

Key Points
Question
What is the prevalence of rare likely pathogenic or pathogenic variants in candidate genes in ethnic minority probands with early-onset atrial fibrillation and genotype-phenotype associations?
Findings
In this cohort study, a family history of atrial fibrillation was noted in 24 (10.6%) African American and Hispanic/Latinx probands with early-onset atrial fibrillation; sequencing identified 16 (7.0%) probands harboring pathogenic (56.2%) or likely pathogenic (43.8%) variants, with most (46.7%) loss-of-function variants in the TTN gene. In 6 families with more than 2 affected members, variants of unknown significance in cardiac ion channels, sarcomeric proteins, and signaling molecules cosegregated with atrial fibrillation.
Meaning
Likely pathogenic or pathogenic variants with most loss-of-function variants in TTN that increase susceptibility to atrial fibrillation were observed in African American and Hispanic/Latinx individuals; these findings may provide insights into the underlying pathophysiologic factors and potential mechanism-based therapies for atrial fibrillation in individuals of minority ethnicity.
Abstract
Importance
Although rare variants in cardiac ion channels, transcription factors, and myocardial structural proteins are associated with early-onset atrial fibrillation (AF) in White individuals of European descent, it remains unclear whether genetic variation also contributes to the cause of AF in those of minority ethnicity.
Objectives
To assess the prevalence of rare and novel pathogenic variants in candidate genes in ethnic minority probands with early-onset AF and determine genotype-phenotype associations.
Design, Setting, and Participants
In this cohort, family-based study, probands of African and Hispanic descent with early-onset AF (defined as AF occurring in individuals aged ≤66 years) prospectively enrolled in a clinical and genetic biorepository underwent sequencing of 60 candidate genes. Recruitment took place from July 1, 2015, to June 30, 2019. Data were analyzed from February 1 to February 28, 2020.
Exposures
Rare and novel variants categorized as pathogenic or likely pathogenic.
Main Outcomes and Measures
The prevalence of rare and novel pathogenic variants in African American and Hispanic/Latinx probands with early-onset AF and genotype-phenotype associations.
Results
Among 227 probands with early-onset AF, mean (SD) age at onset of AF was 51.0 (9.9) years, 132 probands (58.1%) were men, 148 (65.2%) were African American, and 79 (34.8%) were Hispanic/Latinx. A family history of AF was verified in 24 probands with early-onset AF (10.6%). Sequencing 60 candidate genes identified 53 (23 rare and 30 novel) variants with 16 of the 227 (7.0%) probands harboring likely pathogenic (43.8%) or pathogenic (56.2%) variants, with most loss-of-function variants in TTN, the gene encoding the sarcomeric protein titin (46.7%). In 6 families with more than 2 affected members, variants of unknown significance in sodium channel (SCN10A), potassium channel (KCNE5), sarcomeric proteins (MYH6 and TTN), and atrial natriuretic peptide (NPPA) cosegregated with AF.
Conclusions and Relevance
In this study, likely pathogenic and pathogenic variants were identified, with most loss-of-function variants in TTN, that increase susceptibility to early-onset AF in African American and Hispanic/Latinx individuals. These findings provide further understanding toward molecular phenotyping of AF and suggest novel mechanism-based therapeutic approaches for this common arrhythmia in ethnic minority groups.
This cohort study describes comprehensive sequencing of 60 candidate atrial fibrillation genes in African American and Hispanic/Latinx probands to examine the combined prevalence of rare pathogenic variants and assess genotype-phenotype associations.
Introduction
Genetic approaches to the mechanisms of atrial fibrillation (AF), including positional cloning, candidate gene sequencing, and genome-wide association studies, have identified variants in cardiac ion channels, transcription factors, and myocardial structural proteins in White individuals of European descent and provided insights into the underlying pathophysiologic factors.1 It is established that non-White individuals are at a lower risk of developing AF, especially African American and Hispanic/Latinx individuals despite a greater burden of cardiovascular risk factors and poorer outcomes.2,3,4 Over the past 2 decades, several studies have identified rare genetic variants associated with early-onset AF in White individuals, but the contribution of rare pathogenic variants to the source of the arrhythmia in ethnic minority groups remains unclear.5,6,7 Two studies from 2018 reported that probands of African and Hispanic descent with early-onset AF were more likely to have a first-degree relative with AF compared with White individuals.8,9 Herein, we describe comprehensive sequencing of 60 candidate AF genes in African American and Hispanic/Latinx probands to examine the combined prevalence of rare pathogenic variants and assess genotype-phenotype association.
Methods
We recruited patients with early-onset AF and their family members from University of Illinois at Chicago and Jesse Brown Veterans Administration Medical Centers, with all probands self-reported as being of African or Hispanic descent and older than 18 years at the time of enrollment with a history of AF documented by an electrocardiogram, Holter monitor, or implantable loop recorder. Recruitment took place from July 1, 2015, to June 30, 2019. Data were analyzed from February 1 to February 28, 2020. More than 75% of the Hispanic/Latinx cohort are individuals of Mexican descent, which is consistent with the Hispanic population in Chicago. Patients with a history of AF associated with cardiothoracic surgery were excluded. We obtained information on demographics, cardiovascular risk factors, and family history for baseline characteristics with blood drawn for DNA extraction at the time of enrollment as previously reported.8,9 Experienced English- and Spanish-speaking research coordinators obtained a family history and constructed a family pedigree when a proband gave a family history of AF or stroke in a first-degree relative. We then contacted the family member, enrolled them in the registry, and reviewed their medical records, and AF was confirmed by electrocardiogram, Holter monitor, or event recorder.
We obtained written informed consent from all participants under a protocol approved by the University of Illinois at Chicago and the Jesse Brown Veterans Administration institutional review boards. Bilingual research coordinators recruited patients using both English and Spanish consent forms. Participants did not receive financial compensation, and the data were deidentified as stipulated by the institutional review boards. This study followed the Strengthening the Reporting of Observational Studies in Epidemiology (STROBE) reporting guideline for cohort studies.
