

Summary
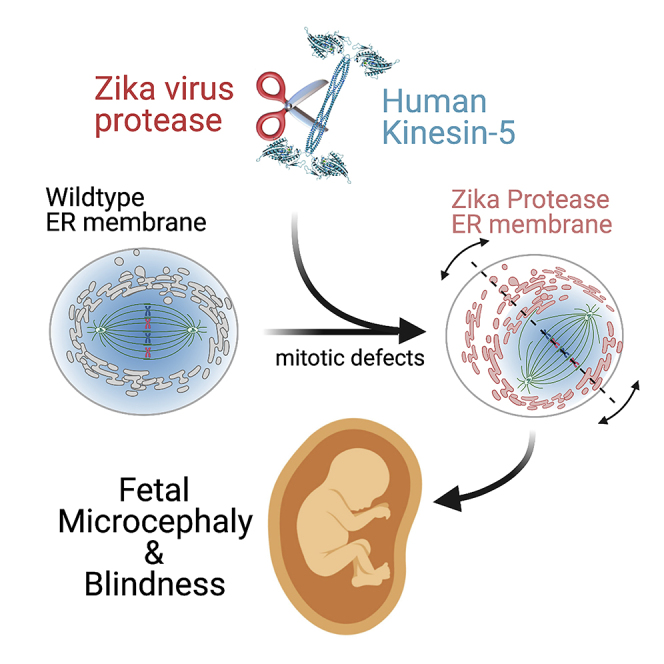
Here we identify human Kinesin-5, Kif11/HsEg5, as a cellular target of Zika protease. We show that Zika NS2B-NS3 protease targets several sites within the motor domain of HsEg5 irrespective of motor binding to microtubules. The native integral ER-membrane protease triggers mitotic spindle positioning defects and a prolonged metaphase delay in cultured cells. Our data support a model whereby loss of function of HsEg5 is mediated by Zika protease and is spatially restricted to the ER-mitotic spindle interface during mitosis. The resulting phenotype is distinct from the monopolar phenotype that typically results from uniform inhibition of HsEg5 by RNAi or drugs. In addition, our data reveal novel inter-organelle interactions between the mitotic apparatus and the surrounding reticulate ER network. Given that Kif11 is haplo-insufficient in humans, and reduced dosage results in microcephaly, we propose that Zika protease targeting of HsEg5 may be a key event in the etiology of Zika syndrome microcephaly.
Subject areas: Developmental Genetics, Virology, Cytoskeleton
Graphical abstract
Highlights
-
•
Zika protease cleavage of Kinesin-5 impairs mitotic progression
-
•
Inter-organelle interactions spatially control Zika proteolysis of Kinesin-5
-
•
Native Zika protease affects mitosis differently than soluble Zika protease
-
•
Zika protease may elicit fetal microcephaly and blindness via Kif11/Kinesin-5
Developmental Genetics; Virology; Cytoskeleton
Introduction
Zika virus has become an emerging global health threat after it was first linked to neurodevelopmental defects, including microcephaly, in the developing fetus during pregnancy in 2016 (Lazear and Diamond, 2016; Mlakar et al., 2016). Transmitted by mosquitos, the virus was endemic to the African continent, but recently spread to South America wherein infection of pregnant mothers quickly became linked to congenital microcephaly and other developmental defects in infants. As a member of the Flavivirus genus, Zika is comprised of a positive single-stranded RNA genome. The RNA is translated, within infected cells, as a single polyprotein that must be cleaved by a combination of host and viral proteases to release three structural and seven non-structural proteins. With this limited set of proteins, Zika must co-opt cellular pathways in order to generate additional virus particles.
The viral polyprotein is expressed as a multi-pass integral protein of the ER membrane within infected cells. Specific loops of the polyprotein within the ER lumen are cleaved by native furin proteases, whereas cytoplasmic-side cleavage sites are cleaved by the virus-encoded protease formed as a heterodimer between nonstructural protein 2B and the N-terminal domain of nonstructural protein 3 (NS2B-NS3). The cytoplasmic-side viral proteins, including the C-terminal helicase domain of NS3 together with ER-modifying NS4 and the RNA-dependent RNA polymerase NS5, together initiate virion replication (Shiryaev et al., 2017; Gruba et al., 2016; Shan et al., 2016; Tsetsarkin et al., 2016; Bera et al., 2007; Mukhopadhyay et al., 2005).
Viruses must hijack a number of cell processes and pathways that are designed to suppress immune responses and support virion replication. Viral proteins directly interact with cellular components, via protein-protein interactions, to exert this control. In the case of Zika, with its limited proteome, the NS2B-NS3 protease is known to cleave host proteins (Golubeva et al., 2020; Hill et al., 2018; Coyaud et al., 2018; Shah et al., 2018; Scaturro et al., 2018), whereas both the NS4 and NS5 proteins collaborate to inhibit the establishment of cellular antiviral state by blocking the interferon (IFN)-α/β pathway (Liang et al., 2016; Dar et al., 2017; Grant et al., 2016; Kumar et al., 2016). Numerous recent studies have documented that Zika virus can impair neurogenesis and interfere with the proliferation of neuronal precursor cells (Ferreira and Garcez, 2019; Yoon et al., 2017; Zhu et al., 2017; Solomon et al., 2016; Hughes et al., 2016; Qian et al., 2016; Ghosh Roy et al., 2014). Furthermore, cell proliferation impacts of the virus are linked to mitotic defects, including aberrant spindle orientation (McDougall et al., 2019). Collectively, these studies argue that negative impacts on neuronal cell proliferation and mitotic machinery by Zika virus is the most likely pathway in the etiology of microcephaly and associated effects.
Kif11 kinesin is a haploinsufficient locus in humans and a known monogenic cause of microcephaly and retinopathies in developing infants (Wang et al., 2020; Rososinski et al., 2019; Güneş et al., 2019; Karjosukarso et al., 2018; Balikova et al., 2016; Hu et al., 2016; Li et al., 2016; Robitaille et al., 2014; Jones et al., 2014; Mirzaa et al., 2014; Ostergaard et al., 2012; Hazan et al., 2012). Due to broad similarities between Zika syndrome and congenital mutations in Kif11 kinesin, we looked for a potential direct link between the two. Kif11 encodes human Kinesin-5 (HsEg5), which is a homotetrameric microtubule motor protein that is required for the assembly of the mitotic spindle (Mann and Wadsworth, 2019; Wojcik et al., 2013). HsEg5 loss of function results in the failure to assemble a bipolar microtubule spindle, metaphase arrest, and catastrophic inability to segregate chromosomes during mitosis (Mann and Wadsworth, 2019; Saeki et al., 2018; Liu et al., 2014; Wojcik et al., 2013; Brust-Mascher et al., 2009; Goshima and Vale, 2005; Cochran et al., 2004; DeBonis et al., 2003, 2004; Mayer et al., 1999; Sawin and Mitchison, 1994; Heck et al., 1993). However, there is also evidence that HsEg5 plays a distinct role in neuronal development and axonal pathfinding (Freund et al., 2016; Kahn et al., 2015; Nadar et al., 2008; Myers and Baas, 2007). The motor also has the ability to modulate microtubule stability in cells (Kim et al., 2019). It is not known which of these functions, together or separately, are responsible for haploinsufficiency of this locus in humans or are causative for Kif11-associated microcephaly and blindness. In this study we focus on the well-characterized requirement for HsEg5 in cell proliferation, rather than on other putative non-mitotic roles for this motor, as its mitotic role is most relevant to the cell proliferation defects thought to directly contribute to Zika microcephaly.
Recent genomic analysis reveals a variety of mutations in Kif11, including C-terminal truncations, and motor domain point mutations that result in potential poison oligomers or loss of an HsEg5 allele are sufficient by themselves to result in both microcephaly and chorioretinopathies in infants (Hu et al., 2016; Jones et al., 2014; Ostergaard et al., 2012). The mechanism by which Zika virus (ZIKV) disrupts neuronal development and results in congenital Zika syndrome is unknown. It is believed that host-pathogen connections that are mediated by viral protein interactions with host proteins result in the disruption in either neuroblast cell division or differentiation. In this regard, the Zika NS2B-NS3 protease complex (ZPrC) has been proposed to be a key player in this mechanism with several studies uncovering numerous critical host protein targets within infected cells. Recent unbiased proteomics studies centered on protein-protein interactions of ZPrC have identified largely non-overlapping sets of host proteins that are either found in association with the protease in cells (Golubeva et al., 2020; Coyaud et al., 2018; Shah et al., 2018; Scaturro et al., 2018) or able to be cleaved by recombinant soluble protease chimera (s-ZPrC) that does not contain the transmembrane components of the NS2B moiety (Li et al., 2019; Hill et al., 2018; Ding et al., 2018; Lennemann and Coyne, 2017). A subset of these suspected targets, including several centriolar proteins, are suspected to be linked to the development of microcephaly in humans (Wen et al., 2019; Wolf et al., 2017). More recently, mitotic septins were found in association with a tagged Zika protease chimera and shown to be cleaved by the protease in vitro (Li et al., 2019; Ferreira and Garcez, 2019). In vitro cleavage of the septins by exogenously expressed protease chimera resulted in the occurrence of binucleate cells that marked the first observation of a mitotic defect caused by the Zika protease in living cells. However, loss of function in septins has not been linked to microcephaly or other developmental neurological defects in human infants. An important caveat of these protease-specific proteomic studies is that they all rely on expression of the tractable recombinant form of the protease comprised of a soluble segment of the NS2B integral membrane protein fused to the globular protease domain of NS3. Although this chimeric construct is widely used for in vitro studies of structure, enzyme kinetics, and specificity (Li et al., 2019; Hill et al., 2018; Lee et al., 2017; Rut et al., 2017; Gruba et al., 2016; Phoo et al., 2016; Lei et al., 2016), being generally expressed in the cytoplasm, they are unlikely to faithfully recapitulate the protein-protein interactions of the native ER-membrane-associated protease.
