

INTRODUCTION
In severe traumatic brain injury (TBI), the relative contribution of diffuse, focal, and/or hypoxic damage from the primary injury, and the extent of secondary postinjury brain damage have the potential to damage both cortical and subcortical control mechanisms of the autonomic nervous system (ANS). In particular, the mix of focal and diffuse injuries for any individual surviving TBI has the potential to produce unique patterns of impaired autonomic control. This autonomic dysfunction, in turn, helps reveal the complex connections of the central autonomic nervous system (CAN).
This chapter presents the anatomic and physiologic structures incorporated into the CAN and describes how TBI may adversely impact upon this. The possible autonomic effects of focal injury post-TBI are highlighted with reference to the literature regarding ischemic stroke. Specifically, unilateral loss of insular cortical function may later result in increased or decreased sympathetic tone, producing arrhythmia and/or myocardial infarction. Studies and case reports in stroke suggest left and right insular cortices exhibit parasympathetic and sympathetic control respectively. More extensive injuries resulting in subarachnoid hemorrhage (SAH) may also produce autonomic dysfunction, possibly related to the release of inflammatory cytokines. Investigators have hypothesized that the acutely elevated intracranial pressure following aneurysmal rupture injures the hypothalamus, resulting in catecholamine release both locally and systemically. This may manifest in the heart as subendocardial hemorrhages or contraction band necrosis or as pulmonary edema in the lungs.
Paroxysmal sympathetic hyperactivity (PSH) provides an example of a clinical correlate of CAN dysfunction. Patients with this syndrome typically display exaggerated vital signs (particularly tachycardia, tachypnea, and hypertension), often in the setting of dystonic posturing. PSH is most frequently encountered in traumatic brain injury (TBI) and is thought to result from severe diffuse axonal injury and/or focal brainstem injuries that functionally disconnect the spinal cord from the CAN.
THE CENTRAL AUTONOMIC NETWORK
The ANS is classically defined in terms of its peripheral connections. In simplified form, general anatomy and physiology textbooks highlight and compare the differences between the sympathetic (“fight or flight”) and parasympathetic (“rest and digest”) arms of the ANS (Guyton and Hall, 2006). These twin arms provide complementary functions through a wide variety of effector organs, the details of which are beyond the scope of this chapter. Instead, this section will focus on the central components of the autonomic nervous system (Benarroch, 1993).
The hypothalamus serves as the main control center of the ANS. The hypothalamus both receives input from higher cortical centers, such as the insula and prefrontal cortex, and transmits information to the brainstem and spinal cord. Injury to either the higher or lower cortical centers may result in autonomic dysfunction.
The paraventricular nucleus is the principal hypothalamic nucleus concerned with autonomic regulation. There are two major cell types located in this nucleus – the magnocellular neurons and the parvocellular neurons. The magnocellular neurons contain oxytocin and vasopressin; they project directly into the posterior pituitary gland. The parvocellular neurons, in contrast, can be divided into two main types: some parvocellular neurons are concerned with anterior pituitary control and neuroendocrine function, while a second group of parvocellular neurons is concerned with central autonomic control (Fig. 34.1). These autonomic parvocellular neurons may be further subdivided into three separate cell types: A, B, and C. Subtypes of parvocellular neurons are characterized by their location and orientation to the third ventricle. Type A neurons are located in the ventral parvocellular nucleus and lie obliquely with respect to the third ventricle. They comprise the majority (52%) of pre-autonomic parvocellular neurons. Type B neurons are located orthogonal to the third ventricle within the posterior parvocellular subnucleus. Type B neurons are characterized by their extensive dendritic branching. Type C neurons are located in both the ventral and posterior subnuclei and are characterized by their concentric dendritic branching. Electrophysiologic studies show different reactions to applied voltage between these cell types, suggesting that voltage-gated channels on these neuronal subtypes are different as well (Stern, 2001; Dougherty, 2013).
Fig. 34.1.
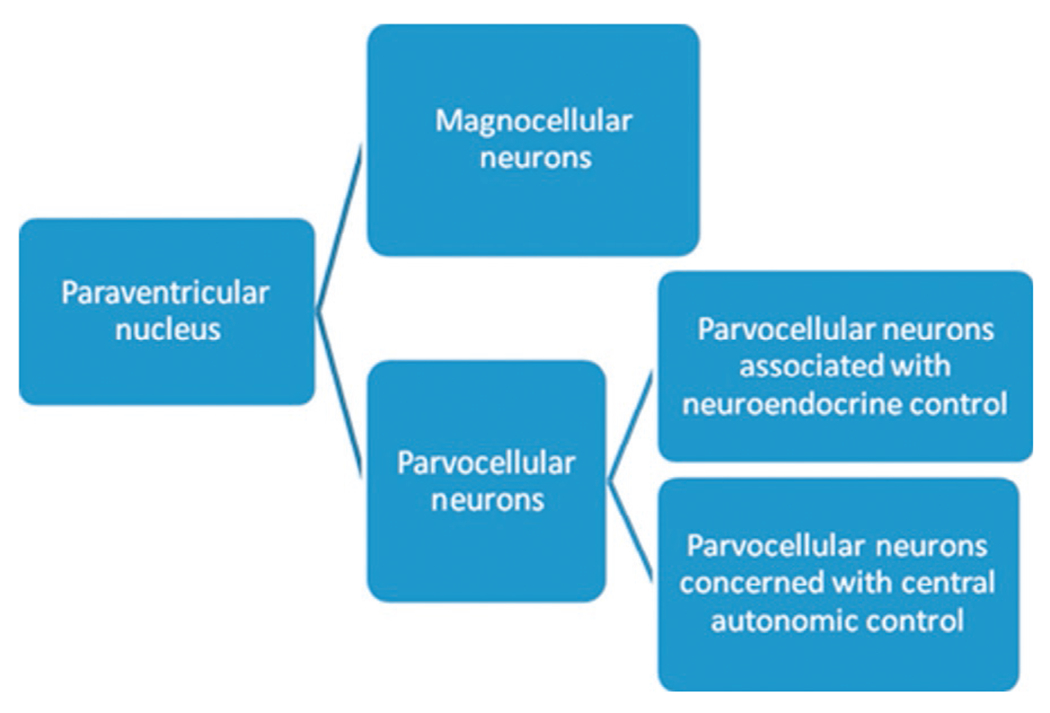
Relationship between paraventricular nuclei and neurons directly associated with central autonomic control.
The preganglionic parvocellular neurons of the hypothalamus can synapse directly onto preganglionic autonomic neurons. These preganglionic neurons then have connections in the dorsal motor nucleus of the vagus nerve, the autonomic relays of the brainstem, and finally onto the intermediolateral area of the spinal cord. The intermediolateral area of the spinal cord has connections into both the sympathetic and parasympathetic systems. Of note, the preganglionic neurons descend ipsilaterally down the brainstem. While the principal transmission of autonomic information is ipsilateral, there are four points of decussation: supramammillary, pontine tegmentum, commissural part of the nucleus of the solitary tract (the major one), and lamina X of the spinal cord. Thus, the efferent tracts of the hypothalamus are rich with multiple connections. Not only can the parvocellular nucleus transmit information to both sides of the brainstem, it also communicates with both the parasympathetic and sympathetic nervous systems (Loewy and Spyer, 1990; Pyner and Coote, 2000; Dougherty, 2013). In addition, three main tracts interconnect the hypothalamus with the rest of the central autonomic nervous system; the medial forebrain bundle (MFB), dorsal longitudinal fasciculus (DLF), and mamillotegmental tract (Fig. 34.2).
Fig. 34.2.