We defined early-onset AF as AF with onset when the participants were 66 years or younger and familial AF as the presence of AF in 2 or more first-degree relatives of probands with early-onset AF. Rare variants were defined as minor allele frequency less than or equal to 0.1% in the Genome Aggregation Database10 with novel variants absent from gnomAD11 (accessed January 5, 2020). If any novel variants were found more than once in our cohort, we only considered variants with a population frequency of less than or equal to 1.0%. Disease-causing variants fulfilled the pathogenic or likely pathogenic standardized criteria proposed by the American College of Medical Genetics and Genomics, the Association for Molecular Pathology, and the Association for Clinical Genomic Science.12,13 Variants were reported on canonical transcripts based on the Ensembl gene tree analysis, and cardiac transcripts were selected based on the highest-expressed isoform in atrial appendage tissue from genotype-tissue expression (GTEx; accessed January 5, 2020).11 All variants under question were required to reside in the exonic regions for both canonical and chosen cardiac transcripts; otherwise, they were excluded from further evaluation.
Sequencing
Blood samples from participants who consented were drawn into 1 vial. The buffy coat layer containing the white blood cells for DNA extraction was collected from the serum. DNA was extracted using a commercially available kit (Gentra Puregene; Qiagen) and samples were stored in a −80 °C freezer. Sequencing was performed on 60 candidate AF genes that have been implicated in mediating AF susceptibility. A custom panel was designed to target all exons from the listed 60 genes (Accel-Amplicon Custom Panel; Swift Biosciences) and was developed in cooperation with the Swift Biosciences team to maximize exon recovery from the genes. The final design consisted of a total of 2449 amplicons, targeting a total of 385 451 base pairs. Sets of 96 samples were pooled for sequencing (NextSeq500 instrument; Illumina). The average depth of sequence was ×1000 and on-target rate of aligned reads was 90%, with coverage uniformity greater than 96%. Demultiplexing of sequence data was performed in the cloud storage environment in Basespace for bioinformatics analysis.
Bioinformatics
Overlapping read lengths were merged into single-end reads using paired-end read merger to avoid overcounting variants and provide a higher stringency for more accurate variant calling.14 We used Trimmomatic to trim reads to ensure that sequences were mapped to the human genome with amplicon primers retained in the reads to improve mapping quality.15 The trimmed reads were mapped to human reference genome hg19 using Burrows-Wheeler alignment maximal exact match, and any alignments that did not match the amplicon region were removed.16 We used Genome Analysis Toolkit (GATK) for realignments around indel regions and GATK HaplotypeCaller to identify variants in the candidate genes.17 The functional annotation of variants was carried out using ANNOVAR.18
Filtering
We used a stepwise filter approach for identifying rare and novel variants in our multiethnic population. First, standardized quality-control thresholds were required to be met with adequate read depths (≥×20), appropriate allelic ratios for heterozygous calls (≥20%) with homozygous alternate calls (≥99%), and homozygous reference calls completely excluded. Genotype quality scores were not available. If reported read depth values did not closely match (>10% difference) with manually calculated read depth values, then these variants were considered uninformative reads and not considered any further. Second, we applied a minor allele frequency less than or equal to 0.1% for rare variants and absent for novel variants from the reference gnomAD database. We used gnomAD (versions 2 and 3) as a reference population for the variants we identified, with almost all of them not previously reported in versions 2 and 3 of gnomAD, indicating that they are novel and not identified in a healthy ethnic-specific population. This was only one of the criteria we used to interpret variants. Third, a filtering strategy focused on identifying missense, stop-gain, stop-loss, frameshift, splice-site, and indel coding variants within the exon regions. Fourth, we used cutoff values from in-silico prediction tools for Genomic Evolutionary Rate Profiling (GERP) greater than or equal to 3.0, Sorting Intolerant from Tolerant (SIFT) less than or equal to 0.05, PolyPhen2 greater than or equal to 0.9, MutationTaster greater than or equal to 0.5, protein variation effect analyzer (PROVEAN) less than or equal to −2.5, and Combined Annotation Dependent Depletion (CADD) greater than or equal to 20. If any variants with more than 1 transcript resulted in multiple in silico scores, we chose the least deleterious value reported to base the imputation. If in silico scores were missing values, we kept these variants because this may have filtered out indels. All variants had a minimum ×20 read depth with allelic balances greater than or equal to 20% for heterozygous and less than or equal to 1% for homozygous between reference and alternative allele differences to be considered an adequate variant call. Fifth, variants were then classified into pathogenic, likely pathogenic, of unknown significance, likely benign, or benign based on American College of Medical Genetics and Genomics, Association for Molecular Pathology, and Association for Clinical Genomic Science criteria for genes with any prior genetic data suggesting a potential link to AF.12,13
Genotyping
All identified rare and novel variants meeting our stringent threshold underwent Sanger sequencing to confirm variant calls from probands as well as enrolled first-degree family members to assess for cosegregation with AF.
The data were analyzed using SAS, version 9.4 (SAS Institute Inc). For continuous variables, distributed variables are reported as means (interquartile range) using an unpaired t test. For categorical variables, frequencies were compared using the Pearson χ2 test and are reported as count (percentage). When comparing multiple groups, analysis of variance and Mann-Whitney and Kruskal-Wallis tests were used for analysis. A post hoc pairwise comparison was applied with Bonferroni correction. For rare variants, comparisons across subpopulations were performed using the Fisher exact test (1-sided for testing enrichment and 2-sided for testing differences). The threshold for P value statistical significance across all analyses was P ≤ .05.
