

Abstract
High-throughput “-omics” analysis may provide a broader and deeper understanding of cancer biology to define prognostic and predictive biomarkers and identify novel therapy targets. In this review we provide an overview of studies where the peritoneal tumor component of peritoneal metastases from colorectal cancer (PM-CRC) and pseudomyxoma peritonei (PMP) were analyzed. Most of the available data was derived from DNA mutation analysis, but a brief review of findings from transcriptomic and protein expression analysis was also performed. Studies reporting genomic analysis of peritoneal tumor samples from 1,779 PM-CRC and 623 PMP cases were identified. The most frequently mutated genes in PM-CRC were KRAS, APC, SMAD4, BRAF, and PIK3CA, while in PMP KRAS, GNAS, FAT4, TGFBR1, TP53 and SMAD3/4 mutations were most commonly identified. Analyses were performed by single-gene analyses and to some extent targeted next-generation sequencing, and a very limited amount of broad explorative data exists. The investigated cohorts were typically small and heterogeneous with respect to the methods used and to the reporting of clinical data. This was even more apparent regarding transcriptomic and protein data, as the low number of cases examined and quality of clinical data would not support firm conclusions. Even for the most frequently mutated genes, the results varied greatly; for instance, KRAS mutations were reported at frequencies between 20–57% in PM-CRC and 38–100% in PMP. Such variation could be caused by random effects in small cohorts, heterogeneity in patient selection, or sensitivity of applied technology. Although a large number of samples have been subjected to analysis, cross-study comparisons are difficult to perform, and combined with small cohorts and varying quality and detail of clinical information, the observed variation precludes useful interpretation in a clinical context. Although omics data in theory could answer questions to aid management decisions in PM-CRC and PMP, the existing data does not presently support clinical implementation. With the necessary technologies being generally available, the main challenge will be to obtain sufficiently large, representative cohorts with adequate clinical data and standardized reporting of results. Importantly, studies where the focus is specifically on peritoneal disease are needed, where the study designs are aligned with clearly defined research questions to allow robust conclusions. Such studies are highly warranted if patients with PM-CRC and PMP are to derive benefit from recent advances in precision cancer medicine.
Keywords: Peritoneal metastasis (PM), colorectal cancer (CRC), pseudomyxoma peritonei (PMP), genomics, transcriptomics
Introduction
Adding “-omics” to a molecular term generally signifies comprehensive or global analysis of a set of molecules, genomics referring to analyses of the genome, transcriptomics to analysis of the transcriptome, and so on, typically driven by the emergence of high-throughput technologies. Importantly, in disease entities where extensive profiling has been performed, integrated analyses are used to combine data from different molecular levels (“multiomic” analysis) (1). Based on more recent advances in high-throughput technologies, a number of additional biological levels may now be interrogated, such as epigenomics, proteomics, metabolomics, and microbiomics, providing the possibility to unravel the complexity of human disease. In the peritoneal metastasis (PM) field, most of the available data is derived from genomic and to some extent transcriptomic analysis using standard technologies, which is therefore the focus of this review. Additionally, although no high-throughput proteomic studies have been identified, we have chosen to also include a small number of available studies reporting on protein expression.
PM is a common metastatic phenomenon associated with several cancerous disease entities, and comprises variable extent of tumor dissemination to the peritoneal surfaces. PM represents a huge burden on patients, their relatives, and society in general, and its treatment poses a substantial therapeutic challenge for health care professionals. Depending on the primary cancer origin and therapeutic opportunities, individual prognosis varies greatly, from 4–8 months median life expectancy from diagnosis of PM from pancreatic cancer (2) to up to 42 months overall survival in resectable PM from colorectal cancer (PM-CRC) after surgical intervention (3). This review will focus on two important peritoneal disease entities that are commonly managed with a surgical treatment strategy, PM-CRC and pseudomyxoma peritonei (PMP). CRC is one of the most frequent cancers in the Western world, with the incidence rising in previous low-incidence countries because of increasing population age and life-style changes (4). Metastatic progression occurs in approximately 50% of cases and is the dominant cause of CRC mortality, with the liver, lungs and peritoneal cavity being the most common metastatic sites (5). PM-CRC is associated with poor prognosis and poor response to systemic chemotherapy compared to other metastatic sites, and thus represents a particular therapeutic challenge (6). Current standard-of-care in resectable cases is cytoreductive surgery (CRS) and hyperthermic intraperitoneal chemotherapy (HIPEC). In contrast to PM-CRC, PMP is a rare cancer that most commonly originates from a ruptured appendiceal mucinous tumor. PMP exhibits an almost exclusive distribution to the peritoneal surfaces and may thus serve as a model disease of peritoneal tumor dissemination. As for PM-CRC, resectable cases are managed with CRS-HIPEC, but long-term outcomes are much more favorable, with up to 50% of cases being cured by the standard treatment with a median overall survival expectancy of 16 years (7). However, in cases where surgery cannot provide long-term disease control, therapeutic options are limited with very poor responses to systemic treatment.
Over the last two decades knowledge derived from omics analyses regarding molecular signaling pathways involved in cancer development, progression and metastasis has contributed to development of new therapeutic concepts in the cancer field. Examples are the success of targeting human epidermal growth factor receptor 2 (HER2) in breast cancer (8), the targeting of tyrosine kinases by imatinib which has completely transformed the outlook for subgroups of patients with gastrointestinal stromal tumors and chronic myeloid leukemia (9), and the emergence of drugs inhibiting immune checkpoint molecules that have shown activity in several cancer entities (10). These biomarker-driven therapies exploit specific dependencies of individual tumors, caused by single molecular aberrations, complex changes in signaling pathways, or dysfunction at the cellular interaction level. The knowledge of how to administer the correct drug to the right patient in the appropriate therapeutic setting requires identification and understanding of the molecular profile/composition of the individual cancer or metastatic site. There are a number of questions pertaining to the clinical management of PM-CRC and PMP where molecular knowledge in theory might contribute useful information, such as predicting prognosis, therapy response and aiding patient selection for surgery. Work to unravel the molecular features of PM is therefore ongoing among research groups across the world, including efforts to characterize PM at the genomic, transcriptomic and protein levels. In this review we have chosen to focus only on reports where the peritoneal components of PM-CRC and PMP are subjected to molecular analysis, which means that details regarding the primary tumor and metastatic locations other than the peritoneum are not included. We will attempt to provide an overview of the current status of knowledge in the field focusing on the clinical utility of omics-derived data, address shortcomings and impediments to utilizing the information that is available today, and point out the most important steps to move the field forward. We present the following article in accordance with the Narrative Review Checklist (available at http://dx.doi.org/10.21037/jgo-20-136).
Methods
Literature search was conducted in February 2020 in PubMed, using individual and combined terms such as peritoneal, carcinomatosis, metastases, PM-CRC, colorectal, pseudomyxoma peritonei, PMP, appendiceal, appendix, neoplasia, cancer, mucinous, molecular, characterization, profiling, sequencing, mutation, gene expression, transcriptome, copy number, proteomics, protein analysis, immunohistochemistry and mass spectrometry. Studies were excluded due to, e.g., very low number of cases or if number of cases was not possible to determine, and is further detailed in the relevant sections.