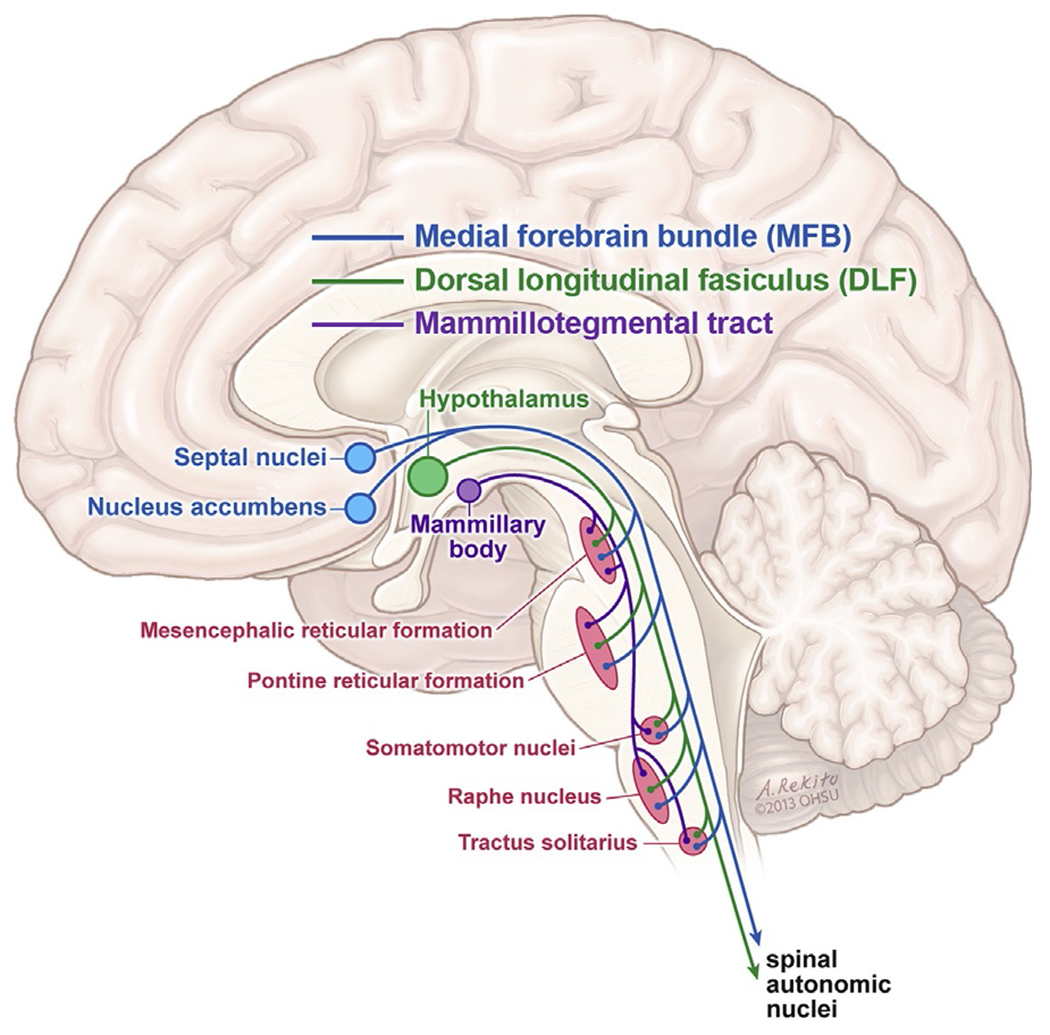
Major nuclei and pathways of the central autonomic network.
The MFB is the most dorsal of these three pathways and is more often thought of for its known association with “addiction.” The amygdala, hippocampus, and prefrontal cortex all connect to the stria terminalis which, in turn, communicates with the paraventricular nucleus of the hypothalamus. The DLF is the primary pathway for the paraventricular nucleus of the hypothalamus, extending from the paraventricular nucleus down into the brainstem. As the DLF travels caudally, it synapses within the mesencephalic reticular formation, pontine reticular formation, and raphe nuclei both ipsilaterally and contralaterally via decussations. The mamillotegmental tract is the most ventrally located, synapsing with the mesencephalic and pontine reticular formations. In this way, all the higher structures mentioned above have potential connections to the paraventricular nucleus of the hypothalamus, allowing them to play an important role in ANS regulation.
Evidence suggests the existence of cortical structures that connect to the hypothalamus and directly influence autonomic control (Loewy and Spyer, 1990; Pyner and Coote, 2000; Spyer and Gourine, 2009). Literature in ischemic stroke implicates the insular cortex in autonomic functions, especially in regard to heart rate and rhythm (Barron et al., 1994; Nagai et al., 2010). Insular cortex damage may result in cardiac arrhythmias, diurnal blood pressure variations, and sleep disordered breathing (Nagai et al., 2010). Another study (Kuriyama et al., 2010) found that hypertension and tachycardia are more likely to occur in patients with putaminal or thalamic strokes, thus implying that these areas are involved with autonomic regulation as well. A definitive pathway between the insula and the central autonomic network has not been described; however, multiple studies with different approaches produce similar results. Animal studies, particularly in rats, imply that the right posterior insula has a role in regulating sympathetic outflow, while the left posterior insula modulates parasympathetic outflow. Human studies corroborate these results; stroke patients with left insular damage are more likely to suffer from sinus tachycardia and arrhythmias (Barron et al., 1994; Oppenheimer, 2006, 2007). Conversely, bradycardia may be induced when the left insula is stimulated, such as intraoperatively during epilepsy surgeries (Oppenheimer, 2006, 2007).
While the human CAN is still poorly understood, there are multiple levels of autonomic regulation between the cortex, diencephalon, and brainstem. It appears probable that the CAN receives contributions from the cortex, thalamus, and diencephalon into the nucleus tractus solitarius, parabrachial nucleus, and even directly onto preganglionic sympathetic neurons. The postganglionic sympathetic neurons then innervate effector organs (e.g., heart, arterial smooth muscle, adrenal glands) (Spyer and Gourine, 2009) and cause hyperadrenergic activity, manifesting with alterations of vital signs (Loewy and Spyer, 1990; Napadow et al., 2008). Thus, it is easy to imagine how severe acute brain injury of either a diffuse or focal nature may result in autonomic dysfunction. Several dysautonomic syndromes have been identified after acute brain injury. The following paragraphs will summarize several common syndromes encountered after diverse brain injuries such as traumatic brain injury (TBI), subarachnoid hemorrhage (SAH), and focal ischemic stroke.
AUTONOMIC DYSFUNCTION AND ISCHEMIC STROKE SYNDROMES
There is compelling evidence that ischemic strokes, particularly insular strokes, may result in cardiac autonomic dysregulation. Autopsy studies have demonstrated that ischemic stroke patients may have subendocardial hemorrhages and myocardial infarction in the absence of coronary atherosclerosis or cardiac disease (Katsanos et al., 2013). Data from Prosser et al. (2007) also support the importance of cardiac causes of morbidity and mortality following ischemic stroke. In a large prospective trial, 846 ischemic stroke patients who also had baseline electrocardiograms (EKGs), cardiac monitoring data, and stroke territory data were followed for 12 weeks postdischarge. Of 180 patients who were deceased after 12 weeks, 35 (19%) of the deaths were identified as cardiac in nature. Notably, patients who suffered a fatal cardiac event were more likely to have had left hemispheric stroke involvement, prolonged QT, premature ventricular beats, and/or one serious, nonfatal cardiac event, often in the first days after the stroke. While this study did not identify a direct mechanism for ischemic stroke resulting in cardiac events, its findings do suggest a connection in humans. Study results imply that it is possible that left insular damage placed patients at higher risk for catecholamine surge and subsequent cardiac mortality in the near future.
While data from human studies provides epidemiologic data to support the ischemic stroke/heart connection, most of the pathophysiologic support comes from animal studies. Experimental right middle cerebral artery (R MCA) occlusion in rats has the effect of increased tyrosine production in the central nucleus of the amygdala; this has the ultimate downstream effect of increased catecholamine production (Ozdemir and Hachinski, 2008). Similarly, right posterior insular ablation in rats results in increased heart rate and blood pressure. These studies all suggest that structures within the right MCA territory, particularly the insula, may have profound effects on the heart. Another study, by Min et al. (2009), highlighted the importance of the left insula in autonomic control. In Min et al.’s study, 24 mice underwent artificial occlusion of the left MCA. Compared to sham mice, the left MCA occlusion mice had significantly decreased left ventricular systolic pressures and contraction band necrosis. Both findings demonstrate that left focal ischemia can result in global heart damage. Hence, animal studies show the importance of both left and right MCA distribution territories in the control of heart rate.
