

Abstract
Angelman syndrome (AS) is a severe neurodevelopmental disorder caused by deletion (~75%) or mutation (~10%) of the ubiquitin E3 ligase A (UBE3A) gene, which encodes a HECT type E3 ubiquitin protein ligase. Although the critical substrates of UBE3A are unknown, previous studies have suggested a critical role of nuclear UBE3A in AS pathophysiology. Here, we investigated to what extent UBE3A missense mutations disrupt UBE3A subcellular localization as well as catalytic activity, stability and protein folding. Our functional screen of 31 UBE3A missense mutants revealed that UBE3A mislocalization is the predominant cause of UBE3A dysfunction, accounting for 55% of the UBE3A mutations tested. The second major cause (29%) is a loss of E3-ubiquitin ligase activity, as assessed in an Escherichia coli in vivo ubiquitination assay. Mutations affecting catalytic activity are found not only in the catalytic HECT domain, but also in the N-terminal half of UBE3A, suggesting an important contribution of this N-terminal region to its catalytic potential. Together, our results show that loss of nuclear UBE3A E3 ligase activity is the predominant cause of UBE3A-linked AS. Moreover, our functional analysis screen allows rapid assessment of the pathogenicity of novel UBE3A missense variants which will be of particular importance when treatments for AS become available.
Introduction
Angelman syndrome (AS, OMIM #105830) is a severe neurodevelopmental disorder characterized by intellectual disability, treatment-refractory epilepsy, motor dysfunction and absence of speech (1). AS results from loss of function of the maternally inherited ubiquitin E3 ligase A (UBE3A) gene on chromosome 15q11.2–q13 (2,3). This may be caused by a number of mechanisms; deletion of the maternally inherited UBE3A gene (~75%), paternal uniparental disomy of chromosome 15 (1–2%), imprinting defects (1–3%) or by UBE3A mutation (~10%) (1). The UBE3A gene encodes a HECT (homologous to E6AP COOH terminus) type ubiquitin protein ligase that ubiquitinates substrate proteins, generally leading to their degradation by the 26S proteasome (4).
Although it is well established that loss of a functional UBE3A protein causes AS, our understanding of how UBE3A affects neuronal function is still very limited. Previous studies have shown that UBE3A is expressed throughout the neuron, such as axons, dendrites and the nucleus (5–9). Given that the loss of UBE3A affects synaptic function, most efforts have focused on its role in the synapse (10,11). However, there is also evidence for a role of UBE3A as a transcriptional co-activator (12,13), and we and others have recently reported that UBE3A staining is highly enriched in the nucleus of mouse and human neurons (6–8,14,15). The subcellular distribution of UBE3A in mouse and human is dictated by the expression of UBE3A isoforms that have distinct localizations. Consistent with a high overall UBE3A immunoreactivity in the nucleus, the most abundantly expressed short UBE3A isoforms in mouse and human [the homologous mouse(m) Isoform 3 and human(h) isoform 1], which constitute ~80% of total UBE3A protein in the brain, are mainly nuclear (14,15). Mice express a second, less abundant (~20% of total UBE3A protein), long isoform (mouse isoform 2) that is localized to the cytosol (14), but in human this isoform (hUBE3A-Iso3) is nuclear due to a mutation in the N-terminus (15). In human, a small pool of cytosolic UBE3A is maintained by the unique, low expressed, isoform hUBE3A-Iso2. Nuclear targeting of UBE3A requires its association with the proteasomal subunit PSMD4 (also known as S5A or Rpn10) (14,16–18). Importantly, selective loss of the nuclear localized mUBE3A-Iso3 in mice, but not the cytosolic mUBE3A-Iso2, results in behavioral and electrophysiological deficits resembling AS mice (14,19). Similarly, a mutation that results in the loss of the most abundantly expressed nuclear isoform in human (hUBE3A-Iso1) also results in an AS phenotype, albeit milder compared with maternal 15q11-13 deletion carriers (20). These data point to a critical role of nuclear UBE3A in AS pathophysiology.
AS is often caused by a deletion of the entire UBE3A gene, but ~ 5% of the patients possess non-truncating missense mutations that result in small changes in the amino acid sequence of the UBE3A protein (21–23). However, most missense variants identified are assessed as a VUS (variant of unknown significance) and determining if, and how, these missense mutations contribute to AS pathogenesis remains a challenge as it often requires time-consuming and labor-intensive biochemical experiments. Consequently, only a limited number of UBE3A missense mutations have been studied in detail (14,18,21,22,24).
Here, we study the functional consequence of 31 different AS-linked UBE3A mutations. Given the importance of nuclear UBE3A for AS-linked pathophysiology, we included a previously established simple transfection assay with GFP (green fluorescent protein)-tagged UBE3A (14) as a primary screen to rapidly assess if AS-linked missense mutations abrogate nuclear localization of UBE3A. By combining the localization assay with robust assays to experimentally address protein stability and catalytic activity of UBE3A missense mutants we provide the most comprehensive functional characterization so far, by studying 28 previously reported and 3 novel UBE3A missense mutations. We show that more than half of these missense mutations result in loss of nuclear UBE3A, emphasizing that disruption of UBE3A nuclear localization is a major cause of AS in patients with UBE3A missense mutations.
Results
The majority of AS-linked missense mutations cause loss of nuclear UBE3A
Currently >100 different missense mutations in the UBE3A gene have been reported in patients with AS in public databases (e.g. ClinVar and LOVD), our own database as well as previous publications [(2,21–31); Fig. 1A]. We selected 31 mutations, 22 of which have not been studied previously, for in depth functional analysis. The selected UBE3A missense mutations are distributed over the entire length of the protein, covering all the previously identified mutational hotspots (22), with 14 mutations in the N-terminal half of UBE3A and 17 mutations in the catalytic HECT domain (Fig. 1A). Although most of the selected UBE3A mutations are associated with a clinical AS phenotype, for 13 of the variants the clinical information is lacking. The human UBE3A-isoform 1 (hUBE3A-Iso1) and mouse UBE3A-isoform 3 (mUBE3A-Iso3) both localize to the nucleus (15) and are highly homologous (96% sequence identity; 97.3% sequence similarity) with most of the amino acid differences found in regions outside the mutational hotspots. Notably, the few amino acid changes that are located within the mutational hotspot regions are predominantly conservative changes (i.e. Ser<>Thr, Leu<>Ile Asp<>Glu) and of all the reported AS-linked missense mutations (>100) there is only one mutation that changes a residue that is not conserved between mouse and human (Fig. 1A; Supplementary Material, Fig. S1). Given the importance of proper nuclear targeting of UBE3A for AS-linked pathophysiology (14), and to avoid possible misinterpretation due to heterologous expression of hUBE3A in mouse neurons, we studied the effect of the hUBE3A mutations in the mouse nuclear mUBE3A-Iso3 isoform. We first assessed the subcellular localization of all mutants by expressing C-terminally GFP-tagged constructs in primary mouse neurons (Fig. 1B). Previously we have shown that tagging of UBE3A at its C-terminus with GFP does not affect the localization of the protein (14). Ectopic expression of these constructs indicated that more than half of the analyzed mutations (17 of 31) showed mislocalization of UBE3A, as evidenced by partial to complete loss of nuclear UBE3A and the appearance of the UBE3A signal in the cytosol (Fig. 2). Mutations affecting UBE3A localization are mainly found in the N-terminal half of the protein (10 of the 14 tested mutations in this region, including the previously analyzed amino-terminal Zn-finger of Ube3a ligase (AZUL) mutants p.Gly20Val and p.Cys21Tyr), but also in the C-terminal HECT domain (7 of the 17 tested HECT mutations, including the previously analyzed p.Gly593Arg (mUBE3A-Iso3 p.Gly590Arg) (Table 1). Given the important role of nuclear UBE3A for normal neurodevelopment in mice and human (14,20) our results strongly suggest that loss of nuclear UBE3A is a major factor underlying AS pathophysiology in patients carrying UBE3A missense mutations.
Figure 1.
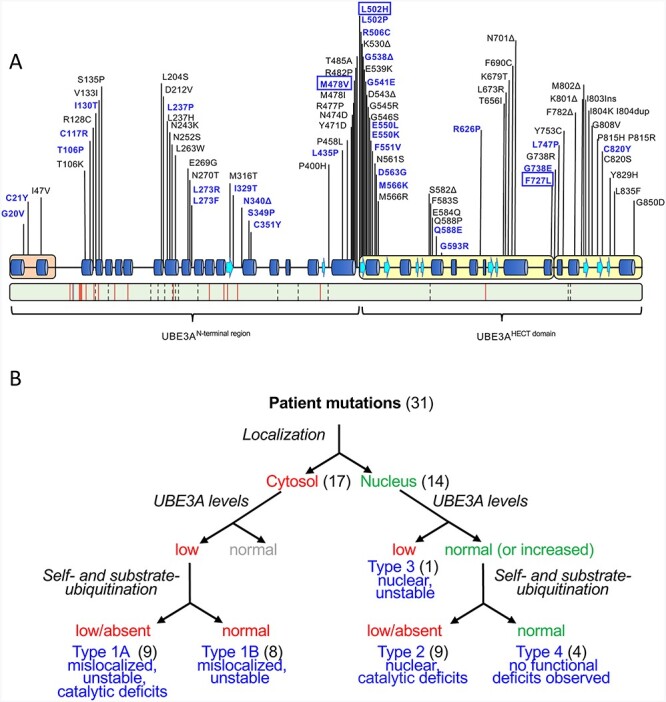
Functional characterization of UBE3A missense mutations linked to AS. (A) Top: schematic representation of human UBE3A-Isoform 1 (accession no.: NP_570853.1) depicting secondary structure elements, functional domains [amino-terminal Zn-finger of Ube3a ligase (AZUL, orange) and C-terminal HECT domain (yellow)] and the location of AS-linked missense mutations. Missense mutations analyzed in this study are shown in blue and include three novel mutations (boxed). Bottom: schematic representation of mouse UBE3A-Isoform 3 (accession no.: NP_001029134.1). Green color depicts sequence identity with human UBE3A-Isoform 1 (96%). The location of non-conservative and conservative amino acid changes are indicated by red lines and by dashed lines, respectively (see Supplementary Material, Fig. S1 for a detailed sequence alignment). (B) Experimental approach used to assess how missense mutations disrupt UBE3A function. Mutations were introduced in nuclear UBE3A C-terminally tagged with GFP and transfected into mouse primary neurons to determine their subcellular localization. More than half of the mutants appeared to be mislocalized to the cytosol (Type 1). All mutants (in the context of UBE3A without any tag) were also expressed in HEK293T cells to assess their stability. All cytosolic UBE3A mutants appeared unstable while the nuclear localized mutants exhibited normal (or increased) UBE3A levels with the exception of one nuclear mutant that showed low protein levels (Type 3). Lastly, catalytic activity was determined for all mutants. Catalytic deficits were observed in both cytosolic UBE3A mutants (Type1A) and nuclear UBE3A mutants (Type 2). Four mutants did not show any functional deficits in these three assays and were classified Type 4, pathogenicity uncertain.