Results
Clinical Characteristics
The study cohort consisted of 227 probands with early-onset AF; of these, 148 (65.2%) were African American, 79 (34.8%) were Hispanic/Latinx, and 132 (58.1%) were men (Table 1). The mean (SD) age at AF onset was 51.0 (9.9) years. There was a verified family history of AF in 24 probands (10.6%) in the cohort, and we found that 56 (24.7%) of the 227 probands were diagnosed with early-onset AF before developing comorbidities. In addition, African American participants were more likely to be obese and have a history of hypertension, coronary artery disease, and congestive heart failure compared with Hispanic/Latinx participants. eTable 1 in the Supplement reports the comparison of the AF-associated comorbidities in the European American, African American, and Hispanic/Latinx cohorts of early-onset AF probands. We found that the multi-ethnic cohort had fewer men, younger age at onset for AF, reduced reported family history of AF, and a greater burden of cardiovascular comorbidities (obstructive sleep apnea, hyperthyroidism, coronary artery disease, valvular heart disease, congestive heart failure, hypertension, type 2 diabetes, and stroke) compared with European American individuals.
Table 1. Baseline Clinical Characteristics of African American, European American, and Hispanic/Latinx Probands With Early-Onset Atrial Fibrillation.
| Category | No. (%) | P value | ||
|---|---|---|---|---|
| Total (n = 227) | African American (n = 148) | Hispanic/Latinx (n = 79) | ||
| Men | 132 (58.1) | 83 (56.1) | 49 (62.0) | .24 |
| Age at onset, mean (SD), y | 51.0 (9.9) | 51.7 (9.4) | 49.8 (10.8) | .16 |
| Atrial fibrillation | ||||
| Paroxysmal | 174 (76.7) | 118 (79.7) | 56 (70.9) | .09 |
| Persistent | 37 (16.3) | 22 (14.9) | 15 (19.0) | .27 |
| Permanent | 16 (7.0) | 8 (5.4) | 8 (10.1) | .15 |
| Family history | 24 (10.6) | 13 (8.8) | 11 (13.9) | .16 |
| BMI, mean (SD) | 34.2 (9.3) | 35.4 (9.8) | 31.8 (7.6) | <.001 |
| Obstructive sleep apnea | 42 (18.5) | 27 (18.2) | 15 (19.0) | .51 |
| Hyperthyroidism | 10 (4.4) | 7 (4.7) | 3 (3.8) | .52 |
| Coronary artery disease | 109 (48.0) | 79 (53.4) | 30 (38.0) | .02 |
| Valve heart disease | 51 (22.5) | 29 (19.6) | 22 (27.8) | .11 |
| Mitral (≥moderate-severe) | 36 (15.9) | 19 (12.8) | 17 (21.5) | .21 |
| Aortic (≥moderate-severe) | 9 (4.0) | 1 (0.7) | 8 (10.1) | <.001 |
| Congestive heart failure | 89 (39.2) | 68 (45.9) | 21 (26.6) | <.001 |
| Hypertension | 154 (67.8) | 116 (78.4) | 38 (48.1) | <.001 |
| Type 2 diabetes | 68 (30.0) | 49 (33.1) | 19 (24.1) | .10 |
| Stroke | 28 (12.3) | 20 (13.5) | 8 (10.1) | .30 |
| Vascular disease | 14 (6.2) | 12 (8.1) | 2 (2.5) | .08 |
| CHA2DS2-VASc score ≥2 | 123 (54.2) | 58 (39.2) | 33 (41.8) | .41 |
| Left ventricular hypertrophy, mean (SD), g/m2 | 64 (28.2) | 41 (27.7) | 23 (29.1) | .88 |
| Intraventricular septum, mean (SD), cm | 1.0 (0.2) | 1.1 (0.2) | 1.0 (0.2) | <.001 |
| Relative wall thickness, mean (SD), cm | 0.42 (0.1) | 0.43 (0.1) | 0.42 (0.1) | .47 |
| LVEF, mean (SD), % | 81 (35.7) | 52 (35.1) | 29 (36.7) | .88 |
| Left atrial diameter, mean (SD), mm | 4.3 (0.8) | 4.3 (0.8) | 4.3 (0.9) | .80 |
Abbreviations: BMI, body mass index (calculated as weight in kilograms divided by height in meters squared); CHA2DS2-VASc, congestive heart failure, hypertension, age, diabetes, stroke, vascular disease, and sex; LVEF, left ventricular ejection fraction.
We reviewed all 227 echocardiograms for the early-onset AF cohort and found that 25 patients (11.0%) displayed a moderate to severe dilated cardiomyopathy (DCM) phenotype. However, only UIC-0358 (TTN-107050G>T; OMIM: 188840) proband carried a pathogenic or likely pathogenic variant and had DCM. We also noted that the remaining patients with reduced left ventricular ejection fraction had a tachycardia-induced cardiomyopathy related to rapid AF.
Variants
Sequencing 60 candidate genes identified 53 variants. We identified 23 rare variants, but no probands with early-onset AF harbored a disease-causing variant. All variants were classified as variants of unknown significance. We sequenced the coding regions of 60 candidate genes and identified likely pathogenic (43.8%) or pathogenic (56.2%) variants in 16 probands (7.0%) of African and Hispanic descent with early-onset AF.
