

Abstract
Pathogenic missense variants in the leucine-rich repeat kinase 2 (LRRK2) gene have been identified through linkage analysis in familial Parkinson disease (PD). Subsequently, other missense variants with lower effect sizes on PD risk have emerged, as well as non-coding polymorphisms (e.g. rs76904798) enriched in PD cases in genome-wide association studies. Here we leverage recent whole-genome sequences from the Accelerating Medicines Partnership-Parkinson’s Disease (AMP-PD) and the Genome Aggregation (gnomAD) databases to characterize novel missense variants in LRRK2 and explore their relationships with known pathogenic and PD-linked missense variants. Using a computational prediction tool that successfully classifies known pathogenic LRRK2 missense variants, we describe an online web-based resource that catalogs characteristics of over 1200 LRRK2 missense variants of unknown significance. Novel high-pathogenicity scoring variants, some identified exclusively in PD cases, tightly cluster within the ROC-COR-Kinase domains. Structure–function predictions support that some of these variants exert gain-of-function effects with respect to LRRK2 kinase activity. In AMP-PD participants, all p.R1441G carriers (N = 89) are also carriers of the more common PD-linked variant p.M1646T. In addition, nearly all carriers of the PD-linked p.N2081D missense variant are also carriers of the LRRK2 PD-risk variant rs76904798. These results provide a compendium of LRRK2 missense variants and how they associate with one another. While the pathogenic p.G2019S variant is by far the most frequent high-pathogenicity scoring variant, our results suggest that ultra-rare missense variants may have an important cumulative impact in increasing the number of individuals with LRRK2-linked PD.
Introduction
Missense variants in LRRK2 are one of the leading genetic causes of neurodegeneration and Parkinson disease (PD) (1,2). Since the discovery of the first missense LRRK2 variants linked to PD in late 2004, seven causal variants are now known (3–7). LRRK2 pathogenic variants have been identified through linkage-based family studies. Genetic linkage studies depend on multiple affected carriers in large well-documented families, with members living long enough where PD manifestation becomes likely (8). Potentially pathogenic de novo mutations and reduced-penetrance variants may not be easily resolved in linkage studies or penetrant high-effect mutations outside of large well-documented families. As larger and more comprehensive genetic sequencing studies are conducted in the general population, as well as different PD cohorts, efforts have shifted toward understanding the potential contribution of novel variants in disease susceptibility.
In a seminal contribution in 2011, Ross et al. (9) investigated LRRK2 variants in an ethnically diverse multicenter study of PD patients and controls and found 90 non-synonymous LRRK2 missense variants. These variants included the p.N551K/p.R1398H protective haplotype, and p.M1646T, p.R1628P and p.G2385R that associate with PD risk. Since then, multiple genetic studies have largely replicated the associations between these variants and PD risk in novel cohorts as well as in meta-analysis (10,11). Apparently unrelated to these missense variants or the known pathogenic LRRK2 missense variants, common non-coding variants at the 5′ end of LRRK2 are positively associated with PD risk in large genome-wide association studies (GWAS), exemplified by rs76904798 (3,12).
Biochemically, known pathogenic LRRK2 missense variants appear to share a similar gain-of-function (GOF) consequence of increasing LRRK2 kinase activity. Increased kinase activity manifests through enhanced self-phosphorylation in cis (i.e. pS1292-LRRK2 autophosphorylation) or in trans Rab-phosphorylation (e.g. pT73-Rab10). However, most pathogenic mutations reside outside of the LRRK2 kinase domain (13–15). Two recent studies focused on the identification of naturally occurring loss-of-function (LOF) LRRK2 variants and their relation to PD and other adverse behavioral phenotypes. Both studies concluded a lack of known effect on disease risk with LOF LRRK2 variants (16), specifically without modification of PD risk or protection from PD in carriers (17).
Given the recent focus on LOF variants in LRRK2, here we hypothesize that large whole-genome sequenced (WGS) cohorts may provide added value in the identification of novel pathogenic missense variants in LRRK2. We further predict that these databases will help clarify how existing PD-linked LRRK2 variants might associate with one another. Using the newly minted beta version of the Accelerating Medicines Partnership-Parkinson Disease (AMP-PD, version 2) cohort, together with the large Genome Aggregation Database (gnomAD, v.2.1.1), we performed a focused investigation of emergent LRRK2 missense variants, both previously annotated as well as novel. We identified an in silico pathogenicity classifier that correctly assigned all known pathogenic LRRK2 mutations to assist in rank-ordering rare and novel LRRK2 missense variants of unknown significance (VUS). We further utilized structure–function predictions in the latest high-resolution LRRK2 structure to prioritize variants that segregated into GOF or LOF prediction categories. Lastly, we provide interactive web-based tools that allow for the rapid interrogation of frequencies, predicted pathogenicity and other attributes corresponding to individual LRRK2 missense variants that might assist ongoing investigations of LRRK2 in future biochemical and biomarker studies. As LRRK2-targeting therapeutics enter clinical trial stages, we predict that a better knowledge of genetic susceptibilities to disease encoded within LRRK2 may better guide precision approaches for disease modification strategies.
Results
Characteristics of the novel AMP-PD cohort
In December 2020 the AMP-PD version 2 (2020 v2_release) released data corresponding to over 9800 participants with WGS information. WGS data corresponding to the LRRK2 gene were available for 9887 participants (Table 1 and Supplementary Material, Table S1). In total, 2844 (28.8%) of these participants had a clinical diagnosis of PD that was surmised from standardized clinical criteria and conformity to National Institute of Neurological Disorders and Stroke common data elements (Fig. 1A) (18). To note, 29% of participants are diagnosed with other neurological conditions in addition to PD (Supplementary Material, Table S2). Of these, 90% are diagnosed with Lewy body dementia (LBD).
Table 1.
AMP-PD participants with WGS
| Cohort | Case (PD) | Control | Other | Totala |
|---|---|---|---|---|
| BioFIND (BF) | 99 | 70 | 3 | 172 |
| Harvard Biomarkers Discovery (HB-PD) | 639 | 531 | 3 | 1173 |
| Lewy Body Dementia Study (LBD) | 0 | 1965 | 2614 | 4579 |
| LRRK2 Cohort Consortium (LCC) | 242 | 357 | 0 | 599 |
| Parkinson’s Disease Biomarkers Program (PDBP) | 851 | 500 | 149 | 1500 |
| Parkinson’s Progression Markers Initiative (PPMI) | 923 | 722 | 128 | 1773 |
| Steady PD Phase 3 | 90 | 1 | 0 | 91 |
| Total | 2844 | 4146 | 2897 | 9887 |
aTotal participants within the gatk_passing_variant
Figure 1.
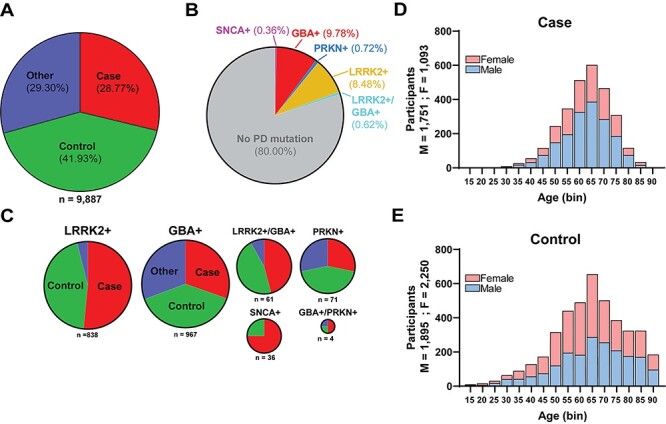
Description of disease-linked missense variants in the first iteration of the AMP-PD cohort. (A) Pie chart depicting diagnosis for 9887 participants that correspond to available WGS data. ‘Case’ indicates a diagnosis of PD. ‘Control’ indicates neurologically healthy participants that were evaluated by movement disorder specialists (see Materials and Methods). ‘Other’ indicates parkinsonism-related neurological disorders including LBD (representing 90% of this category), multiple-system atrophy (MSA), essential tremor, progressive supranuclear palsy (PSP), Alzheimer’s disease, corticobasal degeneration, psychogenic illness, possible prodromal PD (motor and non-motor) and other atypical parkinsonisms (see Supplementary Material, Table S2). (B) Distribution of PD-risk modifying non-synonymous (missense) variants (e.g. disease-linked missense mutations) identified from AMP-PD participants (see Supplementary Material, Table S1 for descriptions of the individual studies combined into the AMP-PD cohort). Subgroups include: LRRK2 variants, ‘LRRK2+’, missense variants include p.G2019S, p.R1441C, p.R1441G and p.R1441H and GBAI variants, GBA+, missense variants include, p.E365K, p.T369M and p.N370S. Smaller subgroups of participants include PD-linked (typically compound heterozygous in disease) missense variants in parkin ‘PRKN+’ that include p.T240M, p.R275W, p.G430D. α-Synuclein ‘SNCA+’ missense variants include p.A53T. ~ 0.6% of AMP-PD participants have both LRRK2 and GBA missense variants associated with PD, ‘LRRK2+/GBA+’, and four participants have PD-associated variants in both GBA and PRKN, ‘GBA+/PRKN+’. (C) Breakdown within the subgroups identified from panel (B) as positive for a disease-linked missense variant according to diagnosis. Color scheme is identical to that in panel (A). (D/E) Age at baseline and sex distribution of cases (D) and controls (E).