Figure 2.
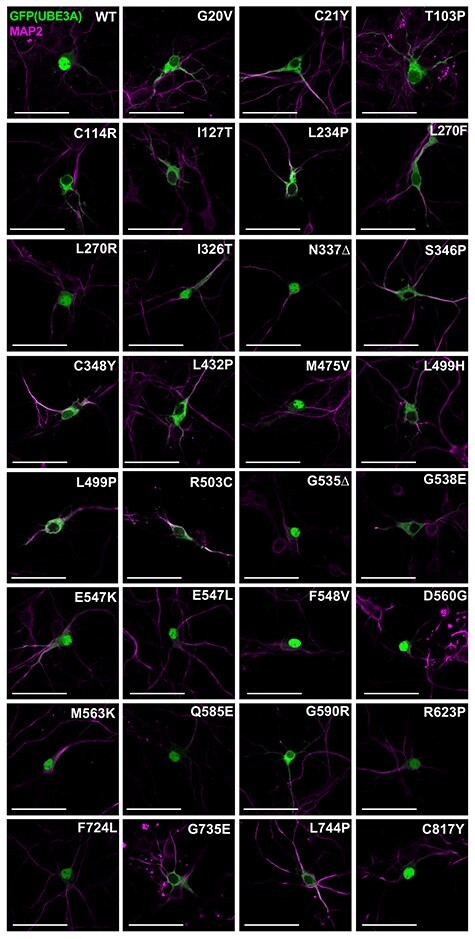
The majority of tested UBE3A missense mutants mislocalize to the cytosol. Patient-linked missense mutations were introduced at the analogous position in mUBE3A-Iso3 tagged C-terminally with GFP. Neurons were transfected at DIV7 with the indicated UBE3A-GFP constructs and were fixed and stained at DIV10. Neurons were stained for MAP2 (pink) and GFP (UBE3A) was visualized by direct fluorescence (green). Scale bar 50 mm.
Table 1.
Functional characteristics and classification of patient-linked UBE3A missense mutants
| Levels | Auto-ubiquitination | RING1B ubiquitination | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| AA position (h-Iso1) | UBE3A mutant | Locali-zation | PSMD4 interaction | % of WT ± SE | P value | % of WT ± SE | P value | % of WT ± SE | P value | N–C interaction | Type | Reference |
| hIso1A; mIso3 (WT) | NB | Yes | 100 | 100 ± 11.1 | 100 ± 11.9 | Yes | ||||||
| 20 | hG20V; mG20V | C | YesC | 12 ± 4.4 | 0.003 | 159 ± 17.4 | 0.022 | 153 ± 6.6 | 0.028 | Yes | 1a | (26) |
| 21 | hC21Y; mC21Y | C | YesC | 41 ± 10.6 | 0.031 | 63 ± 3.1 | 0.090 | 218 ± 8.4 | <0.001 | ND | 1a | (2,21,27) |
| 106 | hT106P; mT103P | C | Yes | 19 ± 10.6 | 0.017 | 78 ± 10.1 | 0.301 | 167 ± 27.8 | 0.027 | No | 1a | (25) |
| 117 | hC117R; mC114R | C | Yes | 20 ± 12.8 | 0.025 | 53 ± 2.1 | 0.041 | 133 ± 12.7 | 0.151 | No | 1b | (23) |
| 130 | hI130T; mI127T | C | YesC | 38 ± 4.0 | 0.004 | 48 ± 2.0 | 0.027 | 132 ± 8.6 | 0.155 | No | 1b | (25) |
| 237 | hL237P; mL234P | C | Yes | 20 ± 5.2 | 0.004 | 129 ± 14.4 | 0.199 | 139 ± 9.8 | 0.091 | ND | 1a | (23) |
| 273 | hL273F; mL270F | C | YesC | 25 ± 0.6 | <0.001 | 85 ± 20.8 | 0.509 | 156 ± 9.9 | 0.027 | ND | 1a | LOVD |
| 273 | hL273R; mL270R | N | ND | 105 ± 9.5 | 0.630 | 85 ± 4.0 | 0.471 | 111 ± 9.5 | 0.607 | ND | 4 | LOVD |
| 329 | hI329T; mI326T | N | YesC | 152 ± 14.3 | 0.070 | 52 ± 3.1 | 0.035 | 182 ± 15.2 | 0.004 | ND | 2 | (28) |
| 340 | hN340Δ; mN337Δ | N | ND | 104 ± 1.1 | 0.066 | 94 ± 10.0 | 0.765 | 100 ± 12.1 | 0.985 | ND | 4D | (24) |
| 349 | hS349P; mS346P | C | Yes | 49 ± 0.8 | <0.001 | 68 ± 16.3 | 0.169 | 43 ± 9.1 | 0.023 | ND | 1b | (29) |
| 351 | hC351Y; mC348Y | C | Yes | 71 ± 5.2 | 0.031 | 35 ± 1.6 | 0.008 | 16 ± 2.9 | 0.003 | ND | 1b | LOVD |
| 435 | hL435P; mL432P | C | Yes | 53 ± 11.6 | 0.057 | 17 ± 0.7 | 0.002 | 22 ± 2.1 | 0.004 | ND | 1b | (23) |
| 478 | hM478V; mM475V | N | ND | 208 ± 13.4 | 0.015 | 14 ± 0.8 | 0.002 | 8 ± 0.5 | 0.001 | ND | 2 | This study |
| 502 | hL502H; mL499H | C | Yes | 9 ± 1.5 | <0.001 | 79 ± 4.1 | 0.314 | 66 ± 4.9 | 0.124 | Yes | 1a | This study |
| 502 | hL502P; mL499P | C | Yes | 15.7 ± 4.5 | 0.003 | 60 ± 10.1 | 0.078 | 57 ± 3.1 | 0.061 | Yes | 1a | LOVD; (21) |
| 506 | hR506C; mR503C | C | Yes | 68 ± 2.5 | 0.006 | 60 ± 4.3 | 0.071 | 53 ± 5.2 | 0.045 | ND | 1b | (49) |
| 535 | hG538Δ; mG535Δ | N | ND | 154 ± 3.3 | 0.004 | 26 ± 5.5 | 0.004 | 25 ± 5.4 | 0.005 | ND | 2 | LOVD; (31) |
| 541 | hG541E; mG538E | C | ND | 45 ± 11.4 | 0.041 | 109 ± 3.2 | 0.671 | 135 ± 14.6 | 0.139 | ND | 1a | LOVD |
| 550 | hE550K; mE547K | N | YesC | 74 ± 16.8 | 0.258 | 5 ± 0.1 | <0.001 | 3 ± 0.4 | 0.001 | ND | 2 | LOVD |
| 550 | hE550L; mE547L | N | ND | 143 ± 10.4 | 0.053 | 13 ± 3.0 | 0.001 | 0.4 ± 2.9 | <0.001 | Yes | 2 | (21,41) |
| 551 | hF551V; mF548V | N | ND | 152 ± 30.8 | 0.232 | 52 ± 0.4 | 0.035 | 66 ± 2.8 | 0.127 | Yes | 2 | LOVD |
| 563 | hD563G; mD560G | N | YesC | 189 ± 26.3 | 0.077 | 72 ± 8.4 | 0.204 | 115 ± 11.0 | 0.495 | Yes | 4 | (30) |
| 566 | hM566K; mM563K | N | YesC | 136 ± 36.1 | 0.429 | 26 ± 3.2 | 0.004 | 45 ± 5.4 | 0.024 | Yes | 2 | (23) |
| 588 | hQ588E; mQ585E | N | ND | 49 ± 2.3 | 0.002 | 96 ± 5.6 | 0.839 | 206 ± 19.5 | 0.001 | ND | 3 | (23) |
| 593 | hG593R; mG590R | C | ReducedC | 40 ± 6.3 | 0.011 | 53 ± 5.6 | 0.041 | 123 ± 8.1 | 0.293 | ND | 1b | (14) |
| 626 | hR626P; mR623P | N | ND | 100 ± 13.5 | 0.976 | 68 ± 10.6 | 0.153 | 91 ± 24.0 | 0.732 | ND | 4 | ClinVar |
| 727 | hF727L; mF724L | N | ND | 85 ± 19.2 | 0.517 | 47 ± 3.7 | 0.024 | 82 ± 9.6 | 0.404 | ND | 2 | This study |
| 738 | hG738E; mG735E | C | Yes | 53 ± 11 | 0.052 | 22 ± 1.1 | 0.003 | 85 ± 3.5 | 0.486 | Reduced | 1b | LOVD |
| 747 | hL747P; mL744P | C | Yes | 26 ± 4.5 | 0.004 | 34 ± 6.1 | 0.008 | 65 ± 5.0 | 0.118 | ND | 1b | LOVD |
| 820 | hC820Y; mC817Y | N | ND | 117 ± 35.2 | 0.675 | 2 ± 0.8 | <0.001 | 0.7 ± 0.3 | <0.001 | ND | 2 | (41) |
AProtein ID: hIso1, NP_570853.1; mIso3, NP_001029134.1.