We identified 30 novel variants, and 17 of these variants were classified as likely pathogenic or pathogenic across 16 probands. In 16 probands with early-onset AF (7.0%), 11 (7.4%) were noted in African American individuals and 5 (6.3%) were observed in Hispanic/Latinx individuals; the remaining 13 variants were classified as variants of unknown significance (Table 2). Six variants (40.0%) encoded for cardiac ion channel proteins, and 3 of them were specific for the cardiac sodium channel gene (SCN5A; OMIM: 600163), 1 L-type calcium channel gene (CACNA1C; OMIM: 114205), and 2 for the potassium channel genes (KCNQ1; OMIM: 607542 and KCNJ11; OMIM: 600937). The most common type of variants were missense (40.0%) and stop-gain (40.0%), followed by frameshift (20.0%). The mean (SD) in silico scores were as follows: GERP++ NR, 5.2 (0.7); GERP++ RS, 4.9 (0.83); SIFT, 0.00 (0.00); Polyphen2 HDIV, 0.99 (0.01); Polyphen2 HVAR, 1.0 (0.01); MutationTaster, 1.0 (0.0); PROVEAN, −4.8 (1.5); and CADD, 39.9 (17.37) (eTable 2 in the Supplement). Comparing the burden of rare variants in the multiethnic subpopulations demonstrated that the reference cohort had a significantly higher number of rare variants than the case cohort (P ≤ .001). This significant finding also held true when we compared the different subpopulations (African American, 1326 rare variants from a total of 6464 variants; P ≤ .001 and Hispanic/Latinx, 1456 rare variants from a total of 6517 variants P ≤ .001) within the reference and case cohorts. Although a low prevalence of rare variants may be related to a cohort enriched with acquired causes of AF, eTable 3 in the Supplement notes that early-onset AF probands with obesity, coronary artery disease, congestive heart failure, and hypertension had a greater prevalence of rare variants.
Table 2. Identified Rare and Novel Variants in African American and Hispanic/Latinx Probands With Early-Onset Atrial Fibrillation.
| Study ID | Race | Chr | Position | Gene | MAF | Transcripta | Exon | Nucleotide | Protein | Variation | PSI | Region | Classification |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| UIC-0066 | H/L | 2 | TTN | 179644802 | 00.00 | ENST00000589042 | 22/363 | 3654T>A | Y1218a | Stop gain | 100 | None | LP (PVS1, PM2) |
| UIC-0357 | AA | 2 | TTN | 179644802 | 00.00 | ENST00000589042 | 22/363 | 3654T>A | Y1218a | Stop gain | 100 | None | LP (PVS1, PM2) |
| UIC-0019 | H/L | 2 | TTN | 179397682 | 00.00 | ENST00000589042 | 358/363 | 103660G>T | E34554a | Stop gain | 100 | M-band | LP (PVS1, PM2) |
| UIC-0160 | H/L | 2 | TTN | 179476320 | 00.00 | ENST00000589042 | 269/363 | 50636delA | N16879fs | Frameshift | 100 | A-band | LP (PVS1, PM2) |
| VA-0090 | AA | 2 | TTN | 179476320 | 00.00 | ENST00000589042 | 269/363 | 50636delA | N16879fs | Frameshift | 100 | A-band | LP (PVS1, PM2) |
| UIC-0119 | AA | 2 | TTN | 179460243 | 00.00 | ENST00000589042 | 295/363 | 57838G>T | E19280a | Stop gain | 100 | A-band | LP (PVS1, PM2) |
| VA-0009 | AA | 2 | TTN | 179396890 | 00.00 | ENST00000589042 | 358/363 | 104452G>T | E34818a | Stop gain | 100 | M-band | LP (PVS1, PM2) |
| UIC-0358 | AA | 2 | TTN | 179393428 | 00.00 | ENST00000589042 | 360/363 | 107050G>T | G35684a | Stop gain | 100 | M-band | LP (PVS1, PM2) |
| UIC-0134 | AA | 3 | SCN5A | 38627224 | 00.00 | ENST00000413689 | 16/28 | 2745C>A | C915a | Stop gain | NA | Extracellular | LP (PVS1, PM2) |
| VA-0070 | AA | 3 | SCN5A | 38601648 | 00.00 | ENST00000413689 | 23/28 | 4235T>G | L1412R | Missense | NA | Pore | LP (PM1, PM2, PP3, BP1) |
| UIC-0093 | H/L | 3 | SCN5A | 38598726 | 00.00 | ENST00000413689 | 24/28 | 4295G>T | R1432M | Missense | NA | Extracellular | LP (PM1, PM2, PP3, PM5, BP1) |
| UIC-0384 | AA | 4 | PITX2 | 111542364 | 00.00 | ENST00000306732 | 2/3 | 367G>C | E123Q | Missense | NA | Homeobox | LP (PM1, PM2, PP3, BP1) |
| UIC-0056 | AA | 4 | PITX2 | 111542357 | 00.00 | ENST00000306732 | 2/3 | 374T>A | I125N | Missense | NA | Homeobox | LP (PM1, PM2, PP3, BP1) |
| UIC-0639 | AA | 4 | PITX2 | 111542357 | 00.00 | ENST00000306732 | 2/3 | 374T>A | I125N | Missense | NA | Homeobox | LP (PM1, PM2, PP3, BP1) |
| UIC-0104 | H/L | 11 | KCNJ11 | 17408936 | 00.00 | ENST00000339994 | 1/1 | 703C>A | Q235K | Missense | NA | Cytoplasmic | LP (PM1, PM2, PP3) |
| UIC-0407 | AA | 11 | KCNQ1 | 2604665 | 00.00 | ENST00000155840 | 7/16 | 922G>T | V308F | Missense | NA | Pore | LP (PM1, PM2, PP3) |
| UIC-0104 | H/L | 12 | CACNA1C | 2794922 | 00.00 | ENST00000347598 | 46/49 | 5741delA | N1914fs | Frameshift | NA | Cytoplasmic | LP (PVS1, PM2) |
Abbreviations: AA, African American; Chr, chromosome; H/L, Hispanic/Latinx; LP, likely pathogenic; MAF, minor allele frequency; NA, not available; PSI, percent spliced-in.
Canonical transcript based on Genome Aggregation Database (gnomAD; accessed January 5, 2020).
We identified 8 probands (3.5%) with TTN variants encoding the sarcomeric protein titin. Because TTN truncating variants are a recognized cause of AF7 and not just DCM, we noted that 25 (11.0%) early-onset AF probands manifest a DCM phenotype by echocardiography (Table 1). However, only UIC-0358 (TTN-107050 G>T) proband carried a likely pathogenic or pathogenic variant and had DCM. We also noted that the remaining patients with reduced left ventricular ejection fraction developed a tachycardia-induced cardiomyopathy associated with rapid ventricular rates during AF. We established a diagnosis of AF-related DCM by reviewing echocardiograms performed before and after treatment of early-onset AF.
