

Abstract
Gene networks for disorders of social behavior provide the mechanisms critical for identifying therapeutic targets and biomarkers. Large behavioral phenotypic effects of small human deletions make the positive sociality of Williams syndrome (WS) ideal for determining transcriptional networks for social dysfunction currently based on DNA variations for disorders such as autistic spectrum disorder (ASD) and schizophrenia (SCHZ). Consensus on WS networks has been elusive due to the need for larger cohort size, sensitive genome-wide detection and analytic tools. We report a core set of WS network perturbations in a cohort of 58 individuals (34 with typical, 6 atypical deletions and 18 controls). Genome-wide exon-level expression arrays robustly detected changes in differentially expressed gene (DEG) transcripts from WS deleted genes that ranked in the top 11 of 12 122 transcripts, validated by quantitative reverse transcription PCR, RNASeq and western blots. WS DEG’s were strictly dosed in the full but not the atypical deletions that revealed a breakpoint position effect on non-deleted CLIP2, a caveat for current phenotypic mapping based on copy number variants. Network analyses tested the top WS DEG’s role in the dendritic spine, employing GeneMANIA to harmonize WS DEGs with comparable query gene-sets. The results indicate perturbed actin cytoskeletal signaling analogous to the excitatory dendritic spines. Independent protein–protein interaction analyses of top WS DEGs generated a 100-node graph annotated topologically revealing three interacting pathways, MAPK, IGF1-PI3K-AKT-mTOR/insulin and actin signaling at the synapse. The results indicate striking similarity of WS transcriptional networks to genome-wide association study-based ASD and SCHZ risk suggesting common network dysfunction for these disorders of divergent sociality.
Introduction
Human complex diseases involving cognition and behavior, such as autistic spectrum disorder (ASD) and schizophrenia (SCHZ), remain a major challenge for translational neuroscience. Regardless of effect size, the risk or causative gene is rarely the adult therapeutic target, and core gene networks are required to identify and link intermediate gene expression traits to active deficits. However, for the vast majority of gene-based risks whether defined by the genome-wide association study (GWAS) or genome copy number variants (CNVs), there are no examples of genes whose expression is correlated with the cognitive and behavioral outcomes of individuals carrying the gene variants. As a result, despite ample research in animal and cellular models (1,2), there are few scalable gene-based outcomes and as yet, no effective treatments. Understanding how CNVs influence human phenotypes associated with ASD and SCHZ may help to define the developmental and adult mechanisms that translate gene dosage into perturbations of genetic pathways that then affect complex traits. In this report, the focus is on scalable downstream transcriptional consequences rather than the non-scalable upstream chromatin regulation (3) to which they may be related. Williams syndrome (WS, OMIM #194050) provides a unique opportunity to understand common, devastating behavioral conditions such as ASD (4), in that its hemizygous deletion of a small number (~28) of genes causes consistent cognitive social–emotional conditions, typically gregarious behavior (5) but also, paradoxically, ASD (6–10). Although a number of deleted WS genes (e.g. GTF2I (encoding TFII-I), GTF2IRD1, LIMK1, FZD9 and BAZ1B) have been implicated by DNA copy number in deficits associated with visual–spatial construction, social approach, myelination, brain structure and electrophysiology (3,11–18), the molecular perturbations that connect genotype to phenotype in WS remain largely unspecified (14). Consequently, the causal mechanisms are unknown and there is no evidence relating gene expression of deleted or non-deleted genes to individual cognitive or behavioral phenotypes.
The initiating event for a network perturbation caused by genomic CNVs is a cis-acting dosage effect at the transcript level. Although specialized mechanisms exist in sex chromosomes, that compensate and alter a gene’s transcript level from a strict relationship to DNA copy number, such dosage compensation is not known in human autosomal aneuploidies. The consequence is that the subtle changes in DNA copy number alter transcript levels of the CNV as well as genome-wide transcripts and result in cognitive and behavioral disorders (19–21). At a genome-wide level, the impact of CNVs on heritable gene expression traits explains only ~20% of the cis-acting transcript variation in human lymphoblastoid cell lines (LCLs) (22), emphasizing the role of trans effects on regulating transcript levels. Similar conclusions derive from genome-wide analyses of multiple tissues from inbred mice suggesting that CNVs contribute both to cis-acting and trans-acting perturbations of regional gene expression (23). However, identifying how thousands of perturbations represent a coherent set of networks has been difficult for at least three reasons, the lack of a target set of loci required by high precision techniques such as quantitative reverse transcription PCR (qRT-PCR), the need for unbiased genome-wide analyses capable of showing the precision necessary to measure the subtle (<2-fold) transcript perturbations associated with CNV’s, and the need for an adequate number of independent samples that share a given CNV. Consequently, in order to understand the biology of WS, alternative approaches that are both robust and quantitative are necessary to determine the core set of genome-wide network disturbances induced by human pathologic CNVs, the goal of this report.
Multiple studies of WS genetic and metabolic network disturbances have revealed a spectrum of differently expressed genes (DEGs) that reflect valid differences between small comparison groups or the effects of single genes in cell culture systems (24–28). Although WS and downstream genes are involved in chromatin dysregulation, they have not converged on a common core of network perturbations, nor have they established the relationship of cellular gene expression to cognitive or behavioral outcomes in single individuals. Therefore, to fill the need for a core set of networks for human social behavior, we increased sensitivity, specificity and reproducibility by increasing cohort size ~3-fold, and utilized exon microarrays followed by qRT-PCR as a sensitive approach for determining dosage effects at genome-wide levels. RNA-seq has become a method of choice for expression analysis but for the goal of the current study in the service of network perturbations, quantitation derived from the well-worked out and highly internally controlled exon arrays used for quantitative gene expression (29), validated by single sample and pooled qRT-PCR, can obviate the technical biases of Log2 comparisons in RNA-seq data for length, GC content and normalization strategies. Finally, to parse the contribution of specific WS genes through core network perturbations to associated phenotypes, networks were determined employing rare humans with partial deletions and phenotypic outcomes. The results indicate a shared perturbation of the networks involving the IGF1-PI3K-AKT-mTOR (insulin) signaling and proteostasis stress pathways, as well as MAPK and actin cytoskeleton signaling in WS, reminiscent of those for ASD. Moreover, genetic analyses reveal a position effect on non-deleted CLIP2 expression, specify genes ABHD11 through CLIP2 as sufficient for actin network dysregulation, and establish critical cohort criteria for rigorous network inferences in neurodevelopmental disorders.
Results
Exon arrays define differentially expressed exons and DEGs in WS
To maximize the ability to identify a common core set of WS networks, we tripled the sample size of previous reports (19,24–27) to 34 WS patients and 18 controls, and utilized a sensitive, quantitative, validated genome-wide approach for transcriptional profiling employing the Affymetrix HuEx-1_0-st-v2 exon arrays (~1.4 million probe sets), and analyzed these at the exon and whole gene levels. The raw data were normalized and background filtered, yielding summary values for 313 717 exons that were included in the initial analysis. When assessed used a moderated t-statistic, genome-wide differential expression at the exon-level yielded a prominent cluster of differentially (under-) differentially expressed exons (DEEs) mapping to the WS region on chromosome 7 (Fig. 1A). Moreover, 95 of the top 100 genome-wide DEEs and 157 of the top 250 DEEs mapped to the WS deletion and flanking regions shown in Figure 1B and C and Table 1 reflecting the sensitivity and consistency of the dataset. Figure 1D shows the heat map of exon-level expression data in each individual WS subject for the 13 DEG’s of typical deletions, confirming previous work (19,30).
Figure 1.
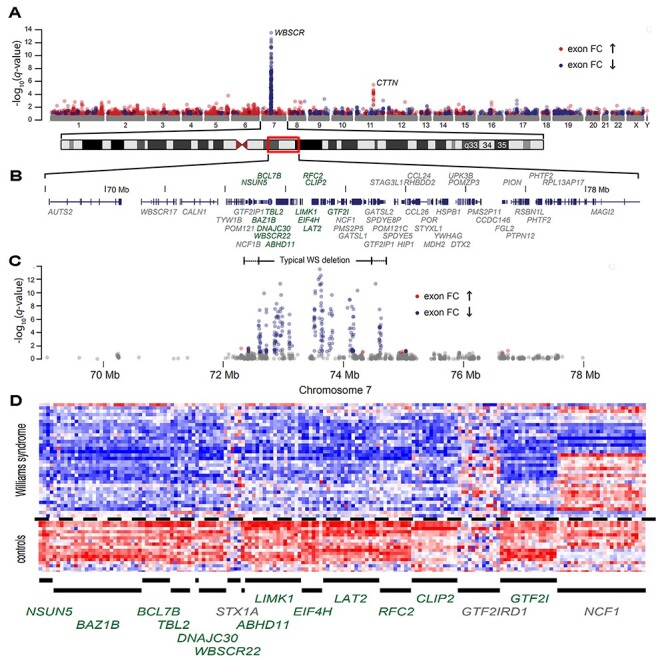
Manhattan plot of genome-wide differential expression in lymphoblastoid cells. (A) −log10q-value (the adjusted P-value using the Benjamini–Hochberg method) by chromosomal location for 304 572 detected exons from 34 WS subjects with typical deletions versus 18 controls. (B) RefSeq track at chr.7: 68 500 000–78 500 000 (Hg18) with differentially expressed transcripts (see Table 1) shown in green. (C) Exon-level differential expression in this chromosome 7 region for 34 WS subjects with typical deletions. The dashed line is at a FDR value of 0.05 (−log10q-value = 1.3), such that the expected proportion of false discoveries in exons above this threshold is controlled to be less than 5%. (D) Individual exon-level relative intensities for 13 differentially expressed genes plus three genes (STX1A, GTF2IRD1 and NCF1) without gene-level evidence of differential expression (see Table 1). Exons are binned in chromosomal order by gene (columns) and by individual (rows) for 18 controls and 34 typical WS deletions.
Table 1.
Differential expression of genes and exons in the WS chromosome region
| Gene symbol | Gene ranka | Fold change (log2) | Gene-level FDR | Top exon rank | # of Exons at FDR < 0.05b |
|---|---|---|---|---|---|
| LAT2 | 1 | −0.83 | 7.58E-14 | 3 | 16 (18) |
| BCL7B | 2 | −0.88 | 5.88E-12 | 8 | 8 (10) |
| LIMK1 | 3 | −0.89 | 9.93E-12 | 14 | 16 (20) |
| CLIP2 | 4 | −0.97 | 2.10E-10 | 19 | 16 (19) |
| GTF2I | 5 | −0.78 | 4.62E-10 | 23 | 38 (65) |
| NSUN5 | 6 | −0.70 | 9.33E-10 | 33 | 9 (34) |
| TBL2 | 7 | −0.82 | 1.47E-09 | 29 | 7 (13) |
| RFC2 | 8 | −0.77 | 3.08E-09 | 38 | 10 (13) |
| BAZ1B | 9 | −0.78 | 3.83E-08 | 24 | 29 (36) |
| WSCR22 | 10 | −0.62 | 1.19E-07 | 55 | 10 (12) |
| EIF4H | 11 | −0.77 | 1.21E-07 | 1 | 9 (13) |
| DNAJC30 | 15 | −0.56 | 4.84E-04 | 274 | 1 (3) |
| ABHD11 | 24 | −0.50 | 4.11E-03 | 317 | 1 (6) |
| MLXIPL | 1263 | −0.25 | 0.26 | 25 641 | 0 (8) |
| VPS37D | 2834 | 0.16 | 0.36 | n.d. | 0 (0) |
| GTF2IRD2 | 3430 | −0.15 | 0.40 | 41 258 | 0 (20) |
| WSCR28 | 4853 | −0.12 | 0.49 | n.d. | 0 (0) |
| ELN | 4868 | −0.09 | 0.50 | 38 429 | 0 (20) |
| CLDN3 | 8798 | 0.06 | 0.70 | 209 408 | 0 (1) |
| WSCR27 | 10 019 | −0.06 | 0.75 | n.d. | 0 (0) |
| GTF2IRD1 | 11 855 | −0.06 | 0.84 | 51 801 | 0 (13) |
| FZD9 | 12 921 | −0.03 | 0.88 | 247 475 | 0 (1) |
| STX1A | 13 880 | 0.03 | 0.91 | 27 391 | 0 (4) |
| TRIM50 | n.d. | n.d. | n.d. | 153 041 | 0 (3) |
| FKBP6 | n.d. | n.d. | n.d. | n.d. | 0 (0) |
| NCF1 | n.d. | n.d. | n.d. | 8017 | 0 (37) |
| CLDN4 | n.d. | n.d. | n.d. | 17 189 | 0 (1) |
a‘Gene Rank’ = gene-level summaries ranked by the log odds of differential expression from 34 WS subjects versus 18 controls; 16 509 core transcripts were included in this ranked list after background filtering.
b‘# of Exons at FDR < 0.05’ = Exon count from RefSeq transcript of detected exons with a Benjamini–Hochberg FDR value < 0.05 with total detected exons for that transcript in parentheses. There were 304 572 total exons detected in this experiment.