BN, nuclear; C, cytosolic
CTested in Avagliano Trezza et al., 2019.
DPathogenicity proven, phenotype markedly milder than classical AS; ND, not determined.
Missense mutations in both the N-terminal region and HECT domain of UBE3A affect catalytic activity
UBE3A mutants may also be affected in their catalytic activity as has been previously shown for a few AS-linked missense mutations in UBE3A [(21,22); Supplementary Material, Table S1]. To assess the catalytic activity of all variants we made use of a previously established bacterial ubiquitination system in which we reconstituted the components of the ubiquitination cascade: E1, E2 (UbcH5c), ubiquitin and UBE3A (E3; wild type or variants). This bacterial ubiquitination assay has been extensively validated in our laboratory (24,32) as well as in other laboratories (33,34), is very robust and shows a high level of specificity. To determine the catalytic activity of UBE3A (and mutants) we assessed both the ability to ubiquitinate itself (called auto-ubiquitination) and its activity towards an established target, RING1B (35,36). Wild type UBE3A exhibits extensive auto- and substrate (RING1B) ubiquitination in this assay while the UBE3A mutant in which the catalytic cysteine is replaced by a tyrosine (p.Cys820Tyr, mUBE3A-iso3 p.Cys817Tyr) shows no auto- or substrate ubiquitination, demonstrating the validity of the assay (Fig. 3A and B). In addition to the variant in which the catalytic cysteine residue is mutated, 10 other mutants in the HECT domain, including the previously reported p.Glu550Leu (mUBE3A-iso3 p.Glu547Leu), showed deficits in catalytic activity as evidenced by either reduced auto-ubiquitination or substrate ubiquitination or both (Fig. 3C,D and Table 1; see Supplementary Material, Figs S2 and S3 for full blots). Depending on the variant, the levels of residual catalytic activity compared with wild type UBE3A varied from 53 to 2% for auto-ubiquitination and from 53 to 0.7% for RING1B ubiquitination. Notably, we identified 7 variants in the N-terminal half of UBE3A that showed deficits in catalytic activity, emphasizing that the region outside the HECT domain of UBE3A contributes to its catalytic potential (21,37,38) (Fig. 3 and Table 1). In addition to the 9 nuclear UBE3A variants with reduced catalytic activity 9 out of the 17 mislocalized UBE3A variants were found to be affected in their catalytic activity as well (Table 1). Interestingly, 6 mutants showed a significant increase in target ubiquitination. Of these only the p.G20V also showed increased auto-ubiquitination while auto-ubiquitination activity of the other mutants was largely unaffected (4 mutants) or reduced (1 mutant). Together, these data indicate that the majority of the AS-linked missense mutations in UBE3A display a mislocalization phenotype or show deficits in catalytic activity.
Figure 3.
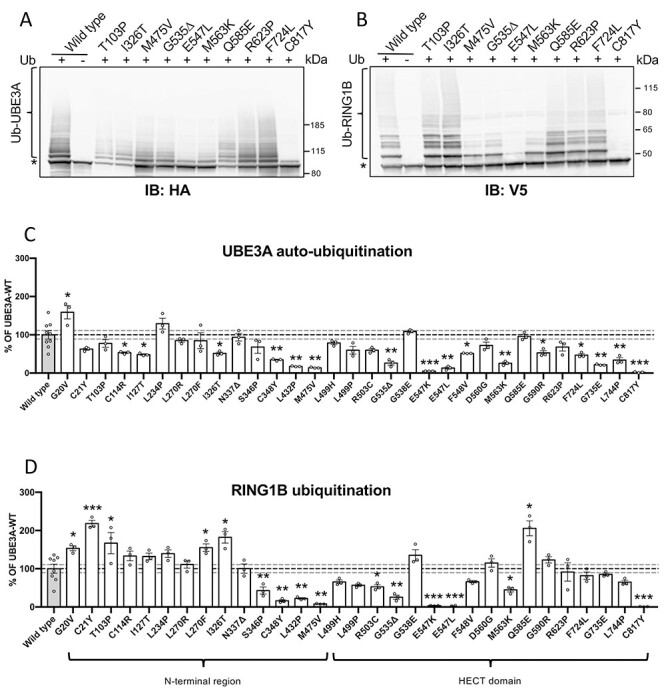
AS-associated missense mutations in both the N-terminal region and HECT domain of UBE3A affect catalytic activity. (A and B) Auto-ubiquitination and target ubiquitination of a representative set of UBE3A mutants and quantification of all mutants (C and D). Escherichia coli cells expressing E1, UbcH5 (E2) and with (+) or without (−) ubiquitin were transfected with a plasmid expressing mouse HA-tagged mUBE3A-iso3 (wild-type or the indicated mutant) and V5-RING1B-I53S, a catalytically inactive RING1B mutant that is unable to ubiquitinate itself. Cell lysates were analyzed by SDS-PAGE and immunoblotting using HA antibody to visualize UBE3A (A) and V5 antibody to visualize RING1B (B). Slower migrating, ubiquitinated UBE3A and RING1B species are indicated by a bracket and the unmodified protein band by an asterisk. Quantification of UBE3A auto-ubiquitination (WT, n = 9; mutants, n = 3) (C) and RING1B ubiquitination (WT, n = 8; mutants, n = 3) (D). Values are shown as the mean percent ± SEM of WT UBE3A. Unpaired Student’s t-test (two-tailed). Significant effects are indicated as *P < 0.05, **P < 0.005 and ***P < 0.001. Dashed grey lines depict 95% confidence interval. See Material and Methods section for detailed quantification and statistical methods.
Mislocalized UBE3A mutants show reduced protein stability independent of their catalytic activity
We were intrigued by the finding that the majority of the tested AS-linked missense mutations caused the mutant UBE3A protein to mislocalize to the cytoplasm. Previous work has shown that nuclear targeting of UBE3A requires association with the proteasomal subunit PSMD4 [also known as Rpn10 or S5a; (14,18)]. In the Avagliano Trezza study, the AS-linked missense mutation p.Gly593Arg (mUBE3A-iso3 p.Gly590Arg), was shown to have strongly reduced interaction with PSMD4 and consequently localized to the cytosol. To investigate if reduced PSMD4 interaction is a major cause of UBE3A mislocalization we assessed the yeast two-hybrid interaction with PSMD4 of the mistargeted UBE3A mutants that were not previously tested in the Avagliano Trezza study (Table 1 and Supplementary Material, Fig. S4). With the exception of the previously reported p. Gly593Arg variant (14), none of the cytosolic UBE3A variants showed a reduction in the strength of interaction with PSMD4 as measured in the yeast 2-hybrid system.
We hypothesized that UBE3A mislocalization could be the result of protein misfolding. Since protein misfolding is typically associated with reduced intrinsic protein stability leading to reduced abundance, we assessed the protein levels of all UBE3A variants. To exclude any possible effect of an epitope tag (either N- or C-terminal) on protein stability or localization, we ectopically expressed untagged UBE3A variants in HEK293T cells, and assessed protein levels by western blotting (Fig. 4, Table 1 and Supplementary Material, Figs S5 and S6). Notably, we found that all mislocalized UBE3A variants exhibited moderately to strongly reduced UBE3A protein levels (significantly reduced in 15 out of 17 mislocalized mutants). To investigate whether these decreased protein levels reflect decreased stability or reduced synthesis, we performed cycloheximide chase experiments. Consistent with previous reports (21,24) the levels of wild type UBE3A did not change over an 8 h. cycloheximide chase period, while protein levels of the UBE3A mutants either moderately or strongly reduced during the chase, suggesting that these mutant proteins are instable and targeted for degradation (Supplementary Material, Fig. S7). Since this includes UBE3A mutants that display strongly reduced catalytic activity (Fig. 3 and Table 1), the degradation of these mutants is unlikely to be induced by auto-ubiquitination.
Figure 4.
