

Abstract
To understand the genetics of steroid-sensitive nephrotic syndrome (SSNS), we conducted a genome-wide association study in 987 childhood SSNS patients and 3,206 healthy controls with Japanese ancestry. Beyond known associations in the HLA-DR/DQ region, common variants in NPHS1-KIRREL2 (rs56117924, P=4.94E-20, odds ratio (OR) =1.90) and TNFSF15 (rs6478109, P=2.54E-8, OR=0.72) regions achieved genome-wide significance and were replicated in Korean, South Asian and African populations. Trans-ethnic meta-analyses including Japanese, Korean, South Asian, African, European, Hispanic and Maghrebian populations confirmed the significant associations of variants in NPHS1-KIRREL2 (Pmeta=6.71E-28, OR=1.88) and TNFSF15 (Pmeta=5.40E-11, OR=1.33) loci. Analysis of the NPHS1 risk alleles with glomerular NPHS1 mRNA expression from the same person revealed allele specific expression with significantly lower expression of the transcript derived from the risk haplotype (Wilcox test p=9.3E-4). Because rare pathogenic variants in NPHS1 cause congenital nephrotic syndrome of the Finnish type (CNSF), the present study provides further evidence that variation along the allele frequency spectrum in the same gene can cause or contribute to both a rare monogenic disease (CNSF) and a more complex, polygenic disease (SSNS).
Keywords: Nephrotic syndrome, glomerulus, pediatric nephrology, podocyte
Introduction
Idiopathic nephrotic syndrome (INS) is the most common glomerular disease in children. The prevalence of INS was reported to be nearly 16 cases per 100,0001. There are substantial ethnic differences in the incidence of INS ranging from two to seven per 100,000 children. The incidence of INS is higher in Asian ancestry compared with European ancestry2. In Japan, the estimated incidence of INS is 6.49 cases per 100,000 children3. Approximately 3% of children with steroid-sensitive nephrotic syndrome (SSNS) have a family history of SSNS4. The pathogenesis of INS is still unclear although more than 50 causative single genes for steroid-resistant nephrotic syndrome (SRNS) have been found, and causative mutations in these genes were identified in approximately 30% of childhood and young adult patients with SRNS5, 6. Recently, six genes associated with Rho-like small GTPase activity were identified as causes of partially-steroid treatable nephrotic syndrome7. EMP2 mutations were identified in SSNS by a combination of linkage analysis and exome sequencing8. However, few patients with SSNS have a mutation in the above genes, indicating alternative genetic architecture might drive the pathogenesis for most patients with SSNS9. Acute infections and insect stings are well-known triggers of the onset and relapse of NS10–13. These findings suggest that genetic and environmental factors are important susceptibility factors for the development of SSNS. To elucidate the genetic susceptibility factors of SSNS in children, four genome-wide association studies (GWASs) in well-characterized case-control series have been performed. HLA-DR/DQ region exhibited the most significant association with disease in European and Japanese populations14–17,=with two non-HLA loci on 4q13.3 (PARM1) and 6q22.1 (CALHM6) also achieving genome-wide significance in only European chidren15, 17. Considering the limited sample sizes in these studies, larger cohorts and international collaborative studies are necessary to identify additional susceptibility factors. In a Japanese population, we performed a discovery GWAS with the largest sample size to date (987 childhood SSNS vs 3,206 controls), followed by an international replication and trans-ethnic meta-analysis (Figure 1).
Figure 1. Flowchart.
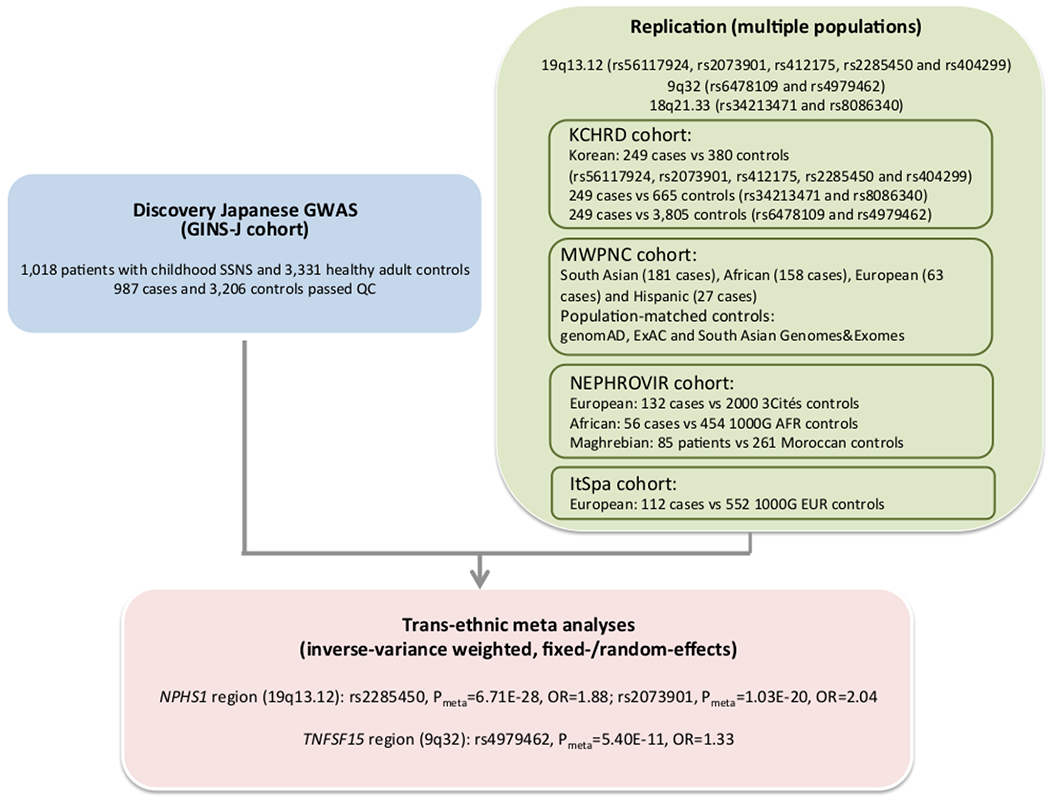
Main outline of the discovery genome-wide association study (GWAS), replication of candidate SNPs outside HLA with multiple populations and trans-ethnic meta-analysis. GINS-J: The Research Consortium on Genetics of Childhood Nephrotic Syndrome in Japan, KCHRD: Korean Consortium of Hereditary Renal Diseases in Children, MWPNC: Midwest Pediatric Nephrology Consortium, NPHROVIR: Children Cohort Nephrosis and Virus, ItSpa: Italian and Spanish cohort.
Results
Subjects
Definitions of INS are shown in Supplementary Table S1. In the discovery stage, 1,018 Japanese patients with childhood-onset SSNS were recruited and 987 cases were included in the association analysis after quality control (QC). Characteristics of the 987 patients are shown in Supplementary Table S2. The male to female ratio was 2.6:1. The median age at onset was 4.0 years (0.4–17.9 years). Renal biopsy was performed in 501 of 987 patients (51%) (minimal change disease [MCD, N=470, 93.8%], focal segmental glomerulosclerosis [FSGS, N=16, 3.2%], diffuse mesangial proliferation [DMP, N=14, 2.8%], focal mesangial proliferation [N=1, 0.2%]). In total, 3,331 healthy Japanese adults were recruited as controls.
Genome-wide SNP-based association analysis
In the discovery stage, 1,018 cases and 3,331 healthy controls were genotyped by ‘Japonica Array’. Whole-genome imputation was performed subsequently using the 2KJPN panel. After data cleaning (Supplementary Figure S1A–E), 987 cases and 3,206 controls with 6,834,340 autosomal single nucleotide variants (SNVs) and short insertions and deletions (INDELs) were retained for association analyses. The power of discovery GWAS exceeded 80% to detect low-frequency variants (minor allele frequency [MAF] >0.5%) with genotypic relative risk (RR) >6.20, or common alleles (MAF ≥5%) with RR >2.05, or variants with an AF=50% conferring RR >1.48 at a significant P-value threshold of 5E-08 under the additive model (Supplementary Figure S2). The inflation factor, λ, was 1.151 for all tested variants. After adjusting for sex and the first four principal components (PC1–4), the λ was 1.048, which decreased to 1.043 when variants in the HLA region (Hg19: chr6: 29,691,116–33,054,976) were excluded (Supplementary Figure S3A–B).
The most significant association was detected in the HLA-DR/DQ region (rs6901541, P-value=2.80E-33, OR=2.49, 95%CI: 2.15–2.89, Figures 2 and 3A). Signals in the NPHS1-KIRREL2 region on 19q13.12 (rs56117924, P-value=4.94E-20, OR=1.90, 95%CI: 1.66–2.18, Figure 3B) and in the TNFSF15 region on 9q32 (rs6478109, P-value=2.54E-08, OR=0.72, 95%CI: 0.64–0.81, Figure 3C) also achieved genome-wide significance (P-value<5E-08). A signal in the TNFRSF11A region on 18q21.33 achieved marginal genome-wide significance (rs34213471, P-value=7.68E-08, OR=1.38, 95%CI: 1.23–1.56, Figure 3D, Supplementary Table S3). Conditional analyses were performed in the novel non-HLA regions. No SNPs showed independent significance after conditioning on the lead SNP in each locus (Supplementary Figures S4–S6). Nine SNPs were chosen for targeted replication and meta-analysis: In the NPHS1-KIRREL2 region, the lead SNP (rs56117924), two potential functional SNPs in linkage disequilibrium (LD) with the lead SNP (rs2073901 and rs2285450) and two SNPs in LD with our lead SNP with sufficient frequency in European populations (rs412175 and rs404299) were selected (Supplementary Figure S7). In the TNFSF15 region, in addition to the lead SNP (rs6478109), rs4979462 (r2=0.55 with rs6478109 in the discovery Japanese set) was selected because it is associated with autoimmune diseases in previous reports. In the TNFRSF11A region, the lead SNP (rs34213471) and SNP in LD with the lead SNP (rs8086340, r2=0.42 with rs34213471 in the discovery Japanese set) were selected. All candidate SNPs were common variants (MAF > 5%) in the Japanese population (Table 1).
Figure 2. Manhattan plot in the discovery GWAS.