We next asked whether differences in genome-wide exon expression were reflected at the gene level. Comparing WS with controls, the results revealed a genome wide distribution of DEGs whose levels were subtly perturbed to the same or a lesser degree as the deleted genes (Fig. 1A and Supplementary Material, Table S3). Of the top 673 non-WS DEGs (the false discovery rate [FDR] < 0.2), 41% were decreased, and 59% were increased in expression. Table 1 shows gene-level differences for 13 of the 27-known WS protein-coding genes are the most consistently decreased DEG’s among the 16 509 detected (gene-level FDR < 0.01). Further, the first 11 DEGs are from the WS region (ranked by moderated t-statistic), despite their modest (<2) fold change. Two further WS region genes, DNAJC30 and ABHD11, ranked in the upper 22 DEGs, while the other 11 (except NCF1) WS genes including STX1A, were among the lowest expressed of the WS genes and showed no evidence for differential expression. As previously shown (19,30), neither GTF2IRD1 nor NCF1, were decreased. Finally, we examined whether array performance could be calibrated for detection by measuring transcript abundance with mRNA sequencing (RNA-Seq) (Supplementary Material, Table S4). We applied a threshold of 10 RNA-Seq reads per transcript as a digital detection filter gene-level summary to derive a conservative list of 12 122 detected transcripts, and used these for differential expression correlated with the WS phenotype (Supplementary Material, Table S3). In summary, analyses at genome wide levels identified the WS deleted genes, their transcripts and exons as the top differentially expressed elements (DEEs 55 out of 313 717; Supplementary Material, Table S2) and DEGs (11 out of 12 122, Supplementary Material, Table S3) validating the sensitivity and specificity of the dataset.
qRT-PCR and western blots validate exon microarray results
To validate the average fold change found in array gene-level summaries, we used qRT-PCR from both unpooled and pooled RNA of the study cohort, followed by western blots to validate altered protein. The results of testing unpooled RNA levels of 34 WS and 18 controls using qRT-PCR are shown (Supplementary Material, Table S3) for 4 WS genes (STX1A, LIMK1, CLIP2 and GTF2I) and 4 of the top non-WS genes (DAPK1, CTTN, FHL3 and DBN1; rank 98, 12, 13 and 20 (Supplementary Material, Table S3)). The results of qRT-PCR showed that all 8 genes differed significantly and were highly correlated with the array results from WS and controls except STX1A (Fig. 2A and B). The limit of arrays as detectors of fold change for genes at low levels of expression determined by both qRT-PCR and RNA-seq is reflected by the ability of the qRT-PCR but not the array, to detect significant decrease of STX1A in WS versus controls (Figs 1D,2A and B and3B; Supplementary Material, Fig. S1). Consequently, despite the superior transcript exon coverage of the arrays versus qRT-PCR, the underlying RNA abundance is a main determinant of fold-change detection. The results of qRT-PCR on pooled RNA consisting of aliquots of 34 WS or 18 controls confirmed the array results for DEG’s for 7 WS genes and 25 non-WS genes (13 decreased and 12 increased expressions; Fig. 2C). The high correlation between individual qRT-PCR measurements and array gene-level summaries strongly support the observed individual variation in array data as an accurate reflection of individual transcript levels within the RNA samples, the few subtle exceptions possibly representing exon specific expression unrelated to the qRT-PCR probe. Finally, to address whether the decreased copy number and transcript levels of WS deleted genes were reflected at the protein level, western blots of WS versus controls were performed. The results revealed decreased (30–60%) protein levels of WS encoded genes (EIF4H, LIMK1, GTF2I/TFII-I, LAT2) in WS (Supplementary Material, Fig. S2). The decreased protein levels suggest a lack of dosage compensation at the protein level in WS. In summary, both qRT-PCR and western blots confirmed the DEG results of the exon microarray as sensitive, accurate reflections of transcript specific network perturbations.
Figure 2.
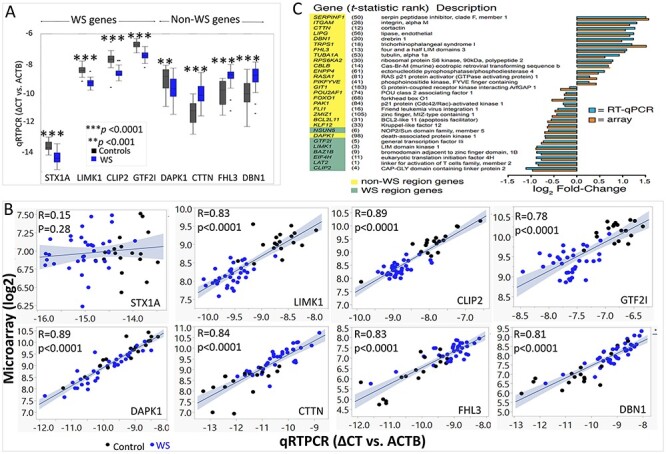
Validation the microarray expression results using qRT-PCR. (A) Relative intensity range for gene-level summaries of qRT-PCR results indicated by boxplots for each gene in control subjects (black) and WS subjects (blue). Genes include 4 WS genes and 4 non-WS genes all show significant difference in WS compared with controls. (B) qRT-PCR of 8 genes (STX1A, LIMK1, CLIP2, GTF2I, DAPK1, CTTN, FHL3, DBN1) in un-pooled 52 subjects (34 WS, blue dots; 18 TC, black dots) except STX1A show highly significant correlations with microarray results. (C) Average fold change from pooled qRT-PCR versus array gene-level summaries of 7 WS genes (green highlight) and 21 genes outside the WS chromosome region (yellow highlight). The rank order of each gene from the gene-level test for differential expression (Supplementary Material, Table S3) is shown in parentheses.
Figure 3.
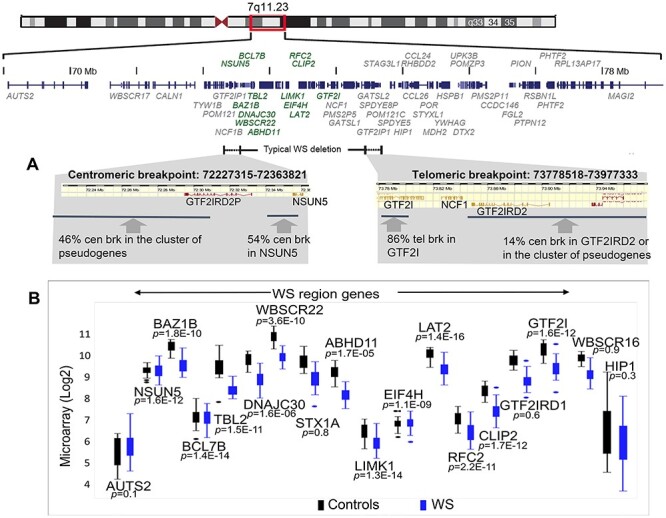
Gene expression level in typical WS is determined largely by the deletion. (A) Precise breakpoints of typical WS are determined by using Illumina DUO SNP and CNV microarray. (B) Individual DEGs of deleted genes in WS versus controls.
DNA CNV breakpoints are reflected by decreased expression without position effects in typical WS
The next question was whether quantitative transcript levels in WS genes were determined largely by CNV or were also affected by cis or trans regulatory effects. To address this, DNA breakpoints were determined in 61 subjects with WS (28 from this report) by using Illumina DUO SNP and CNV microarrays (Fig. 3A, Supplementary Material, Fig. S3) and compared with the transcript levels. The results revealed that 54% of the DNA centromeric breakpoints in WS were within NSUN5, and the remainder were located within the cluster of pseudogenes centromeric to NSUN5. As expected, group comparisons of transcript levels showed significantly decreased expression of NSUN5 in WS (FDR = 9.3E-10), and 76% of WS subjects show >40% decrease in its expression. In the region of the telomeric breakpoint, 86% occurred within GTF2I, as expected (11) and the remaining 14% were within or telomeric to GTF2IRD2. As expected, expression of GTF2I was significantly decreased in WS (FDR = 4.6E-10), and 82% of WS subjects show >40% under-expression of GTF2I due either to nonsense mediated decay or full deletion. In contrast, a bimodal decrease in expression was found for NCF1 or GTF2IRD2 due to the ~97% homology and variable number of their pseudogenes, although long-read sequencing may resolve this. Moreover, using quantitative exon arrays, no position effect was observed in HIP1 or AUTS2 in contrast to previous publications (31,32). In conclusion, comparison of DNA breakpoints and RNA expression in full deletion WS, indicates that gene expression is determined largely by the CNV.
Atypical WS deletion: position effect decreases expression of non-deleted gene transcript for CLIP2, 10 kb telomeric to the breakpoint
The objective of understanding human genetic aneuploidies such as WS (33), is to cut through the largely unknown genetics of human brain development and substrates for behavior, to discover gene-brain-behavior anchors, the vast majority of which currently depend on the association of a DNA gene CNV with a behavior. Studies based on rare partial chromosomal rearrangements neglect the possible effects of unspecified but rearranged regulatory elements on the transcription of non-deleted genes that could erroneously implicate a deleted gene, whereas the phenotype may have been due to a transcriptional perturbation of its non-deleted neighbor. Therefore, to evaluate the possible role of position effects on altered transcription in atypical deletions, exon-level gene expression was evaluated in a family of 6 WS subjects and a small deletion without the typical WS exaggerated approach to strangers. The breakpoints determined with custom high density NimbleGen microarrays of chromosome 7, revealed a 545 Kb deletion that excluded a majority of WS genes (33), shown in Figure 4A. As expected, exon transcription was decreased for deleted genes (ABHD11, LIMK1, EIF4H, LAT2 and RFC2), emphasizing the accurate detection of subtle (<2-fold) expression differences by genome wide arrays. However, decreased expression was also found for CLIP2, the non-deleted gene whose 5’UTR was located ~10 kb distal to the telomeric breakpoint (73 331 254–73 331 770 bp; NCBI36/hg18). The decreased expression suggested that enhancer cis regulatory components (Fig. 4B) for CLIP2 may have been disrupted or deleted. The position effect on CLIP2 in atypical WS emphasizes the importance of determining gene expression in addition to DNA CNV for accurate determination of genetic contributions to phenotype in WS and human neurodevelopmental disorders.
Figure 4.
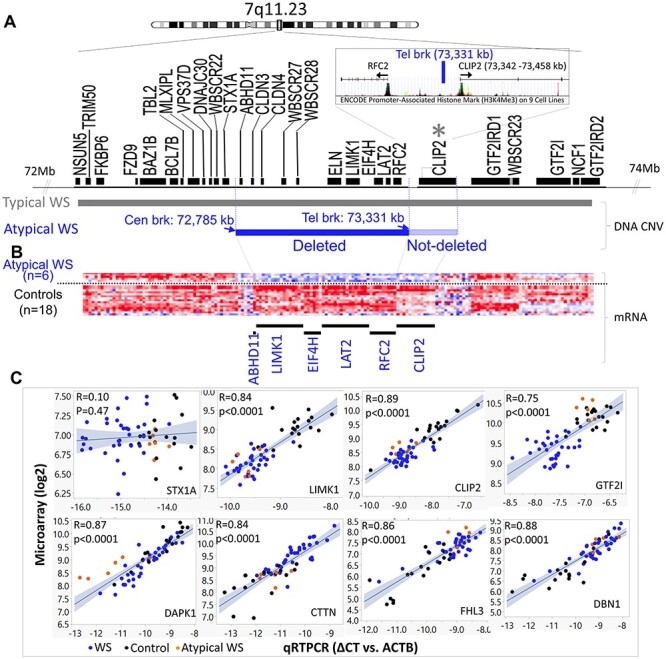
Gene expression level in atypical WS reveals a position effect on a non-deleted gene, CLIP2. (A) The deletion of atypical WS includes genes from ABHD11 to RFC2, which is determined by using custom high density NimbleGen microarrays of chromosome 7. The telomeric breakpoint (73 331 kb) is located ~10 kb upstream of the 5' UTR of CLIP2. (B) Individual exon-level relative intensities for five differentially expressed genes (ABHD11 to RFC2) plus CLIP2, which is not deleted by under-expressed (see Table 1). Exons are binned in chromosomal order by gene (columns) and by individual (rows) for 6 atypical WS and 18 controls. (C) qRT-PCR of 8 genes (STX1A, LIMK1, CLIP2, GTF2I, DAPK1, CTTN, FHL3, DBN1) in un-pooled 58 subjects (34 WS, blue dots; 18 TC, black dots 6 atypical WS, orange dots) except STX1A show highly significant correlations with microarray results.