Approximately 20% of all participants in AMP-PD diagnosed with PD were positive for a missense variant associated with PD in LRRK2, SNCA, GBA or PRKN. These variants are annotated in the ClinVar database as potentially increasing lifetime risk for PD (Fig. 1B). Overall, more cases than controls in AMP-PD have known PD-associated variants in the LRRK2 gene (Fig. 1C). In total, 61 participants were positive for both a GBA and LRRK2 risk variant, represented by an equal number of PD cases and controls in the cohort. The co-occurence of PD-linked GBA and LRRK2 variants thus occurs without an apparent enrichment in the proportion of cases from controls, results consistent with other recent reports in other cohorts (19). Differences in age (unpaired t-test, P < 0.0001) and sex (Chi square: 170.1, P < 0.00001) were noted in the case and control representation in AMP-PD, with the case (average age: 65; 61.6% male) group older on average, with an overrepresentation of males in cases compared with the control group (average age: 67; 45.7% male) (Fig. 1D and E).
Pathogenicity prediction identifies a hotspot in the LRRK2 ROC-COR domain (Ex.30-Ex.37) in AMP-PD participants
In total, 219 distinct non-synonymous missense LRRK2 variants (with respect to reference assembly GRCh38:CM000674.2) were present in AMP-PD participants (Fig. 2A and Supplementary Material, Table S3). In comparison, a previous study detailing exonic LRRK2 variants from Ross et al. study (2011) identified 90 non-synonymous missense variants in an ethnically diverse cohort of 15 540 subjects. Of the 219 variants identified here, only 53 overlapped with the past Ross study. Thus, 166 variants in AMP-PD were not identified in the Ross et al. study, whereas 37 known variants from Ross were not found in AMP-PD. Each variant in AMP-PD WGS data passed rigorous quality control and harmonization standards according to the latest WGS methods (see Materials and Methods). LRRK2 missense variants were divided into known-pathogenic (i.e. high effect size on lifetime penetrance for PD, i.e. odds ratios (ORs) >10), risk variant (i.e. nominal ORs for PD risk <10) and protective (i.e. any measurable effect on reduced risk for PD). As expected, most variants can be classified as VUS. Of the 219 missense variants in LRRK2 in AMP-PD, 44 were identified exclusively in PD cases (Supplementary Material, Table S4), whereas 61 were found exclusively in controls. Of the 44 case-only variants, six were already reported as linked to PD-susceptibility in past studies. Of these six, three are enriched in East Asian populations, p.A419V:0.18 [variant: Rare Exome Variant Ensemble Learner (REVEL) score], p.R1628P:0.55 and p.G2385R:0.044 (9,11,20). Further, the p.Y2189C:0.40 variant had been linked to PD susceptibility in the Arab-Berber population (9). Finally, the last two variants had been described as case-exclusive variants and nominated as pathogenic mutations (p.I2020T:0.98 and N1437H:0.60, (21,22). Unexpectedly, 18 of the 219 missense variants present in the AMP-PD cohort were not annotated in the large (~141 450 individuals) ethnically diverse gnomAD database. Nine of these 18 variants further lack reference numbers in dbSNP, and seven of these nine were found only in PD cases (Supplementary Material, Table S4 and S5).
Figure 2.
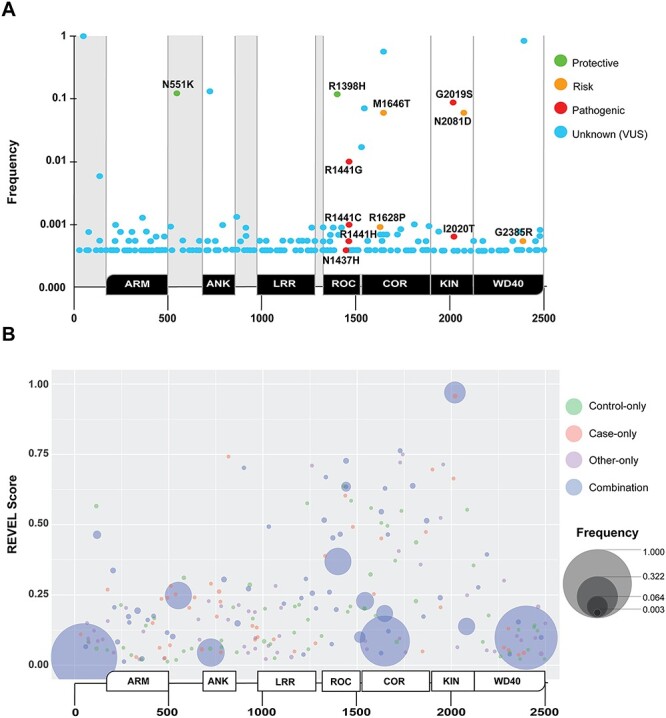
Identification and analysis of missense LRRK2 variants in AMP-PD. Non-synonymous missense variants in LRRK2 with respect to the LRRK2 sequence deposition in reference assembly GRCh38:CM000674.2 (ENSG00000188906). (A) In total, 219 missense LRRK2 variants are aligned with conserved domains in the LRRK2 protein (positioned below the x-axis), and the frequency in which these variants occur in the AMP-PD cohort (PD and control, y-axis, Supplementary Material, Table S3). LRRK2 domain annotation, from N- to C-terminal, are ARM = armadillo, ANK = ankyrin, LRR = leucine-rich repeat, ROC = Rab-like GTPase, COR = C-terminal of ROC, KIN = kinase and WD40 = WD40-repeat. Variants previously annotated in the literature that are associated with risk or susceptibility to PD are indicated as follows: green: suspected protective, yellow: suspected risk and red: pathogenic. Novel variants, or those with an unknown contribution to disease, are indicated as light-blue. (B) Bubble plot aligning all missense variants with protein sequence position and pathogenicity prediction score (y-axis: REVEL score, see Materials and Methods), along with frequency in the AMP-PD cohort (larger bubble is more frequent). Color indicates the diagnosis of participant(s), where green is control-only, salmon is case-only, purple is ‘other’-only and blue is found in a combination of cases, ‘other’ and controls (e.g. the incompletely penetrant pathogenic p.G2019S missense variant). Variants from all participants are included. No removal of PD-linked variant carrying participants was conducted. An interactive browsing tool is available via https://nbviewer.jupyter.org/github/west-lab/LRRK2Figures/blob/master/Interactive%20Figures/AMP-PD_LRRK2_New.html.
The biochemical effects of only a few variants in LRRK2 have been explored (23). To help classify rare missense variants in a manner agnostic of experimental conditions and underlying assumption, the REVEL pathogenicity prediction tool assigns scores to all possible missense variants as likely pathogenic and damaging (REVEL scores from 0.5 to 1.0) to likely benign (scores <0.5) with respect to disease causation. Across thousands of genes associated with human disease, REVEL accuracy has been estimated at ~ 87% for known disease-causative missense variants (24–26). In the case of the LRRK2 gene, we found that the REVEL tool successfully classified all known pathogenic LRRK2 missense variants clearly into the ‘likely pathogenic or damaging’ category (all scores > 0.6). The score distribution included (variant: REVEL score): p.G2019S: 0.97, p.R1441C: 0.73, p.R1441G: 0.64, p.R1441H: 0.64, p.Y1699C: 0.87, p.I2020T: 0.96 and p.N1437H: 0.60 (Fig. 2B). Thirteen novel missense variants (VUS) found in AMP-PD participants exhibit pathogenicity scores >0.6 (Table 2). Notably, p.W817C:0.742, p.I2012T:0.664 (previously described in both PD and healthy controls (27)) and p.G1900E:0.696 occur in PD cases only in AMP-PD. Four variants that surpass the 0.60 REVEL threshold are found in LBD participants only (Table 2). The majority (77%) of the novel variants that passed the empirically derived cutoff of REVEL 0.60 in AMP-PD localized to the ROC or COR domain (Fig. 2B). To visualize the distribution of all missense variants, frequency, position in LRRK2 protein, and REVEL score, we developed an integrated browser available online.
Table 2.