Molecular approaches
The molecular methods used to analyze PM-CRC and PMP samples in the identified studies are generally standard methods, and details are thoroughly explained by experts elsewhere. We will only comment briefly on the main methods that have been applied, focusing on some aspects that may have relevance for the choice of methods in future studies of PM-CRC and PMP.
Genomic analyses
Genomic analyses have been performed using three main groups of methods. For single-gene analysis polymerase chain reaction (PCR)-based assays are commonly used (11). PCR-based methods typically require low to medium amounts of input DNA (25–100 ng) to detect DNA fragments of 200–400 base pairs (bp), with high sample throughput (number of samples that can be analyzed simultaneously), generally at a low cost, high sensitivity (proportion of mutated DNA in a sample can be as low as 0.1–1%) and with simple post-analytic data analysis (12). In some, generally less recent papers, classic Sanger sequencing was also utilized for single-gene analysis (13). This sequencing method requires a medium amount of input DNA (100 ng) for sequencing of one large DNA fragment (500–600 bp) at a time with high sample throughput. The cost is generally low and data analysis is simple, while the major draw-back is the relatively low sensitivity (requires 10–20% mutated DNA in the sample), which precludes analysis of samples with low tumor content. Although the principles of next-generation sequencing (NGS) are similar to Sanger sequencing, new technological platforms now enable massively parallel sequencing of millions of fragments simultaneously per run (14). A number of assay providers have tailored targeted panels specifically to investigate cancer, focusing on actionable mutations, quantifying tumor mutational burden, or broader, more explorative panels. Depending on the proportion of the genome to be analyzed and the desired sensitivity of detection, objective parameters of the method will vary substantially. For instance, for application of a small panel targeting 50–100 cancer-relevant genes, a small amount of input DNA (10–20 ng) will be required, while exome sequencing (sequencing all exons of the genome) may require more than 1 µg of DNA. The desired sequencing depth (the average number of times a particular nucleotide is sequenced per sample) will influence sample throughput, analytical sensitivity and not least the cost of performing NGS. If a small targeted panel is used, high sequencing depth can be obtained for analysis of a reasonable number of samples at an acceptable cost, providing acceptably high sensitivity (1–2% mutated DNA in the sample necessary) to detect mutations even in samples with low tumor content or mutations that are only present in tumor subclones (15). In contrast, if the main aim is to perform broad exploration of mutations in a sample; to achieve similar sensitivity, the per-sample cost will increase and sample through-put decrease dramatically. Common for all NGS strategies is that a down-stream bioinformatics work-flow is required. Customized solutions are to some extent available, particularly from vendors of targeted panels, but nevertheless, bioinformatics competence is needed as part of the research team to ensure correct processing and interpretation of the data. Hence, for genomic analysis, the choice of method will depend to a great extent on the specific aim of the study and available resources with respect to funding and competence.
Transcriptomic analyses
Analysis of the tumor transcriptome, i.e., the RNA molecules present in a tumor sample, presents a greater challenge than genomic analysis (DNA), if not only by the fact that from the ~20,000 human genes, more than 100,000 mRNA transcripts may be generated (16). Although analyses may be performed to quantify single RNA molecules using techniques such as reverse transcriptase PCR assays; for explorative purposes, high-throughput technologies are standard. The studies identified for the purpose of this review have reported transcriptomic information generated by oligonucleotide microarray analysis of RNA isolated from bulk tissue samples. However, unlike somatic mutations, which by definition originate in tumor cells and are identified by comparison with a well-established reference genome, analyses of detected RNA transcripts from bulk tumor samples is more complex. Unlike DNA, which has a stable molecular structure and is present at relatively constant levels, RNA molecules are less stable and the levels vary over time in response to internal and external stimuli. Furthermore, the transcription profile of a tumor sample depends on the cellular composition of the sample. The dominating cell type, often tumor cells, will contribute most to the profile, but a varying proportion of the transcripts will be derived from normal cells, such as fibroblasts, endothelial cells, and infiltrating immune cells. Down-stream transcriptome analysis also represents a challenge, as a plethora of bioinformatics and statistical tools exist to analyze RNA data, and no standardized methods or analysis pipelines exist. Although the added complexity makes interpretation of transcriptomic information from bulk tissue more demanding than genomic data, it also offers the possibility of capturing a more functional picture of tumor biology than genomic information alone, such as down-stream activity in signaling pathways that could be targeted by anti-cancer drugs.
Proteomic analyses
As RNA is translated to proteins, moving closer to the main effector molecules in the cells adds another vast layer of complexity; looking at numbers alone, >1,000,000 proteins have been identified, not including protein modifications (16). High-throughput proteome analysis of bulk tissue has some of the same challenges as were described for transcriptome analysis, but no high-throughput proteomic analyses were applied in the identified studies of PM-CRC and PMP samples. In these studies, standard immunohistochemistry-based (IHC) methods were used to analyze formalin-fixed tissues. Provided that the antibodies used specifically bind the intended target, IHC has the advantage of identifying both the type of cells that expresses the protein in question and the location in the tissue, allowing (semi)quantitative comparison of samples. Quantification of the expression of specific proteins has been implemented in clinical practice as biomarkers for anti-cancer therapies, such as expression of HER2 being strongly predictive of response to anti-HER2 treatment in breast cancer (17). Thus, analysis of protein expression, although potentially complex and challenging, has already proven to be relevant for identification of biomarkers in cancer. However, because the target must be predefined, an important limitation of antibody-based approaches is the low potential for biomarker discovery; for such purposes, high-throughput, explorative methods are needed.
Tumor enrichment strategies
As previously discussed, when analyzing bulk tissue samples, a concern is that the tissue will be variably composed of numerous cell types harboring different genomic, transcriptomic and proteomic profiles. In principle, particularly as immunotherapy is being increasingly implemented in the clinic, analytical “signal” derived from other cell types may be of equal interest, depending on the research questions. Often however, the focus has been on tumor mutation analysis or identification of tumor-specific transcriptomic profiles, and in the identified studies, different tumor enrichment strategies were applied to facilitate such analyses, particularly for the PMP studies. The “simplest” of these is macro dissection of the tissue specimen, where a hematoxylin-eosin (H&E) stained slide is inspected to identify areas of tumor tissue, and the tissue block may be dissected accordingly to enrich the tumor part of the sample. Manual micro dissection dramatically increases the workload, as tumor areas again are selected on an H&E slide that is subsequently used as a guide to dissect serial sections obtained from the same tissue block. With laser capture micro dissection, work load again increases and specialized equipment is needed. With this technique, it is possible to harvest cells of interest or remove undesirable parts of the tissue from slides under direct microscopic guidance. Whichever enrichment method is chosen, the sensitivity of the down-stream method will be crucial, for instance when performing mutation analysis in cases where the amount of tumor DNA is very low. For transcriptomic analysis, another interesting post-analytical “enrichment” opportunity may be considered. Based on identification of transcriptomic profiles of individual cell types, algorithms using expression of cell type-specific markers may be used to predict the relative composition of cells in tissues based on gene expression. This strategy has proven particularly useful for quantification of immune cell infiltration in tumor tissues (18).