Pathway analysis
Three intersecting analyses were used to extract a common set of biological and cellular networks perturbed by the WS deletion: Gene set enrichment analysis (GSEA), Gene ontology (GO) and protein–protein interaction (PPI).
GSEA
To identify specific biological networks informed by the larger difference set, we used the ranked list of 12 122 genes as input for both an absolute GSEA enrichment test (34) and for a standardized GSEA χ2-test that is more appropriate for detecting scale change in gene sets with both up- and down regulated genes (35). As query gene sets with biologic relevance, we used the MSigDB (version 3.0) C2 curated gene sets for canonical pathway and chemical and gene perturbations. As a positive control targeting the deleted genes, we supplemented the target sets with the chromosome 7q11.23 cytogenetic band gene set from the C1 positional database. To capture recent advances related to actin assembly, we used the GeneMANIA tool (36) to integrate comparable genes in the MSigDB gene sets with an unbiased, literature-derived a posteriori gene set describing actin signaling at the glutamatergic synapse (37) along with the top ranked actin-related genes CTTN and DBN1 (Rank 12 and 20). The resulting gene set (37) was seeded and included genes similar to transcripts and proteins involved in actin regulation during brain development, in glutamatergic dendritic spine morphogenesis and in adult postsynaptic dendritic spine function. Because the brain tissue directly related to the behaviors is inaccessible and unknown, the identification of analogous genes with similar roles in comparable networks by GeneMANIA, facilitates harmonizing networks across tissues and there by facilitates identifying possibly cognate pathways shared by brain and adult cell lines. The results of using GSEA to query the 2166 gene sets (MSigDB) (including a posteriori), revealed 30 top enriched gene sets (Supplementary Material, Table S5). A list of the overlapping genes shared by the 30 gene sets is shown in Figure 5A, and reduced the number of clusters to 11 gene sets with a high degree of functional redundancy. This included a posteriori gene set, named ‘actin cytoskeleton form and function at the synapse’ and the ‘Biocarta IGF1 Pathway’ (Fig. 5B; Supplementary Material, Table S6). Unexpectedly, although the transcriptome analyses were performed in immune cells, the most enriched of the actin gene sets harmonized with mechanisms of actin regulation in the dendritic spine, specifically the postsynaptic cytoskeleton of excitatory synapses (Fig. 5C). The hypothesis-based post-synaptic spine mechanism (37) included two WS genes, LIMK1 and CLIP2 in key regulatory positions, an unanticipated clue to a core perturbation of WS. revealed by quantitative differences in transcription.
Figure 5.
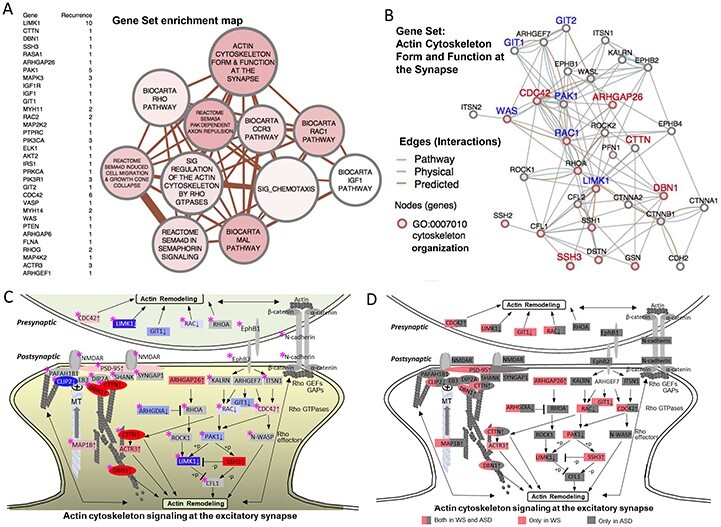
GSEA reveals dysregulation of actin signaling networks. (A) A gene set enrichment list of genes shared by the top 30 gene sets, ranked by their level of differential expression is shown to the left of the network. In the map of the gene sets to the right, nodes represent gene set clusters that result from the χ2-test for gene set enrichment (Supplementary Material, Table S5) and edges represent the gene overlap between sets. Nodes are colored by χ2 values, node size indicates the number of genes in the set and edge thickness indicates number of genes shared between sets. (B) WS DEG (FDR < 0.2) pathway showing physical and predicted interaction network for the top-scoring GSEA gene set, ‘Actin cytoskeleton form and function at the synapse,’ GO term association (2 × 10−13, ‘actin cytoskeleton organization’). (C) WS DEG GSEA gene set (from B) shows unexpected match to the excitatory dendritic spine, capturing systems that regulate cytoskeleton in response to receptor excitation. Diagram is modified from Dillon and Goda (37), updated to include recent evidence for microtubule co-regulation involving WS genes. Node labels identify WS DEGs. Node colors and density indicate WS DEG intensity by rank (FDR < 0.2) (blue = decreased, red = increased), or non-detected as a WS DEG (gray). WS region genes (LIMK1 and CLIP2) are shown with white text in blue shade. Note the growth ends of actin (barbed) and microtubules (plus) form a coregulatory bridge to the NMDAR, which includes WS DEG’s Drebrin 1 (DBN1, rank 20/12122) and cortactin (CTTN, rank 12), major cytoskeletal regulators in adult spines and axonal growth cones (47,48). The bridge complex represents experimentally established components that connect CLIP2 to the NMDAR through PAFAH1B1 (99) and independently through direct MT plus end binding of CLIP2 to EB3 (49), through DIP2A (100), DNB1 and CTTN, to SHANK-PSD95. ACTR3 is a major constituent of the actin branching complex ARP2/3 that is regulated by both CTTN (101), and MAP1B (102) (binds both MT and actin), both of which were significantly over-expressed in WS (Supplementary Material, Table S3). The relationship of WS DEG’s to ASD risk genes (Supplementary Material, Table S11) taken from the literature is noted (pink star), shows a striking synergism, complementarity and overlap, suggesting that WS, caused by a small gene set, may define the excitatory dendritic spine and its gene networks as a common brain substrate for the disturbed behavioral systems involved in approach or avoidant social behaviors seen in WS and ASD, where the large number of genes and networks has precluded identifying these. (D) Duplicate of (C) showing WS genes in pink and ASD in gray or both (half pink and half gray).
GO
We then used GO term enrichment analysis (38) to independently classify the list of perturbed genes broadening the focus beyond the top DEGs related to actin signaling as described above. A conditional hypergeometric test for over-representation of GO terms used the list of 673 genes (FDR < 0.2, excluding the WS region genes) and substantiated the enrichment of genes related to insulin signaling pathways (top three), cell communication and signal transduction (Supplementary Material, Table S7).
PPI
Using the cellular PPI backbone and graphical analysis to filter networks: To further converge on a set of core biological pathways perturbed in WS, we then asked if the functional meaning of the DEG pattern might be captured by querying the cellular networks established by the most direct biochemical property, PPI. Using the Human Protein Reference Database (HPRD), the initial query of the top 114 DEGs (FDR < 0.05), resulted in a direct PPI network containing 12 genes that included three WS region genes (GTF2I, LIMK1 and CLIP2), linked to proteins involved in actin cytoskeleton dynamics and the MAPK pathway (Supplementary Material, Fig. S4). We then expanded the query of HPRD using the top 673 DEGs (FDR < 0.2) plus the WS deleted region genes and retrieved a network of direct PPI defined by the perturbations of this query subset. Two seed genes (SRF and GRB2), known to have PPI with two WS genes (GTF2I and LAT2) (39,40) were included in this HPRD query. The expanded HPRD query yielded a prominent PPI network (Fig. 6) consisting of 100 perturbed genes (nodes) integrated by 137 direct PPI connections (edges) that link WS region genes GTF2I, LAT2 and LIMK1 into a PPI network spanning multiple signal transduction pathways. Functional pathways were assigned to the central PPI network graph by comparisons with graphs of known pathways using a subgraph-matching tool, substructure index-based approximate graph alignment (SAGA) (41) that employs the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways database. The use of graphic annotation (rather than linear gene set overlap) maintains the added information in the topographic relationship of WS genes to constrain more precise pathway identification inherent in the established MSigDB gene sets. The three KEGG pathways with the highest degrees of graphic overlap with the WS PPI nodes were: ‘regulation of actin cytoskeleton,’ ‘insulin signaling pathway’ and ‘MAPK signaling pathways,’ here determined solely by PPI and recapitulated the results from transcriptional analyses. We note that the use of the SAGA tool to graphically annotate the functions of the PPI graph preserves the relationship of the entire set of WS PPI genes, thereby significantly increasing the sensitivity and certainty of the three resulting pathways. This derives from two features; the use of only the PPI subset of DEG’s to generate the graph, as these are determined by the most solid, stringent criterion for inclusion, PPI, and the preservation of information on the interactions of the entire set of gene–protein components. This is in contrast to the standard GSEA or PPI analysis that would compare only the unordered gene sets for overlap and not be able to assign functions to regions of the PPI graph. An illustration of this is seen in comparing the top 114 DEG’s (FDR < 0.05) with the GSEA set that result in the same three pathways but with low probabilities as less information is used. The use of SAGA captured the higher order gene–protein topology and resulted in a stringent, highly significant definition of networks. Taken together, the triad of GSEA, GO and PPI analyses each identified disrupted functions of actin cytoskeletal signaling, of the MAPK and the IGF1-PI3K-AKT-mTOR signaling and proteostasis stress pathways as core consequences of the WS deletion.
Figure 6.
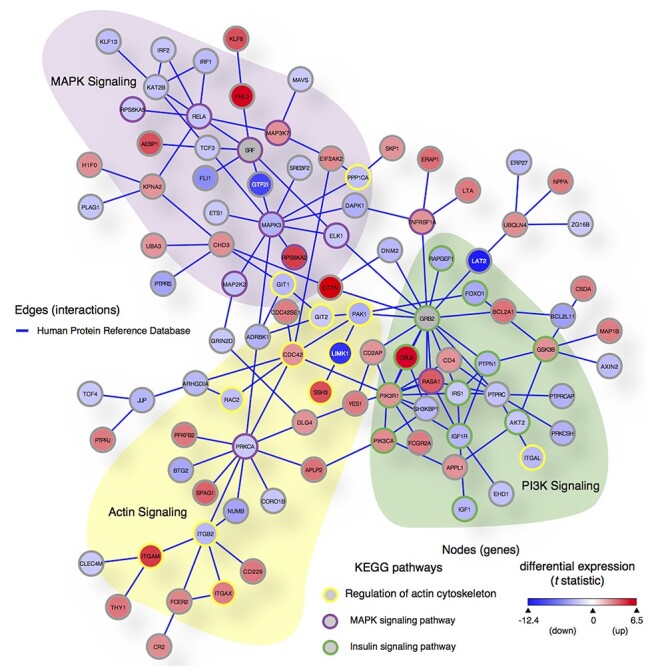
PPI network of DEGs implicates three core networks disturbed in WS; 100 genes (nodes) and 137 direct PPI (edges) from HPRD (release 9) are among the 673 DEGs in WS (FDR < 0.2) plus WS deleted genes and two seed proteins (SRF and GRB2) known to directly interact with two WS genes (GTF2I and LAT2). We used SAGA (41) to query this PPI graph against the KEGG Human pathways database. Node color is scaled by differential expression (scale bar in lower right corner from moderated t statistic between −12.4 and 6.5, from blue to red) and node border is colored by KEGG pathways: purple = MAPK signaling pathway (hsa04010), yellow = regulation of actin cytoskeleton (hsa04810) and green = insulin signaling pathway (hsa04910). Three WS region genes retrieved in this network (GTF2I, LIMK1 and LAT2) are shown with white node labels.