Novel LRRK2 variants from AMP-PD exhibiting pathogenic REVEL score > 0.600
| Protein variant | Revel Score | Case | Control | Other | Total |
|---|---|---|---|---|---|
| p.R1728L | 0.763 | 0 | 2 | 2 | 4 |
| p.W1742L a | 0.750 | 0 | 0 | 1 | 1 |
| p.W817C b | 0.742 | 1 | 0 | 0 | 1 |
| p.R1728H a | 0.721 | 0 | 0 | 1 | 1 |
| p.R1957C a | 0.714 | 0 | 0 | 1 | 1 |
| p.I1260S a | 0.710 | 0 | 0 | 1 | 1 |
| p.E899D | 0.702 | 1 | 0 | 1 | 2 |
| p.G1900E b | 0.696 | 1 | 0 | 0 | 1 |
| p.R1334Q | 0.669 | 1 | 0 | 1 | 2 |
| p.I2012T b | 0.664 | 1 | 0 | 0 | 1 |
| p.L1795F | 0.638 | 3 | 7 | 0 | 10 |
| p.D1429G | 0.636 | 0 | 1 | 0 | 1 |
| p.Q1648R | 0.629 | 1 | 2 | 0 | 3 |
aVariants exclusive to LBD participants
bVariants exclusive to PD participants
LRRK2 missense variant multiplicity in AMP-PD participants
Approximately one-third of the AMP-PD cohort participants have three or more missense variants in LRRK2 (Fig. 3A). We assessed whether any of the known PD-associated LRRK2 missense risk variants (Fig. 2A) were over-represented in PD cases in combination with one another. We identified a reported PD-risk variant, p.M1646T, in all p.R1441G carriers (n = 89). However, the p.M1646T variant was not present in any p.R1441C carriers and not significantly enriched in the pathogenic p.G2019S variant carriers (M1646T population frequency = 0.02, n = 794, Fisher’s exact P > 0.48, Fig. 3B–F, Supplementary Material, Table S6). p.M1646T by itself, more common than p.R1441G, is a reported risk variant that increases likelihood of PD (9).
Figure 3.
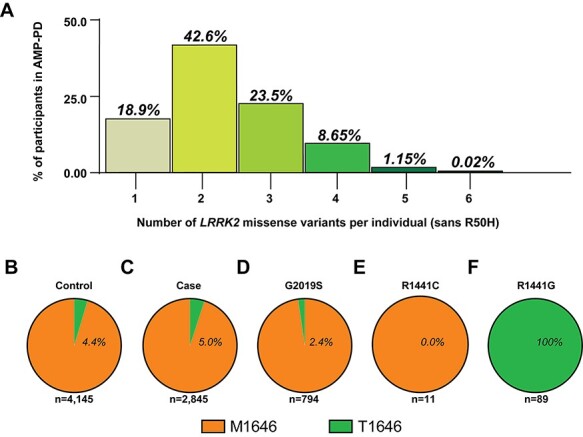
LRRK2 missense variant combinations in AMP-PD participants. (A) Column graph indicating the number of non-synonymous missense variants identified within AMP-PD participants with respect to LRRK2 (reference assembly GRCh38:CM000674.2). All variants included with exception of R50H (rs2256408) (see Materials and Methods). The number of individuals with no LRRK2 variants from consensus (5.2%, sans R50H) is not shown. Pie charts of (B) participants with (percentage of group) or without the p.M1646T missense variant from controls or (C) cases, (D) p.G2019S variant carriers, (E) p.R1441C variant carriers, (E) p.R1441C variant carriers and (F) p.R1441G variant carriers.
In the interrogation of the gnomAD and AMP-PD European cohorts, eight LRRK2 missense variants exist at a population frequency >0.01 (Table 3). Interestingly, four of these eight variants have been reported as independently associated with PD susceptibility (2,9,28). These four variants include the p.N551K/p.R1398H protective haplotype and risk variants p.M1646T and p.N2081D (9,29). Unlike the pathogenic LRRK2 variants mentioned previously, these variants do not have REVEL scores > 0.600. The remaining four common missense variants in LRRK2, p.I723V, p.R1514Q, p.P1542S and p.S1647T, have not previously been associated with PD risk. The largest aggregate of GWAS data identifies common non-coding variants in the 5′ end of the LRRK2 gene, exemplified by rs76904798, associated with increased PD risk [OR = 1.15, confidence interval (CI) 95%: 1.09–1.2] (3). In excluding the carriers of rs76904798 from the AMP-PD European cohort, we found that nearly all carriers of the PD-linked p.R1514Q variant and p.N2081D variant were likewise excluded (90% of p.N2081D and 82% of p.R1514Q, Table 3). Significant decreases in the frequencies of the other common LRRK2 missense variants were not observed in this cohort. These results suggest that the PD linked 5′ non-coding SNPs in LRRK2 drive the association of p.R1514Q and p.N2081D with PD either in full or in part.
Table 3.
Frequencies of common LRRK2 missense variants in cases and controls
| Protein varianta | AMP Case (n = 2615)b | AMP Case Δ 5′ SNP (n = 1953)b | P-valueb |
|---|---|---|---|
| p.Asn551Lys | 0.123 | 0.144 | n.s. |
| p.Ile723Val | 0.163 | 0.193 | n.s. |
| p.Arg1398His | 0.123 | 0.144 | n.s. |
| p.Arg1514Gln | 0.017 | 0.003 | 2.0 × 10 −5 |
| p.Pro1542Ser | 0.046 | 0.050 | n.s. |
| p.Met1646Thr | 0.046 | 0.054 | n.s. |
| p.Ser1647Thr | 0.570 | 0.627 | n.s. |
| p.Asn2081Asp | 0.050 | 0.005 | 8.0 × 10 −9 |
| p.Met2397Thr | 0.900 | 0.892 | n.s. |
aLRRK2 missense variants with frequency > 0.01 in AMP-PD
bFisher’s exact test, comparison of AMP-European case cohort with and without the rs76904798 5′ SNP. Bonferroni corrected. n.s. is P > 0.05
Rare missense LRRK2 variants with high predicted pathogenicity in gnomAD
Through the analysis of known and novel LRRK2 missense variants in AMP-PD, we hypothesized that the REVEL pathogenicity prediction tool may have utility to characterize novel LRRK2 missense variants annotated in the much larger gnomAD database where participants have an uncertain neurological diagnosis or potential for PD risk. We found that the current version of gnomAD (see Materials and Methods) annotated 1259 distinct LRRK2 missense variants (Supplementary Material, Table S7). The number of distinct missense variants in the LRRK2 locus was not significantly more than expected compared with the closest paralogous gene LRRK1 that is not associated with human disease (size corrected, LRRK1: 0.515, LRRK2: 0.489;Chi square: 0.42, P = 0.527).
We developed an interactive browser to visualize the distribution of frequency, position in LRRK2 protein and REVEL score, with binning of variants by genetic ancestry (https://nbviewer.jupyter.org/github/west-lab/LRRK2Figures/blob/master/Interactive%20Figures/BubbleChartRaceFreq.html). Missense variants spread equally across the entirety of the LRRK2 coding domains without apparent bias in distribution (Fig. 4A). Of the 1259 missense variants identified, 10.7% (n = 135) scored above the REVEL ‘likely pathogenic’ threshold we empirically determined based on the known pathogenic LRRK2 variants (i.e. >0.6). In comparison, only 8.68% of total variants of the less-ethnically diverse AMP-PD cohort scored above the 0.6 threshold. Peak clusters of pathogenic scores are localized almost exclusively in the ROC and kinase domains, with intermediate scores dipping in the COR domain and trailing to the WD40 domain. Overall, 109 high-scoring variants in the ROC-COR-Kinase domain were identified, compared with only 30 variants throughout the rest of the protein (Supplementary Material, Table S8). All variants scoring >0.6 in pathogenicity scores were rare, with individual allelic frequencies all <0.001 in gnomAD. The combined allelic frequencies for all rare high-pathogenicity scoring variants in the ROC-COR-Kinase domain totals 0.002 for all variants or ~1 in 500 gnomAD participants. The pathogenic p.G2019S variant, at an allelic frequency of 0.0005, accounts for ~ 26% of this combined frequency, with p.G2019S by far the most common of all high-pathogenicity scoring missense variants in the gnomAD cohort. Thus, ~ 74% of the rare high-pathogenicity scoring variants in the entirety of the gnomAD population are not accounted for by known LRRK2 pathogenic mutations (e.g. p.G2019S or p.R1441G).
Figure 4.