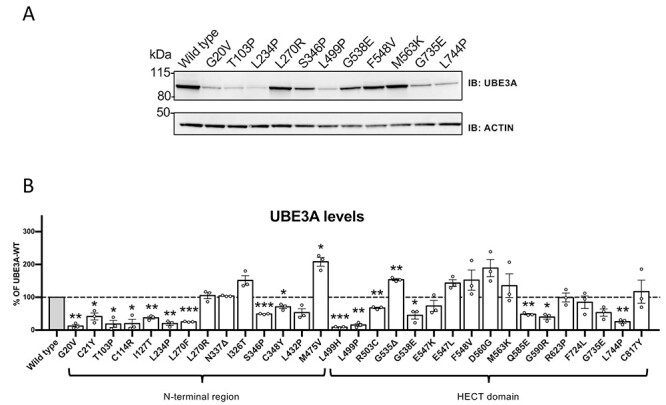
Protein levels of UBE3A missense mutants in HEK293T cells. (A) Protein levels of a representative set of UBE3A mutants and quantification of all mutants (B). HEK293T cells were transiently transfected with untagged UBE3A constructs and protein levels were visualized by anti-UBE3A western blotting using actin as a loading control (A). Quantification of protein levels of all UBE3A variants (n = 3) (B). All data represent mean ± SEM and are normalized against actin. Wild-type (WT) UBE3A levels were set to 100%. One-sample Student’s t-test. Significant effects are indicated as *P < 0.05, **P < 0.005 and ***P < 0.001. See Material and Methods section for detailed quantification and statistical methods.
In contrast to the mislocalized UBE3A mutants, protein levels of 13 nuclear localized UBE3A mutants were either similar to that of wild type UBE3A or even increased. Only the p.Gln588Glu (mUBE3A-iso3 p.Gln585Glu) mutant showed significantly decreased protein levels. With the exception of the p.Asp563Gly mutant (mUBE3A p.Asp560Gly), all the nuclear UBE3A mutants that showed increased UBE3A levels, also showed reduced catalytically activity.
The high correlation between protein instability and mislocalization could suggest that the majority of mislocalized UBE3A variants may not be properly folded. In recent work on the folding of the full-length UBE3A, it was shown that there are intramolecular contacts between the N-terminal region and the HECT domain of UBE3A (39). We confirmed with yeast two-hybrid (Y2H) analysis the physical interaction between the N-terminal half (amino acids 1–491) and the HECT domain (amino acids 492–849) [hereafter referred to as the N–C interaction; Supplementary Material, Fig. S8]. Notably, no Y2H interaction was observed when expressing 2 full-length UBE3A constructs, nor did the N-terminal half interact with full-length UBE3A in our Y2H assay. This suggests that the N–C interaction is an intramolecular interaction that is favored over intermolecular interactions when expressing full-length UBE3A. Deletion analysis revealed that the first 98 amino acids of UBE3A (mUBE3A-Iso3), which includes the AZUL domain, are not required for the N–C interaction. Further N-terminal deletion abolished the N–C interaction as did C-terminal deletion of the fragment. We thus used Y2H interaction assays between the isolated N-terminal half and the isolated HECT domain of UBE3A as a read-out for the ability of UBE3A mutants to form such an N–C interaction. Mutants in the N-terminal UBE3A half were tested against the wild type HECT domain and, conversely, mutants in the UBE3A HECT domain were tested against the wild type N-terminal half (Table 1 and Supplementary Material, Fig. S9). This analysis revealed that, with the exception of the p.Gly20Val mutant in the AZUL domain, all tested UBE3A mutations in the N-terminal half that have a mislocalization phenotype abrogate the N–C interaction. Mislocalized HECT domain mutants were unaffected in their N–C interaction with the exception of the p.Gly738Glu (mUBE3A p. Gly735Glu) mutant that showed reduced N–C interaction. Importantly, none of the tested nuclear UBE3A mutants showed a loss of the N–C interaction. Collectively, these results suggest that loss of intramolecular contacts between the N-terminal half and the HECT domain contributes to mislocalization and instability of some UBE3A mutants.
Discussion
In this study we have functionally annotated 31 AS-linked missense mutations, spread throughout the entire length of the protein and covering all the previously identified mutational hotspots, based on their subcellular localization, stability and catalytic activity. Our functional analyses revealed that pathogenicity of UBE3A missense mutations is predominantly caused by either mislocalization of UBE3A (Type 1, 55%) or loss of catalytic activity of nuclear localized UBE3A (Type 2, 29%), together accounting for 84% of the mutant proteins tested. These results emphasize that UBE3A AS pathophysiology is predominantly caused by loss of nuclear UBE3A E3 ligase activity (Fig. 5 and Table 1).
Figure 5.
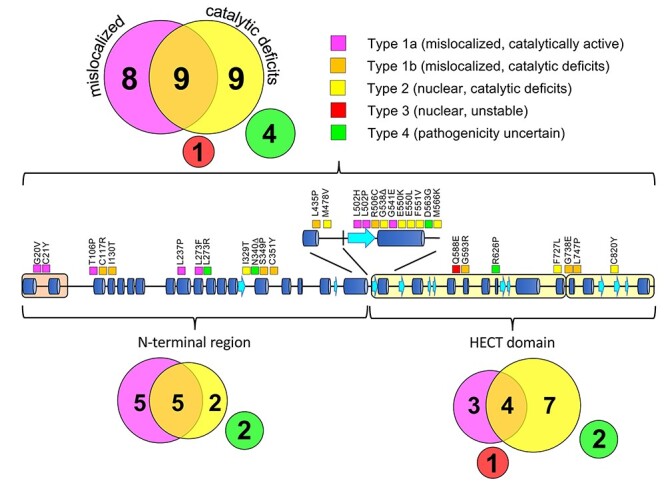
Classification and distribution of AS-linked UBE3A missense mutations. Schematic representation of human UBE3A-Isoform 1 depicting the location of the 31 AS-linked missense mutations analyzed in this study. Each mutation is color-coded indicating the mechanism by which a mutation disrupts UBE3A function:(i) mislocalization of UBE3A with normal catalytic activity (Type 1A, magenta) or reduced catalytic activity (Type 1B, orange), (ii) reduced catalytic activity of nuclear localized UBE3A (Type 2, yellow) or (iii) reduced stability of nuclear localized UBE3A (Type 3, red). UBE3A mutants that showed no functional deficits in our assays are classified as ‘pathogenicity uncertain’ (Type 4, green). The number of mutants in each class is indicated within the colored circles for the full length UBE3A protein (top) and for the UBE3A N-terminal region and HECT domain separately (bottom).
Our functional screen has two major advantages over previous assays. First, we implemented an Escherichia coli in vivo ubiquitination assay instead of an in vitro assay with purified enzymes or an assay that monitors (auto)-ubiquitination in cultured eukaryotic cells. The absence of a ubiquitin-proteasome system in E. coli ensures that (i) the observed ubiquitination in the E. coli assay is dependent on a single E3 ligase and (ii) ubiquitinated proteins (E3 ligase and substrate) are fully stable as E. coli lacks deubiquitinating enzymes and a proteasome. This also allowed us to assess the ubiquitination activity of UBE3A mutants that were unstable when expressed in HEK293T cells. In addition, the ubiquitination assay could be readily adapted to look at selected target ubiquitination (e.g. RING1B).
But probably the biggest advance of our functional screen is the introduction of subcellular localization as primary functional test, which can be readily performed in many laboratories. We showed that this assay is able to predict pathogenicity in ~50% of the mutants tested. Implementing subcellular localization as primary selection criterion, allowed us to re-classify some variants that were previously classified as mutants that are hyperactive and promote self-degradation (22). In that study different mechanisms of protein instability were described. This classification was based on a comparison of protein levels of the UBE3A mutants in the context of catalytically active UBE3A and ligase dead UBE3A (22). Our results show that the mutants that were categorized as intrinsically unstable (8 mutants) and hyperactive self-degraders (2 mutants) in the Yi et al. study, are all mislocalized to the cytosol (Supplementary Material, Table S1). Since we recently showed that nuclear UBE3A is essential for normal neurodevelopment in mice (14), we propose to reclassify all such mutants as Type 1 (mislocalized) mutants, regardless protein stability and E3 ligase activity levels. This classification has the added advantage that this measure is much easier to assess compared with the previous classification, which depends on a careful determination of both UBE3A levels as well as activity levels, to calculate the relative activity per amount of UBE3A present. Of note, the previous study used the cytosolic human isoform 2 for their analysis and, therefore, the effect of the mutations on the localization of UBE3A could not be observed in their study (22).
We have previously shown that the UBE3A–PSMD4 interaction is necessary for nuclear targeting of UBE3A (14). With the exception of the previously characterized p.Gly593Arg variant (14), all mislocalized UBE3A mutants show undisturbed PSMD4 interaction in the yeast 2-hybrid assay (Table 1). Hence, we further investigated the possible underlying mechanism of mislocalization of these mutants. Protein stability experiments showed that most of the mislocalized mutants are also unstable, suggesting that these mutations may affect proper folding of UBE3A. In support of this, we tested a representative set of mislocalized mutants and found that most mutants in the UBE3A N-terminal half, as well as the p.Gly738Glu mutant in the HECT domain, lost the intra-molecular interaction between the N-terminal half and the HECT domain (Table 1). These data suggest that improper folding of UBE3A could underly the mechanism of mislocalization and reduced stability of many of these variants. This observation is in line with our previous results showing that, in addition to the N-terminal Zn-binding AZUL domain, other regions in UBE3A contribute to nuclear localization of the protein (14). Since some of the mislocalized unstable proteins also have catalytic deficits, the reduced stability of these mutants is not strictly related to auto-ubiquitination. Hence, the exact mechanism by which disruption of intramolecular UBE3A folding affects nuclear localization and stability remains to be established.
Type 2 variants (intact nuclear targeting, catalytic deficits) form with 9 mutants (29%) the second largest group of mutants. In contrast to the Type 1 mutants, most of these mutants showed increased protein levels, suggesting increased stability. In general, these mutants showed a strong correlation between increased stability in HEK293T cells and reduced auto-ubiquitination activity in the bacterial ubiquitination system and (6 of the 9 Type 2 mutants). These results are in line with previous observations (22,35) and provide compelling support for the notion that UBE3A levels in vivo are (in part) regulated by UBE3A auto-ubiquitination and subsequent proteasomal degradation (40).