Power simulation analysis
A major challenge contributing to inconsistent gene network determinations across genomic developmental disorders such as WS, has been the difficulty of identifying a sample size sufficient to define genetic variations common to the disorder, versus differences that may exist between small groups of individuals. We therefore performed a simulation to evaluate the consequences that smaller sub-cohort datasets could show on the reliability of DEG’s inferred here (Fig. 7A). The results indicated that while ~9 subjects provided 80% power to detect the top 13 WS genes and 13 were required for 90% power (FDR = 0.2). In contrast, ~20 subjects were needed to detect the top 13 non-WS genes with 60% power (FDR = 0.2). To maintain this power at FDR = 0.01, 5 more subjects are needed for both WS and non-WS genes. A similar analysis described in the methods, of each network below, revealed that capturing 50% of genes in each of the pathways that was identified by the KEGG 2016 pathway analysis through Enrichr (42), converged on 20–25 subjects for 95% power (FDR = 0.2) for each of the three pathways respectively (Fig. 7B). The certainty with which the WS genes progress to top ranks with increasing subject number indicates the need for increased cohort sizes to increase the accuracy and generalizability of WS and non-WS DEG’s reflecting the common CNV in WS. Moreover, the minimal number is related to the inherent variation of the dataset due to technique, mechanism (CNV versus downstream gene) and biological variation. These are also minimal estimates because a simulation analysis using fewer subjects is unlikely to have generated a DEG set supporting the observed PPI network. Consequently, whereas previous network inconsistencies among studies using smaller cohorts found significant group differences, these would have a lower probability of representing the generalizable perturbations of WS and likely explain previous differences. Moreover, in genomic disorders, the power to detect a given network depends on the molecular methods used, the tissue, its internal variation, metabolic state and preparation, and the difference between and inherent variation of genes in WS and controls, making it unrealistic to compare results or to predict size of phenotypic effect or cohort size as commonly required. In contrast, simulation analyses of the sensitivity, consistency and power of a dataset a posteriori as illustrated here, are essential to evaluating rigor and to the ability to generalize results as defining downstream transcriptional networks in WS and other genomic disorders.
Figure 7.
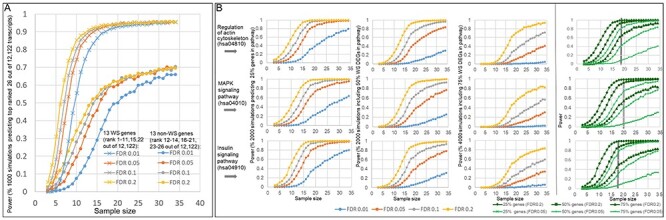
Simulated power curves of sample size. (A) Simulated power curves for detecting differential expression from smaller subsets of the data show that detection of the 13 WS genes (rank 1–11, 15, 22 out of 12 122 transcripts) approaches 90% power at ~9 samples, while detection of the top 13 non-WS genes (rank 12–14, 16–21, 23–26 out of 12 122 transcripts) approaches 60% power at ~20 samples (FDR = 0.2). (B) Simulated power curves for detecting differential expression from smaller subsets of the data show the sample size of detection of 25, 50 or 75% of the WS DEGs (FDR < 0.2) (Supplementary Material, Table S10) picked up by KEGG 2016 pathways (regulation of actin cytoskeleton (hsa04810); MAPK signaling pathway (hsa04010); insulin signaling pathway (hsa04910)). The simulations indicate that to identify the actin signaling pathway, when FDR = 0.2, at least 15 cases of the current dataset would have been needed to have detected at least 25% of the genes with 90% power, and to have detected 50% of the genes with 90% power, at least 20 cases would have been needed. For the insulin signaling pathway, 18 and 25 cases would have been needed to attain this power, and for the MAPK signaling pathway, 16 and 25.
Network analysis in atypical WS: actin signaling perturbation
Finally, we asked whether individuals with atypical deletions could be used to parse the deleted WS genes responsible for particular network perturbations. To examine whether a similar gene subset was enriched or shared by the atypical and full deletions, we used GSEA to compare the DEG expression profile from the 6 atypical WS (query set) with the ‘WS signature gene set’ (target set) derived from the full cohort (114 DEGs, FDR < 0.05). Because the genes deleted in the small (n = 6) cohort are a subset of those deleted in the larger (n = 34), the query is refined to asking whether the leading edge of the atypical DEG profile is enriched for the WS signature gene set, obviating the power of six samples to query the KEGG datasets (Enrichr (42); Supplementary Material, Table S8). Supplementary Material, Fig. S5A shows the enrichment plot indicating which of the 12 122 genes differently expressed in atypical WS (appearing on the x-axis with the highest rank order at the left) were shared with the typical ‘WS signature gene set’ indicated by vertical black bars distributed throughout. The leading edge (55 genes defined by the maximum enrichment score in Supplementary Material, Fig. S5A) for the atypical WS indicates those are shared with the typical WS. Supplementary Material, Fig. S5B shows shared deleted genes (LAT2, LIMK1, RFC2, EIF4H and ABHD11) as well as the non-deleted gene CLIP2 (with a position effect shown in Fig. 4), are all enriched in the left-side of the leading subset, shared with full deletion WS, while non-deleted genes (NSUN5, WBSCR22, BAZ1B, DNAJC30, GTF2I, BCL7B and TBL2) are in the right-side of the trailing edge, not shared. The pathways of genes in the leading and trailing edge sets were mapped using the PANTHER biological pathways and biological process categories (Supplementary Material, Fig. S5B). Taken together, the atypical deletions implicated non-deleted CLIP2 in the cause of phenotype, and narrowed gene candidates for perturbed actin signaling in WS.
Discussion
Augmented query targets refine networks: synergy in the elucidation of cellular disease mechanisms
The analyses of networks perturbed in WS incorporated the results of increased cohort size for full deletion WS, combined with a rare family of six persons with WS and identical smaller deletions, and exon level analyses, indicated a core set of genetic network perturbations associated with WS and implicated fundamental cellular processes including actin cytoskeletal, IGF1-PI3K-AKT-mTOR proteostasis and MAPK pathways. These emerged from a customized use of a sensitive transcriptional difference set to independently query different aspects of cellular and organismal features such as PPI and transcriptional systems represented in established datasets. Analyses were enriched by the application of analytic tools, GeneMANIA that integrates literature-based datasets with existing query targets, and the SAGA graphing tool that utilizes unbiased graph comparisons of known pathways to annotate the complex, function-free graphs that emerge from the PPI interactions predicted by the DEG difference set. It is important to note that, while each KEGG approach detected the three networks in the top ranked pathways (114 genes (FDR < 0.05) ranks 1–3; 686 genes (FDR < 0.2) ranks 1, 4, 36; of the PPI network subset from FDR 0.2 (100 nodes) ranks 9, 4, 8), only the graphical matching tool identified them with high certainty (adjusted P-value < 2E-12) and their interactions unambiguously (Supplementary Material, Table S9). Each of the independent analytic approaches (GSEA, GO, PPI) provided overlapping evidence that indicated a common set of networks for WS, augmented the cellular systems information and in sequential permutation analyses, converged on a generalizable set of core networks perturbed in WS. The results underscore the value of combined copy number and transcriptional analyses of human genomic disorders with large genetic effect behavioral phenotypes, for anchoring the large number of smaller effect DNA risk loci emerging from GWAS studies to cellular networks. In fact, in-depth grasp of stepwise mechanisms will benefit from multiple simultaneous datasets that detect different cellular regulatory features and thereby converge on interacting networks. In particular, these measures of quantitative transcription provide a framework implicating perturbed translational networks, many of which would not be detected as they are regulated largely by phosphorylation as in proteostasis, or by translational mechanisms such as ribosomal efficiency, thereby defining translation as the critical next direction for deciphering mechanisms and defining therapeutic targets.
Position effect in atypical deletions implications for phenotypic maps
The work provides a clue to the importance of going beyond DNA sequence and copy number to intermediate traits necessary for relating adult cellular processes and mechanisms to adult behaviors and for accurately using rare subjects with atypical deletions or duplications. Specifically, the data provide no evidence for position effects on flanking genes in full deletion WS, in contrast to previous reports, but do indicate a significant position effect in the six atypical deletions, that decreased expression of the non-deleted flanking gene, CLIP2, likely through altered function of undefined regulatory sequences upstream of the established promoter. The consequences of undetected position effects, regardless of tissue, could misidentify causative elements, emphasizing the need for functional analyses to evince meaningful genetic contributions to phenotype using developmental disorders. It is notable that the lack of GTF2I deletion in the atypical WS coincides with its position at the hub of the non-disturbed MAPK pathway. The lack of altered expression of insulin pathway genes IGF1, AKT2, IRS1 and FOXO1, in the atypical WS suggests that their deletion may contribute but is still not sufficient to disturb insulin signaling. However, the striking and shared overexpression of the multi-targeting ubiquitin network genes CBLB and BCL2A1 in the same pathway, may combine with genes found only in the full deletions, to drive the insulin network decreases and differential phenotypic dysfunction.
Dataset power to establish common networks
Although it is clear that increasing sample size improves identifying perturbations common to a disorder such as WS, the current large sample size and results provide an approach to evaluate the certainty with which a dataset has identified generalizable networks. For KEGG, this will depend on the size of the gene set and the number of genes needed to identify it as well as on the features of the query set including technical and biological variation. Therefore, increasing sample size helps to approach but does not guarantee accurate definition of perturbed networks representing the larger population, until it exceeds a number after which the DEG set reproducibly includes the genes defining the network. In the current work, the use of a PPI graph to filter the 686 DEG’s (FDR < 0.2) resulted in a total of 100 nodes (DEG’s) of which only 33 were needed to identify the three networks, each annotated by only 10–14 DEGs. The remaining DEG’s in the WS PPI graph were connected but the functional significance of the pattern was not recognized and therefore not annotated by SAGA as matching any currently known gene association in the KEGG MSigDB gene sets. Therefore, although the group of 100 WS PPI DEG’s recognize six further KEGG functional gene sets, the matching DEG’s are not connected by known functional properties in the WS PPI graph nor in the KEGG gene sets. Thus, the WS graph provides evidence for novel PPI of unknown functional significance. Moreover, because the ranks of many nodes fall close to the FDR limit, it is likely that smaller sample size would not have detected the complete WS PPI graph. Therefore, to illustrate the extent to which network identification is dependent on sample size, we simulated the proportions of WS DEGs defining KEGG genes, that would have been detected by smaller sample sizes.
Transcriptional consequences of genomic aneuploidy and CNV: lack of dosage compensation in WS. Is decreased network flexibility a shared CNV consequence?
In WS, the 13 detected deleted genes show decreased transcript levels largely proportional to copy number, viz., a lack of dosage compensation, and a variance (standard error/mean) similar to controls. In contrast, although it would not be unreasonable to expect the downstream genes and disturbed networks to be more variable in WS than controls, they are not and in fact, the opposite is seen for the top 13 non WS DEG’s (Fig. 2A and B; Supplementary Material, Fig. S6). Moreover, this was a property of the overall dataset, wherein 92% of non-WS genes revealed decreased gene variation in WS versus controls, and the extent of the decrease was greatest in genes with greatest between group difference for the top versus bottom 100 DEG ranks. The relationship of DEG rank to observed difference in WS versus control gene variance is not obvious in that this would have led to greater between group variance and a consequent decreased power to detect a DEG. The unexpected decreased gene variance in WS for the top DEG’s may suggest that in controls, genes with greatest variance among individuals may constitute a functional class but regardless, their decreased variation in WS indicates a general blunting of response that may reflect a decreased flexibility of transcriptional networks due to invariant dosed decreases of the deleted genes involved. This blunting may be imagined to result from a subset of tightly dosed transcriptional regulatory mechanisms that act as on–off switches. Taken together, the data and speculation require replication and further study but if consistent across CNV disorders, the hypothesis may provide a common mechanism by which human aneuploidies result in phenotypic deficits.
Significance of human cellular network perturbations for role of excitatory dendritic spines in WS
It is clear that brain gene expression modules are shared properties across tissues (43) and conversely, that expression varies enormously across development and adult brain tissues and functional states. Consequently, the striking network match (No. 1 of ~2000 queried gene sets) between the WS DEG and the MSigDB, actin cytoskeletal signaling at the synapse not unreasonably, suggests that in WS, the actin cytoskeletal signaling network is awry, likely related to a module shared at the synapse, specifically the dendritic spine of excitatory synapses (37) (Fig. 5C). Further, the DEG’s for this network are also disturbed in the six atypical deletions (Fig. 4; Supplementary Material, Fig. S5), suggesting that the WS genes contributing to the perturbed actin cytoskeletal signaling network, includes both those deleted (LAT2, LIMK1) and the non-deleted flanking gene, CLIP2, whose expression is decreased due to the position effect noted above. The involvement of CLIP2 was not implicated by the DNA deletion but only by the transcriptional network analysis. Unexpectedly, previous work has shown a dosage sensitive role for the above WS and non WS genes, in spine regulation. In murine and cellular models (44), decreased CLIP2 results in greater numbers of thin spine protrusions that are also the characteristic features of WS brain, layer V/VI frontal cortex (45,46). The data reported here are distinct from previous work on single WS genes, and by integrating these with their network perturbations, now provide independent transcriptional evidence from WS cells, for the intermediate cellular traits of non-WS genes drebrin (DBN1, rank 20) and cortactin (CTTN, rank 12) that independently indicate perturbed actin cytoskeleton signaling and place the shared module at the post synaptic spine.