While there are multiple important nuclei and cortical structures lying within the MCA territories, studies suggest that the bilateral insular territories influence cardiac function and provide higher autonomic control. The NOMAS study, which followed outcomes in poststroke patients, reported 6.7% of patients suffered fatal cardiac events, with left parietal lobe infarctions more commonly associated with cardiac death (Rincon et al., 2008). Other smaller studies have associated right posterior insular lesions with increased in blood pressure and heart rate and left insular lesions with increased baroreceptor gain (Oppenheimer, 2006, 2007). In humans, intraoperative stimulation of the right insula may cause tachycardia and hypertension, while stimulation of the left insula may cause bradycardia and hypotension. Conversely, baroreceptor stimulation may cause retrograde activation of the right insula (Oppenheimer, 2006; Rincon et al., 2008). Other studies do not support the concept of the effect of cerebral laterality on cardiac function. For example, heart rate variability (HRV) (measured by the R-R interval) has been found to be reduced regardless of stroke laterality (Barron et al., 1994). Reduced HRV is a common feature of elevated catecholamines, indicative of heightened physiologic stress (Van Ravenswaaij-Arts et al., 1993). In this situation, HRV would be expected to be inversely correlated with stroke severity. This contention would appear to be supported by a prospective follow-up study which found that increased norepinephrine levels and insular lesions were associated with worse patient outcomes, as measured by modified Rankin scores and Barthel indices (Sander et al., 2001). Epidemiologic studies, animal studies, and retrospective data all suggest that ischemic strokes may produce cardiac dysfunction syndromes. The aggregate data support the prevailing hypothesis that the right side of the brain controls sympathetic outflow while the left side controls parasympathetic outflow (Praveen Kumar et al., 2012). The data also suggest that there is higher cortical input, particularly from the bilateral insula, that plays a significant role in central autonomic regulation.
Treatment implications for ischemic stroke patients
Unfortunately, there are no good preventive or therapeutic interventions for cardiac complications in ischemic stroke patients. The majority of ischemic stroke patients do not develop serious or life-threatening arrhythmias. Currently, treatment is supportive and the treatments and monitoring modalities listed below are suggestions only (Kasner et al., 2002; Rabinstein, 2007; Badjatia, 2009; Spyer and Gourine, 2009).
Telemetry monitoring. Since telemetry monitoring is noninvasive, painless, and widely available at most hospitals, it should be strongly considered for ischemic stroke patients. Telemetry will help to identify any abnormal heart rhythms and expedite treatments if necessary.
Medication review. QT prolongation was identified in several studies as a risk factor for cardiac arrhythmia in ischemic stroke patients. When prescribing medications that may traditionally prolong QT (e.g., antipsychotics, antibiotics), it is appropriate for the clinician to carefully consider alternatives first. Any medication that has the capacity to prolong QT should be administered for the shortest period possible.
Aggressive management of electrolytes, including hypokalemia and hypomagnesemia, may reduce symptomatic cardiac events. Correction of hypokalemia and hypomagnesemia is easily facilitated in the hospital setting and is usually well tolerated.
SUBARACHNOID HEMORRHAGE-RELATED HYPERADRENERGIC CRISES
In aneurysmal subarachnoid hemorrhage (SAH) patients, inappropriate catecholamine release can also produce a hyperadrenergic state. However, the hyperadrenergic state experienced by SAH patients is usually monophasic, in contrast to the polyphasic, episodic nature of PSH. It can be postulated that the free blood within and around the brain occurring with SAH may adversely affect the control of the CAN, with the end result of sympathetic hyperactivity, manifested as a hyperadrenergic state. An alternate explanation is that the surge in intracranial pressure (ICP) following aneurysm rupture stimulates the hypothalamus and results in an adrenergic surge (Hinson and Sheth, 2012). It is important to keep in mind, however, that these explanations are unproven hypotheses.
Clinical descriptions
Approximately 20% of aneurysmal SAH patients experience cardiac dysfunction (Horowitz et al., 1998; Tung et al., 2004). Manifestations range from an isolated arrhythmia or troponin leak, to global dysfunction resulting in a reduced ejection fraction. About 10% of patients will experience left ventricular dysfunction (Kono et al., 1994). Four percent of patients will suffer cardiac arrhythmias, including atrial fibrillation/flutter, ventricular arrhythmia, junctional rhythms, and supraventricular tachycardias (Frontera et al., 2008).
Cardiac dysfunction resulting from neurologic injury is typically called stressed myocardium. While it can be clinically challenging to distinguish stressed myocardium from coronary artery disease or other primary cardiac disease, there are some clinically distinguishing signs of stressed myocardium. Stressed myocardium is associated with contraction band necrosis on histopathology, thus differentiating it from damage due to ischemic heart disease. Additionally, wall motion abnormalities tend to occur in nonvascular territories (Hinson and Sheth, 2012). SAH canine models have demonstrated cardiac dysfunction as well; experimental animals suffered from hypertension, sinus tachycardia, ventricular tachycardia, AV block, ventricular fibrillation, and sudden death (Gao et al., 2009).
The proposed diagnostic criteria of stressed myocardium are as follows (Wittstein et al., 2005; Stevens and Nyquist, 2007; Hinson and Sheth, 2012):
Acute structural or functional brain disorder
- New onset systolic and/or diastolic left ventricular dysfunction
- global wall motion abnormalities, or
- regional wall motion abnormalities extending beyond one vascular distribution
Partial or complete resolution of left ventricular dysfunction in more than 4 weeks
- At least one of the following:
- no history of congestive heart failure, left ventricular dysfunction or coronary artery disease
- no evidence of myocardial ischemia on myocardial perfusion scan
- absence of angiographic evidence of obstructive coronary disease or of acute plaque rupture.
The pathophysiology of cardiac dysfunction in the SAH patient is not well defined. Many years ago, Hans Selye, a student of Pavlov, performed animal studies to prove that fright and physiologic stressors (e.g., surgery) can induce cardiac damage (Samuels, 2007). Cardiac dysfunction is associated with higher grade SAH; it is more common in patients with Hunt and Hess scores 3–5 (Tung et al., 2004). Studies have also shown that cardiac dysfunction is more common in patients who experience vasospasm. While we can associate cardiac complications in patients with severe hemorrhage, there is no specific neurologic lesion correlated with these findings. Some studies assert that blood in the insula, particularly the right insula, may be more strongly associated with cardiac dysfunction (Hirashima et al., 2001). Serum samplings of catecholamines show that levels are higher in those patients with cardiac complications compared to controls; however, higher grade hemorrhage does not necessarily result in higher level of catecholamines. Hence, it is possible that higher-grade hemorrhage may result in inappropriate catecholamine release and cardiac dysfunction, but there does not seem to be a direct correlation between severity of neurologic injury and level of circulating catecholamines.
There are other complications of SAH that may be associated with adrenergic release. These include acute lung injury, possibly from neurogenic pulmonary edema. Neurogenic pulmonary edema is defined as excess interstitial lung fluid presumed secondary to neurologic insult. Acute lung injury complicates the course of 10% of SAH patients (Di Giugno and Rosa, 1998). Acute lung injury and more specifically, neurogenic pulmonary edema, is more common in higher grade SAH. Posterior circulation aneurysms are also associated with neurogenic pulmonary edema (Inamasu et al., 2012a), and those with Hunt and Hess scores of 4–5 are at particularly high risk (Inamasu et al., 2012b). In Inamasu’s studies, age and gender did not correlate with the development of neurogenic pulmonary edema (Inamasu et al., 2012b).
Current studies do not definitively prove whether neurogenic pulmonary edema is related to the systemic catecholamine release, cardiac dysfunction, or whether there is a completely separate mechanism. For example, Inamasu’s retrospective review of neurogenic pulmonary edema patients demonstrated a high incidence of Takatsubos’s cardiomyopathy with neurogenic pulmonary edema (over 80% of cases); but there were also many cases of isolated Takatsubo’s syndrome without evidence of acute lung injury (Inamasu et al., 2012a). Animal studies suggest that neurogenic pulmonary edema occurs as a result of inflammation, since markers such as C3a, C5b-9, interleukin (IL) 6, and IL-8 are released in canine models (Gao et al., 2009). More recently, a retrospective study suggested that plasma norepinephrine levels correlated with the presence of neurogenic pulmonary edema, suggesting that catecholamine release may be associated with the pathophysiology of neurogenic pulmonary edema (Inamasu et al., 2012b). Hence current data suggest that neurogenic pulmonary edema likely occurs as the result of inflammation and subsequent capillary leak. Definitive pathophysiologic mechanisms are difficult to deduce, however, since the majority of evidence comes from animal studies and retrospective reviews with relatively few patients suffering from neurogenic pulmonary edema. The best current treatment recommendations include admission chest X-ray on all SAH patients. If mechanical ventilation is required, supportive care with low tidal volume ventilation is recommended (Hinson and Sheth, 2012). These issues are discussed in more detail in Chapter 14.
Treatment options
Treatment of cardiac dysfunction in SAH is a clinical challenge. Clinicians must balance limiting hyperadrenergic activity with preserving cerebral perfusion pressure (CPP). For this reason, routine adrenergic blockade (with β-blockers, for example) is not recommended. While there is evidence that α and β blockade may help to prevent cardiac complications, the risk of depressing CPP remains. This risk intensifies if the patient develops vasospasm. In a small retrospective trial, Naidech and colleagues (2005) compared dobutamine to milrinone after SAH. Milrinone was more appropriate for patients with low cardiac output but normal vascular resistance, while dobutamine was more suited to patients with low vascular resistance, particularly systolic blood pressure (SBP) < 90.
