anal Biochem. 2016;05, DOI: 10.4172/2161-1009.1000e161. [Google Scholar]
- Igawa T, Fujiwara M, Tanaka Iet al. . Characterization of bacterial-type phosphoenolpyruvate carboxylase expressed in male gametophyte of higher plants. BMC Plant Biol. 2010;10:200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imanaka H, Yamatsu A, Fukui Tet al. . Phosphoenolpyruvate synthase plays an essential role for glycolysis in the modified Embden-Meyerhof pathway in Thermococcus kodakarensis. Mol Microbiol. 2006;61:898–909. [DOI] [PubMed] [Google Scholar]
- Inoue M, Li X. Highly active and stable oxaloacetate decarboxylase Na+ pump complex for structural analysis. Protein Expr Purif. 2015;115:34–8. [DOI] [PubMed] [Google Scholar]
- Izui K, Matsumura H, Furumoto Tet al. . Phosphoenolpyruvate carboxylase: a new era of structural biology. Annu Rev Plant Biol. 2004;55:69–84. [DOI] [PubMed] [Google Scholar]
- Jetten MSM, Sinskey AJ. Purification and properties of oxaloacetate decarboxylase from Corynebacterium glutamicum. Antonie Van Leeuwenhoek. 1995;67:221–7. [DOI] [PubMed] [Google Scholar]
- Jitrapakdee S, St Maurice M, Rayment Iet al. . Structure, mechanism and regulation of pyruvate carboxylase. Biochem J. 2008;413:369–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jitrapakdee S, Wallace JC. Structure, function and regulation of pyruvate carboxylase. Biochem J. 1999;340:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnsen U, Hansen T, Schönheit P. Comparative analysis of pyruvate kinases from the hyperthermophilic archaea Archaeoglobus fulgidus, Aeropyrum pernix, and Pyrobaculum aerophilum and the hyperthermophilic bacterium Thermotoga maritima: unusual regulatory properties in hyperthermophilic ar. J Biol Chem. 2003;278:25417–27. [DOI] [PubMed] [Google Scholar]
- Jung T, Mack M. Interaction of enzymes of the tricarboxylic acid cycle in Bacillus subtilis and Escherichia coli: a comparative study. FEMS Microbiol Lett. 2018;365, DOI: 10.1093/femsle/fny055. [DOI] [PubMed] [Google Scholar]
- Jurica MS, Mesecar A, Heath PJet al. . The allosteric regulation of pyruvate kinase by fructose-1,6-bisphosphate. Structure. 1998;6:195–210. [DOI] [PubMed] [Google Scholar]
- Kabashima Y, Sone N, Kusumoto Tet al. . Purification and characterization of malate:quinone oxidoreductase from thermophilic Bacillus sp. PS3. J Bioenerg Biomembr. 2013;45:131–6. [DOI] [PubMed] [Google Scholar]
- Kagawa T, Bruno PL. NADP-malate dehydrogenase from leaves of Zea mays: Purification and physical, chemical, and kinetic properties. Arch Biochem Biophys. 1988;260:674–95. [DOI] [PubMed] [Google Scholar]
- Kai Y, Matsumura H, Izui K. Phosphoenolpyruvate carboxylase: three-dimensional structure and molecular mechanisms. Arch Biochem Biophys. 2003;414:170–9. [DOI] [PubMed] [Google Scholar]
- Kather B, Stingl K, van der Rest MEet al. . Another unusual type of citric acid cycle enzyme in Helicobacter pylori: the malate:quinone oxidoreductase. J Bacteriol. 2000;182:3204–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawai S, Suzuki H, Yamamoto Ket al. . Purification and characterization of a malic enzyme from the ruminal bacterium Streptococcus bovis ATCC 15352 and cloning and sequencing of its gene. Appl Environ Microbiol. 1996;62:2692–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kengen SW, de Bok FA, van Loo NDet al. . Evidence for the operation of a novel Embden-Meyerhof pathway that involves ADP-dependent kinases during sugar fermentation by Pyrococcus furiosus. J Biol Chem. 1994;269:17537–41. [PubMed] [Google Scholar]