The results unexpectedly provide evidence and WS as a human genetic model for the emerging role of integrated microtubule-actin signaling in dendritic spine dynamics (Fig. 5C and D). Drebrin and cortactin are major regulators of microtubules in adult spines and axonal growth cones (47,48), components of the bridge linking the plus end of microtubules, the site of CLIP2 and EB3 (49), to F-actin in the spine. Upon NMDAR excitation, these translocate MT to the spine and facilitate signaling to the DLG4/PSD-95 (rank 593) (50) complex mediating membrane response. Moreover, LIMK1 phosphorylates p25/TPPP (tubulin polymerization protein) on serine residues and thereby promotes destabilization of microtubules (51). The expression of p25/TPPP predominantly in myelinating oligodendrocytes (52), implicates both LIMK1 and GTF2I (53) deletion in the abnormal myelin observed in WS. In summary (Fig. 8), together with the literature, the current report suggests dysregulated actin and microtubule signaling mediated by WS genes and downstream effects on PPI and regulatory networks, as a mechanism for disturbed brain development and adult WS brain abnormalities of excitatory dendritic spines and myelin and as substrates and possible therapeutic targets for ongoing adult cognitive and behavioral deficits in WS.
Figure 8.
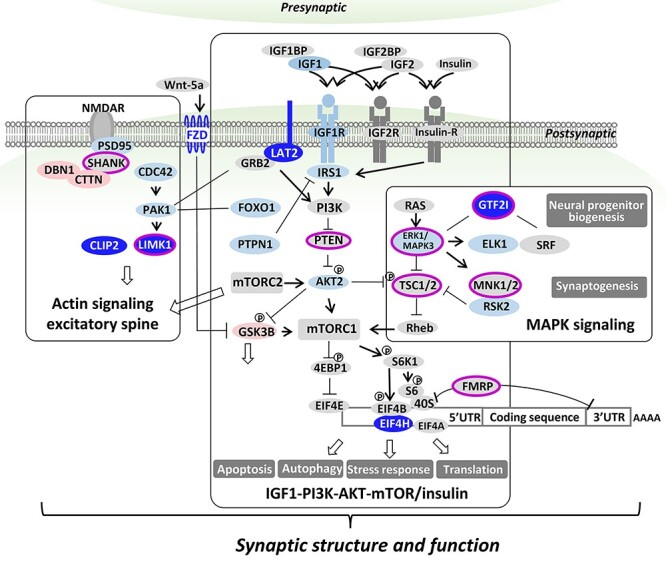
WS core perturbed transcriptional networks echo those for ASD and implicate dendritic spine and synapse. Results indicate three interacting networks that each act at the synapse and include six deleted genes (CLIP2, LIMK1, LAT2, GTF2I, EIF4H, FZD9) that are perturbed in WS: actin signaling at the excitatory dendritic spine (Fig. 5C and D), IGF1-PI3K-AKT-mTOR/insulin and MAPK. These synaptic networks that are involved in brain development and post-natal plasticity influence morphogenesis, proliferation, migration and differentiation, suggesting shared pre- and post-natal mechanisms that affect social brain are involved in WS and ASD. Genes deleted and under-expressed in WS are in dark blue ovals. Downstream DEG’s are light blue or red ovals indicating under- or over-expression, respectively in WS versus TC. Genes associated with ASD are denoted by fuchsia surrounds.
Disturbed IGF1-PI3K-AKT-mTOR signaling at the dendritic spine and synapse in WS and ASD
Our results indicate a role for altered IGF1-PI3K-AKT-mTOR signaling in WS (Fig. 8), although it is unknown how this varies with tissue or development. Nonetheless, there is mounting evidence from rodent models for the role of IGF1-PI3K-AKT-mTOR in synaptic structure or function when hyper- or hypo-activated (54), for MECP2 in spine density (55), for FMRP in accumulation of dendritic proteins and impaired autophagy (56), and for TSC1/TSC2 in impaired dendritic spine pruning and autophagy associated with ASD-like behavioral abnormalities (57). In conclusion, it is not unreasonable to suggest that genes(s) in the WS region may regulate this core network in a dosage-dependent manner, possibly identifying specific genes associated with common cognitive and inverse behavioral features seen in WS compared with ASD. The intriguing similarity of WS networks inferred from transcription, to ASD networks inferred largely from Mendelian and ASD associated DNA based risks, does suggest their involvement in the pathogenesis of social and associated cognitive deficits, but does not establish which genes are critical or proportional for adult deficits, nor whether their downstream functional directions are the same in these disorders of contrasting social behavior. These are important questions that have been focused but not answered by the current results. Therefore, although caution is warranted in that the flip of unpredicted phosphorylation or methylation sites in protein or transcriptional regulation respectively, can reverse functionality (58), elucidating these has been critical for understanding regulation, and may similarly identify gene regulatory mechanisms for social behavior in humans. It is not unreasonable to expect a gene-specific regulatory switch for approach versus avoidant social behavior in the WS region in view of the paradoxical increased risk of ASD in WS as well as the inverse pattern of increased and decreased transcript levels observed in the duplications and deletions (24). However, identifying such genes will require quantitative analyses comparing expression and phenotypes, studies that are currently lacking. Moreover, further phenotypic parsing is necessary to define ASD features such as language production and affect that are quantitatively inverse in WS versus ASD (59) or versus WS region duplications (60). In summary, combining the current results with the literature paints WS as a promising model in which to Garner critical insights into regulatory genetic switches for human social behavior. Further study is warranted to define downstream proteins regulated by mTOR in WS and ASD and determine those that vary with adult cognition and behavior.
Mendelian metabolic disorders of IGF1-PI3K-AKT-mTOR signaling are more prevalent in WS and ASD
In WS, the most common metabolic abnormality, impaired glucose tolerance and insulin resistance are seen in ~75% of adults (61,62) and may be related to dysregulation of the IGF1-PI3K-AKT-mTOR pathway reported here as it regulates insulin signaling and resistance (63,64). Three further syndromes due to single gene mutations that regulate mTOR, Tuberous sclerosis 1 and 2 (TSC1, TSC2) (65) and Rett syndrome (MECP2) (66), also show abnormal glucose tolerance, the latter due to upregulation of PTPN1, also dysregulated in WS. It is unclear but of interest to consider if the positive response in rodents and humans, to inhibiting PTPN1 would support upregulating it in WS as a therapeutic (67). Further, although insulin resistance was found in rodents but not humans with the Fragile X syndrome (mutations of FMRP) (Fig. 8), the results of a preliminary therapeutic trial with metformin, a treatment for insulin resistance, suggested possible response (68). In mice heterozygous for knockout of the tumor suppressor, PTEN, a regulator both of insulin signaling and tumor formation, the associated social deficits are in part rescued by mTOR inhibition (69), implicating deficits of multiple single genes with different mechanisms as related to this network. Finally, despite the multigenic contributions to idiopathic ASD, clinical features related to metabolic syndrome including insulin resistance, obesity, adaptive cellular stress responses and systemic inflammation, are more prevalent (54). In conclusion, although further study is warranted, both WS and genetic disorders associated with ASD, point to dysregulation of the IGF-PI3K-AKT-mTOR pathways as possible metabolic bases not only for insulin resistance, but also for social and cognitive deficits due to developmental and ongoing adult brain dysfunction.
WS core transcriptional networks echo those for ASD and SCHZ: IGF1-PI3K-AKT-mTOR, MAPK and actin signaling
Converging gene-disease associations from GWAS and genomic disorders have implicated excitatory dendritic spine components and IGF1-PI3K-AKT-mTOR (70–74), MAPK (75,76) and actin signaling (77–79) related to proteostasis stress pathways (80,81), in both ASD and SCHZ (82–88). The results reported indicate that the transcriptionally perturbed networks of WS, a single CNV disorder of social dysfunction, are surprisingly similar to those signaling networks related to the spine and proteostasis-stress networks inferred from a subset of the DNA risk alleles of ASD and SCHZ. The association of both WS region deletion and duplication with ASD risk (81,89,90) combines with the current report to suggest a possible common genetic and neurobiological pathogenesis of neuropsychiatric disorders of social interaction for which WS now provides large effect size gene candidates for both the developmental and adult brain substrates. The unexpected coherence of networks for social dysfunctions, one of which results in approach and enjoyment of social interaction, WS, and the other that results in social avoidance, ASD, suggests that a common neural and genetic system may be involved in both, and that the small number of genes deleted in WS may provide critical insights for treatments of ASD, for which hundreds of genes with small effects have obscured clarity of causation. The role of these networks in the dysregulation of hypothalamic oxytocin and vasopressin related to emotion in WS (91), and social behavior in ASD, may provide focus for determining shared mechanisms and therapeutic targets. In contrast to ASD, the defined network perturbations due to a single genetic cause now provide a cohort in which to define the transcripts that scale with cognitive and behavioral phenotypes.
Material and Methods
Subject population
Our population includes 34 WS patients and 18 controls (a subset of the parents who transmitted the normal chromosome 7) (Supplementary Material, Table S1). The subjects and their families are recruited as part of an ongoing research study approved by the Institutional Review Boards at Cedars-Sinai Medical Center, The Salk Institute and The University of Utah. In each case, the diagnosis of WS is determined by medical history, clinical and laboratory evidence and is then confirmed by multicolor FISH (92). All WS subjects completed the WAISR-test (Supplementary Material, Table S1) within 2 years when they donated cells for LCLs generation (30).
Total RNA and DNA preparation from cell lines
Immortalized LCLs from each of the 34 WS patients and 18 normal controls are generated and cultivated under standardized conditions. The cells are grown in RMPI 1640 with 10% FBS, 5% pen/strep, 5% L-glutamine and 0.5% gentamycin. The total RNA is isolated by using the QIAGEN RNeasy Kit. The DNA is isolated by using the QIAGEN DNA kit.
DNA CNV determination
The DNA CNV of subjects of typical WS were determined by using Illumina Human 1 M-Duo microarray analysis. The precise CNVs of subjects of atypical WS were determined by using NimboGen custom designed high density microarray on Chromosome 7.
Exon array expression profiling
Ribosomal RNA was removed from total RNA (1 μg per sample) using the RiboMinus Human/Mouse Transcriptome Isolation Kit (Invitrogen). Biotinylated target was then prepared following the protocol described in Affymetrix GeneChip® Whole Transcript (WT) Sense Target Labeling and Control Reagents (Affymetrix P/N 900652). Hybridization was performed at 45°C for 16 h using 5 μg of biotinylated target with each GeneChip® Human Exon 1.0 ST array (HuEx-1_0-st-v2, Affymetrix). Following hybridization, non-specifically bound material was removed by washing and detection of specifically bound target was performed using the GeneChip Hybridization, Wash and Stain kit, and the GeneChip Fluidics Station 450 (Affymetrix). The arrays were scanned using the GeneChip® Scanner 3000 7G (Affymetrix) and raw data were extracted from the scanned images and analyzed with the Expression Console software package (Affymetrix). Expression Console was used to compute separate gene-level and exon-level signal estimates for the Exon 1.0 ST Array data. Exon-level estimates were derived using the PLIER (pm-gcbg) method after quantile sketch normalization. Probes sets (exons) were filtered for a ‘detected above background’ (DABG) P-value less than 0.05, resulting in 313 707 filtered exons that were considered in this study. Exon-level probe sets annotations were derived from the Affymetrix NetAffx file HuEx-1_0-st-v2 Probeset Annotations, CSV Format, Release 31. Gene-level estimates were derived using the IterPLIER (pm-gcbg) method after quantile sketch normalization. Only genes with core annotation (22 012 genes) were considered in this study. Transcript annotations were derived from the Affymetrix NetAffx file HuEx-1_0-st-v2 Transcript Cluster Annotations, CSV, Release 31.