Here we report the development of a Zika protease toolkit including both an innovative ZPrC expression construct that more closely resembles the native complex in cells and a cellular reporter of active Zika protease. Based on a segment of the Zika genomic RNA sequence, we develop an NS2B-NS3 protease that is natively ER membrane bound by NS2B, but with a C-terminal truncation in NS3 that removes the helicase domain. This protease construct recapitulates the native ER organization of the viral protease complex in cells and exhibits proteolytic activity. Moreover, the ER-bound protease is able to target and cleave a soluble reporter mimic of HsEg5 kinesin. Our data support a mechanism whereby the Zika protease reduces HsEg5 levels within infected cells. Furthermore, we propose the ER-tethered protease mediates a spatially nonuniform proteolysis of HsEg5 in cells. In this regard, we have uncovered novel inter-organellar communication between viral components in the ER membrane and the cytoskeletal mitotic spindle. Taken together, this effect is expected to negatively impact the native functions of HsEg5 during cell division and thereby trigger the downstream impacts on fetal neuronal development that are characteristic of Kif11 haploinsufficiency. This work provides the first molecular and cellular evidence supporting a direct connection between Zika syndrome and Kif11-mediated microcephaly and blindness.
Results
Zika-encoded protease readily cleaves sites within the motor domain of HsEg5
To determine whether the Zika NS2B-NS3 protease complex (ZPrC) could cause cell proliferation defects by directly targeting HsEg5, we first identified potential ZPrC cleavage sites in the motor by searching for matches to its cleavage consensus (Hill et al., 2018; Rut et al., 2017; Gruba et al., 2016). There are ten potential cleavage sites that match the consensus, and they are distributed across all domains (Figure 1) of the motor. In addition, the most preferred sequence of the protease favors a P1-position Arg instead of Lys, which occurs at three positions in HsEg5 concentrated in the motor domain (Figure 1, red sites). Of these sites, only one occurs in a surface loop that is predicted to be readily accessible to the protease (motor domain loop8, Figure 1). We tested whether the catalytic center of HsEg5, comprised of the motor domain, is susceptible to cleavage by ZPrC. Soluble recombinant ZPrC (s-ZPrC) and inactivated S135A-s-ZPrC (Figure S4) were challenged to hydrolyze recombinant HsEg5 motor domain with in vitro assays. After a 24-h incubation, we find by SDS-PAGE that s-ZPrC cleaves the HsEg5 motor domain at a single primary site followed by weaker cleavage targeting at least two additional sites (Figure 2). As previously reported, S135A-s-ZPrC exhibited no detectable protease activity (not shown) (Li et al., 2019; Hill et al., 2018; Shiryaev et al., 2017). To identify the primary site of cleavage, the distinct 20.4 kDa band (Figure 2, cyan arrow) was excised and sequenced by quantitative LC-MS (Figure S1). The primary site of cleavage maps to loop8 of the motor domain at the sequence DPRNKRˆG (Figure 1) as predicted by the consensus. Subsequent partial cleavage of the C-terminal 20.4 kDa fragment releases 9.7 and 10.7 kDa fragments that are visible in the SDS-PAGE (Figure 2, asterisk). These smaller fragments are consistent with the protease targeting Switch 2 of the motor domain with cleavage at GAVDKRˆA. The s-ZPrC construct with C-terminal 29.9 kDa Tag-RFP moiety (Figure S5) shifts the relative position of the 10.7 kDa fragment to 40.5 kDa in SDS-PAGE and thereby identifies it as the C-terminal peptide of HsEg5 motor domain (Figure S2).
Figure 1.

List of potential ZPrC cleavage sites and corresponding secondary structure elements within HsEg5 ranked in order of decreasing likelihood of cleavage
Mass spectrometry analysis confirms in vitro cleavage by ZPrC of the sites marked in red. AMPPNP is shown in the active site in stick view, whereas the primary landmark MT-binding element of the motor domain, alpha helix4, is shown in cyan.
Figure 2.

In vitro hydrolysis of HsEg51-367 motor domain by s-ZPrC
Recombinant soluble s-ZPrC (27 kDa) was incubated with purified HsEg5 motor domain (42 kDa) for 24 h. s-ZPrC exhibited a low level of self-cleavage after 24 h marked primarily by a 23 kDa fragment. After 24 h, HsEg5 was cut into 21.7 kDa N-terminal (red arrow) and 20.4 kDa C-terminal (cyan arrow) fragments. In turn, the 20.4 kDa fragment was incompletely cut into 9.7 kDa and 10.7 kDa fragments (asterisk). The 20.4 kDa fragment was excised for sequencing to identify the primary s-ZPrC cleavage site (Figure 3).
To assess whether or not the primary s-ZPrC cleavage site within the HsEg5 motor domain qualifies as an efficient target for the protease, we measured the kinetics of hydrolysis of a target peptide derived from the loop8 cleavage site relative to known ZPrC target sequences. We chose two labeled peptide control sequences that were derived from known ZPrC cleavage sites within the Zika polyprotein including the NS3-NS4A and NS4B-NS5 junctions and compared kinetic parameters with an HsEg5 loop8-derived labeled peptide of the confirmed cleavage site (Figure 3, panel A). The loop8-derived peptide exhibited comparable kinetic parameters, including Km, kcat, and catalytic specificity (kcat/Km), to the peptides derived from endogenous Zika cut sites (Figure 3, panel B).
Figure 3.

Kinetic parameters of the hydrolysis of p-nitroanilide-labeled peptides by s-ZPrC
(A) Two labeled peptides based on native ZPrC polyprotein cleavage sites were challenged for proteolysis by s-ZPrC: Ac-FAAGKR-pNA, based on the NS3-NS4A junction, and Ac-GLVKRR-pNA, based on the NS4B-NS5 junction. Three replicate sets of data from each peptide were obtained using 0.1 μM enzyme. Error = ± S.D.
(B) The HsEg5 loop8 peptide, Ac-DPRNKR-pNA, exhibited comparable kinetics of hydrolysis to the native-derived peptides. Values are ± S.D. The overall s-ZPrC enzyme kinetics were similar to those already reported in the literature (Hill et al., 2018; Gruba et al., 2016; Lei et al., 2016; Leung et al., 2001).
Although these proteolysis and hydrolysis results find the HsEg5 motor domain to be efficiently targeted by s-ZPrC in vitro, it is possible that the protease may be sterically blocked from access to the loop8 target site during allosteric structural changes known to occur in the motor during cycles of ATP hydrolysis or in the context of motor-microtubule interactions.
We first tested whether continuous low basal ATPase activity of the motor that is elicited with saturating levels of ATP would prevent cleavage by soluble s-ZPrC. We found that the protease was able to efficiently cut the motor domain in this context (Figure 4, ATP lanes), with no reproducible inhibition of proteolysis (not shown). Given that both co-crystal structures and cryo-EM analyses of kinesins bound to tubulin indicate a potential role for the loop8 segment of the motor to interact directly with the microtubule lattice (Peña et al., 2020; Bodey et al., 2009), we sought to test whether this interaction would impact the ability of s-ZPrC to target loop8. After adding Taxol-stabilized microtubules to the reaction plus ATP, we observed no appreciable change in the susceptibility of the motor to cleavage by s-ZPrC (Figure 4, MT + ATP) and no evidence of significant proteolysis of microtubules by the protease. In the presence of ATP, the motor domain is expected to continuously cycle between both low- and high-affinity states and interactions with microtubules, but the weakly hydrolyzable nucleotide analog, AMPPNP, instead traps the motor in a long-lived state that is tightly bound to the microtubule lattice (Chang et al., 2013; van den Wildenberg et al., 2008; Hirose et al., 2006; Alonso et al., 1998; Lockhart and Cross, 1996). We found that when the motor is rigor-bound to microtubules, loop8 remains accessible and readily cleavable by s-ZPrC (Figure 4, MT + AMPPNP). After 24 h, we observed the expected two segments that are released upon cleavage of the loop8 site, as well as lesser amounts of subsequent lower-molecular-weight cleavage products. Taken together, neither the expected motor allosteric structural changes nor motor-microtubule interactions prevented cleavage of the motor at loop8 by soluble s-ZPrC in vitro.
Figure 4.

Microtubule-bound HsEg5 motor domain is not protected from hydrolysis by s-ZPrC
HsEg5 motor domain was incubated with or without taxol-stabilized microtubules and in the presence of either AMPPNP or ATP. HsEg5 motor domain plus ATP in the absence of microtubules was readily hydrolyzed by s-ZPrC (ATP). The addition of microtubules to the motor plus ATP (MT + ATP), which would promote transient motor/microtubule complexes, did not protect the motor from hydrolysis by s-ZPrC. The formation of long-term stable motor/microtubule complexes by AMPPNP (Wang et al., 2017; Gigant et al., 2013) also did not protect the motor from hydrolysis by s-ZPrC. Note that cleavage products attributable to tubulin were not observed over the time span of this experiment.
A fusion protein comprised of the native two-component NS2B-NS3 proteins with an additional C-terminal eGFP localizes selectively to the ER membrane in cells
Prior studies designed to identify and confirm cellular targets of Zika protease commonly utilize a well-characterized chimera fusion between a soluble loop from NS2B transmembrane component and the NS3 protease domain that expresses as a soluble active protease in cells (Yamaoka et al., 2021; De Jesús-González et al., 2020; Li et al., 2019; Hill et al., 2018; Coyaud et al., 2018; Shah et al., 2018; Scaturro et al., 2018). More recent proteomic analyses expressed the NS3 component in the absence of the NS2B subunit (Golubeva et al., 2020; Coyaud et al., 2018). In our hands, the NS2B-NS3 chimera, s-ZPrC, exhibited toxicity in cells and cell death of the majority of transfected cells, similar to that observed in previous reports (not shown) (Arias-Arias et al., 2020; Li et al., 2019). At 24 h post-transfection, HEK293-T cells exhibited a strong metaphase block in cell division. The relative degree of this block is quantified by an abnormal ratio of metaphase to anaphase cells (M/A ratio) in the population. Although control HEK293-T cells transfected with labeled histone, mScarlet-H2A, exhibit an M/A ratio of 2.8 ± 0.3, this ratio shifts to 11.0 ± 1.2 in cells co-transfected with s-ZPrC-eGFP, indicating a strong block to transition out of metaphase during cell division.
We reasoned that expression of the freely soluble variant of the ZPrC that we utilized in our in vitro protease assays (s-ZPrC) would not accurately reflect the biology of the native enzyme in cells, which is compartmentalized and prevented from free diffusion by its integral ER-membrane protein component, NS2B, which is essential for its activity (Hill et al., 2018; Shiryaev et al., 2017; Phoo et al., 2016; Chen et al., 2016; Lei et al., 2016; de la Cruz et al., 2014; Kim et al., 2013; Robin et al., 2009; D'Arcy et al., 2006; Yusof et al., 2000; Falgout et al., 1991; Gupta et al., 2015). To overcome these issues and to investigate the cellular impact of expression of the Zika protease on endogenous HsEg5 function, we developed a transfection construct that expresses the native ER-resident, (two-component), Zika protease that matures in the ER membrane, n-ZPrC, which is identical to the virally expressed version.