- Klaffl S, Eikmanns BJ. Genetic and functional analysis of the soluble oxaloacetate decarboxylase from Corynebacterium glutamicum. J Bacteriol. 2010;192:2604–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knowles VL, Dennis DT, Plaxton WC. Purification of a novel pyruvate kinase from a green alga. FEBS Lett. 1989;259:130–2. [Google Scholar]
- Kobayashi K, Doi S, Negoro Set al. . Structure and properties of malic enzyme from Bacillus stearothermophilus. J Biol Chem. 1989;264:3200–5. [PubMed] [Google Scholar]
- Koendjbiharie JG, Wiersma K, van Kranenburg R. Investigating the Central Metabolism of Clostridium thermosuccinogenes. Atomi H (ed.). Appl Environ Microbiol. 2018;84:e00363–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korla K, Vadlakonda L, Mitra CK. Kinetic simulation of malate-aspartate and citrate-pyruvate shuttles in association with Krebs cycle. J Biomol Struct Dyn. 2015;33:2390–403. [DOI] [PubMed] [Google Scholar]
- Kumar S, Barth A. Phosphoenolpyruvate and Mg2+ Binding to Pyruvate Kinase Monitored by Infrared Spectroscopy. Biophys J. 2010;98:1931–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labrou NE, Clonis YD. Oxaloacetate Decarboxylase from Pseudomonas stutzeri: Purification and Characterization. Arch Biochem Biophys. 1999;365:17–24. [DOI] [PubMed] [Google Scholar]
- Lai H, Kraszewski JL, Purwantini Eet al. . Identification of pyruvate carboxylase genes in Pseudomonas aeruginosa PAO1 and development of a P. aeruginosa-based overexpression system for alpha4- and alpha4beta4-type pyruvate carboxylases. Appl Environ Microbiol. 2006;72:7785–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laivenieks M, Vieille C, Zeikus JG. Cloning, sequencing, and overexpression of the Anaerobiospirillum succiniciproducens phosphoenolpyruvate carboxykinase (pckA) gene. Appl Environ Microbiol. 1997;63:2273–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latorre-Muro P, Baeza J, Armstrong EAet al. . Dynamic Acetylation of Phosphoenolpyruvate Carboxykinase Toggles Enzyme Activity between Gluconeogenic and Anaplerotic Reactions. Mol Cell. 2018;71:718–32..e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao JC, Chao YP, Patnaik R. Alteration of the biochemical valves in the central metabolism of Escherichia coli. Ann N Y Acad Sci. 1994;745:21–34. [DOI] [PubMed] [Google Scholar]
- Lietzan AD, St. Maurice M. Functionally diverse biotin-dependent enzymes with oxaloacetate decarboxylase activity. Arch Biochem Biophys. 2014;544:75–86. [DOI] [PubMed] [Google Scholar]
- Lin M, Turpin DH, Plaxton’ WC. Pyruvate Kinase lsozymes from the Green Alga, Selenastrum minutum’ I. Purification and Physical and Immunological Characterization. 1989;269:219–27. [DOI] [PubMed] [Google Scholar]
- Liu Y, Budelier MM, Stine Ket al. . Allosteric regulation alters carrier domain translocation in pyruvate carboxylase. Nat Commun. 2018;9:1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lochmüller H, Wood HG, Davis JJ. Phosphoenolpyruvate carboxytransphosphorylase. II. Crystallization and properties. J Biol Chem. 1966;241:5678–91. [PubMed] [Google Scholar]