The HECT domain of UBE3A (and of other members of the HECT E3 ligase family) is generally considered as the catalytic unit as this domain contains the catalytic cysteine residue (in the C-lobe of the HECT domain) and the binding site for the E2 enzyme (in the N-lobe of the HECT domain) (41–44). Consistent with this, variants with catalytic deficits (Type 1B and Type 2) were mainly found in the HECT domain (11 mutants). It is notable, however, that we also identified 7 variants outside the HECT domain that affected the catalytic activity of UBE3A (Fig. 5). These data emphasize that the region N-terminal of the HECT domain is probably not only involved in target binding but also contributes to the catalytic potential of UBE3A. In fact, these observations are in line with previous work of Sailer et al. (39) who showed that upon binding of the viral activator E6 UBE3A undergoes structural rearrangements that bring the N-terminal half and the HECT domain into closer proximity and support the biochemical observations that the N-terminal half of UBE3A impacts the catalytic activity of UBE3A (21,37,45). Notably, while our analysis indicates that most mislocalized variants are unstable when expressed in mammalian cells, the stability and expression levels of these mutants in E. coli cells were indistinguishable from wild type UBE3A. Hence, the ubiquitination assays in E. coli cells allowed us to measure E3 ligase activity without confounding issues of protein stability, which greatly facilitates the interpretation and classification of these mutants. This is best illustrated by the two UBE3A variants in the N-terminal domain that were previously shown to be unstable in cells as a consequence of hyperactive auto-ubiquitination, [p.Thr106Pro and p.Ile130Thr; (22)]. These mutants did not show increased auto-ubiquitination in the bacterial ubiquitination assay. Hence, our results indicate that hyperactive auto-ubiquitination is not an intrinsic property of these UBE3A mutants and suggest that in eukaryotic cells additional factors (interacting proteins, posttranslational modifications) are involved in the regulation of auto-ubiquitination.
Notably, 6 mutants (p.Gly20Val, p.Cys21Tyr, p.Thr106Pro, p.Leu273Phe, p.Ile329Thr and p.Gln588Glu) showed significantly increased target (RING1B) ubiquitination. Four of these mutants (p.Gly20Val, p.Cys21Tyr, p.Thr106Pro and p.Leu273Phe) are mislocalized to the cytosol. We therefore think that it is unlikely that the increased target ubiquitination activity is the primary cause for the pathogenicity of these mutants, and therefore we propose to classify these mutants as Type 1 mutants. The two nuclear mutants that show increased target RING1B ubiquitination are quite puzzling. The p.Ile329Thr mutant was shown to be nuclear but despite its increased target ubiquitination it showed significantly reduced auto-ubiquitination activity. We therefore propose to classify this mutant as Type 2 mutant (intact targeting, catalytic deficits). The p.Gln588Glu is notable since the protein is localized in the nucleus but it is the only nuclear mutant that shows significantly decreased protein levels. The decreased levels of the p.Gln588Glu mutant are not accompanied with increased auto-ubiquitination, but target ubiquitination appears to be significantly increased. It is difficult to predict if the increased target ubiquitination can compensate for the reduced protein levels, since we do not know the critical UBE3A targets that cause AS. This mutation was reported in Sadikovic et al. (23) without further genetic or clinical information. It will be interesting to know what the clinical phenotype is of the patient carrying such a hyperactive UBE3A mutation and whether it is different from patients with classical AS. We have classified this mutation as a distinct class: a Type 3 mutant that is nuclear but unstable.
Four UBE3A mutants were assigned to the Type 4 class: pathogenicity uncertain. We would like to emphasize that the absence of a phenotype in our functional analyses does not necessarily imply that the variant is benign. We have previously reported that the UBE3A variant hUBE3A p.Asn340del did not show any deficits in our functional assays, but the clinical data of the eight individuals carrying this variant clearly indicated that the p.Asn340del variant caused (a milder form of) intellectual disability and neurodevelopmental delay untypical for AS (24). Similarly, an individual with the hUBE3A p.Asp563Gly mutation, which was inherited from the proband’s grandfather through his asymptomatic mother, presented with relatively mild clinical phenotypes with respect to development and speech and showed no microcephaly or seizures (30). The fact that we were unable to detect significant changes in our assays in these two cases, could mean that either subtle changes in localization or activity may not be detected in our assays or that some mutations affect an, as of yet unexplored, function of UBE3A that goes undetected in our assays. The significance of the two remaining Type 4 variants (hUBE3A p.Leu273Arg; and p. Arg626Pro) is unclear. These variants were obtained from a public database (LOVD and ClinVar respectively, but the p.Leu273Arg has recently been removed from LOVD). Since there is no clinical data available for these individuals, we should interpret these variants with care, as it is possible that these variants are in fact benign. Notably, while the UBE3A p.Leu273Arg variant was classified as Type 4, the variant in which the same residue (Leu273) is changed to a Phenylalanine (hUBE3A p.Leu273Phe) showed a strong mislocalization and instability phenotype (Type 1). These observations underscore the importance of rigorous functional analysis of each UBE3A variant even if the change is affecting the same residue that has previously been identified as being pathogenic.
While it has been challenging to understand how mutations in the N-terminal domain of UBE3A affect catalytic activity, the available X-ray structure of the UBE3A HECT domain in complex with its cognate E2 UbcH7 has provided insights into the role of the HECT domain in catalysis (41,42). The UBE3A HECT domain consists of two flexibly tethered lobes (the N- and C-lobes), the interface of which forms the catalytic cleft. On the N-lobe portion of the cleft surface three conserved residues, Glu539, Arg506 and Glu550, are involved in a series of tight salt bridges situated close to the active site cysteine (41). Consistent with an important role of these residues in catalysis, AS-associated mutations in Arg506 (mUBE3A-Iso3 R503C) and Glu550 (mUBE3A-Iso3 E547K and mUBE3A-Iso3 E547L) severely reduced UBE3A activity towards itself and the target RING1B. Notably, our results are in line with previous biochemical analysis that showed strongly reduced activity of the p.Glu550Leu variant towards the target HHR23A (21), further validating the E. coli ubiquitination assay. In silico modeling has suggested that deletion of p.Gly538 (mUBE3A-iso3 G535D), a de novo mutation that was found in a patient with AS, affects the salt bridges between Glu539-Arg506 and Arg506-Glu550, and was predicted to reduce catalytic activity (31). We now provide experimental evidence showing that this mutation indeed reduces UBE3A catalytic activity, supporting the notion that p.Gly538del is indeed the cause of AS in this patient. In addition, we identified five other AS-linked mutations in the HECT domain that severely reduced UBE3A catalytic activity. How these HECT domain mutations disrupt catalytic activity remains to be investigated.
In conclusion, our detailed analysis of a large set of AS-linked UBE3A missense mutations shows that either loss of nuclear UBE3A or loss of catalytic activity is the underlying cause of AS in most of these patients. These observations support previous work of our group and others showing that UBE3A has an essential role in the nucleus for which it requires its E3 ubiquitin ligase activity (6,7,14,15,46). Our work not only provides functional insights into known mutations, but also establishes robust assays that can be readily adapted for diagnosis of patients with novel UBE3A missense variants of unknown significance. We envision that a two-step functional analysis involving the localization of the UBE3A variant in the context of the, major, nuclear isoform (hUBE3A-Iso1) and its catalytic activity in the bacterial ubiquitination assay will allow assessment of the pathogenicity of the UBE3A missense variant. Early diagnosis and rapid assessment of the pathogenicity of UBE3A missense variants will be of particular importance when treatments for AS become available.
Materials and Methods
Cell lines
Mouse cortical, primary neurons used for localization studies were isolated from E16.5 male and female wild-type FVB/NHanHsd embryo brains. Cells were cultured on poly-d-lysine coated glass coverslips at 37°C in a 5% CO2 humidified incubator in Neurobasal™ medium (NBM; Thermoscientific, Massachusetts, US 2 110 304) supplemented with 1% penicillin and streptomycin, 1% Glutamax (Invitrogen) and 2% B27 (NBM+++).
HEK293T cells (ATCC) were cultured in DMEM GlutaMAX (Thermo Scientific, 10569010) supplemented with 10% fetal calf serum and 1% penicillin and streptomycin.
Saccharomyces cerevisiae strains used for yeast two-hybrid analysis were Y187 (MATα, ura3-52, his3-200, ade2-101, trp1-901, leu2-3, 112, gal4Δ, met−, gal80Δ, URA3::GAL1UAS-GAL1TATA-lacZ; Takara Bio Europe, Saint-Germain-en-Laye, France) and Y2H Gold (MATa, ura3-52, his3-200, ade2-101, trp1-901, leu2-3, 112, gal4Δ, gal80Δ, met−, LYS2::GAL1UAS-Gal1TATA-His3,GAL2UAS-Gal2TATA-Ade2, URA3::MEL1UAS-Mel1TATA-AUR1-C MEL1; Takara Bio Europe, Saint-Germain-en-Laye, France). Cells were grown at 28°C in rich medium or in minimal glucose medium as previously described (47).
