

Abstract
While the administration of anti‐CD154 mAbs in mice validated the CD40‐CD154 pathway as a target against inflammatory disorders, this approach caused thromboembolism in humans (unrelated to CD40 inhibition) and is expected to predispose to opportunistic infections. There is a need for alternative approaches to inhibit CD40 that avoid these complications. CD40 signals through TRAF2,3 and TRAF6‐binding sites. Given that CD40‐TRAF6 is the pathway that stimulates responses key for cell‐mediated immunity against opportunistic pathogens, we examined the effects of pharmacologic inhibition of CD40‐TRAF2,3 signaling. We used a model of ischemia/reperfusion (I/R)‐induced retinopathy, a CD40‐driven inflammatory disorder. Intravitreal administration of a cell‐penetrating CD40‐TRAF2,3 blocking peptide impaired ICAM‐1 upregulation in retinal endothelial cells and CXCL1 upregulation in endothelial and Müller cells. The peptide reduced leukocyte infiltration, upregulation of NOS2/COX‐2/TNF‐α/IL‐1β, and ameliorated neuronal loss, effects that mimic those observed after I/R in Cd40−/− mice. While a cell‐penetrating CD40‐TRAF6 blocking peptide also diminished I/R‐induced inflammation, this peptide (but not the CD40‐TRAF2,3 blocking peptide) impaired control of the opportunistic pathogen Toxoplasma gondii in the retina. Thus, inhibition of the CD40‐TRAF2,3 pathway is a novel and potent approach to reduce CD40‐induced inflammation, while likely diminishing the risk of opportunistic infections that would otherwise accompany CD40 inhibition.
Keywords: endothelial, muller cell, polymorphonuclear leukocyte, retina, toxoplasma
Abbreviations
- CRALBP
cellular retinaldehyde‐binding protein
- GCL
ganglion cell layer
- IOP
intraocular pressure
- I/R
ischemia/reperfusion
- MPO
myeloperoxidase
- PMN
polymorphonuclear
- ri
retroinverso
- TRAF
TNF receptor‐associated factors
1. INTRODUCTION
The CD40‐CD154 (CD40 ligand) pathway triggers pro‐inflammatory responses and drives various disorders that include inflammatory bowel disease, autoimmunity, graft rejection, and ischemic tissue injury. 1 Blockade of CD40‐CD154 via administration of neutralizing anti‐CD154 mAbs is therapeutically effective in various inflammatory disorders in animals. 1 A similar approach appeared effective in patients with lupus nephritis. 2 However, the mAbs caused thromboembolism, a complication not due to CD40 blockade per se but likely caused by the binding of mAbs to CD154 and FcγRIIa expressed on platelets. 2 , 3 While other approaches that cause global inhibition of the CD40‐CD154 pathway have been explored, 4 they are likely to predispose to opportunistic infections since this pathway is central to protection against numerous pathogens. 5 New methods to block CD40 based on the inhibition of selective signaling downstream of CD40 may avoid thromboembolic side‐effects and reduce the risk of infections.
CD40 signals by recruiting TNF Receptor‐Associated Factors (TRAF). 6 CD40 has a major domain (PxQxT) that directly binds TRAF2 and TRAF3 7 (TRAF3 typically inhibits CD40 signaling), a distal minor domain (SVxE) that binds TRAF2, 8 and a site that binds TRAF6 (PxExxAr/Ac). 7 Studies using cells that express wild‐type CD40 or CD40 with mutations in the TRAF2,3 or TRAF6‐binding sites revealed that both sites induce pro‐inflammatory responses. 9 , 10 While the CD40‐TRAF6‐binding site plays a dominant role in myeloid cells, the effects of these binding sites are largely non‐compensatory in non‐hematopoietic cells explaining why isolated blockade of CD40‐TRAF2,3 or CD40‐TRAF6 signaling markedly reduces pro‐inflammatory responses in these cells. 9 , 10 The TRAF2,3 and TRAF6‐binding sites can also have non‐overlapping effects. In contrast to the CD40‐TRAF2,3‐binding site, the CD40‐TRAF6‐binding site and TRAF6 are essential for responses relevant to cellular immunity: dendritic cell development and maturation, production of IL‐12 by dendritic cells, and induction of nitric oxide‐dependent or autophagy‐mediated anti‐microbial activity. 10 , 11 , 12 , 13 , 14 Studies in transgenic mice revealed that both sites are key to humoral immunity. The CD40‐TRAF6 site mediates the production of high‐affinity antibodies and plasma cell formation. 15 The CD40‐TRAF2,3‐binding site controls isotype switch, 16 although other studies indicate that both sites promote this response. 15 , 17 Finally, germinal center formation requires both CD40‐TRAF2,3, and CD40‐TRAF6 sites. 17 Taken together, it is clear that both sites play important roles in humoral immunity. 18
Studies in transgenic mice whose MHC II+ cells expressed wild‐type CD40 or CD40 with mutations in TRAF‐binding sites revealed that CD40‐TRAF6 signaling in hematopoietic cells promotes vascular inflammation. 19 The role of CD40‐TRAF2,3 signaling was difficult to assess accurately since the expression of the TRAF2,3‐binding site mutant was high 15 raising the possibility that higher CD40 expression facilitated responses downstream of the mutant molecule, minimizing the effect of the mutation. Nevertheless, important studies revealed that the pharmacologic inhibition of CD40‐TRAF6 signaling reduces CD40‐driven inflammation. 20 , 21 However, the fact that marked susceptibility to infections controlled by cellular immunity is the most important clinical feature and leading cause of mortality in patients with defective CD40‐CD154 signaling (X‐linked Hyper IgM syndrome) 5 together with the central role of CD40‐TRAF6 for cellular responses required to control these pathogens raise the concern for opportunistic infections caused by CD40‐TRAF6 inhibition. While a CD40‐TRAF6 inhibitor did not impair survival in a mouse model of sepsis, 20 , 21 CD40 is not required for protection in this model. 22 Thus, these findings do not rule out a risk of opportunistic infections caused by the inhibition of CD40‐TRAF6 signaling.
CD40 in non‐hematopoietic cells is central to the development of inflammation as demonstrated by studies in ischemia/reperfusion (I/R)‐induced retinopathy, diabetic retinopathy, and vascular injury. 23 , 24 , 25 These findings together with the demonstration that CD40‐TRAF2,3 blockade markedly inhibits inflammatory responses in non‐hematopoietic cells 24 , 26 point to the inhibition of CD40‐TRAF2,3 signaling as a new approach to control CD40‐driven inflammation. This approach would likely avoid thrombotic events and reduce the risk of opportunistic infections since it does not rely on anti‐CD154 mAbs and does not impair CD40‐TRAF6 signaling.
CD40 is expressed in retinal cells: endothelial cells, Müller cells, microglia, ganglion cells, and retinal pigment epithelial cells. 27 Given that CD40 is a key driver of retinal inflammation and development of ischemic retinopathies, 23 , 24 , 26 we examined the effects of the pharmacologic inhibition of CD40‐TRAF2,3 signaling in the development of inflammation and retinopathy after retinal ischemia. Cell‐penetrating peptides impair protein‐protein interactions. 28 We showed that a cell‐permeable peptide that consists of the amino acid sequence of the TRAF2,3 site fused with HIV TAT47‐57 to make it cell‐permeable inhibits CD40‐driven inflammatory responses in vitro. 9 , 10 The peptide impairs the CD40‐TRAF2,3 pathway both in human and mouse cells since the amino acid sequence of the TRAF2,3‐binding site is the same in both species. Retro‐inverso (ri) peptides made following reverse amino acid sequence and synthesized with D‐amino acids can have an identical function to those made with L‐amino acids and are resistant to peptidases. 29 We report herein that a ri CD40‐TRAF2,3 blocking peptide markedly impairs inflammation and cell loss in the ganglion cell layer after retinal I/R even if administered after retinal ischemia. In contrast to a CD40‐TRAF6 blocking peptide, the CD40‐TRAF2,3 blocking peptide did not increase susceptibility to retinitis caused by Toxoplasma gondii, an opportunistic pathogen normally controlled by CD40. These studies provide the first evidence that the pharmacologic inhibition of the CD40‐TRAF2,3 pathway effectively controls a CD40‐driven inflammatory disorder without increasing susceptibility to an opportunistic pathogen.
2. MATERIALS AND METHODS
2.1. Animals
C57BL/6 (B6) and Cd40−/− mice (B6 background) were purchased from Jackson Laboratories (Bar Harbor, ME) and bred at Case Western Reserve University. Studies were approved by the Institutional Animal Care and Use Committee of Case Western Reserve University School of Medicine.
2.2. Model of retinal I/R
Retinal ischemia was induced in male mice (25 to 30 g) as described. 24 , 30 The anterior chamber of one eye was cannulated with a 30‐gauge needle. Intra‐ocular pressure (IOP) was maintained at 80 to 90 mm Hg for 90 minutes. The other eye of the same animal was set up as a control. After ischemia, the needle was withdrawn, IOP was normalized, and reflow of the retinal circulation was documented visually. Animals were euthanized 2 days after I/R.