To generate this construct, we excised a portion of the Zika genome DNA that encodes a segment that begins from the native polyprotein cleavage site between NS2A-NS2B and continues into the linker segment separating the NS3 peptidase domain from the downstream helicase domain (Figure 5). Given the predicted structure of the NS2B integral membrane component, based on NMR data of the homologous Dengue virus NS2B (Li et al., 2015), we expect n-ZPrC to insert within the ER membrane with the correct topology. We added a C-terminal eGFP tag to the NS3 component to follow the subcellular localization of the chimeric protein. The n-ZPrC-GFP cassette (Figure S6) was cloned into the transfection vector pcDNA3.1 and driven by the included CMV promoter post-transfection.
Figure 5.

Schematic of the Zika polyprotein showing the segment utilized to create the n-ZPrC-eGFP expression construct
The Zika polyprotein coding region from the NS2A-NS2B processing cleavage site (from residue # 1,369) through to the linker region between the NS3 peptidase domain (residue # 1,683) was synthesized (blue feature, Native NS2B-NS3) in-frame with C-terminal eGFP and cloned into the pcDNA3.1 transfection vector. Expressed under the control of the CMV promoter, the fusion protein exhibited ER-specific subcellular localization.
If n-ZPrC-GFP behaves similarly to the native protease, then it is expected to exhibit several testable characteristics or attributes of the native enzyme. First, we expect n-ZPrC-GFP to exhibit subcellular localization that is restricted to the ER. Although a canonical ER retention signal is not apparent in either the native or n-ZPrC-GFP sequence, the native protease complex exhibits a well-characterized specific localization to, and retention within, the ER membrane (Xing et al., 2020; Mohd Ropidi et al., 2020; Cortese et al., 2017). However, the signal for the ER retention of the native NS2B-NS3 protease complex is unclear. To test the extent of subcellular localization of n-ZPrC-GFP, we looked for colocalization with known ER markers in living cells. We chose two well-characterized markers that localize to the ER. First, we generated a mScarlet-labeled cytochrome b5 reductase chimera. Cytochrome b5 reductase is a well-characterized resident ER integral membrane protein (Borgese et al., 1993; Mitoma and Ito, 1992; Hanlon et al., 2000). Comprised of an exposed cytoplasmic catalytic domain and a transmembrane ER anchor helix (b5anchor), we substituted mScarlet (Bindels et al., 2017) for the catalytic domain of the reductase to generate an ER fluorescent-marker expression-construct in pcDNA3.1 (Figure S7). Cotransfection of mScarlet-b5anchor, together with n-ZPrC-GFP into cultured animal cells, exhibited unambiguous colocalization with no indication of n-ZPrC-GFP localization outside the ER (Figure 6, Video S1). We observed similar results (not shown) upon cotransfection of n-ZPrC-GFP together with an ER-specific cytochrome P450, pCytERM-mScarlet (Bindels et al., 2017). Furthermore, we built an alternative ER-tethered NS2-NS3 protease by adding the soluble chimeric form of the NS2-NS3 protease, as utilized in our in vitro assays, as an N-terminal component of a TagRFP-b5anchor (Figure S5). We also tested this form of the protease, s-ZPrC-TagRFP-ER, for activity against the HsEg5 biosensor (below, Figure S3).
Figure 6.

The subcellular localization of ectopic n-ZPrC-eGFP in HeLa cells is restricted to the endoplasmic reticulum (ER)
HeLa cells were transiently co-transfected with n-ZPrC-eGFP (A, green channel) and mScarlet-b5anchor ER marker (B, red channel) and examined by confocal microscopy. n-ZPrC-eGFP exhibits consistent colocalization with mScarlet-b5anchor and is only found associated with the ER membranes (C, merge).
The cells exhibit discrete ER localization of the protease. Asterisk marks a dividing cell in prolonged metaphase whose spindle appears to undergo abnormal rotations prior to anaphase onset. Time shown in min:sec.
Expression of peptidase-inactive S135A-n-ZPrC-RFP670 in cultured cells does not impact cell-cycle progression
If endogenous HsEg5 is a target of n-ZPrC, we expect to see mitotic abnormalities consistent with the well-characterized essential role for this motor in the morphogenesis of the mitotic spindle upon loss of function (Mann and Wadsworth, 2019; Wojcik et al., 2013; Ferenz et al., 2010; Skoufias et al., 2006; Kapoor et al., 2000; Mayer et al., 1999). To observe the impact of expression of n-ZPrC on cell-cycle progression in live cells, the eGFP moiety on our transfection construct was replaced with the far-red fluorescent RFP670 (Shemetov et al., 2017) to generate n-ZPrC-RFP670. For these experiments, we used HeLa cells that were stably transfected with eGFP-tubulin and transiently with mScarlet-H3 to facilitate live-cell imaging of cell division. In order to interpret any phenotype that we might observe upon expression of n-ZPrC, we first created a peptidase-inactive variant; we mutated Ser135 of the catalytic triad to Ala to generate S135A-n-ZPrC-RFP670. We observed the cells 24–48 h after transfection. This peptidase-inactive variant localizes to the ER as before (Figure 7, Video S2). The cells that are transfected with this construct can be maintained in culture indefinitely and do not exhibit any cell-cycle abnormalities. They exhibit normal timing of mitotic progression with stably oriented mitotic spindles similar to their untransfected counterparts (Figure 7). The M/A ratio of cells transfected with S135A-n-ZPrC-RFP670 was 2.8 ± 0.4 and indistinguishable from untransfected control cells. Similar results were obtained with other cell lines including HEK293T, MCF7, and U2OS cells (not shown).
Figure 7.

Timelapse images of live HeLa cells 24 h after being transfected with S135A-n-ZPrC-RFP670 inactive protease exhibit normal timing and progression through mitosis
Mitotic defects, or delays, are not observed after expression of S135A-n-ZPrC-RFP670. Stable-transfected GFP-Tubulin (green) HeLa cells were transiently transfected with mScarlet-H3 (red) and peptidase-inactive S135A-n-ZPrC-RFP670 (ER network, cyan). These cells all completed mitosis within the normal range of 40–60′. Timelapse confocal data were collected 24 h post-transfection. Scale bar, 25μm, time = mm:ss.
A mitotic cell is observed to divide with normal timing and behavior. Time shown in min:sec.
Expression of n-ZPrC-RFP670 in cultured cells results in prolonged metaphase delay and spindle-positioning defects
Transfection of peptidase-active n-ZPrC-RFP670 resulted in detectable expression after 24 h in HEK293T cells, but with the majority undergoing apoptosis within 72 h post-transfection. Cells with detectable fluorescence from the fluorescent protein reporter are seen in longer term cultures, but it is unclear whether they retain peptidase expression or activity of the n-ZPrC component.
Therefore, we examined mitotic progression in the transfected cells within a 24- to 48-h window. All of the dividing cells expressing this construct experienced a prolonged metaphase delay, often with misaligned chromosomes (Video S3). In contrast to the peptidase-inactive control M/A ratio of approximately 2.9, n-ZPrC-RFP670 transfection resulted in a significant shift in the M/A to 5.0 ± 1.0. This value identifies a moderate delay in progression through anaphase but not as severe as that elicited by expression of the soluble protease chimera (s-ZPrC above, M/A = 11.0 ± 1.2). In addition, in all our long-term timelapse observations (n = 34), the mitotic spindle failed to maintain a stable position and orientation and could be observed to rapidly rotate or move out of position at the onset of anaphase (Figure 8, Videos S4 and S5). These mitotic defects were consistent and reproducible but rarely resulted in mitotic catastrophe, and most cells were nonetheless able to exit mitosis. In contrast, these defects were not observed in long-term timelapse images of cells transfected with the inactive n-S135A-ZPrC-RFP670 variant (n = 10). Very similar impacts were seen in transfected HeLa and MCF7 cells (not shown). However, we were unable to recover any U2OS cells with detectable transient expression of active Zika protease, indicating a range of cell-type specific effects of the protease.
Figure 8.

HeLa cells exhibit prolonged metaphase delay and spindle positioning defects in the presence of native-like n-ZPrC-RFP670
Stable-transfected GFP-tubulin (green) HeLa cells were transiently transfected with mScarlet-H2A (red) and n-ZPrC-RFP670 (cyan). Timelapse confocal data were collected 24 h post-transfection. Also visible in the field of view (lower right) is a mitotic cell that is co-transformed for GFP-tubulin and mScarlet H2A, but with no detectable n-ZPrC-RFP670 fluorescence, and which divides with normal timing and stable spindle orientation. Scale bar, 25μm, time = mm:ss.
A dividing cell expressing chimeric s-ZPrC-TagRFP-ER is blocked at metaphase. For comparison, nearby cells that do not appear to be transfected with the protease are seen entering and completing mitosis in a normal time frame of ∼60’. Time shown in min:sec.
A mitotic cell blocked in metaphase exhibits abnormal spindle movements and positioning. Time shown in min:sec.
HEK293-T cells with Tubulin-GFP (green) were co-transfected with mScarlet-H2A (red) and n-ZPrC-RFP670 (cyan) and observed in timelapse multi-channel confocal microscopy. A mitotic cell blocked in metaphase exhibits abnormal spindle movements and positioning. Time shown in min:sec.
Membrane-bound n-ZPrC can cleave a biosensor mimic of HsEg5 in cells
It is established that Zika protease can target many cellular proteins (Golubeva et al., 2020; Hill et al., 2018; Coyaud et al., 2018; Shah et al., 2018; Scaturro et al., 2018). Therefore, the protease likely impacts more than one mitotic protein in vivo, and the resulting cell division phenotype, including long metaphase delay and spindle positioning defects, is likely a pleiotropic effect caused by partial loss of function of more than one mitotic protein. Because HsEg5 contains a KEN box site recognized by the APC ubiquitin ligase, which targets the motor for degradation by the proteasome following the onset of anaphase (Eguren et al., 2014), it is technically intractable to use standard western blotting approaches to separate and identify specific cleavage products due to cleavage of HsEg5 by n-ZPrC. To overcome this challenge, we designed an expressible, soluble, biosensor protein mimic that produces a fluorescent readout upon selective cleavage of a peptide loop segment (Figures 9, and S8). The ZPrC biosensor is based on a bacterial infrared fluorescent phytochrome protein (IFP, Figure 9) that was originally designed to detect Caspase-3 activity in vitro (To et al., 2015). Inactive IFP is blocked from forming its chromophore by an exposed peptide loop. The loop contains a cleavage consensus that is cleaved by the requisite peptidase, thereby allowing the fluorescent chromophore to form. We modified the loop to contain the primary HsEg5 motor domain cleavage sequence (DPRNKRG, Figures 9, and S8). With the modified loop segment, the HsEg5 biosensor does not exhibit far-red fluorescence when transfected into in HeLa, HEK293T, MCF7, and U2Os cells, indicating its usefulness for surveillance of Zika protease activity.