Cosegregating Variants
None of the early-onset AF probands carrying a disease-causing variant reported a family history of AF. However, in 6 probands (2.6%) with more than 2 affected family members, variants of unknown significance were identified that cosegregated with AF (Table 3). We identified 3 rare variants in genes encoding sodium channel protein type 10 subunit α (SCN10A; OMIM: 604427), voltage-gated potassium channel accessory subunit 5 (KCNE5; OMIM: 300328), and titin (TTN), and 3 novel variants in myosin heavy chain 6 (MYH6; OMIM: 160710 [2 variants]) and natriuretic peptide precursor A (NPPA; OMIM: 108780) that cosegregated with AF (Figure 1 and Figure 2). The mean (SD) read depth for these variants was 509.5 (678.2). The mean (SD) for the novel variants was 102.2 (125.2) and mean (SD) in silico scores were as follows: GERP++ NR, 5.0 (0.7); GERP++ RS, 4.9 (1.0); SIFT, 0.0 (0.0); Polyphen2 HDIV, 1.0 (0.0); Polyphen2 HVAR, 1.0 (0.01); MutationTaster, 1.0 (0.0); PROVEAN, −5.8 (1.4); and CADD, 28.1 (2.7) (eTable 3 in the Supplement).
Table 3. Clinical Characteristics of Familial AF Kindreds Harboring Rare and Novel Gene Variants of Uncertain Significance.
| Study ID | Gene variant | Sex | Symptoms | Age at diagnosis, y | AF type | Comorbidities | Treatment | PR interval, ms | QRS duration, ms | LA size, mm | LVEF, % |
|---|---|---|---|---|---|---|---|---|---|---|---|
| AA 0174 | KCNE5 (R85H) | ||||||||||
| II:1 | M | Palpitations | 48 | Paroxysmal | Hypertension, obstructive apnea, obesity | Rhythm | 162 | 82 | 4.0 | 60 | |
| II:2 | M | Dyspnea | 54 | Persistent | Hypertension | Rhythm | 148 | 96 | 3.8 | 60 | |
| II:4 | F | Fatigue | 59 | Permanent | None | Rate | 178 | 102 | 4.4 | 60 | |
| H/L 0207 | NPPA (D36Y) | ||||||||||
| II:2 | F | Tiredness | 58 | Paroxysmal | Stroke, pacemaker | Rate | 142 | 104 | 3.5 | 60 | |
| II:3 | F | Dyspnea | 65 | Paroxysmal | Stroke | Rate | 138 | 92 | 3.0 | 55 | |
| III:1 | M | Palpitations | 41 | Paroxysmal | None | Rhythm | 158 | 108 | 3.8 | 60 | |
| AA 0281 | SCN10A (R1142C) | ||||||||||
| I:2 | F | Palpitations | 58 | Paroxysmal | Stroke | Rate | 134 | 88 | 3.5 | 60 | |
| II:2 | F | Palpitations | 61 | Paroxysmal | Hypertension | Rhythm (pulmonary vein isolation) | 138 | 90 | 3.8 | 55 | |
| III:2 | M | Dyspnea | 42 | Persistent | Hypothyroidism | Rate | 145 | 110 | 3.9 | 60 | |
| AA 0473 | MYH6 (E1323V) | ||||||||||
| II:2 | F | Fatigue | 59 | Paroxysmal | Hypertension, chronic kidney disease | Rhythm | 127 | 72 | 3.4 | 50 | |
| II:3 | F | Dyspnea | 56 | Paroxysmal | Hypertension | Rhythm | 142 | 78 | 3.6 | 50 | |
| II:6 | M | Palpitations | 61 | Persistent | Hypertension | Rate | 88 | 4.6 | 45 | ||
| H/L 0526 | MYH6 (K1307M) | ||||||||||
| II:2 | M | Palpitations | 62 | Paroxysmal | Type 2 diabetes | Rate | 154 | 88 | 4.4 | 60 | |
| II:3 | F | Fatigue | 58 | Persistent | None | Rate | NA | 104 | NA | 55 | |
| II:6 | M | Palpitations | 61 | Permanent | None | NA | 148 | 94 | NA | 60 | |
| H/L 0606 | TTN (G25630V) | ||||||||||
| I:2 | F | Deceased | 68 | Permanent | Stroke | Rate control | |||||
| II:2 | F | Palpitations | 48 | Paroxysmal | None | Rhythm (pulmonary vein isolation) | 122 | 63 | 3.5 | 60 | |
| III:1 | M | Palpitations | None | 98 | 98 | 3.8 | 60 |
Abbreviations: AF, atrial fibrillation; LA, left atrium; LVEF, left ventricular ejection fraction; NA, not applicable.
Figure 1. Pedigrees of 3 Families Carrying Rare Candidate Atrial Fibrillation (AF) Gene Variants.

Squares indicate male and circles indicate female family members, symbols with a slash mark indicate deceased family members, and arrowheads indicate the probands. Complete solid symbols indicate the presence of AF. Open symbols indicate unaffected members, and half-shading indicates individuals with AF by history. Gray shaded symbols indicate individuals whose status was indeterminate: presence (+) or absence (−) of a rare variant is indicated for persons with DNA samples available for testing.
Figure 2. Pedigrees of 3 Families Carrying Novel Candidate Atrial Fibrillation (AF) Gene Variants.