At present, once cardiac dysfunction is identified, the best course of action is treatment with cardiac support. Milrinone and dobutamine are often suitable agents to ensure systemic perfusion (Naidech et al., 2005).
Acute brain injuries and hyperthermia
Hyperthermia is a pervasive complication of acute brain injury. In the literature, hyperthermia is often defined as temperature > 38.3 °C (Rabinstein and Sandhu, 2007; Scaravilli et al., 2011a). While hyperthermia and fever may be used interchangeably in common parlance, there is a mechanistic difference between the two entities. The term “fever” implies prostaglandins as the underlying cause, typically produced in the context of infection. In contrast, “hyperthermia” indicates an elevation in temperature without specifying an etiology. In many clinical scenarios, hyperthermia may be the first sign of infection. While infections certainly occur in SAH patients, 20–33% of SAH patients with hyperthermia do not have an identifiable infectious cause. Studies of both TBI and SAH patients have shown that significant proportions (33–50%) of both groups experience hyperthermia with an identified infectious source (Rabinstein and Sandhu, 2007; Badjatia, 2009). Noninfectious fevers in SAH patients also share a temporal pattern; it is more likely that fever occurring within 72 hours of the aneurysmal rupture will be attributable to a noninfectious cause (Rabinstein and Sandhu, 2007). Amongst these patients, it is very possible that the observed hyperthermia is representative of autonomic dysfunction. We infer from the epidemiologic data that the brain damage, either from trauma or subarachnoid blood, results in autonomic dysfunction and more specifically, temperature dysregulation. Hyperthermia may result from direct hypothalamic injury; however, it may be a more common consequence of hypothalamic irritation from blood in cerebrospinal fluid. In his review, Badjatia (2009) states that direct damage to the preoptic nucleus of the hypothalamus and pons can result in hyperthermia. Other studies have demonstrated that blood within the cerebrospinal fluid, particular within the ventricles, may irritate hypothalamic thermoregulatory centers and result in fevers.
Control of hyperthermia can be a vexing clinical problem with serious implications. Studies indicate that hyperthermia results in increased neuronal excitotoxicity. Data in rats show fever results in increased neurotransmitter release, increased release of glutamate, activation of heat shock proteins, and increased cellular depolarization in the area surrounding ischemic penumbra (Ginsberg et al., 1992; Rabinstein and Sandhu, 2007). Hence, multiple studies show that damage occurs on the cellular level and may lead to worsened outcomes. Microdialysis studies in humans show increased lactate/pyruvate levels in hyperthermic patients, implying increased metabolic stress (Oddo et al., 2009). Hyperthermia without an underlying infectious cause may confer a worse outcome in SAH patients, which may be related to vasospasm. Vasospasm rates may be higher in patients with noninfectious fever compared to those with fever from an infectious source (Rabinstein and Sandhu, 2007). Rabinstein showed that 12/15 patients with noninfectious fever suffered from vasospasm, compared to 4/11 patients with infectious fever who experienced vasospasm. Although small, the study suggests worse outcomes for patients with noninfectious fever. Other data demonstrate worsened functional outcomes for patients with treatment-refractory fever within the first 10 days following SAH, with increased mortality, severe functional disability, and more severe cognitive impairment. Even in the lowest grade, mildest SAH patients, a single fever during the first 10 days postinjury is associated with overall worsened patient outcomes (Hanafy et al., 2010).
Fever treatments in subarachnoid hemorrhage
At present, available data do not clearly show that aggressive temperature control results in improved outcomes (Broessner et al., 2010). Results of recent studies of fever control are mixed. For example, Badjatia et al. (2010) performed a case control study in which 40 consecutive subarachnoid hemorrhage patients with fever were enrolled. Patients then received advanced fever control with either a surface cooling or intravascular cooling device. These patients were then compared to matched patients who received conventional fever management, consisting of scheduled acetaminophen (paracetamol) with or without the use of a water cooling blanket. Advanced fever control patients were more likely to need mechanical intubation and intravenous sedation, to have longer stays in the ICU, and to undergo tracheostomy when compared to case matched controls. Outcomes were the same for both groups of patients at 3 months. Interestingly, at 12 months postevent, the advanced fever control patients had significantly better outcomes. Badjatia’s study (2010) illustrates how aggressive fever control might result in better long term outcomes, despite the potential for worse short term consequences. The advantages and disadvantages of hyperthermia treatments are listed in Table 34.1.
Table 34.1.
Treatments for hyperthermia
| Treatment | Advantages | Disadvantages |
|---|---|---|
| Acetaminophen (paracetamol) | Appropriate for almost all patients, contraindicated with liver failure | Usually ineffective alone |
| Cooling devices, e.g., fans, ice packs | Low cost, noninvasive, can be used with almost all patients | Usually ineffective for lowering temperature |
| Advanced cooling devices, e.g., Arctic Sun®, intravascular cooling devices | Effective in most patients for lowering temperature | Shivering common and may counteract benefits of cooling |
First-line treatment for fever is acetaminophen. While acetaminophen is safe to administer in most patients (save for those with concomitant liver disease), acetaminophen alone is often ineffective for fever control. Some studies suggest that doses up to 6 g per day maybe needed to see a significant effect on fever. More importantly, the effectiveness of acetaminophen is based on the assumption that the prostaglandin E is functionally driving the underlying hyperthermia. If prostaglandin E is not responsible for the fever or the pathways are dysfunctional, administration of acetaminophen will not lower temperature. NSAIDs may be considered as well, but were not effective for fever control in small studies of stroke patients (Kasner et al., 2002; Scaravilli et al., 2011b). NSAIDs also confer a bleeding risk, thus making it an inappropriate treatment for patients with intracranial hemorrhage or external ventricular drain (EVD) placement.
External cooling devices, including fans and ice packs, have been relatively ineffective at lowering body temperature (Badjatia, 2009). In a case control study, surface cooling devices, which function by circulating chilled water through pads that are applied directly to the patient’s skin, are more effective at lowering temperature than conventional fever control (a combination of acetaminophen and external cooling devices) (Badjatia et al., 2010). Intravascular cooling devices also performed well, with data suggesting superiority over conventional fever control treatment (Badjatia et al., 2010).
While advanced cooling devices, such as surface and intravascular cooling devices are the most effective treatments for lowering temperature, shivering complicates their use. Shivering, which occurs in up to 40% of cooled patients, can result in a two-to threefold increase in oxygen consumption over a patient’s basal rate along with paradoxical lower brain tissue oxygenation. Both these effects would have a negative impact on SAH patients (Badjatia, 2009). Magnesium administration and buspirone administration are considered the first line measure to counteract the effects of shivering. Hypomagnesemia has been linked with both shivering and exacerbation of tissue ischemia; hence administration of magnesium may mitigate several disease processes. Target levels for magnesium, which may be given intravenously in the ICU setting, are 3–4 mg/dL. Buspirone has few side-effects and may also reduce shivering, with its principal limitation being the need for oral administration (Badjatia, 2009). When magnesium and buspirone are insufficient for shiver control, meperidine may be effective. Short-acting sedatives may also be used if other measures are ineffective.
PAROXYSMAL SYMPATHETIC HYPERACTIVITY
Paroxysmal sympathetic hyperactivity (PSH) (Rabinstein, 2007) is characterized by the episodic increase in vital signs, particularly heart rate, blood pressure, respiratory rate, and temperature. In addition to vital sign abnormalities, patients with PSH may exhibit excessive sweating (diaphoresis) and abnormal, nonpurposeful movements. The clinical features tend to occur simultaneously with each other and may occur in response to external stimuli, such as with endotracheal tube suctioning, or occur spontaneously (Perkes et al., 2010).
A challenge to recognizing PSH has been disagreement regarding nomenclature. PSH has been identified by at least 31 different clinical terms in the literature (Perkes et al., 2010, 2011). Among these are terms such as diencephalic autonomic epilepsy (Penfield, 1929), sympathetic storming (Lemke, 2004), autonomic dysfunction syndrome (Rossitch and Bullard, 1988), dysautonomia (Fearnside et al., 1993), and paroxysmal autonomic instability with dystonia (Blackman et al., 2004). Understandably, there have been multiple different diagnostic criteria applied to the syndrome (Table 34.2). The unifying principle underlying all the nomenclature is the episodic nature of the dysregulation of the autonomic nervous system. “PSH” is the preferred term in this text as it is descriptive of the syndrome without obligating an etiology.