Genomic analyses in PM-CRC
The identified literature on genomic analysis of PM-CRC mainly comprises mutation analyses (14 studies), with a subset of samples analyzed for copy number variation (CNV, 4 studies) (Figure 1). Analyses are reported based on single-gene analysis or NGS, and most of the studies report data from moderately sized patient cohorts, typically comprising 50–60 cases, with some exceptions (Table 1). Two larger cohorts, which comprised 397 and 378 cases respectively, applied mainly single-gene mutation analysis, with NGS limited to a smaller subset of cases (n≈60) in one of the studies (19,22). Seven studies (comprising 738 cases) reported data from cohorts composed of PM-CRC only, utilizing mainly single-gene analysis to examine the mutation frequencies of KRAS and BRAF (22-28). The remaining studies have had characterization of metastatic CRC (mCRC) as the main aim, comparing mCRC with primary CRC (pCRC) or comparing metastatic sites, and in a recent review of this topic, the proportion of PM-CRC in such studies was shown to be relatively low, comprising only 4% of the analyzed samples (22). We excluded such studies from analysis if the number of PM-CRC cases included was very low (typically <10 cases) or if it was not possible to derive the number of cases included. Studies that concentrated on associations between mutations identified in pCRC and later development of PM-CRC were also excluded, because this question was not the focus of this review and often, it was not clear if the mutation data was derived from analysis of pCRC or PM-CRC.
Figure 1.
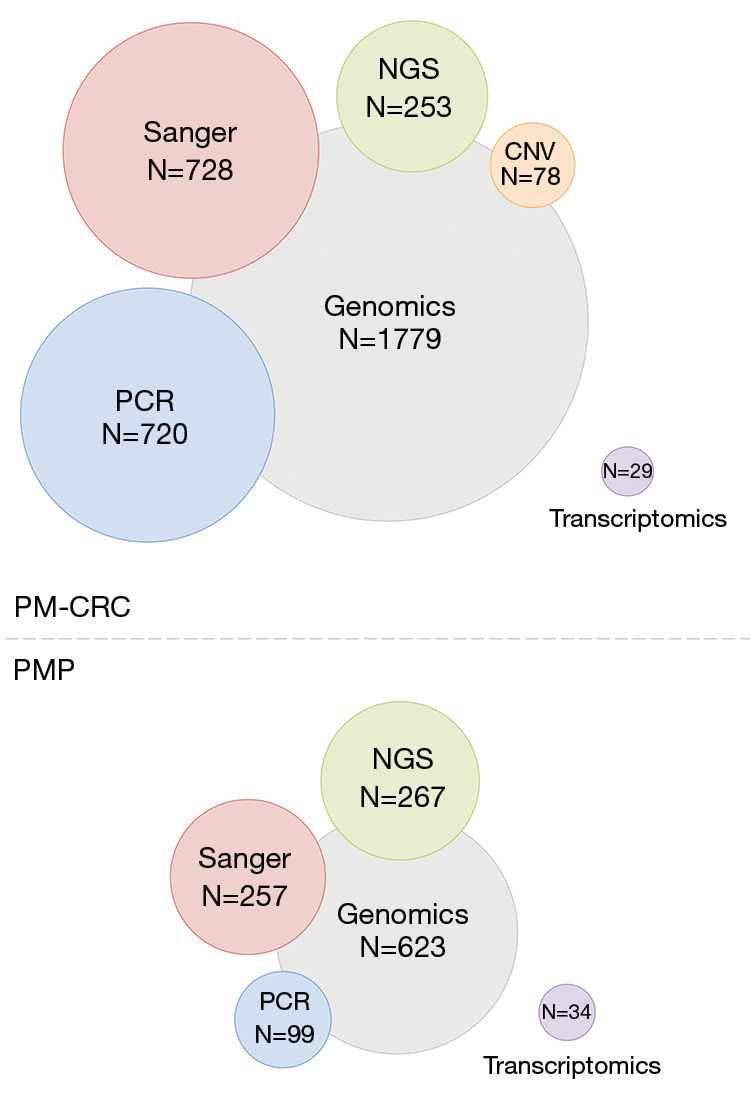
Overview of the number of samples (N) subjected to omics analysis in PM-CRC (upper panel, 22 studies included) and PMP (lower panel, 21 studies included), also showing the distribution between methods used within the field of genomics. PCR, polymerase chain reaction (including Luminex technology and High-Resolution Melting); Sanger, Sanger sequencing, NGS, next generation sequencing, CNV, copy number variation; PM-CRC, peritoneal metastases from colorectal cancer; PMP, pseudomyxoma peritonei.
Table 1. Median frequency of mutated genes found in PM-CRC patient samples in more than one study.
| Mutated genes | Median mutation, frequency (%) [min–max] | Median # samples analyzed [min–max] | References |
|---|---|---|---|
| TP53 | 54 [33–75] | 54 [51–57] | (19,20) |
| RAS, not specified* | 58 | 150 | (21) |
| KRAS | 45 [20–57] | 52 [11–397] | (19,20,22-31) |
| APC | 44 [31–57] | 56 [51–61] | (19,20) |
| SMAD4 | 22 [15–29] | 56 [51–60] | (19,20) |
| BRAF | 15 [6–40] | 59 [11–378] | (19,20,22-25,27-30,32,33) |
| PIK3CA | 13 [9–14] | 58 [51–103] | (19,20,23) |
| FBXW7 | 9 [8–9] | 55 [51–59] | (19,20) |
| GNAS | 4 [2–5] | 55 [51–58] | (19,20) |
| NRAS | 4 [2–5] | 96 [47–378] | (19,22) |
| PTEN | 4 [2–6] | 55 [51–58] | (19,20) |
| JAK3 | 4 [2–6] | 55 [51–59] | (19,20) |
| AKT1 | 3 [2–4] | 55 [51–59] | (19,20) |
| STK11 | 3 [2–4] | 54 [51–56] | (19,20) |
| CTNNB1 | 3 [2–3] | 56 [51–61] | (19,20) |
| HNF1A | 3 [2–4] | 51 [50–51] | (19,20) |
| SMO | 3 [2–4] | 54 [51–57] | (19,20) |
| ERBB2 | 2 [2–2] | 55 [51–58] | (19,20) |
Data from (19) was provided through personal communication. *, frequency of KRAS and NRAS mutations separately were not provided. PM-CRC, peritoneal metastases from colorectal cancer.
The most commonly occurring mutations were reported in the TP53 (median 54%; min-max 33–75%), KRAS (45%; 20–57%), APC (44%; 31–57%), SMAD4 (22%; 15–29%), BRAF (15%; 6–40%), and PIK3CA (13%; 9–14%), but the frequencies were quite variable for most of these genes (Table 1 and for details of individual studies in Table S1). Most of the data was derived from single-gene analysis (81% of samples) and the remaining studies used targeted NGS sequencing panels of different size (14%), with no broad exploratory analysis (exome or whole genome sequencing) being performed. Therefore, in principle, only mutations in predefined genes of known or suspected cancer relevance would be identified by these approaches. Interestingly, in one study where NGS analysis was performed in 51 PM-CRC cases using larger targeted gene panels (covering 341–468 genes) more than 104 gene mutations were reported at a frequency above 2%, although the top most frequently detected mutations were similar to the genes listed in Table 1 (20). Other frequently mutated genes identified in this study were PTPRS (12%), NOTCH3 (10%), FAT1 (10%), RNF43 (10%), FLT4 (10%), and GRIN2A (10%) (a list of other gene mutations detected >2% is available at http://cdn.amegroups.cn/static/application/7378337a6e200b5ef8e47395aec5de9f/JGO-20-136-table.docx). The usefulness of extending the data by application of larger panels remains unclear, since too few samples have been analyzed to provide meaningful insight in a clinical context.