- Long CP, Au J, Sandoval NRet al. . Enzyme I facilitates reverse flux from pyruvate to phosphoenolpyruvate in Escherichia coli. Nat Commun. 2017;8:14316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu M, Zhou L, Stanley WCet al. . Role of the malate–aspartate shuttle on the metabolic response to myocardial ischemia. J Theor Biol. 2008;254:466–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machová I, Snášel J, Dostál Jet al. . Structural and Functional Studies of Phosphoenolpyruvate Carboxykinase from Mycobacterium tuberculosis. Cardona P-J (ed.). PLoS One. 2015;10:e0120682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madern D. Molecular evolution within the L-Malate and L-Lactate dehydrogenase super-family. J Mol Evol. 2002;54:825–40. [DOI] [PubMed] [Google Scholar]
- Malcovati M, Valentini G. [27] AMP- and fructose 1,6-bisphosphate-activated pyruvate kinases from Escherichia coli. Method Enzymol. 1982;90:170–9. [DOI] [PubMed] [Google Scholar]
- Marreiros BC, Calisto F, Castro PJet al. . Exploring membrane respiratory chains. Biochim Biophys Acta - Bioenerg. 2016;1857:1039–67. [DOI] [PubMed] [Google Scholar]
- Matsumura H, Izui K, Mizuguchi K. A novel mechanism of allosteric regulation of archaeal phosphoenolpyruvate carboxylase: a combined approach to structure-based alignment and model assessment. Protein Eng Des Sel. 2006;19:409–19. [DOI] [PubMed] [Google Scholar]
- Matte A, Tari LW, Goldie Het al. . Structure and mechanism of phosphoenolpyruvate carboxykinase. J Biol Chem. 1997;272:8105–8. [DOI] [PubMed] [Google Scholar]
- Matula TI, McDonald IJ, Martin SM. CO2 fixation by malic enzyme in a species of Micrococcus. Biochem Biophys Res Commun. 1969;34:795–802. [DOI] [PubMed] [Google Scholar]
- McCoy JG, Levin EJ, Zhou M. Structural insight into the PTS sugar transporter EIIC. Biochim Biophys Acta - Gen Subj. 2015;1850:577–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinlay JB, Shachar-Hill Y, Zeikus JGet al. . Determining Actinobacillus succinogenes metabolic pathways and fluxes by NMR and GC-MS analyses of 13C-labeled metabolic product isotopomers. Metab Eng. 2007;9:177–92. [DOI] [PubMed] [Google Scholar]
- Mertens E. ATP versus pyrophosphate: glycolysis revisited in parasitic protists. Parasitol Today. 1993;9:122–6. [DOI] [PubMed] [Google Scholar]
- Meyer FM, Gerwig J, Hammer Eet al. . Physical interactions between tricarboxylic acid cycle enzymes in Bacillus subtilis: Evidence for a metabolon. Metab Eng. 2011;13:18–27. [DOI] [PubMed] [Google Scholar]
- Miesel L, Weisbrod TR, Marcinkeviciene JAet al. . NADH dehydrogenase defects confer isoniazid resistance and conditional lethality in Mycobacterium smegmatis. J Bacteriol. 1998;180:2459–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minges A, Höppner A, Groth G. Trapped intermediate state of plant pyruvate phosphate dikinase indicates substeps in catalytic swiveling domain mechanism. Protein Sci. 2017;26:1667–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minárik P, Tomásková N, Kollárová Met al. . Malate dehydrogenases–structure and function. Gen Physiol Biophys. 2002;21:257–65. [PubMed] [Google Scholar]
- Mitsch MJ, Voegele RT, Cowie Aet al. . Chimeric Structure of the NAD(P) + - and NADP + -dependent Malic Enzymes of Rhizobium (Sinorhizobium) meliloti. J Biol Chem. 1998;273:9330–6. [DOI] [PubMed] [Google Scholar]
- Mogi T, Murase Y, Mori Met al. . Polymyxin B Identified as an Inhibitor of Alternative NADH Dehydrogenase and Malate: Quinone Oxidoreductase from the Gram-positive Bacterium Mycobacterium smegmatis. J Biochem. 2009;146:491–9. [DOI] [PubMed] [Google Scholar]