Identification and analysis of missense LRRK2 variants in gnomAD. (A) Bubble plot for missense variants in gnomAD participants (v2.1.1; https://gnomad.broadinstitute.org). The size of the bubble represents the relative frequency of the variant. The x-axis aligns the bubbles across conserved domains in LRRK2. The y-axis indicates the REVEL pathogenicity prediction score (26). LRRK2 domains are: ARM, ANK, LRR, ROC, COR, KIN and WD40. (B) Seven pathogenic LRRK2 variants (red boxes) known to cause PD and the corresponding REVEL scores. (C) Blue boxes highlight seven missense variants found only in PD cases in AMP-PD that were not identified in any participant in gnomAD, with REVEL scores indicated for each of these variants. An integrated browser is available via https://nbviewer.jupyter.org/github/west-lab/LRRK2Figures/blob/master/Interactive%20Figures/BubbleChartRaceFreq.html.
Structural modeling of high-REVEL scoring missense variants in LRRK2
Several of the high-pathogenicity scoring missense variants localize to stretches of the LRRK2 protein that correspond to high-resolution structural information (30). We hypothesized that structural alignments of the novel missense VUS identified in AMP-PD and gnomAD may provide clues as to the possible functional effects of high-scoring pathogenicity variants. In particular, whether a gain or loss of LRRK2 protein function might be predicted. We selected a subset of variants for analysis with the highest REVEL scores that correspond to proposed high-resolution structures.
Within the ROC domain, the highest scoring variants include (variant: REVEL score) p.D1394V:0.977 and p.T1348P:0.848 (Fig. 5A and B). The evolutionary conserved residues T1348 in the p-loop and D1394 in the switch II loop together interact directly with Mg to coordinate GTP positioning through charge interactions (31,32). In aligned models, both the missense variants p.T1348P and p.D1394V might interrupt Mg interactions that might decrease GTP hydrolysis activity (Fig. 5C, (33,34)). Through understanding the biochemical effects of R1441C, decreased GTPase activity might increase LRRK2 kinase activity as well as trans phosphorylation activity in the phosphorylation of Rab substrates (13,14,35). Other ROC and COR domain variants were all outside of known high-resolution structures.
Figure 5.
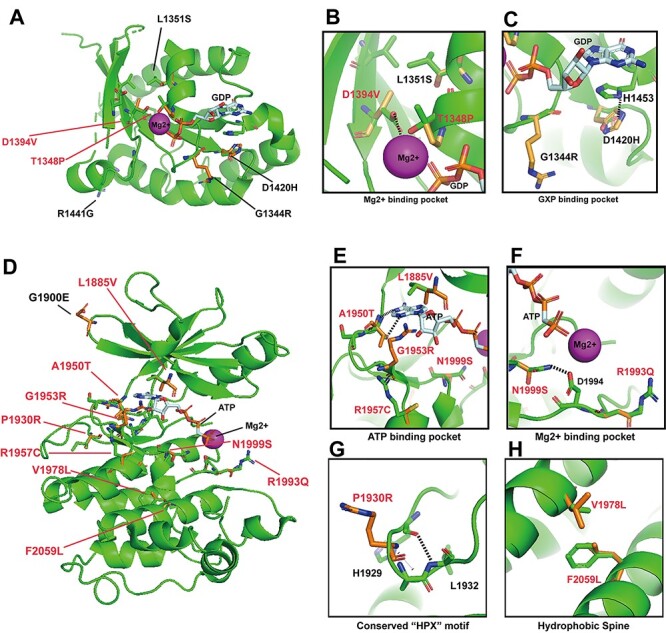
Structural models of high-REVEL LRRK2 missense variants in the ROC and kinase domains suggest both LOF and GOF variants. (A) cryo-EM structure of LRRK2 bound with GDP and Mg2+ (6VNO) (30) was used to model variants from gnomAD predicted to alter protein function (REVEL scores >0.700). ROC variants and their corresponding REVEL scores include: p.D1394V:0.977, p.L1351S:0.806, p.T1348P:0.848, p.G1344R:0.719, p.D1420H:0.786. Red-colored variants are discussed in text. (B) Blow-up model of variants predicted to interact in the Mg-binding pocket. p.T1348P and p.D1394V may interfere with Mg binding. (C) Magnification of the GXP-binding pocket in the ROC-domain. H1453 interacts with the guanosine group in GTPase activity, disrupted by p.D1420H. (D) AppCp and Mg2+ is docked into the LRRK2 cryo-EM structure(6VNO) based on the crystal structure of the G1179S Roco4 Kinase Domain bound to AppCp and Mg2+ from D. discoideum (PDB ID 4F1M) (36). REVEL scores for novel variants include p.L1885V:0.793, p.A1950T:0.866, p.G1953R:0.960, p.R1957C:0.714, p.V1978L:0.809, p.F2059L:0.952, p.R1993Q:0.924, p.N1999S:0.862, p.P1930R:0.882. (E) Blow-up model of the ATP-binding pocket with variants that may affect ATP binding. (F) The Mg-binding pocket in the LRRK2 kinase domain, demonstrating p.N1999S potentially interrupts hydrogen bond interactions (dashed lines) with the highly conserved D1994 residue required for kinase activity (H/YRD motif). p.R1993Q:0.924, another potentially disrupting variant within this loop, is also positioned in the model. (G) Magnification of the structure-altering variant p.P1930R:0.882 that lies within the conserved β-turn segment, likely disrupting bonds with residues L1932 and H1929 (dashed line). (H) Predicted function-altering variants, p.V1978L:0.809 and p.F2059L:0.952, lie on the hydrophobic spine and would likely disrupt hydrophobic interactions critical for overall conformational regulation of the kinase domain.
Within the kinase domain, the highest scoring variants include (variant: REVEL score): p.L1885V:0.793, p.P1930R:0.882, p.A1950T:0.866, p.G1953R:0.984, p.R1957C:0.714, p.V1978L:0.770, p.F2059L:0.952, p.R1993Q:0.924, p.N1999S:0.862 and the case-only AMP-PD variant G1900E:0.696 (Fig. 5D). Residues L1885, A1950, G1953 and R1957 localize within the conserved adenosine binding pocket (36). Substitutions at these residues might alter the binding affinity of ATP. For example, p.A1950 on the hinge motif directly interacts with N1 within ATP through hydrogen bonding (37). p.A1950T might increase affinity by forming an extra hydrogen bond between the hydrogen on the hydroxyl group of threonine and N3 on ATP (Fig. 5E). Further, the N1999 residue may interact with the conserved D1994 residue on the catalytic loop (H/YRD) motif through potential hydrogen bonding (Fig. 5F). The nearby N1999S variant might interrupt hydrogen bonding and affect the catalytic activity through altering the position or the charge distribution of the catalytic D1994 residue. The p.R1993Q substitution might also affect functional positioning of the critical D1994 aspartic acid residue common to most protein kinases (37,38). We predict these variants might activate LRRK2 kinase activity.
In contrast, the p.P1930R variant lies within the conserved β-turn segment in the kinase domain, likely disrupting bonds with residues L1932 and H1929 (dashed line) with a possible deleterious structural effect needed for kinase activity (Fig. 5G). Lastly, V1978 on α-helix 4 and F2059 on α-helix 7 are hydrophobic spine residues that might stabilize the C-lobe of the kinase domain (37). The variants p.V1978L and p.F2059L are thus predicted to increase the overall structural flexibility (Fig. 5H). However, because of a lack of knowledge of how the hydrophobic spine regulates LRRK2 kinase activity, we cannot predict whether these variants might serve in LOF or GOF capacities and thus await functional assessments.
Overall, these structural analyses suggest some high-REVEL scoring LRRK2 missense variants may be destructive with respect to LRRK2 enzymatic function, whereas other missense variants are plausibly activating or otherwise promoting LRRK2 enzymatic function.
Discussion
This study contributes several novel observations and tools that describe LRRK2 missense variant combinations in emergent WGS cohorts. First, we provide an initial characterization of the AMP-PD cohort. Novel missense variants with high pathogenicity scores were identified in AMP-PD, mainly clustered in the ROC and COR domains. Some of these variants were identified exclusively in PD cases, absent from the larger gnomAD cohort (Fig. 6A). These include p.W817C, p.V1330M, p.P1865R, p.G1900E among others. Second, we developed interactive online browsing tools for rapid evaluation of LRRK2 missense variants as they emerge in different sequencing efforts. These tools allow the visualization of a concentration of novel potentially pathogenic variants in the ROC-COR-Kinase domain in LRRK2 (Fig. 6B). Third, we clarify important relationships between LRRK2 variants not previously described. The two most important observations from this analysis include the finding that all carriers of the p.R1441G variant in AMP-PD also carry the disease-linked p.M1646T variant, and nearly all carriers of the risk variant p.N2081D, associated with both Crohn’s disease and PD, are also positive for the 5′ non-coding LRRK2 risk variants identified by GWAS (i.e. rs76904798). Fourth, we are able to nominate several novel and very rare LRRK2 missense variants with high potential for pathogenic GOF. The analysis suggests that although the p.G2019S variant is by far the most frequent of the high-scoring pathogenic variants identified in either AMP-PD or gnomAD cohorts, collectively, cumulative frequencies of other rare variants with very high pathogenicity scores have the potential to significantly add to the number of individuals with high-effect LRRK2 missense variants.