Figure 9.

Design of the HsEg5 biosensor for the in vivo detection of specific ZPrC protease activity
We modified a fluorescent protein reporter of Caspase activity to instead detect ZPrC activity in vivo. We substituted the original caspase cleavage target sequence with HsEg5 loop8 ZPrC target sequence (DPRNKRG) in an exposed loop of the bacterial phytochrome domain of the reporter. Cleavage of this segment permits integration of the biliverdin chromophore (rendered in “stick” cartoon form) into the phytochrome and activates far-red fluorescence. Biosensor fluorescence is not detectably activated by endogenous cellular proteases in the absence of active ZPrC. Continued association of the cleaved phytochrome domains are sustained by the split-GFP moiety.
To determine if endogenous HsEg5 is likely to be among the cellular targets of native ZPrC, we performed experiments with two different ZPrC constructs. First, we co-transfected MCF7 cells with both the HsEg5 biosensor and either peptidase inactive S135A-n-ZPrC-eGFP or active n-ZPrC-eGFP (Figure 10A). We found that all cells that expressed the n-ZPrC-eGFP protease on the ER also exhibited far-red fluorescence from the cytoplasmic HsEg5 biosensor, indicating selective cleavage had occurred to the HsEg5 peptide segment (Figure 10B). All cells transfected with the biosensor alone or co-transfected with the protease inactive S135A-n-ZPrC-eGFP never exhibited far-red fluorescence (not shown). Second, a HeLa-tubulin-GFP cell line was co-transfected with both the HsEg5 biosensor and s-ZPrC-TagRFP-ER expression constructs (Figure S3). In this case, the otherwise soluble s-ZPrC-TagRFP-ER is anchored to the ER membrane through a C-terminal cytochrome b5 reductase ER-retention signal. Cells expressing the HsEg5 biosensor were identified by nucleocytoplasmic eGFP fluorescence. Only HeLa cells expressing both the biosensor and s-ZPrC-TagRFP-ER exhibited far-red fluorescence from the reporter, indicating cleavage of the HsEg5 peptide segment of the cytoplasmic biosensor (Figures S3A–S3C).
Figure 10.

ER-localized n-ZPrC-eGFP can cut the soluble HsEg5 loop8 biosensor mimic in MCF7 cells
(A) The localization of transient co-transfected HsEg5 biosensor-GFP and n-ZPrC-eGFP in live cells by confocal microscopy. Cells expressing the biosensor display nucleocytoplasmic localization of the reporter GFP moiety in contrast to the reticulate ER network marked by n-ZPrC-eGFP. Asterisk marks a cell expressing n-ZPrC-eGFP without detectable nucleocytoplasmic biosensor.
(B) All cells expressing both the HsEg5 biosensor and n-ZPrC protease exhibit cleavage-mediated activation of far-red fluorescence from the biosensor (white signal, n = 100). Cells transfected with either the biosensor alone (n = 100) or co-transfected with protease-inactive S135A-n-ZPrC-eGFP (n = 100) do not exhibit any detectable far-red fluorescence from the biosensor (not shown). Scale bar, 25 μm.
Conversely, cells that were co-transfected with the HsEg5 biosensor together with peptidase-inactive S135A-s-ZPrC-TagRFP-ER never exhibited far-red fluorescence (Figures S3D–S3F). Therefore, these data indicate that soluble cytoplasmic proteins bearing the activation loop consensus, DPRNKRG, likely limited to endogenous HsEg5 kinesin, can be targeted and cleaved by ER-membrane-bound Zika protease in animal cells.
Discussion
Here we investigate whether HsEg5 could directly link Zika virus infection to Zika syndrome microcephaly and blindness. The human HsEg5 gene, Kif11, is a haploinsufficient and essential human gene (Landrum et al., 2018). Partial loss-of-function mutations in Kif11, including both missense changes and nonsense mutation truncations, result in profound microcephaly together with retinopathies (Hu et al., 2016; Jones et al., 2014; Ostergaard et al., 2012). Haploinsufficiency appears to be a hallmark of Kif11, whereas in other organisms, loss-of-function mutations in their respective Kinesin-5 genes instead are recessive. Given that the Zika disease syndrome, which occurs during fetal development, closely matches the disease outcomes of partial loss of HsEg5 function, we tested whether it was feasible for Zika virus to directly cause a HsEg5 partial loss of function.
Recent work has identified the Zika-encoded protease as the viral component that is thought to play a key role in the virus host-pathogen interactions that are responsible for the etiology of human fetal microcephaly in Zika syndrome. Several unbiased proteomics studies that utilized cell expression of the recombinant soluble Zika NS2B-NS3 protease identified a number of putative host protein targets of Zika protease, some of which have indirect genetic links to human microcephaly such as certain centrosomal proteins (Wen et al., 2019; Wolf et al., 2017). Other approaches have looked to identify putative host targets of the Zika protease that are required for cell division, such as septins, which might provide a mechanism by which the virus could halt neuronal proliferation in the developing fetus. Although cleavage of septin by the Zika peptidase can disrupt cell division and thereby theoretically prematurely terminate the development of neuronal cell lineages during fetal development, septin genomic mutations in humans have heretofore not been directly or indirectly linked to occurrences of microcephaly.
Notably, the Zika protease is up to 58% identical to those of related flaviviruses Dengue and West Nile. In addition, they share high structural similarity and partially overlapping substrate selectivity. The high degree of structural and functional similarity between these closely related flaviviruses begs the question; if the Zika protease is responsible for the etiology of microcephaly in the affected fetus, then why this syndrome is not observed with infection by closely related flaviviruses such as Dengue or West Nile? We propose that the unique tropism of the Zika virus for neuronal precursor cells, which rely on asymmetric division, as compared with the other flavivirus relatives, can explain the increased incidence of microcephaly with Zika infection. We observe that, although the protease causes a disruption to the mitotic apparatus that results in activation of a metaphase delay in cultured cells, this delay often resolves with anaphase and successful mitotic exit. However, neuronal precursor cells rely on complex asymmetric segregation of cell fate determinants into daughter cells that is easily disrupted by aberrant spindle positioning (Link et al., 2019; Zhong and Chia, 2008; Arai and Taverna, 2017; Lancaster and Knoblich, 2012; Shitamukai and Matsuzaki, 2012). Therefore, although the mitotic delay caused by the protease is not typically catastrophic, it does disrupt spindle positioning and therefore is capable of terminating the development of neuronal cell lineages (Figure 11).
Figure 11.

Model depicting the potential impact of ER-tethered Zika protease on asymmetric cell division
(A) In a wild-type asymmetric mitosis, the cell division axis is established (dotted line) at metaphase and cell fate determinants (purple and yellow shading) are asymmetrically localized. After anaphase/telophase, the cell fate determinants are partitioned into a neuroblast (purple, NB) and a pluripotent ganglion mother cell (yellow, GMC).
(B) An asymmetric mitosis with Zika protease (red) tethered to the reticulate ER membrane differs from wild-type cell division. The protease is able to cleave targets in close proximity to the ER, including HsEg5 and presumably other targets, and causes the spindle to exhibit abnormal mobility and mis-positioning with respect to the metaphase axis. Subsequent anaphase/telophase shows daughter cells with an aberrant mixture of cell fate determinants, which likely results in the termination of cell differentiation.
In this study, we have asked whether it is possible for the Zika NS2B-NS3 protease to target HsEg5 kinesin in vitro. We show that the chimeric soluble form of the Zika NS2B-NS3 protease can readily attack and hydrolyze a consensus site within the exposed loop8 segment of the HsEg5 motor domain. This site is targeted with comparable kinetics as two native cleavage sites targeted by this protease during the processing of the viral polyprotein on the surface of the ER membrane within infected cells. Furthermore, this site remains available and cleavable by the protease even when the motor is rigor-bound to the microtubule lattice by nonhydrolyzable nucleotide, AMPPNP. Once this primary site is cut, additional cut sites appear to become available for further proteolysis of the motor.
However, within living cells the functional NS2B-NS3 protease is not freely soluble but instead is comprised of a soluble peptidase domain that forms a two-component complex with the integral membrane NS2B protein. Although the NS3 peptidase domain is separable from the NS2B transmembrane domain, it only exhibits protease activity when in complex with NS2B. Being anchored to the outer leaflet of the ER the protease activity of this complex is spatially segregated from other elements of the cytoplasm. To explore the ramifications of spatially restricted Zika protease activity, we developed a novel Zika protease expression construct that includes the complete NS2B integral membrane protein and adjacent NS3 peptidase domain as expressed by the virus. Expression of this construct results in ER-specific localization of the complex and permits native processing of the NS2B-NS3 cleavage site to generate an active protease complex (n-ZPrC) that reflects the native viral protease complex. The reconstitution of protease activity in this complex is confirmed by two lines of evidence. First, we observe a significant increase in the M/A ratio of transfected cells as compared with the transfection of the S135A inactive protease. This indicates activation of the spindle assembly checkpoint in response to spindle organization defects caused by reduced HsEg5 activity. Second, we also observe cleavage-mediated activation of our cytoplasmic soluble Zika protease biosensor, which reports HsEg5 susceptibility to cleavage, and also confirms the correct membrane topology of the n-ZPrC construct.
We find that the soluble Zika protease chimera (s-ZPrC-eGFP, above) results in different impacts on transfected cells than the native ER-resident protease (n-ZPrC-eGFP). In our hands, HEK293-T cells that express s-ZPrC-eGFP exhibit a stronger metaphase block (M/A ratio = 11 ± 1.2) than that elicited by the n-ZPrC-eGFP (5 ± 1.0). We also observe that the majority of cells transfected with the s-ZPrC-eGFP protease succumb to apoptosis within 48 h post-transfection (data not shown), a level of toxicity not seen with the n-ZPrC-eGFP. These data indicate that sequestration of the Zika protease to the ER membrane modifies or limits its impact on the biology of the cell relative to the fully soluble variant. This observation is consistent with our hypothesis that the spatially constrained activity of the native Zika protease is an important facet of its biology.