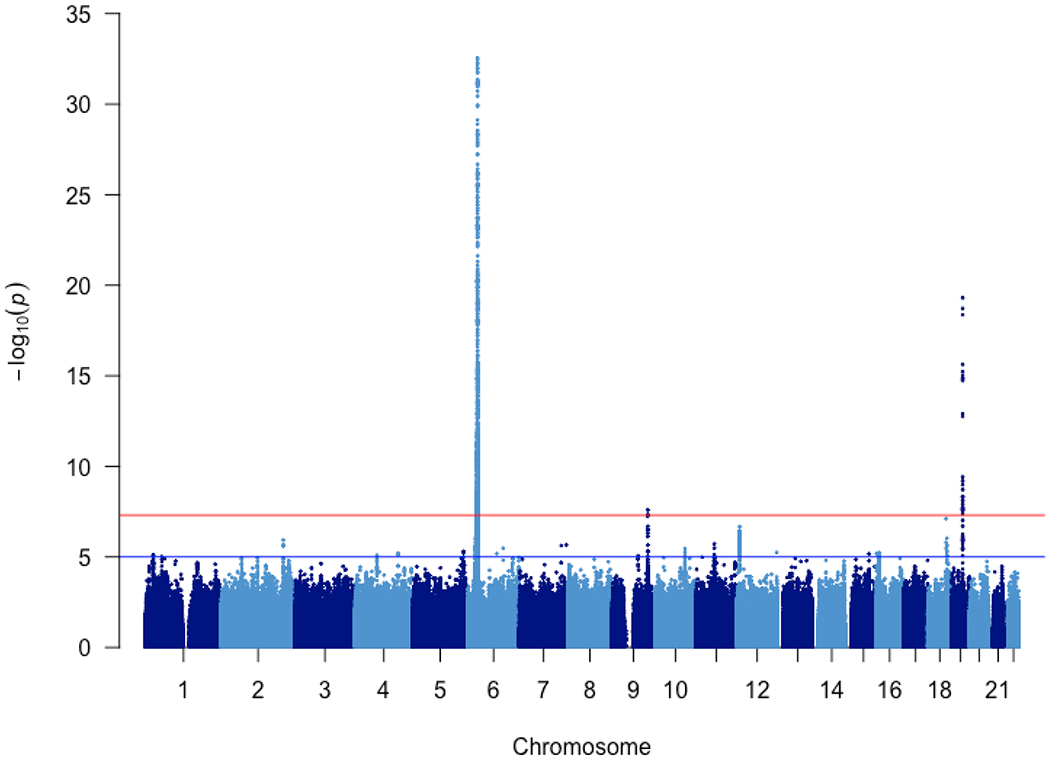
In the discovery stage, 987 patients with childhood SSNS and 3,206 healthy adult controls with 6,834,340 autosomal SNVs and INDELs after whole-genome imputation were included. P-values were calculated using logistic regression and adjusting for sex and PC1-4.
Figure 3. Regional plots of loci with genome-wide significance (P<5E-08) or marginal genome-wide significance in the discovery stage.
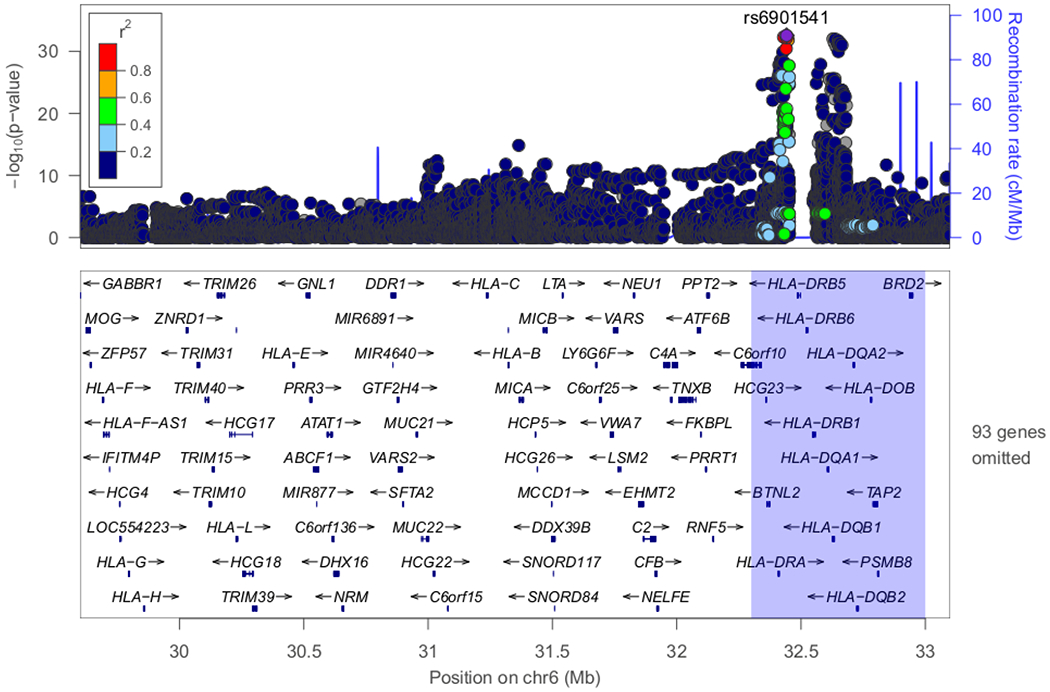
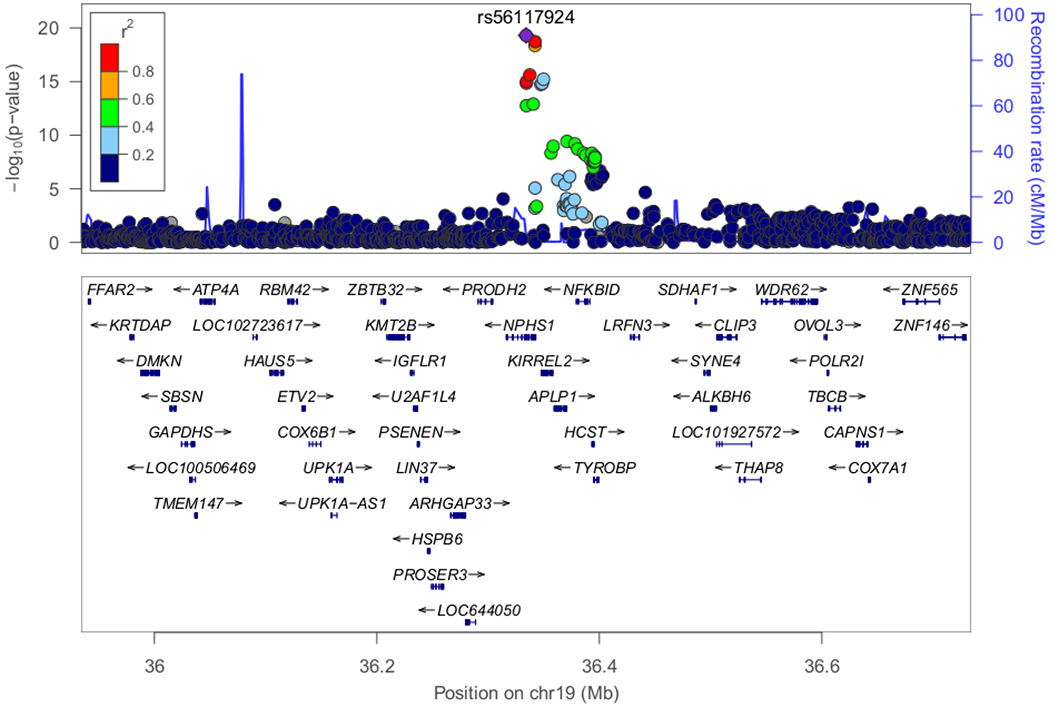
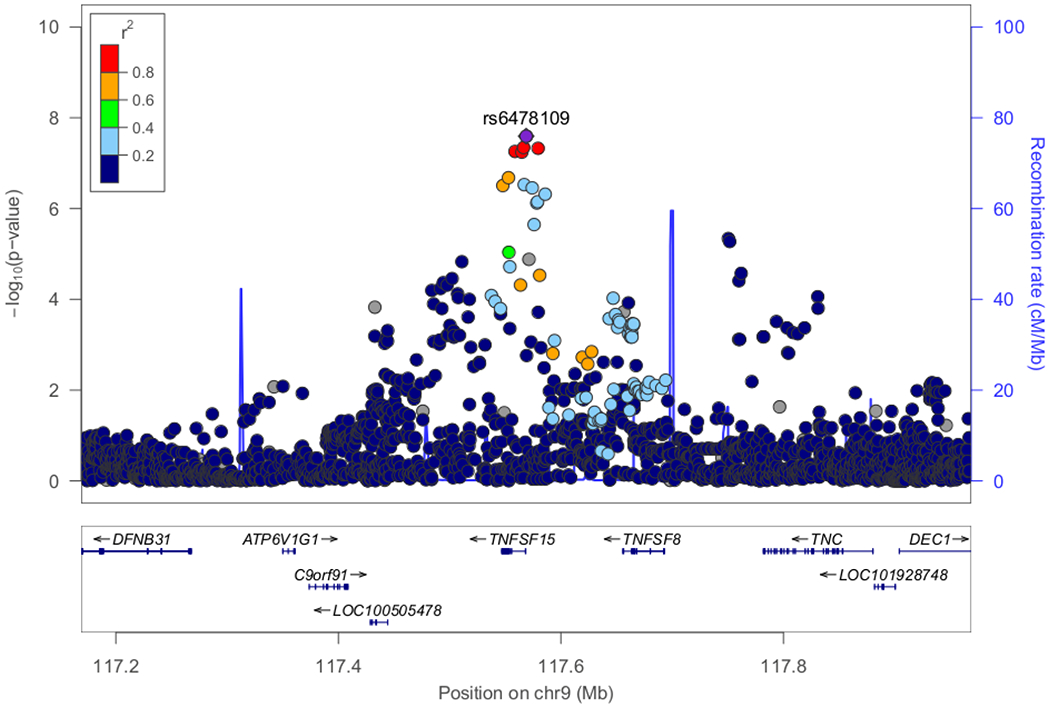
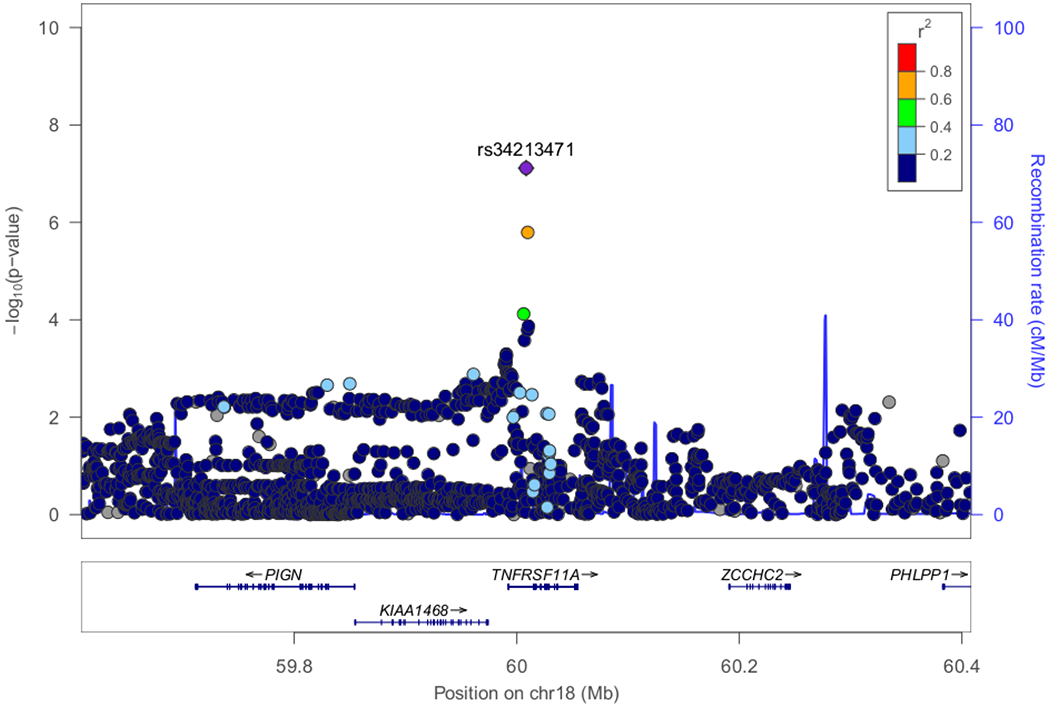
(A) Classical HLA region on chromosome 6 exhibited the most significant association (P-value=2.80E-33). (B) Candidate locus with genome-wide significance on chromosome 19 (P-value=4.94E-20). (C) Candidate locus with genome-wide significance on chromosome 9 (P-value=2.54E-08). (D) Candidate locus with marginal genome-wide significance on chromosome 18 (P=7.68E-08).