2.3. Toxoplasma gondii infection
Female B6 mice (8 to 10 weeks old) were infected with 10 tissue cysts of the ME49 strain of T. gondii. 31 Mouse retinal endothelial cells were challenged with tachyzoites of the RH strain of the parasite and the number of vacuoles and tachyzoites per 100 cells were determined by light microscopy. 31
2.4. Cell‐penetrating peptides
Peptide consisted of the TRAF2,3‐binding site of CD40 that was made cell‐permeable by linking it to the TAT47‐57 cell‐penetrating peptide. 9 , 10 Peptides were synthesized using D‐amino acids following reverse amino acid sequence (ri format). The sequence for the CD40‐TRAF2,3 blocking peptide was NH2‐rrrqrrkkrgy ghlteqvhaatn‐OH. The TAT47‐57 sequence is underlined. The peptide contains a proline (p) to histidine (h) substitution (shown in italics) that appears to enhance affinity to TRAF2. 32 The scrambled peptide NH2‐rrrqrrkkrgy ntqalahtgevh‐OH was used as control. The CD40‐TRAF2,3 blocking peptide was also synthesized with Alexa Fluor 488 conjugated via maleimide coupling to an additional Cys in the N terminus. The sequence for the CD40‐TRAF6 blocking peptide was NH2‐rrrqrrkkrgy demeqadqrr‐OH. The proline to alanine (a) substitution (italics) maintains strong interaction with TRAF6. 32 ri TAT47‐57 cell‐penetrating peptide was used as a control in experiments that compared CD40‐TRAF2,3 and CD40‐TRAF6 blocking peptides. Peptides were manufactured by Biopeptide Co. (San Diego, CA) and were low in endotoxin and >98% pure by HPLC. Peptides were not toxic to Müller and endothelial cells as assessed by alamarBlue cell viability assay (Invitrogen, Carlsbad, CA). Peptides were used at 1 μM in vitro experiments (10 μM in the case of Alexa Fluor 488‐conjugated peptide for the optimal visualization of fluorescence) or a single dose of 1 μg for intravitreal injections studies. In certain experiments, peptides were injected ip (10 μg/kg) 3 hours prior to ip administration of a stimulatory anti‐CD40 mAbs (1C10; 100 μg).
2.5. Histopathology
Histological changes induced by I/R were assessed as described before. 30 Formalin‐fixed, paraffin‐embedded sections were stained with H&E for light microscopy. Cells in the ganglion cell layer (GCL) that were not associated with vessels were counted under 400×. Cells in the GCL positive for β‐III tubulin (ganglion cell marker) and cells in the inner retina and vitreous positive for myeloperoxidase (MPO; highly expressed in PMNs, while cells of monocytic origin are either nonreactive of weakly positive) 33 were counted in retinal sections. Histopathologic evaluation of mice with ocular toxoplasmosis was performed as described. 31
2.6. Immunohistochemistry
Sections were incubated with antibodies against ICAM‐1 (eBioscience, San Diego, CA), CXCL1 (Novus, Littleton, CO), NOS2 (EMD Millipore, Burlington, MA), vimentin (Novus), CRALBP (Proteintech Group, Rosemont, IL), β‐III tubulin (BioLegend) or MPO (Agilent, Santa Clara, CA) followed by incubation with fluorescent secondary antibodies (Jackson ImmunoResearch Laboratories, Inc., West Grove, PA). Sections were also incubated with Tomato lectin (Vector Laboratories, Burlingame, CA) or ApopTag Red, In situ Apoptosis Detection kit (EMD Millipore, Billerica, MA, USA). Frozen sections were used when examining the expression of Alexa Fluor 488‐conjugated peptide. Retinas were analyzed using Olympus FV1200 IX‐83 confocal microscope. Images were processed in Photoshop CC 19.1.1. using similar linear adjustments for all samples.
2.7. Real‐time quantitative PCR
RNA was isolated using the RNeasy kit (QIAGEN). After treating RNA with DNase (Ambion, Austin, TX), cDNA was generated using oligo(dT)12–18 primers and Superscript III reverse transcriptase (Invitrogen). RT‐PCR was performed using SYBR GREEN PCR Master Mix (Applied Biosystems, Foster City, CA) and primers (Table S1). Gene expression was assessed using a 7300 Real‐Time PCR System (Applied Biosystems). Each cDNA sample was run in triplicate. Samples were normalized according to the content of 18S rRNA.
2.8. Retinal cells and CD40 stimulation
Primary human retinal endothelial cells were obtained as described 26 and cultured in the complete medium supplemented with endothelial cell growth supplement from bovine pituitary (15 µg/mL; Sigma Chemical, St Louis, MO) and insulin/transferrin/selenium (Sigma Chemical). Cell identity was confirmed by the incorporation of acetylated low‐density lipoprotein (>90%). Endothelial cells were used between passages 3 to 6. The human Müller cell line MIO M1 (gift from Dr. Gloria Limb; University College London, UK; >95% vimentin+, CRALBP+, and GFAP‐) was also used. Human cells were incubated with multimeric human CD154 to induce CD40 stimulation (1:10 dilution; obtained from Dr. Richard Kornbluth, Multimeric Biotherapeutics Inc., La Jolla, CA), a non‐functional CD154 mutant (T147N) 24 or TNF‐α (PeproTech, Rocky Hill, NJ). Mouse retinal endothelial cells (mREC) were incubated with a stimulatory anti‐mouse CD40 mAbs (1C10; 10 μg/mL).
2.9. Luciferase assay
Peptides were tested as described using the mouse endothelial cells mHEVc that expresses a chimera of the extracellular domain of human CD40 and intracytoplasmic domain of mouse CD40 (hmCD40) with either a mutation that prevents the recruitment of TRAF2,3 (hmCD40 ΔT2,3) or TRAF6 (hmCD40 ΔT6). 10 These cells were transfected with pGL4.32luc2P/NF‐κB‐RE/Hygro vector (Promega, Madison, WI) encoding an NF‐κB response element that drives the luciferase reporter gene luc2P (Photinus pyralis). 13 Cells were pre‐incubated with peptides for 3 hours. followed by stimulation with human CD154. Luciferase activity was assessed using a Steady‐Glo luciferase assay system (Promega) and a luminometer.
2.10. Flow cytometry
Endothelial cells or splenocytes were incubated with conjugated mAbs (Table S2). After fixation with 1% paraformaldehyde, cells were analyzed using an LSR II (Becton Dickinson).
2.11. ELISA
Serum concentrations of IL‐12 p70 were measured by ELISA (BioLegend, San Diego, CA). Anti‐T. gondii IgG was detected in sera by ELISA and the antibody titer was calculated as described. 31
2.12. Statistical analysis
D’Agostino & Pearson omnibus test was used to confirm the normal distribution of the data. Data were analyzed by 2‐tailed Student’s t test or ANOVA. Differences were considered statistically significant at P < .05.
3. RESULTS
3.1. Retro‐inverso (ri) CD40‐TRAF2,3 blocking peptide inhibits CD40‐TRAF2,3 but not CD40‐TRAF6 signaling and inhibits CD40‐induced pro‐inflammatory responses
A ri version of the previously described TAT47‐57 ‐based CD40‐TRAF2,3 blocking peptide 9 , 10 was generated. ri CD40‐TRAF2,3 blocking peptide tagged with Alexa Fluor 488 was used to determine whether it translocates inside cells. Using human Müller cells (the principal glia in the retina) as a model, immunofluorescence studies revealed the intracellular incorporation of the fluorescent peptide after its addition to the culture medium (Figure 1A). Next, we examined whether the ri CD40‐TRAF2,3 blocking peptide inhibited CD40‐TRAF2,3 signaling. We used reporter cell lines that consisted of mouse endothelial cells expressing a human‐mouse CD40 chimera (extracellular human CD40 and intracellular mouse CD40) that signal either through the TRAF2,3‐binding site (hmCD40 ΔT6) or the TRAF6‐binding site (hmCD40 ΔT2,3). 13 These cells express an NF‐κB response element that drives a luciferase reporter gene. 13 The blocking peptide inhibited CD154‐induced NF‐κB activity only in cells that expressed CD40 that signals through the TRAF2,3‐binding site (Figure 1B). In addition, the blocking peptide impaired CD154‐induced activation of JNK, an event largely dependent on CD40‐TRAF2 signaling 34 (Figure S1). Thus, ri CD40‐TRAF2,3 blocking peptide selectively impairs CD40‐TRAF2,3 signaling.
FIGURE 1.