We next examined whether transfected n-ZPrC could target and cleave HsEg5 in cultured cells. Given the technical challenges in measuring a net decrease in HsEg5 due to n-ZPrC during mitosis while in a cellular system in which HsEg5 is naturally degraded at the end of mitosis, we opted to design a soluble surrogate HsEg5 biosensor that could unequivocally report specific cleavage of the loop8 site by n-ZPrC. Based on a published caspase protease biosensor (To et al., 2015), reporter fluorescence in our modified variant is inhibited by a peptide loop bearing the HsEg5 loop8 cleavage site that must be cleaved to permit chromophore formation. Only cells expressing the active ER-associated n-ZPrC are able to elicit fluorescence from the reporter, verifying that the tethered Zika protease can cleave a soluble protein bearing an exposed DPRNKRG cleavage site in living cells. Work by others has established that the ER maintains intimate direct connections, mediated predominately by REEP proteins, to the microtubules comprising the outside of the mitotic spindle (Kumar et al., 2019; Schlaitz, 2014; Schlaitz et al., 2013; Ellenberg et al., 1997). In this context, HsEg5 is extant as both a microtubule-bound form that is performing work throughout the spindle and a cytoplasmic pool of unbound motor. Although the biosensor mimics the unbound cytoplasmic form of HsEg5, our in vitro protease assays show that the Zika protease can cleave motor that is rigor-bound to microtubules as well. Taken altogether, this evidence supports the conclusion that ER-tethered Zika protease targets both free and microtubule-bound HsEg5 in the mitotic spindle.
It is well established that loss of function in HsEg5 in human cells undergoing mitosis results in the catastrophic collapse of the mitotic spindle into a monopolar array (Mann and Wadsworth, 2019; Waitzman and Rice, 2014; Wojcik et al., 2013). Although we observe mitotic defects in cells suffering from the activity of n-ZPrC, we do not observe mitotic cells with monopolar spindles, a common outcome of drug-mediated HsEg5 inhibition. However, both small molecule inhibition and genetic knockdown are thought to cause a uniform and unbiased reduction in HsEg5 activity during mitosis that ultimately result in a monopolar spindle array. We propose that Zika protease targets both soluble and microtubule-bound HsEg5 primarily at the interface between the ER and spindle structures, with the HsEg5 that lies deeper within the spindle unavailable to the ER-resident protease. In this model (Figure 11), ER-tethered Zika protease activity results in a new kind of impact on HsEg5 function, a partial loss of function that is spatially non-uniform within a cell. Our data suggest that spindle bipolarity is maintained by a pool of internal spindle-associated HsEg5 that is inaccessible to the protease. As a result, spindle bipolarity is maintained, but loss of HsEg5 at the periphery of the spindle results in metaphase delay and misorientation of the spindle. Our results support the idea that not all knock-outs are the same; spatially organized downregulation of protein function can result in unanticipated phenotypes. To further test this hypothesis, future experiments capable of investigating the spatial distribution of HsEg5 degradation in this system will need to be developed.
Taken altogether, our data indicate that Zika protease can single-handedly orchestrate the spindle-positioning defects that are observed in this report and also observed by others (McDougall et al., 2019). To our knowledge, HsEg5 is the only identified target of Zika protease with the capability to disrupt the behavior of the mitotic spindle. These data suggest a model by which novel inter-organelle interactions between the ER and mitotic spindle bring Zika protease into close proximity to mediate the proteolysis of microtubule-associated HsEg5 on the spindle. Therefore, we propose a simple model in which asymmetric neuronal stem cell divisions may be sharply affected by spindle misalignment and thereby lead to termination of neuronal lineages and subsequent microcephaly. It is thereby possible that the action of native Zika NS2-BNS3 on HsEg5 kinesin may be a key component of the mechanism responsible for the development of microcephaly seen in Zika syndrome.
Limitations of the study
Given the similarities between Zika fetal syndrome and human Kinesin-5 loss-of-function mutations, we looked for a direct connection between the two. Our experimental systems included in vitro protease assays that utilized a chimeric, soluble, Zika protease and an analysis of the native protease within cultured cells. Although we have uncovered mitotic impacts that could lead to microcephaly within a developing fetus, future experiments need to utilize more complex and developmental neuronal systems to solidify this hypothesis.
Although the preponderance of indirect data, based on our biosensor, support a direct cleavage of Kinesin-5 by the native Zika protease in cultured cells, we found it to be intractable to gain direct evidence of Zika protease cleavage of native Kinesin-5 in living cells. We were unable to unambiguously separate and identify Zika protease-specific cleavage products from among the large number of naturally occurring post-mitotic Kinesin-5 degradation products.
Finally, we argue that the mitotic phenotypes that we observe are caused, at least in part, due to some degree of loss of Kinesin-5 function. However, there is an ever-growing list of putative host targets for Zika protease, and it is possible that the mitotic phenotype we observe is the result of cleavage of more than one host mitotic protein target. Should a functional protease-resistant form of the motor be developed, then it will be possible to tease out the motor's precise role. Furthermore, although the essential mitotic role of Kinesin-5 has been very well characterized, it remains possible that Kinesin-5-associated microcephaly results instead from a novel non-mitotic cellular role for this enzyme during fetal neuronal development.
Resource availability
Lead contact
Information and requests for resources should be directed to and will be fulfilled by the lead contact, Edward Wojcik (ewojci@lsuhsc.edu).
Materials availability
Unique materials in this study, including plasmid constructs, are available from the lead contact, Edward Wojcik, upon request.
Data and code availability
All relevant data are available from the lead contact upon request. There were no custom codes or algorithms used in this study.
Methods
All methods can be found in the accompanying transparent methods supplemental file.
Acknowledgments
We thank the MotorHeads group (LSU School of Medicine; Xavier University) for helpful discussions, and Prof. Sunyoung Kim for manuscript review and helpful suggestions. These studies were supported by funds from the LSU Health Sciences Center and NIGMS R01-GM097350 (S.K. and E.W.).
Author contributions
L.L. performed all experimental procedures. L.L. developed the molecular reagents for this study. M.D. performed data analysis and manuscript editing. J.G. performed mass spectrometry. E.W. contributed live-cell microscopy data and designed and supervised this work. E.W. and M.D. wrote and edited the manuscript.
Declaration of interests
The authors declare no competing interests.
Inclusion and diversity
One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in science.
Published: May 21, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2021.102385.
Supplemental information
References
- Alonso M.C., van Damme J., Vandekerckhove J., Cross R.A. Proteolytic mapping of kinesin/ncd-microtubule interface: nucleotide-dependent conformational changes in the loops L8 and L12. EMBO J. 1998;17:945–951. doi: 10.1093/emboj/17.4.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arai Y., Taverna E. Neural progenitor cell polarity and cortical development. Front. Cell Neurosci. 2017;11:384. doi: 10.3389/fncel.2017.00384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arias-Arias J.L., MacPherson D.J., Hill M.E., Hardy J.A., Mora-Rodríguez R. A fluorescence activatable reporter of flavivirus NS2B-NS3 protease activity enables live imaging of infection in single cells and viral plaques. J. Biol. Chem. 2020 doi: 10.1074/jbc.RA119.011319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balikova I., Robson A.G., Holder G.E., Ostergaard P., Mansour S., Moore A.T. Ocular manifestations of microcephaly with or without chorioretinopathy, lymphedema or intellectual disability (MCLID) syndrome associated with mutations in KIF11. Acta Ophthalmol. 2016;94:92–98. doi: 10.1111/aos.12759. [DOI] [PubMed] [Google Scholar]
- Bera A.K., Kuhn R.J., Smith J.L. Functional characterization of cis and trans activity of the Flavivirus NS2B-NS3 protease. J. Biol. Chem. 2007;282:12883–12892. doi: 10.1074/jbc.M611318200. [DOI] [PubMed] [Google Scholar]
- Bindels D.S., Haarbosch L., van Weeren L., Postma M., Wiese K.E., Mastop M., Aumonier S., Gotthard G., Royant A., Hink M.A. mScarlet: a bright monomeric red fluorescent protein for cellular imaging. Nat. Methods. 2017;14:53–56. doi: 10.1038/nmeth.4074. [DOI] [PubMed] [Google Scholar]
- Bodey A.J., Kikkawa M., Moores C.A. 9-Angström structure of a microtubule-bound mitotic motor. J. Mol. Biol. 2009;388:218–224. doi: 10.1016/j.jmb.2009.03.008. [DOI] [PubMed] [Google Scholar]
- Borgese N., D’Arrigo A., De Silvestris M., Pietrini G. NADH-cytochrome b5 reductase and cytochrome b5 isoforms as models for the study of post-translational targeting to the endoplasmic reticulum. FEBS Lett. 1993;325:70–75. doi: 10.1016/0014-5793(93)81416-w. [DOI] [PubMed] [Google Scholar]