The available studies employed variable approaches to analyze the presence of mutations in a clinical context, and half of the reports were generated from general mCRC cohorts. An important emphasis has been placed on evaluation of associations between mutational profiles and clinicopathological parameters, such as whether the tumor was microsatellite stable or instable, tumor differentiation, and pCRC location (right- or left-sided tumors), thus typically exploring relationships with known prognostic parameters in PM-CRC. Findings of general interest from these studies were that in BRAF-mutated mCRC, the pCRC tended to be located in the right colon, have mucinous differentiation and be associated with PM-CRC development with inferior clinical outcome (overall survival) compared to wild-type cases. In two studies reporting data from PM-CRC specifically, the presence of BRAF mutations was associated with inferior overall survival (after CRS-HIPEC) in a study of 97 cases (25), and with inferior cancer specific survival in a study including 378 cases, which also reported similar associations with KRAS mutation (22). Six studies presented analysis of associations between the presence of single mutations or combinations of mutations in mCRC in general, but no associations between specific mutations in PM-CRC and outcome were reported (20,21,29,32-34).
Analyses of CNV
Only 4 studies were identified reporting on CNV analysis of a total of 78 samples, with the largest study comprising 52 samples (Table 2). Two of the studies identified CNV in large areas of the genome, with genes associated with CRC being present in gained regions of chromosome 1p (REG4 and NOTCH2) and 15q (MAP2K1, SMAD3, SMAD6 and IGF1R). Chromosomal gains in these regions (both chr 1p and 15q or in 15q alone) were associated with poor overall survival (≤2 years) following CRS-HIPEC (35). When comparing CNV in PM-CRC with other CRC locations, gains in chromosomes 5p and 12p were found to be more commonly present in PM-CRC (38). In a study including 12 PM-CRC cases, concordance of CNV findings was reported when comparing pCRC samples with matched mCRC samples, including PM-CRC (36).
Table 2. Chromosome regions of copy number gain and loss in PM-CRC.
| Regions of CNV gains | Regions of CNV loss | # samples analyzed | References |
|---|---|---|---|
| Chr. 1p/q, 2p/q, 5p, 7p/q, 8q, 12p/q, 13q, 15q, 16p and 20p/q | Chr. 1p, 4p/q, 8p, 15q, 17p/q, 18p/q, 22q | 52 | (35) |
| Chr 8* | Chr 8, 18, 20, X* | 12 | (36) |
| Chr 1p | 7 | (37) | |
| Chr 7, 5p, 8q, 12p, 13q, 20 | Chr 4q, 8p, 17p, 18 | 7 | (38) |
PM-CRC, peritoneal metastases from colorectal cancer; Chr, chromosome; CNV, copy number variation. *, compared to matched pCRC.
Genomic analyses in PMP
In PMP, the variation in methods used for mutation detection was similar to that of PM-CRC with results being based on single-gene analysis or NGS (Figure 1). A total of 623 cases from 19 studies were analyzed and the most commonly reported mutations were detected in the KRAS, GNAS, FAT4, TGFBR1, TP53 and SMAD3/4 genes at median frequencies of 78%, 44%, 35%, 21%, 17%, and 16%, respectively (Tables 3,4, and for details of individual studies in Table S2). The studies mostly comprised small cohorts and the variability between reported results was even more pronounced than for PM-CRC. For instance, the lowest detection frequency of KRAS and GNAS mutations were 38% and 17%, respectively, in contrast to other studies reporting mutations in up to 100% of cases for both genes. This rather extreme variation may be explained, in part, by small cohorts not providing representative selection of cases, in addition to varying amount of tumor cells in the analyzed tissues and different sensitivity of the employed detection methods. It is well known that PMP samples, particularly from low-grade tumors, are frequently dominated by large amounts of extracellular mucin, often with few or no visible tumor cells, and consequently, the DNA isolated from such samples will mainly be derived from normal cells. To compensate for this, different sample enrichment strategies were applied to almost 80% of the analyzed samples, such as macro dissection in 7%, manual micro dissection in 64%, and laser capture micro dissection in 8%. However, in cases with very few detectable tumor cells, the input DNA may still have been insufficient for reliable detection of mutations, given that the analytical sensitivity of the methods used varied substantially (between 1% and 20%). This also means that the probability of detecting mutations is likely to be higher in samples with high cellularity, which would suggest that high-grade cases would be over-represented among the cases where mutations were detected and the accuracy of reports regarding low-grade cases would be similarly lower.
Table 3. Median frequency of mutated genes found in PMP patient samples (findings in more than one study).
| Mutated genes | Median mutation, frequency (%) [min–max] | Median # samples analyzed [min–max] | References |
|---|---|---|---|
| KRAS | 78 [38–100] | 18 [5–150] | (25,28,39-55) |
| GNAS | 44 [17–100] | 40 [5–66] | (39,42,43,45-55) |
| FAT4 | 35 [20–30] | 8 [5–10] | (46,53) |
| TGFBR1 | 21 [20–22] | 10 [9–10] | (46,52) |
| TP53 | 17 [5–38] | 16 [5–75] | (41-43,46,49-51,53,54) |
| SMAD3/4 | 16 [3–60] | 19 [5–66] | (42,50,51,54,55) |
| APC | 11 [2–20] | 19 [5–66] | (53-55) |
| ATM | 11 [6–16] | 19 [19–19] | (50,55) |
| FGFR2/3 | 7 [3–20] | 15 [5–40] | (42,43) |
| PIK3CA | 6 [2–10] | 31 [19–66] | (42,54,55) |
| CTNNB1 | 6 [3–10] | 25 [10–40] | (42,46,49) |
| HNF1A | 5 [3–7] | 28 [15–40] | (42,43) |
PMP, pseudomyxoma peritonei.
Table 4. Median frequency of mutated genes found in PMP patient samples (additional mutations found in one study only).
| Identified by only one study | Mutation frequency (%) | # samples analyzed | References |
|---|---|---|---|
| BRAF | 14 | 21 | (55) |
| cMET | 11 | 19 | (55) |
| RB1 | 13 | 8 | (47) |
| RNF43 | 6 | 66 | (54) |
| NRAS | 5 | 21 | (55) |
| MLH1 | 5 | 19 | (55) |
| AKT1 | 5 | 19 | (50) |
| PDGFRA | 3 | 40 | (42) |
| SMO | 3 | 40 | (42) |
| CDH1 | 3 | 40 | (42) |
PMP, pseudomyxoma peritonei.