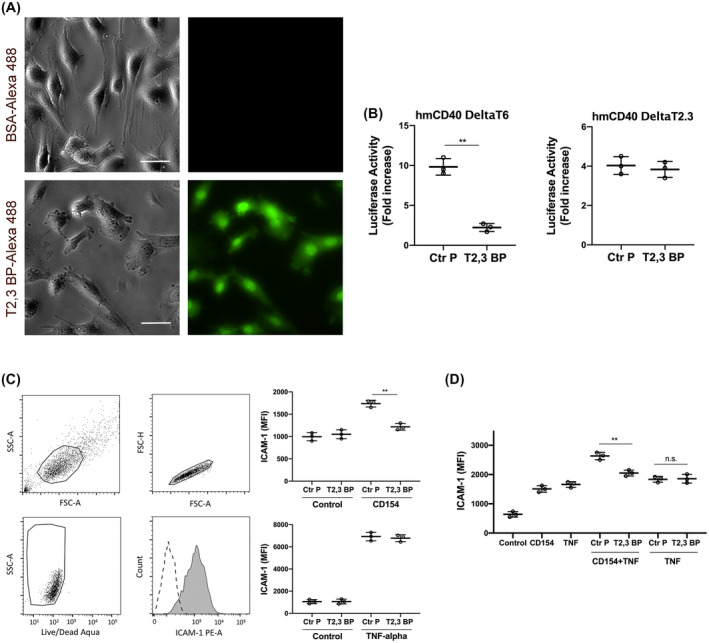
ri CD40‐TRAF2,3 blocking peptide penetrates cells, inhibits CD40‐TRAF2,3 signaling and impairs CD40‐driven ICAM‐1 upregulation. A, Human Müller cells were incubated in medium containing Alexa Fluor 488‐conjugated ri CD40‐TRAF2,3 blocking peptide (T2,3 BP) or Alexa Fluor 488‐conjugated bovine serum albumin (BSA; both at 10 µM) for 3 hours. Scale bar, 50 μm. Original magnification 400×. Images represent fluorescence of unfixed Müller cells after extensive washing of monolayers. B, Mouse endothelial cells (mHEVc) that express an NF‐κB response element that drives the transcription of a luciferase reporter plus either hmCD40 ΔT2,3 or hmCD40 ΔT6 were pre‐incubated with ri control peptide (Ctr P) or ri CD40‐TRAF2,3 blocking peptide (T2,3 BP; both at 1 µM) or medium alone followed by stimulation with human CD154. Data are expressed as fold increase in normalized luciferase activity in cells stimulated with CD154 compared to cells treated with respective peptide in the absence of CD154. C, Human retinal endothelial cells were treated with ri control peptide (Ctr P) or ri CD40‐TRAF2,3 blocking peptide (T2,3 BP; both at 1 µM) followed by stimulation with CD154 or TNF‐α (100 pg/mL) for 24 hours. Expression of ICAM‐1 was assessed by flow cytometry. Dot plot and histogram show gating strategy. ICAM‐1 was analyzed on live cells that did not stain with Aqua LIVE/DEAD kit. D. Human retinal endothelial cells were incubated with or without TNF‐α (30 pg/mL) followed by treatment with peptides and stimulation with CD154. Data shown represent mean ± SD of triplicate samples. Results are representative of three independent experiments. **P < .01 by ANOVA
Human retinal endothelial cells were incubated with control or blocking peptides followed by stimulation with CD154. ri CD40‐TRAF2,3 blocking peptide impaired the upregulation of ICAM‐1 in response to CD154 (Figure 1C). The blocking peptide also impaired CD154‐induced upregulation of ICAM‐1 in endothelial cells previously exposed to TNF‐α, indicating that the inhibition of CD40‐TRAF2,3 signaling impairs pro‐inflammatory responses even in the presence of a co‐existing inflammatory milieu (Figure 1D). In addition, ri CD40‐TRAF2,3 blocking peptide impaired CCL2 secretion induced by CD154 (Figure S2). Taken together, ri CD40‐TRAF2,3 blocking peptide inhibits CD40‐induced pro‐inflammatory responses.
3.2. Intravitreal administration of ri CD40‐TRAF2,3 blocking peptide prior to I/R protects against cell loss in the ganglion cell layer (GCL) and leukocyte infiltration in a manner similar to that observed in Cd40−/− mice
To begin to test the in vivo effect of ri CD40‐TRAF2,3 blocking peptide, we examined whether it translocates into retinal cells. Alexa Fluor 488‐conjugated ri CD40‐TRAF2,3 blocking peptide or non‐fluorescent peptide were injected intravitreally and eyes were collected after 2 days. Figure 2A shows uptake of fluorescent peptides that tended to localize in the inner retina, especially in nuclei located in this region. This is consistent with the pattern of nuclear accumulation reported for TAT. 35 As described for other TAT‐based cell‐penetrating peptides, 36 the ri CD40‐TRAF2,3 blocking peptide did not cause retinal pathology in the injected eye and was not detected in the contralateral eye (Figure 2A). Nuclei that accumulated the peptide included those localized within tomato lectin+ cells (endothelial cells; Figure 2B). In vitro studies revealed that while the peptide accumulated in nuclei, it was also present in the cytoplasm (Figure 1A). Indeed, the peptide was also detected in retinal cytoplasmic processes that co‐expressed CRALBP, indicating that Müller cells were also among those that exhibited peptide accumulation (Figure 2C).
FIGURE 2.
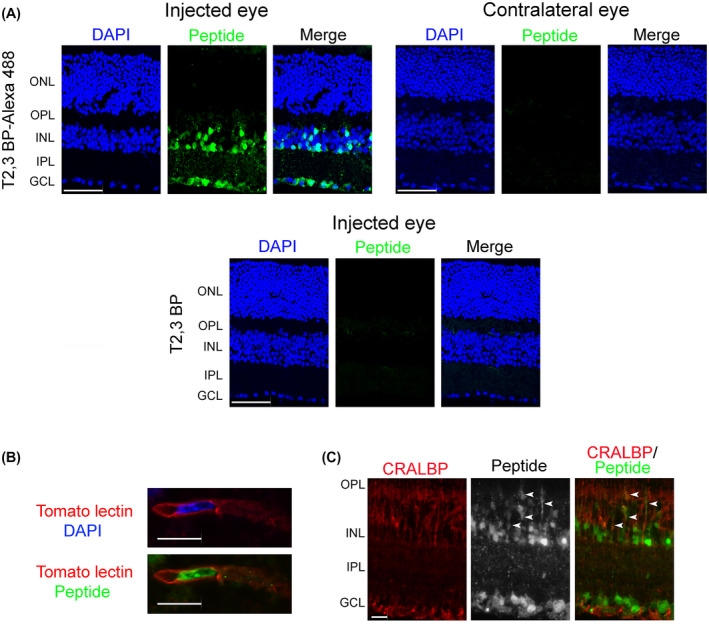
ri CD40‐TRAF2,3 blocking peptide penetrates retinal cells. A, B6 mice received Alexa Fluor 488‐conjugated ri CD40‐TRAF2,3 blocking peptide or non‐fluorescent ri CD40‐TRAF2,3 blocking peptide (both 1 μg) via intravitreal injection of one eye. Injected and contralateral eyes were collected after 48 hours and frozen sections were examined. GCL = Ganglion cell layer; IPL = Inner plexiform layer; INL = Inner nuclear layer. OPL = Outer plexiform layer; ONL = Outer nuclear layer. Scale bar, 50 µm. B, C, Retinas from mice injected with Alexa Fluor 488‐conjugated ri CD40‐TRAF2,3 blocking peptide were stained with DyLight 594 tomato lectin (labels neural endothelial cells, B) or with anti‐CRALBP antibody (labels Müller cells, C). Green fluorescence was detected in cytoplasmic processes that co‐stain with CRALBP (arrowheads). Scale bar 10 µm. Original magnification 600× for panel B and 400× for panel C, Results are representative of three independent experiments
Next, we examined the in vivo effects of the ri CD40‐TRAF2,3 blocking peptide using a model of I/R‐induced retinal injury based on the transient elevation of IOP. 24 , 30 I/R of the retina causes cell loss in the GCL. 24 , 30 , 37 Compared to contralateral non‐ischemic eyes in B6 mice, eyes subjected to I/R developed cell loss in the GCL (Figure 3A). There was a significant attenuation in cell loss in the GCL in eyes from B6 mice that received an intravitreal injection of ri CD40‐TRAF2,3 blocking peptide 1 hour prior to retinal ischemia (Figure 3A). The effect of the blocking peptide was similar to that conferred by the lack of CD40 (Cd40−/− mice) (Figure 3A). Neurons in the GCL include ganglion cells and displaced amacrine cells. Quantification of cells that express β‐III tubulin, a ganglion cell marker, confirmed the protective effects of the ri CD40‐TRAF2,3 blocking peptide on that cell type (Figure 3B). In addition, administration of the ri CD40‐TRAF2,3 blocking peptide also reduced the numbers of cells in the GCL that underwent programmed cell death as assessed by Tunel staining (Figure S3).
FIGURE 3.