Table 34.2.
Past terms and definitions used to describe the clinical phenomena of paroxysmal sympathetic hyperactivity
| Term used/author | Diagnostic criteria |
|---|---|
| Dysautonomia/Baguley et al. | “For the purposes of the study, dysautonomia was defined as simultaneous, paroxysmal increases in at least five out of the seven reported features of dysautonomia(heart rate, respiratory rate, temperature, blood pressure, posturing, dystonia, and sweating), with episodes persisting for at least 2 weeks after injury.” |
| Paroxysmal autonomic instability with dystonia (PAID)/Blackman et al., 2004 | “Within the clinical setting of traumatic brain injury, signs of PAID syndrome include severe brain injury (Rancho Los Amigos level IV), temperature of at least 38.5 °C, pulse of at least 130 beats/min, respiratory rate of at least [20] breaths/min, systolic blood pressure of at least 140 mmHg, agitation, diaphoresis, and dystonia (i.e., rigidity or decerebrate posturing). The duration is at least 1 cycle per day for at least 3 days. Finally, other conditions must be ruled out.” |
| Dysautonomic crises/Fernández-Ortega et al., 2006 | “Dysautonomic crises were defined as short, repeated episodes throughout the day, for no apparent reason, of at least five of the following: tachycardia, tachypnoea, hypertension, profuse sweating, pupillary dilation, reduction in the level of consciousness with muscle rigidity, and a tendency to a decerebration posture.” |
(Adapted from Perkes et al., 2011.)
Incidence, risk factors, and pathophysiology
Estimates of the incidence of PSH vary from 8% to 33% of TBI patients depending on how the syndrome is defined, the research setting, and time postinjury (Perkes et al., 2010; Fernández-Ortega et al., 2012). However, prospective trials in the ICU setting and with more consistent diagnostic criteria indicate that the incidence of PSH that continues for 2 weeks or more is around 10% (Baguley et al., 2007b; Fernández-Ortega et al., 2012). The true incidence may be hard to judge because these patients typically have significant neurologic and systemic injuries; thus, some patients may die from other causes before PSH is identified.
As discussed previously, the CAN involves multiple levels of the central nervous system, all of which are susceptible to disruption following acquired brain injury. However, thus far imaging data have not presented a clear picture of injury patterns underlying PSH. The best studied etiology of PSH is TBI, accounting for 80% of the 349 cases of PSH identified in the literature (Perkes et al., 2010). In a retrospective study in an inpatient rehabilitation setting, PSH risk factors included diffuse axonal injury and younger age (Baguley et al., 1999). More recent computed tomography (CT) data collected by Fernández-Ortega and colleagues (2012) found focal parenchymal lesions were more common in patients with PSH compared to non-PSH matched peers. Another prospective ICU-based study failed to find any association between CT findings and the condition (Baguley et al., 2007b).
In a recent MRI study, Lv and colleagues (2010) found that patients with midbrain and pontine lesions were at higher risk of developing PSH. Their study also suggested that injury to the periventricular white matter, corpus callosum, and deep gray nuclei (e.g., thalamus) were associated with the development of PSH. While these studies suggest that structural lesions maybe associated with PSH, no specific lesion or pattern of lesions has been identified (Fernández-Ortega et al., 2011).
While the majority of patients with PSH in the literature are those surviving TBI, PSH has also been observed in a wide range of other conditions including ischemic stroke, cerebral neoplasm, hydrocephalus, and anoxic brain injury (Perkes et al., 2010; Fernández-Ortega et al., 2012). In addition, more exotic but isolated case studies of PSH following diabetic coma (Baguley and Nott, 2008) and anti-NMDA receptor encephalitis (Hinson et al., 2013) have been reported. The wide diversity of etiologies behind PSH is also problematic for identifying a single underlying pathophysiology.
Rather than require a specific pathology, Baguley and colleagues (2008) have expanded the suggestion that PSH may be part of a “disconnection syndrome,” hypothesizing that preganglionic neurons that project to the brainstem and spinal cord might be “released” from the higher cortical centers (e.g., paraventricular nucleus) after neurologic injury. After such a “release,” sympathetic activity may continue unchecked. This mechanism does not require specific patterns of injury to occur, only that the cumulative cerebral injury produces sufficient damage at the level of the midbrain to reduce inhibitory drive of spinal cord sympathetic neurons (Baguley, 2008a).
The phenomenon of “triggering,” where discrete episodes of PSH follow an observable stimulus such as endotracheal tube suctioning, may also give insights into the syndrome’s pathophysiology (Fig. 34.3) (Baguley et al., 2009b). Such “triggering” bears a marked similarity to the phenomenon of eliciting paroxysms in autonomic dysreflexia as occurs in patients with high thoracic or cervical cord injuries. Initially, patients may experience bradycardia due to loss of supraspinal influence on the distal spinal cord. Over time, the absence of supraspinal inputs causes the dendritic rearborization of neurons distal to the lesion (Rabchevsky, 2006). This regrowth is disorganized, however, and may produce dysfunctional connections between the afferent sensory neurons and sympathetic lateral horn efferents. The poorly reformed connections may therefore lack sufficient inhibitory input, resulting in disproportionate sympathetic discharge to usually benign afferent stimuli. With regards to PSH, some limited data support this link, as the exaggerated sensitivity of the sympathetic discharge in response to non-nociceptive stimuli has been recorded in the early postinjury period (Baguley et al., 2009b) through to 5 years post-trauma (Baguley et al., 2009a).
Fig. 34.3.
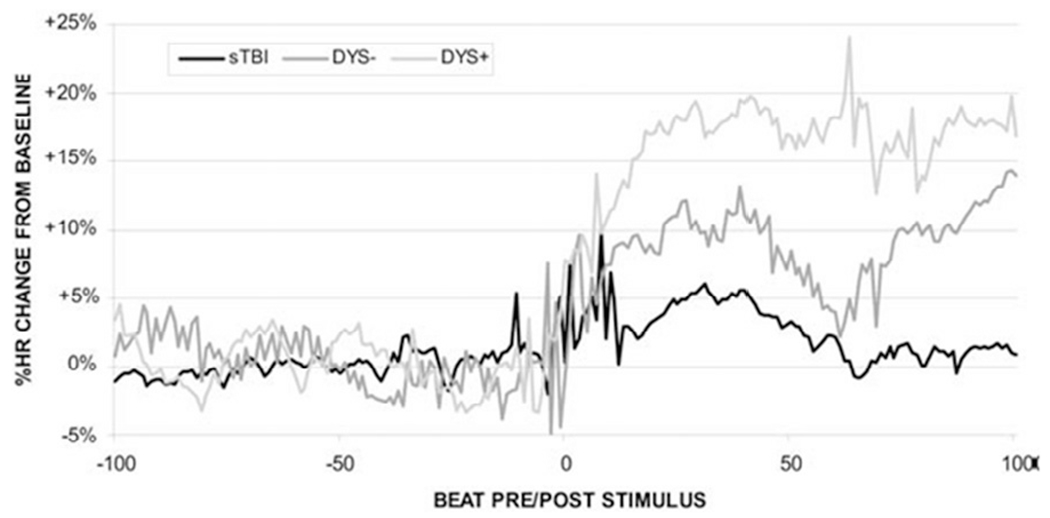
Normalized heart rate (HR) change for 100 beats before and after endotracheal tube (ETT) suctioning in an intensive care unit. Poststimulus HR increased by 2% in the sTBI group, by 8% in the DYS — group, and by 16% in the DYS+group. DYS+, dysautonomic; DYS−, nondysautonomic; sTBI, standard TBI. (Reproduced from Baguley et al., 2009b.)
Clinical descriptions
PSH is typically identified in the hospital setting during the acute phase of recovery from an acquired brain injury. Following TBI, patients commonly develop symptoms of PSH a week after injury, but the syndrome may appear before or after this mark. The first appearance of features may correspond to the weaning of sedation (Baguley et al., 1999). While PSH patients may have multiple vital sign abnormalities, tachycardia and hypertension often manifest first. While paroxysmal increases in heart rate may be extreme, with heart rates up to 190 reported (Baguley et al., 2008), patients will usually have periods of normal heart rates during any 24 hour period (Fig. 34.4). In addition to increased physiologic parameters, abnormal movements usually accompany PSH. Sweating, decerebrate posturing, decorticate posturing, dystonia, rigidity, elevated intracranial pressure, and spasticity may also be found in PSH patients (Turazzi and Bricolo, 1977; Baguley et al., 2006).
Fig. 34.4.