For most of the PMP cases included (~75%), some clinicopathological data was available, but associations between such data and the presence of mutations were reported in only 19% of the cases. Two studies found that tumors with signet ring cell differentiation tended to exhibit wild-type GNAS (39) and KRAS (40), suggesting that the molecular drivers of this tumor entity may be undetermined. No significant differences were identified between high-grade and low-grade disease, mirroring the findings and conclusions of a recent systematic review of somatic alterations in primary appendiceal tumors (56). Conflicting conclusions were drawn regarding mutation status and outcome, as two studies reporting single gene analysis of GNAS and KRAS in (n=55) (39) and of KRAS (n=64) (41) did not observe associations with overall survival. In contrast, a recently reported study using a small targeted NGS panel to analyze 40 PMP cases suggested that GNAS and KRAS mutations were associated with inferior progression-free survival (PFS), and the association was preserved for KRAS mutation in multivariable analysis (42). In 15 recurrent cases of PMP treated with capecitabine and bevacizumab where 20% responded to treatment, GNAS mutation was associated with shorter PFS compared to wild-type cases (43).
Transcriptomic analysis in PM-CRC and PMP
Four studies reported gene expression analysis performed on a total of 29 PM-CRC cases using microarray technology (57-60). Because of the low number of studies available, for descriptive purposes, we chose to include two studies that involved only 4 cases each (59,60). Gene expression profiles of PM-CRC showed high expression of genes involved in WNT signaling (LGR5, RNF43, CTHRC1), FGFR4, CFTR, HOXA9, TNRC9, VIL1, V-MYB, MXRA5 and COL12A1 (57,59,60). Twenty genes located on chromosome arm 5p, a region that is commonly gained in PM-CRC (Table 2), were highly expressed in PM-CRC compared to liver metastases and primary carcinomas (59).
Two of the studies included both PM-CRC and PMP samples, comparing these at the transcriptional level with somewhat conflicting conclusions. One study identified distinctly different gene expression patterns when comparing PM-CRC and PMP (n= 15 and 26, respectively), and suggested that gene expression signatures could predict patient outcome in PMP (57). Cases with poor overall survival were found to have high expression of mucin-related genes, such as MUC-5AC, MUC-2, and TFF1/2. In contrast, a study comparing 6 PM-CRC and 8 PMP cases concluded that the gene expression patterns were quite similar, as only 64 out of 1,090 high variance genes were differentially expressed, while the profile was distinct from 14 liver metastases that were analyzed simultaneously (58). Genes involved in metastasis, angiogenesis, cell cycle regulation, cell proliferation and cell adhesion were distinctly altered between the two metastatic sites. Genes associated with metastasis, such as CDH17, ALCAM, CD2 and CD14 were highly expressed at both sites, while TIMP-2, IGF-1 and HIF1-a (involved in extracellular matrix, growth and angiogenesis) were highly expressed in only in PM-CRC and PMP compared to the liver metastases. Given the low number of samples included in the transcriptome analyses, the findings reported should be interpreted with caution and the available data is insufficient to provide guidance for management of PM-CRC and PMP.
Protein expression analysis in PM-CRC and PMP
In contrast to pCRC and to some extent other mCRC sites, the exploration of proteomic biomarkers in PM-CRC has been very limited. Only one report was identified, where a large number of PM-CRC cases was available (n=465) and protein expression was analyzed using IHC. However, interpreting the reported data was made difficult by a lack of reporting the number of samples analyzed for expression of individual biomarkers (only percentages were given). The main proteomic findings highlighted in this study were high expression of topoisomerase 1 (TOPO 1, in 54% of cases), which is suggested as a predictive biomarker of response to irinotecan; high expression of excision repair cross-complementation group 1 (ERCC1, in 41% of cases) which has been associated with resistance to oxaliplatin, and low expression of thymidylate synthase (TS, in 25% of cases), which is associated with responses to fluoropyrimidine-based therapies. Comparing PM-CRC to pCRC, the investigators found higher expression of TOPO1 and ERCC1, which would point to favoring irinotecan over oxaliplatin for treatment of PM-CRC. The main limitation of the study was the lack of detail with respect to reporting cases analyzed, leaving questions regarding the general validity of the results (19).
Three studies analyzing protein expression in PMP samples using IHC were identified. The number of samples included in each cohort was low (10–66 cases), but some of the findings were quite consistent across the studies for the most frequently analyzed proteins (Table S3). For TOPO1 (63–76% positive cases), ERCC1 (23–44% positive) and TS (8–16% positive), results were qualitatively relatively similar to the results reported for PM-CRC, pointing towards favoring irinotecan and fluoropyrimidines over oxaliplatin in PMP. Available data generally suggest low efficacy of systemic chemotherapy in PMP, and therefore, the clinical utility of these results may seem low. However, the relatively reproducible findings across the three studies for this subset of biomarkers suggest that focused analysis of protein expression could be feasible to validate.
Discussion
The identification and interpretation of literature for this review was challenging because the available datasets were often complex and were not always clearly and consistently defined with respect to type and number of samples that were analyzed. For instance, in several studies, data from PM-CRC was reported together with results from pCRC, other metastatic sites, or even together with PMP cases, and the actual data contribution from each entity was often not distinguishable. Also, the number of cases analyzed by different methods could vary substantially within the same study (for instance combined data from NGS and single-gene mutation analysis), and this information could only be accessed by extensive scrutiny of supplementary files or by direct communication with the authors. This is illustrated by the study by El-Deiry et al., which is an extensive study of almost 7,000 CRC cases, combining single-gene analysis, NGS and IHC to provide a broadly-based descriptive profile of pCRC and mCRC at the genomic and protein levels (19). Although the study included 465 PM-CRC cases, which is the largest cohort reported to date, no clinical data was available, and the number of PM-CRC cases included in analyses of individual parameters varied and was not reported specifically. When comparing findings between studies, incomplete descriptions of the included datasets is a major challenge, and the lack of dedicated studies reporting specifically on PM-CRC and PMP thus represents a major impediment to drawing solid conclusions in this context.
The most robust data was generated from mutation analyses. When comprehensively viewing the results, some data seems to be relatively consistently reported, such as order of the most frequently mutated genes in PM-CRC and PMP, but the large variation reported in gene mutation frequencies are a concern that leads to uncertainty regarding the true rates of even the most commonly detected mutations. When subgroup analyses are subsequently performed to investigate associations with clinicopathological parameters and outcome in the same cohorts, lack of consistency across studies would be expected. Therefore, the utility as to guiding treatment decisions regarding chemotherapy or selection of patients for CRS-HIPEC cannot in our opinion be deduced from these studies. Another important consideration when interpreting omics data in a clinical context is whether the samples analyzed can be considered to be representative of the disease entity in question. The potential bias towards PMP samples with high cellularity being more likely to contribute to results in mutation analysis than low-grade cases was previously mentioned. Another issue, particularly relevant in PM-CRC, is that samples from almost all analyzed cases were collected at the time of surgery, i.e., by definition from potentially resectable patients, while samples from non-resectable patients are rarely available. This is of course almost inevitable, since specimens from patients in a metastatic setting are challenging to come by from both a practical and an ethical perspective. Since only a small proportion of PM-CRC cases are offered curatively intended surgical intervention, molecular findings in samples collected at such procedures may not be representative of the majority of PM-CRC cases, and validation in metastatic cohorts not eligible for surgery should be considered when possible.