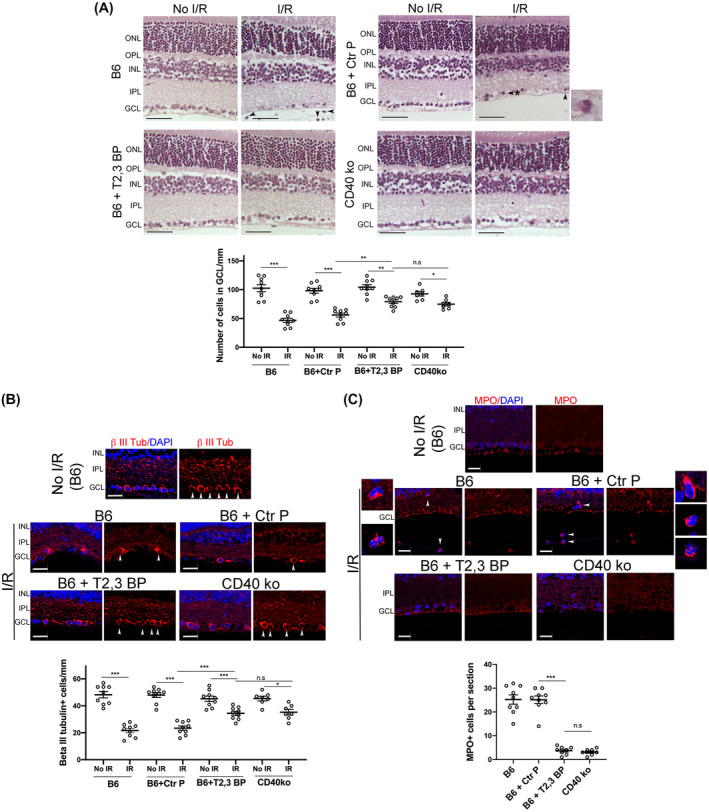
ri CD40‐TRAF2,3 blocking peptide protects against cell loss in the GCL and infiltration by MPO+ leukocytes in retinas subjected to I/R. One eye from each B6 and Cd40−/− mouse was subjected to I/R. Contralateral non‐ischemic eye was used as control. Eyes subjected to I/R in B6 mice were treated intravitreously with or without ri control peptide (Ctr P), ri CD40‐TRAF2,3 blocking peptide (T2,3 BP; both 1 μg) 1 hour prior to an increase in IOP. Eyes were collected 2 days after I/R. A, Cell loss in the GCL is observed in ischemic eyes from B6 mice treated with ri control peptide or vehicle (original magnification 400×). H&E; Scale bar, 50 μm. Eyes from these mice also exhibited PMN infiltration in the inner retina and vitreous (arrowhead). Arrowhead plus asterix identifies a PMN magnified in the inset (original magnification 600×). The graph shows the numbers of cells in the GCL per mm. Horizontal bars represent mean ± SEM (9 mice per group). B, Sections were stained with anti‐β‐III tubulin antibody. Arrowheads identify β‐III tubulin+ cells. Original magnification 400×. Scale bar, 20 μm. The graph shows the numbers of β‐III tubulin+ cells in the GCL per mm. C, Sections were stained with anti‐MPO antibody. MPO+ cells (arrowheads) are magnified in the insets. Number of infiltrating MPO+ leukocytes in the inner retina and vitreous per section. No MPO+ cells were detected in the absence of I/R. GCL = Ganglion cell layer; IPL = Inner plexiform layer; INL = Inner nuclear layer. OPL = Outer plexiform layer; ONL = Outer nuclear layer. *P < .05; **P < .01; ***P < .001 by ANOVA
Leukocytes including polymorphonuclear leukocytes (PMNs) infiltrate the retina since the early stages after I/R, a response important to the pathogenesis of neuronal loss after retinal I/R. 24 , 30 , 37 , 38 Eyes from B6 mice treated with the control peptide developed infiltration in the inner retina and vitreous by leukocytes positive for MPO (strongly expressed in PMNs) (Figure 3A, C). In contrast, ischemic eyes from B6 mice treated with ri CD40‐TRAF2,3 blocking peptide exhibited diminished inflammatory infiltrate, an effect that was similar to that observed in Cd40−/− mice subjected to I/R (Figure 3A, C). Taken together, the ri CD40‐TRAF2,3 blocking peptide effectively protects against I/R‐induced cell loss in the GCL and infiltration by MPO+ leukocytes.
3.3. Intravitreal administration of ri CD40‐TRAF2,3 blocking peptide impairs I/R‐induced upregulation of NOS2, COX‐2, TNF‐α, and IL‐1β
Leukocytes recruited after ischemia express NOS2, COX‐2 and pro‐inflammatory cytokines. 24 , 38 , 39 Moreover, retinopathy induced by I/R causes the upregulation of NOS2, COX‐2, TNFα, and IL‐1β, 38 , 40 , 41 , 42 molecules that are linked to the development of neuronal cell loss. 38 , 41 , 42 , 43 Compared to contralateral non‐ischemic eyes, eyes subjected to I/R after treatment with the control peptide upregulated NOS2, COX‐2, TNF‐α, and IL‐1β mRNA levels (Figure 4). Eyes from B6 mice treated with ri CD40‐TRAF2,3 blocking peptide exhibited impaired upregulation of NOS2, COX‐2, TNF‐α, and IL‐1β, an effect that was similar to that observed in Cd40−/− mice subjected to retinal I/R (Figure 4).
FIGURE 4.
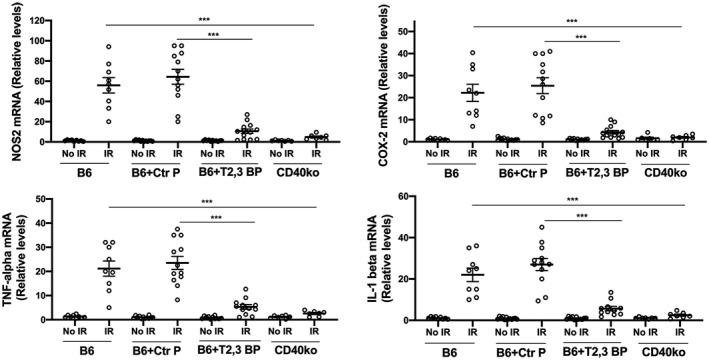
ri CD40‐TRAF2,3 blocking peptide impairs the upregulation of NOS2, COX‐2, TNF‐α, and IL‐1β in retinas subjected to I/R. One eye of each mouse was treated as above and subjected to I/R. Eyes that underwent I/R and non‐ischemic eyes were collected after 2 days. mRNA levels of NOS2, COX‐2, TNF‐α, and IL‐1β were assessed by quantitative real‐time PCR. Samples were normalized according to the content of 18S rRNA and one non‐ischemic eye from a B6 mouse was given an arbitrary value of 1. Data are expressed as fold increase compared to this animal. Horizontal bars represent mean ± SEM (9‐12 mice per group). ***P < .001 by ANOVA
3.4. Intravitreal administration of ri CD40‐TRAF2,3 blocking peptide impairs I/R‐induced ICAM‐1 and CXCL1 upregulation in retinal endothelial cells
We examined the expression of ICAM‐1 and CXCL1, molecules that are central to leukocyte recruitment and that are upregulated after retinal I/R. 24 , 30 Retinas from B6 mice treated with the control peptide and subjected to I/R showed marked upregulation of ICAM‐1 and CXCL1 mRNA levels (Figure 5A). Administration of ri CD40‐TRAF2,3 blocking peptide caused a significant reduction in ICAM‐1 and CXCL1 mRNA levels that were similar to that observed in the retinas of Cd40−/− mice subjected to I/R (Figure 5A). Next, we examined the expression of these inflammatory molecules in retinal endothelial cells. Expression of ICAM‐1 and CXCL1 was increased in retinal endothelial cells from mice that received control peptide (Figure 5B). In agreement with mRNA data, mice treated with the ri CD40‐TRAF2,3 blocking peptide had decreased expression of these molecules in retinal endothelial cells (Figure 5B). Thus, the ri CD40‐TRAF2,3 blocking peptide protected against I/R‐induced upregulation of ICAM‐1 and CXCL1 in retinal endothelial cells.
FIGURE 5.
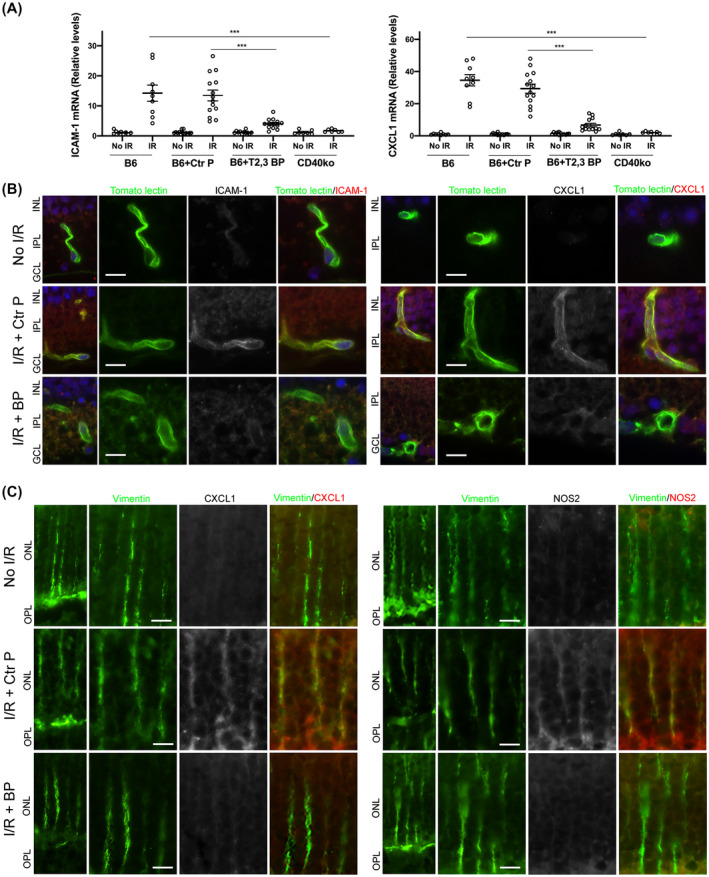
ri CD40‐TRAF2,3 blocking peptide impairs the upregulation of ICAM‐1 and CXCL1 in retinal endothelial cells and upregulation of NOS2 and CXCL1 in Müller cells from retinas subjected to I/R. One eye of each mouse was treated as above and subjected to I/R. Eyes that underwent I/R and non‐ischemic eyes were collected after 2 days. A, mRNA levels of ICAM‐1 and CXCL1 were assessed by quantitative real‐time PCR. One non‐ischemic eye from a B6 mouse was given an arbitrary value of 1 and data are expressed as fold increase compared to this animal. Horizontal bars represent Mean ± SEM (9‐12 mice per group). ***P < .001 by ANOVA. B, Retinal sections were incubated with Tomato Lectin (labels endothelial cells) plus either anti‐ICAM‐1 or anti‐CXCL1 Ab. C, Retinal sections were incubated with anti‐Vimentin Ab (labels Müller cells) plus either anti‐NOS2 or anti‐CXCL1 Ab. Protein expression at the level of Müller cells stalks. Original magnification 600×. GCL, Ganglion cell layer; IPL, inner plexiform layer; INL, inner nuclear layer; OPL, outer plexiform layer; ONL, outer nuclear layer. Scale bar, 10 µm. Six mice/group
3.5. Intravitreal administration of ri CD40‐TRAF2,3 blocking peptide impairs I/R‐induced CXCL1 and NOS2 upregulation in Müller cells
Retinal Müller cells are important for the development of inflammation in the retina. 23 , 44 Moreover, it has been suggested that Müller cells upregulate NOS2 after retinal ischemia. 45 NOS2 upregulation occurred in Müller cells from retinas subjected to I/R and treated with the control peptide (Figure 5C). In addition, Müller cells from these retinas also exhibited increased CXCL1 expression (Figure 5C). Treatment with ri CD40‐TRAF2,3 blocking peptide impaired expression of these molecules in Müller cells from retinas subjected to I/R (Figure 5C). Altogether, Müller cells from ischemic retinas upregulate NOS2 and CXCL1, an effect that was impaired by the ri CD40‐TRAF2,3 blocking peptide.