- Brust-Mascher I., Sommi P., Cheerambathur D.K., Scholey J.M. Kinesin-5-dependent poleward flux and spindle length control in Drosophila embryo mitosis. Mol. Biol. Cell. 2009;20:1749–1762. doi: 10.1091/mbc.e08-10-1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang Q., Nitta R., Inoue S., Hirokawa N. Structural basis for the ATP-induced isomerization of kinesin. J. Mol. Biol. 2013;425:1869–1880. doi: 10.1016/j.jmb.2013.03.004. [DOI] [PubMed] [Google Scholar]
- Chen X., Yang K., Wu C., Chen C., Hu C., Buzovetsky O., Wang Z., Ji X., Xiong Y., Yang H. Mechanisms of activation and inhibition of Zika virus NS2B-NS3 protease. Cell Res. 2016;26:1260–1263. doi: 10.1038/cr.2016.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cochran J.C., Sontag C.A., Maliga Z., Kapoor T.M., Correia J.J., Gilbert S.P. Mechanistic analysis of the mitotic kinesin Eg5. J. Biol. Chem. 2004;279:38861–38870. doi: 10.1074/jbc.M404203200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortese M., Goellner S., Acosta E.G., Neufeldt C.J., Oleksiuk O., Lampe M., Haselmann U., Funaya C., Schieber N., Ronchi P. Ultrastructural characterization of Zika virus replication factories. Cell Rep. 2017;18:2113–2123. doi: 10.1016/j.celrep.2017.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coyaud E., Ranadheera C., Cheng D., Gonçalves J., Dyakov B.J.A., Laurent E.M.N., St-Germain J., Pelletier L., Gingras A.C., Brumell J.H. Global interactomics uncovers extensive organellar targeting by Zika virus. Mol. Cell Proteomics. 2018;17:2242–2255. doi: 10.1074/mcp.TIR118.000800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Arcy A., Chaillet M., Schiering N., Villard F., Lim S.P., Lefeuvre P., Erbel P. Purification and crystallization of dengue and West Nile virus NS2B-NS3 complexes. Acta Crystallogr. F Struct. Biol. Cryst. Commun. 2006;62:157–162. doi: 10.1107/S1744309106001199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dar H.A., Zaheer T., Paracha R.Z., Ali A. Structural analysis and insight into Zika virus NS5 mediated interferon inhibition. Infect. Genet. Evol. 2017;51:143–152. doi: 10.1016/j.meegid.2017.03.027. [DOI] [PubMed] [Google Scholar]
- De Jesús-González L.A., Cervantes-Salazar M., Reyes-Ruiz J.M., Osuna-Ramos J.F., Farfán-Morales C.N., Palacios-Rápalo S.N., Pérez-Olais J.H., Cordero-Rivera C.D., Hurtado-Monzón A.M., Ruíz-Jiménez F. The nuclear Pore complex: a target for NS3 protease of dengue and Zika viruses. Viruses. 2020;12 doi: 10.3390/v12060583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Cruz L., Chen W.N., Graham B., Otting G. Binding mode of the activity-modulating C-terminal segment of NS2B to NS3 in the dengue virus NS2B-NS3 protease. FEBS J. 2014;281:1517–1533. doi: 10.1111/febs.12729. [DOI] [PubMed] [Google Scholar]
- DeBonis S., Simorre J.P., Crevel I., Lebeau L., Skoufias D.A., Blangy A., Ebel C., Gans P., Cross R., Hackney D.D. Interaction of the mitotic inhibitor monastrol with human kinesin Eg5. Biochemistry. 2003;42:338–349. doi: 10.1021/bi026716j. [DOI] [PubMed] [Google Scholar]
- DeBonis S., Skoufias D.A., Lebeau L., Lopez R., Robin G., Margolis R.L., Wade R.H., Kozielski F. In vitro screening for inhibitors of the human mitotic kinesin Eg5 with antimitotic and antitumor activities. Mol. Cancer Ther. 2004;3:1079–1090. [PubMed] [Google Scholar]
- Ding Q., Gaska J.M., Douam F., Wei L., Kim D., Balev M., Heller B., Ploss A. Species-specific disruption of STING-dependent antiviral cellular defenses by the Zika virus NS2B3 protease. Proc. Natl. Acad. Sci. U S A. 2018;115:E6310–E6318. doi: 10.1073/pnas.1803406115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eguren M., Álvarez-Fernández M., García F., López-Contreras A.J., Fujimitsu K., Yaguchi H., Luque-García J.L., Fernández-Capetillo O., Muñoz J., Yamano H. A synthetic lethal interaction between APC/C and topoisomerase poisons uncovered by proteomic screens. Cell Rep. 2014;6:670–683. doi: 10.1016/j.celrep.2014.01.017. [DOI] [PubMed] [Google Scholar]
- Ellenberg J., Siggia E.D., Moreira J.E., Smith C.L., Presley J.F., Worman H.J., Lippincott-Schwartz J. Nuclear membrane dynamics and reassembly in living cells: targeting of an inner nuclear membrane protein in interphase and mitosis. J. Cell Biol. 1997;138:1193–1206. doi: 10.1083/jcb.138.6.1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falgout B., Pethel M., Zhang Y.M., Lai C.J. Both nonstructural proteins NS2B and NS3 are required for the proteolytic processing of dengue virus nonstructural proteins. J. Virol. 1991;65:2467–2475. doi: 10.1128/JVI.65.5.2467-2475.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferenz N.P., Gable A., Wadsworth P. Mitotic functions of kinesin-5. Semin. Cell Dev. Biol. 2010;21:255–259. doi: 10.1016/j.semcdb.2010.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira R.O., Garcez P.P. Dissecting the toxic effects of Zika virus proteins on neural progenitor cells. Neuron. 2019;101:989–991. doi: 10.1016/j.neuron.2019.03.009. [DOI] [PubMed] [Google Scholar]
- Freund R.K., Gibson E.S., Potter H., Dell’Acqua M.L. Inhibition of the motor protein Eg5/kinesin-5 in Amyloid β-mediated impairment of hippocampal long-term potentiation and dendritic spine loss. Mol. Pharmacol. 2016;89:552–559. doi: 10.1124/mol.115.103085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh Roy S., Sadigh B., Datan E., Lockshin R.A., Zakeri Z. Regulation of cell survival and death during Flavivirus infections. World J. Biol. Chem. 2014;5:93–105. doi: 10.4331/wjbc.v5.i2.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gigant B., Wang W., Dreier B., Jiang Q., Pecqueur L., Plückthun A., Wang C., Knossow M. Structure of a kinesin-tubulin complex and implications for kinesin motility. Nat. Struct. Mol. Biol. 2013;20:1001–1007. doi: 10.1038/nsmb.2624. [DOI] [PubMed] [Google Scholar]
- Golubeva V.A., Nepomuceno T.C., Gregoriis G., Mesquita R.D., Li X., Dash S., Garcez P.P., Suarez-Kurtz G., Izumi V., Koomen J. Network of interactions between ZIKA virus non-structural proteins and human host proteins. Cells. 2020;9 doi: 10.3390/cells9010153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goshima G., Vale R.D. Cell cycle-dependent dynamics and regulation of mitotic kinesins in Drosophila S2 cells. Mol. Biol. Cell. 2005;16:3896–3907. doi: 10.1091/mbc.e05-02-0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant A., Ponia S.S., Tripathi S., Balasubramaniam V., Miorin L., Sourisseau M., Schwarz M.C., Sánchez-Seco M.P., Evans M.J., Best S.M. Zika virus targets human STAT2 to inhibit type I interferon signaling. Cell Host Microbe. 2016;19:882–890. doi: 10.1016/j.chom.2016.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruba N., Rodriguez Martinez J.I., Grzywa R., Wysocka M., Skoreński M., Burmistrz M., Łęcka M., Lesner A., Sieńczyk M., Pyrć K. Substrate profiling of Zika virus NS2B-NS3 protease. FEBS Lett. 2016;590:3459–3468. doi: 10.1002/1873-3468.12443. [DOI] [PubMed] [Google Scholar]
- Güneş N., Taşdemir E., Jeffery H., Yetik H., Ostergaard P., Tüysüz B. A novel mutation of KIF11 in a child with 22q11.2 deletion syndrome associated with MCLMR. Mol. Syndromol. 2019;9:266–270. doi: 10.1159/000491568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta G., Lim L., Song J. NMR and MD studies reveal that the isolated dengue NS3 protease is an intrinsically disordered chymotrypsin fold which absolutely requests NS2B for correct folding and functional dynamics. PLoS One. 2015;10:e0134823. doi: 10.1371/journal.pone.0134823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanlon M.R., Begum R.R., Newbold R.J., Whitford D., Wallace B.A. In vitro membrane-inserted conformation of the cytochrome b(5) tail. Biochem. J. 2000;352(Pt 1):117–124. doi: 10.1042/bj3520117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazan F., Ostergaard P., Ozturk T., Kantekin E., Atlihan F., Jeffery S., Ozkinay F. A novel KIF11 mutation in a Turkish patient with microcephaly, lymphedema, and chorioretinal dysplasia from a consanguineous family. Am. J. Med. Genet. A. 2012;158A:1686–1689. doi: 10.1002/ajmg.a.35371. [DOI] [PubMed] [Google Scholar]
- Heck M.M., Pereira A., Pesavento P., Yannoni Y., Spradling A.C., Goldstein L.S. The kinesin-like protein KLP61F is essential for mitosis in Drosophila. J. Cell Biol. 1993;123:665–679. doi: 10.1083/jcb.123.3.665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill M.E., Kumar A., Wells J.A., Hobman T.C., Julien O., Hardy J.A. The unique cofactor region of Zika virus NS2B-NS3 protease facilitates cleavage of key host proteins. ACS Chem. Biol. 2018;13:2398–2405. doi: 10.1021/acschembio.8b00508. [DOI] [PubMed] [Google Scholar]
- Hirose K., Akimaru E., Akiba T., Endow S.A., Amos L.A. Large conformational changes in a kinesin motor catalyzed by interaction with microtubules. Mol. Cell. 2006;23:913–923. doi: 10.1016/j.molcel.2006.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu H., Xiao X., Li S., Jia X., Guo X., Zhang Q. KIF11 mutations are a common cause of autosomal dominant familial exudative vitreoretinopathy. Br. J. Ophthalmol. 2016;100:278–283. doi: 10.1136/bjophthalmol-2015-306878. [DOI] [PubMed] [Google Scholar]