Mutation analysis remains an important pillar in development of precision cancer medicine, although in mCRC, the number of actionable mutations is not high at present. Some of the mutations detected in PM-CRC and PMP samples have been shown to be associated with response or absence of response to specific drugs. For instance, KRAS mutations are associated with lack of response to EGFR inhibition, and mutation analysis has been implemented as a routine diagnostic criterion in many national programs to identify patients likely to benefit from therapy targeting EGFR (61). The BRAFV600E mutation, which is commonly detected in PM-CRC, has been successfully targeted in metastatic melanoma with BRAF inhibitors, but because of adaptive feedback reactivation of MAPK signaling often mediated by EGFR, lack of efficacy has been observed in mCRC. Recently, in a randomized trial investigating combined treatment targeting BRAF (dabrafenib), EGFR (panitumumab), and MEK (trametinib) in BRAFV600E mCRC, the combination treatment was shown to be feasible with a response rate of 20%, although the median PFS was only 4.2 months (62). This study illustrates how detailed biological knowledge may be translated directly into clinical trials in the mCRC field, with potential impact on future patient management. Although most of the current biomarkers in the precision cancer medicine field are genomic, the study also illustrates the key role of complex down-stream signaling in response and resistance to therapy, and biomarkers may in the future be derived from transcriptomic or proteomic characterization of cancer. Extensive research is ongoing within the pharmaceutical industry and academia to increase the repertoire of therapeutic targets and available drugs. To ensure that the PM-CRC and PMP disease entities are well positioned when novel treatment opportunities arise continued efforts to establish detailed characterization of tumors remains of high importance.
Conclusions
Although omics technologies have the potential to substantially impact cancer management, the information available from such approaches has not yet reached a level that can influence management of PM-CRC and PMP. In this review, we have pointed out some of the major hurdles that currently restrict integration of omics data in this setting. Since the technologies needed to address genomics and transcriptomics are now standardized, generally available, and becoming more affordable, the main challenge is not the technology, but obtaining sufficiently large, representative cohorts with adequate clinical data, including follow-up, to allow robust analysis with specific focus on peritoneal disease. Additionally, accurate, transparent, and standardized reporting of sample collection, processing and analysis will be necessary to allow cross-study comparisons. For PM-CRC, the volume in many centers may be large enough to support single-center cohorts, although an advantage could be gained through standardization to facilitate generation of larger combined cohorts across centers. For a rare disease such as PMP, work should be coordinated between centers to increase the number of available cases for analysis and maximize the output from limited resources, a work that is currently ongoing within the EuroPMP COST Action research network (63).
Acknowledgments
The authors thank Dr. Vigdis Nygaard for critical reading of the manuscript. This article is based upon work from COST Action CA17101, the European Network on Pseudomyxoma Peritonei, supported by COST (the European Cooperation in Science and Technology).
Funding: CLA and KGF are fellows of the Norwegian Cancer Society (grant number 4499184 to KF).
Ethical Statement: The authors are accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Open Access Statement: This is an Open Access article distributed in accordance with the Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License (CC BY-NC-ND 4.0), which permits the non-commercial replication and distribution of the article with the strict proviso that no changes or edits are made and the original work is properly cited (including links to both the formal publication through the relevant DOI and the license). See: https://creativecommons.org/licenses/by-nc-nd/4.0/.
Footnotes
Provenance and Peer Review: This article was commissioned by the Guest Editors (Paul H. Sugarbaker and Kurt Van der Speeten) for the focused issue “Intraperitoneal Chemotherapy for Peritoneal Metastases: HIPEC, EPIC, NIPEC, PIPAC and More” published in Journal of Gastrointestinal Oncology. This article has undergone external peer review.
Reporting Checklist: The authors have completed the Narrative Review Checklist. Available at http://dx.doi.org/10.21037/jgo-20-136
Conflicts of Interest: All authors have completed the ICMJE uniform disclosure form (available at http://dx.doi.org/10.21037/jgo-20-136). The focused issue was sponsored by the Peritoneal Surface Oncology Group International (PSOGI). The authors have no other conflicts of interest to declare.
References
- 1.Hasin Y, Seldin M, Lusis A. Multi-omics approaches to disease. Genome Biol 2017;18:83. 10.1186/s13059-017-1215-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tabernero J, Chiorean EG, Infante JR, et al. Prognostic factors of survival in a randomized phase III trial (MPACT) of weekly nab-paclitaxel plus gemcitabine versus gemcitabine alone in patients with metastatic pancreatic cancer. Oncologist 2015;20:143-50. 10.1634/theoncologist.2014-0394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Quenet F, Elias D, Roca L, et al. A UNICANCER phase III trial of hyperthermic intra-peritoneal chemotherapy (HIPEC) for colorectal peritoneal carcinomatosis (PC): PRODIGE 7. J Clin Oncol 2018;36:LBA3503. 10.1200/JCO.2018.36.18_suppl.LBA3503 [DOI] [Google Scholar]
- 4.Arnold M, Sierra MS, Laversanne M, et al. Global patterns and trends in colorectal cancer incidence and mortality. Gut 2017;66:683-91. 10.1136/gutjnl-2015-310912 [DOI] [PubMed] [Google Scholar]
- 5.Van Cutsem E, Cervantes A, Adam R, et al. ESMO consensus guidelines for the management of patients with metastatic colorectal cancer. Ann Oncol 2016;27:1386-422. 10.1093/annonc/mdw235 [DOI] [PubMed] [Google Scholar]
- 6.Franko J, Shi Q, Meyers JP, et al. Prognosis of patients with peritoneal metastatic colorectal cancer given systemic therapy: an analysis of individual patient data from prospective randomised trials from the Analysis and Research in Cancers of the Digestive System (ARCAD) database. Lancet Oncol 2016;17:1709-19. 10.1016/S1470-2045(16)30500-9 [DOI] [PubMed] [Google Scholar]
- 7.Chua TC, Moran BJ, Sugarbaker PH, et al. Early- and long-term outcome data of patients with pseudomyxoma peritonei from appendiceal origin treated by a strategy of cytoreductive surgery and hyperthermic intraperitoneal chemotherapy. J Clin Oncol 2012;30:2449-56. 10.1200/JCO.2011.39.7166 [DOI] [PubMed] [Google Scholar]