3.6. Intravitreal administration of ri CD40‐TRAF2,3 blocking peptide after I/R protects against cell loss in the GCL and retinal inflammatory responses
Control or ri CD40‐TRAF2,3 blocking peptide were administered 90 min post‐retinal ischemia. As shown in Figure 6 the blocking peptide protected against cell loss in the GCL, loss of β‐III tubulin+ cells, recruitment of MPO+ leukocytes, and upregulation of ICAM‐1, CXCL1, NOS2, COX‐2, TNF‐α, and IL‐1β. Thus, ri CD40‐TRAF2,3 blocking peptide is effective in ameliorating the development of inflammation and neuronal loss even if administered after the induction of retinal ischemia.
FIGURE 6.
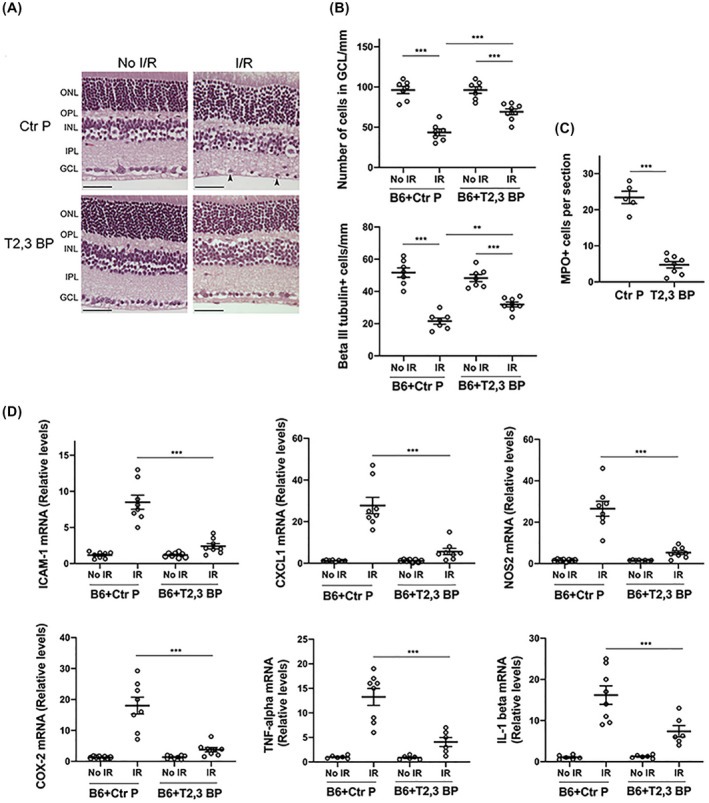
ri CD40‐TRAF2,3 blocking peptide protects against cell loss in the GCL and inflammation when administered after retinal I/R. One eye from each B6 mouse was subjected to I/R. Non‐ischemic eyes were used as controls. Eyes that were subjected to I/R received either ri control peptide or ri CD40‐TRAF2,3 blocking peptide (1 μg) 90 minutes after an increase in IOP. Eyes were collected 2 days after I/R. A, Administration of the blocking peptide protects against cell loss in the ganglion cell layer and infiltration by leukocytes (arrowhead). Original magnification 400×. Scale bar, 50 μm. B, Number of cells in the GCL and β‐III tubulin+ cells per mm. C, Number of MPO+ leukocytes in the inner retina and vitreous per section. D, mRNA levels of ICAM‐1, CXCL1, NOS2, COX‐2, TNF‐α, and IL‐1β were assessed by quantitative real‐time PCR as above. Horizontal bars represent mean ± SEM (6‐9 mice per group). **P < .01; ***P < .001 by ANOVA
3.7. Intravitreal administration of ri CD40‐TRAF6 blocking peptide impairs retinal inflammatory responses induced by I/R but exacerbates an infectious retinitis
Pharmacologic inhibition of CD40‐TRAF6 reduces CD40‐driven inflammation. 20 , 21 However, this pathway also drives responses key for protection against opportunistic pathogens. We generated ri cell‐permeable peptide that consists of the amino acid sequence of the TRAF6‐binding site of mouse CD40 fused with HIV TAT47‐57 to examine its effects in I/R‐induced retinopathy. The ri CD40‐TRAF6 blocking peptide inhibited CD154‐induced NF‐κB activity only in cells that expressed CD40 that signals through the TRAF6‐binding site (Figure S4A). The peptide also inhibited CD40‐driven ICAM‐1 upregulation in mouse retinal endothelial cells (Figure S4B). Intravitreal administration of the ri CD40‐TRAF6 blocking peptide ameliorated the upregulation of pro‐inflammatory molecules in retinas subjected to I/R in a manner similar to that observed after the administration of the CD40‐TRAF2,3 blocking peptide (Figure 7A).
FIGURE 7.
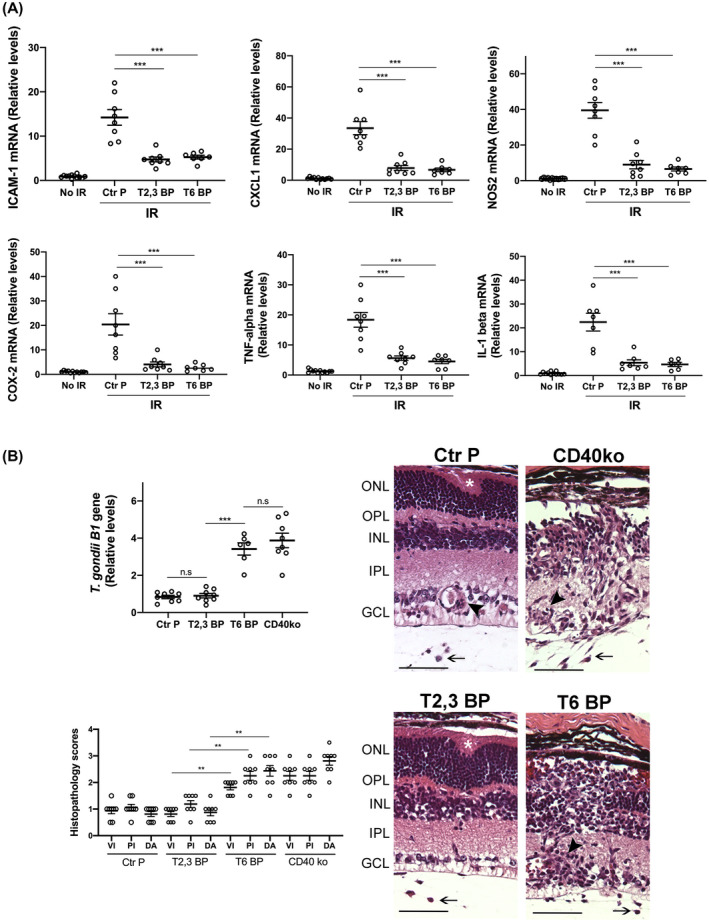
Both the ri CD40‐TRAF2,3 and CD40‐TRAF6 blocking peptides impair retinal expression of inflammatory molecules induced by I/R but the ri CD40‐TRAF6 blocking peptide exacerbates infectious retinitis. A, Eyes subjected to I/R were treated intravitreously with or without ri control peptide (Ctr P), ri CD40‐TRAF2,3 blocking peptide (T2,3 BP) or ri CD40‐TRAF6 blocking peptide (T6 BP; 1 μg) 1 hour prior to an increase in IOP. Eyes were collected 2 days after I/R. mRNA levels of were assessed by quantitative real‐time PCR as above. B, B6 and Cd40−/− mice were infected with T. gondii tissue cysts. B6 mice received peptides intravitreally 4 days after infection. Eyes were collected 14 days post‐infection. Levels of T. gondii B1 gene expression were assessed by real‐time PCR. Eyes from infected B6 mice that received the CD40‐TRAF6 blocking peptide or infected Cd40−/− mice showed prominent disruption of retinal architecture (asterix), perivascular (arrowhead), and vitreal inflammation (arrow). H&E; X200. Bar, 50 μm. Histopathology scores for vitreal inflammation (VI), perivascular inflammation (PV), and disruption of retinal architecture (DA). Graphs represent mean ± SEM of 8 mice per group. **P < .01; ***P < .001 by ANOVA
The CD40‐CD154 pathway is essential for the control of toxoplasmosis in humans and mice. 47 , 48 Toxoplasma gondii infects 1/3 of the world population and is one of the most common causes of infectious retinitis worldwide. 46 Ocular toxoplasmosis can occur either as the reactivation of the chronic (quiescent) phase of infection or as a manifestation of acute infection. 46 We examined the effects of the CD40‐TRAF blocking peptides on CD40‐dependent induction of toxoplasmacidal activity within infected cells, an important mechanism by which CD40 promotes resistance against toxoplasmosis. 48 , 49 In contrast to the CD40‐TRAF2,3 blocking peptide, the CD40‐TRAF6 blocking peptide markedly impaired T. gondii killing induced by CD40 ligation (Figure S4C). These results are in agreement with the effects of the genetic blockade of CD40‐TRAF6 signaling (cells that express CD40 ΔT6) on T. gondii infection. 50 Next, we examined the effects of the peptides in ocular toxoplasmosis. While intravitreal administration of the CD40‐TRAF2,3 blocking peptide to T. gondii‐infected mice had no effect on parasite load and retinal histopathology, both of these parameters were worsened in mice treated with the CD40‐TRAF6 blocking peptide as well as in Cd40−/− mice (Figure 7B). Similar to Cd40−/− mice, the CD40‐TRAF6 blocking peptide did not appear to impair the expression of immune mediators of protection against ocular toxoplasmosis (Figure S5A). In contrast to Cd40−/− mice, neither peptide affected the production of anti‐T. gondii IgG (Figure S5B). Thus, in contrast to the ri CD40‐TRAF6 blocking peptide, the ri CD40‐TRAF2,3 blocking peptide did not impair CD40‐driven toxoplasmacidal activity and did not increase susceptibility to this opportunistic pathogen in the retina.