Plasmid construction and DNA manipulation
Construction of PSMD4 and mouse UBE3A-isoform 3 constructs has been described previously (14). For the generation of the UBE3A point mutations described in this study a standard Quickchange site-directed mutagenesis protocol was employed (Stratagene, La Jolla, US). Point mutations up to amino acid 399 were generated in pSB176, a pRSF-Duet backbone vector (Novagen, Madison, Wisconsin) containing mUBE3A-Iso3 amino acids 1–491 flanked by 5′ AscI site and a 3′ NotI site, and transferred to the relevant plasmids via introduced 5′ AscI site and the 3′ internal BamHI site of UBE3A. Point mutations from position 399 onwards were generated in pSB189, a pRSF-Duet (Novagen, Madison, Wisconsin) backbone vector containing mUBE3A-Iso3 amino acids 399–849 flanked by a 5′ BamHI site and a 3′ NotI site, and transferred to the relevant plasmids using the 5′ internal BamHI site of UBE3A and an introduced 3′ NotI site. For neuronal transfections the UBE3A-GFP constructs were cloned AscI-NotI into pYE969, expressing the gene of interest under a CAG promoter (48). For the stability assay in HEK293T cells untagged UBE3A variants were cloned AscI-NotI in the multiple cloning site of pYE969. For the bacterial ubiquitination assay (33), we designed novel compatible expression vectors that will be described in detail elsewhere (32). The system consists of a generic, polycistronic, plasmid expressing rabbit E1, E2 (UbcH5c) and ubiquitin (pMB313) or a plasmid in which the ubiquitin gene was deleted (pMB314), a plasmid expressing UBE3A (N-terminally tagged with the HA tag) and a plasmid expressing the substrate RING1B- I53S [catalytically inactive variant of RING1B (36) tagged at its N-terminus with the V5 tag; pMB433)]. For the bacterial ubiquitination assay E. coli cells were co-transformed with the three expression constructs. For yeast two-hybrid interaction with PSMD4, UBE3A variants were cloned AscI-NotI in pYR022 (47) and tested against full length PSMD4 cloned in pGADT7 (Takara Bio Europe, Saint-Germain-en-Laye, France) as described in (14). UBE3A deletion constructs were generated by PCR using mUBE3A-Iso3 as a template. For N-terminal fragments a 5′ NcoI site and a 3′ NotI site preceded by a stop codon were introduced, and the resulting fragment were cloned NcoI-NotI in the yeast two-hybrid bait vector pGBKT7 (Takara Bio Europe, Saint-Germain-en-Laye, France). A fragment harboring the UBE3A HECT domain (amino acids 492–849) was constructed by PCR amplification using primers which introduced a 5′ AscI site and a 3′ NotI site. The resulting fragment was cloned AscI-NotI in the yeast two-hybrid prey vector pYR035, which is a derivative of pGADT7 (Takara Bio Europe, Saint-Germain-en-Laye, France) containing a modified multiple cloning site. To assay the interaction between the N-terminus and the HECT domain of mutants (so called N–C interaction) points mutations were introduced either in the N-terminal fragment (amino acids 1–491) or in the HECT domain (amino acids 492–849). A complete list of the plasmids and primers employed in this study can be found in Supplementary Material, Tables S2 and S3. All constructed plasmids were sequence verified with Sanger sequencing (Macrogen Europe). Details of the construction of these plasmids and their full sequences are available upon request.
Transfection of neurons
On DIV7, mouse cortical primary neurons were transferred in NBM supplemented with glutamine (500 μM) and their conditioned medium was temporarily stored at 37°C, 5% CO2. 1.8 ug of the desired constructs were complexed with Lipofectamine 2000 (Invitrogen 11 668-019) and added to the neurons. After 1 h incubation at 37°C in 5% CO2, the medium was replaced by the conditioned medium previously stored. Neurons were fixed at the indicated time points.
Transfection of HEK293T cells
The day before transfection 0.2–0.4 × 106 cells were seeded. On the day of transfection between 1.5 μg (for 1 well of a 12 well plate) and 2 μg (for 1 well of a 6 well plate) of DNA were diluted into DMEM serum free medium (100 μl for 1 well of a 12 well plate and 200 μl for 1 well of a 6 well plate). Polyethylenimine (PEI, Polysciences Inc. 23 966, Bergstrasse, Germany) was added to the DNA mix in a ratio DNA (μg): PEI (μg) of 1:3 and the DNA mix was incubated for 15 min. Fresh DMEM (10% v/v fetal calf serum, 5%v/v P/S) was added to the HEK293T cells and the DNA mix was added drop-wise to each well. The medium was refreshed after 4–6 h to prevent PEI toxicity. Forty-eight hours post-transfection cells were harvested. Cell pellets were taken up in 1X Laemmli sample buffer (0.2 M Tris–HCl [pH 6.8], 1.5% sodium dodecyl sulfate [SDS], 10% glycerol, 1 mm EDTA, 0.004% bromophenol blue) containing protease inhibitor cocktail and cells were lysed by sonication (3 × 4 s, 5 mAmp). Protein lysate (20 μg) was analyzed by SDS-PAGE and western blotting.
Cycloheximide chase assay
Protein stability of UBE3A variants in HEK293T cells was analyzed using a cycloheximide chase assay as described previously (24).
Escherichia coli ubiquitination assay
Escherichia coli strain BL21-GOLD (DE3) [B F−ompT hsdS(rb− mb−) dcm+ Tetr gal λ(DE3) endA Hte] cells were co-transformed with constructs encoding for the requisite components of the ubiquitination cascade and selected on LB agar [1% (w/v) Bacto tryptone, 0.5% (w/v) Bacto yeast extract, 1% (w/v) NaCl, 1.5% agar] containing half the concentration of antibiotics as needed (ampicillin, 25 μg/ml; kanamycin, 15 μg/ml; streptomycin/spectinomycin, 25 μg/ml). Single transformants were re-streaked and grown at 37°C overnight in LB medium supplemented with 2% glucose, 50 mm Tris pH 8 and antibiotics as needed. The following morning culture was diluted to an OD600 of 0.2 and grown at room temperature to an OD600 of 0.7, at which point protein synthesis is induced by adding 0.5 mm Isopropyl β-D-1-thiogalactopyranoside (IPTG, Sigma-Aldrich I6758) while growing overnight at 16°C. Cells were collected in 20 OD600 pellets, and lysed in lysis buffer (50 mm Na-Pi buffer (pH 8.0), 300 mm NaCl, 5% glycerol, 100 μl PMSF (17.4 mg/ml), 100 μl Protease Inhibitor Cocktail (4.28 mg), 10 μl DNase (10 mg/ml), 10 μl RNase (10 mg/ml), 3.5 μl 2-Mercaptoethanol. After lysis cells were sonicated for 6 × 20 s at 5mAmp. Protein amount corresponding to 0.3 OD units per sample was used for western blot analysis.
Yeast two-hybrid assay
Yeast two-hybrid assays were performed as described previously (14) using S. cerevisiae strains Y187 and Y2H Gold. Briefly, Y187 cells transformed with the bait constructs and Y2H Gold cells transformed with the prey constructs, were selected on minimal agar plates lacking tryptophan or leucine, respectively. Following an O.N. (16 h) incubation in selective minimal medium (while shaking, 200 rpm), 2 × 106 cells of each transformed strain (1:1 ratio) were mated in 2X-YPAD for 24 h while shaking (50 rpm) at 28°C. The so obtained diploids were then grown on minimal glucose plates lacking both tryptophan and leucine for 48–72 h. Selected colonies were cultured for 16 h in minimal glucose medium lacking tryptophan and leucine, re-inoculated at an OD600 of 0.2 and grown till an OD600 of 1. Cells were then washed with sterile water and serially diluted so as to plate from 102 to 105 cells/spot on minimal glucose agar lacking tryptophan and leucine, and with or without histidine. Minimal glucose agar plates lacking tryptophan, leucine and histidine were used to determine the strength of interaction. Plates were incubated for 4 days at 28°C before analysis.
Western blot analysis
For western blot analysis ~20 μg of total protein (HEK293T lysates) or 0.3 OD units (bacterial lysates) per sample were separated by SDS-PAGE and transferred onto nitrocellulose membranes. Next, membranes were blocked in TBS (10 mm Tris–HCl [pH 8.0], 150 mm NaCl) containing 5% (w/v) powdered milk for 1 h at room temperature, washed 3 times in TBS-T (TBS with 0.1% Tween-20, Sigma St. Louis, US, P1379) and incubated at 4°C over-night, rotating end-over-end, with the primary antibody dissolved in TBS-T with 2% (w/v) milk solution. The day after, membranes were washed 3 times for 10 min with TBS-T and incubated with the secondary antibody dissolved in TBS-T with 2% (w/v) milk for 1 h. At the end of the incubation, membranes were washed 3 times for 10 min with TBS and analyzed by measuring enhanced chemiluminescence using an AI600 Chemiluminescent Imager. Primary antibodies used for western blotting were: Mouse monoclonal anti-UBE3A (Sigma, St. Louis, US SAB1404508 or Sigma, St. Louis, US E8655), mouse monoclonal anti-V5 horseradish peroxidase (HRP)-conjugated (Thermo Fisher Scientific R961-25), rat monoclonal anti-HA HRP-conjugated (Roche, Basel, Switzerland 12 013 819 001) and mouse monoclonal anti-ACTIN (Millipore, Massachusetts, US MAB1501R). Secondary antibodies used was: goat anti-mouse IgG IgA IgM (H+L) HRP-conjugated (Sigma, St. Louis, US SAB3701048).
Quantification of the western blots
Quantification of proteins levels was done using the ImageJ image-processing software (https://imagej.net/). First, a background correction was done using the background subtraction feature of ImageJ, with a rolling ball radius set to 50 pixels. Next, the intensity of the protein bands was determined by drawing an identical sized ROI (region of interest) around all bands (UBE3A and actin) on the blot. In order to compare UBE3A levels between each sample, the amount of UBE3A was first normalized to the actin signal within the sample, with the ratio UBE3A/actin with respect to the wild-type being set to 1.0.