Figure 6.
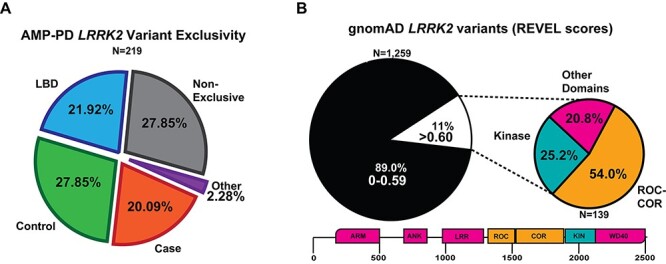
Summary of LRRK2 variant discoveries from both AMP-PD and gnomAD. (A) Pie chart showing the distribution of the 219 LRRK2 variant in case (PD), control, LBD or multiple participants from each group, ‘Non-exclusive’ from AMP-PD. ‘Other’ refers to either MSA-exclusive (N = 2, p.R924C and p.A75G), PSP (N = 1, p.K657I) and prodromal PD (N = 2, p.S1096C and p.T146S). Case-only (PD) variants are listed in Supplementary Material, Table S4. (B) Pie chart showing the portion of LRRK2 variants from gnomAD with less REVEL score ≤0.599 (black) or ≥0.6 (white). Smaller pie chart represents the distribution of location within LRRK2 for variants that are at or exceed the REVEL score of 0.6 in gnomAD (N = 139). List of gnomAD variants > 0.6 are described in Supplementary Material, Table S8.
Both the National Institutes of Health (NIH) and the Michael J. Fox Foundation for Parkinson’s Research (MJFF) have invested into the collection and WGS analysis of different cohorts of participants. The participants in the second release of AMP-PD utilized here are split between Parkinson’s Disease Biomarker Program (PDBP) samples (N = 1500), Lewy Body Disease Study (N = 4579), Steady Phase 3 PD (N = 91) and the MJFF Parkinson’s Progression Markers Initiative samples (N = 1773), LRRK2 Cohort Consortium (N = 599), Harvard Biomarkers Discovery (N = 1173) and 172 participants from BioFIND. Most participants in these series are longitudinally followed, with clinical information updated regularly, and existing biospecimens deposited and available for analysis. As expected, variants in LRRK2 and GBA account for most of the PD cases with known PD-associated missense variants. However, it is worth noting that AMP-PD (version 2, December 2020) is not an accurate representation of the general PD population, since recruitment of some arms of the included cohorts bias toward GBA, LRRK2 and SNCA affected and unaffected carriers. Additionally, it is worth noting that AMP-PD is also enriched in LBD patients, represented just under the frequency of PD cases (26% versus 29% respectively). The 61 participants harboring both LRRK2 and GBA missense variants linked to PD are split equally in numbers between cases and controls, suggesting no dramatic effects on additive susceptibility as recently demonstrated by others (19).
With the assumption of the possible existence of penetrant, high-effect, novel LRRK2 missense variants that are exclusive to PD cases, 16 variants in particular caught our interest in AMP-PD cases because they were absent from the control cohorts as well as the much larger genome sequencing database gnomAD. Of these 16, two variants have elevated REVEL pathogenicity scores equal to or higher than some known pathogenic LRRK2 variants. Three variants of particular interest are within the ROC-COR-Kinase stretch of LRRK2 protein (p.V1330M, p.P1865R and p.G1900E) and a fourth lies in the N-terminal Ankyrin domain (p.W817C). The novel p.P1865R (COR-Kinase, REVEL score 0.47), p.V1330M (ROC, REVEL score 0.39) and p.G1900E (Kinase, REVEL score 0.696) variants are attractive candidates for biochemical analysis.
Allowing for incomplete penetrance or the existence of the variant in the general population (gnomAD), many other variants of interest emerge. Notably, the p.R1334Q (ROC, REVEL score 0.67) variant exceeds pathogenicity scores of several known LRRK2 pathogenic variants. In this vein, the previously recognized PD risk variant p.R1628P (COR, REVEL score 0.55), and the novel p.R1628C variant (REVEL score 0.45) also emerge with borderline pathogenicity scores. Outside of the ROC-COR-Kinase domain, the only variant in a PD case with an elevated pathogenicity score localized to the ANK domain (p.W817C, REVEL score 0.742). We hypothesize that the addition of this cysteine might affect a disulfide bridge that structurally connects the missense variant back to the ROC-COR-kinase stretch of LRRK2 protein, although there are no high-resolution structures available to support or refute this hypothesis. An additional Ankyrin-localized, high-REVEL scoring variant (p.E899D, REVEL score 0.702) was identified in one PD case and one LBD case. Further biochemical analyses of all of these nominated variants seem warranted. Within the conserved ROC and Kinase domains, many high-pathogenicity scoring variants were identified in different races and ethnicities that are not (or poorly) represented in AMP-PD. We predict that a more inclusive approach to future inclusions in AMP-PD will open a wide-range of new variants of interest to understand the biochemical basis of LRRK2-linked disease.
On the basis of population frequencies, the vast majority of PD cases currently identified with pathogenic LRRK2 missense variants carry p.G2019S, or more rarely, p.R1441G. The p.G2019S variant, although incompletely penetrant for lifetime PD susceptibility (1), stands alone in elevated frequency in both AMP-PD and gnomAD, with a near-perfect pathogenicity score (REVEL score 0.97). The p.R1441G variant has been identified within a single shared ~ 5.8 Mb allele that dates to the seventh century in Northern Spain (39,40). Biochemical effects of the p.R1441G variant have been described as different from the R1441C variant in vitro (41) as well as proposed features associated with transgenic mice overexpressing R1441G-LRRK2 (42,43) and knock-in mice (44–46). Through WGS analysis, the shared p.R1441G allele appears to co-harbor the p.M1646T variant in all participants, likely in cis because 89 of 89 p.R1441G carriers are positive for p.M1646T. In contrast, no p.R1441C carriers are positive. We hypothesize that the p.M1646T variant in the ROC domain plays an important part of the biochemical pathogenicity of the p.R1441G missense variant, with the p.M1646 residue conserved between human and mouse. Thus, the mouse knock-in models would lack the p.M1646T variant as found in human carriers. On the basis of recently published cryo-EM structure of LRRK2 (30), the p.R1441 residue exists on the interface between ROC domain and COR domain, and M1646 is on the interface between COR domain and kinase domain. Although the synergic effect is difficult to predict, these residues might involve the interdomain regulation of LRRK2 that can potentially drive the activation of LRRK2 kinase activity. Sequence analysis of the most commonly used LRRK2 expression constructs in PD-related research reveals only one plasmid family widely used in the field harbors a threonine at 1646, with all others harboring the more frequent variant (Supplementary Material, Table S9). While p.T1646 would be correct for studying the p.R1441G variant, it would likely be incorrect for other known LRRK2 missense variants. Besides the variation at 1646, nearly all other LRRK2-expressing plasmids used in the field have the same sequence composition, despite the majority of subjects analyzed by WGS showing more than one missense LRRK2 variant in conserved domains. We hypothesize that combinations of LRRK2 missense variants may influence the biochemical effects of both known and novel pathogenic LRRK2 variants. Future biochemical studies, and allelic sequencing efforts, will be required to test this hypothesis.
The p.N2081D variant has been suggested to confer shared risk effects on both Crohn’s disease (CD) and PD (29). However, the p.N2081D variant, as well as the p.R1514Q variant, occur almost exclusively with participants analyzed here that already carry the potent risk allele in the LRRK2 promoter, rs76904798 (3,12). The potential contribution of p.N2081D and p.R1514Q towards the linkage of rs76904798 to PD therefore warrants further investigation. In contrast, the p.M1646T COR-domain missense variant has been shown to be enriched in PD cases with a substantial effect size (OR 1.43) and does not appear to be enriched in the shared rs76904798 risk haplotype, at least in the cohorts evaluated here (2,9,28). The breadth of genetic analysis and size of the PD case cohort needed to untangle which factors, if not both, (PD-associated risk variant rs76904798 or LRRK2-coding variants) are driving association with disease fell beyond the limits of this study. However, it is important to stress that we provide some of the first evidence of linkage between a PD-associated coding variant and a non-coding variant, suggesting caution in the interpretation of how an individual missense variant may interact with PD risk.