3.8. The ri CD40‐TRAF6 blocking peptide but not the ri CD40‐TRAF2,3 blocking peptide impairs CD40‐driven systemic IL‐12 production and dendritic cell activation
While T. gondii can induce expression of effector molecules of cellular immunity even in the absence of CD40, this molecule directly induces IL‐12 production and dendritic cell activation, responses that likely promote protection against other opportunistic pathogens. We began to examine the effects of the blocking peptides in these responses using a model of systemic administration of a stimulatory anti‐CD40 mAbs. As previously reported, ip administration of anti‐CD40 mAbs induced serum levels of IL‐12 p70 (Figure S6A). This response was inhibited by ip administration of ri CD40‐TRAF6 blocking peptide but not the ri CD40‐TRAF2,3 blocking peptide (Figure S6A). The anti‐CD40 mAbs also upregulated CD80 and CD86 in splenic dendritic cells, but did not increase the high‐level basal expression of MHC II (Figure S6B). CD40‐driven CD80 and CD86 upregulation were inhibited only by the administration of ri CD40‐TRAF6 blocking peptide (Figure S6C). Thus, the ri CD40‐TRAF2,3 blocking peptide does not appear to impair CD40‐dependent systemic IL‐12 p70 production and dendritic cell activation.
4. DISCUSSION
Studies in mice indicate that CD40 is a molecular target for the treatment of various inflammatory and neurodegenerative disorders. However, there is a need to find new therapeutic approaches to inhibit CD40 signaling in humans since the administration of neutralizing anti‐CD154 mAbs was problematic due to the development of thromboembolic events. Moreover, generalized inhibition of CD40 signaling or inhibition of CD40‐TRAF6 signaling is expected to increase the risk of infections caused by pathogens controlled by cell‐mediated immunity. We present herein the first evidence that inflammation can be controlled by pharmacologic inhibition of CD40‐TRAF2,3 signaling. Moreover, in contrast to the inhibition of CD40‐TRAF6 signaling, inhibition of CD40‐TRAF2,3 signaling had no detectable effect on susceptibility to the opportunistic pathogen T. gondii and did not appear to alter IL‐12 p70 production and dendritic cell activation. These findings suggest that the pharmacologic inhibition of CD40‐TRAF2,3 signaling will be less likely to increase susceptibility to opportunistic infections than global blockade of CD40 or inhibition of CD40‐TRAF6.
Using a model of retinal I/R, we report that a ri cell‐penetrating CD40‐TRAF2,3 blocking peptide markedly diminished ICAM‐1 and CXCL1 upregulation and reduced leukocyte infiltration yielding a phenotype similar to that observed in Cd40−/− mice subjected to retinal I/R. The in vivo effects of the ri blocking peptide correlate with the in vitro studies showing that CD40‐TRAF2,3 signaling is crucial for adhesion molecule and chemokine upregulation in a broad range of cells. 9 , 10 In addition, given that infiltrating leukocytes express NOS2, COX‐2, and pro‐inflammatory cytokines, the reduction in leukocyte recruitment likely contributes to the reduced expression of molecules linked to neural cell death. Taken together, these results indicate that the inhibition of CD40‐TRAF2,3 after ischemia protected against inflammation and achieved neuro‐protection.
CD40 is an important driver of inflammation and pathology not only in retinal ischemia but also in ischemic injury of the brain and other organs. 51 , 52 Like in the retina, ischemia in these organs leads to CD40‐driven upregulation of chemokines and adhesion molecules, promoting leukocyte recruitment and tissue injury. 24 , 51 , 52 While the recruitment of leukocyte subsets such as macrophages, microglia, and lymphocytes can play either a detrimental role (impair blood‐brain barrier integrity, aggravate neuron injury, secrete pro‐inflammatory cytokines) or a protective role (phagocytose cell debris, express anti‐inflammatory cytokines and neuroprotectants) after cerebral ischemia, 53 several studies support the deleterious role of infiltrating PMN. 54 Indeed, PMNs can be key mediators of injury after I/R 37 , 54 and blockade of ICAM‐1 or CXCL1 protects against inflammation and organ injury after I/R. 55 , 56 Whereas it remains to be determined whether approaches to impair inflammatory responses can yield beneficial effects in patients with ischemic stroke, strategies that target adhesion molecules or chemokines have been found to be effective against other inflammatory disorders. 57 , 58 The fundamental role of adhesion molecule and chemokine upregulation in inflammatory disorders supports that pharmacologic blockade of CD40‐TRAF2,3 will likely control CD40‐driven inflammatory disorders besides those triggered by ischemia.
Müller cells acquire expression of pro‐inflammatory molecules 44 that appear to include NOS2 in retinopathies. 45 The present work indicates that Müller cells express NOS2 and CXCL1 in the ischemic retina. Moreover, pharmacologic inhibition of the CD40‐TRAF2,3 pathway reduces the expression of these molecules in Müller cells. Of relevance, in vitro studies in Müller cells that express CD40 with mutations in CD40‐TRAF2,3‐binding site or Müller cells treated with a CD40‐TRAF2,3 blocking peptide revealed that the CD40‐TRAF2,3 pathway is a major inducer of pro‐inflammatory responses in Müller cells. 9
The need to find novel approaches to treat CD40‐driven inflammatory disorders together with the central role of TRAFs as mediators of the effects of CD40 emphasizes the importance of examining the inhibition of CD40‐TRAF signaling for potential therapeutic applications. Prior important studies centered on the effects of the inhibition of CD40‐TRAF6 signaling. 19 , 20 , 21 Our studies uncovered that CD40‐TRAF2,3 signaling is critical to the development of inflammation and neuronal cell loss. Moreover, inhibition of the CD40‐TRAF2,3 pathway did not increase susceptibility to an opportunistic pathogen. This finding is important since, whereas general inhibition of CD40 signaling (blocking anti‐CD154 or anti‐CD40 mAbs), inhibition of CD40‐TRAF2,3 or CD40‐TRAF6 signaling may impair antibody production, opportunistic pathogens rather than pathogens controlled by humoral immunity are the most important cause of death in patients without functional CD40‐CD154 signaling. 5 In contrast to the blockade of CD40‐TRAF2,3, the inhibition of CD40 or CD40‐TRAF6 signaling would impair effector mechanisms that control opportunistic pathogens. Of note, a pharmacologic inhibitor of CD40‐TRAF6 was reported not to affect antigen‐driven T cell proliferation. 21 However, those studies were performed using previously matured dendritic cells, thus likely bypassing CD40‐induced dendritic cell activation, a key function of CD40 during dendritic cell‐T cell interaction.