Table 1.
SNPs selected from candidate loci in the discovery GWAS and replication of candidate SNPs in multiple populations
| CHR | SNP | BP | A1 | A2 | Related gene(s) | Discovery GWAS and Replication | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Consortium | Population | N of cases | N of controls | A1_Cases | A1_Controls | OR (95% CI) | P-value | |||||||
| 19 | rs56117924 | 36334182 | A | G | NPHS1 | intron variant | GINS-J | Discovery Japanese GWAS | 987 | 3206 | 0.27 | 0.17 | 1.90 (1.66-2.18) | 4.94E-20 |
| KCHRD | Korean | 247 | 378 | 0.25 | 0.17 | 1.70 (1.26-2.27) | 4.19E-04 | |||||||
| MWPNC | South Asian | 181 | 176 | 0.04 | 0.03 | 1.34 (0.57-3.27) | 4.68E-01 (NS) | |||||||
| African | 158 | 4345 | 0.23 | 0.14 | 1.76 (1.32-2.31) | 3.20E-05 | ||||||||
| European | 63 | 7711 | 0.02 | 0.00 | 19.10 (2.07-85.83) | 6.39E-03 (NS) | ||||||||
| Hispanic | 27 | 424 | 0.02 | 0.02 | 1.21 (0.03-8.36) | 5.81E-01 (NS) | ||||||||
| NPHROVIR | African | 56 | 454 | 0.20 | 0.17 | 1.19 (0.66-2.16) | 5.62E-01 (NS) | |||||||
| European | / | / | / | / | / | / | ||||||||
| Maghrebian | 85 | 261 | 0.05 | 0.03 | 1.53 (0.50-4.63) | 4.55E-01 (NS) | ||||||||
| ItSpa | European | / | / | / | / | / | / | |||||||
| 19 | rs2073901 | 36334485 | A | G | NPHS1 | synonymous codon | GINS-J | Discovery Japanese GWAS | 962 | 3171 | 0.15 | 0.08 | 1.93 (1.62-2.29) | 1.77E-13 |
| KCHRD | Korean | 249 | 370 | 0.16 | 0.08 | 2.09 (1.44-3.04) | 1.10E-04 | |||||||
| MWPNC | South Asian | 181 | 8255 | 0.04 | 0.01 | 3.62 (1.87-6.45) | 1.49E-04 | |||||||
| African | 158 | 5202 | 0.01 | 0.01 | 1.36 (0.27-4.16) | 4.91E-01 (NS) | ||||||||
| European | 62 | 33361 | 0.00 | 0.00 | 4.15 (0.26-67.45) | 1 (NS) | ||||||||
| Hispanic | 27 | 5789 | 0.00 | 0.00 | 8.49 (0.50-145.19) | 1 (NS) | ||||||||
| NPHROVIR | African | 56 | 454 | 0.02 | 0.01 | 2.98 (0.45-19.88) | 2.60E-01 (NS) | |||||||
| European | / | / | / | / | / | / | ||||||||
| Maghrebian | / | / | / | / | / | / | ||||||||
| ItSpa | European | / | / | / | / | / | / | |||||||
| 19 | rs412175 | 36342103 | C | T | NPHS1, KIRREL2 | intron variants upstream variant 2KB | GINS-J | Discovery Japanese GWAS | 961 | 3157 | 0.25 | 0.15 | 1.93 (1.67-2.23) | 4.25E-19 |
| KCHRD | Korean | 247 | 374 | 0.25 | 0.15 | 1.91 (1.41-2.60) | 3.55E-05 | |||||||
| MWPNC | South Asian | 181 | 176 | 0.17 | 0.08 | 2.39 (1.45-4.02) | 2.77E-04 | |||||||
| African | 158 | 4330 | 0.50 | 0.44 | 1.26 (1.00-1.59) | 4.38E-02 (NS) | ||||||||
| European | 63 | 7692 | 0.04 | 0.03 | 1.24 (0.39-2.99) | 6.07E-01 (NS) | ||||||||
| Hispanic | 27 | 423 | 0.06 | 0.06 | 0.94 (0.18-3.06) | 1 (NS) | ||||||||
| NPHROVIR | African | 56 | 454 | 0.56 | 0.46 | 1.49 (0.91-2.44) | 1.12E-01 (NS) | |||||||
| European | 132 | 2000 | 0.04 | 0.04 | 0.97 (0.46-2.06) | 9.37E-01 (NS) | ||||||||
| Maghrebian | 85 | 261 | 0.18 | 0.15 | 0.93 (0.49-1.76) | 8.14E-01 (NS) | ||||||||
| ItSpa | European | 112 | 552 | 0.05 | 0.03 | 1.13 (0.49-2.60) | 7.68E-01 (NS) | |||||||
| 19 | rs2285450 | 36342267 | A | G | NPHS1, KIRREL2 | synonymous codon upstream variant 2KB | GINS-J | Discovery Japanese GWAS | 963 | 3163 | 0.25 | 0.15 | 1.93 (1.68-2.23) | 1.91E-19 |
| KCHRD | Korean | 246 | 379 | 0.25 | 0.15 | 1.98 (1.46-2.70) | 1.32E-05 | |||||||
| MWPNC | South Asian | 181 | 7983 | 0.04 | 0.01 | 3.31 (1.80-5.66) | 1.27E-04 | |||||||
| African | 158 | 4750 | 0.18 | 0.13 | 1.45 (1.06-1.96) | 1.18E-02 (NS) | ||||||||
| European | 63 | 30946 | 0.01 | 0.00 | 4.89 (0.12-28.41) | 1.88E-01 (NS) | ||||||||
| Hispanic | 27 | 5377 | 0.00 | 0.01 | 1.25 (0.08-20.38) | 1 (NS) | ||||||||
| NPHROVIR | African | 56 | 454 | 0.18 | 0.16 | 1.20 (0.64-2.25) | 5.79E-01 (NS) | |||||||
| European | / | / | / | / | / | / | ||||||||
| Maghrebian | 85 | 261 | 0.06 | 0.02 | 2.13 (0.72-6.25) | 1.71E-01 (NS) | ||||||||
| ItSpa | European | / | / | / | / | / | / | |||||||
| 19 | rs404299 | 36349752 | A | G | KIRREL2 | missense variant (Ala to Thr) | GINS-J | Discovery Japanese GWAS | 987 | 3206 | 0.16 | 0.09 | 2.00 (1.69-2.36) | 6.01E-16 |
| KCHRD | Korean | 248 | 374 | 0.16 | 0.09 | 1.88 (1.32-2.69) | 4.99E-04 | |||||||
| MWPNC | South Asian | 181 | 8245 | 0.13 | 0.05 | 2.95 (2.11-4.06) | 1.56E-12 | |||||||
| African | 158 | 5203 | 0.43 | 0.38 | 1.20 (0.95-1.52) | 1.10E-01 (NS) | ||||||||
| European | 63 | 33348 | 0.03 | 0.04 | 0.87 (0.23-2.28) | 1 (NS) | ||||||||
| Hispanic | 27 | 5789 | 0.06 | 0.03 | 2.03 (0.40-6.33) | 1.96E-01 (NS) | ||||||||
| NPHROVIR | African | 56 | 454 | 0.50 | 0.39 | 1.53 (0.95-2.47) | 7.85E-02 (NS) | |||||||
| European | 132 | 2000 | 0.04 | 0.04 | 0.98 (0.46-2.09) | 9.60E-01 (NS) | ||||||||
| Maghrebian | 85 | 261 | 0.14 | 0.12 | 0.78 (0.38-1.64) | 5.15E-01 (NS) | ||||||||
| ItSpa | European | 112 | 552 | 0.05 | 0.03 | 1.34 (0.57-3.14) | 5.07E-01 (NS) | |||||||
| 9 | rs4979462 | 117567013 | T | C | TNFSF15 | intron variant | GINS-J | Discovery Japanese GWAS | 987 | 3206 | 0.53 | 0.47 | 1.34 (1.20-1.49) | 2.96E-07 |
| KCHRD | Korean | 248 | 3805 | 0.38 | 0.35 | 1.18 (0.98-1.43) | 8.78E-02 (NS) | |||||||
| MWPNC | South Asian | 180 | 176 | 0.09 | 0.06 | 1.56 (0.84-2.96) | 1.30E-01 (NS) | |||||||
| African | 147 | 4343 | 0.41 | 0.32 | 1.47 (1.15-1.87) | 1.36E-03 | ||||||||
| European | 62 | 7716 | 0.03 | 0.01 | 2.45 (0.65-6.54) | 8.84E-02 (NS) | ||||||||
| Hispanic | 27 | 422 | 0.24 | 0.23 | 1.06 (0.51-2.06) | 8.70E-01 (NS) | ||||||||
| NPHROVIR | African | 56 | 454 | 0.46 | 0.36 | 1.65 (1.03-2.65) | 3.87E-02 (NS) | |||||||
| European | 132 | 2000 | 0.01 | 0.01 | 0.80 (0.11-6.10) | 8.33E-01 (NS) | ||||||||
| Maghrebian | 85 | 261 | 0.06 | 0.07 | 0.56 (0.18-1.72) | 3.08E-01 (NS) | ||||||||
| ItSpa | European | 112 | 552 | 0.02 | 0.02 | 2.20 (0.52-9.32) | 2.84E-01 (NS) | |||||||
| 9 | rs6478109 | 117568766 | A | G | TNFSF15 | upstream variant 2KB | GINS-J | Discovery Japanese GWAS | 987 | 3206 | 0.32 | 0.39 | 0.72 (0.64-0.81) | 2.54E-08 |
| KCHRD | Korean | 248 | 3805 | 0.38 | 0.48 | 0.65 (0.54-0.79) | 1.09E-05 | |||||||
| MWPNC | South Asian | 176 | 176 | 0.21 | 0.24 | 0.84 (0.58-1.21) | 3.24E-01 (NS) | |||||||
| African | 154 | 4349 | 0.07 | 0.11 | 0.60 (0.37-0.94) | 2.24E-02 (NS) | ||||||||
| European | 59 | 7694 | 0.28 | 0.33 | 0.79 (0.51-1.20) | 2.62E-01 (NS) | ||||||||
| Hispanic | 26 | 423 | 0.27 | 0.22 | 1.29 (0.63-2.50) | 4.31E-01 (NS) | ||||||||
| NPHROVIR | African | 56 | 454 | 0.07 | 0.10 | 0.69 (0.29-1.68) | 4.17E-01 (NS) | |||||||
| European | 132 | 2000 | 0.29 | 0.33 | 0.97 (0.71-1.33) | 8.44E-01 (NS) | ||||||||
| Maghrebian | 85 | 261 | 0.16 | 0.18 | 0.58 (0.31-1.10) | 9.43E-02 (NS) | ||||||||
| ItSpa | European | 112 | 552 | 0.31 | 0.32 | 1.26 (0.83-1.93) | 2.69E-01 (NS) | |||||||
| 18 | rs8086340 | 60006978 | C | G | TNFRSF11A | intron variant | GINS-J | Discovery Japanese GWAS | 986 | 3199 | 0.43 | 0.46 | 0.81 (0.73-0.91) | 2.66E-04 |
| KCHRD | Korean | 247 | 648 | 0.37 | 0.41 | 0.87 (0.70-1.08) | 1.92E-01 (NS) | |||||||