- Hughes B.W., Addanki K.C., Sriskanda A.N., McLean E., Bagasra O. Infectivity of immature neurons to Zika virus: a link to congenital Zika syndrome. EBioMedicine. 2016;10:65–70. doi: 10.1016/j.ebiom.2016.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones G.E., Ostergaard P., Moore A.T., Connell F.C., Williams D., Quarrell O., Brady A.F., Spier I., Hazan F., Moldovan O. Microcephaly with or without chorioretinopathy, lymphoedema, or mental retardation (MCLMR): review of phenotype associated with KIF11 mutations. Eur. J. Hum. Genet. 2014;22:881–887. doi: 10.1038/ejhg.2013.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahn O.I., Sharma V., González-Billault C., Baas P.W. Effects of kinesin-5 inhibition on dendritic architecture and microtubule organization. Mol. Biol. Cell. 2015;26:66–77. doi: 10.1091/mbc.E14-08-1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapoor T.M., Mayer T.U., Coughlin M.L., Mitchison T.J. Probing spindle assembly mechanisms with monastrol, a small molecule inhibitor of the mitotic kinesin, Eg5. J. Cell Biol. 2000;150:975–988. doi: 10.1083/jcb.150.5.975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karjosukarso D.W., Cremers F.P.M., van Nouhuys C.E., Collin R.W.J. Detection and quantification of a KIF11 mosaicism in a subject presenting familial exudative vitreoretinopathy with microcephaly. Eur. J. Hum. Genet. 2018;26:1819–1823. doi: 10.1038/s41431-018-0243-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim C.D., Kim E.D., Liu L., Buckley R.S., Parameswaran S., Kim S., Wojcik E.J. Small molecule allosteric uncoupling of microtubule depolymerase activity from motility in human Kinesin-5 during mitotic spindle assembly. Sci. Rep. 2019;9:19900. doi: 10.1038/s41598-019-56173-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y.M., Gayen S., Kang C., Joy J., Huang Q., Chen A.S., Wee J.L., Ang M.J., Lim H.A., Hung A.W. NMR analysis of a novel enzymatically active unlinked dengue NS2B-NS3 protease complex. J. Biol. Chem. 2013;288:12891–12900. doi: 10.1074/jbc.M112.442723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A., Hou S., Airo A.M., Limonta D., Mancinelli V., Branton W., Power C., Hobman T.C. Zika virus inhibits type-I interferon production and downstream signaling. EMBO Rep. 2016;17:1766–1775. doi: 10.15252/embr.201642627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar D., Golchoubian B., Belevich I., Jokitalo E., Schlaitz A.L. REEP3 and REEP4 determine the tubular morphology of the endoplasmic reticulum during mitosis. Mol. Biol. Cell. 2019;30:1377–1389. doi: 10.1091/mbc.E18-11-0698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lancaster M.A., Knoblich J.A. Spindle orientation in mammalian cerebral cortical development. Curr. Opin. Neurobiol. 2012;22:737–746. doi: 10.1016/j.conb.2012.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landrum M.J., Lee J.M., Benson M., Brown G.R., Chao C., Chitipiralla S., Gu B., Hart J., Hoffman D., Jang W. ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018;46:D1062–D1067. doi: 10.1093/nar/gkx1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazear H.M., Diamond M.S. Zika virus: new clinical syndromes and its emergence in the western hemisphere. J. Virol. 2016;90:4864–4875. doi: 10.1128/JVI.00252-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H., Ren J., Nocadello S., Rice A.J., Ojeda I., Light S., Minasov G., Vargas J., Nagarathnam D., Anderson W.F. Identification of novel small molecule inhibitors against NS2B/NS3 serine protease from Zika virus. Antivir. Res. 2017;139:49–58. doi: 10.1016/j.antiviral.2016.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei J., Hansen G., Nitsche C., Klein C.D., Zhang L., Hilgenfeld R. Crystal structure of Zika virus NS2B-NS3 protease in complex with a boronate inhibitor. Science. 2016;353:503–505. doi: 10.1126/science.aag2419. [DOI] [PubMed] [Google Scholar]
- Lennemann N.J., Coyne C.B. Dengue and Zika viruses subvert reticulophagy by NS2B3-mediated cleavage of FAM134B. Autophagy. 2017;13:322–332. doi: 10.1080/15548627.2016.1265192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung D., Schroder K., White H., Fang N.X., Stoermer M.J., Abbenante G., Martin J.L., Young P.R., Fairlie D.P. Activity of recombinant dengue 2 virus NS3 protease in the presence of a truncated NS2B co-factor, small peptide substrates, and inhibitors. J. Biol. Chem. 2001;276:45762–45771. doi: 10.1074/jbc.M107360200. [DOI] [PubMed] [Google Scholar]
- Li H., Saucedo-Cuevas L., Yuan L., Ross D., Johansen A., Sands D., Stanley V., Guemez-Gamboa A., Gregor A., Evans T. Zika virus protease cleavage of host protein septin-2 mediates mitotic defects in neural progenitors. Neuron. 2019;101:1089–1098.e4. doi: 10.1016/j.neuron.2019.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J.K., Fei P., Li Y., Huang Q.J., Zhang Q., Zhang X., Rao Y.Q., Li J., Zhao P. Identification of novel KIF11 mutations in patients with familial exudative vitreoretinopathy and a phenotypic analysis. Sci. Rep. 2016;6:26564. doi: 10.1038/srep26564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y., Li Q., Wong Y.L., Liew L.S., Kang C. Membrane topology of NS2B of dengue virus revealed by NMR spectroscopy. Biochim. Biophys. Acta. 2015;1848:2244–2252. doi: 10.1016/j.bbamem.2015.06.010. [DOI] [PubMed] [Google Scholar]
- Liang Q., Luo Z., Zeng J., Chen W., Foo S.S., Lee S.A., Ge J., Wang S., Goldman S.A., Zlokovic B.V. Zika virus NS4A and NS4B proteins deregulate Akt-mTOR signaling in human fetal neural stem cells to inhibit neurogenesis and induce autophagy. Cell Stem Cell. 2016;19:663–671. doi: 10.1016/j.stem.2016.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Link N., Chung H., Jolly A., Withers M., Tepe B., Arenkiel B.R., Shah P.S., Krogan N.J., Aydin H., Geckinli B.B. Mutations in ANKLE2, a ZIKA virus target, disrupt an asymmetric cell division pathway in Drosophila neuroblasts to cause microcephaly. Dev. Cell. 2019;51:713–729.e6. doi: 10.1016/j.devcel.2019.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L., Richard J., Kim S., Wojcik E.J. Small molecule screen for candidate antimalarials targeting Plasmodium Kinesin-5. J. Biol. Chem. 2014;289:16601–16614. doi: 10.1074/jbc.M114.551408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lockhart A., Cross R.A. Kinetics and motility of the Eg5 microtubule motor. Biochemistry. 1996;35:2365–2373. doi: 10.1021/bi952318n. [DOI] [PubMed] [Google Scholar]
- Mann B.J., Wadsworth P. Kinesin-5 regulation and function in mitosis. Trends Cell Biol. 2019;29:66–79. doi: 10.1016/j.tcb.2018.08.004. [DOI] [PubMed] [Google Scholar]
- Mayer T.U., Kapoor T.M., Haggarty S.J., King R.W., Schreiber S.L., Mitchison T.J. Small molecule inhibitor of mitotic spindle bipolarity identified in a phenotype-based screen. Science. 1999;286:971–974. doi: 10.1126/science.286.5441.971. [DOI] [PubMed] [Google Scholar]
- McDougall W.M., Perreira J.M., Hung H.F., Vertii A., Xiaofei E., Zimmerman W., Kowalik T.F., Doxsey S., Brass A.L. Viral infection or IFN-α alters mitotic spindle orientation by modulating Pericentrin levels. iScience. 2019;12:270–279. doi: 10.1016/j.isci.2019.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirzaa G.M., Enyedi L., Parsons G., Collins S., Medne L., Adams C., Ward T., Davitt B., Bicknese A., Zackai E. Congenital microcephaly and chorioretinopathy due to de novo heterozygous KIF11 mutations: five novel mutations and review of the literature. Am. J. Med. Genet. A. 2014;164A:2879–2886. doi: 10.1002/ajmg.a.36707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitoma J., Ito A. The carboxy-terminal 10 amino acid residues of cytochrome b5 are necessary for its targeting to the endoplasmic reticulum. EMBO J. 1992;11:4197–4203. doi: 10.1002/j.1460-2075.1992.tb05513.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mlakar J., Korva M., Tul N., Popović M., Poljšak-Prijatelj M., Mraz J., Kolenc M., Resman Rus K., Vesnaver Vipotnik T., Fabjan Vodušek V. Zika virus associated with microcephaly. N. Engl. J. Med. 2016;374:951–958. doi: 10.1056/nejmoa1600651. [DOI] [PubMed] [Google Scholar]
- Mohd Ropidi M.I., Khazali A.S., Nor Rashid N., Yusof R. Endoplasmic reticulum: a focal point of Zika virus infection. J. Biomed. Sci. 2020;27:27. doi: 10.1186/s12929-020-0618-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhopadhyay S., Kuhn R.J., Rossmann M.G. A structural perspective of the flavivirus life cycle. Nat. Rev. Microbiol. 2005;3:13–22. doi: 10.1038/nrmicro1067. [DOI] [PubMed] [Google Scholar]
- Myers K.A., Baas P.W. Kinesin-5 regulates the growth of the axon by acting as a brake on its microtubule array. J. Cell Biol. 2007;178:1081–1091. doi: 10.1083/jcb.200702074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadar V.C., Ketschek A., Myers K.A., Gallo G., Baas P.W. Kinesin-5 is essential for growth-cone turning. Curr. Biol. 2008;18:1972–1977. doi: 10.1016/j.cub.2008.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostergaard P., Simpson M.A., Mendola A., Vasudevan P., Connell F.C., van Impel A., Moore A.T., Loeys B.L., Ghalamkarpour A., Onoufriadis A. Mutations in KIF11 cause autosomal-dominant microcephaly variably associated with congenital lymphedema and chorioretinopathy. Am. J. Hum. Genet. 2012;90:356–362. doi: 10.1016/j.ajhg.2011.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peña A., Sweeney A., Cook A.D., Topf M., Moores C.A. Structure of microtubule-trapped human kinesin-5 and its mechanism of inhibition revealed using cryoelectron microscopy. Structure. 2020;28:450–457.e5. doi: 10.1016/j.str.2020.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phoo W.W., Li Y., Zhang Z., Lee M.Y., Loh Y.R., Tan Y.B., Ng E.Y., Lescar J., Kang C., Luo D. Structure of the NS2B-NS3 protease from Zika virus after self-cleavage. Nat. Commun. 2016;7:13410. doi: 10.1038/ncomms13410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian X., Nguyen H.N., Song M.M., Hadiono C., Ogden S.C., Hammack C., Yao B., Hamersky G.R., Jacob F., Zhong C. Brain-region-specific organoids using mini-bioreactors for modeling ZIKV exposure. Cell. 2016;165:1238–1254. doi: 10.1016/j.cell.2016.04.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robin G., Chappell K., Stoermer M.J., Hu S.H., Young P.R., Fairlie D.P., Martin J.L. Structure of West Nile virus NS3 protease: ligand stabilization of the catalytic conformation. J. Mol. Biol. 2009;385:1568–1577. doi: 10.1016/j.jmb.2008.11.026. [DOI] [PubMed] [Google Scholar]