Cell‐penetrating peptides have been used in studies that address fundamental biology questions, and in pre‐clinical as well as clinical trials. 59 , 60 , 61 Given that TAT47‐57 ‐based CD40‐TRAF2,3 blocking peptide inhibits CD40‐TRAF2,3 signaling and CD40‐dependent pro‐inflammatory responses in vitro, 10 , 13 we generated a ri peptide as an initial pharmacologic approach to block CD40‐TRAF2,3 signaling in vivo. Intravitreal administration of this peptide may become an attractive approach for the treatment of ocular disorders (eg, ischemic retinopathies, diabetic retinopathy, and perhaps glaucoma) since TAT47‐57 ‐based ri cell‐penetrating peptides are good tools for transporting molecules into retinal cells 29 , 36 and they appear to persist in the retina for at least 14 days after a single intravitreal injection. 29 Moreover, the CD40‐TRAF2,3 blocking peptide did not impair the control of T. gondii in the retina, an important finding with given the high prevalence of chronic T. gondii infection worldwide.
In summary, the studies herein uncovered the pharmacologic inhibition of the CD40‐TRAF2,3 pathway as a novel approach to control CD40‐driven inflammation. This work opens up the possibility of therapeutic approaches to treat CD40‐driven inflammatory disorders based on pharmacologic blockade of CD40‐TRAF2,3 signaling either by the administration of a CD40‐TRAF2,3 blocking peptide or a small molecule inhibitor of CD40‐TRAF2,3 signaling. This approach is likely to be effective in a broad range of CD40‐mediated diseases given that blockade of this pathway restricts fundamental inflammatory responses.
CONFLICT OF INTEREST
Drs. Carlos S. Subauste and M. Cecilia Subauste have a patent on the CD40‐TRAF2,3 blocking peptide. The other authors have no conflicts of interest to declare.
AUTHOR CONTRIBUTIONS
C. S. Subauste and M. C. Subauste designed the research; J‐A. C. Portillo, J‐S. Yu, S. Hansen, and T.S. Kern performed the research; J‐A. C. Portillo, J‐S. Yu, and S. Hansen analyzed the data; C. S. Subauste wrote the paper.
Supporting information
Supplementary Material
ACKNOWLEDGMENTS
We thank Dr. Jonathan Sears for performing intravitreal injections, Dr. Yalitza Lopez Corcino for infecting mice with T. gondii, and Dr. Richard Kornbluth for providing CD154. We are grateful to Catherine Doller for expert tissue processing for histopathology.
Portillo J‐AC, Yu J‐S, Hansen S, Kern TS, Subauste MC, Subauste CS. A cell‐penetrating CD40‐TRAF2,3 blocking peptide diminishes inflammation and neuronal loss after ischemia/reperfusion. The FASEB Journal. 2021;35:e21412. 10.1096/fj.201903203RR
Funding information
This work was supported by NIH grants EY019250 (C.S.S), EY018341 (C.S.S), P30 EY11373, and Crohn’s and Colitis Foundation of America (Litwin Pioneers Program) grant 529250 (C.S.S.).
REFERENCES
- 1. Peters AL, Stunz LL, Bishop GA. CD40 and autoimmunity: the dark side of a great activator. Semin Immunol. 2009;21:293‐300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Boumpas DT, Furie R, Manzi S, et al. A short course of BG9588 (anti‐CD40 ligand antibody) improves serologic activity and decreases hematuria in patients with proliferative lupus glomerulonephritis. Arthritis Rheum. 2003;48:719‐727. [DOI] [PubMed] [Google Scholar]
- 3. Kawai T, Andrews D, Colvin RB, Sachs DH, Cosimi B. Thromboembolic complications after treatment with monoclonal antibody against CD40 ligand. Nat Med. 2000;6:114. [DOI] [PubMed] [Google Scholar]
- 4. Oura T, Yamashita K, Suzuki T, et al. Long‐term hepatic allograft acceptance based on CD40 blockade by ASKP1240 in nonhuman primates. Am J Transplant. 2012;12:1740‐1754. [DOI] [PubMed] [Google Scholar]
- 5. Levy J, Espanol‐Boren T, Thomas C, et al. Clinical spectrum of X‐linked hyper‐IgM syndrome. J Pediatr. 1997;131:47‐54. [DOI] [PubMed] [Google Scholar]
- 6. Bishop GA, Hostager BS, Brown KD. Mechanisms of TNF receptor‐associated factor (TRAF) regulation in B lymphocytes. J Leuk Biol. 2002;72:19‐23. [PubMed] [Google Scholar]
- 7. Pullen SS, Miller HG, Everdeen DS, Dang TT, Crute JJ, Kehry MR. CD40‐tumor necrosis factor receptor‐associated factor (TRAF) interactions: regulation of CD40 signaling through multiple TRAF binding sites and TRAF hetero‐oligomerization. Biochemistry. 1998;37:11836‐11845. [DOI] [PubMed] [Google Scholar]
- 8. Lu LF, Cook WJ, Lin LL, Noelle RJ. CD40 signaling through a newly identified tumor necrosis factor receptor‐associated factor 2 (TRAF2) binding site. J Biol Chem. 2003;278:45414‐45418. [DOI] [PubMed] [Google Scholar]
- 9. Portillo J‐A, Schwartz I, Zarini S, et al. Pro‐inflammatory responses induced by CD40 in retinal endothelial and Muller cells are inhibited by blocking CD40‐TRAF2,3 or CD40‐TRAF6 signaling. Invest Ophthalmol Vis Sci. 2014;55:8590‐8597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Portillo J‐AC, Greene JA, Schwartz I, Subauste MC, Subauste CS. Blockade of CD40‐TRAF2,3 or CD40‐TRAF6 interactions is sufficient to impair pro‐inflammatory responses in human aortic endothelial cells and human aortic smooth muscle cells. Immunology. 2015;144:21‐33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kobayashi T, Walsh PT, Walsh MC, et al. TRAF6 is a critical factor for dendritic cell maturation and development. Immunity. 2003;19:353‐363. [DOI] [PubMed] [Google Scholar]
- 12. Mackey MF, Wang Z, Eichelberg K, Germain RN. Distinct contributions of different CD40 TRAF binding sites to CD154‐induced dendritic cell maturation and IL‐12 secretion. Eur J Immunol. 2003;33:779‐789. [DOI] [PubMed] [Google Scholar]
- 13. Portillo J‐AC, Feliciano LM, Subauste MC, Heinzel FP, Subauste CS. CD40 and tumor necrosis factor‐a co‐operate to up‐regulate nitric oxide synthase expression in macrophages. Immunology. 2012;135:140‐150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Subauste CS, Andrade RM, Wessendarp M. CD40‐TRAF6 and autophagy‐dependent anti‐microbial activity in macrophages. Autophagy. 2007;3:245‐248. [DOI] [PubMed] [Google Scholar]
- 15. Ahonen CL, Manning EM, Erickson LD, et al. The CD40‐TRAF6 axis controls affinity maturation and the generation of long‐lived plasma cells. Nat Immunol. 2002;3:451‐456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jabara HH, Laouini D, Tsitsikov E, et al. The binding site for TRAF2 and TRAF3 but not for TRAF6 is essential for CD40‐mediated immunoglobulin class switching. Immunity. 2002;17:265‐276. [DOI] [PubMed] [Google Scholar]
- 17. Yasui T, Muraoka M, Takaoka‐Shichijo Y, et al. Dissection of B cell differentiation during primary immune responses in mice with altered CD40 signals. Int Immunol. 2002;14:319‐329. [DOI] [PubMed] [Google Scholar]
- 18. Bishop GA. The multifaceted roles of TRAFS in the regulation of B‐cell function. Nat Rev Immunol. 2004;4:775‐786. [DOI] [PubMed] [Google Scholar]
- 19. Lutgens E, Lievens D, Beckers L, et al. Deficient CD40‐TRAF6 signaling in leukocytes prevents atherosclerosis by skewing the immune response toward an antiinflammatory profile. J Exp Med. 2010;207:391‐404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zarzycka B, Seijkens T, Nabuurs SB, et al. Discovery of small molecule CD40‐TRAF6 inhibitors. J Chem Inf Model. 2015;55:294‐307. [DOI] [PubMed] [Google Scholar]
- 21. Seijkens TTP, van Tiel CM, Kusters PJH, et al. Targeting CD40‐induced TRAF6 signaling in macrophages reduces atherosclerosis. J Am Coll Cardiol. 2018;71:527‐542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gold JA, Parsey M, Hoshino Y, et al. CD40 contributes to lethality in acute sepsis: in vivo role for CD40 in innate immunity. Infect Immun. 2003;71:3521‐3528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Portillo J‐AC, Lopez Corcino Y, Miao Y, et al. CD40 in retinal Muller cells induces P2X7‐dependent cytokine expression in macrophages/microglia in diabetic mice and development of early experimental diabetic retinopathy in mice. Diabetes. 2017;66:483‐493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Portillo J‐AC, Van Grol J, Zheng L, et al. CD40 mediates retinal inflammation and neuro‐vascular degeneration. J Immunol. 2008;181:8719‐8726. [DOI] [PubMed] [Google Scholar]