| MWPNC | South Asian | 181 | 176 | 0.31 | 0.43 | 0.53 (0.39-0.73) | 4.66E-05 | |||||||
| African | 158 | 4342 | 0.43 | 0.45 | 0.94 (0.74-1.19) | 5.93E-01 (NS) | ||||||||
| European | 63 | 7695 | 0.45 | 0.44 | 1.04 (0.72-1.49) | 8.46E-01 (NS) | ||||||||
| Hispanic | 27 | 422 | 0.37 | 0.36 | 1.03 (0.55-1.88) | 9.22E-01 (NS) | ||||||||
| NPHROVIR | African | 56 | 454 | 0.45 | 0.43 | 1.07 (0.66-1.73) | 7.99E-01 (NS) | |||||||
| European | 132 | 2000 | 0.44 | 0.46 | 0.76 (0.57-1.03) | 8.07E-02 (NS) | ||||||||
| Maghrebian | 85 | 261 | 0.50 | 0.52 | 1.09 (0.69-1.73) | 7.01E-01 (NS) | ||||||||
| ItSpa | European | 112 | 552 | 0.48 | 0.45 | 1.23 (0.83-1.82) | 2.87E-01 (NS) | |||||||
| 18 | rs34213471 | 60008436 | A | C | TNFRSF11A | intron variant | GINS-J | Discovery Japanese GWAS | 987 | 3206 | 0.36 | 0.31 | 1.38 (1.23-1.56) | 7.68E-08 |
| KCHRD | Korean | 247 | 658 | 0.31 | 0.28 | 1.16 (0.92-1.47) | 2.08E-01 (NS) | |||||||
| MWPNC | South Asian | 172 | 176 | 0.23 | 0.16 | 1.54 (1.04-2.28) | 2.49E-02 (NS) | |||||||
| African | 146 | 4357 | 0.01 | 0.01 | 0.74 (0.09-2.80) | 1 (NS) | ||||||||
| European | 60 | 7717 | 0.03 | 0.07 | 0.48 (0.13-1.27) | 1.41E-01 (NS) | ||||||||
| Hispanic | 26 | 424 | 0.06 | 0.10 | 0.52 (0.10-1.67) | 2.75E-01 (NS) | ||||||||
| NPHROVIR | African | 56 | 454 | 0.02 | 0.01 | 2.50 (0.33-19.04) | 3.77E-01 (NS) | |||||||
| European | 132 | 2000 | 0.08 | 0.06 | 1.26 (0.69-2.28) | 4.52E-01 (NS) | ||||||||
| Maghrebian | 85 | 261 | 0.06 | 0.01 | 4.07 (0.99-16.68) | 5.10E-02 (NS) | ||||||||
| ItSpa | European | 112 | 552 | 0.04 | 0.05 | 0.65 (0.281.53) | 3.25E-01 (NS) | |||||||
CHR: Chromosome. A1: Minor alleles in discovery Japanese sample set (effect alleles). A2: Major alleles in discovery Japanese sample set (reference alleles). A1_Cases: Allele frequency of A1 in cases. A1_Controls: Allele frequency of A1 in controls. OR: Odds ratio. 95% CI: 95% confidence interval. GINS-J: The Research Consortium on Genetics of Childhood Idiopathic Nephrotic Syndrome in Japan. KCHRD: Korean Consortium of Hereditary Renal Diseases in Children. MWPNC: Midwest Pediatric Nephrology Consortium. NEPHROVIR: Children Cohort Nephrosis and Virus. ItSpa: Italian and Spanish cohort.
Replication and trans-ethnic meta-analyses of candidate SNPs in non-HLA regions
Replication of nine candidate SNPs was conducted in multiple populations including Korean, South Asian, African, European, Hispanic and Maghrebian (Table 1). P<0.05/9=5.55E-03 was considered as significant threshold. The genome-wide significant signals in NPHS1-KIRREL2 region were replicated in Korean (rs2285450, P=1.32E-05, OR=1.98; rs412175, P=3.55E-05, OR=1.91; rs2073901, P=1.10E-04, OR=2.09; rs56117924, P=4.19E-04, OR=1.70 and rs404299, P=4.99E-04, OR=1.88), South Asian (Midwest Pediatric Nephrology Consortium, MWPNC) (rs404299, P=1.56E-12, OR=2.95; rs2285450, P=1.27E-04, OR=3.31; rs2073901, P=1.49E-04, OR=3.62; rs412175, P=2.77E-04, OR=2.39) and African (MWPNC) datasets (rs56117924, P=3.20E-05, OR=1.76). Candidate SNPs in TNFSF15 region were replicated in the Korean (rs6478109, P=1.09E-05, OR=0.65) and African (MWPNC) datasets (rs4979462, P=1.36E-03, OR=1.47). The association in TNFRSF11A region was replicated in South Asians (MWPNC) (rs8086340, P=4.66E-05, OR=0.53). None of the nine candidate SNPs achieved statistical significance in European, Hispanic (MWPNC), African (NEPHROVIR) or Maghrebian (NEPHROVIR) cohorts after multiple corrections. The limited power for detecting moderate associations in the replication stage should be taken into consideration (Supplementary Table S4).
Trans-ethnic meta-analyses of the 9 SNPs including discovery and replication sample sets confirmed significant associations of the NPHS1-KIRREL2 (rs2285450, Pmeta=6.71E-28, OR=1.88; rs2073901, Pmeta=1.03E-20, OR=2.04) and TNFSF15 regions (rs4979462, Pmeta=5.40E-11, OR=1.33), but not the association of the TNFRSF11A locus (Table 2).
Table 2.
Trans-ethnic meta-analysis of candidate SNPs.
| CHR | SNP | BP | Effect allele | P_value | OR (95% CI) | P_heterogeneity | I2 | Model |
|---|---|---|---|---|---|---|---|---|
| 19 | rs56117924 | 36,334,182 | A | 2.73E-07 | 1.76 (1.42-2.18) | 7.51E-02 | 45.66 | Random-effect |
| 19 | rs2073901 | 36,334,485 | A | 1.03E-20 | 2.04 (1.76-2.38) | 4.09E-01 | 2.04 | Fixed-effect |
| 19 | rs412175 | 36,342,103 | C | 6.21E-05 | 1.52 (1.24-1.87) | 1.58E-02 | 55.80 | Random-effect |
| 19 | rs2285450 | 36,342,267 | A | 6.71E-28 | 1.88 (1.68-2.11) | 1.58E-01 | 33.85 | Fixed-effect |
| 19 | rs404299 | 36,349,752 | A | 8.64E-04 | 1.56 (1.20-2.03) | 1.21E-04 | 72.92 | Random-effect |
| 9 | rs4979462 | 117,567,013 | T | 5.40E-11 | 1.33 (1.22-1.44) | 5.16E-01 | 0.00 | Fixed-effect |
| 9 | rs6478109 | 117,568,766 | A | 7.86E-04 | 0.78 (0.68-0.90) | 7.01E-02 | 43.21 | Random-effect |
| 18 | rs8086340 | 60,006,978 | C | 4.58E-02 | 0.88 (0.77-1.00) | 3.28E-02 | 50.57 | Random-effect |
| 18 | rs34213471 | 60,008,436 | A | 5.61E-02 | 1.22 (0.99-1.50) | 9.63E-02 | 39.23 | Random-effect |
SNP: Single nucleotide polymorphism. BP: Physical position according to Hg19. CHR: Chromosome.
OR: Odds ratio. 95% CI: 95% confidence interval. P heterogeneity: P-value of Cochran Q test for heterogeneity. I2: Percentage of total variation across studies due to heterogeneity. Model: Meta-analysis method used. Fixed-effect: Inverse-variance method based on a fixed-effects model. Random-effect: Inverse-variance method based on a random-effects model
Gene-based analysis in the discovery stage
In a gene-based test, 6,834,340 autosomal variants were mapped to 18,644 protein-coding genes. Seventy-one genes achieved genome-wide significance (P<0.05/18,644=2.68E-06) (Supplementary Figure S8 and Supplementary Table S5). NPHS1 (Pgene=6.29E-18) and KIRREL2 (Pgene=7.79E-14) exhibited the most significant associations with disease outside the HLA region. In gene-set analysis, MHC class II protein complex (P-Bonferroni=1.22E-03, Beta=1.50), MHC class II receptor activity (P-Bonferroni=6.17E-03, Beta=1.61), luminal side of membrane (P-Bonferroni=1.97E-02, Beta=0.75) and innate immune response (P-Bonferroni=1.97E-02, Beta=0.17) were significantly associated with the disease (Supplementary Tables S6 and S7).
Post-GWAS analysis of the NPHS1-KIRREL2 locus
Given that it was the most significant peak after the HLA region, that NPHS1 and KIRREL2 have key roles in podocyte biology and NPHS1 is one of the most common Mendelian nephrotic syndrome genes, we focused our post-GWAS analysis on risk variants at this locus (19q13.12; 36.2-36.6Mb). Given the biologic proximity of DNA to mRNA and availability of existing paired genomic and biopsy-derived kidney transcriptomic data from participants in the Nephrotic Syndrome Study Network (NEPTUNE) study18, 19, we chose to investigate the relationship between the risk alleles at this locus and local mRNA expression.