- Molenaar D, van der Rest ME, Drysch Aet al. . Functions of the membrane-associated and cytoplasmic malate dehydrogenases in the citric acid cycle of Corynebacterium glutamicum. J Bacteriol. 2000;182:6884–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molenaar D, van der Rest ME, Petrovic S. Biochemical and genetic characterization of the membrane-associated malate dehydrogenase (acceptor) from Corynebacterium glutamicum. Eur J Biochem. 1998;254:395–403. [DOI] [PubMed] [Google Scholar]
- Mullinax TR, Mock JN, McEvily AJet al. . Regulation of mitochondrial malate dehydrogenase. Evidence for an allosteric citrate-binding site. J Biol Chem. 1982;257:13233–9. [PubMed] [Google Scholar]
- Müller V. Bacterial Fermentation. Encyclopedia of Life Sciences. Chichester, UK: John Wiley & Sons, Ltd, 2008. [Google Scholar]
- Nantapong N, Kugimiya Y, Toyama Het al. . Effect of NADH dehydrogenase-disruption and over-expression on respiration-related metabolism in Corynebacterium glutamicum KY9714. Appl Microbiol Biotechnol. 2004;66:187–93. [DOI] [PubMed] [Google Scholar]
- Narayanan BC, Niu W, Han Yet al. . Structure and Function of PA4872 from Pseudomonas aeruginosa, a Novel Class of Oxaloacetate Decarboxylase from the PEP Mutase/Isocitrate Lyase Superfamily†,‡. 2007, DOI: 10.1021/BI701954P. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narindrasorasak S, Bridger WA. Phosphoenolypyruvate synthetase of Escherichia coli: molecular weight, subunit composition, and identification of phosphohistidine in phosphoenzyme intermediate. J Biol Chem. 1977;252:3121–7. [PubMed] [Google Scholar]
- Netzer R, Krause M, Rittmann Det al. . Roles of pyruvate kinase and malic enzyme in Corynebacterium glutamicum for growth on carbon sources requiring gluconeogenesis. Arch Microbiol. 2004;182:354–63. [DOI] [PubMed] [Google Scholar]
- Ng SK, Wong M, Hamilton IR. Properties of oxaloacetate decarboxylase from Veillonella parvula. J Bacteriol. 1982;150:1252–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen C., Saier M. Phylogenetic analysis of the putative phosphorylation domain in the pyruvate kinase of Bacillus stearothermophilus. Res Microbiol. 1995;146:713–9. [DOI] [PubMed] [Google Scholar]
- Noor E, Eden E, Milo Ret al. . Central carbon metabolism as a minimal biochemical walk between precursors for biomass and energy. Mol Cell. 2010;39:809–20. [DOI] [PubMed] [Google Scholar]
- Noor E, Haraldsdóttir HS, Milo Ret al. . Consistent estimation of gibbs energy using component contributionsBeard DA (ed). PLoS Comput Biol. 2013;9:e1003098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Leary B, Park J, Plaxton WC. The remarkable diversity of plant PEPC (phosphoenolpyruvate carboxylase): recent insights into the physiological functions and post-translational controls of non-photosynthetic PEPCs. Biochem J. 2011;436:15–34. [DOI] [PubMed] [Google Scholar]
- O'Leary B, Rao SK, Kim Jet al. . Bacterial-type Phosphoenolpyruvate Carboxylase (PEPC) functions as a catalytic and regulatory subunit of the novel class-2 PEPC complex of vascular plants. J Biol Chem. 2009;284:24797–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberholzer AE, Bumann M, Schneider Pet al. . Crystal structure of the phosphoenolpyruvate-binding enzyme I-Domain from the Thermoanaerobacter tengcongensis PEP: sugar phosphotransferase system (PTS). J Mol Biol. 2005;346:521–32. [DOI] [PubMed] [Google Scholar]
- Ohta S, Ishida Y, Usami S. Expression of cold-tolerant pyruvate, orthophosphate dikinase cDNA, and heterotetramer formation in transgenic maize plants. Transgenic Res. 2004;13:475–85. [DOI] [PubMed] [Google Scholar]