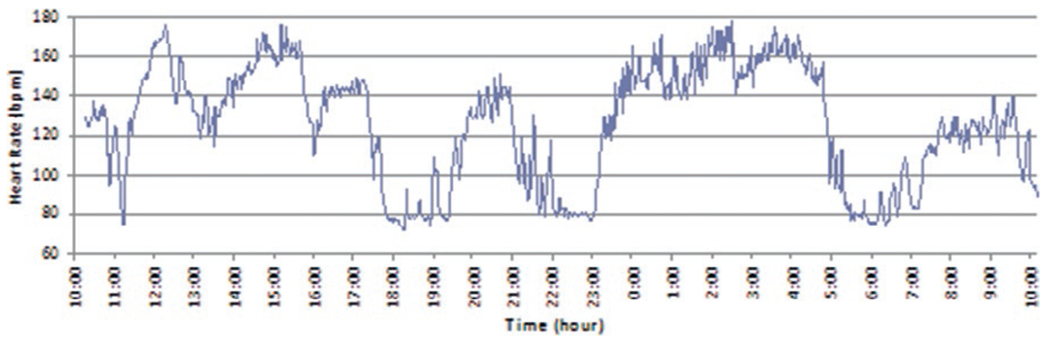
Heart rate over time in a patient with paroxysmal sympathetic hyperactivity following traumatic brain injury. Note the periods of tachycardia contrasted against a background of normal heart rate. (From Baguley (2008), with permission from Jaypee Brothers Medical Publishers Pty. Ltd., India.)
It has been suggested that PSH is best considered as a spectral disorder (Baguley et al., 2007b). One-quarter to one-third of patients exhibit the syndrome for 7 days. In contrast, around 8–14% of patients will have symptoms longer than 7 days, continuing into rehabilitation in an estimated 5% of patients (Baguley et al., 2007b). The patients with longer duration of symptoms often have worse outcomes, manifesting as prolonged swallowing abnormalities, longer comas, longer post-traumatic amnesia, longer hospital admissions, and increased healthcare costs (Baguley et al., 1999, 2007b).
The most severely affected patients will sustain symptoms of PSH such as excessive diaphoresis for many months after their injury (Baguley et al., 2007b; Dolce et al., 2008). The cessation of diaphoresis is one clinical marker used to define the transition from acute to chronic PSH (Baguley et al., 2006; Hinson et al., 2012). There may be other subtle signs of PSH that persist months after injury. Baguley et al. (2006) demonstrated that abnormalities of heart rate variability may persist in PSH patients for 14 months postinjury. Remarkably, a follow-up study showed a propensity for abnormal heart rate variability in response to nociception a mean of 5 years postinjury (Baguley et al., 2009a).
Paroxysmal sympathetic hyperactivity management
After the diagnosis of PSH has been made, patients may warrant treatment based on the severity of their clinical features. This is important, as circumstantial evidence suggests that undermanaged PSH produces unnecessary secondary morbidity (Baguley et al., 2004). Clinicians also worry that unchecked sympathetic hyperactivity can perpetuate arrhythmias, hypertensive crises, and end organ damage. Unfortunately, the management of PSH symptoms is challenging as there is only level III research available to guide management (Baguley, 2008b), and the multiple agents that have been utilized are reported to show varying degrees of success (Baguley et al., 2008; Perkes et al., 2010; Masel, 2011).
At present, symptomatic management is the mainstay of therapy for PSH (Perkes et al., 2010). Sedatives and opioids, especially morphine, may be helpful in reducing PSH episodes. One small, open-label trial of four patients suggested that gabapentin, an agonist of the a28 subunit of a voltage-dependent calcium channel, may be useful in symptomatic management (Baguley et al., 2007a). Retreatment with sedatives prior to known procedures may be helpful in managing planned stimulation (Lemke, 2007). While evidence supports intrathecal baclofen in PSH management, there are many disadvantages to its use (Becker et al., 2000; Turner, 2003; Anderson et al., 2004; Senno et al., 2004). Implanting intrathecal pumps is invasive and expensive. Furthermore, the syndrome is often self-limiting, thus the placement of a permanent device may not be justified. Table 34.3 sets out the uses and efficacy of pharmacologic treatments for PSH.
Table 34.3.
Pharmacologic treatments for paroxysmal sympathetic hyperactivity*
| Neurotransmitter class | Drug | Unhelpful | Beneficial |
|---|---|---|---|
| Opiate agonist | Morphine | 5 | 7 |
| Methadone | 2 | ||
| GABA A agonist | Diazepam | 2 | |
| Midazolam | 2 | ||
| Clonazepam | 3 | 1 | |
| Lorazepam | 3 | 1 | |
| GABA B agonist | Baclofen (oral) | 4 | |
| Baclofen (intrathecal) | 7 | ||
| α Antagonist | Clonidine | 5 | 3 |
| β Antagonist | Propanolol | 4 | 8 |
| Labetalol | 1 | 1 | |
| Metoprolol | 2 | ||
| Dopamine agonist | Carbi/levodopa | 1 | 2 |
| Bromocriptine | 4 | 7 | |
| Dopamine antagonist | Chlorpromazine | 2 | |
| Other | Gabapentin | 1 | |
| Dantrolene | 5 | 1 | |
| Phenobarbital | 4 | ||
| Phenytoin | 3 | ||
| Carbamazepine | 1 | ||
| Thorazine | 1 | ||
| Propofol | 1 | ||
| Acetaminophen (paracetamol) | 1 |
indicative treatment reports from published literature, generated from the number of papers referencing the use and efficacy of each medication. (Adapted from Supplementary Table 2, in Perkes et al., 2010.)
Other less invasive medical options are available. β-Blockers may be used to help blunt tachycardia and are considered a first-line agent for the treatment of PSH. Propranolol and labetalol have the most success in terms of efficacy; metoprolol seems to be less useful in the treatment of PSH (Salim et al., 2008). Propranolol has also been shown to decrease catecholamine levels in PSH (Feibel et al., 1981; Robertson et al., 1983). a Antagonists, such as clonidine and dexmedetomidine may also help to reduce adrenergic crises. Gabapentin maybe useful in reducing sympathetic tone in the spinal cord and thus helping to restore the autonomic balance (Baguley et al., 2007a; Baguley and Nott, 2008). Bromocriptine may also have efficacy in PSH treatment (Russo and O’Flaherty, 2000) but may be more useful as an adjunctive agent. Bromocriptine is a dopamine agonist and has fallen out of favor in recent years for two reasons. First, it is sporadically effective for PSH and other medications, such as β-blockers are typically more reliably effective. Second, it has multiple side-effects, including psychosis, which limits its utility in the ICU setting.
CONCLUSION
The central autonomic network is a complex system of nuclei and pathways providing communication from the cortex, deep gray matter, and brainstem to sympathetic neurons of the spinal cord. Autonomic dysfunction of any type may occur following traumatic brain injury, with the complexity of CAN pathways and nuclei showing variable susceptibility to damage and abnormal autonomic control. In this regard, this chapter presents ischemic stroke data as a model of how focal injuries may produce autonomic dysregulation and presents non-TBI-related SAH as an example of a disease process producing a predictable pattern of hyperadrenergic activity. The syndrome of PSH, representing a common clinical end point of excessive sympathetic hyperactivity resulting from multiple etiologies, is discussed along with diagnosis and management suggestions. Overall, these syndromes suggest that while manifestations of the hyperadrenergic state are many, dysfunction arising from damage to CAN centers and connections may be thought of as a disturbed balance of excitatory:inhibitory inputs to the ANS effector organs.