We first tested the hypothesis that these risk SNPs altered expression of nearby genes as a cis expression quantitative trait locus (cis-eQTL). However, there was no strong evidence that were significant eQTLs for NPHS1 or KIRREL2 in kidney or other tissues (Supplementary Table S8). We also found it to be unlikely that the two synonymous variants (rs2285450, c.294 C>T; rs2073901, c.2223C>T) in NPHS1 affected the secondary structure using RNAsnp Web Server (https://rth.dk/resources/rnasnp/).
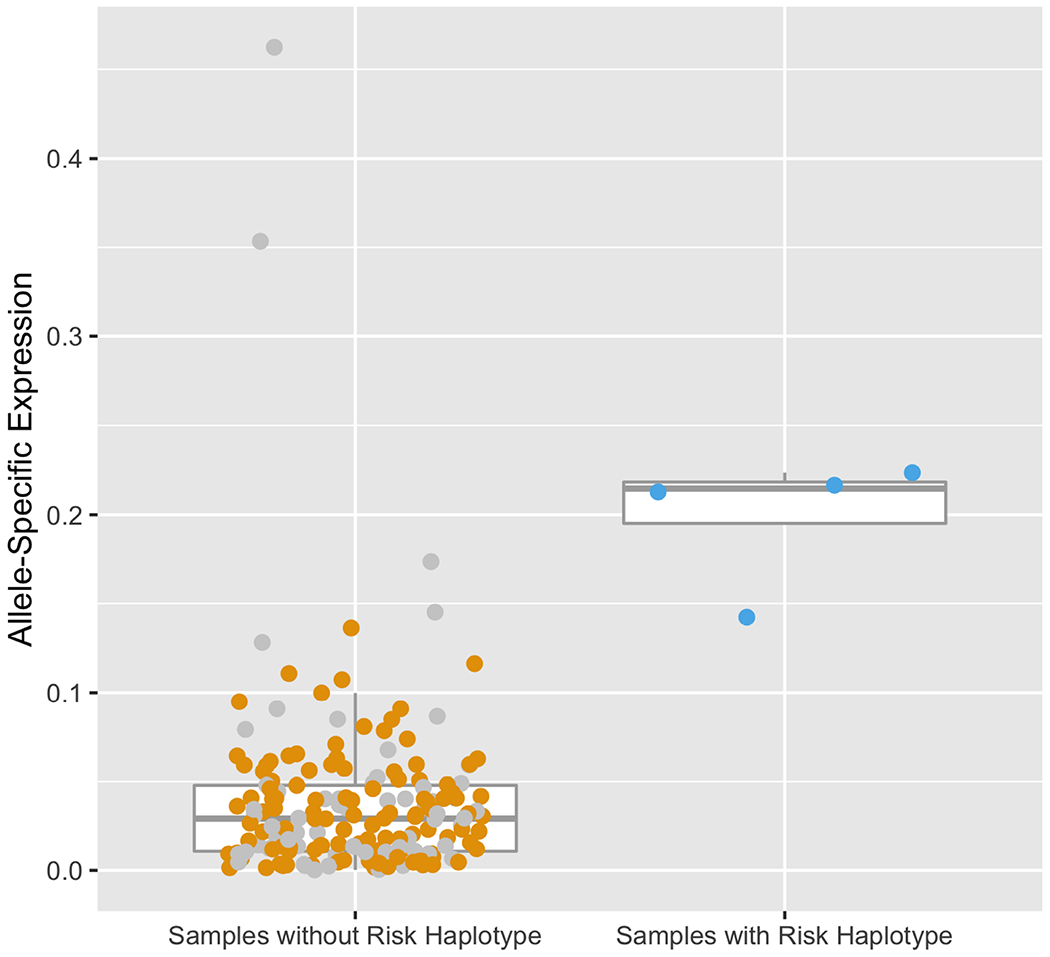
We then turned our attention to studying the impact of the chromosome 19, five-SNP risk haplotype on NPHS1 transcriptional regulation. To do this, we compared NPHS1 expression between four NEPTUNE participants harboring the full five NPHS1 risk haplotype with 183 participants without the full risk haplotype. The four participants with the risk haplotype did not have a significant difference in NPHS1 expression (Wilcox test p=0.39, Figure 4A). With no evidence that this risk haplotype harbors an eQTL for glomerular NPHS1, we hypothesized that the NPHS1 transcript derived from the risk haplotype would be differentially expressed compared to that from the reference haplotype (allele specific expression [ASE]) as ASE has been reported to be associated with susceptibility to diseases including inflammatory bowel disease, autism spectrum disorder and alcohol use disorders20–22. We used phASER to quantify haplotype abundance, then quantified the “magnitude of ASE” as the degree of deviation from the expected 1:1 ratio of expression from each chromosome |0.5 − (haplotype A/ total count)|. In patients with the risk haplotype, haplotype A harbors all five NPHS1 risk variants, in patients without the risk haplotype, haplotype A is randomly selected from one of their two haplotypes. We found significantly lower expression of the NPHS1 transcript from the risk haplotype (Table 3). The magnitudes of ASE in the four risk haplotype samples were 0.14, 0.21, 0.22, and 0.22, whereas the median of the other 183 patients was 0.03 (IQR:0.01-0.05, Wilcox test for difference in ASE p=9.3E-4, Figure 4B).
Figure 4. Glomerular NPHS1 mRNA expression in NEPTUNE cohort.
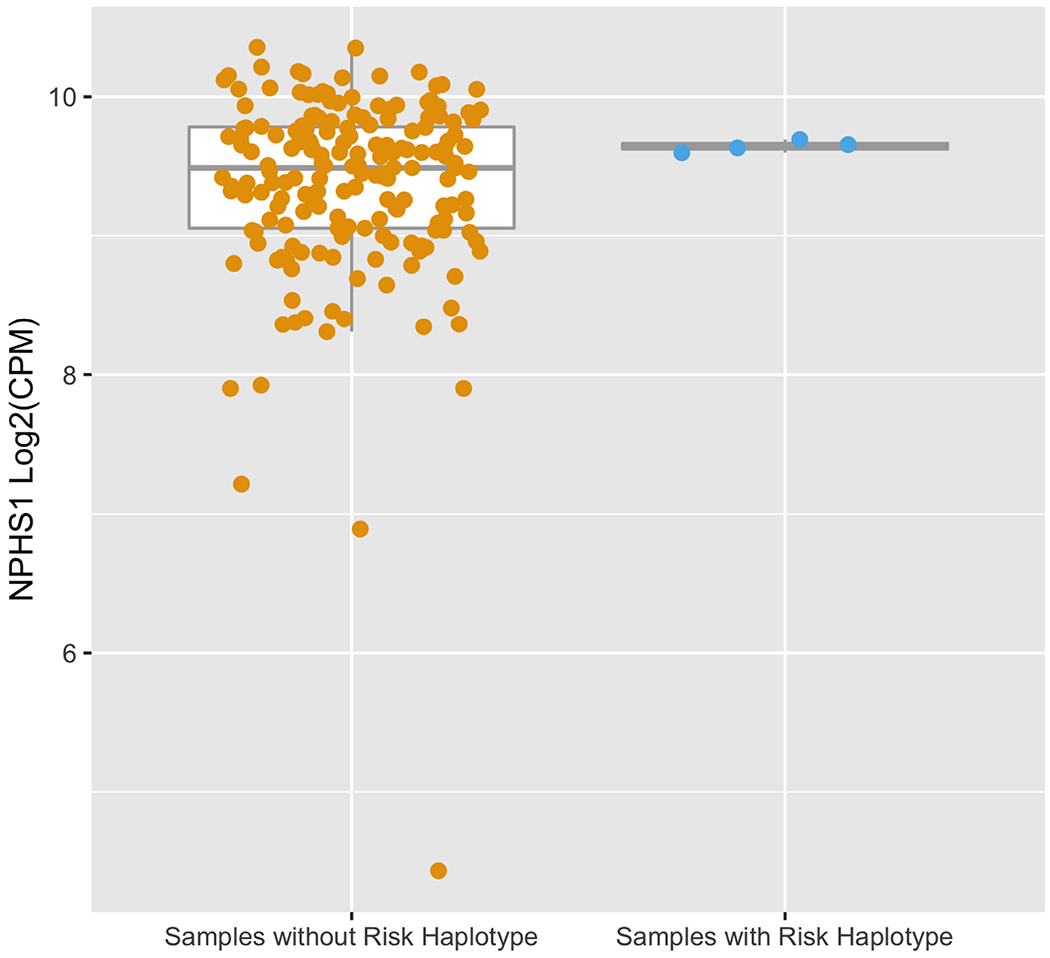
(A) NPHS1 fragments per kilobase million (FPKM) expression comparing samples withand without NPHS1 risk haplotype. Samples with the NPHS1 risk haplotype do not show significantly different expression levels (Wilcox test p=0.39).
(B) Allele-specific expression (ASE) comparing samples with and without NPHS1 risk haplotype. ASE = |0.5 − (haplotype A / total reads)|. In patients with the risk alleles, haplotype A harbors all five NPHS1 risk variants, in patients without the risk haplotype, haplotype A is randomly selected from one of their two haplotypes. Sample with less than two heterozygous SNPs or in the bottom 10% of total counts are indicated in gray. Samples with the NPHS1 risk haplotype show significant allele specific expression with lower expression of the risk haplotype (Wilcox test p=9.3E-4).
Table 3.
NPHS1 allele specific expression for samples harboring risk haplotype
| Patient* | Risk Haplotype Counts | Total Counts | Risk Haplotype/Total Counts | Magnitude of allele specific expression |
|---|---|---|---|---|
| 1 | 3006 | 10466 | 0.287 | 0.213 |
| 2 | 4823 | 13484 | 0.358 | 0.142 |
| 3 | 826 | 2914 | 0.283 | 0.217 |
| 4 | 2135 | 7723 | 0.276 | 0.224 |
Order of samples follows figure 4B from left to right
HLA fine-mapping
HLA association analysis in the present study confirmed our previous findings with more significant p values16. Details of HLA fine mapping are shown in Supplementary Material (Supplementary Table S9–S20, Supplementary Figure S9A–G). HLA-DRB1*08:02-DQB1*03:02 was the most significant susceptibility haplotype (Pc=1.16E-22, OR=3.38), with a more significant and stronger association than HLA-DRB1*08:02 (Pc=2.60E-22, OR=2.66) or HLA-DQB1*03:02 (Pc=2.59E-10, OR=1.71) alone. HLA-DRB1*13:02-DQB1*06:04 was the most significant protective haplotype (Pc=1.63E-16, OR=0.18). One individual in the case group (1/870=0.11%) and 15 healthy controls (15/2,903=0.52%) were heterozygotes for the susceptible haplotype (HLA-DRB1*08:02-DQB1*03:02) and the protective haplotype (HLA-DRB1*13:02-DQB1*06:04), suggesting a dominant effect of the protective haplotype over the susceptible haplotype although the difference was not statistically significant (P=0.14, Supplementary Table S20).