- Olson DG, Hörl M, Fuhrer Tet al. . Glycolysis without pyruvate kinase in Clostridium thermocellum. Metab Eng. 2016;39:169–80. [DOI] [PubMed] [Google Scholar]
- Oria-Hernández J, Riveros-Rosas H, Ramírez-Sílva L. Dichotomic phylogenetic tree of the pyruvate kinase family: K+ -dependent and -independent enzymes. J Biol Chem. 2006;281:30717–24. [DOI] [PubMed] [Google Scholar]
- Park JH, Lee SY. Metabolic pathways and fermentative production of L-aspartate family amino acids. Biotechnol J. 2010;5:560–77. [DOI] [PubMed] [Google Scholar]
- Park JO, Rubin SA, Xu Y-Fet al. . Metabolite concentrations, fluxes and free energies imply efficient enzyme usage. Nat Chem Biol. 2016;12:482–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel HM, Kraszewski JL, Mukhopadhyay B. The phosphoenolpyruvate carboxylase from Methanothermobacter thermautotrophicus has a novel structure. J Bacteriol. 2004;186:5129–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel H V, Vyas KA, Savtchenko Ret al. . The monomer/dimer transition of enzyme I of the Escherichia coli phosphotransferase system. J Biol Chem. 2006;281:17570–8. [DOI] [PubMed] [Google Scholar]
- Pawluk A, Scopes RK, Griffiths-Smith K. Isolation and properties of the glycolytic enzymes from Zymomonas mobilis. The five enzymes from glyceraldehyde-3-phosphate dehydrogenase through to pyruvate kinase. Biochem J. 1986;238:275–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters-Wendisch PG, Wendisch VF, Paul Set al. . Pyruvate carboxylase as an anaplerotic enzyme in Corynebacterium glutamicum. Microbiology. 1997;143:1095–103. [DOI] [PubMed] [Google Scholar]
- Pickl A, Johnsen U, Schonheit P. Fructose Degradation in the Haloarchaeon Haloferax volcanii Involves a Bacterial Type Phosphoenolpyruvate-Dependent Phosphotransferase System, Fructose-1-Phosphate Kinase, and Class II Fructose-1,6-Bisphosphate Aldolase. J Bacteriol. 2012;194:3088–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pircher H, von Grafenstein S, Diener Tet al. . Identification of FAH domain-containing protein 1 (FAHD1) as oxaloacetate decarboxylase. J Biol Chem. 2015;290:6755–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plaxton WC, Smith CR, Knowles VL. Molecular and regulatory properties of leucoplast pyruvate kinase from Brassica napus (rapeseed) suspension cells. Arch Biochem Biophys. 2002;400:54–62. [DOI] [PubMed] [Google Scholar]
- Plaxton WC. Molecular and immunological characterization of plastid and cytosolic pyruvate kinase isozymes from castor-oil-plant endosperm and leaf. Eur J Biochem. 1989;181:443–51. [DOI] [PubMed] [Google Scholar]
- Pudlik AM, Lolkema JS. Mechanism of citrate metabolism by an oxaloacetate decarboxylase-deficient mutant of Lactococcus lactis IL1403. J Bacteriol. 2011;193:4049–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeves RE. A new enzyme with the glycolytic function of pyruvate kinase. J Biol Chem. 1968;243:3202–4. [PubMed] [Google Scholar]
- Repizo GD, Blancato VS, Mortera Pet al. . Biochemical and Genetic Characterization of the Enterococcus faecalis Oxaloacetate Decarboxylase Complex. Appl Environ Microbiol. 2013;79:2882–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivero LA, Concepción JL, Quintero-Troconis Eet al. . Trypanosoma evansi contains two auxiliary enzymes of glycolytic metabolism: Phosphoenolpyruvate carboxykinase and pyruvate phosphate dikinase. Exp Parasitol. 2016;165:7–15. [DOI] [PubMed] [Google Scholar]