References
- Anderson VL, Ahmed G, Duraski SA et al. (2004). Alternative treatment in the management of combined hyperadrenergia and spasticity in the adult with a severe traumatic brain injury: case report. Arch Phys Med Rahabil 85: e15. [Google Scholar]
- Badjatia N (2009). Hyperthermia and fever control in brain injury. Crit Care Med 37: S250–S257. [DOI] [PubMed] [Google Scholar]
- Badjatia N, Fernandez L, Schmidt JM et al. (2010). Impact of induced normothermia on outcome after subarachnoid hemorrhage. Neurosurg 66: 696–701. [DOI] [PubMed] [Google Scholar]
- Baguley IJ (2008a). The excitatory:inhibitory model (EIR model): an integrative explanation of acute autonomic overactivity syndromes. Med Hypotheses 70: 26–35. [DOI] [PubMed] [Google Scholar]
- Baguley IJ (2008b). Autonomic complications following central nervous system injury. Sem Neurol 28: 716–725. [DOI] [PubMed] [Google Scholar]
- Baguley I (2008c). Dysautonomia following traumatic brain injury. In Nayyar V (Ed.) Critical Care Update 2008, pp. 155–165. [Google Scholar]
- Baguley IJ, Nott MT (2008). Quantitating the efficacy of gabapentin in a novel case of dysautonomia. Neurorehabil Neural Repair 22: 570–571. [Google Scholar]
- Baguley IJ, Nichols JL, Felmingham KL et al. (1999). Dysautonomia after traumatic brain injury: a forgotten syndrome? J Neurol Neurosurg Psychiatr 67: 39–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baguley IJ, Cameron ID, Green AM et al. (2004). Pharmacological management of dysautonomia following traumatic brain injury. Brain Inj 18: 409–417. [DOI] [PubMed] [Google Scholar]
- Baguley IJ, Heriseanu RE, Felmingham KL et al. (2006). Dysautonomia and heart rate variability following severe traumatic brain injury. Brain Inj 20: 437–444. [DOI] [PubMed] [Google Scholar]
- Baguley IJ, Heriseanu RE, Gurka JA et al. (2007a). Gabapentin in the management of dysautonomia following severe traumatic brain injury: a case series. J Neurol Neurosurg Psychiatr 78: 539–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baguley IJ, Slewa-Younan S, Heriseanu RE et al. (2007b). The incidence of dysautonomia and its relationship with autonomic arousal following traumatic brain injury. Brain Inj 21: 1175–1181. [DOI] [PubMed] [Google Scholar]
- Baguley IJ, Heriseanu RE, Cameron ID et al. (2008). A critical review of the pathophysiology of dysautonomia following traumatic brain injury. Neurocrit Care 8: 293–300. [DOI] [PubMed] [Google Scholar]
- Baguley IJ, Heriseanu RE, Nott MT et al. (2009a). Dysautonomia following severe traumatic brain injury: evidence of persisting over-responsiveness to afferent stimuli. Am J Phys Rehab Med 88: 615–622. [DOI] [PubMed] [Google Scholar]
- Baguley IJ, Nott MT, Slewa-Younan S et al. (2009b). Diagnosing dysautonomia after acute traumatic brain injury: evidence for overresponsiveness to afferent stimuli. Arch Phys Med Rehabil 90: 580–586. [DOI] [PubMed] [Google Scholar]
- Barron SA, Rogovski Z, Hemli J (1994). Autonomic consequences of cerebral hemisphere infarction. Stroke 25: 113–116. [DOI] [PubMed] [Google Scholar]
- Becker R, Benes L, Sure U et al. (2000). Intrathecal baclofen alleviates autonomic dysfunction in severe brain injury. J Clin Neurosci 7: 316–319. [DOI] [PubMed] [Google Scholar]
- Benarroch EE (1993). The central autonomic network: functional organization, dysfunction, and perspective. Mayo Clin Proc 68: 988–1001. [DOI] [PubMed] [Google Scholar]
- Blackman JA, Patrick PD, Buck ML et al. (2004). Paroxysmal autonomic instability with dystonia after brain injury. Arch Neurol 61: 321–328. [DOI] [PubMed] [Google Scholar]
- Broessner G, Lackner P, Fischer M et al. (2010). Influence of prophylactic, endovascularly based normothermia on inflammation in patients with severe cerebrovascular disease: a prospective, randomized trial. Stroke 41: 2969–2972. [DOI] [PubMed] [Google Scholar]
- Di Giugno G, Rosa G (1998). Neurogenic pulmonary edema during subarachnoid hemorrhage. Minerva Anestesiol 64: 229–230. [PubMed] [Google Scholar]
- Dolce G, Quintieri M, Serra S et al. (2008). Clinical signs and early prognosis in vegetative state: a decisional tree, data-mining study. Brain Inj 22: 617–623. [DOI] [PubMed] [Google Scholar]
- Dougherty P (2013). Neuroscience online, an electronic textbook for the neurosciences. University of Texas Medical School at Houston; Available at, http:/nba.uth.tmc.edu/neuroscience/s4/chapter03.html (Accessed March13, 2013). [Google Scholar]
- Fearnside MR, Cook RJ, McDougall P et al. (1993). The Westmead Head Injury Project outcome in severe head injury. A comparative analysis of pre-hospital, clinical and CT variables. Br J Neurosurg 7: 267–279. [DOI] [PubMed] [Google Scholar]
- Feibel JH, Baldwin CA, Joynt RJ (1981). Catecholamine-associated refractory hypertension following acute intracranial hemorrhage: control with propranolol. Ann Neurol 9: 340–343. [DOI] [PubMed] [Google Scholar]
- Fernandez-Ortega JF, Prieto-Palomino MA, Munoz-Lopez A, Lebron-Gallardo M, Cabrera-Ortiz H, Quesada-Garcia G (2006). Prognostic influence and computed tomography findings in dysautonomic crises after traumatic brain injury. Journal of Trauma-Injury Infection & Critical Care 61: 1129–1133. [DOI] [PubMed] [Google Scholar]
- Fernández-Ortega JF, Prieto-Palomino MA, Quesada-García G et al. (2011). Findings in the magnetic resonance of paroxysmal sympathetic hyperactivity. J Neurotrauma 28: 1327–1328. [DOI] [PubMed] [Google Scholar]
- Fernández-Ortega JF, Prieto-Palomino MA, Garcia-Caballero M et al. (2012). Paroxysmal sympathetic hyperactivity after traumatic brain injury: clinical and prognostic implications. J Neurotrauma 29: 1364–1370. [DOI] [PubMed] [Google Scholar]
- Frontera JA, Parra A, Shimbo D et al. (2008). Cardiac arrhythmias after subarachnoid hemorrhage: risk factors and impact on outcome. Cerebrovasc Dis 26: 71–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao C, Liu X, Shi ZH (2009). Relationship between sympathetic nervous activity and inflammatory response after subarachnoid hemorrhage in a perforating canine model. Auton Neurosci 147: 70–74. [DOI] [PubMed] [Google Scholar]
- Ginsberg MD, Sternau LL, Globus MY et al. (1992). Therapeutic modulation of brain temperature: relevance to ischemic brain injury. Cerebrovasc Brain Metab Rev 4: 189–225. [PubMed] [Google Scholar]
- Guyton AC, Hall JE (2006). The autonomic nervous system and the adrenal medulla. In: Guyton AC, Hall JE (Eds.), Textbook of Medical Physiology. 11th edn. Saunders, Philadelphia: p. 758, ch. 60. [Google Scholar]
- Hanafy KA, Morgan Stuart R, Fernandez L et al. (2010). Cerebral inflammatory response and predictors of admission clinical grade after aneurysmal subarachnoid hemorrhage. J Clin Neurosci 17: 22–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinson HE, Sheth KN (2012). Manifestations of the hyperadrenergic state after acute brain injury. Curr Op in Crit Care 18: 139–145. [DOI] [PubMed] [Google Scholar]
- Hinson HE, Takahashi C, Altowaijri G et al. (2013). Anti-NMDA receptor encephalitis with paroxysmal sympathetic hyperactivity: an under-recognized association? Clin Auton Res 23: 109–111. [DOI] [PubMed] [Google Scholar]
- Hirashima Y, Takashima S, Matsumura N et al. (2001). Right sylvian fissure subarachnoid hemorrhage has electrocardiographic consequences. Stroke 32: 2278–2281. [DOI] [PubMed] [Google Scholar]
- Horowitz MB, Willet D, Keffer J (1998). The use of cardiac troponin-I (cTnI) to determine the incidence of myocardial ischemia and injury in patients with aneurysmal and presumed aneurysmal subarachnoid hemorrhage. Acta Neurochir (Wien) 140: 87–93. [DOI] [PubMed] [Google Scholar]
- Inamasu J, Nakatsukasa M, Mayanagi K et al. (2012a). Subarachnoid hemorrhage complicated with neurogenic pulmonary edema and takotsubo-like cardiomyopathy. Neurol Med Chir (Tokyo) 52: 49–55. [DOI] [PubMed] [Google Scholar]
- Inamasu J, Sugimoto K, Yamada Y et al. (2012b). The role of catecholamines in the pathogenesis of neurogenic pulmonary edema associated with subarachnoid hemorrhage. Acta Neurochir (Wien) 154: 2179–2184. [DOI] [PubMed] [Google Scholar]