Variance explained by all autosomal variants in the current study
The observed heritability of childhood SSNS explained by genome-wide variants was estimated to be 41.1% (±14.0%). The heritability at the liability scale was 14.8% (±5.0%), assuming a population prevalence of 0.016%. Considering the strong association of the HLA region, the observed heritability was estimated to be 31.6% (±13.6%), and became 11.4% (±4.9%) after transformation when variants on chromosome 6 were excluded. Common variants (MAF > 5%) explained 88-90% of the disease heritability (Supplementary Table S21, Supplementary Figure S10).
Discussion
The present Japanese GWAS with the largest sample size to date and replication in multiple continental populations identified common variants in the NPHS1 and TNFSF15 regions as new susceptibility factors for childhood SSNS. Post-GWAS analysis of the Chromosome 19 locus identified a potential transcriptional mechanism by which the NPHS1 risk haplotype may contribute to disease. Finally, the larger sample size empowered additional fine-mapping of the previously implicated HLA loci16. Altogether, these findings markedly expand and improve our understanding of the genetic background of childhood SSNS. They identify nephrin, the proinflammatory cytokine TNF super-family member 15 (TNFSF15) and their associated molecules as new targets for biologic inquiry to better understand SSNS and for potentially therapeutic development. Finally, the association with common variants in the NPHS1 locus provide another example that Mendelian kidney disease genes can harbor susceptibility variants for a more common multifactorial disease (SSNS).
Rare mutations in NPHS1 cause congenital nephrotic syndrome of the Finnish type, a rare monogenic nephrotic syndrome, which is steroid resistant and has a poor renal prognosis23. Surprisingly, the present study revealed that variants in NPHS1 are associated with susceptibility to SSNS.
While NPHS1 has never been implicated in genome-wide scans for SSNS, a candidate association study between this gene’s variants and SSNS in East Asian patients supports our current findings. In our present study, one of the significant SNPs in the NPHS1 locus was the synonymous variant rs2285450 (c.294 C>T) in exon 3. Previously, Sun et al. reported a higher frequency of the rs2285450 minor allele in Singapore Chinese sporadic NS patients than in controls (20% vs 13%, P=0.025) and showed that the risk allele resulted in a decreased ability to inhibit TRPC6 currents in HEK293-M1 cells via patch clamp, which might account for susceptibility to proteinuria24, 25. This study provides independent support for rs2285450 as a risk factor for SSNS and suggests an alternative mechanism for susceptibility to NS requiring further inquiry.
Using paired human genetic and glomerular transcriptomic data, we did not observe differences in total NPHS1 expression as a function of this risk haplotype. However, RNA-seq data allowed observation of significant ASE resulting in lower NPHS1 expression from the haplotype harboring the risk alleles (Supplementary Figure S11). And a number of the risk variants at this locus were synonymous exonic changes in NPHS1. We therefore focused more on NPHS1 and didn’t go further to discuss the role of KIRREL2 in potential disease mechanisms. Future work should include (1) validating this observation in additional patients with genotyping and kidney expression data and (2) gaining a mechanistic understanding of how this ASE of NPHS1 is contributing to or causing the association with SSNS that we observed. Potential hypotheses for dysfunction include 1) increased burden on the cell to produce the necessary amount of nephrin from the reference chromosome leading to cellular stress and increased susceptibility to injury, 2) potentially dysfunctional nephrin protein produced from the risk haplotype (e.g. via differential post-translational modifications) resulting in an abnormal glomerular filtration barrier.
Genetic polymorphisms at the 9q32 locus are linked with several autoimmune and inflammatory diseases26–29. The most likely disease-causing gene within 9q32 is TNFSF15, which encodes TNFSF15. In the present study, the significant association of rs4979462 was replicated independently and further strengthened by a trans-ethnic meta-analysis. Previously, Hitomi et al. identified rs4979462 as a functional variant by in vitro functional analysis using luciferase assay and electrophoretic mobility shift assay. Super-shift assay clarified that the risk allele (T) of rs4979462 generated a novel NF-1 binding site30. In addition, several reports have shown that the risk allele of rs4979432 affects the expression level of TNFSF15 mRNA27, 30. Further studies are required to confirm the association of TNFSF15 with SSNS in children.
In the present study, the variants in NPHS1 and TNFSF15 loci were identified and replicated mainly in the East Asian (Japanese and Korean) and South Asian populations, in which a higher incidence of disease has been reported. In contrast, the frequencies of risk alleles of candidate SNPs in NPHS1 were rare in people of European ancestry (Table 1). These findings may partially explain the epidemiological difference among populations from the perspective of disease-associated polymorphisms. This is further illustrated by the absence of significant signals in our Japanese dataset in the CALHM6/FAM26F and PARM1 regions, which were detected in homogeneous cohort of European ancestry15, 17.
With the predominant contribution of HLA-DR/DQ genes, gene sets of MHC class II protein complex and MHC class II receptor activity were strongly associated with the disease. Innate immune response was also identified as a significant gene set with a moderate effect. The innate immune system including the activation of professional cells (antigen presenting cells and B cells/Toll-like receptors), as well as the adaptive immune system, in which HLA molecules play a crucial role, are involved in the disease process and response to treatment31, 32.
Around 90% of the heritability of childhood SSNS was contributed by common variants, although variants with relatively rare allele frequencies (MAF: 0.5%–5%) were also taken into consideration. However, a large proportion of the disease heritability remains unaccounted for. Other disease-associated variants in non-HLA regions might be identified by future genome-wide studies with larger sample sizes, especially for patients of European and other non-East Asian ancestries, as disease associated polymorphisms in non-HLA regions are still largely unknown.
In the present study, the sample sizes in replication cohorts with different ancestries have limited the power for the replication of signals with smaller effect sizes. Population stratification might exist in the replication stage due to lack of adjustment. Moreover, batch effects might have occurred between cases and controls when allele frequencies from public databases were utilized as population-matched controls.
In summary, the discoveries here provide new SSNS loci. The NPHS1 locus in particular, provides a provocative new concept for the disease’s pathogenesis linking and reinforcing an emerging paradigm in nephrology; that in certain genes harboring Mendelian variants, more common alleles can increase the susceptibility to multifactorial, polygenic diseases, as previously demonstrated for UMOD, COL4A3 and NPHS223, 33–39. Finally, it again emphasizes the importance of performing genomic research in diverse populations. By doing so here, we made discoveries of specific SNPs likely more impactful to East and South Asian population while also identifying genes and loci that may important in all populations, but to which we would be statistically blind if using a European discovery cohort.
Methods
Supplementary information is available at Kidney International’s website.
Samples
In the discovery stage, 1,018 Japanese patients diagnosed as childhood SSNS (onset age <18 years) were recruited. Patients with a history of steroid resistance during follow-up were excluded. Overall, 3,331 Japanese healthy adults were recruited as controls. All participants provided written informed consent.
Genotyping and whole-genome imputation in the discovery stage
In the discovery stage, 1,018 cases and 3,331 controls were genotyped using the Affymetrix ‘Japonica Array’40Nineteen samples were excluded by low call rate (<97%) during genotype calling process. Nine controls with ambiguous sex were excluded. Then whole-genome imputation was performed with IMPUTE441(ver. 2.3.1) using a phased reference panel of 2,036 healthy Japanese individuals (2KJPN panel). After whole-genome imputation, there were 22,049,786 autosomal SNVs and short INDELs with info score >0.5. QC was conducted using the following threshold: individual missing rate <3%, SNV/INDEL call rate ≥97%, MAF ≥0.5% and HWE P-value ≥0.0001 in healthy controls. Identical-by-Descent test was performed using a threshold of Pi-hat >0.1875. PCA was performed using GCTA (Version 1.26.0)42 for cases, controls, and HapMap Phase III data (113 CEU, 113 YRI, 84 CHB, and 86 JPT), samples identified as outliers were excluded (Supplementary Figure S1A–E).
Genome-wide association analyses in the discovery stage
Genome-wide association analyses and SNV-based conditional analyses were conducted using logistic regression, adjusting for sex and the first four principal components (PC1 to PC4) by PLINK 1.9. R package “qqman” was utilized to generate Manhattan and QQ plots. Regional plots were generated using Locuszoom (1000 Genomes Nov 2014 ASN was used as LD reference)43
Gene-based test and gene-set analysis
Gene-based test and gene-set analysis were performed by MAGMA v1.644 (implemented through FUMA45). The Genotype-Tissue Expression (GTEx) database46 and the NephVS eQTL Browser (NephQTL)19ere utilized to find eQTLs affecting the expression of various genes in various tissues and kidney-specific tissues.
Replication of candidate SNPs
Replication was carried out in multiple populations. Participants in the Korean dataset were recruited from the South Korea. The MWPNC cohort included 181 patients of South Asian ancestry recruited from the USA and Sri Lanka, 158 patients of African ancestry recruited from the USA and Nigeria, and 63 European and 27 Hispanic patients recruited from the USA. The NEPHROVIR cohort included 132 European, 56 African and 85 Maghrebian children with SSNS, which were recruited in the Paris area, 2000 European controls from 3 Cités, and 454 controls from the 1000G African cohort and 261 Moroccan controls were used as population-matched controls, respectively. The ItSpa cohort comprised 112 European patients from Italy and Spain and 552 controls from the 1000G European cohort. Detailed information on the data sets of each population is shown in the Supplementary Methods and Table 1.
In the replication stage, logistic regression was performed in the Korean dataset using individual genotype data. P-values were calculated by Pearson’s chi-square test or Fisher’s exact test in MWPNC cohort and public databases. For cell=0 in the Chi-square test, 0.5 was added to each of the four cells to calculate the odds ratio (ORs), standard error (SE) and 95% confidence interval (95% CI). In the NEPHROVIR and ItSpa datasets, association analyses were carried out under additive model.
Trans-ethnic meta-analysis
Trans-ethnic meta-analysis was conducted using the inverse-variance method based on fixed- or random-effects models by “META” (https://mathgen.stats.ox.ac.uk/genetics_software/meta/meta.html). Heterogeneity was considered when the Cochran Q test had a P-value <0.10. Pmeta<5E-08 was considered the genome-wide significant threshold for the meta-analysis.