- Ronimus RS, Morgan HW. Distribution and phylogenies of enzymes of the Embden-Meyerhof-Parnas pathway from archaea and hyperthermophilic bacteria support a gluconeogenic origin of metabolism. Archaea. 2003;1:162593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz-Ballesta I, Baena G, Gandullo Jet al. . New insights into the post-translational modification of multiple phosphoenolpyruvate carboxylase isoenzymes by phosphorylation and monoubiquitination during sorghum seed development and germination. J Exp Bot. 2016;67:3523–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saavedra-Lira E, Ramirez-Silva L, Perez-Montfort R. Expression and characterization of recombinant pyruvate phosphate dikinase from Entamoeba histolytica. Biochim Biophys Acta - Protein Struct Mol Enzymol. 1998;1382:47–54. [DOI] [PubMed] [Google Scholar]
- Saavedra E, Encalada R, Vázquez Cet al. . Control and regulation of the pyrophosphate-dependent glucose metabolism in Entamoeba histolytica. Mol Biochem Parasitol. 2019;229:75–87. [DOI] [PubMed] [Google Scholar]
- Sakai H, Suzuki K, Imahori3 K. Purification and properties of pyruvate kinase from Bacillus stearothermophilus. J Biochem. 1986;99:1157–67. [DOI] [PubMed] [Google Scholar]
- Sakuraba H, Ohshima T. Novel energy metabolism in anaerobic hyperthermophilic archaea: a modified Embden-Meyerhof pathway. J Biosci Bioeng. 2002;93:441–8. [DOI] [PubMed] [Google Scholar]
- Sakuraba H, Utsumi E, Kujo Cet al. . An AMP-Dependent (ATP-Forming) Kinase in the Hyperthermophilic ArchaeonPyrococcus furiosus:Characterization and Novel Physiological Role. Arch Biochem Biophys. 1999;364:125–8. [DOI] [PubMed] [Google Scholar]
- Sakuraba H, Utsumi E, Schreier HJet al. . Transcriptional regulation of phosphoenolpyruvate synthase by maltose in the hyperthermophilic archaeon, Pyrococcus furiosus. J Biosci Bioeng. 2001;92:108–13. [DOI] [PubMed] [Google Scholar]
- Sauer U, Eikmanns BJ. The PEP—pyruvate—oxaloacetate node as the switch point for carbon flux distribution in bacteria: We dedicate this paper to Rudolf K. Thauer, Director of the Max-Planck-Institute for Terrestrial Microbiology in Marburg, Germany, on the occasion of his 65th. FEMS Microbiol Rev. 2005;29:765–94. [DOI] [PubMed] [Google Scholar]
- Schaftingen E, Opperdoes FR, Hers H-G. Stimulation of Trypanosoma brucei pyruvate kinase by fructose 2,6-bisphosphate. Eur J Biochem. 1985;153:403–6. [DOI] [PubMed] [Google Scholar]
- Scheibe R, Jacquot J-P. NADP regulates the light activation of NADP-dependent malate dehydrogenase. Planta. 1983;157:548–53. [DOI] [PubMed] [Google Scholar]
- Schramm A, Siebers B, Tjaden Bet al. . Pyruvate kinase of the hyperthermophilic crenarchaeote Thermoproteus tenax: physiological role and phylogenetic aspects. J Bacteriol. 2000;182:2001–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schöcke L, Weimer PJ. Purification and characterization of phosphoenolpyruvate carboxykinase from the anaerobic ruminal bacterium Ruminococcus flavefaciens. Arch Microbiol. 1997;167:289–94. [DOI] [PubMed] [Google Scholar]
- Scrutton MC, Taylor BL. Isolation and characterization of pyruvate carboxylase from Azotobacter vinelandii OP. Arch Biochem Biophys. 1974;164:641–54. [DOI] [PubMed] [Google Scholar]
- Sender PD, Martín MG, Peirú Set al. . Characterization of an oxaloacetate decarboxylase that belongs to the malic enzyme family. FEBS Lett. 2004;570:217–22. [DOI] [PubMed] [Google Scholar]