- Kasner SE, Wein T, Piriyawat P et al. (2002). Acetaminophen for altering body temperature in acute stroke: a randomized clinical trial. Stroke 33: 130–134. [DOI] [PubMed] [Google Scholar]
- Katsanos AH, Korantzopoulos P, Tsivgoulis G et al. (2013). Electrocardiographic abnormalities and cardiac arrhythmias in structural brain lesions. Int J Cardiol 167: 328–334. [DOI] [PubMed] [Google Scholar]
- Kono T, Morita H, Kuroiwa T et al. (1994). Left ventricular wall motion abnormalities in patients with subarachnoid hemorrhage: neurogenic stunned myocardium. J Am Coll Cardiol 24: 636–640. [DOI] [PubMed] [Google Scholar]
- Kuriyama N, Mizuno T, Niwa F et al. (2010). Autonomic nervous dysfunction during acute cerebral infarction. Neurol Res 32: 821–827. [DOI] [PubMed] [Google Scholar]
- Lemke DM (2004). Riding out the storm: sympathetic storming after traumatic brain injury. J Neurosci Nurs 36: 4–9. [PubMed] [Google Scholar]
- Lemke DM (2007). Sympathetic storming after severe traumatic brain injury. Crit Care Nurse 27: 30–37. [PubMed] [Google Scholar]
- Loewy AD, Spyer KM (1990). Central autonomic pathways. In: Loewy AD, Spyer KM (Eds.), Central Regulation of Autonomic Functions, Oxford University Press, Oxford, pp. 88–100. [Google Scholar]
- Lv LQ, Hou LJ, Yu MK (2010). Prognostic influence and magnetic resonance imaging findings in paroxysmal sympathetic hyperactivity after severe traumatic brain injury. J Neurotrauma 27: 1945–1950. [DOI] [PubMed] [Google Scholar]
- Masel BE (2011). Hyperbaric oxygen therapy for traumatic brain injury: still an enigma. Arch Phys Med Rehabil 92: 1519–1521. [DOI] [PubMed] [Google Scholar]
- Min J, Farooq MU, Greenberg E et al. (2009). Cardiac dysfunction after left permanent cerebral focal ischemia: the brain and heart connection. Stroke 40: 2560–2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagai M, Hoshide S, Kario K (2010). The insular cortex and cardiovascular system: a new insight into the brain-heart axis. J Am Soc Hypertens 4: 174–182. [DOI] [PubMed] [Google Scholar]
- Naidech A, Du Y, Kreiter KT et al. (2005). Dobutamine versus milrinone after subarachnoid hemorrhage. Neurosurgery 56: 21–26. [DOI] [PubMed] [Google Scholar]
- Napadow V, Dhond R, Conti G et al. (2008). Brain correlates of autonomic modulation: combining heart rate variability with fMRI. Neuroimage 42: 169–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oddo M, Frangos S, Milby A et al. (2009). Induced normothermia attenuates cerebral metabolic distress in patients with aneurysmal subarachnoid hemorrhage and refractory fever. Stroke 40: 1913–1916. [DOI] [PubMed] [Google Scholar]
- Oppenheimer S (2006). Cerebrogenic cardiac arrhythmias. Clin Auton Res 16: 6–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oppenheimer S (2007). Cortical control of the heart. Cleve Clin J Med 74 (S1): S27–S29. [DOI] [PubMed] [Google Scholar]
- Ozdemir O, Hachinski V (2008). Brain lateralization and sudden death: its role in the neurogenic heart syndrome. J Neurol Sci 268: 6–11. [DOI] [PubMed] [Google Scholar]
- Penfield W (1929). Diencephalic autonomic epilepsy. Arch Neurol Psychiatry 22: 358. [Google Scholar]
- Perkes IE, Baguley IJ, Nott MT et al. (2010). A review of paroxysmal sympathetic hyperactivity after acquired brain injury. Ann Neurol 68: 126–135. [DOI] [PubMed] [Google Scholar]
- Perkes IE, Menon DK, Nott MT et al. (2011). Paroxysmal sympathetic hyperactivity after acquired brain injury: a review of diagnostic criteria. Brain Inj 25: 925–932. [DOI] [PubMed] [Google Scholar]
- Praveen Kumar A, Babu E, Subrahmanyam D (2012). Cerebrogenic tachyarrhythmia in acute stroke. J Neurosci Rural Practice 3: 204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prosser J, MacGregor L, Lees KR et al. (2007). Predictors of early cardiac morbidity and mortality after ischemic stroke. Stroke 38: 2295–2302. [DOI] [PubMed] [Google Scholar]
- Pyner S, Coote JH (2000). Identification of branching paraventricular neurons of the hypothalamus that project to the rostroventrolateral medulla and spinal cord. Neuroscience 100: 549–556. [DOI] [PubMed] [Google Scholar]
- Rabchevsky AG (2006). Segmental organization of spinal reflexes mediating autonomic dysreflexia after spinal cord injury. Prog Brain Res 152: 265–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabinstein AA (2007). Paroxysmal sympathetic hyperactivity in the neurological intensive care unit. Neurol Res 29: 680–682. [DOI] [PubMed] [Google Scholar]
- Rabinstein AA, Sandhu K (2007). Non-infectious fever in the neurological intensive care unit: incidence, causes and predictors. J Neurol Neurosurg Psychiat 78: 1278–1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rincon F, Dhamoon M, Moon Y et al. (2008). Stroke location and association with fatal cardiac outcomes: Northern Manhattan Study (NOMAS). Stroke 39: 2425–2431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson CS, Clifton GL, Taylor AA et al. (1983). Treatment of hypertension associated with head injury. J Neurosurg 59: 455–460. [DOI] [PubMed] [Google Scholar]
- Rossitch E Jr, Bullard DE (1988). The autonomic dysfunction syndrome: aetiology and treatment. Br J Neurosurg 2: 471–478. [DOI] [PubMed] [Google Scholar]
- Russo RN, O’Flaherty S (2000). Bromocriptine for the management of autonomic dysfunction after severe traumatic brain injury. J Paediatr Child Health 36: 283–285. [DOI] [PubMed] [Google Scholar]
- Salim A, Hadjizacharia P, Brown C et al. (2008). Significance of troponin elevation after severe traumatic brain injury. J Trauma 64: 46–52. [DOI] [PubMed] [Google Scholar]
- Samuels MA (2007). The brain-heart connection. Circulation 116: 77–84. [DOI] [PubMed] [Google Scholar]
- Sander D, Winbeck K, Klingelhöfer J et al. (2001). Prognostic relevance of pathological sympathetic activation after acute thromboembolic stroke. Neurology 57: 833–838. [DOI] [PubMed] [Google Scholar]
- Scaravilli V, Tinchero G, Citerio G (2011a). The participants in the international multi-disciplinary consensus conference on the critical care management of subarachnoid hemorrhage, fever management in SAH. Neurocrit Care 15: 287–294. [DOI] [PubMed] [Google Scholar]
- Scaravilli V, Tinchero G, Citerio G (2011b). Fever management in SAH. Neurocrit Care 15: 287–294. [DOI] [PubMed] [Google Scholar]
- Senno R, Anderson V, Ahmed G et al. (2004). Intrathecal baclofen administration in the management of hyperadrenergic state in an adult with severe anoxic brain injury. Arch Phys Med Rahabil 85: e15. [Google Scholar]
- Spyer KM, Gourine AV (2009). Chemosensory pathways in the brainstem controlling cardiorespiratory activity. Phil Trans R Soc B Biol Sci 364: 2603–2610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern JE (2001). Electrophysiological and morphological properties of pre-autonomic neurones in the rat hypothalamic paraventricular nucleus. J Physiol (Lond) 537: 161–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens RD, Nyquist PA (2007). The systemic implications of aneurysmal subarachnoid hemorrhage. J Neurol Sci 261: 143–156. [DOI] [PubMed] [Google Scholar]
- Tung P, Kopelnik A, Banki N et al. (2004). Predictors of neurocardiogenic injury after subarachnoid hemorrhage. Stroke 35: 548–551. [DOI] [PubMed] [Google Scholar]
- Turazzi S, Bricolo A (1977). Acute pontine syndromes following head injury. Lancet 2: 62–64. [DOI] [PubMed] [Google Scholar]
- Turner MS (2003). Early use of intrathecal baclofen in brain injury in pediatric patients. Acta Neurochir Suppl 87: 81–83. [DOI] [PubMed] [Google Scholar]
- Van Ravenswaaij-Arts CM, Kollée LA, Hopman JC et al. (1993). Heart rate variability. Ann Intern Med 118:436–447. [DOI] [PubMed] [Google Scholar]
- Wittstein IS, Thiemann DR, Lima JAC et al. (2005). Neurohumoral features of myocardial stunning due to sudden emotional stress. N Engl J Med 352: 539–548. [DOI] [PubMed] [Google Scholar]