Allele-specific expression (ASE) analysis in the NEPTUNE cohort
Total RNA from glomerular biopsies and 30X whole genome sequencing was performed on 269 and 625 samples from the Nephrotic Syndrome Study Network Consortium (NEPTUNE18, see supplemental data for more detail). Variants on chromosome 19 where filtered, removing MAF < 0.0001, indels, QUAL < 20, and missing > 10%. Variants were phased with Eagle v2.4.147 on the Michigan Imputation Server48 using the 1,000 Genomes Phase 3 reference panel49. Samples harboring all five chromosome 19 risk alleles (rs56117924, rs2073901, rs412175, rs2285450 and rs404299) were identified. Gene quantification (Log2CPM) was calculated with the edgeR TMM-normalization method50. The bam files were subset to NPHS1 and the surrounding 1KB intergenic region (chr19: 36,315,274-36,343,895). The NPHS1 bam files and phased WGS were input into phASER51 to perform haplotype phasing. Due to a high recombination hotspot within NPHS1, we found inconsistent phasing of rs2071347 (NPHS1, exon 26) when comparing different reference panels and methods, and thus removed it from downstream analyses. We used phASER to calculate haplotype-specific expression by summing RNA-seq counts across all heterozygous SNPs. Samples with less than 20 total reads across NPHS1 were removed. For the 187 remaining samples, we calculated ASE as |0.5 − (haplotype A / total reads)|. In patients with the risk haplotype, haplotype A harbors all five NPHS1 risk variants, in patients without the risk haplotype, haplotype A is randomly selected from one of their two haplotypes. We then compared ASE and gene expression for samples with and without the risk haplotype with a Wilcoxon rank-sum test in R. The total reads and number of supporting heterozygous SNPs varied across samples. Samples with lower power to detect ASE, less than two heterozygous SNPs or in the bottom 10% of total reads, were nonetheless included for completeness and are indicated as gray points in corresponding figures.
HLA fine-mapping
Methods for HLA imputation and HLA genotyping are shown in the Supplementary Methods.
Power calculation
The power of discovery GWAS (987 cases and 3,206 controls) was calculated using the R package “CATS”52. Assuming a disease prevalence of 0.016%, study power was calculated separately under the additive model for variants with an allele frequency of 0.5%, 5% and 50% with a significance threshold α=5E-08 (Supplementary Figure S2). The Bioinformatics Institute’s Online Sample Size Estimator (OSSE, http://osse.bii.a-star.edu.sg/index.php) was used to estimate the power of replication for each candidate SNP.
Heritability estimates
The disease heritabilities explained by genome-wide variants were estimated using GCTA41, 53 (GCTA-LDMS method), assuming a disease prevalence of 0.016% in the Japanese population. All variants that passed QC procedure after whole-genome imputation were included in the analysis. Variants were grouped as common variants (MAF>5%) or uncommon variants (0.5%<MAF<=5%) when making genetic relationship matrixes (GRMs) during the calculation. Details are shown in “Supplementary Methods”.
Supplementary Material
Acknowledgements
We thank all patients who participated in this study and their families, and Ms. Yoshimi Nozu and Ms. Ming Juan Ye for their technical assistance. We also thank Minoru Nakamura, Hitoshi Okazaki and Mika Matsuhashi for their support in functional studies. This work was supported by: the Japan Agency for Medical Research and Development (AMED) under grant number JP17km0405108h0005 to Kazumoto Iijima, Kenji Ishikura, Kandai Nozu, and Katsushi Tokunaga, and JP17km0405205h0002 and 18km0405205h0003 to Katsushi Tokunaga and Masao Nagasaki; and by the Japan Society for the Promotion of Science (JSPS) under Grant-in-Aid for Scientific Research fostering Joint International Research (B) 18KK0244 to Kazumoto Iijima, Yuki Hitomi, Tomoko Horinouchi, China Nagano, and Kandai Nozu. Part of this study was funded by a European Research Council grant ERC-2012-ADG_20120314 (grant agreement 322947) and Agence Nationale pour la Recherche “Genetransnephrose” grant ANR-16-CE17-004-01 to Pierre Ronco. Rasheed Gbadegesin is supported by National Institutes of Health/National Institutes of Diabetes and Digestive and Kidney Diseases (NIH/NIDDK) grants 5R01DK098135 and 5R01DK094987, Doris Duke Charitable Foundation Clinical Scientist Development Award 2009033, and Duke Health Scholars award. Matthew G. Sampson is supported by a National Institute of Health grant (R01-DK108805). The Nephrotic Syndrome Study Network Consortium (NEPTUNE; U54-DK-083912) is a part of the National Center for Advancing Translational Sciences (NCATS). The Rare Disease Clinical Research Network (RDCRN) was supported through a collaboration between the Office of Rare Diseases Research (ORDR), NCATS, and the National Institute of Diabetes, Digestive, and Kidney Diseases. The RDCRN is an initiative of the ORDR of NCATS. Additional funding and/or programmatic support for this project was provided by the University of Michigan, NephCure Kidney International, and the Halpin Foundation. The NEPHROVIR cohort was supported by two grants to Georges Deschénes from the Programme Hospitalier de Recherche Clinique: grants PHRC 2007-AOM07018 and PHRC 2011-AOM11002. The NEPHROVIR network is coordinated by the Pediatric Nephrology Unit of Robert Debré Hospital, the “Unité de Recherche Clinique de l’Est Parisien,” and the “Délégation de la Récherche Clinique de la Région Ile-de-France.” Marina Vivarelli was supported by the Associazione per la Cura del bambino Nefropatico ONLUS (Organizzazione Non Lucrativa di Utilità Sociale). We thank J. Ludovic Croxford, PhD, from Edanz Group (www.edanzediting.com/ac) for editing a draft of this manuscript.
Summarized data of variants in the discovery GWAS are available through the Japan National Bioscience Database Center database (Research ID: hum0126.v2.imp-gwas.v1, https://humandbs.biosciencedbc.jp/en/hum0126-v2).
Sources of support that require acknowledgment
This work was supported by: the Japan Agency for Medical Research and Development (AMED) under grant number JP17km0405108h0005 to Kazumoto Iijima, Kenji Ishikura, Kandai Nozu, and Katsushi Tokunaga, and JP17km0405205h0002 and 18km0405205h0003 to Katsushi Tokunaga and Masao Nagasaki; and by the Japan Society for the Promotion of Science (JSPS) under Grant-in-Aid for Scientific Research fostering Joint International Research (B) 18KK0244 to Kazumoto Iijima, Yuki Hitomi, Tomoko Horinouchi, China Nagano, and Kandai Nozu. Part of this study was funded by a European Research Council grant ERC-2012-ADG_20120314 (grant agreement 322947) and Agence Nationale pour la Recherche “Genetransnephrose” grant ANR-16-CE17-004-01 to Pierre Ronco. Rasheed Gbadegesin is supported by National Institutes of Health/National Institutes of Diabetes and Digestive and Kidney Diseases (NIH/NIDDK) grants 5R01DK098135 and 5R01DK094987, Doris Duke Charitable Foundation Clinical Scientist Development Award 2009033, and Duke Health Scholars award. Matthew G. Sampson is supported by a National Institute of Health grant (R01-DK108805). The Nephrotic Syndrome Study Network Consortium (NEPTUNE; U54-DK-083912) is a part of the National Center for Advancing Translational Sciences (NCATS). The Rare Disease Clinical Research Network (RDCRN) was supported through a collaboration between the Office of Rare Diseases Research (ORDR), NCATS, and the National Institute of Diabetes, Digestive, and Kidney Diseases. The RDCRN is an initiative of the ORDR of NCATS. Additional funding and/or programmatic support for this project was provided by the University of Michigan, NephCure Kidney International, and the Halpin Foundation. The NEPHROVIR cohort was supported by two grants to Georges Deschénes from the Programme Hospitalier de Recherche Clinique: grants PHRC 2007-AOM07018 and PHRC 2011-AOMU002. The NEPHROVIR network is coordinated by the Pediatric Nephrology Unit of Robert Debré Hospital, the “Unité de Recherche Clinique de l’Est Parisien,” and the “Délégation de la Recherche Clinique de la Région Ile-de-France.” Marina Vivarelli was supported by the Associazione per la Cura del bambino Nefropatico ONLUS (Organizzazione Non Lucrativa di Utilità Sociale).
Disclosure
MGS has received consulting fees from Maze Therapeutics. AS has received travel support from Biofem Pharmaceuticals. GD has received consulting fees from Chiesi and Biocodex, a lecture fee from Alnhylam and travel support from Sanofi. MV has received consulting fees from Achillion Pharmaceuticals and Gentium-Jass, a lecture fee from Sanofi and a grant from Alexion Pharmaceuticals. KIi has received consulting fees from Zenyaku Kogyo. All the other authors declared no competing interests.