- Robitaille J.M., Gillett R.M., LeBlanc M.A., Gaston D., Nightingale M., Mackley M.P., Parkash S., Hathaway J., Thomas A., Ells A. Phenotypic overlap between familial exudative vitreoretinopathy and microcephaly, lymphedema, and chorioretinal dysplasia caused by KIF11 mutations. JAMA Ophthalmol. 2014;132:1393–1399. doi: 10.1001/jamaophthalmol.2014.2814. [DOI] [PubMed] [Google Scholar]
- Rososinski A., Tran T., Galvin J., Patel C., Fung A.T. New findings from multimodal fundus imaging over 3 years of a patient with microcephaly, chorioretinopathy, and Kif11 mutation. Retin. Cases Brief Rep. 2019;13:79–83. doi: 10.1097/ICB.0000000000000538. [DOI] [PubMed] [Google Scholar]
- Rut W., Zhang L., Kasperkiewicz P., Poreba M., Hilgenfeld R., Drąg M. Extended substrate specificity and first potent irreversible inhibitor/activity-based probe design for Zika virus NS2B-NS3 protease. Antivir. Res. 2017;139:88–94. doi: 10.1016/j.antiviral.2016.12.018. [DOI] [PubMed] [Google Scholar]
- Saeki E., Yasuhira S., Shibazaki M., Tada H., Doita M., Masuda T., Maesawa C. Involvement of C-terminal truncation mutation of kinesin-5 in resistance to kinesin-5 inhibitor. PLoS One. 2018;13:e0209296. doi: 10.1371/journal.pone.0209296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawin K.E., Mitchison T.J. Microtubule flux in mitosis is independent of chromosomes, centrosomes, and antiparallel microtubules. Mol. Biol. Cell. 1994;5:217–226. doi: 10.1091/mbc.5.2.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scaturro P., Stukalov A., Haas D.A., Cortese M., Draganova K., Płaszczyca A., Bartenschlager R., Götz M., Pichlmair A. An orthogonal proteomic survey uncovers novel Zika virus host factors. Nature. 2018;561:253–257. doi: 10.1038/s41586-018-0484-5. [DOI] [PubMed] [Google Scholar]
- Schlaitz A.L. Microtubules as key coordinators of nuclear envelope and endoplasmic reticulum dynamics during mitosis. Bioessays. 2014;36:665–671. doi: 10.1002/bies.201400022. [DOI] [PubMed] [Google Scholar]
- Schlaitz A.-L., Thompson J., Wong C., Yates J., Heald R. REEP3/4 ensure endoplasmic reticulum clearance from metaphase chromatin and proper nuclear envelope architecture. Dev. Cell. 2013;26:315–323. doi: 10.1016/j.devcel.2013.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah P.S., Link N., Jang G.M., Sharp P.P., Zhu T., Swaney D.L., Johnson J.R., Von Dollen J., Ramage H.R., Satkamp L. Comparative flavivirus-host protein interaction mapping reveals mechanisms of dengue and Zika virus pathogenesis. Cell. 2018;175:1931–1945.e18. doi: 10.1016/j.cell.2018.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan C., Xie X., Muruato A.E., Rossi S.L., Roundy C.M., Azar S.R., Yang Y., Tesh R.B., Bourne N., Barrett A.D. An infectious cDNA clone of Zika virus to study viral virulence, mosquito transmission, and antiviral inhibitors. Cell Host Microbe. 2016;19:891–900. doi: 10.1016/j.chom.2016.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shemetov A.A., Oliinyk O.S., Verkhusha V.V. How to increase brightness of near-infrared fluorescent proteins in mammalian cells. Cell Chem. Biol. 2017;24:758–766.e3. doi: 10.1016/j.chembiol.2017.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiryaev S.A., Farhy C., Pinto A., Huang C.T., Simonetti N., Elong Ngono A., Dewing A., Shresta S., Pinkerton A.B., Cieplak P. Characterization of the Zika virus two-component NS2B-NS3 protease and structure-assisted identification of allosteric small-molecule antagonists. Antivir. Res. 2017;143:218–229. doi: 10.1016/j.antiviral.2017.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shitamukai A., Matsuzaki F. Control of asymmetric cell division of mammalian neural progenitors. Dev. Growth Differ. 2012;54:277–286. doi: 10.1111/j.1440-169X.2012.01345.x. [DOI] [PubMed] [Google Scholar]
- Skoufias D.A., DeBonis S., Saoudi Y., Lebeau L., Crevel I., Cross R., Wade R.H., Hackney D., Kozielski F. S-trityl-L-cysteine is a reversible, tight binding inhibitor of the human kinesin Eg5 that specifically blocks mitotic progression. J. Biol. Chem. 2006;281:17559–17569. doi: 10.1074/jbc.M511735200. [DOI] [PubMed] [Google Scholar]
- Solomon I.H., Milner D.A., Folkerth R.D. Neuropathology of Zika virus infection. J. Neuroinfect Dis. 2016;7:1–6. doi: 10.4172/2314-7326.1000220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- To T.L., Piggott B.J., Makhijani K., Yu D., Jan Y.N., Shu X. Rationally designed fluorogenic protease reporter visualizes spatiotemporal dynamics of apoptosis in vivo. Proc. Natl. Acad. Sci. U S A. 2015;112:3338–3343. doi: 10.1073/pnas.1502857112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsetsarkin K.A., Kenney H., Chen R., Liu G., Manukyan H., Whitehead S.S., Laassri M., Chumakov K., Pletnev A.G. A full-length infectious cDNA clone of Zika virus from the 2015 epidemic in Brazil as a genetic platform for studies of virus-host interactions and vaccine development. MBio. 2016;7 doi: 10.1128/mBio.01114-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Wildenberg S.M., Tao L., Kapitein L.C., Schmidt C.F., Scholey J.M., Peterman E.J. The homotetrameric kinesin-5 KLP61F preferentially crosslinks microtubules into antiparallel orientations. Curr. Biol. 2008;18:1860–1864. doi: 10.1016/j.cub.2008.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waitzman J.S., Rice S.E. Mechanism and regulation of kinesin-5, an essential motor for the mitotic spindle. Biol. Cell. 2014;106:1–12. doi: 10.1111/boc.201300054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W., Cantos-Fernandes S., Lv Y., Kuerban H., Ahmad S., Wang C., Gigant B. Insight into microtubule disassembly by kinesin-13s from the structure of Kif2C bound to tubulin. Nat. Commun. 2017;8:70. doi: 10.1038/s41467-017-00091-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Smallwood P.M., Williams J., Nathans J. A mouse model for Kinesin Family Member 11 (Kif11)-associated familial exudative vitreoretinopathy. Hum. Mol. Genet. 2020 doi: 10.1093/hmg/ddaa018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen F., Armstrong N., Hou W., Cruz-Cosme R., Obwolo L.A., Ishizuka K., Ullah H., Luo M.H., Sawa A., Tang Q. Zika virus increases mind bomb 1 levels, causing degradation of pericentriolar material 1 (PCM1) and dispersion of PCM1-containing granules from the centrosome. J. Biol. Chem. 2019;294:18742–18755. doi: 10.1074/jbc.RA119.010973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojcik E.J., Buckley R.S., Richard J., Liu L., Huckaba T.M., Kim S. Kinesin-5: cross-bridging mechanism to targeted clinical therapy. Gene. 2013;531:133–149. doi: 10.1016/j.gene.2013.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf B., Diop F., Ferraris P., Wichit S., Busso C., Missé D., Gönczy P. Zika virus causes supernumerary foci with centriolar proteins and impaired spindle positioning. Open Biol. 2017;7:160231–160239. doi: 10.1098/rsob.160231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing H., Xu S., Jia F., Yang Y., Xu C., Qin C., Shi L. Zika NS2B is a crucial factor recruiting NS3 to the ER and activating its protease activity. Virus Res. 2020;275:197793. doi: 10.1016/j.virusres.2019.197793. [DOI] [PubMed] [Google Scholar]
- Yamaoka Y., Matsunaga S., Jeremiah S.S., Nishi M., Miyakawa K., Morita T., Khatun H., Shimizu H., Okabe N., Kimura H. Zika virus protease induces caspase-independent pyroptotic cell death by directly cleaving gasdermin D. Biochem. Biophys. Res. Commun. 2021;534:666–671. doi: 10.1016/j.bbrc.2020.11.023. [DOI] [PubMed] [Google Scholar]
- Yoon K.J., Song G., Qian X., Pan J., Xu D., Rho H.S., Kim N.S., Habela C., Zheng L., Jacob F. Zika-virus-encoded NS2A disrupts mammalian cortical neurogenesis by degrading adherens junction proteins. Cell Stem Cell. 2017;21:349–358.e6. doi: 10.1016/j.stem.2017.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yusof R., Clum S., Wetzel M., Murthy H.M., Padmanabhan R. Purified NS2B/NS3 serine protease of dengue virus type 2 exhibits cofactor NS2B dependence for cleavage of substrates with dibasic amino acids in vitro. J. Biol. Chem. 2000;275:9963–9969. doi: 10.1074/jbc.275.14.9963. [DOI] [PubMed] [Google Scholar]
- Zhong W., Chia W. Neurogenesis and asymmetric cell division. Curr. Opin. Neurobiol. 2008;18:4–11. doi: 10.1016/j.conb.2008.05.002. [DOI] [PubMed] [Google Scholar]
- Zhu Z., Gorman M.J., McKenzie L.D., Chai J.N., Hubert C.G., Prager B.C., Fernandez E., Richner J.M., Zhang R., Shan C. Zika virus has oncolytic activity against glioblastoma stem cells. J. Exp. Med. 2017;214:2843–2857. doi: 10.1084/jem.20171093. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The cells exhibit discrete ER localization of the protease. Asterisk marks a dividing cell in prolonged metaphase whose spindle appears to undergo abnormal rotations prior to anaphase onset. Time shown in min:sec.
A mitotic cell is observed to divide with normal timing and behavior. Time shown in min:sec.
A dividing cell expressing chimeric s-ZPrC-TagRFP-ER is blocked at metaphase. For comparison, nearby cells that do not appear to be transfected with the protease are seen entering and completing mitosis in a normal time frame of ∼60’. Time shown in min:sec.
A mitotic cell blocked in metaphase exhibits abnormal spindle movements and positioning. Time shown in min:sec.
HEK293-T cells with Tubulin-GFP (green) were co-transfected with mScarlet-H2A (red) and n-ZPrC-RFP670 (cyan) and observed in timelapse multi-channel confocal microscopy. A mitotic cell blocked in metaphase exhibits abnormal spindle movements and positioning. Time shown in min:sec.
Data Availability Statement
All relevant data are available from the lead contact upon request. There were no custom codes or algorithms used in this study.