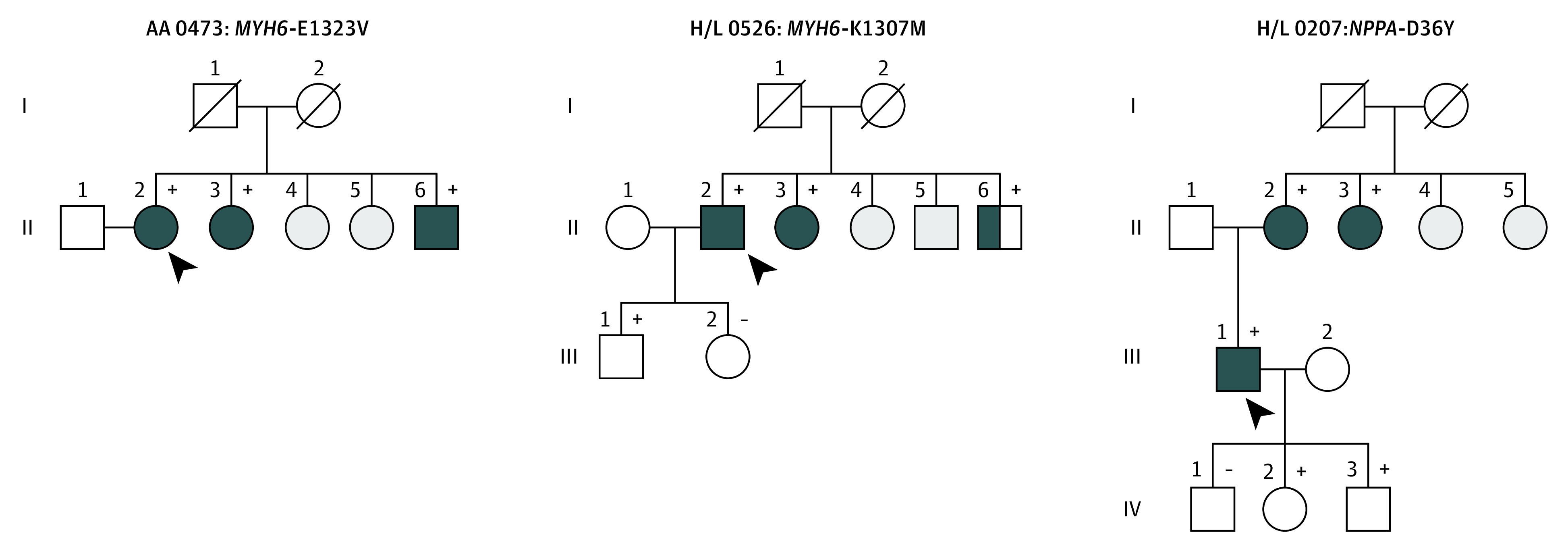
Squares indicate male and circles indicate female family members, respectively, and symbols with a slash mark deceased family members. Arrowheads indicate the probands. Complete solid symbols indicate the presence of AF. Open symbols indicate unaffected members, and half-shading indicates individuals with AF by history. Gray shaded symbols indicate individuals whose status was indeterminate: presence (+) or absence (−) of a rare variant is indicated for persons with DNA samples available for testing.
Discussion
We sequenced the coding regions of 60 candidate genes and identified likely pathogenic (43.8%) or pathogenic (56.2%) variants in 16 probands (7.0%) of African and Hispanic descent with early-onset AF, with most (46.7%) loss-of-function variants in the TTN gene. In addition, 6 novel and rare variants encoding cardiac sodium (SCN10A) and potassium channels (KCNE5), myocardial structural proteins (MYH6 and TTN), and signaling molecules (NPPA) cosegregated with AF in multiple affected family members. Although most ethnic minority probands with early-onset AF did not harbor rare variants, our findings not only represent progress toward the molecular phenotyping of AF but also identify novel mechanism-based therapeutic approaches for this common arrhythmia in ethnic minority individuals.
We identified rare variants associated with AF in a small percentage (7.0%) of ethnic minority probands with early-onset AF. This percentage is lower than that found in European White individuals with early-onset AF, of whom approximately 20% carried very rare variants.19 There are several potential explanations for why a minority of probands of self-reported African American and Hispanic/Latinx ethnicity carried rare gene variants. Because our ethnic cohort, especially African American individuals, had a greater burden of AF-associated comorbidities, one reason may relate to a population enriched with acquired causes. Our probands did not have lone AF. However, we noted that that probands with early-onset AF with comorbidities had a greater prevalence of rare variants, suggesting that these may be risk alleles rather than mendelian disease-associated variants (eTable 3 in the Supplement). Nonetheless, a study across race/ethnicity populations supports the concept of the 2-hit hypothesis in which susceptibility to AF is dependent not only on carrying a pathogenic variant associated with AF, but also on a second hit, such as an established AF risk factor (eg, obesity or hypertension) or common genetic variants on chromosome 4q25.20 Other potential explanations for why a minority of African American and Hispanic/Latinx probands with early-onset AF harbored rare variants include the failure to sequence genes not yet identified that are involved in the pathogenesis of early-onset AF; sequencing only the coding regions of candidate genes; rare variants with a minor allele frequency less than or equal to 0.1% and missing variants or combinations of variants with intermediate effects; and the challenges of correctly phenotyping participants with AF, which can be asymptomatic.