Quantification of the ubiquitination assays was done similarly using ImageJ. The intensity of the ubiquitin-modified protein bands was determined by drawing a ROI around all bands that appear above the unmodified protein band. Background correction was done by subtracting the intensity in the same-sized ROI measured in the neighboring no-ubiquitin (-Ub) control lane. Similarly, unmodified protein band was quantified by drawing a ROI around the protein band at the anticipated molecular weight. The intensity ratio ubiquitin-modified: unmodified was used as measure for the activity of the different mutants tested. Wild type activity-ratios were set to 100%.
Fluorescence microscopy
The following fixation and staining protocols were applied to cortical/hippocampal neurons:
After medium aspiration, cells plated on glass coverslips were incubated with 0.5 ml of 4% PFA (paraformaldehyde)/4% Sucrose at room temperature for 10 min. Fixed cells were washed 3 times for 5 min in PBS at room temperature. Coverslips were incubated over-night at 4°C with Guinea pig polyclonal anti-MAP2 (1:500, Synaptic Systems, Göttingen, Germany, 188 004) dissolved in GDB solution (0.1% (w/v) gelatin, 0.3% (v/v) Triton X-100, 450 mm NaCl, 16 mm phosphate buffer pH 7.4). The day after coverslips were washed 3 times for 5 min in PBS at room temperature and incubated 1 h at room temperature with Anti-Guinea Pig Alexa 647 (1:200, Jackson ImmunoResearch Labs Europe Ltd, Cambridgeshire, UK, 706-605-148) dissolved in GDB solution. Coverslips were washed 2 times for 5 min in PBS and incubated in a PB (phosphate buffer, 0.2 M)-DAPI (4′,6-diamidino-2-phenylindole, 300 nM final concentration, Thermo Fisher Scientific D3571) solution for 10 min at room temperature if required. Coverslips were rinsed briefly in milliQ water and mounted on glass slides using MOWIOL.
Statistics
Statistical analyses regarding the stability and ubiquitination assays were done using GraphPad Prism (version 8.0.0 for Windows, GraphPad Software, San Diego, California, USA). For analysis of the ubiquitination assays (Fig. 3), individual unpaired two-tailed Student’s t-tests were done to compare wild type to mutants, and a one-sample Student’s t-test was used for analysis of protein levels (Fig. 4), with theoretical mean set to 100, where P < 0.05 was considered significant. All data are presented as mean ± SEM.
Study approval
All animal experiments were conducted in accordance with the European Commission Council Directive 2010/63/EU (CCD approval AVD101002016791) and approved by the local ethics committee (Instantie voor Dierenwelzijn; IVD).
Supplementary Material
Acknowledgements
The authors are grateful to the following individuals and groups: M. Elgersma, C. de Konink and M. Aghadavoud Jolfaei for colony management and genotyping; I. Wallaard for setting up mouse primary neuron cultures. Isabella Zampeta for providing an IF picture for the p.Leu747Pro mutant; and A. van Esbroeck and M. Elgersma for reviewing of the manuscript.
Conflict of Interest statement. The authors declare no competing interests.
Functional testing of identified UBE3A variants can be requested by contacting us at info@functionalgenomics.nl.
Funding
This work was supported by a ZonMw TOP (40-00812-98-16045) and grants from the Angelman Syndrome Alliance and Angelman Syndrome Foundation to B.D. and Y.E.
Authors’ Contributions
BD, YE and SB conceptualized the study and designed the methodology. SB, MP, IdG and JvdB generated UBE3A constructs. SB performed confocal imaging experiments. SB and IdG performed stability assays and Y2H. MP performed ubiquitination assays. HH and MW contributed to patient recruitment, genotyping, and phenotype and clinical data collection and analyses. BD and YE supervised the research. BD, YE and SB wrote the first draft of the manuscript. All authors reviewed the manuscript. YE and BD acquired funding.
References
- 1. Buiting, K., Williams, C. and Horsthemke, B. (2016) Angelman syndrome — insights into a rare neurogenetic disorder. Nat. Rev. Neurol., 12, 584–593. [DOI] [PubMed] [Google Scholar]
- 2. Matsuura, T., Sutcliffe, J.S., Fang, P., Galjaard, R.J., Jiang, Y.-H., Benton, C.S., Rommens, J.M. and Beaudet, A.L. (1997) De novo truncating mutations in E6-AP ubiquitin-protein ligase gene (UBE3A) in Angelman syndrome. Nat. Genet., 15, 74–77. [DOI] [PubMed] [Google Scholar]
- 3. Kishino, T., Lalande, M. and Wagstaff, J. (1997) UBE3A/E6-AP mutations cause Angelman syndrome. Nat. Genet., 15, 70–73. [DOI] [PubMed] [Google Scholar]
- 4. Scheffner, M. and Kumar, S. (2014) Mammalian HECT ubiquitin-protein ligases: biological and pathophysiological aspects. Biochim. Biophys. Acta, 1843, 61–74. [DOI] [PubMed] [Google Scholar]
- 5. Dindot, S.V., Antalffy, B.A., Bhattacharjee, M.B. and Beaudet, A.L. (2008) The Angelman syndrome ubiquitin ligase localizes to the synapse and nucleus, and maternal deficiency results in abnormal dendritic spine morphology. Hum. Mol. Genet., 17, 111–118. [DOI] [PubMed] [Google Scholar]
- 6. Burette, A.C., Judson, M.C., Li, A.N., Chang, E.F., Seeley, W.W., Philpot, B.D. and Weinberg, R.J. (2018) Subcellular organization of UBE3A in human cerebral cortex. Mol. Autism., 9, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Burette, A.C., Judson, M.C., Burette, S., Phend, K.D., Philpot, B.D. and Weinberg, R.J. (2016) Subcellular organization of UBE3A in neurons. J. Comp. Neurol., 525, 233–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sirois, C.L., Bloom, J.E., Fink, J.J., Gorka, D., Keller, S., Germain, N.D., Levine, E.S. and Chamberlain, S.J. (2020) Abundance and localization of human UBE3A protein isoforms. Hum. Mol. Genet., 29, 3021–3031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Miao, S., Chen, R., Ye, J., Tan, G.H., Li, S., Zhang, J., Jiang, Y.-H. and Xiong, Z.Q. (2013) The Angelman syndrome protein Ube3a is required for polarized dendrite morphogenesis in pyramidal neurons. J. Neurosci., 33, 327–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Greer, P.L., Hanayama, R., Bloodgood, B.L., Mardinly, A.R., Lipton, D.M., Flavell, S.W., Kim, T.-K., Griffith, E.C., Waldon, Z., Maehr, R.et al. (2010) The Angelman syndrome protein Ube3A regulates synapse development by ubiquitinating arc. Cell, 140, 704–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Pignatelli, M., Piccinin, S., Molinaro, G., Di Menna, L., Riozzi, B., Cannella, M., Motolese, M., Vetere, G., Catania, M.V., Battaglia, G.et al. (2014) Changes in mGlu5 receptor-dependent synaptic plasticity and coupling to homer proteins in the hippocampus of Ube3A hemizygous mice modeling Angelman syndrome. J. Neurosci., 34, 4558–4566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Nawaz, Z., Lonard, D.M., Smith, C.L., Lev-Lehman, E., Tsai, S.Y., Tsai, M.J. and O'Malley, B.W. (1999) The Angelman syndrome-associated protein, E6-AP, is a coactivator for the nuclear hormone receptor superfamily. Mol. Cell. Biol., 19, 1182–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ramamoorthy, S. and Nawaz, Z. (2008) E6-associated protein (E6-AP) is a dual function coactivator of steroid hormone receptors. Nucl. Recept. Signal., 6, e006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Avagliano Trezza, R.A., Sonzogni, M., Bossuyt, S.N.V., Zampeta, F.I., Punt, A.M., Van den Berg, M., Rotaru, D.C., Koene, L.M.C., Munshi, S.T., Stedehouder, J.et al. (2019) Loss of nuclear UBE3A causes electrophysiological and behavioral deficits in mice and is associated with Angelman syndrome. Nat. Neurosci., 22, 1235–1247. [DOI] [PubMed] [Google Scholar]