- 8.Loibl S, Gianni L. HER2-positive breast cancer. Lancet 2017;389:2415-29. 10.1016/S0140-6736(16)32417-5 [DOI] [PubMed] [Google Scholar]
- 9.Iqbal N, Iqbal N. Imatinib: a breakthrough of targeted therapy in cancer. Chemother Res Pract 2014;2014:357027. 10.1155/2014/357027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Luoh SW, Flaherty KT. When Tissue Is No Longer the Issue: Tissue-Agnostic Cancer Therapy Comes of Age. Ann Intern Med 2018;169:233-9. 10.7326/M17-2832 [DOI] [PubMed] [Google Scholar]
- 11.Starostik P. Clinical mutation assay of tumors: new developments. Anticancer Drugs 2017;28:1-10. 10.1097/CAD.0000000000000427 [DOI] [PubMed] [Google Scholar]
- 12.Dong L, Wang S, Fu B, et al. Evaluation of droplet digital PCR and next generation sequencing for characterizing DNA reference material for KRAS mutation detection. Sci Rep 2018;8:9650. 10.1038/s41598-018-27368-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sanger F, Nicklen S, Coulson AR. DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci U S A 1977;74:5463-7. 10.1073/pnas.74.12.5463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Slatko BE, Gardner AF, Ausubel FM. Overview of Next-Generation Sequencing Technologies. Curr Protoc Mol Biol 2018;122:e59. 10.1002/cpmb.59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Petrackova A, Vasinek M, Sedlarikova L, et al. Standardization of Sequencing Coverage Depth in NGS: Recommendation for Detection of Clonal and Subclonal Mutations in Cancer Diagnostics. Front Oncol 2019;9:851. 10.3389/fonc.2019.00851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gerszten RE, Wang TJ. The search for new cardiovascular biomarkers. Nature 2008;451:949-52. 10.1038/nature06802 [DOI] [PubMed] [Google Scholar]
- 17.Wolff AC, Hammond ME, Schwartz JN, et al. American Society of Clinical Oncology/College of American Pathologists guideline recommendations for human epidermal growth factor receptor 2 testing in breast cancer. Arch Pathol Lab Med 2007;131:18-43. 10.5858/2007-131-18-ASOCCO [DOI] [PubMed] [Google Scholar]
- 18.Finotello F, Trajanoski Z. Quantifying tumor-infiltrating immune cells from transcriptomics data. Cancer Immunol Immunother 2018;67:1031-40. 10.1007/s00262-018-2150-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.El-Deiry WS, Vijayvergia N, Xiu J, et al. Molecular profiling of 6,892 colorectal cancer samples suggests different possible treatment options specific to metastatic sites. Cancer Biol Ther 2015;16:1726-37. 10.1080/15384047.2015.1113356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yaeger R, Chatila WK, Lipsyc MD, et al. Clinical Sequencing Defines the Genomic Landscape of Metastatic Colorectal Cancer. Cancer Cell 2018;33:125-36.e3. 10.1016/j.ccell.2017.12.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Passot G, Kim BJ, Glehen O, et al. Impact of RAS Mutations in Metastatic Colorectal Cancer After Potentially Curative Resection: Does Site of Metastases Matter? Ann Surg Oncol 2018;25:179-87. 10.1245/s10434-017-6141-7 [DOI] [PubMed] [Google Scholar]
- 22.Schneider MA, Eden J, Pache B, et al. Mutations of RAS/RAF Proto-oncogenes Impair Survival After Cytoreductive Surgery and HIPEC for Peritoneal Metastasis of Colorectal Origin. Ann Surg 2018;268:845-53. 10.1097/SLA.0000000000002899 [DOI] [PubMed] [Google Scholar]
- 23.Sasaki Y, Hamaguchi T, Yamada Y, et al. Value of KRAS, BRAF, and PIK3CA Mutations and Survival Benefit from Systemic Chemotherapy in Colorectal Peritoneal Carcinomatosis. Asian Pac J Cancer Prev 2016;17:539-43. 10.7314/APJCP.2016.17.2.539 [DOI] [PubMed] [Google Scholar]
- 24.Massalou D, Benizri E, Chevallier A, et al. Peritoneal carcinomatosis of colorectal cancer: novel clinical and molecular outcomes. Am J Surg 2017;213:377-87. 10.1016/j.amjsurg.2016.03.008 [DOI] [PubMed] [Google Scholar]
- 25.Graf W, Cashin PH, Ghanipour L, et al. Prognostic Impact of BRAF and KRAS Mutation in Patients with Colorectal and Appendiceal Peritoneal Metastases Scheduled for CRS and HIPEC. Ann Surg Oncol 2020;27:293-300. 10.1245/s10434-019-07452-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gillern SM, Chua TC, Stojadinovic A, et al. KRAS status in patients with colorectal cancer peritoneal carcinomatosis and its impact on outcome. Am J Clin Oncol 2010;33:456-60. 10.1097/COC.0b013e3181b4b160 [DOI] [PubMed] [Google Scholar]
- 27.Morgan Z, Chow BE, Strong EA, et al. RAS Mutation Status Confers Prognostic Relevance in Patients Treated With Cytoreductive Surgery and Hyperthermic Intraperitoneal Chemotherapy for Colorectal Cancer. J Surg Res 2019;240:130-5. 10.1016/j.jss.2019.02.050 [DOI] [PubMed] [Google Scholar]
- 28.Green DE, Jayakrishnan TT, Hwang M, et al. Immunohistochemistry - microarray analysis of patients with peritoneal metastases of appendiceal or colorectal origin. Front Surg 2015;1:50. 10.3389/fsurg.2014.00050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kawazoe A, Shitara K, Fukuoka S, et al. A retrospective observational study of clinicopathological features of KRAS, NRAS, BRAF and PIK3CA mutations in Japanese patients with metastatic colorectal cancer. BMC Cancer 2015;15:258. 10.1186/s12885-015-1276-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fujiyoshi K, Yamamoto G, Takahashi A, et al. High concordance rate of KRAS/BRAF mutations and MSI-H between primary colorectal cancer and corresponding metastases. Oncol Rep 2017;37:785-92. 10.3892/or.2016.5323 [DOI] [PubMed] [Google Scholar]
- 31.Gattenlohner S, Etschmann B, Kunzmann V, et al. Concordance of KRAS/BRAF Mutation Status in Metastatic Colorectal Cancer before and after Anti-EGFR Therapy. J Oncol 2009;2009:831626. 10.1155/2009/831626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yaeger R, Cercek A, Chou JF, et al. BRAF mutation predicts for poor outcomes after metastasectomy in patients with metastatic colorectal cancer. Cancer 2014;120:2316-24. 10.1002/cncr.28729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cremolini C, Di Bartolomeo M, Amatu A, et al. BRAF codons 594 and 596 mutations identify a new molecular subtype of metastatic colorectal cancer at favorable prognosis. Ann Oncol 2015;26:2092-7. 10.1093/annonc/mdv290 [DOI] [PubMed] [Google Scholar]