- Shirahashi K, Hayakawa S, Sugiyama T. Cold lability of pyruvate, orthophosphate dikinase in the maize leaf. Plant Physiol. 1978;62:826–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slamovits CH, Keeling PJ. Pyruvate-phosphate dikinase of oxymonads and parabasalia and the evolution of pyrophosphate-dependent glycolysis in anaerobic eukaryotes. Eukaryot Cell. 2006;5:148–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyer JR, Jeter RM. Characterization of phosphoenolpyruvate synthase mutants in Salmonella typhimurium. Arch Microbiol. 1989;153:26–32. [DOI] [PubMed] [Google Scholar]
- Solomons JTG, Johnsen U, Schönheit Pet al. . 3-Phosphoglycerate Is an Allosteric Activator of Pyruvate Kinase from the Hyperthermophilic Archaeon Pyrobaculum aerophilum. Biochemistry. 2013;52:5865–75. [DOI] [PubMed] [Google Scholar]
- Somavanshi R, Ghosh B, Sourjik V. Sugar Influx Sensing by the Phosphotransferase System of Escherichia coli. Stock A (ed.). PLoS Biol. 2016;14:e2000074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spahich NA, Vitko NP, Thurlow LRet al. . Staphylococcus aureus lactate- and malate-quinone oxidoreductases contribute to nitric oxide resistance and virulence. Mol Microbiol. 2016;100:759–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St Maurice M, Reinhardt L, Surinya KHet al. . Domain architecture of pyruvate carboxylase, a biotin-dependent multifunctional enzyme. Science. 2007;317:1076–9. [DOI] [PubMed] [Google Scholar]
- Studer R, Dahinden P, Wang W-Wet al. . Crystal structure of the carboxyltransferase domain of the oxaloacetate decarboxylase Na+ pump from Vibrio cholerae. J Mol Biol. 2007;367:547–57. [DOI] [PubMed] [Google Scholar]
- Sundaram TK, Cazzulo JJ, Kornberg HL. Synthesis of pyruvate carboxylase from its apoenzyme and (+)-biotin in Bacillus stearothermophilus. Mechanism and control of the reaction. Biochem J. 1971;122:663–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suye S-I, Okada Y, Funada Aet al. . Purification and properties of malic enzyme from Pseudomonas diminuta IFO-13182. J Ferment Bioeng. 1992;73:343–7. [Google Scholar]
- Taillefer M, Rydzak T, Levin DBet al. . Reassessment of the Transhydrogenase/Malate Shunt Pathway in Clostridium thermocellum ATCC 27405 through Kinetic Characterization of Malic Enzyme and Malate Dehydrogenase. Nojiri H (ed.). Appl Environ Microbiol. 2015;81:2423–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi-Íñiguez T, Aburto-Rodríguez N, Vilchis-González ALet al. . Function, kinetic properties, crystallization, and regulation of microbial malate dehydrogenase. J Zhejiang Univ B. 2016;17:247–61. [Google Scholar]
- Takeya M, Hirai MY, Osanai T. Allosteric inhibition of phosphoenolpyruvate carboxylases is determined by a single amino acid residue in Cyanobacteria. Sci Rep. 2017;7:41080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao X, Yang Z, Tong L. Crystal structures of substrate complexes of malic enzyme and insights into the catalytic mechanism. Structure. 2003;11:1141–50. [DOI] [PubMed] [Google Scholar]
- Thakker C, Martínez I, San K-Yet al. . Succinate production in Escherichia coli. Biotechnol J. 2012;7:213–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tjaden B, Plagens A, Dorr Cet al. . Phosphoenolpyruvate synthetase and pyruvate, phosphate dikinase of Thermoproteus tenax: key pieces in the puzzle of archaeal carbohydrate metabolism. Mol Microbiol. 2006;60:287–98. [DOI] [PubMed] [Google Scholar]