Our current understanding of how LRRK2 pathogenic variants affect LRRK2 enzymatic function are largely on the basis of the G2019S and R1441C pathogenic variants. With this understanding, and high-resolution structural models of the ROC and kinase domains, we could probe a number of novel variants for similarity or divergence in what known pathogenic variants do to LRRK2 structure and function. Whereas GOF missense variants (with respect to LRRK2 kinase activity) link LRRK2 variants with pathogenicity (47), REVEL scores do not specifically predict LOF or GOF, only likely disease-linked association. Structurally, we found evidence that some variants might impart a GOF in LRRK2 kinase activity, as well as other variants that might impart LOF and therefore be unlikely to contribute to disease risk. Given the complexity of post-translational modifications occurring in LRRK2 that include transient phosphorylation, as well as protein interactors like 14-3-3 proteins, the functional effects of particular variants may be difficult to assign. Ultimately, in-depth biochemical dissection and (perhaps more importantly) LRRK2-activity associated biomarkers in patients will help confirm or refute our predictions. As LRRK2-targeting therapeutics march closer to efficacy trials in the clinic, studies that identify all the patients that might best benefit may be a key aspect to successful clinical trials. We predict that WGS cohorts and subsequent analyses like those performed here will be important in these efforts.
Materials and Methods
Identification of missense variants and participant information in AMP-PD
Access to the AMP PD Knowledge Platform data was granted via amp-pd.org/register-for-amp-pd instructions including submission, approval and adherence with the AMP PD Data Use Agreement. Duke Institutional Review Board (DUHS IRB Pro00104464) determined that our research protocol meets the definition of not involving human subjects (per: 45 CFR 46.102(f), 21 CFR 56.102(e) and 21 CFR 812.3(p)) and satisfies Privacy Rules (per: 45CFR164.514).
Following approval of the AMP-PD data use agreement and Tier 2 access, the Terra cloud platform and Py3-WGS-Query Variants notebook was used to query missense variants in LRRK2 that passed quality control metrics (gatk_passing_variants). Additional queries were conducted for SNCA, PRKN and GBA in these participants. Participant IDs were used to query diagnosis, sex and age at baseline from BigQuery under the resource: amp-pd-research/2020_v2release_1218. From our query we identified 9887 participants with WGS in LRRK2, subsequent analysis of SNCA, GBA, PRKN and separate query for 5’SNP (rs76904798) found no participant IDs outside of the initial group of 9887. This suggests QC-passing genomic data available in AMP-PD release 2 totals to 9887 participants.
AMP-PD WGS data collection and harmonization was conducted on the GRCh38DH Human Genome reference. AMP-PD represents a partnership with Data Tecnica International and NIH/NIA/LNG to manage WGS samples and quality metrics for inclusion using preset criterion for contamination, minimum read coverage, outliers and missingness (https://amp-pd.org/whole-genome-data). Samples were matched in clinically reported sex and genetic ancestry and screened for participant duplications. Samples containing discrepancies were discarded. Samples that did not pass quality metrics were excluded in entirety and not included in the Py3-WGS-Query Variants notebook. PD-associated variants in SNCA, GBA and PRKN missense variants were determined on the basis of current ClinVar status. Variants included: SNCA: p.A53T (VCV000014007.1), GBA: p.N370S (VCV000004290.16), p.E326K (VCV000199044.8), p.T369M (VCV000093447.8), PRKN: p.R275W (VCV000007050.10), p.G430D (VCV000356016.5), p.T240M (VCV000007054.5). To note, WGS analysis may not capture all GBA variants therefore AMP-PD may not annotate all possible GBA carriers.
R50H was omitted from the common missense variants analysis throughout. This variant was omitted because the corresponding variant frequency for each variant fit the description of the major allele at the given nucleotide and is likely inaccurately annotated in the consensus sequence. R50H was observed in 100% of AMP-PD participants.
Variant ancestry was determined through the gnomAD v2.1.1 database that provides details on coding variants from > 141 000 individuals. Allele frequency, allele number and homozygous count are described for the entire group as well as eight different racial/ethnic groups: African, Ashkenazi Jewish, European (Finnish), European (non-Finnish), East Asian, South Asian, Latino and Other (https://gnomad.broadinstitute.org/blog/2018-10-gnomad-v2-1/). If a variant was found in multiple racial or ethnic groups, they were categorized as ‘multiracial’ variants. gnomAD comparison of variation percentage was performed through a comparison of missense variant calls for each locus divided by the number of residues.
In silico pathogenicity prediction
The REVEL open-source tool was used to calculate pathogenicity scores that were manually curated (24–26). REVEL combines 18 different pathogenicity prediction models each weighted in the overall score on the basis of the correlative strength (includes eight conservation scoring tools and eight functional scoring tools). Scores range from 0.000 to 1.000, with >0.500 categorized as ‘likely pathogenic’ or damaging, and <0.500 as ‘likely benign’ or tolerant.
Structural modeling
The LRRK2 structural model was generated on the basis of the cryo-EM structure of LRRK2 bound with GDP and Mg (6VNO). The AppCp bound kinase domain model is generated by docking AppCp and Mg2+ in to the cryo-EM structure on the basis of the crystal structure of the G1179S Roco4 Kinase Domain bound to AppCp and Mg2+ from Dictyostelium discoideum (PDB ID 4F1M). Human LRRK2 peptide sequences were identified by sequence alignment using SPEM (Sequence and Secondary Structure Profile Enhanced Multiple Alignment, Laboratory of Biophysics and Bioinformatics, University of Buffalo) as previously described (Liu 2014). The model of the LRRK2 kinase domain carrying the indicated variants was generated using the Mutagenesis plugin in PyMol (2.3.2).
Statistics
Statistical comparison of age between PD and control was determined using unpaired t-test Graphpad Prism Version 8. Chi square statistic to compare sex differences between PD and control groups was determined by Graphpad Prism Version 8. P-values for population frequency comparisons were calculated using Fisher’s exact test with multiple testing corrections. CIs, z-statistics and P-values were determined using MedCalc Version 19.5.3.
Supplementary Material
Acknowledgements
AMP PD Knowledge Platform provided data used for this manuscript; for more information and ongoing updates for the AMP PD platform, visit https://www.amp-pd.org. AMP PD—a public–private partnership—is managed by the Foundation for the National Institutes of Health and funded by Celgene, GSK, the Michael J. Fox Foundation for Parkinson’s Research (MJFF), the National Institute of Neurological Disorders and Stroke (NINDS), Pfizer, Sanofi and Verily. The AMP PD WGS platform (as of December 2020 version release) provided data from BioFIND, Parkinson’s Disease Biomarker Program (PDBP), Harvard Biomarkers Discovery, Lewy Body Dementia Study, LRRK2 Cohort Consortium (LCC), Steady PD Phase 3 and Parkinson’s Progression Markers Initiative (PPMI). BioFIND is sponsored by the MJFF with support from the NINDS. The BioFIND Investigators have not participated in reviewing the data analysis or content of the manuscript. For up-to-date information on the study, visit michaeljfox.org/biofind. PDBP consortium is supported by the NINDS at the National Institutes of Health (NIH). A full list of PDBP investigators can be found at https://pdbp.ninds.nih.gov/policy. The PDBP Investigators have not participated in reviewing the data analysis or content of the manuscript. PPMI—a public–private partnership—is funded by the MJFF and funding partners, which include a consortium of industry, non-profit organizations and private individuals (list the full names of all of the PPMI funding partners found at www.ppmi-info.org/fundingpartners). The PPMI Investigators have not participated in reviewing the data analysis or content of the manuscript. For up-to-date information on the study, visit www.ppmi-info.org. Genome Sequencing in Lewy Body Dementia and Neurologically Healthy Controls: A Resource for the Research Community was generated by the International LBD Genomics Consortium (iLBDGC), under the co-directorship by Dr Bryan J. Traynor and Dr Sonja W. Scholz from the Intramural Research Program of the U.S. NIH. The iLBDGC Investigators have not participated in reviewing the data analysis or content of the manuscript. For a complete list of contributors, please see: bioRxiv 2020.07.06.185066; doi:https://doi.org/10.1101/2020.07.06.185066. The LCC was created to assemble and study groups of people with and without Parkinson’s disease who carry mutations in the LRRK2 gene. The LCC is coordinated and funded by The MJFF. The investigators within the LCC contributed to the design and implementation of the LCC and/or provided data and/or collected biospecimens but did not necessarily participate in the analysis or writing of this report. The full list of LCC investigators can be found at www.michaeljfox.org/lccinvestigators. STEADY-PD III is a 36-month, Phase 3, parallel group, placebo-controlled study of the efficacy of isradipine 10 mg daily in 336 participants with early Parkinson’s disease that was funded by the NINDS and supported by the MJFF for Parkinson’s Research and the Parkinson’s Study Group. The STEADY-PD III Investigators have not participated in reviewing the data analysis or content of the manuscript. The full list of STEADY PD III investigators can be found at: https://clinicaltrials.gov/ct2/show/NCT02168842.
Conflict of Interest statement. A.W. is a member of the Scientific Advisory Board for the Michael J. Fox Foundation and an active consultant for eScapeBio, Inc., and Neuro23, Inc., and has received research support unrelated to this study from Biogen/Idec and eScapeBio, Inc. All other authors have declared that no conflict of interest exists.