- 15. Zampeta, F.I., Sonzogni, M., Niggl, E., Lendemeijer, B., Smeenk, H., deVrij, F.M.S., Kushner, S.A., Distel, B. and Elgersma, Y. (2020) Conserved UBE3A subcellular distribution between human and mice is facilitated by non-homologous isoforms. Hum. Mol. Genet., 12, 3032–3043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lee, S.Y., Ramirez, J., Franco, M., Lectez, B., Gonzalez, M., Barrio, R. and Mayor, U. (2013) Ube3a, the E3 ubiquitin ligase causing Angelman syndrome and linked to autism, regulates protein homeostasis through the proteasomal shuttle Rpn10. Cell. Mol. Life Sci., 71, 2747–2758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tomaić, V. and Banks, L. (2015) Angelman syndrome-associated ubiquitin ligase UBE3A/E6AP mutants interfere with the proteolytic activity of the proteasome. Cell Death Dis., 6, e1625–e1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kühnle, S., Martínez-Noël, G., Leclere, F., Hayes, S.D., Harper, J.W. and Howley, P.M. (2018) Angelman syndrome–associated point mutations in the Zn2+-binding N-terminal (AZUL) domain of UBE3A ubiquitin ligase inhibit binding to the proteasome. J. Biol. Chem., 293, 18387–18399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rotaru, D.C., Woerden, G.M.V., Wallaard, I. and Elgersma, Y. (2018) Adult Ube3a gene reinstatement restores the electrophysiological deficits of prefrontal cortex layer 5 neurons in a mouse model of Angelman syndrome. J. Neurosci., 38, 8011–8030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sadhwani, A., Sanjana, N.E., Willen, J.M., Calculator, S.N., Black, E.D., Bean, L.J.H., Li, H. and Tan, W.-H. (2018) Two Angelman families with unusually advanced neurodevelopment carry a start codon variant in the most highly expressed UBE3Aisoform. Am. J. Med. Genet. A, 176, 1641–1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cooper, E.M., Hudson, A.W., Amos, J., Wagstaff, J. and Howley, P.M. (2004) Biochemical analysis of Angelman syndrome-associated mutations in the E3 ubiquitin ligase E6-associated protein. J. Biol. Chem., 279, 41208–41217. [DOI] [PubMed] [Google Scholar]
- 22. Yi, J.J., Berrios, J., Newbern, J.M., Snider, W.D., Philpot, B.D., Hahn, K.M. and Zylka, M.J. (2015) An autism-linked mutation disables phosphorylation control of UBE3A. Cell, 162, 795–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sadikovic, B., Fernandes, P., Zhang, V.W., Ward, P.A., Miloslavskaya, I., Rhead, W., Rosenbaum, R., Gin, R., Roa, B. and Fang, P. (2014) Mutation update for UBE3A variants in Angelman syndrome. Hum. Mutat., 35, 1407–1417. [DOI] [PubMed] [Google Scholar]
- 24. Geerts-Haages, A., Bossuyt, S.N.V., Besten, I., Bruggenwirth, H., Burgt, I., Yntema, H.G., Punt, A.M., Brooks, A., Elgersma, Y., Distel, B.et al. (2020) A novel UBE3A sequence variant identified in eight related individuals with neurodevelopmental delay, results in a phenotype which does not match the clinical criteria of Angelman syndrome. Mol. Genet. Genomic Med., 8, e1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rapakko, K., Kokkonen, H. and Leisti, J. (2004) UBE3A gene mutations in finnish Angelman syndrome patients detected by conformation sensitive gel electrophoresis. Am. J. Med. Genet. A, 126A, 248–252. [DOI] [PubMed] [Google Scholar]
- 26. Mueller, O.T. and Coovadia, A. (2008) Gene symbol: UBE3A. Disease: Angelman syndrome. Hum. Genet., 124, 304. [PubMed] [Google Scholar]
- 27. Fang, P., Lev-Lehman, E., Tsai, T.F., Matsuura, T., Benton, C.S., Sutcliffe, J.S., Christian, S.L., Kubota, T., Halley, D.J., Meijers-Heijboer, H.et al. (1999) The spectrum of mutations in UBE3A causing Angelman syndrome. Hum. Mol. Genet., 8, 129–135. [DOI] [PubMed] [Google Scholar]
- 28. Camprubí, C., Guitart, M., Gabau, E., Coll, M.D., Villatoro, S., Oltra, S., Roselló, M., Ferrer, I., Monfort, S., Orellana, C.et al. (2009) Novel UBE3A mutations causing Angelman syndrome: different parental origin for single nucleotide changes and multiple nucleotide deletions or insertions. Am. J. Med. Genet. A, 149A, 343–348. [DOI] [PubMed] [Google Scholar]
- 29. Malzac, P., Webber, H., Moncla, A., Graham, J.M., Jr., Kukolich, M., Williams, C., Pagon, R.A., Ramsdell, L.A., Kishino, T. and Wagstaff, J. (1998) Mutation analysis of UBE3A in Angelman syndrome patients. Am. J. Hum. Genet., 62, 1353–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bai, J.-L., Qu, Y.-J., Jin, Y.-W., Wang, H., Yang, Y.L., Jiang, Y.W., Yang, X.Y., Zou, L.P. and Song, F. (2014) Molecular and clinical characterization of Angelman syndrome in Chinese patients. Clin. Genet., 85, 273–277. [DOI] [PubMed] [Google Scholar]
- 31. Horsthemke, B., Wawrzik, M., Groß, S., Lich, C., Sauer, B., Rost, I., Krasemann, E., Kosyakova, N., Liehr, T., Weise, A.et al. (2011) Parental origin and functional relevance of a de novo UBE3A variant. Eur. J. Med. Genet., 54, 19–24. [DOI] [PubMed] [Google Scholar]
- 32. Avagliano Trezza, R.A., Punt, A.M., Mientjes, E., Berg, M., Zampeta, F.I., Graaf, I.J., Weegen, Y., Demmers, J.A.A., Elgersma, Y. and Distel, B. (2021) Mono-ubiquitination of Rabphilin 3A by UBE3A serves a non-degradative function. Sci. Rep., 11, 3007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Keren-Kaplan, T., Attali, I., Motamedchaboki, K., Davis, B.A., Tanner, N., Reshef, Y., Laudon, E., Kolot, M., Levin-Kravets, O., Kleifeld, O.et al. (2011) Synthetic biology approach to reconstituting the ubiquitylation cascade in bacteria. EMBO J., 31, 378–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Li, C., Han, T., Guo, R., Chen, P., Peng, C., Prag, G. and Hu, R. (2020) An integrative synthetic biology approach to interrogating cellular ubiquitin and ufm signaling. Int. J. Mol. Sci., 21, 4231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Nuber, U., Schwarz, S.E. and Scheffner, M. (1998) The ubiquitin-protein ligase E6-associated protein (E6-AP) serves as its own substrate. Eur. J. Biochem., 254, 643–649. [DOI] [PubMed] [Google Scholar]
- 36. Zaaroor-Regev, D., Bie, P.de, Scheffner, M., Noy, T., Shemer, R., Heled, M., Stein, I., Pikarsky, E. and Ciechanover, A. (2010) Regulation of the polycomb protein Ring1B by self-ubiquitination or by E6-AP may have implications to the pathogenesis of Angelman syndrome. Proc. Natl. Acad. Sci. U. S. A., 107, 6788–6793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kobayashi, F., Nishiuchi, T., Takaki, K. and Konno, H. (2018) Ubiquitin chain specificities of E6AP E3 ligase and its HECT domain. Biochem. Biophys. Res. Commun., 496, 686–692. [DOI] [PubMed] [Google Scholar]
- 38. Huibregtse, J.M., Scheffner, M. and Howley, P.M. (1993) Localization of the E6-AP regions that direct human papillomavirus E6 binding, association with p53, and ubiquitination of associated proteins. Mol. Cell. Biol., 13, 4918–4927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sailer, C., Offensperger, F., Julier, A., Kammer, K.-M., Walker-Gray, R., Gold, M.G., Scheffner, M. and Stengel, F. (2018) Structural dynamics of the E6AP/UBE3A-E6-p53 enzyme-substrate complex. Nat. Commun., 9, 4441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bie, P. and Ciechanover, A. (2011) Ubiquitination of E3 ligases: self-regulation of the ubiquitin system via proteolytic and non-proteolytic mechanisms. Cell Death Differ., 18, 1393–1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Huang, L., Kinnucan, E., Wang, G., Beaudenon, S., Howley, P.M., Huibregtse, J.M. and Pavletich, N.P. (1999) Structure of an E6AP-UbcH7 complex: insights into ubiquitination by the E2-E3 enzyme cascade. Science, 286, 1321–1326. [DOI] [PubMed] [Google Scholar]
- 42. Lorenz, S. (2018) Structural mechanisms of HECT-type ubiquitin ligases. Biol. Chem., 399, 127–145. [DOI] [PubMed] [Google Scholar]
- 43. Kim, H.C. and Huibregtse, J.M. (2009) Polyubiquitination by HECT E3s and the determinants of chain type specificity. Mol. Cell. Biol., 29, 3307–3318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wang, M. and Pickart, C.M. (2005) Different HECT domain ubiquitin ligases employ distinct mechanisms of polyubiquitin chain synthesis. EMBO J., 24, 4324–4333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Mortensen, F., Schneider, D., Barbic, T., Sladewska-Marquardt, A., Kühnle, S., Marx, A. and Scheffner, M. (2015) Role of ubiquitin and the HPV E6 oncoprotein in E6AP-mediated ubiquitination. Proc. Natl. Acad. Sci. U. S. A., 112, 9872–9877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hokayem, J.E. and Nawaz, Z. (2013) E6AP in the brain: one protein, dual function multiple diseases. Mol. Neurobiol., 49, 827–839. [DOI] [PubMed] [Google Scholar]
- 47. Gkourtsa, A., Burg, J.van den, Avula, T., Hochstenbach, F. and Distel, B. (2016) Binding of a proline-independent hydrophobic motif by the Candida albicans Rvs167-3 SH3 domain. Microbiol. Res., 190, 27–36. [DOI] [PubMed] [Google Scholar]
- 48. Reijnders, M.R.F., Kousi, M., vanWoerden, G.M., Klein, M., Bralten, J., Mancini, G.M.S., vanEssen, T., Proietti-Onori, M., Smeets, E.E.J., vanGastel, M.et al. (2017) Variation in a range of mTOR-related genes associates with intracranial volume and intellectual disability. Nat. Commun., 8, 1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Baumer, A., Balmer, D. and Schinzel, A. (1999) Screening for UBE3A gene mutations in a group of Angelman syndrome patients selected according to non-stringent clinical criteria. Hum. Genet., 105, 598–602. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.