RNA-Seq
The standard Illumina mRNA-Seq protocol with random hexamer priming (mRNA-Seq Sample Prep Kit RS-100-0801, Illumina, San Diego, CA) was used to build libraries from 1 μg of total RNA for single-read sequencing on the Illumina Cluster Station and Genome Analyzer. The adaptor ligated library was size selected in the ~200 bp range by separation and extraction from a 4% agarose. The library was PCR amplified by Phusion DNA polymerase (Stratagene), and purified by Qiaquick PCR purification kit (Qiagen). Each library was quantified with an Agilent Bioanalyzer and was loaded into its own single Illumina flow cell lane, producing an average of 15 million clusters per lane. 36-mer sequence reads were generated on the Illumina Genome Analyzer IIx using the 36-cycle Sequencing Kit (FC-104-4002). Sequence alignments were generated with ELAND (Illumina). ELAND result files from matching Illumina 36-mer sequence data against reference sequences were parsed with a custom-written Perl script.
qRT-PCR
TaqMan Gene Expression Assays, designed for human gene transcripts (Applied Biosystems, Foster City, CA), were used for quantifying gene expression in this report. The assay mixture consists of a TaqMan® MGB probe (labeled with FAM™ dye) and unlabeled PCR primers for a specific human gene. All the Taqman expression primer sets were commercially available from ABI, and matched the Affymetrix chip at exon level. First, pooled total RNAs from 34 WS patients or 18 typical controls were reverse transcribed into single strand cDNAs using the SuperScript III Frist-Strand Synthesis system for RT-PCR (Life Technologies, USA) and used as templates for qRT-PCR of 8 WS genes (LIMK1, CLIP2, EIF4H, GTF2I, BAZ1B, NSUN5, LAT2 and STX1A), and 22 top under- or over-expressed non-WS genes (under-expressed: FOXO1, BCL2L11, GIT1, ZMIZ1, KLF12, DAPK1, FLI1, POU2AF1 and PAK1; over-expressed: CTTN, FHL3, SERPINF1, LIPG, RPS6KA2, CBLB, TRPS1, DBN1, ITGAM, PIKFYVE, TUBA1A, ENPP4 and RASA1). Then, the un-pooled total RNAs from 34 WS patients or 18 typical controls were used to apply qRT-PCR of 8 genes (STX1A, DAPK1, LIMK1, GTF2I, CTTN, DBN1, CLIP2 and FHL3). Three genes (ACTB, PPIA and TBP) were used as endogenous controls throughout all the qRT-PCR batches. The qRT-PCRs were performed on Applied Biosystems 7900HT system (USA) and the data were analyzed using SDS 2.3.
Western blotting
Cellular protein extracts were prepared by a single-step lysis method. The harvested cells from 6 WS patient- and five control-derived LCLs were suspended in SDS-PAGE sample buffer (Laemmli sample buffer; Bio-Rad, Cat# 161-0737) and then boiled for 5 min. Equal amounts of the extracts were used for western blot analyses to determine the levels of proteins using the antibodies listed below. Extracts were separated using SDS-PAGE and then transferred to membranes. After the transfer, membranes were blocked using 4% skim milk in 0.1% Tween-20/PBS for 2 h. After blocking the membranes were incubated with primary antibody overnight in 4% skim milk in 0.1% Tween 20/PBS at 4°C. After the primary antibody incubation, several washes were performed using 0.1% Tween 20/PBS and then the corresponding secondary antibody conjugated with HRP was added. The secondary antibody was incubated with the membrane for 2 h in 4% skim milk in 0.1% Tween 20/PBS at 4°C. After the secondary antibody incubation and several more washes with 0.1% Tween 20/PBS signals were detected using Immobilon Western Chemiluminescent HRP Substrate (Millipore, Cat# WBKLSO500). To ensure equal loading of each sample, the membranes were then re-probed with β-actin conjugated with HRP. The primary antibodies were used at a concentration of 1:5000. The primary antibodies used were: EIF4H Rabbit mAb (cell signaling, Cat# 3469), LIMK1 Rabbit polyclonal (cell signaling, Cat# 3842), NTAL/LAB Rabbit mAb (cell signaling, Cat# 11986), GTF2I/TFII-I Rabbit polyclonal (cell signaling, Cat# 4562), monoclonal anti-β-Actin−peroxidase (clone AC-15; 1:50000; Sigma-Aldrich, A3854). Secondary antibody used was Peroxidase-conjugated AffiniPure goat anti-rabbit IgG (H + L) (1:5000; Jackson ImmunoResearch Laboratories; Cat# 111-035-144).
Statistical analysis
Signal estimates from Affymetrix expression console processing were corrected for batch effects using the ComBat algorithm implemented in the R package (93). Batch-corrected exon-level and gene-level signal estimates were processed for differential expression analysis using the functions ‘lmFit,’ ‘eBayes’ and ‘topTable’ implemented in the software package Limma for the R computing environment. The empirical Bayes approach was used to calculate the moderated t-statistic, and the FDR was controlled using Benjamini and Hochberg’s method as implemented within ‘topTable.’ GO term enrichment analysis used the hypergeometric-based tests on the gene-level summaries ranked by their moderated t-statistic, performed by the GOstats package from Bioconductor with a P-value cutoff of 0.01. GSEA used the C2 collection of canonical pathways and literature gene sets, version 3.0 from the Molecular Signatures Database (MSigDB), supplemented with the CHR7Q11 gene set from the C1 collection, and an a posteriori literature gene set ‘Actin cytoskeleton form and function at the synapse’ derived from Dillon and Goda (37) is also included. A list of 12 022 genes (Supplementary Material, Table S3) was used, and the rank order was by the absolute value of the moderated t-statistic. GSEA using the algorithm of Subramanian et al. (34), was used with the pre-ranked gene list, and the algorithm of Irizarry et al. (35), used a standard χ2 test was also used for detecting changes in scale. Direct PPI networks were constructed using the Disease Association Protein–Protein Link Evaluator, DAPPLE version 0.17 within the GenePattern tool suite (https://genepattern.broadinstitute.org) (94) and by manual evaluation of direct PPI from the HPRD, release 9 (downloaded from www.hprd.org) (95).
Power simulation analysis
Simulated power curves of sample size were performed as: This involved selecting the set of 13 differentially expressed WS genes (rank 1–11, 15, 22 out of 12 122 transcripts) and 13 differentially expressed non-WS genes (rank 12–14, 16–21, 23–26 out of 12 122 transcripts) (defined by the 0.006% FDR threshold for differential expression, that was generated using 34 WS samples versus 18 controls) from the gene-level summaries (12 122 detected transcripts). Next, for n = 3–34 (WS samples) and for n = 3–18 (controls), n samples were drawn from each group (matched sample size from 3 to 18, and controls were fixed at 18 for 19–34 WS samples). Differential expression analysis was performed using the Limma functions ‘lmFit,’ ‘eBayes’ and ‘topTable’ cohort subset. Following this, genes at 0.01, 0.05, 0.1 and 0.2 FDR levels were selected and the above analysis of sub-cohorts was iterated 1000 times. Power was defined as the proportion of iterations for each sub cohort, that yielded the target gene set within the FDR level selected. The power was averaged over the 1000 iterations for the top 13 DEG’s deleted in WS and the top 13 downstream non-deleted DEG’s and graphed. In addition, by query KEGG 2016 database through Enrichr (42) using the 686 DEGs (FDR < 0.2), we identified 20, 14 and 11 genes (referred as positive genes hereafter) in three pathways respectively (Regulation of actin cytoskeleton (hsa04810); MAPK signaling pathway (hsa04010); insulin signaling pathway (hsa04910)). We investigated what is the power to detect 25, 50 or 75% of those genes having different expression between case and control groups should, we had smaller sample size than we have used for the analysis, i.e. 34 cases plus 18 controls. Briefly, n (3 ≤ n ≤ 34) subjects are sampled from the 34 cases and 18 controls respectively with replacement. When n > 18, only 18 subjects were sampled from the control group. A linear regression was fitted for each gene and moderated t-statistics, and log-odds of differential expression were calculated by empirical Bayes moderation of the standard errors towards a common value (96). P-values were adjusted by Benjamini and Hochberg method (97) and the top genes with P-values smaller than 0.2 are identified. If 25, 50 or 75% genes of the positive genes are identified as the top genes, we call it a success. The power was defined as the probability of success in 2000 simulations. R package limma was used for the analysis (98).
Supplementary Material
Acknowledgements
Power Analyses were conducted by the University of Utah CCTS Population Health Research Core, Jian Ying in consultation with Julio C. Facelli, University of Utah Biomedical Informatics. We would like to acknowledge the acquisition of blood samples by core B (P01 NICHD033113) and LCLs culture by Tal Tirosh-Wagner. We would like to thank all WS individuals and their families.
Contributor Information
Li Dai, Center for Integrated Neuroscience and Human Behavior, Brain Institute, Department of Pediatrics, University of Utah, Salt Lake City, UT 84112, USA.
Robert B Weiss, Department of Human Genetics, University of Utah, Salt Lake City, UT 84112, USA.
Diane M Dunn, Department of Human Genetics, University of Utah, Salt Lake City, UT 84112, USA.
Anna Ramirez, Center for Integrated Neuroscience and Human Behavior, Brain Institute, Department of Pediatrics, University of Utah, Salt Lake City, UT 84112, USA.
Sharan Paul, Department of Neurology, University of Utah, Salt Lake City, UT 84112, USA.
Julie R Korenberg, Center for Integrated Neuroscience and Human Behavior, Brain Institute, Department of Pediatrics, University of Utah, Salt Lake City, UT 84112, USA; Department of Neurology, University of Utah, Salt Lake City, UT 84112, USA.
Funding
National Institute of Child Health and Human Development Grant (P01 NICHD033113 to J.R.K and Ursula Bellugi), Utah Science Technology and Research Initiative (USTAR) to J.R.K.
Conflict of Interest Statement. The authors declare no conflict of interest.