- Tolentino R, Chastain C, Burnell J. Identification of the amino acid involved in the regulation of bacterial pyruvate, orthophosphate dikinase and phosphoenolpyruvate synthetase. Adv Biol Chem. 2013;3:12–21. [Google Scholar]
- Tong L. Structure and function of biotin-dependent carboxylases. Cell Mol Life Sci. 2013;70:863–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tronconi MA, Andreo CS, Drincovich MF. Chimeric structure of plant malic enzyme family: different evolutionary scenarios for NAD- and NADP-dependent isoforms. Front Plant Sci. 2018;9:565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tronconi MA, Fahnenstich H, Gerrard Weehler MCet al. . Arabidopsis NAD-malic enzyme functions as a homodimer and heterodimer and has a major impact on nocturnal metabolism. Plant Physiol. 2008;146:1540–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valentini G, Chiarelli L, Fortin Ret al. . The allosteric regulation of pyruvate kinase. J Biol Chem. 2000;275:18145–52. [DOI] [PubMed] [Google Scholar]
- van der Rest ME, Frank C, Molenaar D. Functions of the Membrane-Associated and Cytoplasmic Malate Dehydrogenases in the Citric Acid Cycle of Escherichia coli. J Bacteriol. 2000;182:6892–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voegele RT, Mitsch MJ, Finan TM. Characterization of two members of a novel malic enzyme class. Biochim Biophys Acta - Protein Struct Mol Enzymol. 1999;1432:275–85. [DOI] [PubMed] [Google Scholar]
- Wang S, Huang H, Moll Jet al. . NADP+ reduction with reduced ferredoxin and NADP+ reduction with NADH are coupled via an electron-bifurcating enzyme complex in Clostridium kluyveri. J Bacteriol. 2010;192:5115–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber G. Regulation of pyruvate kinase. Adv Enzyme Regul. 1969;7:15–40. [DOI] [PubMed] [Google Scholar]
- Weiss MC, Sousa FL, Mrnjavac Net al. . The physiology and habitat of the last universal common ancestor. Nat Microbiol. 2016;1:16116. [DOI] [PubMed] [Google Scholar]
- Xu X, Shi H, Gong Xet al. . Structural insights into sodium transport by the oxaloacetate decarboxylase sodium pump. Elife. 2020;9:1–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y-F, Amador-Noguez D, Reaves MLet al. . Ultrasensitive regulation of anapleurosis via allosteric activation of PEP carboxylase. Nat Chem Biol. 2012;8:562–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada T, Carlsson J. Glucose-6-phosphate-dependent pyruvate kinase in Streptococcus mutans. J Bacteriol. 1975;124:562–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Kalhan SC, Hanson RW. What Is the metabolic role of phosphoenolpyruvate carboxykinase? J Biol Chem. 2009;284:27025–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying S, Hill AT, Pyc Met al. . Regulatory phosphorylation of bacterial-type PEP carboxylase by the Ca2+-dependent protein kinase RcCDPK1 in developing castor oil seeds. Plant Physiol. 2017;174:1012–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeczycki TN, Maurice MS, Attwood PV. Inhibitors of pyruvate carboxylase. Open Enzym Inhib J. 2010;3:8–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong W, Morgan HP, Nowicki MWet al. . Pyruvate kinases have an intrinsic and conserved decarboxylase activity. Biochem J. 2014;458:301–11. [DOI] [PubMed] [Google Scholar]
- Zhou J, Olson DG, Argyros Daet al. . Atypical glycolysis in Clostridium thermocellum. Appl Environ Microbiol. 2013;79:3000–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.