We identified 8 probands (3.5%) of African and Hispanic descent with TTN variants encoding the sarcomeric protein titin. This prevalence may in part reflect a higher burden of cardiovascular risk factors, especially in African American individuals, creating a substrate for atrial myopathy and re-entrant AF.21 Titin acts as a molecular scaffold for sarcomeric assembly and directly interacts with actin and myosin filaments to provide stability during the contraction and relaxation phases of the cardiac cycle.22 Variants in TTN have been associated with dilated and hypertrophic cardiomyopathy, neuromuscular disorders, and, most recently, early-onset AF.7,23,24,25 Thus, another possible explanation for the high frequency of TTN variants in the early-onset AF cohort in the present study is that AF is associated with DCM. However, echocardiographic characterization of the probands carrying TTN revealed that 11.0% of the cohort had evidence of DCM and only 1 kindred (UIC-0358) harbored a likely pathogenic or pathogenic variant (Table 2). UIC-0358 proband is better classified as a TTN cardiomyopathy because there was evidence of DCM by echocardiography. However, at this time, UIC-0357 proband, who also carries a pathogenic or likely pathogenic variant, is not classified as a TTN cardiomyopathy because there is no evidence of DCM. The underlying mechanisms by which these variants cause AF remain unclear, and further research directed at functionally characterizing TTN loss-of-function variants in vitro and in vivo is required. An improved understanding of the pathophysiologic mechanisms by which TTN variants cause AF will also enable a more mechanism-based approach to AF therapy in African American and Hispanic/Latinx individuals with early-onset AF because current antiarrhythmic drug therapy targets cardiac ion channels. We also identified 2 new variants in MYH6 that cosegregated with AF in multiple family members, further supporting the association between myocardial integrity and the development of AF.26,27,28,29
Our findings have several potential clinical implications. First, an improved understanding of the underlying genetic mechanisms by which pathogenic variations, especially those in TTN, cause AF may identify a new therapeutic target for the arrhythmia across race/ethnicity because current antiarrhythmic therapy is limited and directed at cardiac ion channels. Second, our findings have potential implications for the screening and assessment of African American and Hispanic/Latinx individuals with early-onset AF. Cascade screening will identify family members at increased risk for developing AF. Third, although gene sequencing identified pathogenic variants in only a small percentage of participants, there was a high prevalence of TTN variants in probands of self-reported African American and Hispanic/Latinx ethnicity. However, studies focused on determining the utility of sequencing candidate genes encoding myocardial structural proteins like TTN are needed, especially in individuals with AF onset before age 40 years.30
Limitations
This study has some limitations. First, there is great diversity in Hispanic/Latinx ancestry, especially in gnomAD. However, more than 75% of the Hispanic probands with early-onset AF were of Mexican descent, as previously described.9 Race/ethnicity was self-reported rather than determined by genetic ancestry. Second, we did not perform linkage analysis and quantify cosegregation with informative meiosis because of the limited number of multiethnic kindreds. However, the likelihood of misclassifying variants was low given the stringent criteria we used to classify them. Third, we sequenced 60 candidate genes implicated in the pathogenesis of AF. Despite this extensive target panel of genes, additional candidate genes remain unknown. Fourth, even though we sequenced the coding region of most of the cardiac ion channel proteins, signaling molecules, and transcription factors, the calcium auxiliary subunit channel genes or an expanded list of myocardial structural protein genes were not screened owing to size restriction in the gene panel. This lack of screening may underestimate the true prevalence of pathogenic variants seen in individuals of ethnic minority with early-onset AF. Fifth, despite good overall coverage across 60 candidate genes, approximately 4% per gene was uncovered in the exonic regions. It is thus possible that we missed a small number of variants residing in these challenging regions. Sixth, genome-wide association studies have identified a large number of AF loci that reside in noncoding regions. Although our study did not assess common genetic variation as an etiologic factor associated with early-onset AF in individuals of ethnic minority, the chromosome 4q25 locus has been associated with increased susceptibility to AF in Hispanic/Latinx individuals.9 Seventh, our filtering threshold used a minor allele frequency less than or equal to 0.1% to identify rare variants. We did not assess the frequency of less rare variants or combinations of variants in the cause of early-onset AF in African American and Hispanic/Latinx individuals. Eighth, formal gene to disease associations were not curated in a stringent way, and future studies will need to do this to determine which genes should be considered for genetic testing in clinical practice when applying these diagnostic criteria.
Conclusions
To our knowledge, this is one of the first studies to sequence candidate genes in ethnic minority probands with early-onset AF. We identified likely pathogenic and pathogenic variants with most loss-of-function variants in TTN that increase susceptibility to AF in African American and Hispanic/Latinx individuals. Our findings not only represent progress toward molecular phenotyping of AF but also identify novel mechanism-based therapeutic approaches for AF in ethnic minority populations.
eTable 1. Baseline Clinical Characteristics of African American, European American and Hispanic/Latinx Probands With Early-Onset Atrial Fibrillation (EOAF)
eTable 2. In-Silico Scores for Pathogenic or Likely Pathogenic Variants in African American and Hispanic/Latinx Probands With EOAF
eTable 3. In-Silico Scores for Co-Segregating Variants of Unknown Significance in African American and Hispanic/Latinx Probands With EOAF
References
- 1.Fatkin D, Santiago CF, Huttner IG, Lubitz SA, Ellinor PT. Genetics of atrial fibrillation: state of the art in 2017. Heart Lung Circ. 2017;26(9):894-901. doi: 10.1016/j.hlc.2017.04.008 [DOI] [PubMed] [Google Scholar]