References
- 1. De Rubeis, S., He, X., Goldberg, A.P., Poultney, C.S., Samocha, K., Cicek, A.E., Kou, Y., Liu, L., Fromer, M., Walker, S.et al. (2014) Synaptic, transcriptional and chromatin genes disrupted in autism. Nature, 515, 209–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Willsey, A.J., Morris, M.T., Wang, S., Willsey, H.R., Sun, N., Teerikorpi, N., Baum, T.B., Cagney, G., Bender, K.J., Desai, T.A.et al. (2018) The psychiatric cell map initiative: a convergent systems biological approach to illuminating key molecular pathways in neuropsychiatric disorders. Cell, 174, 505–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lalli, M.A., Jang, J., Park, J.H., Wang, Y., Guzman, E., Zhou, H., Audouard, M., Bridges, D., Tovar, K.R., Papuc, S.M.et al. (2016) Haploinsufficiency of BAZ1B contributes to Williams syndrome through transcriptional dysregulation of neurodevelopmental pathways. Hum. Mol. Genet., 25, 1294–1306. [DOI] [PubMed] [Google Scholar]
- 4. Barak, B. and Feng, G. (2016) Neurobiology of social behavior abnormalities in autism and Williams syndrome. Nat. Neurosci., 19, 647–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Meyer-Lindenberg, A., Kohn, P., Mervis, C.B., Kippenhan, J.S., Olsen, R.K., Morris, C.A. and Berman, K.F. (2004) Neural basis of genetically determined visuospatial construction deficit in Williams syndrome. Neuron, 43, 623–631. [DOI] [PubMed] [Google Scholar]
- 6. Malenfant, P., Liu, X., Hudson, M.L., Qiao, Y., Hrynchak, M., Riendeau, N., Hildebrand, M.J., Cohen, I.L., Chudley, A.E., Forster-Gibson, C.et al. (2012) Association of GTF2i in the Williams-Beuren syndrome critical region with autism spectrum disorders. J. Autism Dev. Disord., 42, 1459–1469. [DOI] [PubMed] [Google Scholar]
- 7. Mervis, C.B., Dida, J., Lam, E., Crawford-Zelli, N.A., Young, E.J., Henderson, D.R., Onay, T., Morris, C.A., Woodruff-Borden, J., Yeomans, J.et al. (2012) Duplication of GTF2I results in separation anxiety in mice and humans. Am. J. Hum. Genet., 90, 1064–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sakurai, T., Dorr, N.P., Takahashi, N., McInnes, L.A., Elder, G.A. and Buxbaum, J.D. (2011) Haploinsufficiency of Gtf2i, a gene deleted in Williams syndrome, leads to increases in social interactions. Autism Res., 4, 28–39. [DOI] [PubMed] [Google Scholar]
- 9. Schweiger, J.I. and Meyer-Lindenberg, A. (2017) Common variation in the GTF2I gene: a promising neurogenetic mechanism for affiliative drive and social anxiety. Biol. Psychiatry, 81, 175–176. [DOI] [PubMed] [Google Scholar]
- 10. Swartz, J.R., Waller, R., Bogdan, R., Knodt, A.R., Sabhlok, A., Hyde, L.W. and Hariri, A.R. (2017) A common polymorphism in a Williams syndrome gene predicts amygdala reactivity and extraversion in healthy adults. Biol. Psychiatry, 81, 203–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dai, L., Bellugi, U., Chen, X.N., Pulst-Korenberg, A.M., Jarvinen-Pasley, A., Tirosh-Wagner, T., Eis, P.S., Graham, J., Mills, D., Searcy, Y.et al. (2009) Is it Williams syndrome? GTF2IRD1 implicated in visual-spatial construction and GTF2I in sociability revealed by high resolution arrays. Am. J. Med. Genet. A, 149A, 302–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Frangiskakis, J.M., Ewart, A.K., Morris, C.A., Mervis, C.B., Bertrand, J., Robinson, B.F., Klein, B.P., Ensing, G.J., Everett, L.A., Green, E.D.et al. (1996) LIM-kinase1 hemizygosity implicated in impaired visuospatial constructive cognition. Cell, 86, 59–69. [DOI] [PubMed] [Google Scholar]
- 13. Mills, D.L., Dai, L., Fishman, I., Yam, A., Appelbaum, L.G., St George, M., Galaburda, A., Bellugi, U. and Korenberg, J.R. (2013) Genetic mapping of brain plasticity across development in Williams syndrome: ERP markers of face and language processing. Dev. Neuropsychol., 38, 613–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kopp, N., McCullough, K., Maloney, S.E. and Dougherty, J.D. (2019) Gtf2i and Gtf2ird1 mutation do not account for the full phenotypic effect of the Williams syndrome critical region in mouse models. Hum. Mol. Genet., 28, 3443–3465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Osso, L.A. and Chan, J.R. (2019) A surprising role for myelin in Williams syndrome. Nat. Neurosci., 22, 681–683. [DOI] [PubMed] [Google Scholar]
- 16. Roy, A.L. (2017) Pathophysiology of TFII-I: old guard wearing new hats. Trends Mol. Med., 23, 501–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kopp, N.D., Nygaard, K.R., Liu, Y., McCullough, K.B., Maloney, S.E., Gabel, H.W. and Dougherty, J.D. (2020) Functions of Gtf2i and Gtf2ird1 in the developing brain: transcription, DNA binding and long-term behavioral consequences. Hum. Mol. Genet., 29, 1498–1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ramirez, V.T., Ramos-Fernandez, E., Henriquez, J.P., Lorenzo, A. and Inestrosa, N.C. (2016) Wnt-5a/Frizzled9 receptor Signaling through the Galphao-Gbetagamma complex regulates dendritic spine formation. J. Biol. Chem., 291, 19092–19107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Collette, J.C., Chen, X.N., Mills, D.L., Galaburda, A.M., Reiss, A.L., Bellugi, U. and Korenberg, J.R. (2009) William's syndrome: gene expression is related to parental origin and regional coordinate control. J. Hum. Genet., 54, 193–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Merla, G., Howald, C., Henrichsen, C.N., Lyle, R., Wyss, C., Zabot, M.T., Antonarakis, S.E. and Reymond, A. (2006) Submicroscopic deletion in patients with Williams-Beuren syndrome influences expression levels of the nonhemizygous flanking genes. Am. J. Hum. Genet., 79, 332–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Torres, E.M., Williams, B.R. and Amon, A. (2008) Aneuploidy: cells losing their balance. Genetics, 179, 737–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Stranger, B.E., Forrest, M.S., Dunning, M., Ingle, C.E., Beazley, C., Thorne, N., Redon, R., Bird, C.P., deGrassi, A., Lee, C.et al. (2007) Relative impact of nucleotide and copy number variation on gene expression phenotypes. Science, 315, 848–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Henrichsen, C.N., Vinckenbosch, N., Zollner, S., Chaignat, E., Pradervand, S., Schutz, F., Ruedi, M., Kaessmann, H. and Reymond, A. (2009) Segmental copy number variation shapes tissue transcriptomes. Nat. Genet., 41, 424–429. [DOI] [PubMed] [Google Scholar]
- 24. Adamo, A., Atashpaz, S., Germain, P.L., Zanella, M., D'Agostino, G., Albertin, V., Chenoweth, J., Micale, L., Fusco, C., Unger, C.et al. (2015) 7q11.23 dosage-dependent dysregulation in human pluripotent stem cells affects transcriptional programs in disease-relevant lineages. Nat. Genet., 47, 132–141. [DOI] [PubMed] [Google Scholar]
- 25. Antonell, A., Vilardell, M. and Perez Jurado, L.A. (2010) Transcriptome profile in Williams-Beuren syndrome lymphoblast cells reveals gene pathways implicated in glucose intolerance and visuospatial construction deficits. Hum. Genet., 128, 27–37. [DOI] [PubMed] [Google Scholar]
- 26. Chailangkarn, T., Trujillo, C.A., Freitas, B.C., Hrvoj-Mihic, B., Herai, R.H., Yu, D.X., Brown, T.T., Marchetto, M.C., Bardy, C., McHenry, L.et al. (2016) A human neurodevelopmental model for Williams syndrome. Nature, 536, 338–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Henrichsen, C.N., Csardi, G., Zabot, M.T., Fusco, C., Bergmann, S., Merla, G. and Reymond, A. (2011) Using transcription modules to identify expression clusters perturbed in Williams-Beuren syndrome. PLoS Comput. Biol., 7, e1001054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Khattak, S., Brimble, E., Zhang, W., Zaslavsky, K., Strong, E., Ross, P.J., Hendry, J., Mital, S., Salter, M.W., Osborne, L.R.et al. (2015) Human induced pluripotent stem cell derived neurons as a model for Williams-Beuren syndrome. Mol. Brain, 8, 77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Love, M.I., Hogenesch, J.B. and Irizarry, R.A. (2016) Modeling of RNA-seq fragment sequence bias reduces systematic errors in transcript abundance estimation. Nat. Biotechnol., 34, 1287–1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gao, M.C., Bellugi, U., Dai, L., Mills, D.L., Sobel, E.M., Lange, K. and Korenberg, J.R. (2010) Intelligence in Williams syndrome is related to STX1A, which encodes a component of the presynaptic SNARE complex. PLoS One, 5, e10292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gheldof, N., Witwicki, R.M., Migliavacca, E., Leleu, M., Didelot, G., Harewood, L., Rougemont, J. and Reymond, A. (2013) Structural variation-associated expression changes are paralleled by chromatin architecture modifications. PLoS One, 8, e79973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Merla, G., Ucla, C., Guipponi, M. and Reymond, A. (2002) Identification of additional transcripts in the Williams-Beuren syndrome critical region. Hum. Genet., 110, 429–438. [DOI] [PubMed] [Google Scholar]
- 33. Hoeft, F., Dai, L., Haas, B.W., Sheau, K., Mimura, M., Mills, D., Galaburda, A., Bellugi, U., Korenberg, J.R. and Reiss, A.L. (2014) Mapping genetically controlled neural circuits of social behavior and visuo-motor integration by a preliminary examination of atypical deletions with Williams syndrome. PLoS One, 9, e104088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Subramanian, A., Tamayo, P., Mootha, V.K., Mukherjee, S., Ebert, B.L., Gillette, M.A., Paulovich, A., Pomeroy, S.L., Golub, T.R., Lander, E.S.et al. (2005) Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U. S. A., 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Irizarry, R.A., Wang, C., Zhou, Y. and Speed, T.P. (2009) Gene set enrichment analysis made simple. Stat. Methods Med. Res., 18, 565–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mostafavi, S., Ray, D., Warde-Farley, D., Grouios, C. and Morris, Q. (2008) GeneMANIA: a real-time multiple association network integration algorithm for predicting gene function. Genome Biol., 9, S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Dillon, C. and Goda, Y. (2005) The actin cytoskeleton: integrating form and function at the synapse. Annu. Rev. Neurosci., 28, 25–55. [DOI] [PubMed] [Google Scholar]
- 38. Falcon, S. and Gentleman, R. (2007) Using GOstats to test gene lists for GO term association. Bioinformatics, 23, 257–258. [DOI] [PubMed] [Google Scholar]
- 39. Grueneberg, D.A., Henry, R.W., Brauer, A., Novina, C.D., Cheriyath, V., Roy, A.L. and Gilman, M. (1997) A multifunctional DNA-binding protein that promotes the formation of serum response factor/homeodomain complexes: identity to TFII-I. Genes Dev., 11, 2482–2493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Koonpaew, S., Janssen, E., Zhu, M. and Zhang, W. (2004) The importance of three membrane-distal tyrosines in the adaptor protein NTAL/LAB. J. Biol. Chem., 279, 11229–11235. [DOI] [PubMed] [Google Scholar]
- 41. Tian, Y., McEachin, R.C., Santos, C., States, D.J. and Patel, J.M. (2007) SAGA: a subgraph matching tool for biological graphs. Bioinformatics, 23, 232–239. [DOI] [PubMed] [Google Scholar]
- 42. Kuleshov, M.V., Jones, M.R., Rouillard, A.D., Fernandez, N.F., Duan, Q., Wang, Z., Koplev, S., Jenkins, S.L., Jagodnik, K.M., Lachmann, A.et al. (2016) Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res., 44, W90–W97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. McKenzie, A.T., Wang, M., Hauberg, M.E., Fullard, J.F., Kozlenkov, A., Keenan, A., Hurd, Y.L., Dracheva, S., Casaccia, P., Roussos, P.et al. (2018) Brain cell type specific gene expression and co-expression network architectures. Sci. Rep., 8, 8868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Osborne, L.R. (2010) Animal models of Williams syndrome. Am. J. Med. Genet. C Semin. Med. Genet., 154C, 209–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lew, C.H., Brown, C., Bellugi, U. and Semendeferi, K. (2017) Neuron density is decreased in the prefrontal cortex in Williams syndrome. Autism Res., 10, 99–112. [DOI] [PubMed] [Google Scholar]
- 46. Lew, C.H., Groeniger, K.M., Bellugi, U., Stefanacci, L., Schumann, C.M. and Semendeferi, K. (2018) A postmortem stereological study of the amygdala in Williams syndrome. Brain Struct. Funct., 223, 1897–1907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ketschek, A., Spillane, M., Dun, X.P., Hardy, H., Chilton, J. and Gallo, G. (2016) Drebrin coordinates the actin and microtubule cytoskeleton during the initiation of axon collateral branches. Dev. Neurobiol., 76, 1092–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Adams, G., Jr., Zhou, J., Wang, W., Wu, H., Quan, J., Liu, Y., Xia, P., Wang, Z., Zhou, S., Jiang, J.et al. (2016) The microtubule plus end tracking protein TIP150 interacts with cortactin to steer directional cell migration. J. Biol. Chem., 291, 20692–20706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Komarova, Y., Lansbergen, G., Galjart, N., Grosveld, F., Borisy, G.G. and Akhmanova, A. (2005) EB1 and EB3 control CLIP dissociation from the ends of growing microtubules. Mol. Biol. Cell, 16, 5334–5345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sweet, E.S., Previtera, M.L., Fernandez, J.R., Charych, E.I., Tseng, C.Y., Kwon, M., Starovoytov, V., Zheng, J.Q. and Firestein, B.L. (2011) PSD-95 alters microtubule dynamics via an association with EB3. J. Neurosci., 31, 1038–1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Acevedo, K., Li, R., Soo, P., Suryadinata, R., Sarcevic, B., Valova, V.A., Graham, M.E., Robinson, P.J. and Bernard, O. (2007) The phosphorylation of p25/TPPP by LIM kinase 1 inhibits its ability to assemble microtubules. Exp. Cell Res., 313, 4091–4106. [DOI] [PubMed] [Google Scholar]