Contributor Information
Nicole Bryant, Duke Center for Neurodegeneration and Neurotherapeutics Research, Departments of Pharmacology and Cancer Biology, and Neurology, Duke University, Durham, NC 27710 USA.
Nicole Malpeli, Duke Center for Neurodegeneration and Neurotherapeutics Research, Departments of Pharmacology and Cancer Biology, and Neurology, Duke University, Durham, NC 27710 USA.
Julia Ziaee, Duke Center for Neurodegeneration and Neurotherapeutics Research, Departments of Pharmacology and Cancer Biology, and Neurology, Duke University, Durham, NC 27710 USA.
Cornelis Blauwendraat, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Bethesda, MD 20892, USA.
Zhiyong Liu, Duke Center for Neurodegeneration and Neurotherapeutics Research, Departments of Pharmacology and Cancer Biology, and Neurology, Duke University, Durham, NC 27710 USA.
Andrew B West, Duke Center for Neurodegeneration and Neurotherapeutics Research, Departments of Pharmacology and Cancer Biology, and Neurology, Duke University, Durham, NC 27710 USA.
Funding
Michael J. Fox Foundation and National Institutes of Health/National Institutes of Neurological Disorders and Stroke (P50 NS108675, R01 NS064934); Intramural Research Program of the National Institutes of Health/National Institute on Aging.
References
- 1. Trinh, J., Guella, I. and Farrer, M.J. (2014) Disease penetrance of late-onset parkinsonism: a meta-analysis. JAMA Neurol., 71, 1535–1539. [DOI] [PubMed] [Google Scholar]
- 2. Heckman, M.G., Soto-Ortolaza, A.I., Aasly, J.O., Abahuni, N., Annesi, G., Bacon, J.A., Bardien, S., Bozi, M., Brice, A., Brighina, L.et al. (2013) Population-specific frequencies for LRRK2 susceptibility variants in the Genetic Epidemiology of Parkinson’s Disease (GEO-PD) Consortium. Mov. Disord., 28, 1740–1744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Nalls, M.A., Blauwendraat, C., Vallerga, C.L., Heilbron, K., Bandres-Ciga, S., Chang, D., Tan, M., Kia, D.A., Noyce, A.J., Xue, A.et al. (2019) Identification of novel risk loci, causal insights, and heritable risk for Parkinson’s disease: a meta-analysis of genome-wide association studies. Lancet Neurol., 18, 1091–1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Paisán-Ruíz, C., Jain, S., Evans, E.W., Gilks, W.P., Simón, J., van derBrug, M., López de Munain, A., Aparicio, S., Gil, A.M., Khan, N.et al. (2004) Cloning of the gene containing mutations that cause PARK8-linked Parkinson’s disease. Neuron, 44, 595–600. [DOI] [PubMed] [Google Scholar]
- 5. Zimprich, A., Biskup, S., Leitner, P., Lichtner, P., Farrer, M., Lincoln, S., Kachergus, J., Hulihan, M., Uitti, R.J., Calne, D.B.et al. (2004) Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron, 44, 601–607. [DOI] [PubMed] [Google Scholar]
- 6. Dächsel, J.C. and Farrer, M.J. (2010) LRRK2 and Parkinson disease. Arch. Neurol., 67, 542–547. [DOI] [PubMed] [Google Scholar]
- 7. Kachergus, J., Mata, I.F., Hulihan, M., Taylor, J.P., Lincoln, S., Aasly, J., Gibson, J.M., Ross, O.A., Lynch, T., Wiley, J.et al. (2005) Identification of a novel LRRK2 mutation linked to autosomal dominant parkinsonism: evidence of a common founder across European populations. Am. J. Hum. Genet., 76, 672–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Greenblatt, M.S. (2015) Sequence variants of uncertain significance. Sequence variants of uncertain significance. Surg. Oncol. Clin. N. Am. (2015), 24, 833–846. [DOI] [PubMed] [Google Scholar]
- 9. Ross, O.A., Soto-Ortolaza, A.I., Heckman, M.G., Aasly, J.O., Abahuni, N., Annesi, G., Bacon, J.A., Bardien, S., Bozi, M., Brice, A.et al. (2011) Association of LRRK2 exonic variants with susceptibility to Parkinson’s disease: a case-control study. Lancet Neurol., 10, 898–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wu, X., Tang, K.-F., Li, Y., Xiong, Y., Shen, L., Wei, Z., Zhou, K., Niu, J., Han, X., Yang, L.et al. (2012) Quantitative assessment of the effect of LRRK2 exonic variants on the risk of Parkinson’s disease: a meta-analysis. Quantitative assessment of the effect of LRRK2 exonic variants on the risk of Parkinson’s disease: a meta-analysis. Parkinsonism Relat. Disord. (2012), 18, 722–730. [DOI] [PubMed] [Google Scholar]
- 11. Gopalai, A.A., Lim, S.-Y., Chua, J.Y., Tey, S., Lim, T., Mohamed, I.N., Tan, A.H., Eow, G.B., Abdul Aziz, Z., Puvanarajah, S.D.et al. (2014) LRRK2 G2385R and R1628P mutations are associated with an increased risk of Parkinson’s disease in the Malaysian population. Biomed. Res. Int., 2014, 867321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Nalls, M.A., Pankratz, N., Lill, C.M., Do, C.B., Hernandez, D.G., Saad, M., DeStefano, A.L., Kara, E., Bras, J., Sharma, M.et al. (2014) Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson’s disease. Nat. Genet., 46, 989–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Steger, M., Tonelli, F., Ito, G., Davies, P., Trost, M., Vetter, M., Wachter, S., Lorentzen, E., Duddy, G., Wilson, S.et al. (2016) Phosphoproteomics reveals that Parkinson’s disease kinase LRRK2 regulates a subset of Rab GTPases. Elife, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. West, A.B., Moore, D.J., Choi, C., Andrabi, S.A., Li, X., Dikeman, D., Biskup, S., Zhang, Z., Lim, K., Dawson, V.L.et al. (2007) Parkinson’s disease-associated mutations in LRRK2 link enhanced GTP-binding and kinase activities to neuronal toxicity. Hum. Mol. Genet., 16, 223–232. [DOI] [PubMed] [Google Scholar]
- 15. Sheng, Z., Zhang, S., Bustos, D., Kleinheinz, T., Le Pichon, C.E., Dominguez, S.L., Solanoy, H.O., Drummond, J., Zhang, X., Ding, X.et al. (2012) Ser1292 autophosphorylation is an indicator of LRRK2 kinase activity and contributes to the cellular effects of PD mutations. Sci. Transl. Med., 4, 164ra161. [DOI] [PubMed] [Google Scholar]
- 16. Whiffin, N., Armean, I.M., Kleinman, A., Marshall, J.L., Minikel, E.V., Goodrich, J.K., Quaife, N.M., Cole, J.B., Wang, Q., Karczewski, K.J.et al. (2020) The effect of LRRK2 loss-of-function variants in humans. Nat. Med., 26, 869–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Blauwendraat, C., Reed, X., Kia, D.A., Gan-Or, Z., Lesage, S., Pihlstrøm, L., Guerreiro, R., Gibbs, J.R., Sabir, M., Ahmed, S.et al. (2018) Frequency of loss of function variants in LRRK2 in Parkinson disease. JAMA Neurol., 75, 1416–1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Rosenthal, L.S., Drake, D., Alcalay, R.N., Babcock, D., Bowman, F.D., Chen-Plotkin, A., Dawson, T.M., Dewey, R.B., Jr., German, D.C., Huang, X.et al. (2016) The NINDS Parkinson’s disease biomarkers program. Mov. Disord., 31, 915–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Omer, N., Giladi, N., Gurevich, T., Bar-Shira, A., Gana-Weisz, M., Goldstein, O., Kestenbaum, M., Cedarbaum, J.M., Orr-Urtreger, A., Mirelman, A.et al. (2020) A possible modifying effect of the G2019S mutation in the LRRK2 gene on GBA Parkinson’s disease. Mov. Disord., 35, 1249–1253. [DOI] [PubMed] [Google Scholar]
- 20. Fu, X., Zheng, Y., Hong, H., He, Y., Zhou, S., Guo, C., Liu, Y., Xian, W., Zeng, J., Li, J.et al. (2013) LRRK2 G2385R and LRRK2 R1628P increase risk of Parkinson’s disease in a Han Chinese population from Southern Mainland China. LRRK2 G2385R and LRRK2 R1628P increase risk of Parkinson’s disease in a Han Chinese population from southern mainland China. Parkinsonism Relat. Disord. (2013), 19, 397–398. [DOI] [PubMed] [Google Scholar]
- 21. Funayama, M., Hasegawa, K., Ohta, E., Kawashima, N., Komiyama, M., Kowa, H., Tsuji, S. and Obata, F. (2005) An LRRK2 mutation as a cause for the parkinsonism in the original PARK8 family. Ann. Neurol., 57, 918–921. [DOI] [PubMed] [Google Scholar]