- 34.Datta J, Smith JJ, Chatila WK, et al. Coaltered Ras/B-raf and TP53 Is Associated with Extremes of Survivorship and Distinct Patterns of Metastasis in Patients with Metastatic Colorectal Cancer. Clin Cancer Res 2020;26:1077-85. 10.1158/1078-0432.CCR-19-2390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Enblad M, Graf W, Terman A, et al. Gains of Chromosome 1p and 15q are Associated with Poor Survival After Cytoreductive Surgery and HIPEC for Treating Colorectal Peritoneal Metastases. Ann Surg Oncol 2019;26:4835-42. 10.1245/s10434-019-07923-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mekenkamp LJ, Haan JC, Israeli D, et al. Chromosomal copy number aberrations in colorectal metastases resemble their primary counterparts and differences are typically non-recurrent. PLoS One 2014;9:e86833. 10.1371/journal.pone.0086833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Thorstensen L, Qvist H, Heim S, et al. Evaluation of 1p losses in primary carcinomas, local recurrences and peripheral metastases from colorectal cancer patients. Neoplasia 2000;2:514-22. 10.1038/sj.neo.7900111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Diep CB, Teixeira MR, Thorstensen L, et al. Genome characteristics of primary carcinomas, local recurrences, carcinomatoses, and liver metastases from colorectal cancer patients. Mol Cancer 2004;3:6. 10.1186/1476-4598-3-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Singhi AD, Davison JM, Choudry HA, et al. GNAS is frequently mutated in both low-grade and high-grade disseminated appendiceal mucinous neoplasms but does not affect survival. Hum Pathol 2014;45:1737-43. 10.1016/j.humpath.2014.04.018 [DOI] [PubMed] [Google Scholar]
- 40.Davison JM, Choudry HA, Pingpank JF, et al. Clinicopathologic and molecular analysis of disseminated appendiceal mucinous neoplasms: identification of factors predicting survival and proposed criteria for a three-tiered assessment of tumor grade. Mod Pathol 2014;27:1521-39. 10.1038/modpathol.2014.37 [DOI] [PubMed] [Google Scholar]
- 41.Shetty S, Thomas P, Ramanan B, et al. Kras mutations and p53 overexpression in pseudomyxoma peritonei: association with phenotype and prognosis. J Surg Res 2013;180:97-103. 10.1016/j.jss.2012.10.053 [DOI] [PubMed] [Google Scholar]
- 42.Pietrantonio F, Perrone F, Mennitto A, et al. Toward the molecular dissection of peritoneal pseudomyxoma. Ann Oncol 2016;27:2097-103. 10.1093/annonc/mdw314 [DOI] [PubMed] [Google Scholar]
- 43.Pietrantonio F, Berenato R, Maggi C, et al. GNAS mutations as prognostic biomarker in patients with relapsed peritoneal pseudomyxoma receiving metronomic capecitabine and bevacizumab: a clinical and translational study. J Transl Med 2016;14:125. 10.1186/s12967-016-0877-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zauber P, Berman E, Marotta S, et al. Ki-ras gene mutations are invariably present in low-grade mucinous tumors of the vermiform appendix. Scand J Gastroenterol 2011;46:869-74. 10.3109/00365521.2011.565070 [DOI] [PubMed] [Google Scholar]
- 45.Nishikawa G, Sekine S, Ogawa R, et al. Frequent GNAS mutations in low-grade appendiceal mucinous neoplasms. Br J Cancer 2013;108:951-8. 10.1038/bjc.2013.47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Alakus H, Babicky ML, Ghosh P, et al. Genome-wide mutational landscape of mucinous carcinomatosis peritonei of appendiceal origin. Genome Med 2014;6:43. 10.1186/gm559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu X, Mody K, de Abreu FB, et al. Molecular profiling of appendiceal epithelial tumors using massively parallel sequencing to identify somatic mutations. Clin Chem 2014;60:1004-11. 10.1373/clinchem.2014.225565 [DOI] [PubMed] [Google Scholar]
- 48.Sio TT, Mansfield AS, Grotz TE, et al. Concurrent MCL1 and JUN amplification in pseudomyxoma peritonei: a comprehensive genetic profiling and survival analysis. J Hum Genet 2014;59:124-8. 10.1038/jhg.2013.132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hara K, Saito T, Hayashi T, et al. A mutation spectrum that includes GNAS, KRAS and TP53 may be shared by mucinous neoplasms of the appendix. Pathol Res Pract 2015;211:657-64. 10.1016/j.prp.2015.06.004 [DOI] [PubMed] [Google Scholar]
- 50.Nummela P, Saarinen L, Thiel A, et al. Genomic profile of pseudomyxoma peritonei analyzed using next-generation sequencing and immunohistochemistry. Int J Cancer 2015;136:E282-9. 10.1002/ijc.29245 [DOI] [PubMed] [Google Scholar]
- 51.Noguchi R, Yano H, Gohda Y, et al. Molecular profiles of high-grade and low-grade pseudomyxoma peritonei. Cancer Med 2015;4:1809-16. 10.1002/cam4.542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Saarinen L, Nummela P, Thiel A, et al. Multiple components of PKA and TGF-beta pathways are mutated in pseudomyxoma peritonei. PLoS One 2017;12:e0174898. 10.1371/journal.pone.0174898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pengelly RJ, Rowaiye B, Pickard K, et al. Analysis of Mutation and Loss of Heterozygosity by Whole-Exome Sequencing Yields Insights into Pseudomyxoma Peritonei. J Mol Diagn 2018;20:635-42. 10.1016/j.jmoldx.2018.05.002 [DOI] [PubMed] [Google Scholar]
- 54.Tokunaga R, Xiu J, Johnston C, et al. Molecular Profiling of Appendiceal Adenocarcinoma and Comparison with Right-sided and Left-sided Colorectal Cancer. Clin Cancer Res 2019;25:3096-103. 10.1158/1078-0432.CCR-18-3388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gleeson EM, Feldman R, Mapow BL, et al. Appendix-derived Pseudomyxoma Peritonei (PMP): Molecular Profiling Toward Treatment of a Rare Malignancy. Am J Clin Oncol 2018;41:777-83. 10.1097/COC.0000000000000376 [DOI] [PubMed] [Google Scholar]
- 56.Stein A, Strong E, Clark Gamblin T, et al. Molecular and Genetic Markers in Appendiceal Mucinous Tumors: A Systematic Review. Ann Surg Oncol 2020;27:85-97. 10.1245/s10434-019-07879-7 [DOI] [PubMed] [Google Scholar]
- 57.Levine EA, Blazer DG, 3rd, Kim MK, et al. Gene expression profiling of peritoneal metastases from appendiceal and colon cancer demonstrates unique biologic signatures and predicts patient outcomes. J Am Coll Surg 2012;214:599-606; discussion 606-7. 10.1016/j.jamcollsurg.2011.12.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Varghese S, Burness M, Xu H, et al. Site-specific gene expression profiles and novel molecular prognostic factors in patients with lower gastrointestinal adenocarcinoma diffusely metastatic to liver or peritoneum. Ann Surg Oncol 2007;14:3460-71. 10.1245/s10434-007-9557-7 [DOI] [PubMed] [Google Scholar]
- 59.Kleivi K, Lind GE, Diep CB, et al. Gene expression profiles of primary colorectal carcinomas, liver metastases, and carcinomatoses. Mol Cancer 2007;6:2. 10.1186/1476-4598-6-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tan F, Liu F, Liu H, et al. CTHRC1 is associated with peritoneal carcinomatosis in colorectal cancer: a new predictor for prognosis. Med Oncol 2013;30:473. 10.1007/s12032-013-0473-3 [DOI] [PubMed] [Google Scholar]
- 61.Lièvre A, Bachet JB, Le Corre D, et al. KRAS mutation status is predictive of response to cetuximab therapy in colorectal cancer. Cancer Res 2006;66:3992-5. 10.1158/0008-5472.CAN-06-0191 [DOI] [PubMed] [Google Scholar]
- 62.Corcoran RB, Andre T, Atreya CE, et al. Combined BRAF, EGFR, and MEK Inhibition in Patients with BRAF(V600E)-Mutant Colorectal Cancer. Cancer Discov 2018;8:428-43. 10.1158/2159-8290.CD-17-1226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.EuroPMP.com. EuroPMP [cited 2020.02.25]. Available online: https://europmp.eu/