- 52. Fu, M.M., McAlear, T.S., Nguyen, H., Oses-Prieto, J.A., Valenzuela, A., Shi, R.D., Perrino, J.J., Huang, T.T., Burlingame, A.L., Bechstedt, S.et al. (2019) The golgi outpost protein TPPP nucleates microtubules and is critical for myelination. Cell, 179, 132–146 e114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Barak, B., Zhang, Z., Liu, Y., Nir, A., Trangle, S.S., Ennis, M., Levandowski, K.M., Wang, D., Quast, K., Boulting, G.L.et al. (2019) Neuronal deletion of Gtf2i, associated with Williams syndrome, causes behavioral and myelin alterations rescuable by a remyelinating drug. Nat. Neurosci., 22, 700–708. [DOI] [PubMed] [Google Scholar]
- 54. Rivell, A. and Mattson, M.P. (2019) Intergenerational metabolic syndrome and neuronal network hyperexcitability in autism. Trends Neurosci., 42, 709–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Jiang, M., Ash, R.T., Baker, S.A., Suter, B., Ferguson, A., Park, J., Rudy, J., Torsky, S.P., Chao, H.T., Zoghbi, H.Y.et al. (2013) Dendritic arborization and spine dynamics are abnormal in the mouse model of MECP2 duplication syndrome. J. Neurosci., 33, 19518–19533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Yan, J., Porch, M.W., Court-Vazquez, B., Bennett, M.V.L. and Zukin, R.S. (2018) Activation of autophagy rescues synaptic and cognitive deficits in fragile X mice. Proc. Natl. Acad. Sci. U. S. A., 115, E9707–E9716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Tang, G., Gudsnuk, K., Kuo, S.H., Cotrina, M.L., Rosoklija, G., Sosunov, A., Sonders, M.S., Kanter, E., Castagna, C., Yamamoto, A.et al. (2014) Loss of mTOR-dependent macroautophagy causes autistic-like synaptic pruning deficits. Neuron, 83, 1131–1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Sweatt, J.D. (2019) The epigenetic basis of individuality. Curr. Opin. Behav. Sci., 25, 51–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Ridley, E., Riby, D.M. and Leekam, S.R. (2020) A cross-syndrome approach to the social phenotype of neurodevelopmental disorders: focusing on social vulnerability and social interaction style. Res. Dev. Disabil., 100, 103604. [DOI] [PubMed] [Google Scholar]
- 60. Mervis, C.B., Klein-Tasman, B.P., Huffman, M.J., Velleman, S.L., Pitts, C.H., Henderson, D.R., Woodruff-Borden, J., Morris, C.A. and Osborne, L.R. (2015) Children with 7q11.23 duplication syndrome: psychological characteristics. Am. J. Med. Genet. A, 167, 1436–1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Cherniske, E.M., Carpenter, T.O., Klaiman, C., Young, E., Bregman, J., Insogna, K., Schultz, R.T. and Pober, B.R. (2004) Multisystem study of 20 older adults with Williams syndrome. Am. J. Med. Genet. A, 131, 255–264. [DOI] [PubMed] [Google Scholar]
- 62. Palacios-Verdu, M.G., Segura-Puimedon, M., Borralleras, C., Flores, R., Del Campo, M., Campuzano, V. and Perez-Jurado, L.A. (2015) Metabolic abnormalities in Williams-Beuren syndrome. J. Med. Genet., 52, 248–255. [DOI] [PubMed] [Google Scholar]
- 63. Blagosklonny, M.V. (2019) Fasting and rapamycin: diabetes versus benevolent glucose intolerance. Cell Death Dis., 10, 607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Mao, Z. and Zhang, W. (2018) Role of mTOR in glucose and lipid metabolism. Int. J. Mol. Sci., 19, 2043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Sareen, C.K., Ruvalcaba, R.H., Scotvold, M.J., Mahoney, C.P. and Kelley, V.C. (1972) Tuberous sclerosis. Clinical, endocrine, and metabolic studies. Am. J. Dis. Child., 123, 34–39. [PubMed] [Google Scholar]
- 66. Cooke, D.W., Naidu, S., Plotnick, L. and Berkovitz, G.D. (1995) Abnormalities of thyroid function and glucose control in subjects with Rett syndrome. Horm. Res., 43, 273–278. [DOI] [PubMed] [Google Scholar]
- 67. Krishnan, N., Krishnan, K., Connors, C.R., Choy, M.S., Page, R., Peti, W., Van Aelst, L., Shea, S.D. and Tonks, N.K. (2015) PTP1B inhibition suggests a therapeutic strategy for Rett syndrome. J. Clin. Invest., 125, 3163–3177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Dy, A.B.C., Tassone, F., Eldeeb, M., Salcedo-Arellano, M.J., Tartaglia, N. and Hagerman, R. (2018) Metformin as targeted treatment in fragile X syndrome. Clin. Genet., 93, 216–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Huang, W.C., Chen, Y. and Page, D.T. (2019) Genetic suppression of mTOR rescues synaptic and social behavioral abnormalities in a mouse model of Pten haploinsufficiency. Autism Res., 12, 1463–1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Chen, J., Alberts, I. and Li, X. (2014) Dysregulation of the IGF-I/PI3K/AKT/mTOR signaling pathway in autism spectrum disorders. Int. J. Dev. Neurosci., 35, 35–41. [DOI] [PubMed] [Google Scholar]
- 71. Ganesan, H., Balasubramanian, V., Iyer, M., Venugopal, A., Subramaniam, M.D., Cho, S.G. and Vellingiri, B. (2019) mTOR signalling pathway - a root cause for idiopathic autism? BMB Rep., 52, 424–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Heavner, W.E. and Smith, S.E.P. (2020) Resolving the synaptic versus developmental dichotomy of autism risk genes. Trends Neurosci., 43, 227–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Sato, A. (2016) mTOR, a potential target to treat autism Spectrum disorder. CNS Neurol. Disord. Drug Targets, 15, 533–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Zhang, J., Zhang, J.X. and Zhang, Q.L. (2016) PI3K/AKT/mTOR-mediated autophagy in the development of autism spectrum disorder. Brain Res. Bull., 125, 152–158. [DOI] [PubMed] [Google Scholar]
- 75. Rosina, E., Battan, B., Siracusano, M., Di Criscio, L., Hollis, F., Pacini, L., Curatolo, P. and Bagni, C. (2019) Disruption of mTOR and MAPK pathways correlates with severity in idiopathic autism. Transl. Psychiatry, 9, 50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Vithayathil, J., Pucilowska, J. and Landreth, G.E. (2018) ERK/MAPK signaling and autism spectrum disorders. Prog. Brain Res., 241, 63–112. [DOI] [PubMed] [Google Scholar]
- 77. Griesi-Oliveira, K., Suzuki, A.M., Alves, A.Y., Mafra, A., Yamamoto, G.L., Ezquina, S., Magalhaes, Y.T., Forti, F.L., Sertie, A.L., Zachi, E.C.et al. (2018) Actin cytoskeleton dynamics in stem cells from autistic individuals. Sci. Rep., 8, 11138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Joensuu, M., Lanoue, V. and Hotulainen, P. (2018) Dendritic spine actin cytoskeleton in autism spectrum disorder. Prog. Neuropsychopharmacol. Biol. Psychiatry, 84, 362–381. [DOI] [PubMed] [Google Scholar]
- 79. Yan, Z., Kim, E., Datta, D., Lewis, D.A. and Soderling, S.H. (2016) Synaptic actin dysregulation, a convergent mechanism of mental disorders? J. Neurosci., 36, 11411–11417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Costa-Mattioli, M. and Walter, P. (2020) The integrated stress response: from mechanism to disease. Science, 368, eaat5314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Basilico, B., Morandell, J. and Novarino, G. (2020) Molecular mechanisms for targeted ASD treatments. Curr. Opin. Genet. Dev., 65, 126–137. [DOI] [PubMed] [Google Scholar]
- 82. Penzes, P., Cahill, M.E., Jones, K.A., VanLeeuwen, J.E. and Woolfrey, K.M. (2011) Dendritic spine pathology in neuropsychiatric disorders. Nat. Neurosci., 14, 285–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Fromer, M., Pocklington, A.J., Kavanagh, D.H., Williams, H.J., Dwyer, S., Gormley, P., Georgieva, L., Rees, E., Palta, P., Ruderfer, D.M. et al. (2014) De novo mutations in schizophrenia implicate synaptic networks. Nature, 506, 179–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Fromer, M., Roussos, P., Sieberts, S.K., Johnson, J.S., Kavanagh, D.H., Perumal, T.M., Ruderfer, D.M., Oh, E.C., Topol, A., Shah, H.R. et al. (2016) Gene expression elucidates functional impact of polygenic risk for schizophrenia. Nat. Neurosci., 19, 1442–1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Gusev, A., Mancuso, N., Won, H., Kousi, M., Finucane, H.K., Reshef, Y., Song, L., Safi, A., Schizophrenia Working Group of the Psychiatric Genomics, C., McCarroll, S. et al. (2018) Transcriptome-wide association study of schizophrenia and chromatin activity yields mechanistic disease insights. Nat. Genet., 50, 538–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Hall, L.S., Medway, C.W., Pain, O., Pardinas, A.F., Rees, E.G., Escott-Price, V., Pocklington, A., Bray, N.J., Holmans, P.A., Walters, J.T.R. et al. (2020) A transcriptome-wide association study implicates specific pre- and post-synaptic abnormalities in schizophrenia. Hum. Mol. Genet., 29, 159–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Kirov, G., Pocklington, A.J., Holmans, P., Ivanov, D., Ikeda, M., Ruderfer, D., Moran, J., Chambert, K., Toncheva, D., Georgieva, L. et al. (2012) De novo CNV analysis implicates specific abnormalities of postsynaptic signalling complexes in the pathogenesis of schizophrenia. Mol. Psychiatry, 17, 142–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Lin, J.R., Cai, Y., Zhang, Q., Zhang, W., Nogales-Cadenas, R. and Zhang, Z.D. (2016) Integrated Post-GWAS Analysis Sheds New Light on the Disease Mechanisms of Schizophrenia. Genetics, 204, 1587–1600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Mervis, C.B., Morris, C.A., Klein-Tasman, B.P., Velleman, S.L. and Osborne, L.R. (1993) 7q11.23 duplication syndrome. In Pagon, R.A., Adam, M.P., Ardinger, H.H., Wallace, S.E., Amemiya, A., Bean, L.J.H., Bird, T.D., Ledbetter, N., Mefford, H.C., Smith, R.J.H. and Stephens, K. (eds), GeneReviews(R) [Internet], University of Washington, Seattle, WA. 1993–2017. [Google Scholar]
- 90. Sanders, S.J., Ercan-Sencicek, A.G., Hus, V., Luo, R., Murtha, M.T., Moreno-De-Luca, D., Chu, S.H., Moreau, M.P., Gupta, A.R., Thomson, S.A.et al. (2011) Multiple recurrent de novo CNVs, including duplications of the 7q11.23 Williams syndrome region, are strongly associated with autism. Neuron, 70, 863–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Dai, L., Carter, C.S., Ying, J., Bellugi, U., Pournajafi-Nazarloo, H. and Korenberg, J.R. (2012) Oxytocin and vasopressin are dysregulated in Williams syndrome, a genetic disorder affecting social behavior. PLoS One, 7, e38513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Korenberg, J.R., Dai, L., Bellugi, U., Jarvinen-Pasley, A., Mills, D.L., Galaburda, A., Reiss, A. and Pober, B. (2008) Deletion of 7q11.23 Genes and Williams syndrome. In Inborn Errors of Development: The Molecular Basis of Clinical Disorders of Morphogenesis, pp. 1544–1552.
- 93. Johnson, W.E., Li, C. and Rabinovic, A. (2007) Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics, 8, 118–127. [DOI] [PubMed] [Google Scholar]
- 94. Rossin, E.J., Lage, K., Raychaudhuri, S., Xavier, R.J., Tatar, D., Benita, Y., International Inflammatory Bowel Disease Genetics Constortium, Cotsapas, C. and Daly, M.J. (2011) Proteins encoded in genomic regions associated with immune-mediated disease physically interact and suggest underlying biology. PLoS Genet., 7, e1001273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Prasad, R., Longley, M.J., Sharief, F.S., Hou, E.W., Copeland, W.C. and Wilson, S.H. (2009) Human DNA polymerase theta possesses 5'-dRP lyase activity and functions in single-nucleotide base excision repair in vitro. Nucleic Acids Res., 37, 1868–1877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Smyth, G.K. (2004) Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat. Appl. Genet. Mol. Biol., 3, 1–25. [DOI] [PubMed] [Google Scholar]
- 97. Benjamini, Y. and Hochberg, Y. (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B, 57, 289–300. [Google Scholar]
- 98. Ritchie, M.E., Phipson, B., Wu, D., Hu, Y., Law, C.W., Shi, W. and Smyth, G.K. (2015) Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res., 43, e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Coquelle, F.M., Caspi, M., Cordelieres, F.P., Dompierre, J.P., Dujardin, D.L., Koifman, C., Martin, P., Hoogenraad, C.C., Akhmanova, A., Galjart, N.et al. (2002) LIS1, CLIP-170's key to the dynein/dynactin pathway. Mol. Cell. Biol., 22, 3089–3102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Ma, J., Zhang, L.Q., He, Z.X., He, X.X., Wang, Y.J., Jian, Y.L., Wang, X., Zhang, B.B., Su, C., Lu, J.et al. (2019) Autism candidate gene DIP2A regulates spine morphogenesis via acetylation of cortactin. PLoS Biol., 17, e3000461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Uruno, T., Liu, J., Zhang, P., Fan, Y., Egile, C., Li, R., Mueller, S.C. and Zhan, X. (2001) Activation of Arp2/3 complex-mediated actin polymerization by cortactin. Nat. Cell Biol., 3, 259–266. [DOI] [PubMed] [Google Scholar]
- 102. Ishikawa, Y., Okada, M., Honda, A., Ito, Y., Tamada, A., Endo, N. and Igarashi, M. (2019) Phosphorylation sites of microtubule-associated protein 1B (MAP 1B) are involved in axon growth and regeneration. Mol. Brain, 12, 93. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.