- 2.Marcus GM, Alonso A, Peralta CA, et al. ; Candidate-Gene Association Resource (CARe) Study . European ancestry as a risk factor for atrial fibrillation in African Americans. Circulation. 2010;122(20):2009-2015. doi: 10.1161/CIRCULATIONAHA.110.958306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gbadebo TD, Okafor H, Darbar D. Differential impact of race and risk factors on incidence of atrial fibrillation. Am Heart J. 2011;162(1):31-37. doi: 10.1016/j.ahj.2011.03.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lipworth L, Okafor H, Mumma MT, et al. Race-specific impact of atrial fibrillation risk factors in Blacks and Whites in the southern community cohort study. Am J Cardiol. 2012;110(11):1637-1642. doi: 10.1016/j.amjcard.2012.07.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lubitz SA, Yin X, Lin HJ, et al. ; AFGen Consortium . Genetic risk prediction of atrial fibrillation. Circulation. 2017;135(14):1311-1320. doi: 10.1161/CIRCULATIONAHA.116.024143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Weng LC, Choi SH, Klarin D, et al. Heritability of atrial fibrillation. Circ Cardiovasc Genet. 2017;10(6):e001838. doi: 10.1161/CIRCGENETICS.117.001838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Choi SH, Weng LC, Roselli C, et al. ; DiscovEHR study and the NHLBI Trans-Omics for Precision Medicine (TOPMed) Consortium . Association between titin loss-of-function variants and early-onset atrial fibrillation. JAMA. 2018;320(22):2354-2364. doi: 10.1001/jama.2018.18179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Alzahrani Z, Ornelas-Loredo A, Darbar SD, et al. Association between family history and early-onset atrial fibrillation across racial and ethnic groups. JAMA Netw Open. 2018;1(5):e182497. doi: 10.1001/jamanetworkopen.2018.2497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chalazan B, Mol D, Sridhar A, et al. Genetic modulation of atrial fibrillation risk in a Hispanic/Latino cohort. PLoS One. 2018;13(4):e0194480. doi: 10.1371/journal.pone.0194480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Karczewski KJ, Francioli LC, Tiao G, et al. ; Genome Aggregation Database Consortium . The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581(7809):434-443. doi: 10.1038/s41586-020-2308-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.GTEx Consortium . The genotype-tissue expression (GTEx) project. Nat Genet. 2013;45(6):580-585. doi: 10.1038/ng.2653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Richards S, Aziz N, Bale S, et al. ; ACMG Laboratory Quality Assurance Committee . Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405-424. doi: 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Amendola LM, Jarvik GP, Leo MC, et al. Performance of ACMG-AMP variant-interpretation guidelines among nine laboratories in the Clinical Sequencing Exploratory Research Consortium. Am J Hum Genet. 2016;98(6):1067-1076. doi: 10.1016/j.ajhg.2016.03.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang J, Kobert K, Flouri T, Stamatakis A. PEAR: a fast and accurate Illumina Paired-End reAd mergeR. Bioinformatics. 2014;30(5):614-620. doi: 10.1093/bioinformatics/btt593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30(15):2114-2120. doi: 10.1093/bioinformatics/btu170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010;26(5):589-595. doi: 10.1093/bioinformatics/btp698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McKenna A, Hanna M, Banks E, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20(9):1297-1303. doi: 10.1101/gr.107524.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38(16):e164. doi: 10.1093/nar/gkq603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Weeke P, Parvez B, Blair M, et al. Candidate gene approach to identifying rare genetic variants associated with lone atrial fibrillation. Heart Rhythm. 2014;11(1):46-52. doi: 10.1016/j.hrthm.2013.10.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ritchie MD, Rowan S, Kucera G, et al. Chromosome 4q25 variants are genetic modifiers of rare ion channel mutations associated with familial atrial fibrillation. J Am Coll Cardiol. 2012;60(13):1173-1181. doi: 10.1016/j.jacc.2012.04.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sajeev JK, Kalman JM, Dewey H, Cooke JC, Teh AW. The atrium and embolic stroke: myopathy not atrial fibrillation as the requisite determinant? JACC Clin Electrophysiol. 2020;6(3):251-261. doi: 10.1016/j.jacep.2019.12.013 [DOI] [PubMed] [Google Scholar]
- 22.Labeit S, Gautel M, Lakey A, Trinick J. Towards a molecular understanding of titin. EMBO J. 1992;11(5):1711-1716. doi: 10.1002/j.1460-2075.1992.tb05222.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gerull B, Gramlich M, Atherton J, et al. Mutations of TTN, encoding the giant muscle filament titin, cause familial dilated cardiomyopathy. Nat Genet. 2002;30(2):201-204. doi: 10.1038/ng815 [DOI] [PubMed] [Google Scholar]
- 24.Gerull B. Between disease-causing and an innocent bystander: the role of titin as a modifier in hypertrophic cardiomyopathy. Can J Cardiol. 2017;33(10):1217-1220. doi: 10.1016/j.cjca.2017.07.010 [DOI] [PubMed] [Google Scholar]
- 25.Hackman P, Vihola A, Haravuori H, et al. Tibial muscular dystrophy is a titinopathy caused by mutations in TTN, the gene encoding the giant skeletal-muscle protein titin. Am J Hum Genet. 2002;71(3):492-500. doi: 10.1086/342380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Holm H, Gudbjartsson DF, Sulem P, et al. A rare variant in MYH6 is associated with high risk of sick sinus syndrome. Nat Genet. 2011;43(4):316-320. doi: 10.1038/ng.781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gudbjartsson DF, Holm H, Sulem P, et al. A frameshift deletion in the sarcomere gene MYL4 causes early-onset familial atrial fibrillation. Eur Heart J. 2017;38(1):27-34. doi: 10.1093/eurheartj/ehw379 [DOI] [PubMed] [Google Scholar]
- 28.Orr N, Arnaout R, Gula LJ, et al. A mutation in the atrial-specific myosin light chain gene (MYL4) causes familial atrial fibrillation. Nat Commun. 2016;7:11303. doi: 10.1038/ncomms11303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thorolfsdottir RB, Sveinbjornsson G, Sulem P, et al. A missense variant in PLEC increases risk of atrial fibrillation. J Am Coll Cardiol. 2017;70(17):2157-2168. doi: 10.1016/j.jacc.2017.09.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Goodyer WR, Dunn K, Caleshu C, et al. Broad genetic testing in a clinical setting uncovers a high prevalence of titin loss-of-function variants in very early onset atrial fibrillation. Circ Genom Precis Med. 2019;12(11):e002713. doi: 10.1161/CIRCGEN.119.002713 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
eTable 1. Baseline Clinical Characteristics of African American, European American and Hispanic/Latinx Probands With Early-Onset Atrial Fibrillation (EOAF)
eTable 2. In-Silico Scores for Pathogenic or Likely Pathogenic Variants in African American and Hispanic/Latinx Probands With EOAF
eTable 3. In-Silico Scores for Co-Segregating Variants of Unknown Significance in African American and Hispanic/Latinx Probands With EOAF