- 25. Song Z, Jin R, Yu S, Nanda A, Granger DL, Li G. Crucial role of CD40 signaling in vascular wall cells in neointima formation and vascular remodeling after vascular interventions. Atheroscler Thromb Vasc Biol. 2012;32:50‐64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Portillo J‐AC, Greene JA, Okenka G, et al. CD40 promotes the development of early diabetic retinopathy. Diabetologia. 2014;57:2222‐2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Portillo J‐AC, Okenka G, Kern TS, Subauste CS. Identification of primary retinal cells and ex vivo identification of pro‐inflammatory molecules in retinal cells using flow cytometry. Mol Vis. 2009;15:1383‐1389. [PMC free article] [PubMed] [Google Scholar]
- 28. Prive GG, Melnick A. Specific peptides for the therapeutic targeting of oncogenes. Curr Opin Gen Dev. 2006;16:71‐77. [DOI] [PubMed] [Google Scholar]
- 29. Schorderet D, d'Alleves Manzi V, Canola K, et al. D‐TAT transporter as an ocular peptide delivery system. Clin Exp Ophthalmol. 2005;33:628‐635. [DOI] [PubMed] [Google Scholar]
- 30. Zheng L, Gong B, Hatala DA, Kern TS. Retinal ischemia and reperfusion causes capillary degeneration: similarities to diabetes. Invest Ophthalmol Vis Sci. 2007;48:361‐367. [DOI] [PubMed] [Google Scholar]
- 31. Portillo J‐AC, Van Grol J, Saffo S, et al. CD40 in endothelial cells restricts neural tissue invasion by Toxoplasma gondii . Infect Immun. 2019;87:e00868‐18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Pullen SS, Dang TTA, Crute JJ, Kehry MR. CD40 signaling through Tumor Necrosis Factor Receptor‐associated Factors (TRAFs). Binding site specificity and activation of down‐stream pathways by distinct TRAFs. J Biol Chem. 1999;274:14246‐14254. [DOI] [PubMed] [Google Scholar]
- 33. Pinkus GS, Pinkus JL. Myeloperoxidase: a specific marker for myeloid cells in paraffin sections. Mod Pathol. 1991;4:733‐741. [PubMed] [Google Scholar]
- 34. Sutherland CL, Krebs DL, Gold MR. An 11‐amino acid sequence in the cytoplasmic domain of CD40 is sufficient for activation of c‐Jun N‐terminal kinase, activation of MAPKAP kinase‐2, phosphorylation of I kappa B alpha, and protection of WEHI‐231 cells from anti‐IgM‐induced growth arrest. J Immunol. 1999;162:4720‐4730. [PubMed] [Google Scholar]
- 35. Vives E, Brodin P, Lebleu B. A truncated HIV‐1 Tat protein basic domain rapidly translocates through the plasma membrane and accumulates in the cell nucleus. J Biol Chem. 1997;272:16010‐16017. [DOI] [PubMed] [Google Scholar]
- 36. Touchard E, Omri S, Naud MC, et al. A peptide inhibitor of c‐Jun N‐terminal kinase for the treatment of endotoxin‐induced uveitis. Invest Ophthalmol Vis Sci. 2010;51:4683‐4693. [DOI] [PubMed] [Google Scholar]
- 37. Tsujikawa A, Ogura Y, Hiroshiba N, et al. Retinal ischemia‐reperfusion injury attenuated by blocking of adhesion molecules of vascular endothelium. Invest Ophthalmol Vis Sci. 1999;40:1183‐1190. [PubMed] [Google Scholar]
- 38. Neufeld AH, Kawai S, Das S, et al. Loss of retinal ganglion cells following retinal ischemia: the role of inducible nitric oxide synthase. Exp Eye Res. 2002;75:521‐528. [DOI] [PubMed] [Google Scholar]
- 39. Botchkina GI, Meistrell ME 3rd, Botchkina IL, Tracey KJ. Expression of TNF and TNF receptors (p55 and p75) in the rat brain after focal cerebral ischemia. Mol Med. 1997;3:765‐781. [PMC free article] [PubMed] [Google Scholar]
- 40. Choi JS, Kim D, Hong YM, Mizuno S, Joo CK. Inhibition of nNOS and COX‐2 expression by lutein in acute retinal ischemia. Nutrition. 2006;22:668‐671. [DOI] [PubMed] [Google Scholar]
- 41. Berger S, Savitz SI, Nijhawan S, et al. Deleterious role of TNF‐alpha in retinal ischemia‐reperfusion injury. Invest Ophthalmol Vis Sci. 2008;49:3605‐3610. [DOI] [PubMed] [Google Scholar]
- 42. Yoneda S, Tanihara H, Kido N, et al. Interleukin‐1 beta mediates ischemic injury in the rat retina. Exp Eye Res. 2001;73:661‐667. [DOI] [PubMed] [Google Scholar]
- 43. Hangai M, Yoshimura N, Hiroi K, Mandai M, Honda Y. Inducible nitric oxide synthase in retinal ischemia‐reperfusion injury. Exp Eye Res. 1996;63:501‐509. [DOI] [PubMed] [Google Scholar]
- 44. Gerbardinger C, Biarnes Costa M, Coulombe MC, Toth I, Hoehn T, Grosu P. Expression of acute‐phase response proteins in retinal Muller cells in diabetes. Invest Ophthalmol Vis Sci. 2005;46:349‐357. [DOI] [PubMed] [Google Scholar]
- 45. Kobayashi M, Kuroiwa T, Shimokawa R, Okeda R, Tokoro T. Nitric oxide synthase expression in ischemic rat retinas. Jpn J Ophthalmol. 2000;44:235‐244. [DOI] [PubMed] [Google Scholar]
- 46. Butler NJ, Furtado JM, Winthrop KL, Smith JR. Ocular toxoplasmosis II: clinical features, pathology and management. Clin Exp Ophthalmol. 2013;41:95‐108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Subauste CS, Wessendarp M, Sorensen RU, Leiva L. CD40 ‐ CD40 ligand interaction is central to cell‐mediated immunity against Toxoplasma gondii: Patients with hyper IgM syndrome have a defective type‐1 immune response which can be restored by soluble CD40L trimer. J Immunol. 1999;162:6690‐6700. [PubMed] [Google Scholar]
- 48. Reichmann G, Walker W, Villegas EN, et al. The CD40/CD40 ligand interaction is required for resistance to toxoplasmic encephalitis. Infect Immun. 2000;68:1312‐1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Portillo J‐AC, Okenka G, Reed E, et al. The CD40‐autophagy pathway is needed for host protection despite IFN‐γ‐dependent immunity and CD40 induces autophagy via control of p21 levels. PLoS One. 2010;5:e14472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Andrade RM, Wessendarp M, Portillo J‐AC, et al. TRAF6 signaling downstream of CD40 primes macrophages to acquire anti‐microbial activity in response to TNF‐a. J Immunol. 2005;175:6014‐6021. [DOI] [PubMed] [Google Scholar]
- 51. Ishikawa M, Vowinkel T, Stokes KY, et al. CD40/CD40 ligand signaling in mouse cerebral microvasculature after focal ischemia/reperfusion. Circulation. 2005;111:1690‐1696. [DOI] [PubMed] [Google Scholar]
- 52. de Ramon L, Ripoll E, Merino A, et al. CD154‐CD40 T‐cell co‐stimulation pathway is a key mechanism in kidney ischemia‐reperfusion injury. Kidney Int. 2015;88:538‐549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Jian Z, Liu R, Zhu X, et al. The involvement and therapy target of immune cells after ischemic stroke. Front Immunol. 2019;10:2167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Hermann DM, Kleinschnitz C, Gunzer M. Role of polymorphonuclear neutrophils in the reperfused ischemic brain: insights from cell‐type‐specific immunodepletion and fluorescence microscopy studies. Ther Adv Neurol Disord. 2018;11:1756286418798607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Yamazaki T, Seko Y, Tamatani T, et al. Expression of intercellular adhesion molecule‐1 in rat heart with ischemia/reperfusion and limitation of infarct size by treatment with antibodies against cell adhesion molecules. Am J Pathol. 1993;143:410‐418. [PMC free article] [PubMed] [Google Scholar]
- 56. Miura M, Fu X, Zhang Q‐W, Remick DG, Fairchild RL. Neutralization of Groα and macrophage inflammatory protein‐2 attenuates renal ischemia/reperfusion injury. Am J Pathol. 2001;159:2137‐2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Zundler S, Becker E, Weidinger C, Siegmund B. Anti‐adhesion therapies in inflammatory bowel disease‐molecular and clinical aspects. Front Immunol. 2017;8:891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Trivedi PJ, Adams DH. Chemokines and chemokine receptors as therapeutic targets in inflammatory bowel disease; pitfalls and promise. J Crohns Colitis. 2018;12:S641‐S652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Guidotti G, Brambilla L, Rossi D. Cell‐penetrating peptides: from basic research to clinics. Trends Pharmacol Sci. 2017;38:406‐424. [DOI] [PubMed] [Google Scholar]
- 60. Tejeda GS, Esteban‐Ortega GM, San Antonio E, Vidaurre OG, Diaz‐Guerra M. Prevention of excitotoxicity‐induced processing of BDNF receptor TrkB‐FL leads to stroke neuroprotection. EMBO Mol Med. 2019;11:e9950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Bates E, Bode C, Costa M et al. Intracoronary KAI‐9803 as an adjunct to primary percutaneous coronary intervention for acute ST‐segment elevation myocardial infarction. Circulation. 2008;117:886‐896. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material