Footnotes
Publisher's Disclaimer: This is a PDF file of an article that has undergone enhancements after acceptance, such as the addition of a cover page and metadata, and formatting for readability, but it is not yet the definitive version of record. This version will undergo additional copyediting, typesetting and review before it is published in its final form, but we are providing this version to give early visibility of the article. Please note that, during the production process, errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Noone DG, Iijima K, Parekh R. Idiopathic nephrotic syndrome in children. Lancet. 2018;39:61–74. doi: 10.1016/S0140-6736(18)30536-1. [DOI] [PubMed] [Google Scholar]
- 2.Chanchlani R, Parekh RS. Ethnic Differences in Childhood Nephrotic Syndrome. Front Pediatr. 2016;4:39. doi: 10.3389/fped.2016.00039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kikunaga K, Ishikura K, Terano C, et al. High incidence of idiopathic nephrotic syndrome in East Asian children: a nationwide survey in Japan (JP-SHINE study). Clin Exp Nephrol. 2017;21:651–657. doi: 10.1007/s10157-016-1319-z. [DOI] [PubMed] [Google Scholar]
- 4.White RH The familial nephrotic syndrome. I. A European survey. Clin Nephrol. 1973;1:215–219. [PubMed] [Google Scholar]
- 5.Lane BM, Cason R, Esezobor CI, et al. Genetics of Childhood Steroid Sensitive Nephrotic Syndrome: An Update. Front Pediatr 2019;7:8. doi: 10.3389/fped.2019.00008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sadowski CE, Lovric S, Ashraf S, et al. A single-gene cause in 29.5% of cases of steroid-resistant nephrotic syndrome. J Am Soc Nephrol. 2015;26:1279–1289. doi: 10.1681/ASN.2014050489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ashraf S, Kudo H, Rao J, et al. Mutations in six nephrosis genes delineate a pathogenic pathway amenable to treatment. Nat Commun. 2018;9:1960. doi: 10.1038/s41467-018-04193-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gee HY, Ashraf S, Wan X, et al. Mutations in EMP2 cause childhood-onset nephrotic syndrome. Am J Hum Genet. 2014;94:884–890. doi: 10.1016/j.ajhg.2014.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dorval G, Gribouval O, Martinez-Barquero V, et al. Clinical and genetic heterogeneity in familial steroid-sensitive nephrotic syndrome. Pediatr Nephrol. 2018;33:473–483. doi: 10.1007/s00467-017-3819-9. [DOI] [PubMed] [Google Scholar]
- 10.MacDonald NE, Wolfish N, McLaine P, et al. Role of respiratory viruses in exacerbations of primary nephrotic syndrome. J Pediatr. 1986;108:378–382. [DOI] [PubMed] [Google Scholar]
- 11.Takahashi S, Wada N, Murakami H, et al. Triggers of relapse in steroid-dependent and frequently relapsing nephrotic syndrome. Pediatr Nephrol. 2007;22:232–236. doi: 10.1007/s00467-006-0316-y [DOI] [PubMed] [Google Scholar]
- 12.Gulati A, Sinha A, Sreenivas V, et al. Daily corticosteroids reduce infection-associated relapses in frequently relapsing nephrotic syndrome: a randomized controlled trial. Clin J Am Soc Nephrol. 2011;6:63–69. doi: 10.2215/CJN.01850310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tareyeva IE, Nikolaev AJ, Janushkevitch TN. Nephrotic syndrome induced by insect sting. Lancet. 1982;2:825. [DOI] [PubMed] [Google Scholar]
- 14.Gbadegesin RA, Adeyemo A, Webb NJ, et al. HLA-DQA1 and PLCG2 Are Candidate Risk Loci for Childhood-Onset Steroid-Sensitive Nephrotic Syndrome. J Am Soc Nephrol. 2015;26:1701–1710. doi: 10.1681/ASN.2014030247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Debiec H, Dossier C, Letouzé E, et al. Transethnic, Genome-Wide Analysis Reveals Immune-Related Risk Alleles and Phenotypic Correlates in Pediatric Steroid-Sensitive Nephrotic Syndrome. J Am Soc Nephrol. 2018;29:2000–2013. doi: 10.1681/ASN.2017111185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jia X, Horinouchi T, Hitomi Y, et al. Strong Association of the HLA-DR/DQ Locus with Childhood Steroid-Sensitive Nephrotic Syndrome in the Japanese Population. J Am Soc Nephrol. 2018;29:2189–2199. doi: 10.1681/ASN.2017080859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dufek S, Cheshire C, Levine AP, et al. Genetic Identification of Two Novel Loci Associated with Steroid-Sensitive Nephrotic Syndrome. J Am Soc Nephrol 2019;30:1375–1384. doi: 10.1681/ASN.2018101054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gadegbeku CA, Gipson DS, Holzman LB, et al. Design of the Nephrotic Syndrome Study Network (NEPTUNE) to evaluate primary glomerular nephropathy by a multidisciplinary approach. Kidney Int. 2013;83:749–756. doi: 10.1038/ki.2012.428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gillies CE, Putler R, Menon R, et al. An eQTL Landscape of Kidney Tissue in Human Nephrotic Syndrome. Am J Hum Genet. 2018;103:232–244. doi: 10.1016/j.ajhg.2018.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Richard AC, Peters JE, Savinykh N, et al. Reduced monocyte and macrophage TNFSF15/TL1A expression is associated with susceptibility to inflammatory bowel disease. PLoS Genet. 2018;14(9):e1007458. doi: 10.1371/journal.pgen.1007458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee C, Kang EY, Gandal MJ, et al. Profiling allele-specific gene expression in brains from individuals with autism spectrum disorder reveals preferential minor allele usage. Nat Neurosci. 2019;22(9):1521–1532. doi: 10.1038/s41593-019-0461-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rao X, Thapa KS, Chen AB, et al. Allele-specific expression and high-throughput reporter assay reveal functional genetic variants associated with alcohol use disorders. Mol Psychiatry. 2019. September 2. doi: 10.1038/s41380-019-0508-z. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.KestilÄ M, Lenkkeri U, MÄnnikkö M, et al. Positionally cloned gene for a novel glomerular protein--nephrin--is mutated in congenital nephrotic syndrome. Mol Cell. 1998;1:575–582. [DOI] [PubMed] [Google Scholar]
- 24.Winn MP, Conlon PJ, Lynn KL, et al. A mutation in the TRPC6 cation channel causes familial focal segmental glomerulosclerosis. Science. 2005;308(5729):1801–1804. doi: 10.1126/science.1106215. [DOI] [PubMed] [Google Scholar]
- 25.Sun ZJ, Ng KH, Liao P, et al. Genetic Interactions Between TRPC6 and NPHS1 Variants Affect Posttransplant Risk of Recurrent Focal Segmental Glomerulosclerosis. Am J Transplant. 2015;15:3229–3238. doi: 10.1111/ajt.13378. [DOI] [PubMed] [Google Scholar]
- 26.Wang XM, Tu JC. TNFSF15 is likely a susceptibility gene for systemic lupus erythematosus. Gene. 2018;670:106–113. doi: 10.1016/j.gene.2018.05.098. [DOI] [PubMed] [Google Scholar]
- 27.Sun Y, Irwanto A, Toyo-Oka L, et al. Fine-mapping analysis revealed complex pleiotropic effect and tissue-specific regulatory mechanism of TNFSF15 in primary biliary cholangitis, Crohn’s disease and leprosy. Sci Rep. 2016;6:31429. doi: 10.1038/srep31429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Richard AC, Peters JE, Savinykh N, et al. Reduced monocyte and macrophage TNFSF15/TL1A expression is associated with susceptibility to inflammatory bowel disease. PLoS Genet. 2018;14:e1007458. doi: 10.1371/journal.pgen.1007458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang J, Wang X, Fahmi H, et al. Role of TL1A in the pathogenesis of rheumatoid arthritis. J Immunol. 2009;183(8):5350–5357. doi: 10.4049/jimmunol.0802645. [DOI] [PubMed] [Google Scholar]
- 30.Hitomi Y, Kawashima M, Aiba Y, et al. Human primary biliary cirrhosis-susceptible allele of rs4979462 enhances TNFSF15 expression by binding NF-1. Hum Genet. 2015;134:737–747. doi: 10.1007/s00439-015-1556-3. [DOI] [PubMed] [Google Scholar]
- 31.Colucci M, Corpetti G, Emma F, et al. Immunology of idiopathic nephrotic syndrome. Pediatr Nephrol. 2018;33:573–584. doi: 10.1007/s00467-017-3677-5. [DOI] [PubMed] [Google Scholar]
- 32.Bertelli R, Bonanni A, Di Donato A, et al. Regulatory T cells and minimal change nephropathy: in the midst of a complex network. Clin Exp Immunol. 2016;183:166–174. doi: 10.1111/cei.12675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Eckardt KU, Alper SL, Antignac C, et al. Kidney Disease: Improving Global Outcomes. Autosomal dominant tubulointerstitial kidney disease: diagnosis, classification, and management--A KDIGO consensus report. Kidney Int. 2015;88:676–683. doi: 10.1038/ki.2015.28. [DOI] [PubMed] [Google Scholar]
- 34.Rampoldi L. et al. The rediscovery of uromodulin (Tamm-Horsfall protein): from tubulointerstitial nephropathy to chronic kidney disease. Kidney Int. 2011;80:338–347. doi: 10.1038/ki.2011.134. [DOI] [PubMed] [Google Scholar]
- 35.Scolari F, Izzi C, Ghiggeri GM. Uromodulin: from monogenic to multifactorial diseases. Nephrol Dial Transplant. 2015;30:1250–1256. doi: 10.1093/ndt/gfu300. [DOI] [PubMed] [Google Scholar]
- 36.Devuyst O, Pattaro C. The UMOD Locus: Insights into the Pathogenesis and Prognosis of Kidney Disease. J Am Soc Nephrol. 2018;29:713–726. doi: 10.1681/ASN.2017070716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang Y, Zhang J, Zhao Y, et al. COL4A3 Gene Variants and Diabetic Kidney Disease in MODY. Clin J Am Soc Nephrol. 2018;13:1162–1171. doi: 10.2215/CJN.09100817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tsukaguchi H, Sudhakar A, Le TC, et al. NPHS2 mutations in late-onset focal segmental glomerulosclerosis: R229Q is a common disease-associated allele. J Clin Invest. 2002;110:1659–1666. doi: 10.1172/JCI16242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Machuca E, Hummel A, Nevo F, et al. Clinical and epidemiological assessment of steroid-resistant nephrotic syndrome associated with the NPHS2 R229Q variant. Kidney Int. 2009;75:727–35. doi: 10.1038/ki.2008.650. [DOI] [PubMed] [Google Scholar]
- 40.Kawai Y, Mimori T, Kojima K, et al. Japonica array: improved genotype imputation by designing a population-specific SNP array with 1070 Japanese individuals. J Hum Genet. 2015;60:581–587. doi: 10.1038/jhg.2015.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bycroft C, Freeman C, Petkova D, et al. Genome-wide genetic data on ~500,000 UK Biobank participants. Preprint at https://www.biorxiv.org/content/early/2017/07/20/166298. doi: 10.1101/166298 [DOI] [Google Scholar]
- 42.Yang J, Lee SH, Goddard ME, et al. GCTA: a tool for genome-wide complex trait analysis. Am J Hum Genet. 2011;88:76–82. doi: 10.1016/j.ajhg.2010.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pruim RJ, Welch RP, Sanna S, et al. LocusZoom: regional visualization of genome-wide association scan results. Bioinformatics. 2010;26:2336–7. doi: 10.1093/bioinformatics/btq419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.de Leeuw CA, Mooij JM, Heskes T, et al. MAGMA: generalized gene-set analysis of GWAS data. PLoS Comput Biol. 2015;11:e1004219. doi: 10.1371/journal.pcbi.1004219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Watanabe K, Taskesen E, van Bochoven A, et al. Functional mapping and annotation of genetic associations with FUMA. Nat Commun. 2017;8:1826. doi: 10.1038/s41467-017-01261-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.GTEx Consortium. The Genotype-Tissue Expression (GTEx) project. Nat Genet. 2013;45:580–585. doi: 10.1038/ng.2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Loh PR, Danecek P, Palamara PF, et al. Reference-based phasing using the Haplotype Reference Consortium panel. Nat Genet. 2016;48:1443–1448. doi: 10.1038/ng.3679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Das S, Forer L, Schönherr S, et al. Next-generation genotype imputation service and methods. Nat Genet. 2016;48:1284–1287. doi: 10.1038/ng.3656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.1000 Genomes Project Consortium, Auton A, Brooks LD, Durbin RM, et al. A global reference for human genetic variation. Nature. 2015;52:68–74. doi: 10.1038/nature15393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Robinson MD, Oshlack A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010;11:R25. doi: 10.1186/gb-2010-11-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Castel SE, Mohammadi P, Chung WK, et al. Rare variant phasing and haplotypic expression from RNA sequencing with phASER. Nat Commun. 2016;7:12817. doi: 10.1038/ncomms12817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Skol AD, Scott LJ, Abecasis GR, et al. Joint analysis is more efficient than replication-based analysis for two-stage genome-wide association studies. Nat Genet. 2006;38:209–213. doi: 10.1038/ng1706. [DOI] [PubMed] [Google Scholar]
- 53.Yang J, Bakshi A, Zhu Z, et al. Genetic variance estimation with imputed variants finds negligible missing heritability for human height and body mass index. Nat Genet. 2015;47:1114–20. doi: 10.1038/ng.3390. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.