- 22. Aasly, J.O., Vilariño-Güell, C., Dachsel, J.C., Webber, P.J., West, A.B., Haugarvoll, K., Johansen, K.K., Toft, M., Nutt, J.G., Payami, H.et al. (2010) Novel pathogenic LRRK2 p.Asn1437His substitution in familial Parkinson’s disease. Mov. Disord., 25, 2156–2163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. West, A.B. (2015) Ten years and counting: moving leucine-rich repeat kinase 2 inhibitors to the clinic. Mov. Disord., 30, 180–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tian, Y., Pesaran, T., Chamberlin, A., Fenwick, R.B., Li, S., Gau, C., Chao, E.C., Lu, H., Black, M.H. and Qian, D. (2019) REVEL and BayesDel outperform other in silico meta-predictors for clinical variant classification. Sci. Rep., 9, 12752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ganakammal, S.R. and Alexov, E. (2019) Evaluation of performance of leading algorithms for variant pathogenicity predictions and designing a combinatory predictor method: application to Rett syndrome variants. Peer J, 7, e8106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ioannidis, N.M., Rothstein, J.H., Pejaver, V., Middha, S., McDonnell, S.K., Baheti, S., Musolf, A., Li, Q., Holzinger, E., Karyadi, D.et al. (2016) REVEL: an ensemble method for predicting the pathogenicity of rare missense variants. Am. J. Hum. Genet., 99, 877–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tomiyama, H., Li, Y., Funayama, M., Hasegawa, K., Yoshino, H., Kubo, S., Sato, K., Hattori, T., Lu, C., Inzelberg, R.et al. (2006) Clinicogenetic study of mutations in LRRK2 exon 41 in Parkinson’s disease patients from 18 countries. Mov. Disord., 21, 1102–1108. [DOI] [PubMed] [Google Scholar]
- 28. Simón-Sánchez, J., Schulte, C., Bras, J.M., Sharma, M., Gibbs, J.R., Berg, D., Paisan-Ruiz, C., Lichtner, P., Scholz, S.W., Hernandez, D.G.et al. (2009) Genome-wide association study reveals genetic risk underlying Parkinson’s disease. Nat. Genet., 41, 1308–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hui, K.Y., Fernandez-Hernandez, H., Hu, J., Schaffner, A., Pankratz, N., Hsu, N., Chuang, L., Carmi, S., Villaverde, N., Li, X.et al. (2018) Functional variants in the gene confer shared effects on risk for Crohn’s disease and Parkinson's disease. Sci. Transl. Med., 10, eaai7795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Deniston, C.K., Salogiannis, J., Mathea, S., Snead, D.M., Lahiri, I., Matyszewski, M., Donosa, O., Watanabe, R., Böhning, J., Shiau, A.K.et al. (2020) Structure of LRRK2 in Parkinson’s disease and model for microtubule interaction. Nature, 588, 344–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Deng, J., Lewis, P.A., Greggio, E., Sluch, E., Beilina, A. and Cookson, M.R. (2008) Structure of the ROC domain from the Parkinson’s disease-associated leucine-rich repeat kinase 2 reveals a dimeric GTPase. Structure of the ROC domain from the Parkinson’s disease-associated leucine-rich repeat kinase 2 reveals a dimeric GTPase. Proc. Natl. Acad. Sci. (2008), 105, 1499–1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Liu, Z., Mobley, J.A., DeLucas, L.J., Kahn, R.A. and West, A.B. (2016) LRRK2 autophosphorylation enhances its GTPase activity. FASEB J., 30, 336–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Scheidig, A.J., Burmester, C. and Goody, R.S. (1999) The pre-hydrolysis state of p21(ras) in complex with GTP: new insights into the role of water molecules in the GTP hydrolysis reaction of ras-like proteins. Structure, 7, 1311–1324. [DOI] [PubMed] [Google Scholar]
- 34. Knihtila, R., Holzapfel, G., Weiss, K., Meilleur, F. and Mattos, C. (2015) Neutron crystal structure of RAS GTPase puts in question the protonation state of the GTP γ-phosphate. J. Biol. Chem., 290, 31025–31036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Biosa, A., Trancikova, A., Civiero, L., Glauser, L., Bubacco, L., Greggio, E. and Moore, D.J. (2013) GTPase activity regulates kinase activity and cellular phenotypes of Parkinson’s disease-associated LRRK2. Hum. Mol. Genet., 22, 1140–1156. [DOI] [PubMed] [Google Scholar]
- 36. Gilsbach, B.K., Ho, F.Y., Vetter, I.R., vanHaastert, P.J.M., Wittinghofer, A. and Kortholt, A. (2012) Roco kinase structures give insights into the mechanism of Parkinson disease-related leucine-rich-repeat kinase 2 mutations. Proc. Natl. Acad. Sci. U. S. A., 109, 10322–10327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Liu, Z., Galemmo, R.A., Jr., Fraser, K.B., Moehle, M.S., Sen, S., Volpicelli-Daley, L.A., DeLucas, L.J., Ross, L.J., Valiyaveettil, J., Moukha-Chafiq, O.et al. (2014) Unique functional and structural properties of the LRRK2 protein ATP-binding pocket. J. Biol. Chem., 289, 32937–32951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Manning, G. (2002) The protein kinase complement of the human genome. The protein kinase complement of the human genome. Science (2002), 298, 1912–1934. [DOI] [PubMed] [Google Scholar]
- 39. Mata, I.F., Hutter, C.M., González-Fernández, M.C., dePancorbo, M.M., Lezcano, E., Huerta, C., Blazquez, M., Ribacoba, R., Guisasola, L.M., Salvador, C.et al. (2009) Lrrk2 R1441G-related Parkinson’s disease: evidence of a common founding event in the seventh century in Northern Spain. Neurogenetics, 10, 347–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mata, I.F., Taylor, J.P., Kachergus, J., Hulihan, M., Huerta, C., Lahoz, C., Blazquez, M., Guisasola, L.M., Salvador, C., Ribacoba, R.et al. (2005) LRRK2 R1441G in Spanish patients with Parkinson’s disease. Neurosci. Lett., 382, 309–311. [DOI] [PubMed] [Google Scholar]
- 41. Jaleel, M., Nichols, R.J., Deak, M., Campbell, D.G., Gillardon, F., Knebel, A. and Alessi, D.R. (2007) LRRK2 phosphorylates moesin at threonine-558: characterization of how Parkinson’s disease mutants affect kinase activity. Biochem. J., 405, 307–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Li, Y., Liu, W., Oo, T.F., Wang, L., Tang, Y., Jackson-Lewis, V., Zhou, C., Geghman, K., Bogdanov, M., Przedborski, S.et al. (2009) Mutant LRRK2(R1441G) BAC transgenic mice recapitulate cardinal features of Parkinson’s disease. Nat. Neurosci., 12, 826–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Dorval, V., Mandemakers, W., Jolivette, F., Coudert, L., Mazroui, R., De Strooper, B. and Hébert, S.S. (2014) Gene and MicroRNA transcriptome analysis of Parkinson’s related LRRK2 mouse models. PLoS One, 9, e85510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Liu, H.-F., Lu, S., Ho, P.W.-L., Tse, H., Pang, S.Y., Kung, M.H., Ho, J.W., Ramsden, D.B., Zhou, Z. and Ho, S. (2014) LRRK2 R1441G mice are more liable to dopamine depletion and locomotor inactivity. Ann. Clin. Transl. Neurol., 1, 199–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ito, G., Katsemonova, K., Tonelli, F., Lis, P., Baptista, M.A.S., Shpiro, N., Duddy, G., Wilson, S., Ho, P.W., Ho, S.et al. (2016) Phos-tag analysis of Rab10 phosphorylation by LRRK2: a powerful assay for assessing kinase function and inhibitors. Biochem. J., 473, 2671–2685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Steger, M., Diez, F., Dhekne, H.S., Lis, P., Nirujogi, R.S., Karayel, O., Tonelli, F., Martinez, T.N., Lorentzen, E., Pfeffer, S.R.et al. (2017) Systematic proteomic analysis of LRRK2-mediated Rab GTPase phosphorylation establishes a connection to ciliogenesis. Elife, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. West, A.B., Moore, D.J., Biskup, S., Bugayenko, A., Smith, W.W., Ross, C.A., Dawson, V.L. and Dawson, T.M. (2005) Parkinson’s disease-associated mutations in leucine-rich repeat kinase 2 augment kinase activity. Proc. Natl. Acad. Sci. U. S. A., 102, 16842–16847. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.